
Metal porphyrins (M = Ti, Fe, Co, Ni, Cu, or Zn) as potential catalysts for the oxidation of CO by N2O: insight from DFT calculations†
Shutao
Wang‡
 b,
Zhao
Liu‡
a,
Yanjie
Ye
b,
Xu
Meng
a,
Pengcheng
Yang
a,
Zhengze
Zhang
b,
Yifeng
Qiu
*c and
Junqiang
Lei
*a
b,
Zhao
Liu‡
a,
Yanjie
Ye
b,
Xu
Meng
a,
Pengcheng
Yang
a,
Zhengze
Zhang
b,
Yifeng
Qiu
*c and
Junqiang
Lei
*a
aFirst Hospital of Lanzhou University, Lanzhou University, 222 South Tianshui Road, Lanzhou 730000, P. R. China. E-mail: leijq2011@126.com
bState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou 730000, P. R. China
cCollege of Chemistry and Chemical Engineering, Northwest Normal University, 967 East Anning Road, Lanzhou 730070, P. R. China. E-mail: qyfnet@nwnu.edu.cn
First published on 16th November 2022
Abstract
The oxidation of CO by N2O over M–porphyrin (M = Ti, Fe, Co, Ni, Cu, and Zn) catalysts has been investigated via density functional theory calculations. The whole reaction process is divided into two steps: the catalytic decomposition of N2O that breaks the N–O bond resulting in O–M active species, and the carbon atoms of the CO molecule reaction with O–M to form CO2. For the rate-controlled step of the reaction, the porphyrins of different metal centers appear in different positions. The barrier height of N2O decomposition on Ti–porphyrin is 3.8 kcal mol−1, and the barrier height of CO oxidation is 21.9 kcal mol−1. The rate-controlled step appears in the process of oxidation of CO. However, for Fe–porphyrin, the barrier height of N2O decomposition is 24.2 kcal mol−1, and the barrier height of CO oxidation is 11.4 kcal mol−1. The rate-controlled step appears in the process of N2O decomposition. For the catalytic decomposition of N2O, the Ti–porphyrin has a low activation energy barrier, which may be due to the smaller gap between the highest occupied molecular orbital (HOMO) of the metal porphyrin and the lowest unoccupied molecular orbital (LUMO) of N2O for Ti–porphyrin compared to Fe–porphyrin.
Introduction
Nitrous oxide (N2O) and carbon monoxide (CO) are both potent greenhouse gases that contribute to global warming and damage the ozone layer,1 and they mainly come from the industrial production and incomplete combustion of automobile fuel. Moreover, carbon monoxide (CO) poisoning is a common disease that endangers people's health. The main mechanism of poisoning is that the binding force of carbon monoxide and hemoglobin is much higher than that of oxygen, resulting in tissue hypoxia, and the aerobic respiratory chain of cells being seriously affected or even blocked.2 At present, there is no specific drug for the treatment of carbon monoxide poisoning. Therefore, it is urgent to develop effective CO harmless treatment methods. The accepted method for these two harmful gases is to convert them into less harmful nitrogen (N2) and carbon dioxide (CO2),3 and the specific equation involved is N2O + CO = N2 + CO2. However, for this reaction, the high activation energy barrier makes it difficult to carry out at room temperature.4 Therefore, it is necessary to develop economic, environmentally friendly, highly reactive, and selective catalysts.To overcome the reaction energy barrier, numerous catalysts, such as transition metal (TM) cations (Fe+, Os+, Ir+, Pt+), have been attempted for activation reactions.5 However, the metal ions in the gas phase are mainly used to study the mechanism in the laboratory, which is difficult to be applied in the chemical industry.6 Loading TM ions onto oxides is also a widely studied type of catalyst, but this reduces the utilization of TM ions and requires higher reaction temperatures.3f,7 As a common catalyst, precious metals, such as Pt8 and Ag,9 have been studied in detail for the reduction treatment of N2O. However, precious metals are difficult to be applied on a large scale because of their high price, low natural content, and high toxicity.4a Porphyrins easily form ordered monolayers with two axial coordination through self-assembly, which can be used as catalytic active centers. As a functional site, metal porphyrins can be easily anchored on a variety of carrier bases, so they are used in many applications, such as photovoltaic materials, field-response materials, and catalysts.10 The coordination between the gas molecules and the metal centers of porphyrins can lead to measurable changes in the electronic properties and color of porphyrins.10b,11 This phenomenon is due to the interaction between the metal center of porphyrin and some small gas molecules such as NO, CO, and O2.12 Therefore, an important application of metal porphyrins is gas sensing. Moreover, metal porphyrins can be fixed as solid phase catalysts and can be widely used to catalyze the reduction of carbon dioxide13 and nitrogen oxides14 as well as the oxidation of hydrocarbons and alcohols.15
Although metal porphyrins have the excellent performance mentioned above, only the adsorption capacity of N2O has been reported.10d,13a Recently, Ehara et al. reported a direct catalytic decomposition of N2O on Ti–porphyrins and found that it has good catalytic activity and can be used as a potential catalyst for the direct catalytic decomposition of N2O.16 However, despite the support of the above outstanding reports, the activation energy of N2O catalytic decomposition on other transition metal porphyrins has not been studied, which is very important for the direct catalytic decomposition and selective reduction of N2O. In addition, metal porphyrin is an excellent catalyst for the direct catalytic decomposition of N2O and other applications. However, using it as a catalyst for N2O oxidation of CO into CO2 has not been reported.
Therefore, density functional theory (DFT) was employed to calculate the mechanism of catalytic decomposition of N2O and oxidation of CO to CO2 by N2O on metal porphyrins, as well to study the adsorption, transition state and relative energy. The optimum metal porphyrin catalysts in Ti, Fe, Co, Ni, Cu, and Zn were screened by comparing the barrier height of the N2O catalytic decomposition. DFT calculation shows that the activation energy barrier of the N2O decomposition catalyzed by Ti–porphyrin and Fe–porphyrin is lower among these metal porphyrins. Therefore, the whole reaction of CO oxidation by N2O on these two metal porphyrins were studied. In addition, the molecular frontier orbital, and charge transfer of these two metal porphyrins and N2O were also analyzed as rate constants using classical transition-state theory (TST), in an attempt to understand the source of the high catalytic activity of these two metal porphyrins. This work elucidates the mechanism of the N2O oxidation of CO to CO2 on metal porphyrins, and theoretically identifies suitable catalysts for experiments providing a detailed case for the theoretical design of catalyst.
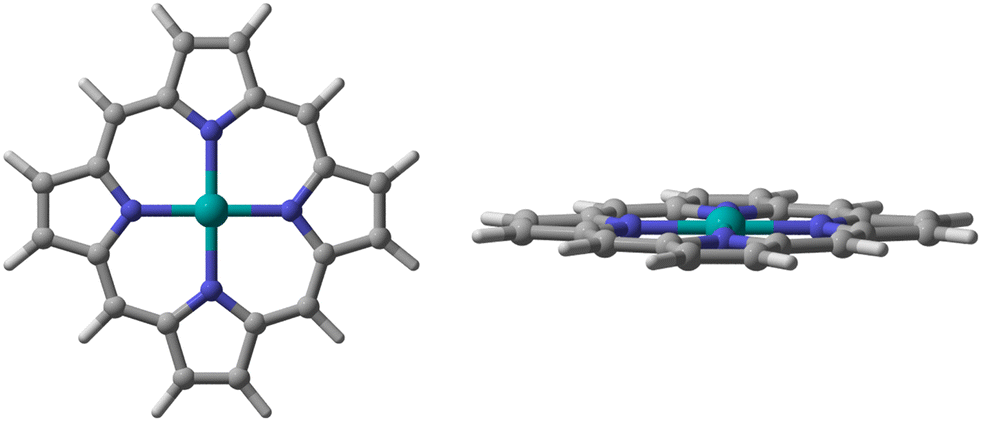 | ||
| Fig. 1 The metal porphyrin model used in this study. | ||
Models and method
We chose a metal porphyrin model with 38 atoms as the research object (Fig. 1). All atoms can relax freely, including the metal porphyrins and the small molecules. For the spin state of the metal center, Ehara et al. screened the most stable spin state of each metal porphyrin by investigating the total energies of isolated metal porphyrins.16 All calculations were performed with the M06-L17 density functional and the def2-TZVP basis sets18 for the metal atoms, N atoms and small molecules attached to metal porphyrins. In addition, the 6-31G(d,p) basis set19 was used for calculatioins for the C and H atoms of metal porphyrins. To verify and confirm the transition states, frequency calculations were performed at the same level of theory to classify the stationary points along the reaction coordinates. In the frequency calculation, zero-point energy (ZPE) was used for energy correction. We also calculated the rate constants using the classical transition-state theory (TST) according to the following equation;where k is the reaction rate, kB is Boltzmann's constant, h is Planck's constant, T is the absolute temperature (298.15 K), R is the universal gas constant, ΔG is the free energy difference between the adsorption complex and transition states. Mulliken charge was used because the dispersion function is not involved in the whole calculation. All calculations were carried out using the Gaussian 09 suite of programs, revision D01.20 All 3D images were drawn using CYL-View.21
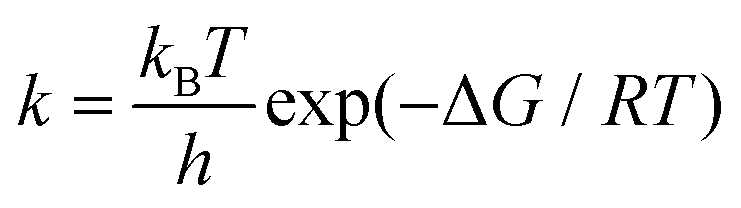
Results and discussion
N2O Decomposition over M–porphyrin (M = Ti, Fe, Co, Ni, Cu, or Zn)We first consider the adsorption of reactants N2O and CO on the M–porphyrin (as shown in Table S1 and Fig. S1, ESI†). The CO molecule can adsorb on the M–porphyrins in two different configurations in which either a C or O atom binds to the metal active site. Regardless of the metal center, the adsorption by the C atom of CO is always stronger than the adsorption by the O atom, which may be due to the negative partial charge located on the C atom. The case of co-adsorption of N2O and CO on bare Ti and Fe atoms shows that only CO has adsorption behavior on metal atoms, while N2O does not. By comparing the adsorption energy of N2O and CO (both ways), we found that CO adsorbs more strongly than N2O. Subsequently, CO may obstruct the active site of M–porphyrins for N2O adsorption and N–O bond breaking. Although one possibility proposed by Sombat Ketrat et al.,3e the N2O reaction should be allowed to break the N–O bond and form active O, followed by the CO oxidation reaction. However, we suggest that the strong adsorption of CO at the M–porphyrin results in the occupation of the active site, which may block the breaking of the N–O bond in N2O to form the M–O complex, and this cannot be ignored.
Next, we explored the reaction mechanism of N2O decomposition over M–porphyrin (M = Ti, Fe, Co, Ni, Cu, or Zn), as shown in Fig. 2. The selection of a suitable catalyst is based on the barrier height of N2O direct catalytic decomposition. In the adsorption complex formed by each metal porphyrin and N2O, the structure of N2O significantly changed, the O–N bonds have an elongation from 0.07 Å to 0.69 Å. This indicates that the coordination action of the metal active center of M–porphyrin can activate the O–N bond in N2O. Relative to others, adsorption energy of −8.8 kcal mol−1 was provided by Ti–porphyrin (triplet) and −2.0 kcal mol−1 was provided by Fe–porphyrin (triplet), while other M–porphyrin exhibited adsorption energies in the range of 0.3 to 1.9 kcal mol−1. For the activation energy barrier, Ti–porphyrin had an activation energy barrier of 3.7 kcal mol−1 and Fe–porphyrin had an activation energy barrier of 24.2 kcal mol−1, while others exhibited higher activation energy barrier in the range of 38.3 to 58.9 kcal mol−1. This indicates that the catalytic activity of Ti–porphyrin is the best of all and Fe–porphyrin has the lowest barrier height. Therefore, Ti–porphyrin and Fe–porphyrin were selected as the catalysts for the subsequent reactions.
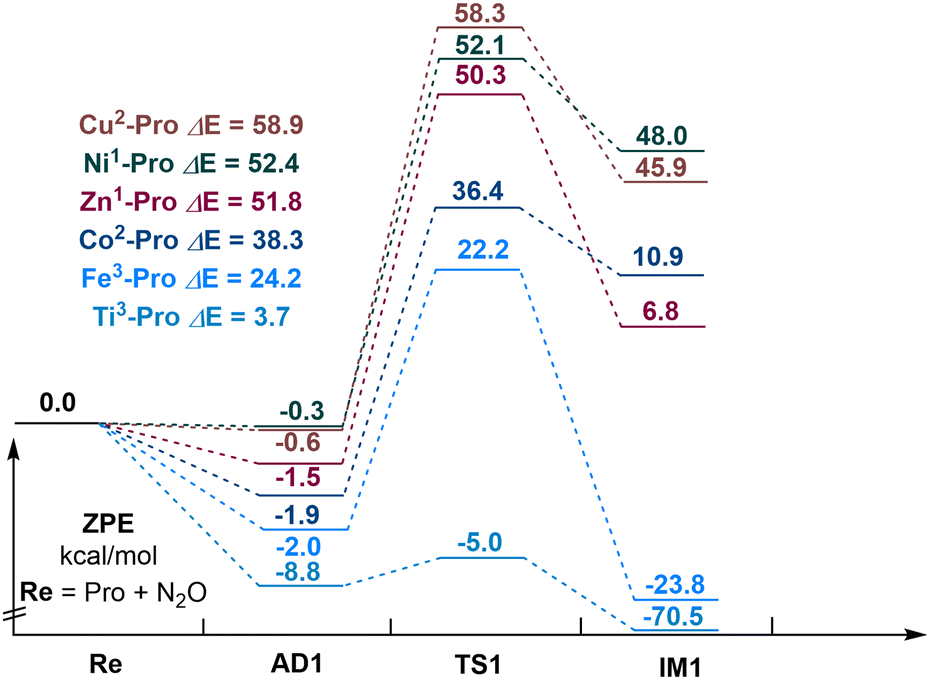 | ||
| Fig. 2 Energy profile of N2O decomposition over M–porphyrin. | ||
Reaction mechanism of CO oxidation by N2O on Ti–porphyrin
Subsequently, we have fully studied the reaction mechanism of CO oxidation by N2O on Ti–porphyrin and Fe–porphyrin. The reaction energy profile of N2O direct decomposition over Ti–porphyrin consists of N2O adsorption, N–O bond scission, and N2 desorption shown in Fig. 3. AD1Ti is the complex form of the N2O adsorbed to the Ti–porphyrin by an O atom. The distance of the Ti–O bond is 2.26 Å, O–N bond has a certain extent of elongation, changing from 1.18 Å to 1.19 Å. The calculated adsorption energy is −8.8 kcal mol−1. The O–N in TS1Ti further elongates to 1.25 Å, unlike adsorbed complex, N–N length changes from 1.12 Å to 1.14 Å. N2O is no longer a straight-line molecule, ∠ON1N2 going from a straight angle to 151.4° and having the angle between Ti–O and the porphyrin plane as 90.0°. Moreover, the Ti–O bond changed from 2.26 Å to 2.02 Å where as TS1Ti has one imaginary frequency of 441.28i cm−1 has an activation energy of 3.8 kcal mol−1 which corresponds to AD1Ti. The Ti active center extracts an oxygen atom from the adsorbed N2O to form IM1Ti, which contains Ti–O active species and N2. After the TS1Ti, the dissociation complex in the IM1Ti formation process was an exothermic process of 67.5 kcal mol−1, where the Ti–O bond was shortened from 2.02 Å to 1.62 Å, and the length of the O–N bond was elongated to 3.29 Å. Compared with the transition state, the Ti–O bond in the decomposition complex has formed, and there is almost no interaction between Ti–O active species and N2, the desorption of N2 requires 2.1 kcal mol−1.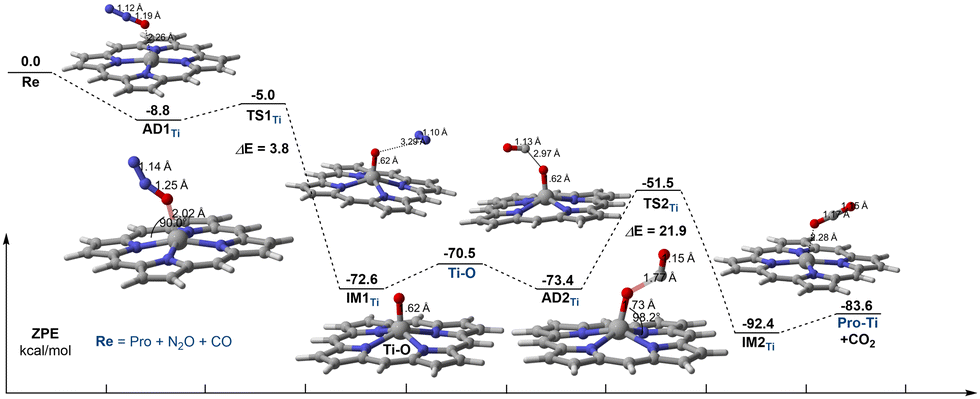 | ||
| Fig. 3 Energy profile and selected geometric parameters of transition states and intermediates involved in the oxidation of CO by N2O on Ti–porphyrin. | ||
For the oxidation of CO by N2O on Ti–porphyrin, the adsorption of CO on the Ti–O active material is weaker than N2O on Ti–porphyrin with the adsorption energy of −2.9 kcal mol−1, which is an exothermic process. In the adsorption complex named AD2Ti, the distance between the C atom in CO and the O atom of the Ti–O active substance is 2.97 Å, and the CO with O atom forms an angle of 92.0°. The TS2Ti with a frequency of 982.22i cm−1 has been found. In this transition state, the Ti–O bond was elongated from 1.62 Å to 1.73 Å with the distance between the C atom and the reactive oxygen species reduced from 2.97 Å to 1.77 Å, the C–O bond in CO extends from the original distance of 1.13 Å to 1.15 Å, where the CO with the reactive oxygen species forms an angle of 115.2°. In addition, the Ti–O bond forms a 98.2° angle with the porphyrin plane. This step is required to overcome the activation energy barrier of 21.9 kcal mol−1. In IM2Ti, the C–O bond of the CO2 was formed, from 1.77 Å to 1.17 Å, close to CO2. The desorption energy of CO2 is 8.8 kcal mol−1, higher than 2.1 kcal mol−1, which is from the desorption of N2. It is possible that CO2 is generated in this step, but N2 escapes much more easily than CO2. The oxidation of CO by O–Ti–porphyrin has a higher activation energy barrier than the direct catalytic decomposition of N2O; therefore, the part of oxidation of CO is the rate-controlled step of the whole reaction, with the barrier height of 21.9 kcal mol−1.
In summary, during the decomposition of N2O on Ti–porphyrin, the N2O can be easily attached to the Ti atom, and the reaction needs to overcome a low activation energy barrier of 3.8 kcal mol−1. However, the activation energy barrier for the subsequent oxidation of CO to CO2 is 21.9 kcal mol−1, which is higher than the direct catalytic decomposition of N2O. Therefore, the oxidation of CO is the rate-control step of the whole reaction.
Reaction mechanism of CO oxidation by N2O on Fe–porphyrin
As shown in Fig. 4, for Fe–porphyrin, N2O is adsorbed to the Fe atom through the O atom to form the adsorption complex AD1Fe. The adsorption energy of N2O on Fe–porphyrin is −2.0 kcal mol−1, which is higher than that on Ti–porphyrin. The Fe–O distance is 2.69 Å, and there is no significant change in O–N and N–N distances, which are 1.18 Å and 1.13 Å respectively. Unlike on Ti–porphyrin, the O–N in TS1Fe elongates to 1.48 Å, with no change in the N–N bond. N2O is no longer a straight-line molecule, ∠ON1N2 goes from a straight angle to 135.1°and the angle between the Fe–O and the porphyrin plane is 93.9°. Moreover, Fe–O bond has changed from 2.69 Å to 1.86 Å. The TS1Fe with one imaginary frequency of 708.38i cm−1 has an activation energy of 24.2 kcal mol−1, which is higher than Ti–porphyrin. The Fe active center extracts an O atom from the adsorbed N2O to form IM1Fe, which contains Fe–O active species and N2. The dissociation complex IM1Fe formation process after the TS1Fe was an exothermic process of 46.0 kcal mol−1, the Fe–O bond was shortened from 1.86 Å to 1.62 Å, and the length of the O–N bond elongated to 3.18 Å. Compared with the transition state, the Fe–O bond in the decomposition complex has formed, and there is almost no interaction between Fe–O active species and N2, the desorption of N2 requires 2.3 kcal mol−1, which is comparable to the desorption of N2 on Ti–porphyrin.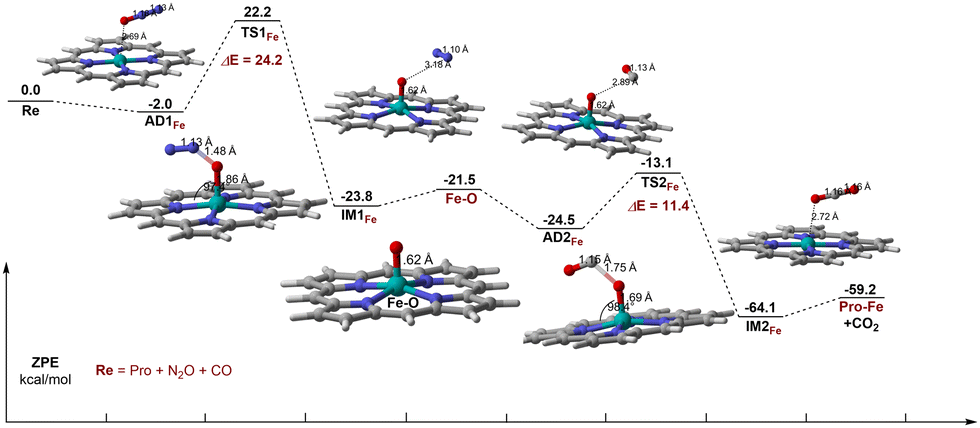 | ||
| Fig. 4 Energy profile and selected geometric parameters of transition states and intermediates involved in the oxidation of CO by N2O on Fe–porphyrin. | ||
Unlike on Ti–porphyrin, for the oxidation of CO by O–Fe–porphyrin, the adsorption of CO on the Fe–O active material is stronger than N2O on Fe–porphyrin and the adsorption energy is −3.0 kcal mol−1, which is an exothermic process. In the adsorption complex named AD2Fe, the distance between the C atom in CO and the reactive oxygen species of the Fe–O active substance is 2.89 Å, and the CO and reactive oxygen species form an angle of 92.2°. TS2Fe with a frequency of 982.22i cm−1 has been found. In the transition state, the Fe–O bond was elongated from 1.62 Å to 1.69 Å. The distance between the C atom and the reactive oxygen species is reduced from 2.89 Å to 1.75 Å, the C–O bond in CO extends from the original 1.13 Å to 1.15 Å and the CO and reactive oxygen species form an angle of 117.7°. Besides these, the Fe–O bond forms a 98.4° angle with the porphyrin plane. This step requires overcoming the activation energy barrier of 11.4 kcal mol−1, which is lower than Ti–porphyrin. In IM2Fe, the C–O bond of CO2 has formed, from 1.75 Å to 1.16 Å, close to CO2. The desorption energy of CO2 is 4.9 kcal mol−1, higher than 2.3 kcal mol−1, which is from the desorption of N2. Although the desorption energy of CO2 is slightly higher than N2, they both are relatively low, indicating that desorption is not difficult.
Different from Ti–porphyrin, CO oxidation is no longer a rate-controlling step on Fe–porphyrin, and the barrier height of N2O decomposition is significantly higher than that of CO oxidation. Therefore, in the reaction of Fe–porphyrin, N2O decomposition is the rate-controlling step of the whole reaction, and the barrier height is 24.2 kcal mol−1.
The energy profile of CO oxidation by N2O on Ti–porphyrin and Fe–porphyrin are placed over each other in Fig. 5. As shown in the figure, for Ti–porphyrin, CO oxidation is the reaction rate control step, the barrier height is 21.9 kcal mol−1; on the contrary, for Fe–porphyrin, the decomposition of N2O is the rate-controlling step of the whole reaction, and the barrier height is 24.2 kcal mol−1. The barrier height of N2O catalyzed decomposition on Ti–porphyrin is 2.3 kcal mol−1, which is lower than Fe–porphyrin (24.2 kcal mol−1), and both are significantly lower than 33.08 kcal mol−1 obtained from Cu-ZSM-5 by Zhang et al.22 The barrier height for CO oxidation on O–Fe–porphyrin is 11.4 kcal mol−1, which is lower than that on O–Ti–porphyrin (21.9 kcal mol−1), and both are lower than 135.5 kcal mol−1 from B11N12Al+ and 165.1 kcal mol−1 from B11N12+ obtained by Xiufang Hou et al.,3c and 25.4 kcal mol−1 from Ge–Gr4a and 25.4 kcal mol−1 from PN4-Gr obtained by Mehdi D. Esrafili.23 Furthermore, we studied the direct catalytic decomposition of the second N2O on O–Ti–porphyrin and O–Fe–porphyrin, and found that for O–Ti–porphyrin, the breaking of the N–O bond of the second N2O needs to overcome the barrier height of 38.8 kcal mol−1. This is much higher than the 3.8 kcal mol−1 required for the first N2O decomposition and 21.9 kcal mol−1 required for CO oxidation on O–Ti–porphyrin. For the O–Fe–porphyrin, the breaking of the N–O bond in the second N2O needs to overcome the barrier height of 74.2 kcal mol−1, which is much higher than the 24.2 kcal mol−1 required for the first N2O decomposition, and much higher than the 11.4 kcal mol−1 required for CO oxidation in O–Fe–porphyrin. Therefore, the reaction of N2O on M–porphyrin tends to stay in O–M–porphyrin rather than a further reaction to produce O–O–M–porphyrin (as shown in Fig. S6 and S7, ESI†).
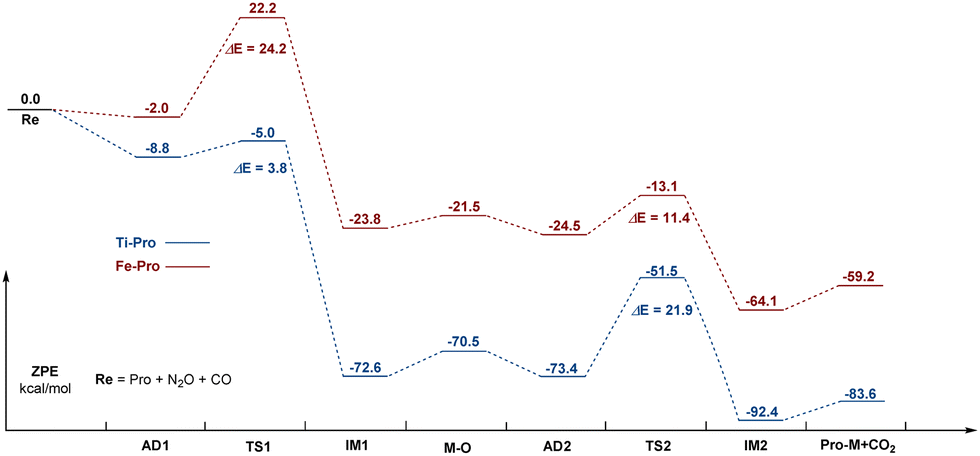 | ||
| Fig. 5 Energy profile of the catalytic oxidation of CO by N2O on Ti–porphyrin and Fe–porphyrin. | ||
Activation energies and reaction rates
Moreover, from the series of activation energies, we can also calculate the reaction rates of the N–O breaking step and C–O forming step (shown in Tables 1 and 2) using simple transition-state theory (TST). These show the reaction rates of the N–O breaking step to be in the order Ti–porphyrin > Fe–porphyrin > Co–porphyrin > Ni–porphyrin > Zn–porphyrin > Cu–porphyrin. The reaction rates of the C–O forming step are in the order O–Fe–porphyrin > O–Ti–porphyrin. The results correlate with the Irving–Williams series.Energy levels and shapes of the frontier molecular orbitals
We further studied the HOMO orbital distribution and the energy levels of Ti–porphyrin and Fe–porphyrin, and the LUMO orbital distribution of N2O molecules, as shown in Fig. 6. The results show that the HOMO energy of Ti–porphyrin is −82.8 kcal mol−1 and that of Fe–porphyrin is −113.3 kcal mol−1. The energy difference between HOMO of Ti–porphyrin and LUMO of N2O is 60.6 kcal mol−1; however, the energy difference between the HOMO of Fe–porphyrin and LUMO of N2O is 91.1 kcal mol−1; Ti–porphyrin and N2O have a small energy gap, but the energy gap of Fe–porphyrin and N2O is bigger than Ti–porphyrin. This could explain why Ti–porphyrin is more active than Fe–porphyrin in the catalytic decomposition of N2O into N2 and O–M active substances. Moreover, we did the same analysis for the oxidation process of CO and found that the energy difference between the HOMO of O–Fe–porphyrin and LUMO of CO was 7.8 kcal mol−1 lower than O–Ti–porphyrin.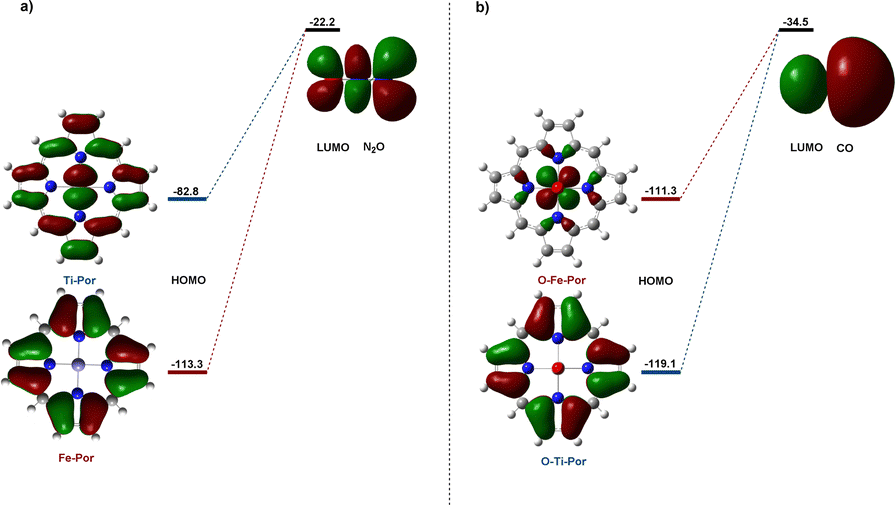 | ||
| Fig. 6 (a) Energy levels and shapes of the frontier molecular orbitals of M–porphyrin and N2O; (b) energy levels and shapes of the frontier molecular orbitals of O–M–porphyrin and CO (energies in kcal mol−1). | ||
Conclusions
In conclusion, DFT calculations have been performed to investigate the mechanism of CO oxidation by N2O on M–porphyrin (M = Ti, Fe, Co, Ni, Cu, and Zn). The reaction mechanism began with the breaking of the N–O bond in adsorbed N2O to form the M–O active species intermediate. The O atom of M–O active species then reacts with the CO molecule via the C–O bond formation to produce CO2. For the reaction on Ti–porphyrin, the activation energy barrier height of N2O decomposition is 3.8 kcal mol−1, and the energy barrier of CO oxidation by Ti–O active species to CO2 is 21.9 kcal mol−1, so the CO oxidation process is the rate-control step of the whole reaction. For the reaction on Fe–porphyrin, the activation energy barrier height of N2O decomposition is 24.2 kcal mol−1, and the energy barrier of CO oxidation by O–Fe active species to CO2 is 11.4 kcal mol−1, so the N2O decomposition process is the speed control step for the whole reaction. The rate constant between the adsorbed complex and the transition state of the N–O bond fracture and C–O bond formation processes is extracted (by transition state theory). Finally, the distribution and energy levels of the HOMO of M–porphyrin and LUMO of N2O were studied. It is found that the low energy gap corresponds to the low activation energy barrier, which could explainmore active Ti–porphyrin being more active than Fe–porphyrin in the catalytic decomposition of N2O into N2 and O–M active substances. A harmless catalyst for the treatment of N2O and CO has been screened in theory, which will provide valuable guidance for experimental research.Author contributions
S.-T. Wang conceived the scientific research project. S.-T. Wang and Z. Liu completed the main content of this study. Y.-J. Ye performed the drawing of pictures in this paper. Y.-J. Ye, X. Meng and P.-C. Yang completed data collation and proofreading. Z.-Z. Zhang is responsible for maintaining the computing cluster. S.-T. Wang, Z. Liu, Y.-F. Qiu and J.-Q. Lei wrote the original manuscript. Y.-F. Qiu and J.-Q. Lei completed review and proofreading of the manuscript. J.-Q. Lei revised the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 81960323). This work was carried out in Shanxi Supercomputing Center of China, and the calculations were performed in Tianhe-2. We thank associate professor Hong Gao (Lanzhou University) for helpful discussion.References
- (a) S. J. Hall and P. A. Matson, Nature, 1999, 400, 152–155 CrossRef CAS; (b) B. A. Hungate, J. S. Dukes, M. R. Shaw, Y. Luo and C. B. Field, Science, 2003, 302, 1512–1513 CrossRef CAS; (c) L. Li, J. Xu, J. Hu and J. Han, Environ. Sci. Technol., 2014, 48, 5290–5297 CrossRef CAS.
- K. Ning, Y. Y. Zhou, N. Zhang, X. J. Sun, W. W. Liu and C. H. Han, Med. Gas Res., 2020, 10, 30–36 CrossRef CAS.
- (a) K.-Q. Tran, P. Kilpinen and N. Kumar, Appl. Catal., B, 2008, 78, 129–138 CrossRef CAS; (b) M. Konsolakis, ACS Catal., 2015, 5, 6397–6421 CrossRef CAS; (c) X. Hou, F. Fu, C. Bai and G. Lei, Comput. Theor. Chem., 2020, 1171, 112660 CrossRef CAS; (d) G. Fan, Q. Wang, H. Xu, X. Wang, X. Tu and X. Chu, Appl. Surf. Sci., 2021, 544, 148766 CrossRef; (e) S. Ketrat, T. Maihom, S. Wannakao, M. Probst, S. Nokbin and J. Limtrakul, Inorg. Chem., 2017, 56, 14005–14012 CrossRef CAS; (f) F. Kapteijn, J. RodriguezMirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- (a) M. D. Esrafili and E. Vessally, Surf. Sci., 2018, 667, 105–111 CrossRef CAS; (b) M. M. Kappes and R. H. Staley, J. Am. Chem. Soc., 1981, 103, 1286–1287 CrossRef CAS.
- (a) M. M. Kappes and R. H. Staley, J. Am. Chem. Soc., 2002, 103, 1286–1287 CrossRef; (b) V. Blagojevic, G. Orlova and D. K. Bohme, J. Am. Chem. Soc., 2005, 127, 3545–3555 CrossRef CAS PubMed; (c) L. N. Wang, X. N. Li, L. X. Jiang, B. Yang, Q. Y. Liu, H. G. Xu, W. J. Zheng and S. G. He, Angew. Chem., Int. Ed., 2018, 57, 3349–3353 CrossRef CAS PubMed; (d) S. Debnath, H. Knorke, W. Schollkopf, S. Zhou, K. R. Asmis and H. Schwarz, Angew. Chem., Int. Ed., 2018, 57, 7448–7452 CrossRef CAS; (e) B. C. Sweeny, S. G. Ard, N. S. Shuman and A. A. Viggiano, ChemPhysChem., 2018, 19, 2835–2838 CrossRef CAS; (f) X. Sun, S. Zhou, L. Yue, C. Guo, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2019, 58, 3635–3639 CrossRef CAS; (g) X.-L. Xu, E. Yang, J.-Q. Li, Y. Li and W.-K. Chen, ChemCatChem, 2009, 1, 384–392 CrossRef CAS.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS.
- M. Jabłońska and R. Palkovits, Catal. Sci. Technol., 2016, 6, 7671–7687 RSC.
- (a) P. Granger, P. Malfoy, P. Esteves, L. Leclercq and G. Leclercq, J. Catal., 1999, 187, 321–331 CrossRef CAS; (b) J. Arenas-Alatorre, A. Gomez-Cortes, M. Avalos-Borja and G. Diaz, J. Phys. Chem. B, 2005, 109, 2371–2376 CrossRef CAS PubMed.
- (a) P. Giese, H. Kirsch, M. Wolf and C. Frischkorn, J. Phys. Chem. C, 2011, 115, 10012–10018 CrossRef CAS; (b) X. Wei, X. F. Yang, A. Q. Wang, L. Li, X. Y. Liu, T. Zhang, C. Y. Mou and J. Li, J. Phys. Chem. C, 2012, 116, 6222–6232 CrossRef CAS.
- (a) J. Harvey, Coord. Chem. Rev., 2003, 247, 1–19 CrossRef CAS; (b) W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS; (c) F. Scandola, C. Chiorboli, A. Prodi, E. Iengo and E. Alessio, Coord. Chem. Rev., 2006, 250, 1471–1496 CrossRef CAS; (d) N. Cheng, C. Kemna, S. Goubert-Renaudin and A. Wieckowski, Electrocatalysis, 2012, 3, 238–251 CrossRef CAS.
- (a) Y. Sivalingam, G. Magna, G. Pomarico, A. Catini, E. Martinelli, R. Paolesse and C. Di Natale, J. Mater. Chem., 2012, 22, 20032–20037 RSC; (b) J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655–4665 CrossRef CAS PubMed; (c) L. L. Li and E. W. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC; (d) G. Giancane and L. Valli, Adv. Colloid Interface Sci., 2012, 171–172, 17–35 CrossRef CAS PubMed.
- (a) I. M. Wasser, H. W. Huang, P. Moenne-Loccoz and K. D. Karlin, J. Am. Chem. Soc., 2005, 127, 3310–3320 CrossRef CAS; (b) K. Kano, Y. Itoh, H. Kitagishi, T. Hayashi and S. Hirota, J. Am. Chem. Soc., 2008, 130, 8006–8015 CrossRef CAS; (c) O. N. Chen, S. Groh, A. Liechty and D. P. Ridge, J. Am. Chem. Soc., 1999, 121, 11910–11911 CrossRef CAS; (d) A. Abdurahman and T. Renger, J. Phys. Chem. A, 2009, 113, 9202–9206 CrossRef CAS PubMed; (e) A. L. Tsai, V. Berka, E. Martin and J. S. Olson, Biochemistry, 2012, 51, 172–186 CrossRef CAS PubMed; (f) T. Karpuschkin, M. M. Kappes and O. Hampe, Angew. Chem., Int. Ed., 2013, 52, 10374–10377 CrossRef CAS.
- (a) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed; (b) K. Leung, I. M. Nielsen, N. Sai, C. Medforth and J. A. Shelnutt, J. Phys. Chem. A, 2010, 114, 10174–10184 CrossRef CAS PubMed; (c) C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- (a) J. T. G. a J. S. Roman, N. Phougat, P. Vasudevan, N. K. Jha and D. K. Bandhopadhyay, Transition Met. Chem., 2003, 28, 838–847 CrossRef; (b) S. Saito, H. Ohtake, N. Umezawa, Y. Kobayashi, N. Kato, M. Hirobe and T. Higuchi, Chem. Commun., 2013, 49, 8979–8981 RSC.
- (a) J. P. Collman, M. Marrocco, C. M. Elliott and M. Lher, J. Electroanal. Chem., 1981, 124, 113–131 CrossRef CAS; (b) B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS; (c) S. I. Murahashi, T. Naota and N. Komiya, Tetrahedron Lett., 1995, 36, 8059–8062 CrossRef CAS; (d) Y. E. Wang, Res. Chem. Intermed., 2006, 32, 235–251 CrossRef CAS; (e) L. Liu, M. Yu, B. B. Wayland and X. Fu, Chem. Commun., 2010, 46, 6353–6355 RSC.
- P. Maitarad, S. Namuangruk, D. Zhang, L. Shi, H. Li, L. Huang, B. Boekfa and M. Ehara, Environ. Sci. Technol., 2014, 48, 7101–7110 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2000, 22, 976–984 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- C. Y. Legault, CYLVIEW, 1.0b, Université de Sherbrooke, 2009, https://www.cylview.org..
- C. R. Gao, J. W. Li, J. Zhang and X. L. Sun, J. Catal., 2020, 10, 646 CAS.
- M. D. Esrafili, Chem. Phys. Lett., 2018, 708, 94–99 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04440d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |