
Bz-8HQ: a novel supramolecular fluorochrome exhibiting multiple stimuli-responsiveness†
Lamia A.
Siddig
a,
Rukayat
Bojesomo
a,
Mohammad A
Khasawneh
 a,
Abdelouahid
Samadi
a,
Alejandro Perez
Paz
*a,
Haythem A.
Saadeh
*ab and
Na’il
Saleh
*a
a,
Abdelouahid
Samadi
a,
Alejandro Perez
Paz
*a,
Haythem A.
Saadeh
*ab and
Na’il
Saleh
*a
aDepartment of Chemistry, College of Science, United Arab Emirates University, Al Ain, 15551, United Arab Emirates. E-mail: n.saleh@uaeu.ac.ae; aperez@uaeu.ac.ae; h.saadeh@uaeu.ac.ae
bDepartment of Chemistry, School of Science, The University of Jordan, Amman 11942, Jordan
First published on 18th November 2021
Abstract
A novel multifunctional fluorescent molecule (Bz-8HQ) has been designed and synthesized by the linkage of benzimidazole (Bz) and 8-hydroxyquinoline (8HQ). The synthesis was straightforward and involved reacting (piperazin-1-yl)-1H-benzo[d]imidazole with 5-(chloromethyl)quinolin-8-ol, which gives Bz-8HQ in a high yield. The structure of Bz-8HQ was subsequently characterized through different spectroscopic techniques such as 1H-NMR, 13C-NMR and high-resolution mass spectrometry. Due to the two distinct binding sites, Bz-8HQ exhibits responsiveness to multiple stimuli, including pH, cucurbit[7]uril (CB7) and cadaverine (CAD). The response of Bz-8HQ toward these perturbations was investigated by 1H-NMR spectroscopy and UV-visible absorption titration experiments. At pH 2.9 both the Bz and 8HQ moieties are protonated while at pH 6.5 both become deprotonated. The pKa values of the Bz and 8HQ groups were determined to be 6.2 and 2.5, respectively. The 1H-NMR titrations of Bz-8HQ with CB7 (0–4.4 equivalents) showed a preferential encapsulation of the Bz group at up to 2.9 equivalents. At 4.4 equivalents of CB7, both Bz and 8HQ sites were encapsulated. Upon the competitive titration with 0.5 equivalents of CAD, the Bz part was partially displaced by CAD but the 8HQ part remained fully encapsulated by CB7. The fluorescence titration of Bz-8HQ with HCl and CB7 showed that the energy transfer increases on decreasing the pH or increasing the concentration of CB7. The multicolor photoluminescence revealed the sensitivity and selectivity of the fluorescent probe. Bz-8HQ exhibits photoinduced electron transfer (PET) and fluorescence resonance energy transfer (FRET). The ability to modulate PET and FRET in fluorescent probes, such as Bz-8HQ, will be a valuable tool in future biological and medical imaging applications.
1. Introduction
Photoinduced electron transfer (PET) is ubiquitous in many biophysical and catalytic processes.1–5 For several decades, the transfer of electrons in the excited state from donor to acceptor molecules has been used in several applications, including artificial photosynthesis,6 solar cells,7 electroluminescent devices,8 logic gates,9 and chemical sensing,9–12 just to name a few. Fluorescence resonance energy transfer (FRET) has also been utilized in the design of fluorescent probes. Probes with FRET modulation can be used in several fields such as bio-imaging13,14,47 and chemical sensing.15,16The use of chromophores that respond to a single input is widespread. Some researchers have attempted to use a single chromophore for generating various responsive signals, which can be switched on demand upon simultaneous triggering with multiple inputs. Few examples, however, are found for systems having responsiveness for more than two inputs. Such chromophores were applied as logic gates,9 artificial proteomic tools17 and molecular switches.18 The present work demonstrates the ability to switch the photoluminescence signal of a single chromophore upon responding to multiple inputs through the use of both PET and FRET mechanisms. To this end, we carefully engineered a novel chromophore (Bz-8HQ) for generating regenerable signals in response to multiple inputs. Bz-8HQ consists of the union of benzimidazole (Bz)10,44,46 and 8-hydroxyquinoline (8HQ)19 moieties linked via a piperazine bridge. The synthesis of Bz-8HQ is straightforward, high yielding and does not necessitate multistep syntheses of complicated structures. Our selection of the Bz and 8HQ chromophores was motivated because of their use as chemosensors in several reports in the literature, which comes from the ability to modulate their photophysical properties through different mechanisms including PET10,20–22 and fluorescence resonance energy transfer (FRET).13–16,47 It is expected to modulate the photoluminescence (PL) signals of the novel Bz-8HQ molecule upon responding to multiple inputs, namely molecular receptors (host macrocycles), HCl, and chemical competitors (model drugs).
We chose the cucurbit[7]uril (CB7) macrocycle as molecular receptor due to its high binding affinity to a wide range of guest molecules and its ability to tune the acid-dissociation constant (Ka) of many guest molecules.23–26 It also forms switchable host–guest complexes27 driven by several bioactive chemical stimuli (such as cadaverine) in aqueous media.28 This ligand-CB7 binding is driven by non-covalent interactions (NCIs) such as hydrogen bonding, electrostatic, cation–π and anion–π, hydrophobic, van der Waals/dispersion interactions, and aromatic effects.29 CB7 provides a hydrophobic cavity for the binding of hydrophobic groups or molecules, whereas its portal carbonyl groups can bind positively charged groups, especially cations and ammonium groups.23,27 Moreover, for chemical sensor applications, it was observed that CB7 upon encapsulation of dyes intervenes with the intramolecular PET or FRET processes30,31 between the donor and acceptor moieties.26,32,33 These singular hosting properties of CB7 (ultrahigh binding affinity, host-induced pKa shifts, competitive displacement,34 and PET and FRET modulation) make it suitable for encapsulation of Bz-8HQ and it was chosen for this study. A few reports in the literature have pursued a similar approach of using cucurbiturils and related macrocycles. For example, Xin-long Ni and his co-workers recently investigated the CB8-induced pKa shift of acridine dyes to create PET-based fluorescence response signals for octreotide (a model drug) in an aqueous medium.35 In our approach, however, we use commercially available components for crafting a new constitutional chromophore (Bz-8HQ) that displays various colored emissions with different intensities in response to three inputs (pH, drugs, and CB7) as we discuss below.
2. Results and discussion
2.1. Synthesis of the Bz-8HQ chromophore
The synthesis of the Bz-8HQ chromophore is outlined in Scheme 1.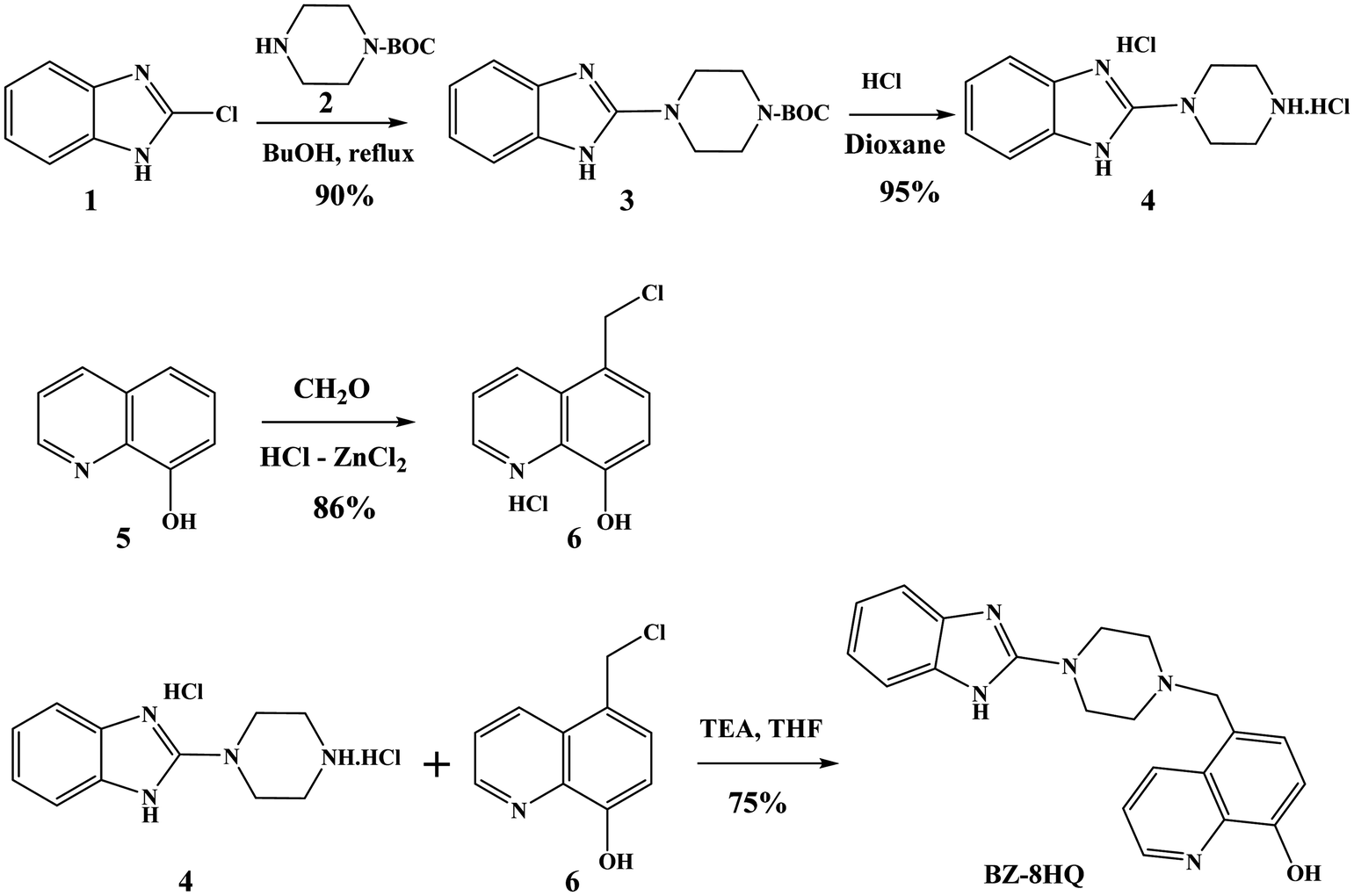 | ||
| Scheme 1 Synthesis of the novel Bz-8HQ chromophore. | ||
The benzimidazole chromophore was obtained by reacting 2-chlorobenzimidazole 1 with protected piperazine 2 to give compound 3, which upon treatment with HCl in dioxane gave the corresponding benzimidazole–piperazine 4 in a high yield (∼91%). The 1H-NMR spectrum shows the benzimidazole aromatic protons at 7.35–7.45 ppm and the piperazine protons at 3.45–3.55 ppm. The 8-hydroxiquinoline chromophore was prepared by chloromethylation of 5 with formaldehyde and ZnCl2–HCl to give compound 6 as a salt in a high yield. The 1H-NMR spectrum shows the quinoline aromatic protons at 7.18–9.06 ppm and the methylene protons at 4.87 ppm. Reacting compound 4 with 6 in the presence of excess triethylamine (TEA) gave the corresponding hybrid chromophore Bz-8HQ. The structure of the Bz-8HQ was confirmed by 1H- and 13C-NMR spectra and mass spectrometry (Fig. S1, ESI†). The 1H-NMR spectrum shows the aromatic protons of benzimidazole at 6.90 and 7.35 ppm and the 8-hydroxyquinoline protons in the range of 7.00–8.86 ppm. The aliphatic region has the methylene protons at 3.83 ppm and the piperazine protons at 2.55 and 3.45 ppm. The benzimidazole N–H and the 8-hydroxyquinoline O–H protons resonated at 11.34 and 9.76 ppm, respectively. Finally, the 13C-NMR spectrum shows the expected number of different carbons for Bz-8HQ and high-resolution mass spectrometry confirms the exact mass![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ion ratio as shown in the ESI.†
:ion ratio as shown in the ESI.†
2.2. The effects of adding HCl, CB7 and CAD on the UV-Vis absorption and NMR properties of Bz-8HQ
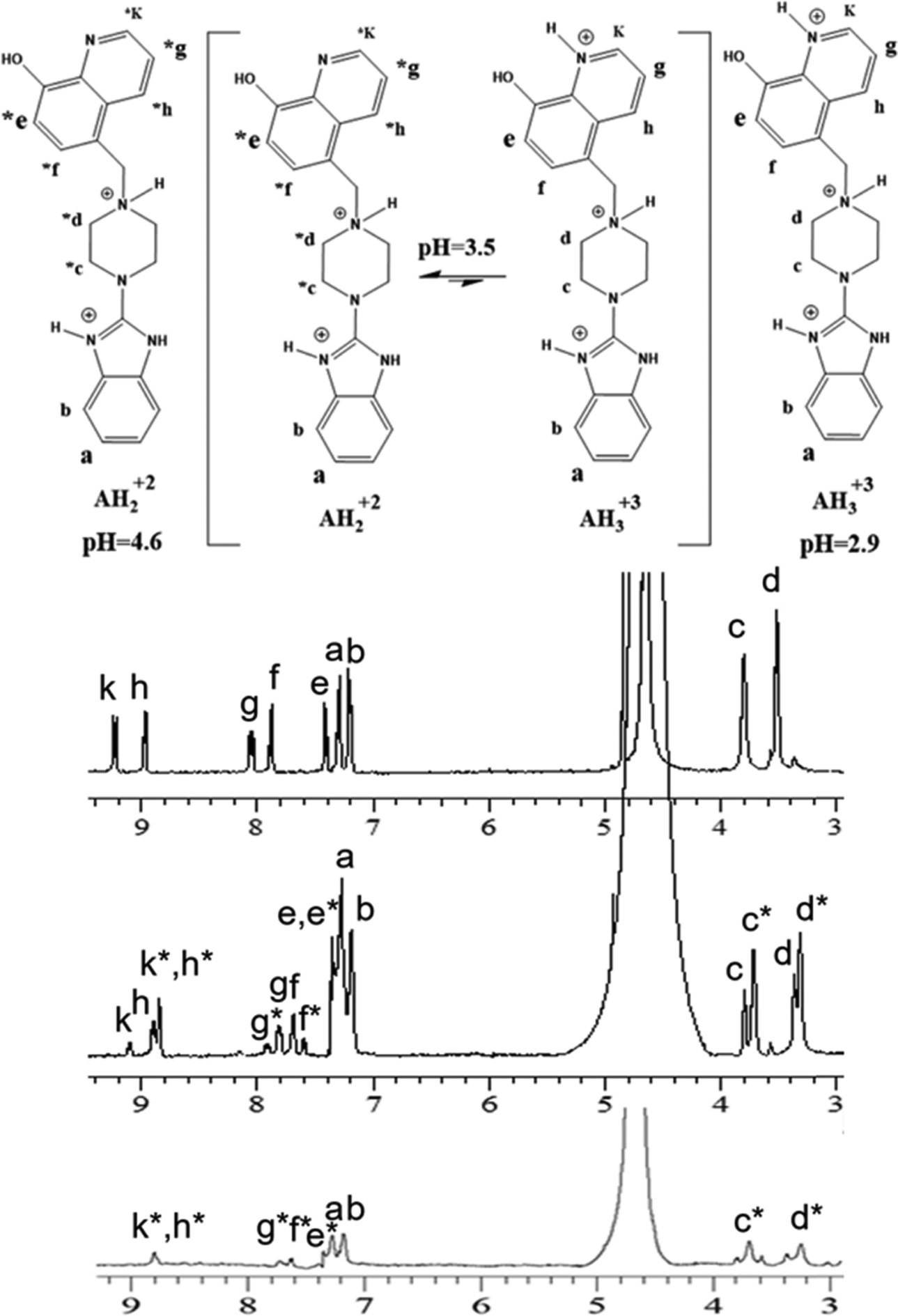 | ||
| Fig. 1 1H-NMR (400 MHz) of Bz-8HQ [1 mM] in D2O at pD 2.9 (top), pD 3.5 (middle) and pD 4.6 (bottom). Bz-8HQ is abbreviated as “A” in figures hereafter. | ||
The 1H-NMR showed that both bi- and tri- protonated forms of Bz-8HQ coexist in this pD range, with the former being the dominant species. The formation of bi-protonated Bz-8HQ was evident from the appearance of upfield peaks corresponding to quinoline protons (e*, f*, h*, g* and k*) and piperazine protons (c* and d*) but the benzimidazole protons (a and b) remained the same. At higher pD (∼4.6), 1H-NMR supports the presence of bi-protonated Bz-8HQ as the dominant species in solution.
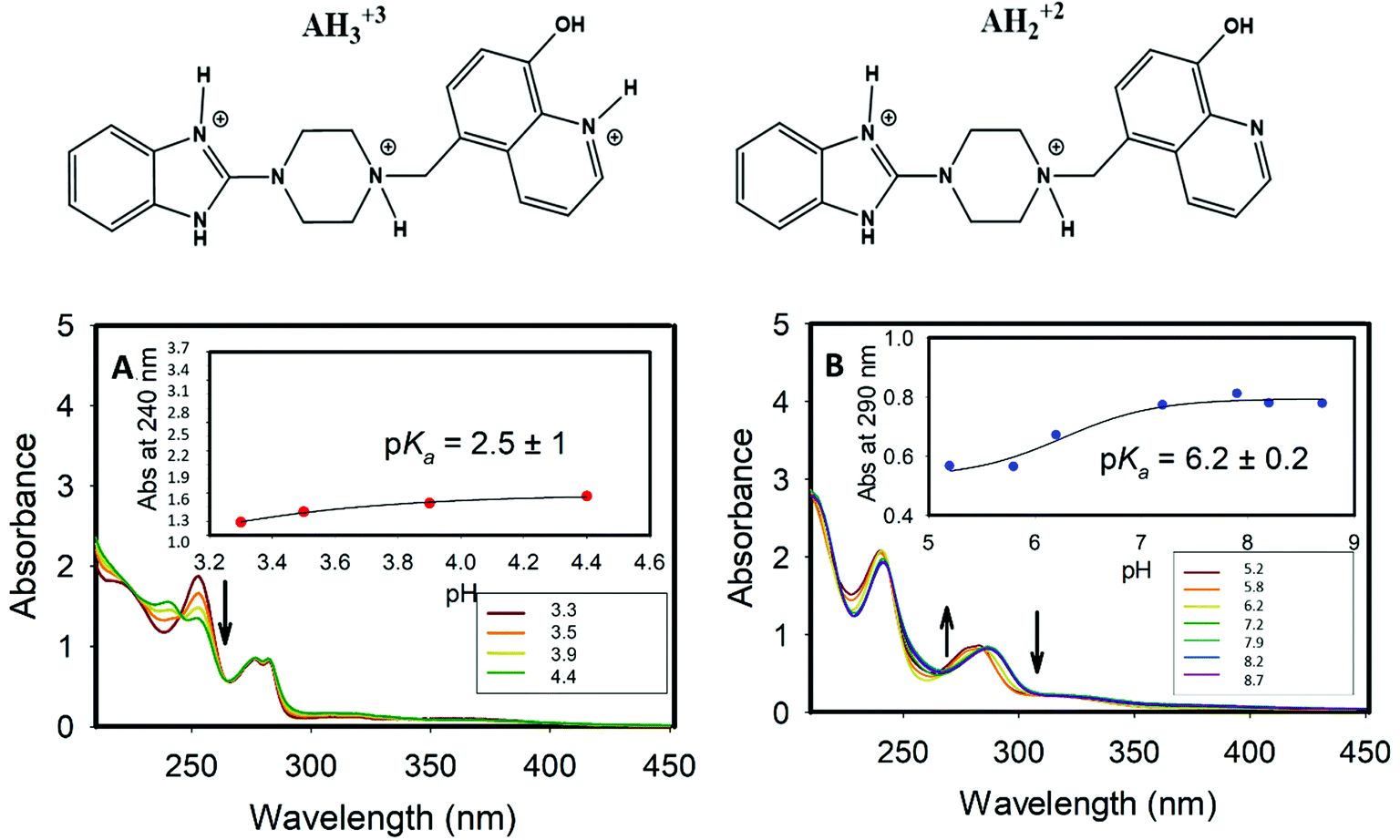 | ||
| Fig. 2 The UV-Vis spectra of Bz-8HQ. (A): in acid media. The arrow indicates the decrease of peak intensity at ∼260 nm as pH increases along the series 3.3, 3.5, 3.9 and 4.4. (B): at intermediate pH values. The arrows indicate the variation of peak intensities as the pH changes in the series 5.2, 5.8, 6.2, 7.2, 7.9, 8.2 and 8.7. The insets show the pKa values extracted from the inflection point of the sigmoidal curve fitted to the experimental absorbances at a fixed wavelength. | ||
For the deprotonated Bz-8HQ species (A−), we removed the proton from the hydroxyl group of 8HQ due to its lower pKa value in water (∼9.9) compared to the proton attached to the imidazole nitrogen atom (pKa ∼ 14.5). For the mono-protonated species (AH+), the structure with the proton on the imidazole nitrogen atom is 2.94 kcal mol−1 more stable than the configuration with the protonated piperazine nitrogen atom next to the methylene group. This energy difference increases up to 7.12 kcal mol−1 for the gas-phase protomers. For the bi-protonated Bz-8HQ (AH22+), DFT calculations predict two quasi-energetic protomers (labeled as #1 and #2 in Fig. 3). In implicit water, the preferred protomer (#1) places one proton on the nitrogen sites of imidazole and piperazine moieties, and no proton on the nitrogen site of 8HQ. Our quantum calculations in implicit water show that protomer #1 is about 1.09 kcal mol−1 more stable than protomer #2, which places one proton on the nitrogen sites of the imidazole and 8HQ fragments. This finding is a bit surprising since physical intuition suggests that placing protons on the endpoints of the molecule (as in protomer #2) reduces the Coulomb repulsion between like charges and lowers the total energy. Indeed, a similar calculation in the gas phase confirms this intuition, with protomer #1 being now 21.29 kcal mol−1 less stable than protomer #2. Thus, the dielectric properties of the solvent seem to play a key role here in screening the two nearby protons in protomer #1. Nonetheless, at this level of theory, the energy difference of 1.09 kcal mol−1 is small compared to the room thermal energy (∼0.6 kcal mol−1), and in aqueous solution both bi-protonated forms are expected to co-exist with an estimated equilibrium constant at 298 K of Keq = [#2]/[#1] ∼ exp[−ΔE/(RT)] = 0.16 for the proton transfer equilibrium #1 ⇔ #2.
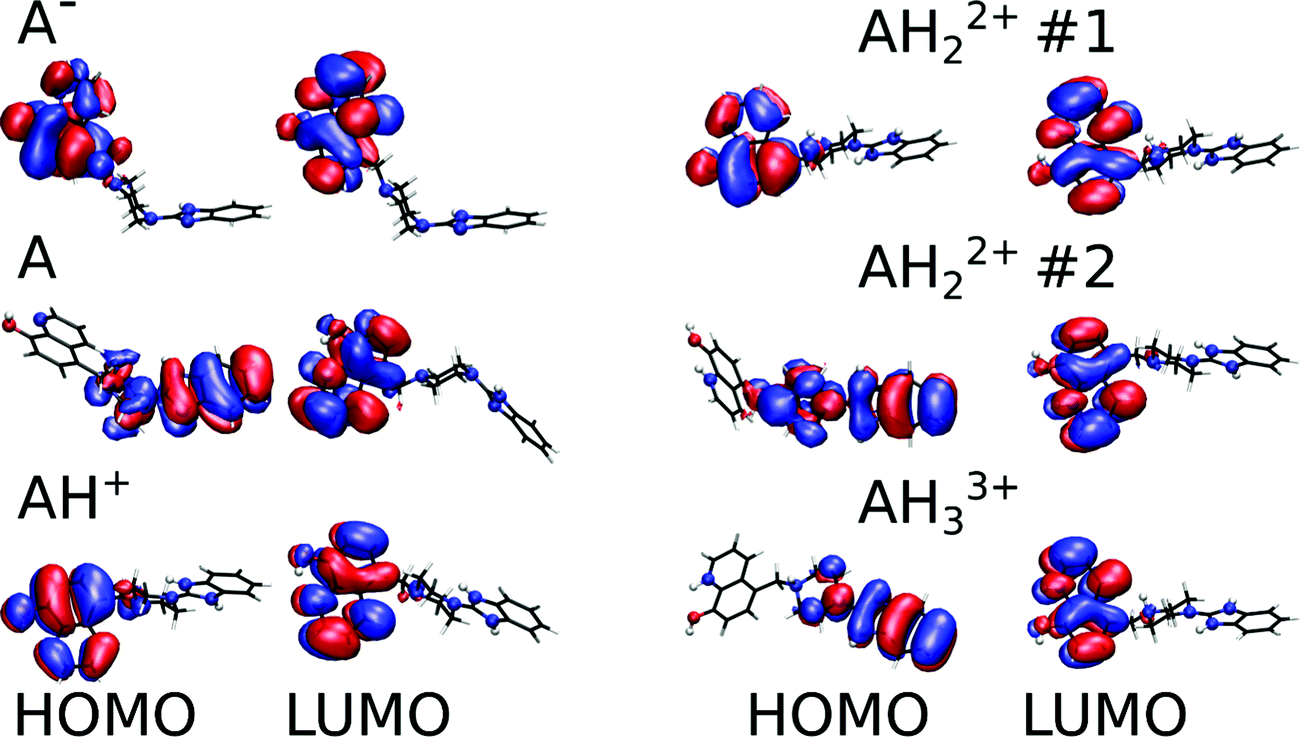 | ||
| Fig. 3 Relaxed structures and corresponding frontier molecular orbitals of the main protonation states of Bz-8HQ (abbreviated as “A” in legend) computed at the B3LYP-D3/def2-TZVP level in implicit water. The blue and red spheres represent the N and O atoms, respectively, while the rest is in wireframe representation for clarity. Isosurface representations (±0.017 a.u.) of the HOMO and LUMO orbitals are shown with the red (blue) regions representing positive (negative) values. | ||
Fig. 3 shows the relaxed conformations together with an isosurface representation of the frontier molecular orbitals. Our calculations show that all the lowest occupied molecular orbitals (LUMOs) have π* character and are consistently localized on the 8HQ part. The highest occupied molecular orbitals (HOMOs) also reside on the 8HQ part for all protomers except for the neutral, bi-protonated #2, and tri-protonated species where the HOMO is delocalized on the π system of the imidazole and piperazine parts. Thus, for the neutral compound, the 8HQ moiety is expected to act as an electron donor during a PET process to the acceptor part, which is located on the imidazole and piperazine parts. Interestingly, it was reported that the 8HQ molecule can undergo photoinduced intramolecular proton transfer in water solution leading to a non-fluorescent tautomer.36 In implicit water, our calculated electronic Kohn–Sham HOMO–LUMO gaps are 3.49, 3.71, 4.29, 4.32, 3.51 and 3.46 eV for the anionic, neutral, singly protonated, #1, #2, and tri-protonated forms of Bz-8HQ, respectively. Simulated UV-Visible spectra of these protomers are shown in Fig. S7 (ESI†).
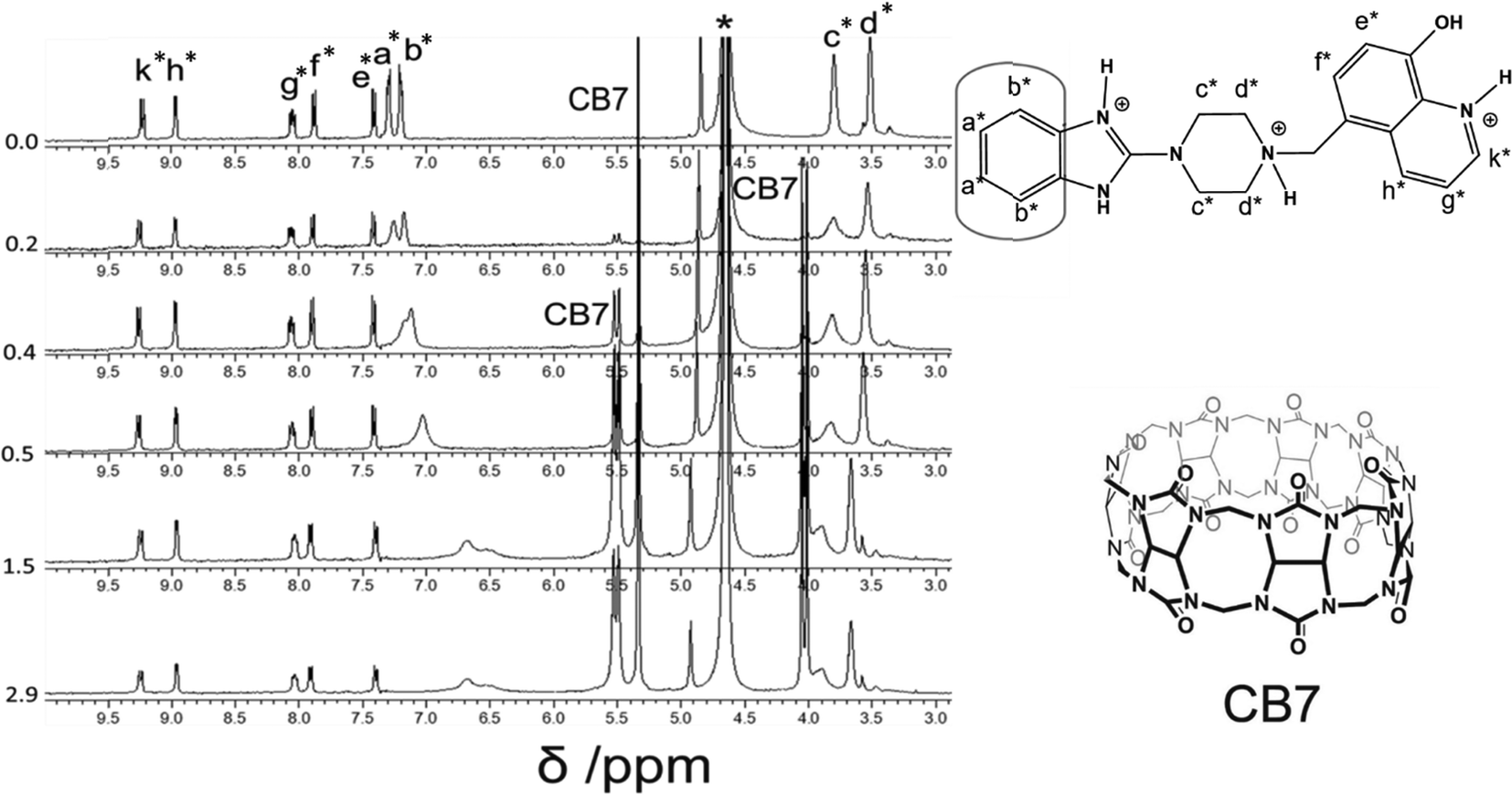 | ||
| Fig. 4 1H-NMR (400 MHz) titration of Bz-8HQ [1 mM] with CB7 (0–2.9 equivalents) in D2O at pD 2.9. | ||
However, when four equivalents of CB7 are added, both the Bz and 8HQ units were encapsulated in the CB7 cavity as manifested by the maximum shift of peaks a and b down to 6.5 and 6.3 ppm, respectively, as well as the onset of shifts and broadening to lower ppm of 8HQ protons (e–k). The amount of the shift depends on the monitored proton, which confirms the selectivity of this host–guest system. The large upfield shifts for protons e and k are due to the preferable interactions of hydroxyl protons and N atoms with the portal carbonyl oxygens of CB7.
In the aliphatic region (Fig. 6), broadening of protons c with concomitant shifts to higher ppm (along with protons d) suggests the shuttling of CB7 along the Bz-piperazine stalk even when 3 equivalents of CB7 are added per one molecule of Bz-8HQ.31,40 When we increased the concentration of CB7 up to 4 equivalents, the amount of shift and broadening to higher ppm of the d and c peaks is increased, whereas the methylene (CH2) peak shifts from 4.90 to lower ppm and becomes hidden under the solvent peak at 4.65 ppm. This NMR pattern is consistent with the engulfing of the 8HQ group by CB7 as discussed above.
 | ||
| Fig. 5 1H-NMR of the aromatic region. Binding of Bz-8HQ/CB7 (0–4.4 equivalents) with cadaverine (CAD) (0.5 equivalents) in D2O at pD 2.9. | ||
The partial replacement of the Bz unit by CAD is also confirmed by the broadening and shifting to lower ppm of the CAD protons in this region before and after mixing with Bz-8HQ/CB7 complexes from 1.3, 1.5, and 2.8 ppm to 1.2, 1.3 and 2.5 ppm, respectively. The results also support the partial inclusion of CAD by CB7 when compared to full encapsulation of CAD by CB7 as illustrated in Fig. 7 upon the titration of CAD by CB7 (0–1.2 equivalents). In the latter result, the CAD peaks were shifted to lower ppm from 1.3, 1.5, and 2.8 ppm to 0.4, 0.7, and 2.1 ppm, respectively, upon full encapsulation in CB7. The lack of interaction between CAD and Bz-8HQ was also confirmed in Fig. 8 using a similar titration method.
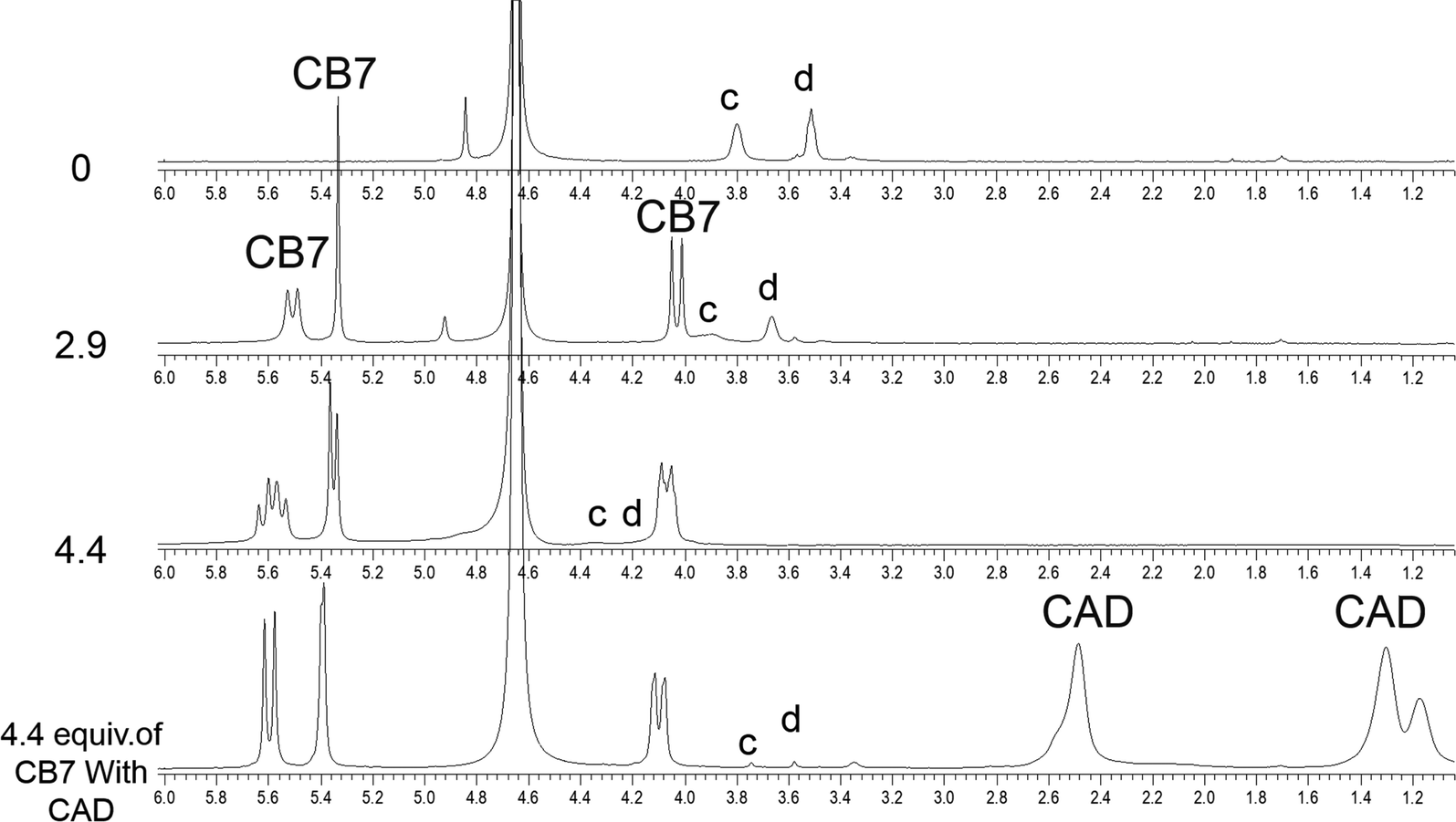 | ||
| Fig. 6 1H-NMR spectra of the aliphatic region of Bz-8HQ after the addition of CB7 (0–4.4 equivalents), followed by the addition of CAD (0.5 equivalents) in D2O at pD 2.9. | ||
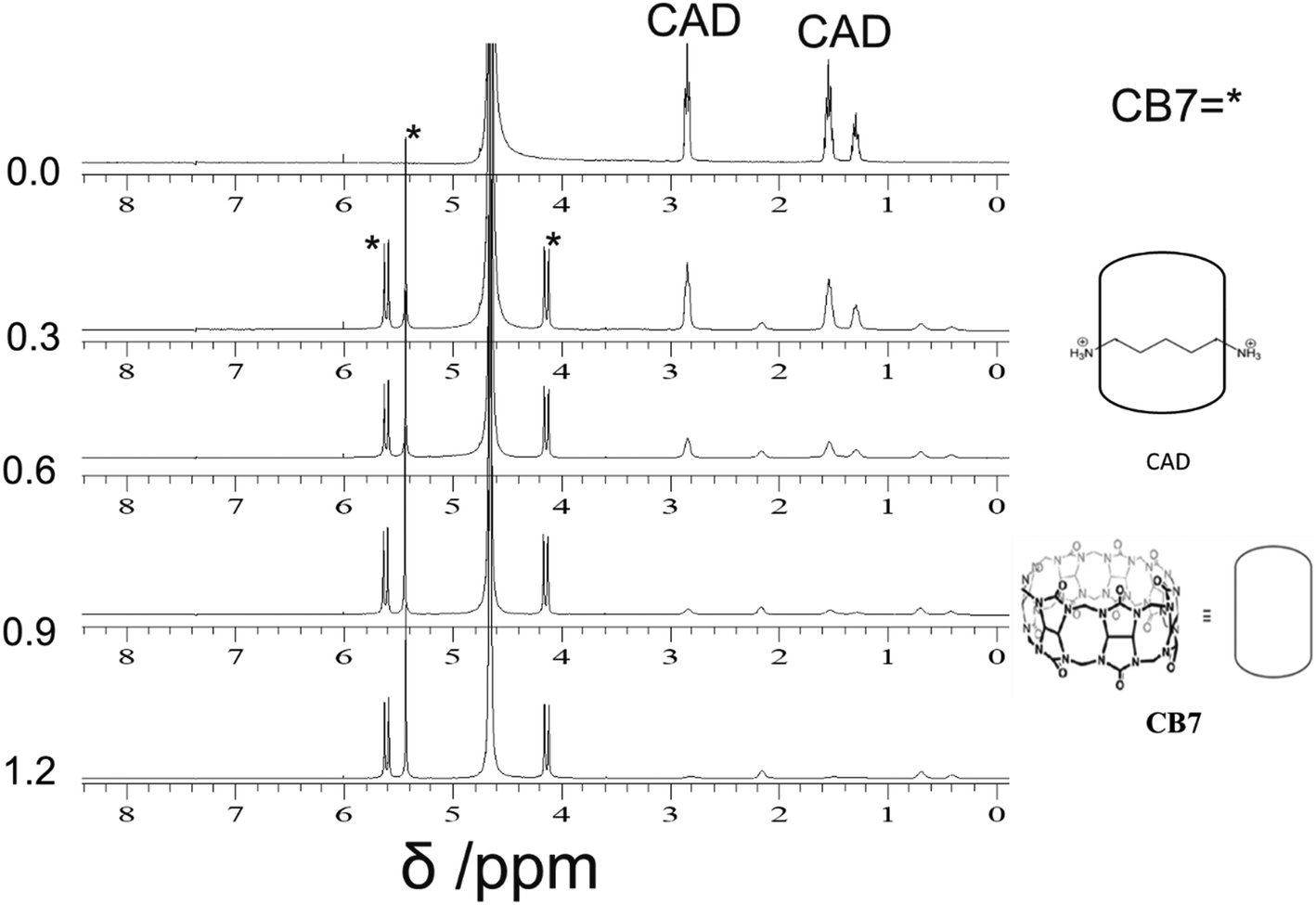 | ||
| Fig. 7 1H-NMR (400 MHz) titration of CAD [2 mM] with CB7 (0–1.2 equivalents) in D2O at pD 4.6. | ||
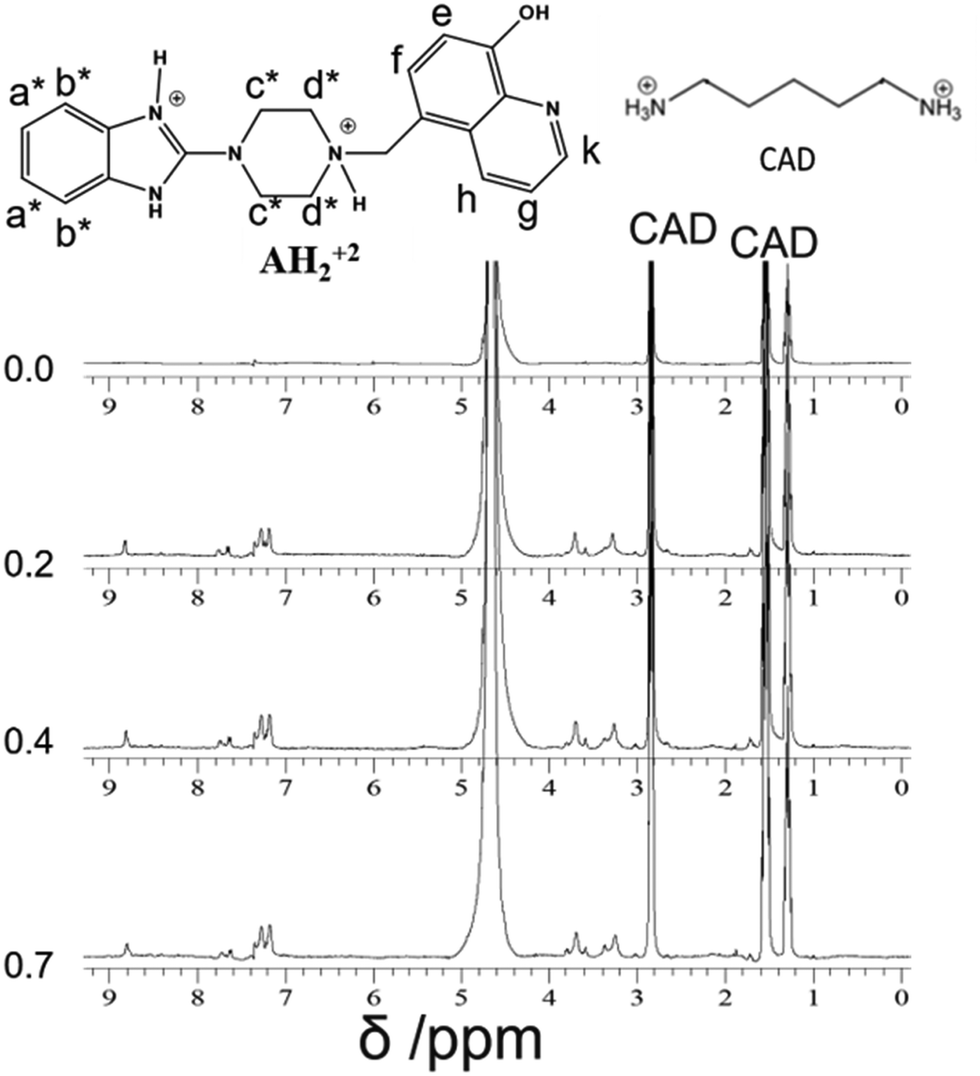 | ||
| Fig. 8 1H-NMR (400 MHz) titration of CAD [2 mM] with Bz-8HQ (0–0.7 equivalents) in D2O at pD 4.6. | ||
Fig. 7 illustrates the changes in the NMR spectra of CAD upon the addition of different equivalents of CB7. The peaks of CAD appear at 1.3, 1.5 and 2.8 ppm. Upon the addition of CB7, the CAD peaks disappeared, and new peaks appeared around 4.1, 5.4 and 5.6 ppm, which represent the CB7 peaks. The results support the full encapsulation of CAD in the cavity of CB7.
Fig. 8 shows the NMR spectra of CAD upon the addition of different equivalents of Bz-8HQ. The peaks of CAD appear at 1.3, 1.5 and 2.8 ppm, and upon the addition of Bz-8HQ new peaks appear between (3–4) ppm and (7–9) ppm, which represent that the Bz-8HQ peaks and the CAD peaks remain the same. The results confirm that no interaction occurs between CAD and Bz-8HQ.
2.3. Quantum calculations of the host–guest interaction
To better understand the intermolecular guest–host interaction, we performed density functional theory (DFT) calculations on these compounds in the gas phase. The molecular electrostatic potential map of CB7 at the B3LYP-D3/def2-TZVP level is shown in Fig. 9 (left), which reveals the high affinity of the portal carbonyl oxygens (red domains) towards cations. Therefore, electrostatic arguments predict that any molecular cation would preferentially place its positive charges (or electropositive atoms) near CB7 carbonyl oxygens.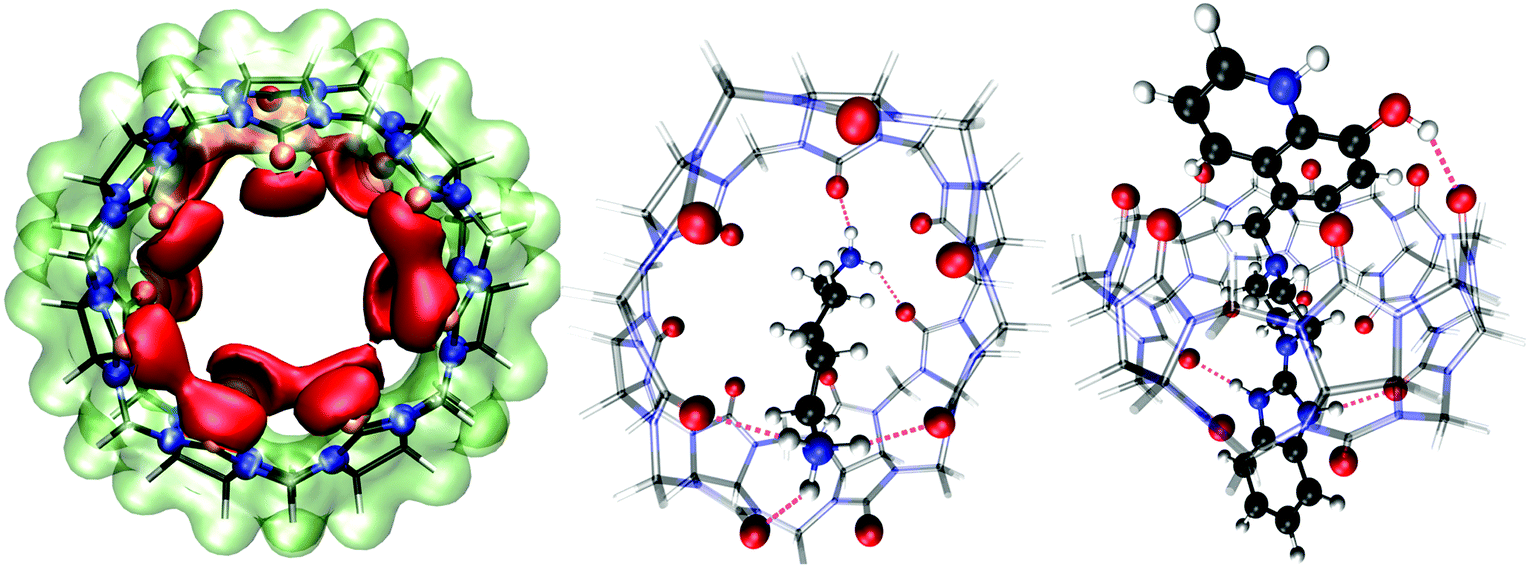 | ||
| Fig. 9 Gas-phase DFT calculations of the host (CB7) and guest molecules (CAD and Bz-8HQ). Left: Electrostatic potential energy map of cucurbit[7]uril (CB7) at the B3LYP-D3/def2-TZVP level. The isosurface value was ±0.09 a.u., with the red (lime) color representing regions of attraction (repulsion) toward a positive test charge. Middle: Relaxed geometry of the complex of CB7 with bi-protonated cadaverine (CAD). Right: Optimized complex of the CB7 macrocycle with bi-protonated Bz-8HQ (protomer #2 in Fig. 3). The black, white, red and blue spheres represent the C, H, O, and N atoms, respectively. CB7 is shown as a transparent wireframe representation with the oxygen atoms highlighted as red spheres. Intermolecular hydrogen bonds are indicated by the violet dashes. All geometry relaxations of the host-ligand complexes are at the BLYP-D3/def2-TZVP level and perspective representation was used in the rendering. | ||
We carried out structural relaxations of the complexes between the host macrocycle (CB7) and the bi-protonated guest molecules (CAD and Bz-8HQ) at the BLYP-D3/def2-TZVP level of theory. Fig. 9 (middle) shows the optimized structure of the CB7/CAD complex. Upon encapsulation, CB7 features a marked oval distortion due to the formation of five hydrogen bonds of mean length ∼2 Å (violet dashes) between the portal carboxylic oxygens and the hydrogen atoms of the CAD ammonium terminations. We find that a single CAD molecule places its nonpolar alkyl chain (of ∼5 Å length) diagonally across the hydrophobic cavity of CB7 (of width ∼4.0 Å and diameter ∼10 Å). This orientation of CAD is optimal because it maximizes hydrogen bonding between CAD terminal ammonium cations with the portal carbonyl oxygens of the CB7 macrocycle. Our DFT calculations predict that the binding energy (defined as BE = ECB7 + Eligand − ECB7/ligand, with all geometries relaxed) of the CB7/CAD complex in a vacuum is 151.17 kcal mol−1 (6.56 eV) indicating irreversible binding and the formation of a very stable complex in the gas phase.
We also optimized the geometry of the complex between CB7 and bi-protonated Bz-8HQ (protomer #2 in Fig. 3), which is the dominant form of Bz-8HQ at pH 4.6 of our experiments and is expected to exhibit the maximum affinity toward CB7 due to its bi-cationic charged state. The relaxed structure (Fig. 9, right) shows that the guest molecule maximizes hydrogen bonding between its imidazole N–H and O–H hydrogens with CB7 carbonyl oxygens. CB7 is now less distorted and preserves better its ring-like shape than when CAD is encapsulated. Bi-protonated Bz-8HQ traverses CB7 and establishes three hydrogen bonds or two less than the five hydrogen bonds for the case of CAD. Our DFT calculations predict a binding energy with CB7 of 123.75 kcal mol−1 (5.37 eV), which is 27.42 kcal mol−1 (1.19 eV) lower than with CAD compound. Since we considered the bi-protonated form of Bz-8HQ, its mono-protonated and neutral species are expected to have even less affinity toward CB7. This explains the partial replacement of Bz-8HQ species by CAD observed during our competitive titration experiment, see Fig. 5 and 6.
2.4. Classical MD simulations of the host–guest complexes in explicit water
We performed classical molecular dynamics (MD) simulations of the CB7-guest complexes. First, we tested the force fields for the gas phase complexes to see whether the binding energies were consistent with our ab initio calculations. Our force-field binding enthalpy for CAD in CB7 is −186.7 kcal mol−1 whereas for the bi-protonated Bz-8HQ molecule it is −154.4 kcal mol−1, which are consistent with our DFT findings in the gas phase (see section 2.3). This indicates that the semiempirical force field parameters for the CB7-ligand interaction are reasonable.In explicit TIP3P water at 298.15 K and 1 bar, the computed binding Gibbs free energies (within the normal mode entropy approximation) are −7.04 ± 2.61 kcal mol−1 for the CAD molecule and −21.23 ± 1.98 kcal mol−1 for bi-protonated Bz-8HQ. Experimental binding affinities between CB7 and CAD in water under room conditions were recently reported by Sinn and co-workers.41 They reported two experimental values for the quantity −pKbinding = log(Kbinding) = 8.37 and 8.64, which correspond to standard binding Gibbs free energies at T = 298.15 K of ΔG°binding = −RTln(Kbinding) = −11.4 and −11.8 kcal mol−1, respectively. Our computed value from MD calculations for the binding of CAD with CB7 (−7.04 ± 2.61 kcal mol−1) is in reasonable agreement with Sinn's experimental values and suggests that our computational approach is correct. To the best of our knowledge, no calculated binding affinity from MD simulations for the CB7/CAD complex was reported in the literature. We remark that the protonation state of CAD (i.e., the pH of the medium) can have an important impact on its binding energy with CB7 but this is beyond our study.
The dominant energy stabilization in the ligand–receptor complex is electrostatics, mainly the formation of hydrogen bonds between amino hydrogens of the ligand and carbonyl oxygens of CB7. For the CAD molecule, our MD calculations show that there is a larger solvation free energy penalty (ΔG°solv = 163.9 kcal mol−1, endergonic) than for the bi-protonated Bz-8HQ molecule (ΔG°solv = 116.0 kcal mol−1). To understand this better, we performed a cluster analysis on the equilibrated MD trajectories to get representative conformations of the ligand–host complexes. The resulting average conformations of the ligand–receptor complexes look very similar to the gas phase ones where we can see that CAD is totally engulfed by CB7 (Fig. 9, middle). Since CAD is a small molecule, there are some water molecules in the CB7 cavity as well (see ESI† animation). However, part of the bi-protonated Bz-8HQ lies outside of CB7 and still interacts with the solvent molecules via hydrogen bonds, which explains its lower solvation free energy penalty.
Both ligands interact with CB7 during most of the time. Rarely, the ligands are seen to depart from the CB7 host in our equilibrated 16 ns-long MD simulations. The probability for these departure events is larger for CAD (see ESI† animation). This is consistent with our computational findings of a lower binding affinity of CAD than for bi-protonated Bz-8HQ.
These findings agree (to some extent) with our competitive experiments, which show that CAD only partially (but not totally) displaces Bz-8HQ in binding CB7. This suggests that, in actual experiments, the simultaneous interaction of CB7 with both ligands could be taking place, and we have four host–ligand combinations corresponding to the homo and hetero ligand bindings. In competitive experiments, we think that bi-protonated Bz-8HQ is not totally engulfed by CB7 (i.e., it interacts more weakly) and it can be partially displaced by CAD, which fits in CB7 better than Bz-8HQ. Indeed, it was reported that small molecules with aliphatic chains without aromatic cores (such as CAD) encapsulate better in CB7 than larger aromatic molecules (such as Bz-8HQ) due to a reduction in steric clashes.42 Thus, in our competitive binding experiments, we think a concomitant interaction of both ligands with CB7 is more favorable energetically. A more detailed account of the CB7-ligand binding would necessitate its own study (e.g., considering the different initial conditions, conformations, and protonation states) that is beyond the scope of this work.
2.5. Stimuli emission responsiveness of Bz-8HQ to the addition of HCl, CB7 and CAD.
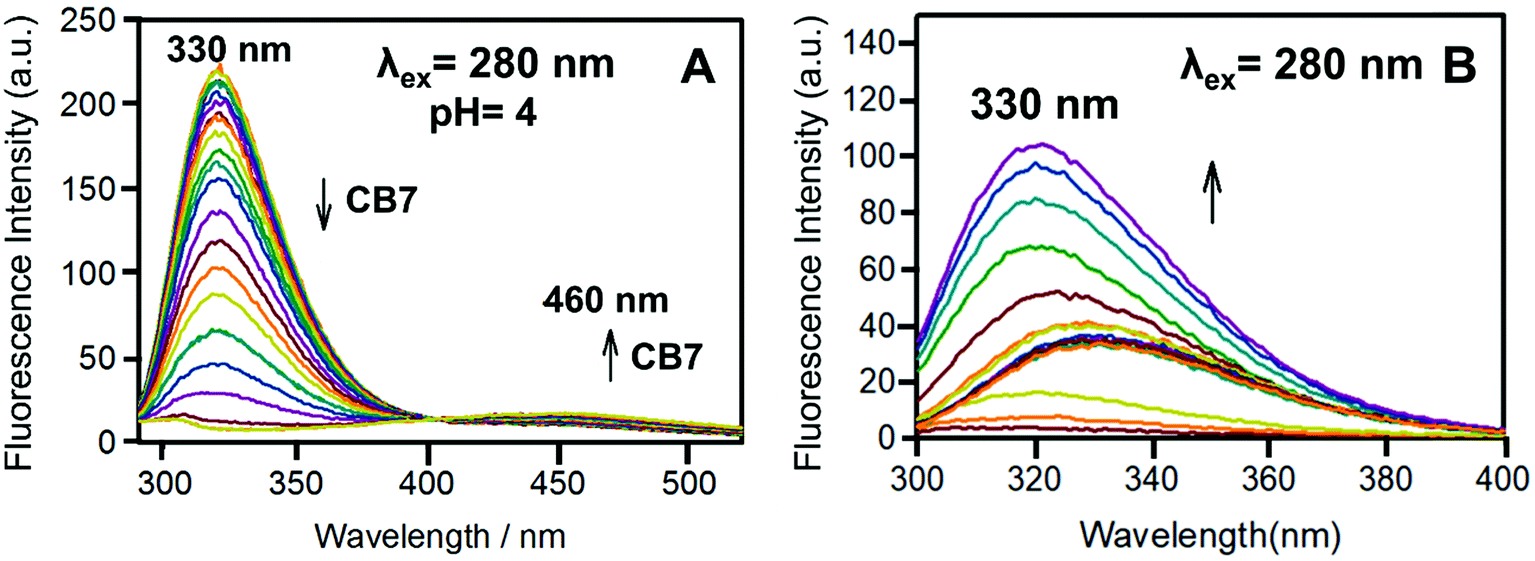 | ||
| Fig. 10 (A) The binding titration of Bz-8HQ with CB7 at pH 4. The left arrow indicates the decrease in the emission at 330 nm, while the right arrow indicates the increase in the emission at 460 nm upon the addition of CB7. (B) The fluorescence titration of Bz-8HQ from pH 3.4 to 11.9. The arrow indicates the increase in the emission at 330 nm upon increasing the pH. | ||
Fig. 11 shows the change in the emission of the Bz-8HQ/CB7 complex [32.1 μM] for Bz-8HQ and 500 μM for CB7 with the addition of different concentrations of CAD [0–500 μM] at a fixed pH of 4. Upon the addition of CAD, the emission peak at around 330 nm increased, while the emission around 460 nm decreased. Collectively, the energy transfer will decrease by adding CAD.
 | ||
| Fig. 11 The fluorescence intensity of CAD [500 μM] with the complex of Bz-8HQ and CB7 at pH 4. The arrow indicates the increase in the emission peak at ∼330 nm upon the addition of CAD. | ||
Fig. S4 (ESI†) shows the change in the emission of Bz-8HQ [31.2 μM] as a function of the added HCl. Noticeably, the emission dramatically increases along with the addition of H+ until it reaches pH 7.62 (Fig. S4B, ESI†). This can be attributed to the protonation of the Bz group, which suppresses the PET quenching.10 On the other hand, when the concentration of H+ increases from pH 7.62 to 3.13, the emission significantly decreases because of the collisional quenching.
Fig. S5 (ESI†) shows the changes in the emission of the complex of Bz-8HQ [31.2 μM] with CB7 [500], the emission at the beginning increases as the pH increases from 3.3 to 7.6, then it starts to decrease from pH 7.6 to 11. The concurrent effects of CB7, Cl− and H+ caused a periodical increase and decrease in the signals, as described in the text.
Fig. 12A shows the effect of binding to CB7 (0–500 μM) on the emission spectra of Bz-8HQ [31.2 μM] at pH 4.6. The fluorescent Bz-8HQ has an emission around 460 nm, and upon the addition of CB7 the emission increased and slightly shifted to 470 nm. This is because at pH 4.6 the extent of protonation of the Bz group enhances inside the cavity of CB7. Thus, the CB7 engulfing suppresses the PET process and restores the 8HQ's blue emission.
 | ||
| Fig. 12 (A) The binding titration of Bz-8HQ with CB7 (0–500 μM) at pH 4.6. The arrow indicates the intensity increase at 470 nm upon the addition of CB7. (B) The binding titration of Bz-8HQ with cadaverine (0–500 μM) at pH 4.6. Arrow shows the decrease in intensity around 460 nm with the addition of CAD. | ||
Fig. 12B shows the changes in the emission of Bz-8HQ at pH 4.6 with the gradual addition of CAD to the Bz-8HQ. We find that the emission at 460 nm was quenched because of an intermolecular PET between the amine group in CAD and the 8HQ group.43 The mechanism of quenching through the intramolecular proton transfer from the HQ group to the nitrogen atom of CAD cannot be excluded.43
Fig. S6 (ESI†) shows the variation of emission spectra of the Bz-8HQ [32.1 μM] and CB7 [500 μM] complex upon the addition of different increments of CAD [0–500 μM] at fixed pH 4.6. The results support the conclusion made from the absorption measurements. Fig. S6A (ESI†) shows that upon the addition of (0–30 μL) of CAD, slight changes occur in the emission profile (Fig. S3, ESI†). However, Fig. S6B (ESI†) shows that with the addition of (34–60 μL) of protonated CAD, the drug starts to replace the Bz unit from the CB7 cavity. This suppressed the intermolecular PET quenching between CAD and 8HQ and the intramolecular PET quenching between residual neutral Bz and 8HQ. Overall, we observed a significant increase in emission. Fig. S6C (ESI†) shows that with the addition of excess (65–1600 μL) of CAD, the emission slightly decreases because of the CAD-assisted intermolecular PET quenching.
3. Conclusions
A novel fluorophore (Bz-8HQ) was synthesized by linking benzimidazole and 8-hydroxyquinoline molecules via a piperazine bridge. The responsiveness of Bz-8HQ to multiple analytes (pH, cucurbit[7]urils (CB7) and the chemical competitor cadaverine) was investigated through various spectroscopic titration techniques, including UV-VIS absorption, fluorescence, and NMR. The Bz-8HQ fluorophore switched off the PET and switched on the FRET upon the addition of CB7, emitting blue color. These results show that the multicolor photoluminescence response upon synergetic modulation of the PET and FRET mechanisms of Bz-8HQ is remarkable and it represents a promising molecule for the development of future fluorescent sensors for diverse chemical and biomedical applications.45Author contributions
LA prepared the dye, conducted all the characterization experiments and wrote the initial manuscript. RB helped in collecting part of the spectroscopic measurements. HS designed the dye. HS, MAK, and AS supervised its preparation. APP conducted and analyzed the computational calculations and wrote the final manuscript. NS created the idea and supervised the NMR competitive binding and photochemistry experiments. All the authors gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the research program in the United Arab Emirates University for financial grants UPAR (2018) 31S141 and UPAR (2020) 31S392. This work used the “Al Ain” computer workstation of UAE University, which was purchased under an internal start-up grant No. 31S410.Notes and references
- W. Zhanga, X. Liua, L. Yana, G. Zhub, Z. Wanga and Y. Yang, Photo-Induced Intermolecular Electron Transfer-Effect of Acceptor Molecular Structures, Chin. J. Chem. Phys., 2018, 772–778, DOI:10.1063/1674-0068/31/cjcp1807171.
- S. S. Souza, M. L’Episcopia, C. Severini, V. Udhayakumar and N. W. Lucchi, Photo-Induced Electron Transfer Real-Time PCR for Detection of Plasmodium Falciparum Plasmepsin 2 Gene Copy Number, Antimicrob. Agents Chemother., 2018, 62(8), 1–4, DOI:10.1128/AAC.00317-18.
- S. Fukuzumi, Nanocarbons as Electron Donors and Acceptors in Photoinduced Electron-Transfer Reactions, ECS J. Solid State Sci. Technol., 2017, 6(6), 3055–3061, DOI:10.1149/2.0061706jss.
- A. Pantazis, K. Westerberg, T. Althoff, J. Abramson and R. Olcese, Harnessing Photoinduced Electron Transfer to Optically Determine Protein Sub-Nanoscale Atomic Distances, Nat. Commun., 2018, 9(1), 4738, DOI:10.1038/s41467-018-07218-6.
- T. Hossen and K. Sahu, Photo-Induced Electron Transfer or Proton-Coupled Electron Transfer in Methylbipyridine/Phenol Complexes: a Time-Dependent Density Functional Theory Investigation, J. Phys. Chem. A, 2019, 123(38), 8122–8129, DOI:10.1021/acs.jpca.9b06274.
- M. Skaisgirski, C. B. Larsen, C. Kerzig and O. S. Wenger, Stepwise Photoinduced Electron Transfer in a Tetrathiafulvalene-Phenothiazine-Ruthenium Triad, Eur. J. Inorg. Chem., 2019,(39–40), 4256–4262, DOI:10.1002/ejic.201900453.
- D. Escudero, Revising Intramolecular Photoinduced Electron Transfer (PET) from First-Principles, Acc. Chem. Res., 2016, 49(9), 1816–1824, DOI:10.1021/acs.accounts.6b00299.
- D. Singh, V. Nishal, S. Bhagwan, R. K. Saini and I. Singh, Electroluminescent Materials: metal Complexes of 8-Hydroxyquinoline – A Review, Mater. Des., 2018, 156, 215–228, DOI:10.1016/j.matdes.2018.06.036.
- J. Andréasson and U. Pischel, Molecules for Security Measures: from Keypad Locks to Advanced Communication Protocols, Chem. Soc. Rev., 2018, 47(7), 2266–2279, 10.1039/C7CS00287D.
- E. Horak, P. Kassal and I. Murković Steinberg, Benzimidazole as a Structural Unit in Fluorescent Chemical Sensors: the Hidden Properties of a Multifunctional Heterocyclic Scaffold, Supramol. Chem., 2018, 30(10), 838–857, DOI:10.1080/10610278.2017.1403607.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Luminescent Chemosensors for Transition Metal Ions, Coord. Chem. Rev., 2000, 205(1), 59–83, DOI:10.1016/S0010-8545(00)00242-3.
- B. Valeur and I. Leray, Design Principles of Fluorescent Molecular Sensors for Cation Recognition, Coord. Chem. Rev., 2000, 205(1), 3–40, DOI:10.1016/S0010-8545(00)00246-0.
- K. Kikuchi, Design, Synthesis and Biological Application of Chemical Probes for Bio-Imaging, Chem. Soc. Rev., 2010, 39(6), 2048–2053, 10.1039/B819316A.
- S. Goswami, S. Maity, A. C. Maity, A. K. Maity, A. K. Das and P. Saha, A FRET-Based Rhodamine–Benzimidazole Conjugate as a Cu2+-Selective Colorimetric and Ratiometric Fluorescence Probe That Functions as a Cytoplasm Marker, RSC Adv., 2014, 4(12), 6300–6305, 10.1039/C3RA46280C.
- D. T. Quang and J. S. Kim, Fluoro- and Chromogenic Chemodosimeters for Heavy Metal Ion Detection in Solution and Biospecimens, Chem. Rev., 2010, 110(10), 6280–6301, DOI:10.1021/cr100154p.
- Y. Wang, K. Jiang, J. Zhu, L. Zhang and H. Lin, A FRET-Based Carbon Dot–MnO2 Nanosheet Architecture for Glutathione Sensing in Human Whole Blood Samples, Chem. Commun., 2015, 51(64), 12748–12751, 10.1039/C5CC04905A.
- K. M. Park, J. Murray and K. Kim, Ultrastable Artificial Binding Pairs as a Supramolecular Latching System: a Next Generation Chemical Tool for Proteomics, Acc. Chem. Res., 2017, 50(3), 644–646, DOI:10.1021/acs.accounts.6b00645.
- N. Basílio, L. Cruz, V. de Freitas and F. Pina, A Multistate Molecular Switch Based on the 6,8-Rearrangement in Bromo-Apigeninidin Operated with PH and Host–Guest Inputs, J. Phys. Chem. B, 2016, 120(29), 7053–7061, DOI:10.1021/acs.jpcb.6b03694.
- R. T. Bronson, M. Montalti, L. Prodi, N. Zaccheroni, R. D. Lamb, N. K. Dalley, R. M. Izatt, J. S. Bradshaw and P. B. Savage, Origins of “on-off” Fluorescent Behavior of 8-Hydroxyquinoline Containing Chemosensors, Tetrahedron, 2004, 60(49), 11139–11144, DOI:10.1016/j.tet.2004.08.062.
- Z. Liu, Y. Qi, C. Guo, Y. Zhao, X. Yang, M. Pei and G. Zhang, Novel Fluorescent Sensors Based on Benzimidazo[2,1-a]Benz[de]Isoquinoline-7-One-12-Carboxylic Acid for Cu2+, RSC Adv., 2014, 4(100), 56863–56869, 10.1039/C4RA12242A.
- K. Velmurugan, S. Mathankumar, S. Santoshkumar, S. Amudha and R. Nandhakumar, Specific Fluorescent Sensing of Aluminium Using Naphthalene Benzimidazole Derivative in Aqueous Media, Spectrochim. Acta, Part A, 2015, 139, 119–123, DOI:10.1016/j.saa.2014.11.103.
- H. Goh, Y. G. Ko, T. K. Nam, A. Singh, N. Singh and D. O. Jang, A Benzimidazole-Based Fluorescent Chemosensor for Cu2+ Recognition and Its Complex for Sensing H2PO4− by a Cu2+ Displacement Approach in Aqueous Media, Tetrahedron Lett., 2016, 57(39), 4435–4439, DOI:10.1016/j.tetlet.2016.08.074.
- D. Das, K. I. Assaf and W. M. Nau, Applications of Cucurbiturils in Medicinal Chemistry and Chemical Biology, Front. Chem., 2019, 7, 619, DOI:10.3389/fchem.2019.00619.
- Z. Li, S. Sun, F. Liu, Y. Pang, J. Fan, F. Song and X. Peng, Large Fluorescence Enhancement of a Hemicyanine by Supramolecular Interaction with Cucurbit[6]uril and Its Application as Resettable Logic Gates, Dyes Pigm., 2012, 93, 1401–1407, DOI:10.1016/j.dyepig.2011.10.005.
- U. Pischel, V. D. Uzunova, P. Remón and W. M. Nau, Supramolecular Logic with Macrocyclic Input and Competitive Reset, Chem. Commun., 2010, 46(15), 2635–2637, 10.1039/B927595A.
- N. Saleh, Y. Al-Soud, L. Al-Kaabi, I. Ghosh and W. Nau, A Coumarin-Based Fluorescent PET Sensor Utilizing Supramolecular pKa Shifts, Tetrahedron Lett., 2011, 52, 5249–5254 CrossRef CAS.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Cucurbituril-Based Molecular Recognition, Chem. Rev., 2015, 115(22), 12320–12406, DOI:10.1021/acs.chemrev.5b00341.
- N. Al-Dubaili, K. El-Tarabily and N. Saleh, Host–Guest Complexes of Imazalil with Cucurbit[8]uril and β-Cyclodextrin and Their Effect on Plant Pathogenic Fungi, Sci. Rep., 2018, 8(1), 2839, DOI:10.1038/s41598-018-21156-9.
- K. I. Assaf and W. M. Nau, Cucurbiturils: from Synthesis to High-Affinity Binding and Catalysis, Chem. Soc. Rev., 2015, 44(2), 394–418, 10.1039/C4CS00273C.
- S. Zhang, K. I. Assaf, C. Huang, A. Hennig and W. M. Nau, Ratiometric DNA Sensing with a Host–Guest FRET Pair, Chem. Commun., 2019, 55(5), 671–674, 10.1039/C8CC09126A.
- A. Sohail, M. A. Alnaqbi and N. Saleh, Alginate/Cucurbit[7]uril/Dequalinium-Based Supramolecular Carbohydrates: modulation of FRET Signals by Temperature Control, Macromolecules, 2019, 52(22), 9023–9031, DOI:10.1021/acs.macromol.9b01788.
- A. Singh, W.-T. Yip and R. L. Halterman, Fluorescence-On Response via CB7 Binding to Viologen–Dye Pseudorotaxanes, Org. Lett., 2012, 14(16), 4046–4049, DOI:10.1021/ol300963c.
- M. A. Alnajjar, J. Bartelmeß, R. Hein, P. Ashokkumar, M. Nilam, W. M. Nau, K. Rurack and A. Hennig, Rational Design of Boron–Dipyrromethene (BODIPY) Reporter Dyes for Cucurbit[7]uril, Beilstein J. Org. Chem., 2018, 14, 1961–1971, DOI:10.3762/bjoc.14.171.
- A. Barba-Bon, Y.-C. Pan, F. Biedermann, D.-S. Guo, W. M. Nau and A. Hennig, Fluorescence Monitoring of Peptide Transport Pathways into Large and Giant Vesicles by Supramolecular Host–Dye Reporter Pairs, J. Am. Chem. Soc., 2019, 141(51), 20137–20145, DOI:10.1021/jacs.9b09563.
- T. Yin, S. Zhang, M. Li, C. Redshaw and X.-L. Ni, Macrocycle Encapsulation Triggered Supramolecular pKa Shift: a Fluorescence Indicator for Detecting Octreotide in Aqueous Solution, Sens. Actuators, B, 2018, 281, DOI:10.1016/j.snb.2018.10.136.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, Excited-State Processes in 8-Hydroxyquinoline: photoinduced Tautomerization and Solvation Effects, J. Phys. Chem. B, 1997, 101(39), 7786–7793, DOI:10.1021/jp971293u.
- A. J. Selinger and D. H. Macartney, Cucurbit[7]uril Complexations of Good’s Buffers, RSC Adv., 2017, 7(67), 42513–42518, 10.1039/C7RA08865E.
- Y. Cao, S. Paudel, X. Zhang, K. M. Kim and S. H. Cheon, Synthesis and Evaluation of Arylpiperazine-Reverse Amides as Biased Dopamine D3 Receptor Ligands, Bioorg. Med. Chem., 2015, 23(17), 5264–5272, DOI:10.1016/j.bmc.2015.07.072.
- Y.-M. Zhang, Y. Chen, Z.-Q. Li, N. Li and Y. Liu, Quinolinotriazole-β-Cyclodextrin and Its Adamantanecarboxylic Acid Complex as Efficient Water-Soluble Fluorescent Cd2+ Sensors, Bioorg. Med. Chem., 2010, 18(4), 1415–1420, DOI:10.1016/j.bmc.2010.01.024.
- E. Y. Chernikova, S. V. Tkachenko, O. A. Fedorova, A. S. Peregudov, I. A. Godovikov, N. E. Shepel, S. Minkovska, A. Kurutos, N. Gadjev, T. G. Deligeorgiev and Y. V. Fedorov, Multistep Assembling via Intermolecular Interaction between (bis)Styryl Dye and Cucurbit[7]uril: spectral Effects and Host Sliding Motion, Dyes Pigm., 2016, 131, 206–214, DOI:10.1016/j.dyepig.2016.04.013.
- S. Sinn, J. Krämer and F. Biedermann, Teaching Old Indicators Even More Tricks: binding Affinity Measurements with the Guest-Displacement Assay (GDA), Chem. Commun., 2020, 56(49), 6620–6623, 10.1039/D0CC01841D.
- A. T. Fenley, N. M. Henriksen, H. S. Muddana and M. K. Gilson, Bridging Calorimetry and Simulation through Precise Calculations of Cucurbituril–Guest Binding Enthalpies, J. Chem. Theory Comput., 2014, 10(9), 4069–4078, DOI:10.1021/ct5004109.
- J. Cho, J. Kwak, J. Oh, D. Kim and S.-Y. Lee, Photoinduced Intermolecular Electron Transfer Mediated by the Colloidal Tyrosyl Bolaamphiphile Assembly, ChemPhysChem, 2018, 19(5), 643–650, DOI:10.1002/cphc.201701099.
- A.-L. Li, Z.-L. Wang, W.-Y. Wang, Q.-S. Liu, Y. Sun and W. Gu, A novel dehydroabietic acid-based turn-on fluorescent probe for the detection of bisulfite and its application in live-cell and zebrafish imaging, New J. Chem., 2021, 45, 16822–16832, 10.1039/D1NJ02959B.
- A. Zheng, H. Liu, C. Peng, X. Gao, K. Xu and B. Tang, A mitochondria-targeting near-infrared fluorescent probe for imaging hypochlorous acid in cells, Talanta, 2021, 226, 122152, DOI:10.1016/j.talanta.2021.122152.
- P. Krawczyk, Optimizing the optical and biological properties of 6-(1H-benzimidazole)-2-naphthalenol as a fluorescent probe for the detection of thiophenols: a theoretical study, RSC Adv., 2020, 10, 24374, 10.1039/d0ra04835f.
- D. Zhang, A. Liu, R. Ji, J. Dong and Y. Ge, A mitochondria-targeted and FRET-based ratiometric fluorescent probe for detection of SO2 derivatives in water, Anal. Chim. Acta, 2019, 1055, 133–139, DOI:10.1016/j.aca.2018.12.042.
Footnote |
| † Electronic supplementary information (ESI) available: Animation of an 8 ns-long MD trajectory of the CAD/CB7 complex in explicit water with 10 ps between snapshots (only the relevant nearby water molecules are shown for clarity), and Cartesian coordinates of all relaxed structures used in the calculations. See DOI: 10.1039/d1nj04998d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |