
A molecular electron density theory study of the higher-order cycloaddition reactions of tropone with electron-rich ethylenes. The role of the Lewis acid catalyst in the mechanism and pseudocyclic selectivity†
Luis R.
Domingo
 *a and
Patricia
Pérez
b
*a and
Patricia
Pérez
b
aDepartment of Organic Chemistry, University of Valencia, Dr Moliner 50, 46100 Burjassot, Valencia, Spain. E-mail: luisrdomingo@gmail.com
bUniversidad Andres Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Computational and Theoretical Chemistry Group, Av. República 498, 8370146, Chile
First published on 29th November 2021
Abstract
The higher-order cycloaddition reactions of tropone with nucleophilic ethylenes, in the absence and presence of Lewis acid (LA) catalysts, have been studied within Molecular Electron Density Theory (MEDT) at the ωB97X-D/6-311G(d,p) and B3LYP-D3BJ/6-311G(d,p) computational levels. The strong electrophilic character of tropone, enhanced by the presence of LAs, allows its participation in polar cycloaddition reactions of reverse electron density flux (REDF) towards nucleophilic ethylenes. Analysis of the Parr functions indicates that the C2 and the C4 positions of tropone are the most electrophilic centers. These polar higher-order cycloaddition reactions take place via a non-concerted two-stage one-step or a two-step mechanism, yielding only one cycloadduct via a total regio and pseudocyclic selectivity. The present MEDT study allows establishing that these higher-order cycloaddition reactions are kinetically controlled by nucleophilic/electrophilic interactions taking place at the polar transition state structures (TSs). LAs not only accelerate the reaction and make it completely regioselective but also determine the pseudocyclic selectivity yielding exclusively [4+2] or [8+2] cycloadducts, which depends on a series of weak attractive/repulsive intramolecular electronic interactions present at the corresponding diastereoisomeric TSs.
1. Introduction
One of the most important classes of organic reactions is the cycloaddition reactions permitting the construction of any cyclic compound with high stereo and regioisomeric features.1,2 Cyclic compounds, from the smaller cyclopropane 3 to higher membered cyclic compounds, can be easily obtained via cycloaddition reactions (see Scheme 1). The more common cycloaddition reactions are the [4+2] cycloaddition or Diels-Alder3 reactions and the [3+2] cycloaddition reactions.4 Cycloaddition reactions are classified by the expression [m+n], where m and n are the numbers of centers of the reagents involved in the cycloaddition reaction. | ||
| Scheme 1 Formation of three – six membered cyclic compounds by cycloaddition reactions. | ||
While the n = 2 component should be an ethylene derivative, usually an electrophilic ethylene, the m component may vary from a carbene, m = 1, to a conjugated diene, m = 4. Interestingly, the three-atom-components (TACs) 7 participating in [3+2] cycloaddition reactions, m = 3, can present a diversity of electronic structures such as pseudoradical, carbenoid, or zwitterionic ones.5
The construction of higher cyclic compounds by use of conjugated trienes such as the tropone 11, a carbonyl derivative of cycloheptatriene 12, opens the possibility of several modes of cycloadditions such as the [4+2], [4+3], [4+4], [6+2] even [8+2] ones. In these higher-order cycloaddition reactions, which involve three or more double bonds allowing for the construction of medium-size rings,6 together with the feasible regio and stereoisomeric possibilities present in the reactions given in Scheme 1, the formation of different constitutional isomeric [m′+n′′] cycloadducts (CA) is feasible. Thus, the reaction of tropone 11 with dimethylfulvene 13, provides the formation of three different CAs (see Scheme 2).7,8 While CAs 14 and 15 are a pair of constitutional isomers; the major product 16 is obtained via a domino reaction involving the second addition of tropone 11 to CA 15. Thus, higher-order cycloaddition reactions can occur through different reaction paths involving different molecular mechanisms.
 | ||
| Scheme 2 Reaction of tropone 11 with dimethylfulvene 13. | ||
Cycloaddition reactions can be classified as non-polar and polar reactions.9 While non-polar cycloaddition reactions do not take place easily in the laboratory, the feasibility of cycloaddition reactions is dependent on the polar character of the reaction; when more polar the reaction, more rapid is.9 The polar character of the cycloaddition reactions depends on the electrophilic/nucleophilic interactions taking place at the transition state structures (TSs).5,9 In this sense, the use of conceptual DFT (CDFT) indices,10,11 as the electrophilicity ω12 and nucleophilicity N13 indices, have become powerful tools to study polar cycloaddition reactions. Besides, analysis of the Parr functions14 characterizing the most electrophilic and nucleophilic centers of a molecule allows the study of the regio and chemoselectivity in polar cycloaddition reactions.
In 1969, Woodward and Hoffmann classified a series of organic reactions involving unsaturated hydrocarbon compounds as “pericyclic”.15 They proposed that in these reactions, “all first order changes in bonding relationships take place in concert on a closed curve”.15 While the widely accepted “pericyclic mechanism” was never quantum chemically verified, many recent theoretical studies of “pericyclic reactions” based on the quantum chemical analysis of the bonding changes along the reaction paths have proven that they are non-concerted but sequential, and none in a “closed curve”, but symmetrically along a molecular plane.16 The name of pseudocyclic reaction was recently proposed to categorize these relevant organic reactions via pseudocyclic TSs.17
Unlike non-polar cycloaddition reactions taking place via low asynchronous TSs, many polar cycloaddition reactions involving asymmetric electrophilic species occur via highly asynchronous pseudocyclic TSs18 associated with a non-concerted two-stage one-step mechanism.19 In this mechanism, the formation of the second single bond begins at the end of the reaction path when the first single bond is already practically formed.19 This non-concerted mechanism proposed in 200819 contradicts Houk's definition made in 1995, which stated that “along a two-stage mechanism the formation of two bonds takes place in separate but overlapping processes”, thus proposing the name of “asynchronous concerted process”.20
The highly asynchronous TSs associated with these polar cycloaddition reactions are characterized by a two-center interaction between the most electrophilic and the most nucleophilic centers of the interacting species. The other two centers involved in the ring closure process do not participate in the first stage of the reaction. Consequently, several constitutional isomeric [m+n] CAs can be obtained along the second stage of the reaction after formation of several diastereoisomeric TSs.
Houk et al. defined in 1970 the “periselectivity” concept as the selective formation of one of the “thermally allowed pericyclic reaction products”.21,22 However, as the “pericyclic mechanism” has been ruled out,16 the term “periselectivity” has lost its meaning. To provide a more precise definition of the selectivity in the formation of constitutional isomers resulting from competitive pseudocyclic reactions, the “pseudocyclic selectivity” concept was recently proposed,23 and it is very frequently found in higher-order cycloaddition reactions.
Tropone 11 participates in higher-order cycloaddition reactions towards ethylene and diene derivatives yielding a variety of isomeric constitutional products (see Scheme 2). Thus, in 1969, Kitahara et al.24 reported the cycloaddition reactions of tropone 11 with enamines 17, one of the most nucleophilic ethylenes (see Scheme 3). Interestingly, depending on the structure of enamines 17, two different constitutional isomers, a [4+2] CA 19 or a [8+2] CA 20, were experimentally obtained with high yield.24 The authors proposed a mechanism for these polar cycloaddition reactions involving a zwitterionic species 18 (see Scheme 3).
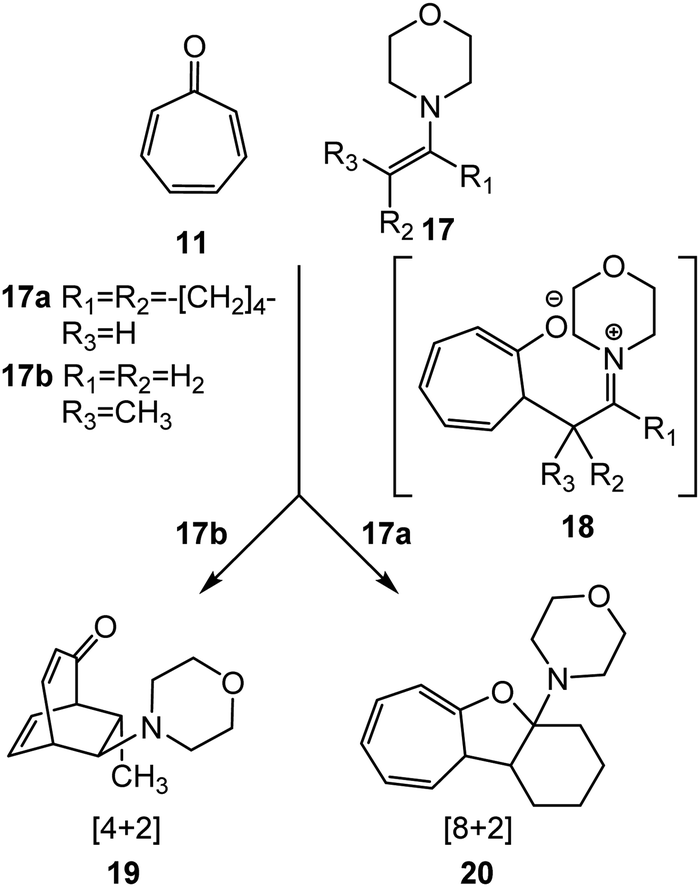 | ||
| Scheme 3 Cycloaddition reactions of tropone 11 with enamines 17. | ||
Polar cycloaddition reactions involving electrophilic unsaturated compounds are accelerated in the presence of Lewis acid (LA) catalysts.18 Coordination of the LA to the electrophilic species, usually a carbonyl derivative, notably increases the electrophilic character of the corresponding LA complex, thus increasing the reaction rate markedly due to the increase of the global electron density transfer25 (GEDT) at the TS, which plays a decisive role in the reaction rate.26
In 2009, Yamamoto et al. reported the LA catalyzed cycloaddition reactions of electrophilic tropone 11 with nucleophilic ethylenes 21 and 24 (see Schemes 4 and 5).27 Thus, when tropone 11 was treated with ethyl vinyl ether 21, in the presence of B(C6F5)3 as LA catalyst, a mixture of the two regioisomeric [4+2] CAs 22 and 23 were obtained.
 | ||
| Scheme 4 LA catalyzed cycloaddition reaction of tropone 11 with ethyl vinyl ether 21. | ||
 | ||
| Scheme 5 Pseudocyclic selectivities promoted by LA catalyzed cycloaddition reactions of tropone 11 with ketene diethyl acetal 24. | ||
When the ketene diethyl acetal 24, a more nucleophilic ethylene than 21, was used in the presence of B(C6F5)3 as LA catalyst, only the [4+2] cycloadduct 25 was obtained (see Scheme 5). Interestingly, the use of other LA catalysts such as BF3·OEt2 yielded the constitutional isomeric [8+2] CA 26.27 This finding indicated that the LAs not only accelerate the cycloaddition reactions, but they can also change pseudocyclic selectivity of the reaction.
Higher-order cycloaddition reactions of tropone 11 with ethylenes have been less theoretically studied than those involving high conjugated systems as dimethylfulvene 13 (see Scheme 2).6,7,28–33 The Woodward–Hoffmann's symmetry rules,15 and the Frontier Molecular Orbital34 (FMO) theory have been widely used to explain the course of higher-order cycloaddition reactions.28,32,33 In 2016, Domingo proposed the Molecular Electron Density Theory35 (MEDT) to study organic chemical reactivity. MEDT proposes that changes of electron density in an organic reaction, not MO interactions such as the FMO theory proposed,34 are responsible for the organic chemical reactivity. Within MEDT, a series of quantum chemical tools developed at the end of the last century, such as CDFT indices,12,13 Atom-in-Molecules36 (AIM) or Electron Localization Function37 (ELF), which permit the analysis of the molecular electron density, are used to study the structure and reactivity in Organic Chemistry.
Given the high regio and pseudocyclic selectivities found in the higher-order cycloaddition reactions of tropone 11 with nucleophilic ethylenes as 17, 21, and 24, in absence24 and the presence of LA catalysts,27 the higher-order cycloaddition reactions of tropone 11 with the cyclic ketene acetal (CKA) 27, in the absence and the presence of LA catalysts, is herein studied for the first time within MEDT35 (see Scheme 6). Note that while the [8+2] CA 29 is a constitutional isomer of the [4+2] CA 28, the [4+2] CA 30 is a regioisomer resulting from the nucleophilic attach on the C4 position of tropone 11. Rigid CKA 27 was chosen as a computational model of the experimental ketene diethyl acetal 24 used by Yamamoto et al.27 in order to avoid the different conformational possibilities resulting of the free C–C single bond rotation of the two ethyl chains present at diethyl acetal 24.
 | ||
| Scheme 6 Cycloaddition reactions of tropone 11 with CKA 27. The new single bonds in the CAs are represented in pink colors. | ||
2. Results and discussion
2.1. Analysis of the GS electronic structures of the reagents
ELF37,38 permits a quantitative characterization of the electron density distribution in a molecule, establishing a correlation between the electronic structure and the reactivity. Consequently, an ELF topological analysis of the electronic structure of tropone 11, tropone:BF3 complex 31, and CKA 27 was first performed. The ELF basin attractor positions and the most relevant valence basin populations of 11, 31 and 27, are shown in Fig. 1.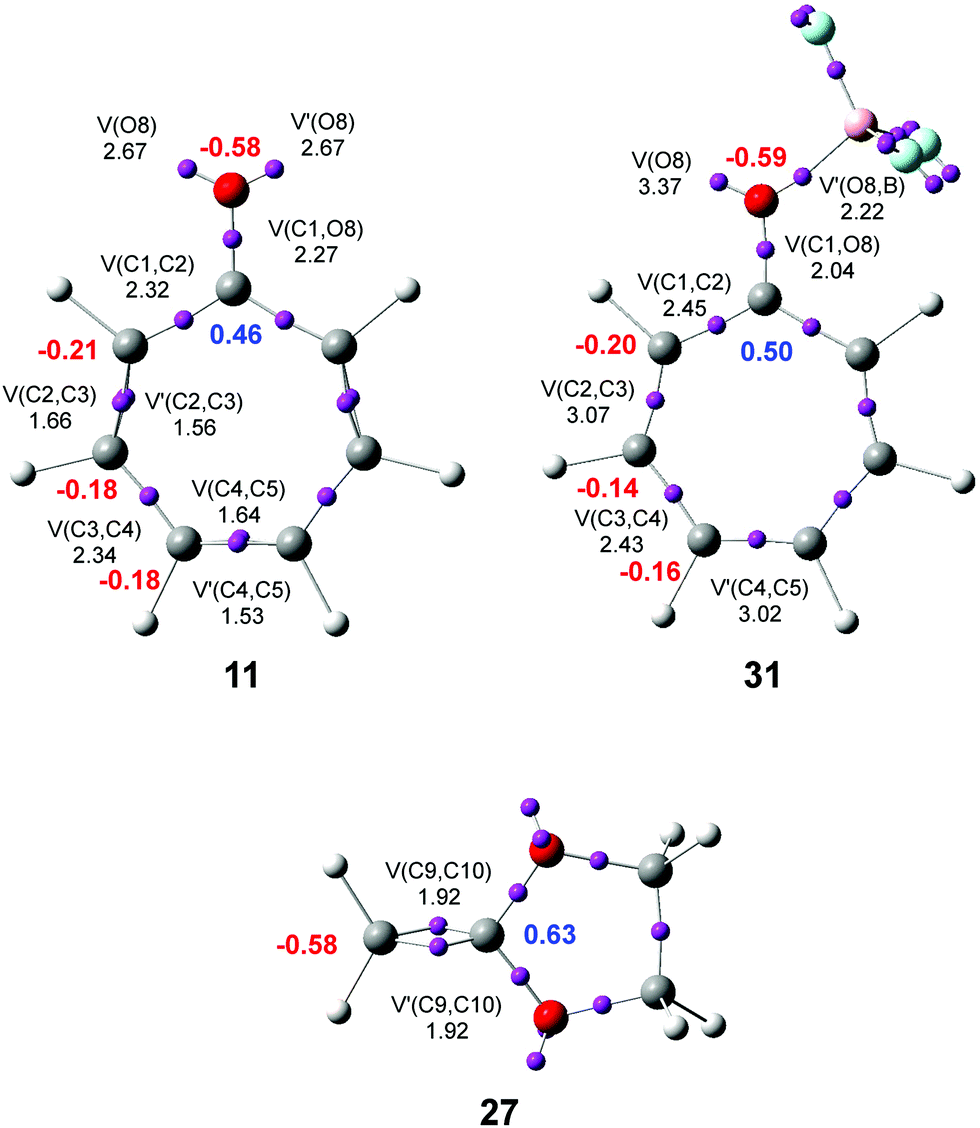 | ||
| Fig. 1 ELF basin attractor positions with the most relevant valence basin populations and the natural atomic charges of tropone 11, tropone:BF3 complex 31, and CKA 27. Valence basin populations and natural atomic charges are given in the average number of electrons e. Negative charges are colored in red, and positive charges in blue. | ||
ELF of tropone 11 shows the presence of one V(C1,C2) disynaptic basin, integrating 2.32 e, two disynaptic basins, V(C2,C3) and V′(C2,C3), integrating a total of 3.22 e, one V(C3,C4) disynaptic basin, integrating 2.34 e, two disynaptic basins, V(C4,C5) and V′(C4,C5), integrating a total of 3.17 e, one V(C1,O8) disynaptic basin, integrating 2.27 e, and two monosynaptic basins, V(O8) and V′(O8), integrating 2.67 e each one. Note that the C1–C7, C7–C6, and C6–C5 bonding regions of tropone 11 and tropone:BF3 complex 31 are symmetrical to the C1–C2, C2–C3, and C3–C4 ones.
While the V(C1,C2) and V(C3,C4) disynaptic basins are associated with Ci–Cj single bonds, the V(C2,C3), V′(C2,C3), V(C4,C5) and V′(C4,C5) disynaptic basins are associated with depopulated Ci–Cj double bonds. These behaviors are a consequence of the cyclic conjugation of the three Ci–Cj double bonds with the carbonyl C1 carbon in tropone 11. The V(C1,O8) disynaptic basin is associated with a significantly depopulated carbonyl C–O double bond, while the two V(O8) and V′(O8) monosynaptic basins are associated with the non-bonding electron density present at the O8 oxygen. These behaviors show the strong polarization of the electron density of the carbonyl C1–O8 group towards the highly electronegative O8 oxygen.
ELF of tropone:BF3 complex 31 shows the presence of three disynaptic basins, V(C1,C2), V(C2,C3), and V(C3,C4), integrating 2.45, 3.07 and 2.43 e, respectively, one V(C1,O8) disynaptic basin, integrating 2.04 e, one V(O8,B) disynaptic basin, integrating 2.22 e, and one V(O8) monosynaptic basins, integrating 3.37 e. At this LA complex, while the C2–C3 and C4–C5 double bonds have diminished their populations concerning tropone 11, the C1–C2 and C3–C4 single bonds have increased. These changes are a consequence of the increase of the conjugation at LA complex 31 due to the rise of the polarization of the carbonyl C1–O8 bond. On the order hand, the V(C1,O8) disynaptic basin population indicates that the C1–O8 bonding region has experienced slight depopulations, 0.23 e, induced by the new O8–B single bond.
ELF of CKA 27 shows the presence of two disynaptic basins, V(C9,C10) and V′(C9,C10), integrating a total of 3.84 e, associated with the C9–C10 double bond.
Finally, the natural atomic charges of tropone 11, tropone:BF3 complex 31, and CKA 27, obtained by a natural population analysis39,40 (NPA) were studied (see Fig. 1). Except for C1 carbon of tropone 11, which presents a high positive charge, +0.46 e, due to the strong polarization of the carbonyl C–O bond, the other six carbons of tropone 11 are negatively charged, between −0.18 to −0.22 e, as a consequence of the more electronegative character of the carbon than the hydrogens bound to them. The carbonyl O8 oxygen atom is highly negatively charged, −0.58 e. Coordination of the LA BF3 to the carbonyl O8 oxygen of tropone 11 enhances the polarization of the carbonyl C–O bond; therefore, the positive charge of the C1 carbon is increased to +0.50 e. This behavior decreases the negative charge of the other carbons slightly, between 0.01 and 0.04 e (see Fig. 1). The presence of the two oxygen in CKA 27 strongly polarizes the C9–C10 double bond so that while the ethylene C9 carbon is negatively charged by −0.58 e, the disubstituted C10 carbon is positively charged by +0.63.
It is worth emphasizing that the most electrophilic centers of tropone 11 and tropone:BF3 complex 31 are the C2 and C7 carbons (see later), which are the two carbons more negatively charged, −0.21 (11) and −0.20 (31) e. This finding supports Domingo's proposal made in 2014 that the local electrophilic/nucleophilic behaviors of organic molecules are not simply caused by charges.41,42 The most electrophilic center of a molecule is the one that accepts the highest amount of electron density resulting from the nucleophilic/electrophilic interactions taking place along the approach of the two reagents.41 Interesting, in some cases, as in tropone 11 and LA complex 31, the most electrophilic center is one negatively charged.41
2.2. Analysis of the CDFT reactivity indices of the reagents
Many studies devoted to polar reactions have shown that the analysis of the reactivity indices defined within the CDFT10,11 is a powerful tool to understand the reactivity in polar reactions. The CDFT indices were calculated at the B3LYP/6-31G(d) computational level since it was used to establish the electrophilicity and nucleophilicity scales.11 The global reactivity indices, namely, the electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and global nucleophilicity N, for the reagents involved in the reactions are gathered in Table 1.| μ | η | ω | N | |
|---|---|---|---|---|
| Tropone:B(C6F5)332 | −4.99 | 2.63 | 4.74 | 2.82 |
| Tropone:BF331 | −5.43 | 4.41 | 3.34 | 1.49 |
| Tropone 11 | −4.28 | 4.20 | 2.18 | 2.75 |
| Cycloheptatriene 12 | −3.28 | 5.02 | 1.07 | 3.33 |
| Ketene diethyl acetal 24 | −2.45 | 7.22 | 0.41 | 3.06 |
| Cyclic ketene acetal 27 | −1.94 | 7.30 | 0.26 | 3.53 |
The electronic chemical potentials10,43μ of the ketene acetals, μ = −1.94 (27) and −2.45 (24) eV, are higher than those of tropone 11, μ = −4.28 eV, and the tropone:LA complexes 31 and 32, μ = −5.43 (31) and −4.99 (32) eV (see Table 1). Consequently, along a polar reaction, the flux of the electron density will take place from ketene acetals 24 and 27 to tropone 11 and the tropone:LA complexes 31 and 32, these cycloaddition reactions being classified as reverse electron density flux (REDF).44
The electrophilicity ω12 and nucleophilicity N13 indices of cycloheptatriene 12, 1.07 and 3.33 eV, respectively, classify it as a moderate electrophile and a strong nucleophile within the electrophilicity and nucleophilicity scales.11 The electrophilicity ω index of tropone 11 is 2.18 eV, being classified as a strong electrophile within the electrophilicity scale. In turn, the nucleophilicity N index of tropone 11 is 2.75 eV, being classified as a moderate nucleophile within the nucleophilicity scale. Consequently, the inclusion of the carbonyl group in tropone 11 significantly increases its electrophilic character and decreases its nucleophilic character considerably concerning cycloheptatriene 12. This behavior allows the participation of tropone 11 as an electrophile in polar cycloadditions of REDF.
Coordination of the LAs to the O8 oxygen of tropone 11 increases the electrophilicity ω index of tropone:LA complexes to 3.34 (31) and 4.74 (32) eV, being classified as strong electrophiles. Interestingly, while the nucleophilicity N index of tropone:BF3 complex 31 decreases to 1.49 eV, being classified as a marginal nucleophile, that of tropone:B(C6F5)3 complex 33 slightly increases to 2.82 eV, respectively, being classified as moderate nucleophile.
On the other hand, the electrophilicity ω index of the ketene acetals 24 and 27, ω = 0.41 and 0.26 eV, respectively, classifies them as marginal electrophiles. In contrast, their high nucleophilicity N index, N = 3.06 and 3.53 eV, classifies them as strong nucleophiles. Note that the strain present in the CKA 27 increases its nucleophilic character concerning the experimental ketene acetal 24.
Consequently, along a polar cycloaddition reaction tropone 11 and the corresponding LA complex 31 and 32 will act as strong electrophiles, and the ketene acetals 24 and 27 as strong nucleophiles.
Along a polar reaction involving non-symmetric species, the most favorable reaction path involves the two-center interaction between the most electrophilic and the most nucleophilic centers of the reagents.45 Many studies have shown that the analysis of the electrophilic Pk+ and nucleophilic Pk− Parr functions,13 resulting from the excess of spin electron density gathered via the GEDT,25 is one of the most accurate and insightful tools for the study of the local reactivity in polar and ionic processes. Hence, the Pk+ electrophilic Parr functions of tropone 11 and tropone:BF3 complex 31, and Pk− nucleophilic Parr functions of CKA 27 are analyzed (see Fig. 2).
 | ||
| Fig. 2 3D representations of the Mulliken atomic spin densities of the radical anions of tropone 11 and the tropone:BF3 complex 31, and the radical cation of CKA 27, and the electrophilic Pk+ Parr functions of tropone 11 and the tropone:BF3 complex 31 and the nucleophilic Pk− Parr functions of CKA 27. | ||
Analysis of the electrophilic Pk+ Parr functions at tropone 11 indicates that symmetric C2 and C7 carbons are the most electrophilicity centers, Pk+ = 0.37, followed by the symmetric C4 and C5 carbons, Pk+ = 0.23. Coordination of the BF3 LA to the O8 oxygen of tropone 11 does not substantially modify the electrophilic Pk+ Parr functions of the tropone:BF3 complex 31 (see Fig. 2).
On the other hand, the non-substituted carbon of CKA 27 is the most nucleophilic center of this species. Pk− = 0.73. Note that the two oxygens of CKA 27 are only slightly nucleophilically activated Pk− = 0.14. (see Fig. 2).
Consequently, along the nucleophilic attacks of CKA 27 to tropone 11 or to the tropone:BF3 complex 31, the most favorable two-center interaction will take place between the C9 carbon of CKA 27 and the C2 carbon of tropone 11 or the LA complex 31.
2.3. Study of the cycloaddition reaction of tropone 11 with CKA 27
First, the non-catalyzed cycloaddition reaction of tropone 11 with CKA 27 was studied. Three competitive reaction paths were explored: (i) reaction path A, which is associated with a formal [4+2] cycloaddition, begins through the nucleophilic attack of CKA 27 on the C2 carbon of tropone 11, and ends by the ring-closure at the C5 carbon; (ii) reaction path B, which is associated with a formal [8+2] cycloaddition, also begins through the nucleophilic attack of CKA 27 on the C2 carbon of tropone 11, but ends by the ring-closure at the carbonyl O8 oxygen; and (iii) reaction path C, which is associated with a formal [4+2] cycloaddition, begins through the nucleophilic of CKA 27 on the C4 carbon of tropone 11, and finishes by the ring-closure at the C7 carbon. Reaction paths A and B are related to two pseudocyclic isomeric reaction paths, while reaction paths A and C are related to two regioisomeric reaction paths. Note that the three reaction paths begin by the nucleophilic attack of CKA 27 on the two most electrophilic centers of tropone 11, the C2 and C4 carbons. Analysis of the stationary points associated with the three reaction paths indicates that while the formation of formal [4+2] CA 28 takes place through a two-step mechanism, the formation of formal [8+2] CA 29 and the formal [4+2] CA 30 takes place through a one-step mechanism (see Scheme 7). The relative energies, in gas phase and DCM, are given in Table 2, while the total energies are given in Table S1 in ESI.†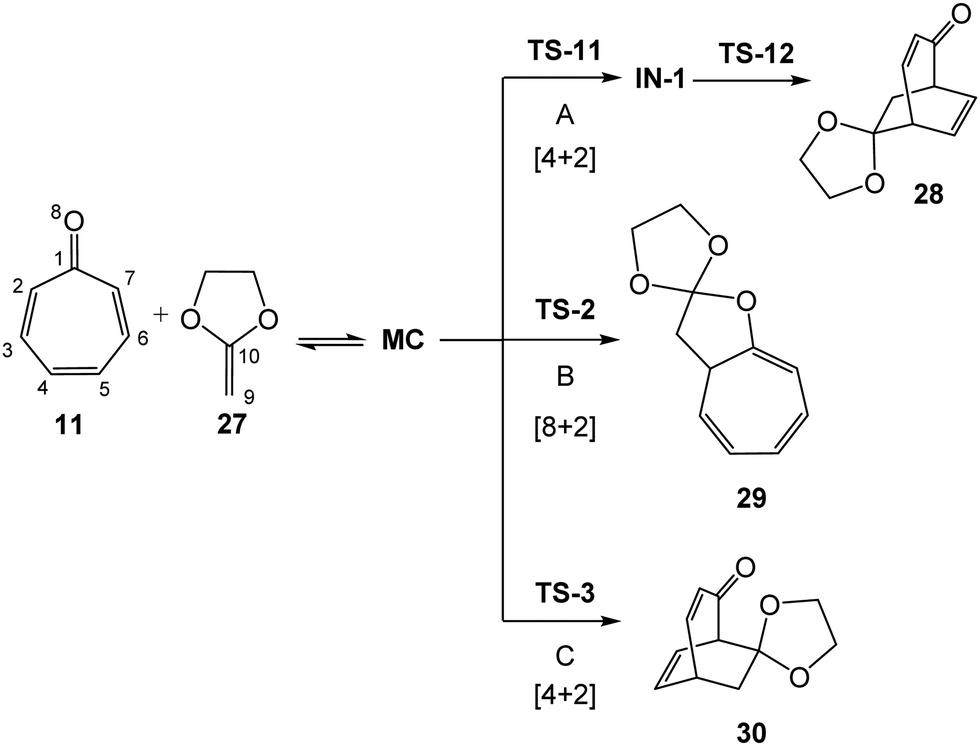 | ||
| Scheme 7 Competitive reaction paths associated with the cycloaddition reaction of tropone 11 with CKA 27. | ||
| ωB97X-D | B3LYP-D3BJ | ||
|---|---|---|---|
| Gas phase | DCM | ||
| MC | −6.6 | −6.0 | −6.1 |
| TS-11 | 13.2 | 12.1 | 10.1 |
| IN-1 | 12.7 | 6.1 | 8.8 |
| TS-12 | 13.4 | 7.1 | 9.1 |
| TS-2 | 11.6 | 13.3 | 9.8 |
| TS-3 | 15.9 | 15.5 | 14.6 |
| 28 | −28.9 | −26.2 | −17.5 |
| 29 | −30.0 | −25.9 | −18.2 |
| 30 | −29.6 | −26.6 | −17.7 |
Analysis of the potential energy surface (PES) permits characterizing a series of molecular complexes (MCs) in an early stage of the cycloaddition reaction in which weak intermolecular interactions already link the two reagents. The most stable MC, located 6.6 kcal mol−1 below the separated reagents, was selected (see Table 2). The geometry of MC is shown in Fig. S1 in ESI.† The distance between the two frameworks at MC is ca. 3.3 Å.
The gas phase activation energies associated with the nucleophilic attacks of CKA 27 to tropone 11 are found between 11.6 and 15.9 kcal mol−1, the reaction being exothermic by more than 29 kcal mol−1. Some appealing conclusions can be obtained from the energy results given in Table 2: (i) the most favorable reaction path corresponds to the [8+2] reaction path B, associated with the attack of CKA 27 on the C2 position of tropone 11via the TS-2, 11.6 kcal mol−1; (ii) in gas phase, this cycloaddition reaction presents a high [8+2] pseudocyclic selectivity18 as TS-11 is found 1.6 kcal mol−1 above TS-2. Note that TS-11 and TS-2 are two stereoisomeric pseudocyclic TSs yielding two constitutional isomeric CAs; (iii) this cycloaddition reaction presents total regioselectivity as TS-3 is found 4.3 kcal mol−1 above TS-2. Note that TS-2 and TS-3 comes the nucleophilic attach to the C2 and C4 position of tropone 11; (iv) in gas phase, the PES at the TSs and intermediate involved in the reaction path A is very flat; TS11, IN-1, and TS12 are found in the short range of energies of 0.7 kcal mol−1; and (v) formation of CAs 28–30 presents a very similar exothermic character, ca. 30 kcal mol−1.
Inclusion of solvent effects of DCM stabilizes all species between 6 and 15 kcal mol−1. Interestingly, the more stabilized species are the TSs due to their polar character (see later). Due to its zwitterionic character, IN-1 is located 6.0 kcal mol−1 below TS-11. TS-11 is 1.2 kcal mol−1 more stabilized than TS-2; consequently, in DCM, there is a change of the pseudocyclic selectivity as the formation of [4+2] CA 28viaTS-11 is 1.2 kcal mol−1 lesser in energy than the formation of [8+2] CA 29viaTS-2. It is worth mentioning that, in solution, the cycloaddition reaction presents total regioselectivity as TS-3 is found 3.4 kcal mol−1 above TS-11.
All stationary points associated with the cycloaddition reaction of tropone 11 with CKA 27 were subsequently optimized at the B3LYP-D3BJ/6-311G(d,p) computational level in DCM. The relative energies as given in Table 2, while the total energies are given in Table S1 in ESI.† The B3LYP-D3BJ calculations decrease the activation energy of the most favorable reaction path B viaTS-2 to 9.8 kcal mol−l, and decreases the exothermic character of the reaction to −18.2 kcal mol−l. However, the more appealing result by using the B3LYP-D3BJ calculations is that the cycloaddition reaction becomes slightly [8+2] pseudocyclic selective as TS-11 associated with the formal [4+2] cycloaddition mode A, is found 0.3 kcal mol−1 above TS-2 associated with the formal [8+2] cycloaddition mode B, and completely regioselective as TS-3 associated with the formal [4+2] cycloaddition mode C, is found 4.8 kcal mol−1 above TS-2.
The geometries of the TSs involved in the cycloaddition reaction of tropone 11 with CKA 27 are given in Fig. 3. At the TSs involved in the nucleophilic attack of CKA 27 to tropone 11, the C–C distances involving the most nucleophilic C9 carbon of CKA 27, 1.865 Å (TS-11), 1.917 Å (TS-2), and 1.877 Å (TS-3), are shorter than those involving the C10 carbon, 3.748 Å (TS-11), 2.808 Å (TS-2) and 3.393 Å (TS-3). These distances at TS-2 and TS-3 indicate that these TSs are associated with a highly asynchronous C–C single bond formation process. At TS-12, while the C2–C9 distance, 1.616 Å, indicates that the C2–C9 single bond is practically formed, the C5–C10 distance, 2.961 Å, suggests the formation of the C5–C10 single bond is very delayed. Note that the formation of the C–C single bonds takes place in the short-range of 2.0–1.9 Å.25 The very high asynchronicity found at TS-2 and TS-3 in the C–C single bond formation found at the two computational levels indicates that the B and C reaction paths are associated with a non-concerted two-stage one-step mechanism.19 Note the great similitude between the C–C distances of the regioisomeric TS-11 and TS-3. In DCM, the distances between the two interacting centers increase slightly. The electronic stabilization of the TSs makes them slightly earlier. Despite that, no remarkable geometrical changes are observed.
 | ||
| Fig. 3 ωB97X-D/6-311G(d,p) Geometries of the TSs involved in the cycloaddition reaction of tropone 11 with CKA 27. Distances in DCM are given in parentheses, and the B3LYP-D3BJ/6-311G(d,p) distances in DCM are given in square brackets. Distances are given in Angstroms. | ||
The B3LYP-D3BJ/6-311G(d,p) parameters in DCM are also given in Fig. 3. The B3LYP-D3BJ geometries of the TSs are very similar to those obtained using the ωB97X-D functional. The C2–C9 distances are slightly shorter, indicating that the B3LYP-D3BJ calculations yield TSs slightly more advanced.
TS-11 and TS-2 are a pair of diastereoisomeric TSs resulting from two different approach modes of the ethylene framework to the tropone one along with the C2–C9 single bond formation (see Fig. 4). The main geometrical difference between them is the C1–C2–C9–C10 dihedral angle values, −71.9 degrees at TS-11 and 41.3 degrees at TS-2 (see Fig. 4). Interestingly, the different geometries of these diastereoisomeric TSs are determined by the alternate conformation of the two interacting trigonal planar C2 and C9 carbons along with the two approach modes. Two constitutionals isomeric CAs related with two formal [4+2] and [8+2] cycloaddition reactions, 28 and 29, are obtained along the two competitive reaction paths. The selectivity involved in these two competitive reaction paths was recently defined as pseudocyclic selectivity,23 as the two constitutional isomeric CAs come from two stereoisomeric pseudocyclic TSs.
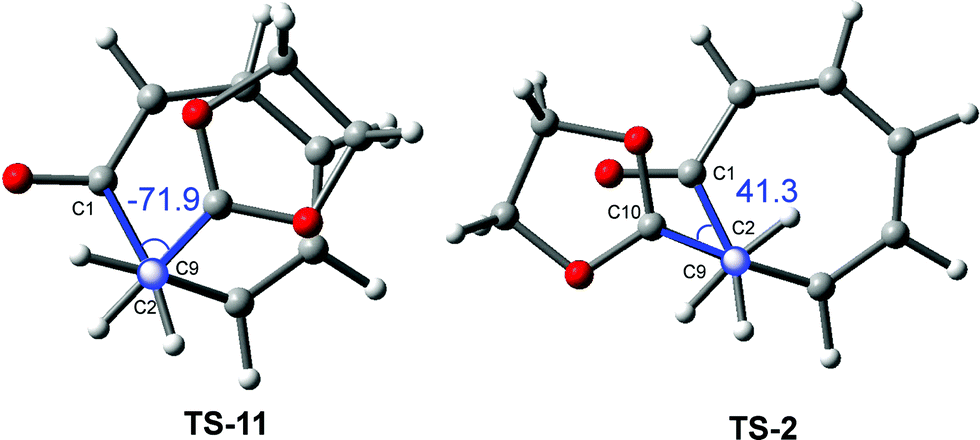 | ||
| Fig. 4 View of TS-11 and TS-2 through the C2–C9 single bond formation. The C1–C2–C9–C10 dihedral angles are given in degrees. | ||
Analysis of GEDT at TSs and the intermediate associated with the three reaction paths permits assessment of the polar character of this cycloaddition reaction. GEDT values lower than 0.05 e correspond with non-polar processes, while values higher than 0.20 e correspond with polar processes. The GEDT values at the TSs and intermediate are 0.50 e at TS-11, 0.66 e at IN-1, 0.64 e at TS-12, 0.47 e at TS-2, and 0.44 e at TS-3. Consequently, this cycloaddition reaction has a very high polar character, with the electron density fluxing from CKA 27 to tropone 11; the reaction is classified as REDF,44 in complete agreement with the CDFT analysis. Along the first step of the two-step mechanism, the GEDT increases until it reaches the maximum value at intermediate IN-1, 0.66 e, while along the second step, there is a decrease of the GEDT because of a retro-donation process. Note that zwitterionic IN-1 corresponds with the zwitterionic intermediate 18 proposed by Kitahara et al. in the cycloaddition reactions of tropone 11 with enamines 17 (see 18 in Scheme 3).24 Interestingly, the GEDT at TS-11, 0.50 e, is slightly higher than that at TS-2, 0.47; this behavior explains that in DCM, the reaction path A will be the most favorable at the ωB97X-D/6-311G(d,p) level.
Finally, to assert the nucleophilic ethylene CKA 27 as a valid computational model of the experimental ketene diethyl acetal 24, the three competitive reaction paths shown in Scheme 7 for the cycloaddition reaction of tropone 11 with ketene diethyl acetal 24 was studied in gas phase at the ωB97X-D/6-311G(d,p). Total and relative energies of the stationary points are shown in Table S2 in ESI,† while the TS geometries are shown in Fig. S2 in ESI.† A comparison of the relative energies given in Tables S1 and S2 in ESI,† and between the TS geometries given in Fig. 3 and Fig. S2 in ESI,† shows a high degree of similarity between the two cycloadditions involving ketene acetals 24 and 27, thus asserting the cyclic ketene acetal as a reliable computational model of the experimental one.
2.4. Study of the LA BF3 catalyzed cycloaddition reaction of tropone 11 with the CKA 27
Similar to the non-catalyzed reaction, three reaction paths, A, B, and C, yielding the [4+2], [8+2], and [4+2] CAs 33, 34, and 35, respectively, for the cycloaddition reaction of tropone:BF3 complex 31 with CKA 27 were studied (see Scheme 8). Analysis of the stationary points associated with the three reaction paths indicates that they take place through a one-step mechanism (see Scheme 8). Relative energies, in gas phase and DCM, are given in Table 3, while the total electronic energies are given in Table S3 in ESI.† | ||
| Scheme 8 Competitive reaction paths associated with the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. | ||
| ωB97X-D | B3LYP-D3BJ | ||
|---|---|---|---|
| Gas phase | DCM | ||
| MC-BF | −13.5 | −8.9 | −9.5 |
| TS-BF-1 | 4.2 | 3.2 | 1.2 |
| TS-BF-2 | −4.6 | 1.2 | −2.2 |
| TS-BF-3 | 1.2 | 3.1 | 1.2 |
| 33 | −25.0 | −19.5 | −10.6 |
| 34 | −21.3 | −17.3 | −11.5 |
| 35 | −25.9 | −20.0 | −10.8 |
Analysis of the PES permits characterizing a series of MCs in the first stage of the reaction. The most stable of them, MC-BF, which is found 13.5 kcal mol−1 below the separated reagents, was selected (see Scheme 8). The geometry of MC-BF is shown in Fig. S1 in ESI.† The distance between the two frameworks at MC-BF is ca. 3.4 Å.
The relative energies of the TSs with respect to the separated reagents are found between −4.6 to 4.2 kcal mol−1, the reaction being exothermic by more than 21 kcal mol−1. Some appealing conclusions can be obtained from the relative energies given in Table 3: (i) the most favorable TS-BF-2, associated with the formal [8+2] cycloaddition mode, is located on the PES 4.6 kcal mol−1 below the separated reagents. This behavior is a consequence of the augmented electrophilic character of LA complex 31 with respect to that of tropone 11. However, if the formation of MC-BF is considered, TS-BF-2 presents activation energy of 8.9 kcal mol−1. Note that in gas phase, the activation barrier associated with TS-2 with respect MC is 18.2 kcal mol−1 (see Table 2); (ii) this LA catalyzed cycloaddition reaction is completely pseudocyclic selective as TS-BF-1, associated with the [4+2] cycloaddition mode A, is found 8.8 kcal mol−1 above TS-BF-2; (iii) this LA catalyzed cycloaddition reaction is completely regioselective as TS-BF-3, associated with the formal [4+2] cycloaddition mode C, is found 5.8 kcal mol−1 above TS-BF-2; both regio and pseudocyclic selectivities are in complete agreement with the experimental outcomes for the BF3 catalyzed cycloaddition reaction of ketene diethyl acetal 24 (see Scheme 5).27
The inclusion of solvent effect of DCM stabilizes all stationary points between 11 and 18 kcal mol−1 because of the polar character of all species involved in this reaction. Now, the reagents are more stabilized than TS-BF-2, and consequently, their relative energy becomes positive by 1.2 kcal mol−l (see Table 3). Accordingly, in DCM, the activation energy associated with the most favorable reaction path B related to the LA catalyzed process viaTS-BF-2 is 10.9 kcal mol−l lower in energy than that associated with the most favorable reaction path A associated with the non-catalyzed process viaTS-11. Like the non-catalyzed reaction, the poor solvation of TS-BF-2 with respect to the other two TSs markedly diminishes the selectivities. Despite this poor solvation, the reaction path B continues being the most favorable one.
All stationary points associated with LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 were subsequently optimized at the B3LYP-D3BJ/6-311G(d,p) computational level in DCM. The total and relative energies as given in Table 3, while the total energies are given in Table S3 in ESI.† The B3LYP-D3BJ calculations decrease the activation energy with respect MC-BF to 7.3 kcal mol−l. However, the more appealing result by using the B3LYP-D3BJ calculations is that this cycloaddition reaction becomes completely [8+2] pseudocyclic selective as TS-BF-1 associated with the formal [4+2] cycloaddition mode A, is found 3.4 kcal mol−1 above TS-BF-2 associated with the formal [8+2] cycloaddition mode B, and completely regioselective as TS-BF-3 associated with the formal [4+2] cycloaddition mode C, is found 3.4 kcal mol−1 above TS-BF-2, in complete agreement with the experimental results found in the BF3 catalyzed cycloaddition reaction of ketene diethyl acetal 24 (see Scheme 5).27 A comparative analysis of the ωB97X-D and the B3LYP-D3BJ relative energies in DCM shows a similar trend in both pseudocyclic selectivity and regioselectivity.
The ωB97X-D/6-311G(d,p) thermodynamic data of the LA BF3 catalyzed cycloaddition reaction of tropone 31 with CKA 27 were analyzed. The total and relative enthalpies, entropies, and Gibbs free energies are given in Table S4 in ESI.† A representation of the enthalpy and Gibbs free energy profiles associated with the three competitive reaction paths is given in Fig. 5. Inclusion of the thermal corrections to the electronic energies in DCM only increases the relative enthalpies by between 0.8 and 1.0 kcal mol−1 (see Table S4 in ESI†). A low incidence has in the relative enthalpies of the TSs, which increase between 0.6 and 1.0 kcal mol−1 with respect to the electronic energies in DCM. Considering the activation enthalpies, TS-BF-1 and TS-BF-3 are found ca. 1.8 kcal mol−1 higher in enthalpy than the TS-BF-2. Inclusion of the thermal corrections and entropies to enthalpies increases the relative Gibbs free energies by between 9.0 and 13.3 kcal mol−1 as a consequence of the unfavorable activation entropies associated with these bimolecular processes, which are found in the range −32.8 and −58.6 cal mol−1 K−1. Formation of MC-BF is endergonic by only 1.3 kcal mol−1. The activation Gibbs free energy associated with the LA BF3 catalyzed cycloaddition reaction of tropone 31 with CKA 27viaTS-BF-2 rises to 14.5 kcal mol−1, while the formation of CA 34 is endergonic by only −1.9 kcal mol−1. Considering the Gibbs free energies associated with TS-BF-1, TS-BF-2 and TS-BF-3 given in Fig. 5 the following relationship between the three isomeric CA can be estimated at 0 °C by using the Eyring–Polanyi equation:46 5.0 (34): 87.9 (35): 7.2 (36). Interestingly, when the Gibbs free energies were computed at the B3LYP-D3BJ/6-311G(d,p) computational level in DCM (see Table S5 in ESI†), a relationship of 0.0 (34): 99.9 (35): 0.0 (36) can be established, in complete agreement with the experimental outcomes found in the BF3 catalyzed cycloaddition reaction of ketene diethyl acetal 24.27
 | ||
| Fig. 5 ωB97X-D/6-311G(d,p) enthalpy, in blue, ΔH in kcal mol−1, and Gibbs free energy, in red, ΔG in kcal mol−1, profiles, in DCM at 0 °C, for the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. | ||
The ωB97X-D/6-311G(d,p) geometries of the TSs involved in the BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 are given in Fig. 6. At the TSs involved in the nucleophilic attack of CKA 27 to tropone:BF3 complex 31, the C–C distances involving the most nucleophilic C9 center of CKA 27, 2.051 Å (TS-BF-1), 2.125 Å (TS-BF-2), and 2.052 Å (TS-BF-3), are shorter than that involving the substituted C10 center, 4.048 Å (TS-BF-1), 2.912 Å (TS-BF-2) and 3.679 Å (TS-BF-3). These distances indicate that these TSs correspond with a high asynchronous single bond formation process associated with a non-concerted two-stage one-step mechanism.19 At the three TSs, the shorter C–C distance, which is higher than 2.05 Å, indicates that the corresponding C–C single bond has not begun yet.25 In DCM, the C2–C9 distances involving the most nucleophilic C9 center of CKA 27 are slightly increased, indicating that the TSs are somewhat earlier. Despite that, the inclusion of solvent effects does not produce any remarkable geometrical change. The B3LYP-D3BJ/6-311G(d,p) parameters in DCM are also given in Fig. 6. The B3LYP-D3BJ geometries of the TSs are very similar to those obtained using the ωB97X-D functional. The C2–C9 distances are slightly shorter, indicating that the B3LYP-D3BJ functional yields TSs slightly more advanced.
 | ||
| Fig. 6 ωB97X-D/6-311G(d,p) Geometries of the TSs involved in the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. Distances in DCM are given in parentheses, and the B3LYP-D3BJ/6-311G(d,p) distances in DCM are given in square brackets. Distances are given in Angstroms. | ||
The GEDT values at the three TSs are 0.40 e at TS-BF-1, 0.39 e at TS-BF-2, and 0.36 e at TS-3. Newly, these very high values indicate that this LA BF3 catalyzed cycloaddition reaction has a very high polar character, being classified as REDF.
2.5. Effects of the nature of the LA catalyst on the [4+2]/[8+2] pseudocyclic selectivity
Yamamoto et al.27 showed that the pseudocyclic selectivity yielding the constitutional isomeric [4+2] and [8+2] CAs depends on the nature of the LA used in the catalyzed cycloaddition reaction (see Scheme 5). Thus, contrary to the BF3 catalysts, which provided the [8+2] CA 26 with a yield >99%, the B(C6F5)3 catalyst provided the [4+2] CA 25 with a yield >99%.27 To evidence the change of the [4+2]/[8+2] pseudocyclic selectivity with the change of the LA used in the reaction, the competitive pseudocyclic selective A and B reaction paths associated with the B(C6F5)3 LA catalyzed cycloaddition reaction of tropone 11 with CKA 27 were studied (see Scheme 9). Relative energies, in gas phase and DCM, are given in Table 4. Total electronic energies are shown in Table S6 in ESI.†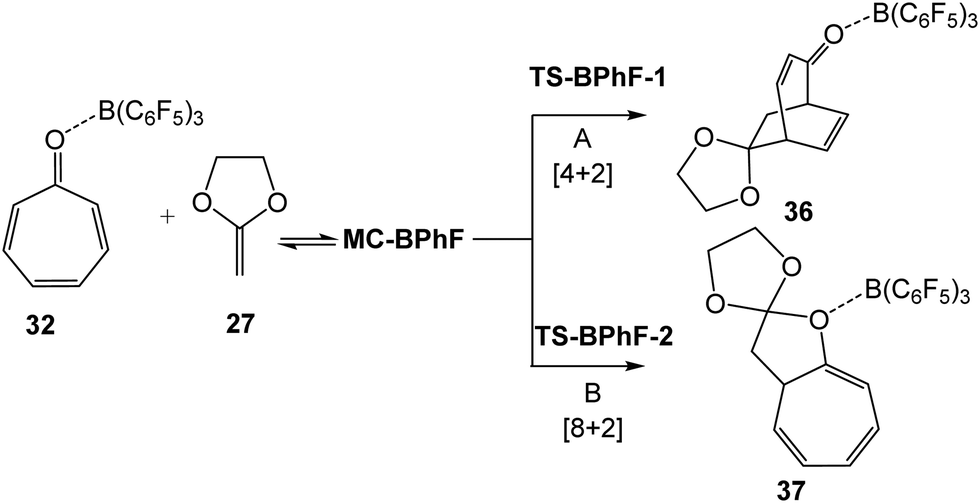 | ||
| Scheme 9 Competitive [4+2] and [8+2] reaction paths associated with the LA B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. | ||
| Gas phase | DCM | |
|---|---|---|
| MC-BPhF | −14.9 | −10.6 |
| TS-BPhF-1 | −2.4 | −1.0 |
| TS-BPhF-2 | 0.5 | 5.4 |
| 36 | −28.8 | −23.0 |
| 37 | −18.0 | −8.8 |
As can be seen, the relative energies of the stationary points involved in the B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 are closer to those found in the BF3 LA catalyzed cycloaddition reaction (see Tables 3 and 4). The most favorable TS-BPhF-1 associated with the B(C6F5)3 LA catalyzed cycloaddition reaction is 2.4 kcal mol−1 below the separated reagents. Still, if the formation of the corresponding MC-BPhF is considered, the barrier becomes positive by 12.5 kcal mol−1. Interestingly, a change of the pseudocyclic selectivity is observed by using the B(C6F5)3 LA catalyst; now the reaction path A yielding the [4+2] CA 36 is the more favorable one as TS-BPhF-1 is found 2.9 kcal mol−1 below TS-BPhF-2, in complete agreement with the experimental outcomes.27
Inclusion of solvent effects of DCM provokes similar changes in the relative energies than those found in the BF3 catalyzed reaction (see Tables 3 and 4). In DCM, TS-BPhF-1 is found 6.4 kcal mol−1 below TS-BPhF-2, showing a complete pseudocyclic selectivity.
The thermodynamic data associated with the stationary points of the B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 are given in Table S7 in ESI.† The inclusion of thermal corrections and entropies to the electronic energies shows similar trend in relative enthalpies and Gibbs free energies than those found in gas and solution phases for the BF3 LA catalyzed cycloaddition reaction (see Table 3 and Table S7 in ESI†). This LA catalyzed cycloaddition reaction is totally [4+2] pseudocyclic selective as TS-BPhF-1 is found 6.8 kcal mol−1 below TS-BPhF-2 in Gibbs free energy; formation of [4+2] cycloadducts 36 being exergonic by 6.0 kcal mol−1 (see Table S7 in ESI†).
The gas phase geometries of the TSs involved in the pseudocyclic selectivity of the B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 are given in Fig. 7. A comparison of the two TSs associated with the reaction paths A and B with those of the BF3 catalyzed reaction given in Fig. 6 shows a great similitude between them. The shorter C2–C9 distance at the two stereoisomeric TSs is found in the short range of 2.07–2.11 Å. Note that these TSs form one pair of diastereoisomeric TSs resulting from a pseudo C2–C9 rotation. The inclusion of solvent effects of DCM does not produce any remarkable geometrical changes (see Fig. 7).
 | ||
| Fig. 7 ωB97X-D/6-311G(d,p) Geometries of the TSs involved in the LA B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. Distances are given in Angstroms. Distances in DCM are given in parentheses. | ||
The GEDT values compute at the two gas phase TSs are 0.45 e at TS-BPhF-1, and 0.41 e at TS-BPhF-2. These high values, which are similar to those found in the LA BF3 catalyzed cycloaddition reaction, account for the high polar character of this B(C6F5)3 catalyzed cycloaddition reaction.
2.6. What is the origin of the pseudocyclic selectivity in higher-order cycloaddition reactions?
The two more common selectivities in cycloaddition reactions are the regio and endo/exo stereoselectivity. While the regioselectivity can implicate energy differences between the two regioisomeric TSs until 20 kcal mol−1, stereoselectivity implicates energy differences of less than 4 kcal mol−1. Endo stereoselectivity has been explained within the FMO theory by “secondary orbital interactions” (SOI) present at the endo TSs, a concept introduced in 1983 by Gleiter and Bohm to explain the regio- and stereoselectivity in Diels–Alder reactions.47 Today, SOI continues to be used to explain endo stereoselectivity in cycloaddition reactions.48 In 1999, Domingo proposed that the favorable electrostatic interactions existing in the endo zwitterionic TSs are responsible for the endo selectivity in polar Diels–Alder reactions.49 In 2000, Salvatella et al. proposed that a combination of well-known mechanisms, such as solvent effects, steric interactions, hydrogen bonds (HBs), electrostatic forces, and others, can be invoked instead of SOI in the endo/exo selectivity of Diels–Alder reactions.50Even though the products coming from a pseudocyclic selectivity are constitutional isomers, do not keep in some cases any structural relationship,23 the pseudocyclic TSs participating in this kind of selectivity have a diastereoisomeric relationship, that in many cases can be related with a pseudo-rotation along the axis formed by the two interacting centers (see Section 2.3). Thus, similar to the endo/exo stereoselectivity, the pseudocyclic selectivity results from a series of weak attractive/repulsive intramolecular interactions present at the diastereoisomeric TSs, as charge-charge, dipole–dipole, formation of HBs, and/or van der Waals interactions, no easy of dissecting.
Fig. 8 shows the map of the molecular electrostatic potential (MEP) of the diastereoisomeric pseudocyclicTS-BF-1 and TS-BF-2 involved in the BF3 catalyzed higher-order cycloaddition reaction of tropone 11 with CKA 27 (see Scheme 8). As shown in the MEP of TS-BF-2, the two –CH2CH2– methylenes of cyclic CKA 27, which are positively charged (blue region), are positioned over two of the fluorines of the BF3 catalyst, which are negatively charged (red region), while at the diastereoisomeric TS-BF-1 they are far (see Fig. 8). The favorable electrostatic interactions present at TS-BF-2 are similar to those appearing between the end of the endo zwitterionic TSs participating in polar Diels–Alder reactions.49
 | ||
| Fig. 8 ωB97X-D/6-311G(d,p) Map of the MEP of the diastereoisomeric pseudocyclicTS-BF-1 and TS-BF-2 involved in the BF3 catalyzed higher-order cycloaddition reaction of tropone 11 with CKA 27, yielding the formation of the constitutional isomers [4+2] CA 33 and the [8+2] CA 34, respectively. | ||
On the other hand, analysis of the geometry of TS-BF-2 shows that one hydrogen of each one of the two -CH2CH2- methylenes of cyclic CKA 27 is positioned in direction to one of the fluorines the BF3 catalyst (see Fig. 9). The short H–F distances at this TSs, 2.13 and 2.22 Å, points to the possibility of forming two intramolecular HBs, which are not feasible at TS-BF-1.
 | ||
| Fig. 9 ωB97X-D/6-311G(d,p) geometry and NCI attractive isosurfaces associated with the density overlap at TS-BF-2, showing the presence of the two intramolecular H-F HBs. Distances between the two interacting atoms are given in Angstroms, while a dotted red circle highlights green attractive NCI surfaces associated with the two HBs. | ||
To illustrate the presence of the two HBs at TS-BF-2, a topological analysis of the non-covalent interactions51 (NCI) taking place at this TS was performed (see Fig. 9). As can be seen, the presence of two NCI green attractive surfaces, which are perpendicular to the H–F interatomic line, accounts for the presence of the two intramolecular HBs at TS-BF-2. The formation of these HBs causes additional stability at TS-BF-2 to the electrostatic interactions shown by analysis of the MEP of the two diastereoisomeric TSs.
These favorable electronic interactions present at TS-BF-2 account for the fact that in gas phase this TS is found 8.8 kcal mol−1 below in energy than TS-BF-1 at ωB97X-D/6-311G(d,p) level (see Table 3), thus explaining the origin of pseudocyclic selectivity in this higher-order cycloaddition reaction, yielding [8+2] CA 34viaTS-BF-2 (see Schemes 5 and 8).
However, these analyses are not so simple in many cycloaddition reactions as several attractive/repulsive weak interactions simultaneously can be present at some TSs. Note that in non-polar Diels–Alder reactions, the repulsive steric interactions appearing at the endo TSs are responsible for the exo stereoselectivity.49
Thus, Fig. 10 shows the map of the MEP of the diastereoisomeric TS-BPhF-1 and TS-BPhF-2, involved in the B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27 (see Scheme 9). As shown in the MEP of TS-BPhF-1, the fluorines of the B(C6F5)3 catalyst are less negatively charged than at the BF3 catalyst (see TS-BF-1 in Fig. 8 and TS-BPhF-1 in Fig. 10), thus diminishing the attractive electrostatic interactions at TS-BPhF-2. In addition, the approach of the two -CH2CH2- methylenes of cyclic CKA 27 to the bulk B(C6F5)3 catalyst at TS-BPhF-2 can increase the repulsive steric interactions at this TS, thus increasing its energy with respect to that of TS-BPhF-1. Thus, the unfavorable steric interactions present at TS-BPhF-2 could explain the change of pseudocyclic selectivity observed in this B(C6F5)3 catalyzed cycloaddition reaction. Note that the bulk B(C6F5)3 catalyst is the only one that experimentally causes a complete change of the pseudocyclic selectivity with respect to that of the BF3 catalyzed cycloaddition reaction of tropone 11 with ketene diethyl acetal 24 (see Scheme 5).27
 | ||
| Fig. 10 ωB97X-D/6-311G(d,p) Map of the MEP of the diastereoisomeric pseudocyclicTS-BPhF-1 and TS-BPhF-2 involved in the B(C6F5)3 catalyzed cycloaddition reaction of tropone 11 with CKA 27, yielding the formation of the constitutional isomers [4+2] CA 36 and the [8+2] CA 37, respectively. | ||
2.7. ELF comparative analysis of the TSs involved in the BF3 LA catalyzed reaction of tropone 11 with CKA 27
Finally, the electronic structure of the TSs involved in the BF3 LA catalyzed reaction of tropone 11 with CKA 27 was analyzed by an ELF topological analysis of the electron density. The ELF valence basin populations at TS-BF-1, TS-BF-2, and TS-BF-3 are given in Table 5, while the corresponding ELF basin attractor positions are represented in Fig. 11.| 31 | 27 | TS-BF-1 | TS-BF-2 | TS-BF-3 | |
|---|---|---|---|---|---|
| V(C1,C2) | 2.45 | 2.36 | 2.40 | 2.41 | |
| V(C2,C3) | 3.04 | 2.48 | 2.63 | 3.18 | |
| V(C3,C4) | 2.43 | 3.05 | 3.00 | 2.32 | |
| V(C4,C5) | 3.02 | 2.57 | 2.59 | 2.45 | |
| V(C5,C6) | 2.43 | 2.92 | 2.90 | 3.06 | |
| V(C6,C7) | 3.04 | 2.69 | 2.68 | 2.66 | |
| V(C1,C7) | 2.45 | 2.96 | 2.93 | 2.94 | |
| V(C7,O8) | 2.04 | 1.82 | 1.76 | 1.77 | |
| V(C9,C10) | 1.92 | 2.88 | 2.93 | 2.87 | |
| V'(C9,C10) | 1.92 | ||||
| V(C2) | 0.09 | ||||
| V(C4) | 0.04 | ||||
| V(C9) | 0.62 | 0.54 | 0.71 |
 | ||
| Fig. 11 ELF basin attractor positions at TS-BF-1, TS-BF-2, and TS-BF-3. | ||
Analysis of the ELF of the electronic structure of three TSs shows a great similitude between them. They present a V(9) monosynaptic basin, integrating between 0.54–0.71 e, at the most nucleophilic C9 carbon of CKA 27. In addition, TS1-BF-1 and TS1-BF-3 show a residual V(C2) and V(C4) monosynaptic basins, integrating less of 0.1 e, at the electrophilic C2 and C4 carbons of LA complex 31. The presence of these monosynaptic basins, which are associated with the pseudoradical centers demanded by the subsequent C–C single bond formation,25 together with the absence of any V(C2[4],C9) disynaptic basin, indicates that the formation of the new C2[4]–C9 single bonds does not have begun yet at any TS. In addition, the low population of the V(C2[4]) monosynaptic basins at TS1-BF-1 and TS1-BF-3, and the absence of any V(C2) monosynaptic basin at TS-BF-2 indicate that the formation of the new C2[4]–C9 single bonds will take place mainly by donation of the electron density of the V(9) monosynaptic basin present at the three TSs.
The most remarkable topological change found at these TSs with respect to separated reagents is that the two disynaptic basins, V(C9,C10) and V′(C9,C10), present at CKA 27 have merged into a one V(C9,C10) disynaptic basins after losing an amount of electron density of 0.96 e. A part of this electron density is used to create the new V(9) monosynaptic basins, and another part is involved in the GEDT process taking place in these polar processes.
On the order hand, the C–C and C–O bond in tropone:BF3 complex 31 also experience changes in their electron density populations. At the three TSs, the tropone C–C and C–O bonding regions are characterized by only one disynaptic basin. Only changes in the corresponding population concerning those at tropone:BF3 complex 31 are observed. At the three TSs, the V(C7,O8) disynaptic basins have been depopulated by ca. 0.2 e due to a higher polarization of the carbonyl C7–O8 bond.
At the regioisomeric TS-BF-3, associated with the nucleophilic attack on the C4 carbon of tropone 11, the disynaptic basins associated with the C4–C5, C5–C6, C6–C7, and C1–C7 bonding regions are the more affected; alternatively, while ones lose ca 0.5 e, the others gain 0.5 e. On the other hand, the diastereoisomeric TS-BF-1 and TS-BF-2, associated with the nucleophilic attack on the C2 carbon of tropone 11, present a very similar ELF electronic structure; except for the C1–C2 and C2–C3 bonds, all other Ci–Cj bonds of the tropone framework experience similar changes in electron density (see the populations of V(Ci,Cj) disynaptic basins in Table 5). The only remarkable change is the presence of the V(C2) monosynaptic basin at TS-BF-1, integrating less of 0.1 e, as a consequence of its slightly more advanced character that TS-BF-2. Note that the C1–C2 distance at these TSs is 2.051 and 2.125 Å, respectively (see Fig. 5). Consequently, the energy difference between the stereoisomeric TS-BF-1 and TS-BF-2, which is responsible for the pseudocyclic selectivity,23 can be associated to the weak intramolecular interactions present at the diastereoisomeric TSs.
4. Conclusions
The higher-order cycloaddition reactions of tropone 11 with CKA 27, a nucleophilic ethylene, in the absence and presence of BF3 and B(C6F5)3 LA catalysts, have been studied within MEDT at the ωB97X-D/6-311G(d,p) and B3LYP-D3BJ/6-311G(d,p) computational levels to understand the reaction mechanisms, and the regio and pseudocyclic selectivity experimentally observed in these higher-order cycloaddition reactions.ELF analysis of tropone 11 shows that it has the structure of cycloheptatriene 12, showing some electronic delocalization between the triene system and the carbonyl group. Coordination of the BF3 LA to the O8 oxygen of tropone 11 increases the strong polarization of the carbonyl C1–O8 group, thus, increasing the electronic delocalization of the triene system of tropone:BF3 complex 31. The electrophilicity ω index of tropone 11 indicates that this species is a strong electrophile participating in polar cycloaddition reactions of REDF toward nucleophilic ethylenes as CKA 27 and ketene diethyl acetal 24. Formation of tropone:LA complexes considerably increases their electrophilic character, favoring the corresponding polar cycloaddition reactions. Analysis of the electrophilic Pk+ Parr functions of tropone 11 and tropone:BF3 complex 31 indicates that the C2 and the C4 positions of tropone are the most electrophilic centers, in complete agreement with experimental regioselectivity observed.24,27
Study of the cycloaddition reaction of tropone 11 with CKA 27 shows that this reaction is regio and pseudocyclic selective, yielding the [4+2] CA 28 through a stepwise mechanism, which an activation energy of 12.1 kcal mol−1 in DCM. The high exothermic character of the reaction, −26.2 kcal mol−1, makes it irreversible. A change of the pseudocyclic selectivity is observed at the B3LYP-D3BJ/6-311G(d,p) computational level.
Analysis of the geometrical parameters indicates that the TSs are associated with the nucleophilic attack of CKA 27 on the C2 or C4 electrophilic positions of tropone 11. Analysis of the geometries of TS-11 and TS-2, which are responsible for the pseudocyclic selectivity yielding [4+2] or [8+2] CAs, respectively, shows that they have a stereoisomeric relationship, i.e., they are one pair of diastereomers resulting from a formal pseudo C2–C9 single bond rotation. Analysis of the GEDT at the TSs points out the high polar character of these cycloaddition reactions of REDF.
Coordination of the BF3 LA to tropone 11 accelerates the reaction through a non-concerted two-stage one-step mechanism.16 This LA catalyzed cycloaddition is completely regio and pseudocyclic selective yielding only the [8+2] CA 34. Interestingly, the change of the BF3 by the B(C6F5)3 LA catalyst changes the pseudocyclic selectivity, yielding the [4+2] CA 36 as the only product, in complete agreement with the experimental selectivities.27
The present MEDT study allows establishing that the higher-order cycloaddition reactions of tropone 11 toward electrophilic ethylenes, in the absence and presence of LAs, is kinetically controlled by the nucleophilic/electrophilic interactions taking place at the polar TSs. LAs not only accelerate the reaction and make the reaction completely regioselective but also determine the pseudocyclic selectivity, which depends on the nature of the LA catalyst, yielding exclusively only [4+2] or [8+2] CAs. Similar to the endo/exo stereoselectivity, pseudocyclic selectivity is determined by a series of weak attractive/repulsive intramolecular electronic interactions present at the diastereoisomeric TSs. Analysis of these higher-order cycloaddition reactions of tropone 11 indicates that the energies obtained by using the B3LYP-D3BJ/6-311G(d,p) calculations are found better than the ωB97X-D/6-311G(d,p) ones for the study of the experimentally observed selectivities.
Computational details
The B3LYP52,53 and ωB97X-D54 functionals, together with the standard 6-311G(d,p)55 basis set, which includes d-type polarization for second row elements and p-type polarization functions for hydrogens, were used throughout this MEDT study. Dispersion energies by using the B3LYP functional were considered by using the Grimme's dispersion-corrected version56 with Becke–Johnson damping,57 B3LYP-D3BJ. The TSs were characterized by the presence of only one imaginary frequency. The Berny method was used in optimizations.58,59 The intrinsic reaction coordinate60 (IRC) calculations were performed to establish the unique connection given between the TSs and the corresponding minima.61,62 Solvent effects of DCM were taken into account by full optimization of the gas phase structures at the same computational level using the polarizable continuum model (PCM)63,64 in the framework of the self-consistent reaction field (SCRF).65–67 Values of ωB97X-D/6-311G(d,p) enthalpies, entropies and Gibbs free energies in DCM were calculated with standard statistical thermodynamics at 318.15 K and 1 atm,55 by PCM frequency calculations at the solvent optimized structures.The GEDT25 values were computed by using the equation GEDT(f) = Σqf, where q are the natural charges39,40 of the atoms belonging to one of the two frameworks (f) at the TS geometries. Global and local CDFT indices10,11 were calculated by using the equations given in ref. 11.
The Gaussian 16 suite of programs was used to perform the calculations.68 ELF37 analyses of the ωB97X-D/6-311G(d,p) monodeterminantal wavefunctions were done by using the TopMod69 package with a cubical grid of step size of 0.1 Bohr. Molecular geometries and ELF basin attractors were visualized by using the GaussView program.70 The topological analysis of the NCI44 was performed with the NCIplot program.71
Author contributions
L. R. D. Data curation, formal analysis, funding acquisition, investigation, supervision, writing – original draft and writing – review & editing. P. P. Data curation, funding acquisition, investigation, writing – original and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work has been supported by the Ministry of Science and Innovation (MICINN) of the Spanish Government, project PID2019-110776GB-I00 (AEI/FEDER, UE) and by FONDECYT – Chile through Project No. 1180348. L. R. D. also acknowledges to Cooperación Internacional (Fondecyt No. 1180348) by continuous support.References
- W. Carruthers, Some Modern Methods of Organic Synthesis. Second edn, Cambridge University Press, Cambridge, 1978 Search PubMed.
- W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, 1990 Search PubMed.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- R. Huisgen, Proc. Chem. Soc., 1961, 357 Search PubMed.
- M. Ríos-Gutiérrez and L. R. Domingo, Eur. J. Org. Chem., 2019, 267–282 CrossRef.
- J. H. Rigby, Higher-Order Cycloaddition Reactions, Organic Synthesis: Stereoselective Preparation of 7- to 10-Membered Ring Systems, John Wiley & Sons Inc., Weinheim, 2008 Search PubMed.
- K. N. Houk, L. J. Luskus and N. S. Bhacca, J. Am. Chem. Soc., 1970, 92, 6392–6394 CrossRef CAS.
- N. S. Bhacca, L. J. Luskus and K. N. Houk, J. Chem. Soc. D, 1971, 109–111 RSC.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576–3583 RSC.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez, B. Silvi and P. Pérez, Eur. J. Org. Chem., 2018, 1107–1120 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez, E. Chamorro and P. Pérez, ChemistrySelect, 2016, 1, 6026–6039 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2020, 25, 2535 CrossRef CAS.
- L. R. Domingo, J. A. Sáez, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed.
- K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81–90 CrossRef CAS.
- K. N. Houk, L. J. Luskus and N. S. Bahacca, J. Am. Chem. Soc., 1970, 92, 6392–6394 CrossRef CAS.
- K. N. Houk, J. Sims, C. R. Watts and L. J. Luskus, J. Am. Chem. Soc., 1973, 95, 7301–7315 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2018, 23, 1913 CrossRef.
- M. Oda, M. Funamizu and Y. Kitahara, Chem. Commun., 1969, 737–738 RSC.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Tetrahedron, 2017, 73, 1718–1724 CrossRef CAS.
- P. Li and H. Yamamoto, J. Am. Chem. Soc., 2009, 131, 16628–16629 CrossRef CAS.
- S. Yamabe, Y. Nishihara and T. Minato, J. Phys. Chem. A, 2002, 106(19), 4980–4987 CrossRef CAS.
- A. R. Rivero, I. Fernandez and M. A. Sierra, Org. Lett., 2013, 15, 4928–4931 CrossRef CAS.
- P. Yu, T. Q. Chen, Z. Yang, C. Q. He, A. Patel, Y. Lam, C.-Y. Liu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 8251–8258 CrossRef CAS PubMed.
- L. C. Yang, Y. N. Wang and R. Liu, Nat. Chem., 2020, 12, 860–868 CrossRef CAS PubMed.
- D. McLeod, M. K. Thøgersen, N. I. Jessen, K. A. Jørgensen, C. S. Jamieson, X.-S. Xue, K. N. Houk, F. Liu and R. Hoffmann, Acc. Chem. Res., 2019, 52, 3488–3501 CrossRef CAS PubMed.
- X. Chen, M. K. Thøgersen, L. Yang, R. F. Lauridsen, X.-S. Xue, K. A. Jørgensen and K. N. Houk, J. Am. Chem. Soc., 2021, 143, 934–944 CrossRef CAS.
- K. Fukui, Molecular Orbitals in Chemistry, Physics, and Biology, New York, 1964 Search PubMed.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Claredon Press, Oxford, UK, 1990 Search PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Sáez, RSC Adv., 2012, 2, 1334–1342 RSC.
- L. R. Domingo and P. Pérez, J. Org. Chem., 2020, 85, 13121–13132 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, RSC Adv., 2020, 10, 15394–15405 RSC.
- M. J. Aurell, L. R. Domingo, P. Pérez and R. Contreras, Tetrahedron, 2004, 60, 11503–11509 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- R. Gleiter and M. C. Bohm, Pure Appl. Chem., 1983, 55, 237–244 CAS.
- M. Ramirez, D. Svatunek, F. Liu, N. K. Garg and K. N. Houk, Angew. Chem., Int. Ed., 2021, 60, 14989–14997 CrossRef CAS.
- L. R. Domingo, M. Arnó and J. Andrés, J. Org. Chem., 1999, 64, 5867–5875 CrossRef CAS.
- J. I. García, J. A. Mayoral and L. Salvatella, Acc. Chem. Res., 2000, 33, 658–664 CrossRef.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Hehre, L. Radom, P. V. R. Schleyer and J. Pople. Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- D. G. A. Smith, L. A. Burns, K. Patkowski and C. D. Sherrill, J. Phys. Chem. Lett., 2016, 7, 2197–2203 CrossRef CAS.
- H. B. Schlegel, J. Comput. Chem., 1982, 2, 214–218 CrossRef.
- H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions-A Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theo. Comput., 2011, 7, 625–632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figure with the ωB97X-D/6-311G(d,p) geometries of the MCs MC and MC-BF. Table with the ωB97X-D/6-311G(d,p) and B3LYP-D3BJ/6-311G(d,p) total and relative energies, in gas phase and DCM, of the stationary points involved in the cycloaddition reaction of tropone 11 with CKA 27. Table with the ωB97X-D/6-311G(d,p) and B3LYP-D3BJ/6-311G(d,p) total and relative energies, in gas phase and DCM, of the stationary points involved in the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. Table with the ωB97X-D/6-311G(d,p) total enthalpies and relative entropies and Gibbs free energies for the stationary points involved in the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. Table with the B3LYP-D3BJ/6-311G(d,p) total enthalpies and relative entropies and Gibbs free energies for the stationary points involved in the LA BF3 catalyzed cycloaddition reaction of tropone 11 with CKA 27. Table with the ωB97X-D/6-311G(d,p) gas phase total and relative energies of the stationary points involved in the B(C6F5)3 LA catalyzed cycloaddition reactions of tropone 11 with CKA 27. ωB97XD/6-311G(d,p) gas phase computed total energies, unique imaginary frequency, and Cartesian coordinates of the stationary points involved the non-catalyzed and LA catalyzed cycloaddition reactions of tropone 11 with CKA 27, and those of the non-catalyzed and cycloaddition reaction of tropone 11 with ketene diethyl acetal 24. See DOI: 10.1039/d1nj04962c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |