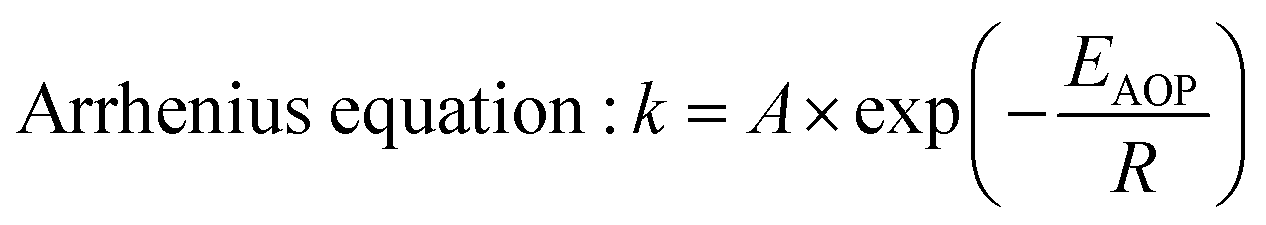DOI:
10.1039/D1NJ04516D
(Paper)
New J. Chem., 2022,
46, 359-369
Effect of preparation conditions and Co–Pi groups as a noble metal-free redox mediator and hole extractor to boost the photoelectrochemical water oxidation for 1D nanorod α-Fe2O3†
Received
20th September 2021
, Accepted 21st November 2021
First published on 22nd November 2021
Abstract
Photoelectrochemical water splitting for hydrogen production is an ideal method for solar energy utilization. Herein, α-Fe2O3 was prepared by a hydrothermal process and the precursor concentration, calcination temperature and hydrothermal time were optimized. The Co–Pi groups were deposited on the surface by two-step dipping process. The preparation conditions and loading capacity of CoPi were optimized. SEM showed that the morphology of α-Fe2O3 was short nanorods. XPS indicated that CoPi was successfully deposited on the surface of Fe2O3. TEM and XRD showed that CoPi was amorphous. The photocurrent density of Fe2O3/CoPi increased to 1.95 mA cm−2 at 1.23 VRHE, which was 1.91 times that of bare Fe2O3. After CoPi modification, the initial potential shifted negatively by 70 mV. It could be inferred that CoPi acted as an effective water oxidation co-catalyst to reduce the activation energy of the oxygen evolution reaction. The chopping I–t curve showed that the transient anodic spike of Fe2O3 decreased after CoPi deposition. This indicated that CoPi acted as a hole extractor and redox mediator to reduce the charge accumulation on the Fe2O3 surface. Through calculation, the ABPE of Fe2O3/Co–Pi was 2.1 times that of Fe2O3. The charge injection efficiency of Fe2O3/Co–Pi at 1.23 VRHE was 1.6 times that of Fe2O3. This study presented a richer understanding on the functions and promotional effects of the preparation conditions and CoPi groups for Fe2O3 photoanodes.
Introduction
As a superior process of artificial photosynthesis, photoelectrochemical (PEC) water splitting has become a promising route toward sustainable hydrogen production and solar energy storage.1,2 However, the efficiency of PEC water splitting is largely limited by water oxidation, which requires high-performance photoanodes to improve the conversion efficiency of solar energy. N-type semiconductors, such as TiO2,3–6 BiVO4,7,8 and α-Fe2O3,9–11 with band gaps of ∼3.2 eV, ∼2.4 eV and ∼2.1 eV, respectively, have been researched. From the relationship between the semiconductor band gap and light absorption wavelength (Eg (eV) = 1240/λ (nm)), it could be found that the maximum light absorption wavelength of α-Fe2O3 would reach 590 nm.12 Moreover, α-Fe2O3 is a non-toxic, stable, and low-cost photoanode material. Its outstanding advantages have attracted a lot of attention in recent years. There are many preparation processes for Fe2O3, such as the hydrothermal process, precipitation process, thermal decomposition process, sol–gel process, and others. The most common process is hydrothermal. The advantage of using this process to prepare Fe2O3 nanorod arrays on FTO conductive substrates is that the process is easy to control. In addition, due to the large exposed area, the one-dimensional nanorod-like structure facilitates electron transport. This contributes to the charge separation in the process of photoelectrolysis of water. However, α-Fe2O3 has a short hole diffusion distance (2–4 nm) and carrier lifetime (10−12 s), which could lead to the easy recombination of electrons and holes, thereby reducing the efficiency of PEC water splitting.13 Under simulated sunlight, the theoretical photocurrent density of α-Fe2O3 is about 13 mA cm−2 at 1.23 VRHE.14 However, the actual photocurrent density is much lower than the theoretical value. It is important to increase the photocurrent density of α-Fe2O3, as well as reduce the overpotential of the water oxidation reaction during the PEC water splitting.
The sluggish oxygen production reaction rate limits the widespread use of α-Fe2O3. Therefore, many studies are focused on overcoming the reaction energy barrier and increasing the charge injection efficiency by introducing a co-catalyst to accelerate the water oxidation reaction. Common cocatalysts used to modify α-Fe2O3 include BiVO4,15 IrO2,16 Rh2O3,17 Co–Pi,18–21 Co3O4,22 CoOx,23 NiFeOx24 and transition metal (Fe, Co, Ni) oxyhydroxide.25,26 Co–Pi is a superior OER co-catalyst, which has the advantages of abundant sources, no pollution, and low preparation cost. The processes of modifying Co–Pi groups on α-Fe2O3 mainly include the electrodeposition process,21 light-assisted electrodeposition process,27 and immersion process. The thickness of the Co–Pi deposition should be moderate. Too thick will hinder the light absorption of α-Fe2O3, while too thin will not be enough to improve the photoelectric performance. When using the electrochemical process to deposit Co–Pi groups, it is necessary to consider whether the applied voltage will cause electrical corrosion of the α-Fe2O3 photoanode.
Herein, the α-Fe2O3 photoanode was synthesized on the surface of FTO by the hydrothermal process, and the precursor concentration, calcination temperature and hydrothermal time were optimized. Then, CoPi groups were deposited on the surface of α-Fe2O3 through an impregnation process. SEM showed that the morphology of Fe2O3 prepared by hydrothermal process is a short nanorod. As the calcination temperature increased, the thickness of Fe2O3 also increased. XPS detected Co and P elements in the sample. It indicated that CoPi was successfully deposited on the surface of Fe2O3. There was no peak of CoPi in the XRD of Fe2O3/CoPi, and there was no lattice fringe of CoPi in the TEM of Fe2O3/CoPi. These results indicated that the CoPi groups on the Fe2O3/CoPi photoanode were amorphous. It was found that the bare Fe2O3 photoelectrode had the best photoelectric performance when the Fe precursor concentration, hydrothermal reaction time, and calcination temperature were 6 mM, 4 h, and 600 °C, respectively. The photocurrent density of the optimized Fe2O3 photoelectrode at 1.23 VRHE was 1.02 mA cm−2, which was about 29 times (0.035 mA cm−2) higher than that of the sample without condition exploration. The CoPi groups were supported on the Fe2O3 surface through the dipping process, and the optimal immersion times of the electrode in the Co and Pi precursor solutions were both 6 h. The photocurrent density of the optimized Fe2O3/CoPi at 1.23 VRHE was 1.95 mA cm−2, which was 1.91 times that of the bare sample. The initial potential of Fe2O3/CoPi was 0.78 VRHE, which was 70 mV lower than that of the bare Fe2O3. According to the law of conservation of energy, it could be inferred that CoPi reduced the activation energy of OER and accelerated the water oxidation reaction. Therefore, CoPi was an effective OER co-catalyst. The chopping I–t curve showed that the transient anodic spike of Fe2O3 decreased after CoPi deposition. It indicated that the charge accumulation on the Fe2O3/CoPi surface was reduced. EIS showed that after CoPi modification, the electrode/electrolyte interface resistance of Fe2O3 decreased. This showed that CoPi could change the interface performance. Combining the chopping I–t curve and EIS analysis, it could be concluded that CoPi could be used as a hole extractant and redox mediator to promote charge injection on the surface of Fe2O3. Through calculation, the applied bias photon-to-current efficiency (ABPE) of the Fe2O3/Co–Pi photoanode was 0.17% (2.1 times that of the bare Fe2O3). The charge injection efficiency (ηinj) of the Fe2O3/Co–Pi photoanode at 1.23 VRHE was 1.6 times that of Fe2O3. In short, the optimization of the preparation conditions of Fe2O3 and the modification of the Co–Pi groups as a non-noble metal catalyst and hole extractor effectively promoted the water oxidation reaction, which was beneficial to the design and synthesis of future photoelectrodes.
Experimental section
Materials
Acetone, ferric nitrate nonahydrate, cobalt nitrate hexahydrate, potassium hydroxide, dodecahydrate and sodium phosphate were purchased from Sinopharm Chemical Reagent Co., Ltd. Anhydrous ethanol and ammonium fluoride were obtained from Xilong Science Co., Ltd. Sodium sulfite was purchased from Macleans Chemical Reagent Co., Ltd. FTO conductive glass was obtained from Opivite Company. All of the chemicals were of analytical grade, and deionized (DI) water was used for all of the experiments.
Preparation of photoelectrodes
Fabrication of the α-Fe2O3 electrode.
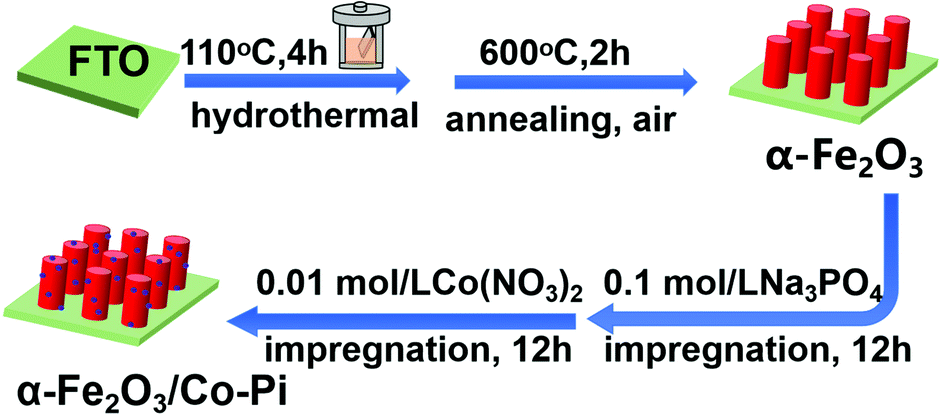
First, the FTO conductive glass (3 cm × 1 cm) was cleaned ultrasonically with an ethanol–acetone mixture (Vethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vacetone = 1:1) and deionized water for 30 minutes. Then, they were dried in an oven at 60 °C. A 35 mL precursor solution containing 6 mM Fe(NO3)3·9H2O and 0.1 M NH4F was prepared, and then stirred with a magnet for 10 min to fully dissolve the solids. Second, the FTO conductive glass was put into a polytetrafluoroethylene liner containing 35 mL of the precursor solution, and the reactor was transferred to an oven. The oven was heated at 110 °C for 4 hours. After the hydrothermal deposition, the FTO substrate was taken out and rinsed with ethanol and deionized water. Finally, the sample obtained by the hydrothermal process was put into a tubular resistance furnace for air atmosphere annealing. After annealing at 500 °C for 2 h, the substrate became red-brown. The first two steps in Fig. 1 are a schematic diagram of the preparation process of α-Fe2O3.
:Vacetone = 1:1) and deionized water for 30 minutes. Then, they were dried in an oven at 60 °C. A 35 mL precursor solution containing 6 mM Fe(NO3)3·9H2O and 0.1 M NH4F was prepared, and then stirred with a magnet for 10 min to fully dissolve the solids. Second, the FTO conductive glass was put into a polytetrafluoroethylene liner containing 35 mL of the precursor solution, and the reactor was transferred to an oven. The oven was heated at 110 °C for 4 hours. After the hydrothermal deposition, the FTO substrate was taken out and rinsed with ethanol and deionized water. Finally, the sample obtained by the hydrothermal process was put into a tubular resistance furnace for air atmosphere annealing. After annealing at 500 °C for 2 h, the substrate became red-brown. The first two steps in Fig. 1 are a schematic diagram of the preparation process of α-Fe2O3.
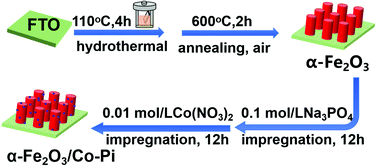 |
| | Fig. 1 Schematic diagram of the preparation process of α-Fe2O3 and α-Fe2O3/Co–Pi photoelectrodes. | |
Preparation of the α-Fe2O3/Co–Pi photoelectrode
First, 0.1 mol L−1 sodium phosphate (Na3PO4) and 0.01 mol L−1 cobalt nitrate (Co(NO3)2)solution were prepared. After that, the as-prepared α-Fe2O3 nanorod was placed into 0.1 mol L−1 Na3PO4 solution for immersion of 6 h. The non-conductive side was close to the inner wall of the beaker; simultaneously, stirring occurred during immersion. After the first step of immersion, the sample was taken out and blow dried with cold air. In the next step, the treated sample was immersed in the 0.01 mol L−1 Co(NO3)2 solution for 3 h, 6 h, 9 h, and 12 h to form Fe2O3/Co–Pi. Then, the sample was taken out and dried with cold air. Finally, the sample was rinsed with deionized water, and dried at 60 °C. The last two steps in Fig. 1 are a schematic diagram of the preparation process of Co–Pi deposition.
Characterizations
Field emission scanning electron microscope (FESEM) and energy dispersive X-ray spectroscopy (EDS).
In order to obtain the morphology and element distribution of the samples, SEM and EDS tests were performed on a field emission scanning electron microscope model (Nova Nano SEM 230). All samples were not sprayed with gold when the front view was taken. The test voltage was 8 kV. Since the conductivity of the FTO substrate was not good enough, all samples were sprayed with gold for 1 minute when the cross-sectional view was taken.
Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM)
In order to observe the morphology and lattice fringes of the sample, the TEM test was carried out in a JEOL JEM 2100 transmission electron microscope. The powder was scraped gently from the substrate with a knife. The powder was ultrasonically dispersed with ethanol for 10 minutes. The sample was then dropped on a micro-mesh copper net, dried and observed through an electron microscope.
X-ray diffraction (XRD)
The phase analysis of the sample was carried out by XRD test. The model of the polycrystalline diffractometer was DY5261/Xpert3, the test voltage was 40 kV, the current was 40 mA, the radiation source was Cu Kα, and the scanning range was 5–80°.
Raman spectrum
In order to obtain the lattice vibration information of the sample, the Raman measurement was performed on an Invia Reflex laser Raman spectrometer with an excitation wavelength of 532 nm.
X-ray photoelectron spectroscopy (XPS)
The XPS study was used to obtain the element and valence information of the sample, and the instrument was an X-ray photoelectron spectrometer (Thermo Fischer, ESCALAB Xi+, USA). Among them, the vacuum degree of the analysis chamber was 8 × 10−10 Pa, the excitation source was Alka rays (hν = 1486.6 eV), the working voltage was 12 kV, and the filament current was 6 mA. All of the data were calibrated with the standard binding energy (C 1s = 284.80 eV).
Ultraviolet visible absorption (UV-vis) and diffuse reflectance spectrum
The absorbance (A) and reflectance (R) of the sample were tested on a Lambda 750 ultraviolet-visible-near-infrared photometer in the diffuse reflection mode. The scanning range was 200–800 nm. Through the integrating sphere of the UV-vis spectrophotometer, the spectral curve of the reflectance of the sample against the wavelength should be measured first, and then the (Fhv)2 − (hv) curve could be calculated by the following equations. The calculated equations were as follows:where R is the reflectivity, h is the Planck constant (6.626 × 10−34 J s), c is the speed of light (3 × 108 m s−1), λ is the incident light wavelength (nm), and e is the electron (1.6 × 10−19), and n is determined by the inter-band transition form. For α-Fe2O3, the value of n is 2. After calculation, the relationship (Fhν) versus (hν) as plotted to obtain a curve and a tangent line was drawn to the curve. The intersection of the tangent and the abscissa is the band gap (Eg) of the sample.
PEC measurements
The PEC measurements were carried out in a self-made H-shaped electrolytic cell equipped with a quartz glass plate, and the tests used a three-electrode system. The reference electrode was a Hg/HgO electrode, and the light was irradiated from the back of the FTO. The counter electrode was a Pt electrode, and the working electrode was a FTO conductive glass with deposited materials. The light area of the working electrode was corrected to 1 cm2, and the electrolyte was 1 M KOH (pH = 13.6) solution. All data were recorded on a CHI750E electrochemical workstation and online computer. The conversion relationship between the reversible hydrogen electrode potential and the Hg/HgO electrode (4) was as follows:| | | ERHE = EHg/Hg + 0.098 + 0.59pH. | (4) |
Linear sweep voltammetry (LSV) test
The test was conducted in a three-electrode system with a 300 W PLS SXE300C xenon lamp as the light source. The light intensity was adjusted to 100 mW cm−2 with a filter. The scanning voltage range was 0.3–1.8 VRHE with a scanning speed of 10 mV s−1.
Chopping Chronoamperometry (I–t) test
Chopping I–t curves were measured with an external potential of 1.23 VRHE, under chopped illumination. The illumination parameters during the test were as follows: first light for 40 seconds, then shading for 40 seconds, and so on until 400 seconds.
Electrochemical impedance spectroscopy (EIS) test
The electrochemical impedance test was carried out under simulated sunlight, with an applied voltage of 1.23 VRHE. The test frequency was 1–106 Hz, and the disturbance was 5 mV. The obtained spectrum was fitted with Zview software.
Flat band potential (Mott–Schottky) test
The test was performed in the dark state, and the test frequency (f) was 104 Hz. The carrier density Nd could be calculated by the Mott–Schottky equation:| | | Nd = (2/eε0ε1)|d(1/C2)dV|. | (5) |
where e is the charge of the electrons (1.602 × 10−19), ε0 is the vacuum dielectric constant (8.854 × 10−12 CV−1 m−1), and εr is the semiconductor dielectric constant (α-Fe2O3 takes 80 CV−1 m−1).
Applied bias photon-to-current efficiency (ABPE)
The test conditions were the same as the linear voltammetry scan under illumination. The calculation formula of ABPE was as follows:| | | ABPE = Jphoto(1.23VRHE − Vapp)/Plight. | (6) |
where Jphoto the photocurrent density, Vapp the applied voltage (relative to reversible hydrogen), and Plight the light intensity (100 mW cm−2).
Charge separation efficiency (ηsep) and charge injection efficiency (ηinj)
The test was carried out in a three-electrode system. The electrolyte was a 1 M KOH solution containing 0.5 M Na2SO3 under light conditions. The values of ηsep and ηinj were obtained by the following eqn (7) and (8):where JNa2SO3 is the photocurrent density when a hole scavenger (Na2SO3) is added to the electrolyte. Assuming APCE = 100%, the Jabs of Fe2O3 is calculated to be 11.4 mA cm−2 by estimating the overlap area between the UV-vis absorption spectrum and the AM 1.5G solar spectrum.28
Results and discussion
Physical and chemical characterization
SEM.
Fig. 2 is the FESEM image of α-Fe2O3 calcined at various temperatures of 500 °C, 550 °C, and 600 °C, as well as α-Fe2O3/Co–Pi photoanodes. Fig. 2a shows the FESEM image of α-Fe2O3 calcined at 500 °C. It could be found that a dense film composed of aggregated short nanorods was formed on the surface of the FTO substrate with some small gaps. This might be attributed to the composite of other small units in the film. The diameter of the short nanorods ranged from 40 to 90 nm, with an average diameter of 60 nm. The inset in Fig. 2a shows that the film thickness was 190 nm. Fig. 2b shows the FESEM image of α-Fe2O3 calcined at 550 °C. It was found that hematite was still composed of short nanorods (∼40 nm), and the film thickness increased to 230 nm. Fig. 2c indicates that after calcination at 600 °C, the surface of α-Fe2O3 was smoother. The average diameter of α-Fe2O3 calcined at 600 °C was 30 nm with a film thickness of 320 nm. Fig. S1 (ESI†) is an enlarged SEM image of the cross-section of the Fe2O3 photoanode calcined at 600 °C. It can be seen from the dotted frame that Fe2O3 was in the shape of short nanorods. Combined with Fig. 2a–c, it was found that as the calcination temperature increased, the thickness of the Fe2O3 film increased. Fig. 2d shows the FESEM image of the α-Fe2O3/Co–Pi photoanode. It indicated that the morphology of hematite remained unchanged after the modification of Co–Pi groups. The EDS scanning spectra of α-Fe2O3 and α-Fe2O3/Co–Pi photoanodes are shown in Fig. S2 (ESI†). It showed that all samples had peaks of F, Si, Sn, and Ca elements, which were attributed to the FTO conductive glass. The peaks of the Fe and O elements appeared among the α-Fe2O3 and α-Fe2O3/Co–Pi photoanodes. It indicated that Fe2O3 was successfully prepared on the FTO substrate. There was no obvious peak of Co and P elements for the Fe2O3/Co–Pi photoanode. That was mainly because only a small amount of Co–Pi groups could be deposited on the surface of Fe2O3 by the immersion process. The scanning depth of EDS had reached the bottom glass layer instead of the surface sample layer, so the surface Co–Pi was not scanned. Table S1 (ESI†) is the contents of the corresponding elements in the α-Fe2O3 and α-Fe2O3/Co–Pi photoanodes obtained from EDS. It showed that the contents of F, Si, Sn, Ca, O, and Fe in α-Fe2O3 were 3.10 wt%, 7.47 wt%, 54.03 wt%, 4.09 wt%, 6.21 wt%, and 25.08 wt%, respectively. The contents of F, Si, Sn, Ca, O, and Fe in α-Fe2O3/Co–Pi were 3.13 wt%, 8.32 wt%, 57.17 wt%, 4.37 wt%, 5.97 wt%, and 21.04 wt%, respectively.
 |
| | Fig. 2 FESEM images of α-Fe2O3 calcined at various temperatures of 500, 550, and 600 °C, as well as α-Fe2O3/Co–Pi photoanodes, (a) 500 °C, (b) 550 °C, (c) 600 °C, (d) α-Fe2O3/Co–Pi, and the inset is a cross-sectional view. | |
TEM.
TEM and HRTEM were used to further explore the effect of Co–Pi groups on α-Fe2O3. Fig. 3a is the TEM image with 50000× magnification of Fe2O3, which showed that Fe2O3 was rod-shaped and the nanorods were tightly connected. Fig. S3 (ESI†) is the TEM image with 50000× magnification of Fe2O3 with or without a dashed frame. It could be seen from the dotted frame that Fe2O3 had the morphology of short nanorods. The result was consistent with the analyses of FESEM. Fig. 3b is the TEM image with 50000× magnification of Fe2O3/Co–Pi. In this image, the red circle showed some small particles on the top of the nanorods. The particles were densely distributed, with a particle size of about 10 nm. Fig. 3c is the HRTEM image of bare α-Fe2O3. There was only one lattice fringe with a lattice spacing of 0.25 nm in it, which corresponded to the (110) crystal plane of α-Fe2O3.29 In the HRTEM diagram of α-Fe2O3/Co–Pi shown in Fig. 3d, the red dotted line indicated that an amorphous Co–Pi layer was formed on the edge of α-Fe2O3.30 It showed that Co–Pi was successfully deposited on the surface of α-Fe2O3. It could be found that the thickness of the amorphous Co–Pi layer was only ∼2 nm, which inferred that Co and P could not be detected in the EDS measurements.
 |
| | Fig. 3 (a) TEM image with 50000× magnification of α-Fe2O3. (b) TEM image with 50000× magnification of α-Fe2O3/Co–Pi. (c) HRTEM image of α-Fe2O3, and (d) HRTEM image of α-Fe2O3/Co–Pi. | |
XRD and Raman.
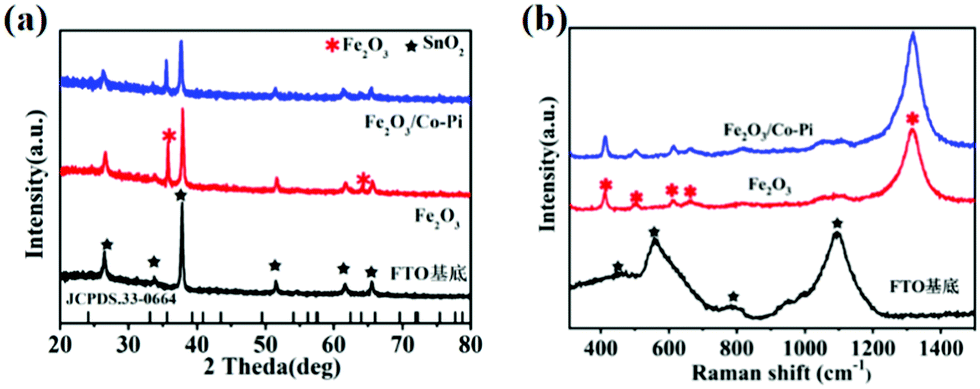
XRD measurements were conducted to obtain the phase information of all samples. The XRD spectra for the FTO substrate, Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C, as well as Fe2O3/Co–Pi photoanodes are shown in Fig. S4 (ESI†) and Fig. 4a. Fig. S4 (ESI†) shows that the hematite calcined at various temperatures of 500, 525, 550, 575, and 600 °C had the characteristic diffraction peaks of SnO2 (JCPDS No. 46-1088) from the FTO substrate. The diffraction peaks located at 35.7° and 64.1° corresponded to the (110) and (330) crystal planes of α-Fe2O3 (JCPDS No. 33-0664), respectively. As was shown in Fig. 4a, after the modification of the Co–Pi groups, there was no new diffraction peak in the XRD pattern of Fe2O3/Co–Pi. It indicated that Co–Pi was amorphous, which was consistent with the HRTEM results. Raman spectroscopy could obtain the lattice vibration information of the samples. The Raman spectra for the FTO substrate, Fe2O3, and Fe2O3/Co–Pi photoanodes are shown in Fig. 4b. The Raman spectrum of the FTO substrate had four peaks located at 451, 557, 788, and 1090 cm−1, which were attributed to the peaks of SnO2. After Fe2O3 deposition on the surface of the substrate by the hydrothermal process, the vibration intensity of the FTO substrate became weaker. There were obvious Raman peaks of hematite, located at 413, 503, 613, 663, and 1318 cm−1. The positions of these peaks were close to the standard peaks of α-Fe2O3.31 The peaks at 413 and 613 cm−1 were attributed to the Eg vibration mode of hematite, and the peak at 613 cm−1 was attributed to the A1g vibration mode of hematite. At 667 cm−1, an infrared (IR) active longitudinal optical (LO) Eu mode could be detected. It was caused by disorder in the α-Fe2O3 lattice.32 The peak at 1320 cm−1 was derived from the dispersion of the two magnons, which was produced by the interaction of the two magnons in the antiparallel closing direction of the spins.31 The above results indicated that α-Fe2O3 was successfully synthesized without any other impurities. After the deposition of Co–Pi, there was no new Raman peak in Fe2O3/Co–Pi. It indicated that Co–Pi has not entered the lattice of Fe2O3.
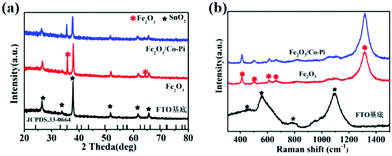 |
| | Fig. 4 (a) XRD pattern and (b) Raman spectra of the FTO substrate, bare Fe2O3, and Fe2O3/Co–Pi photoanodes, SnO2 (black five-pointed star), α-Fe2O3 (red snowflake). | |
XPS.
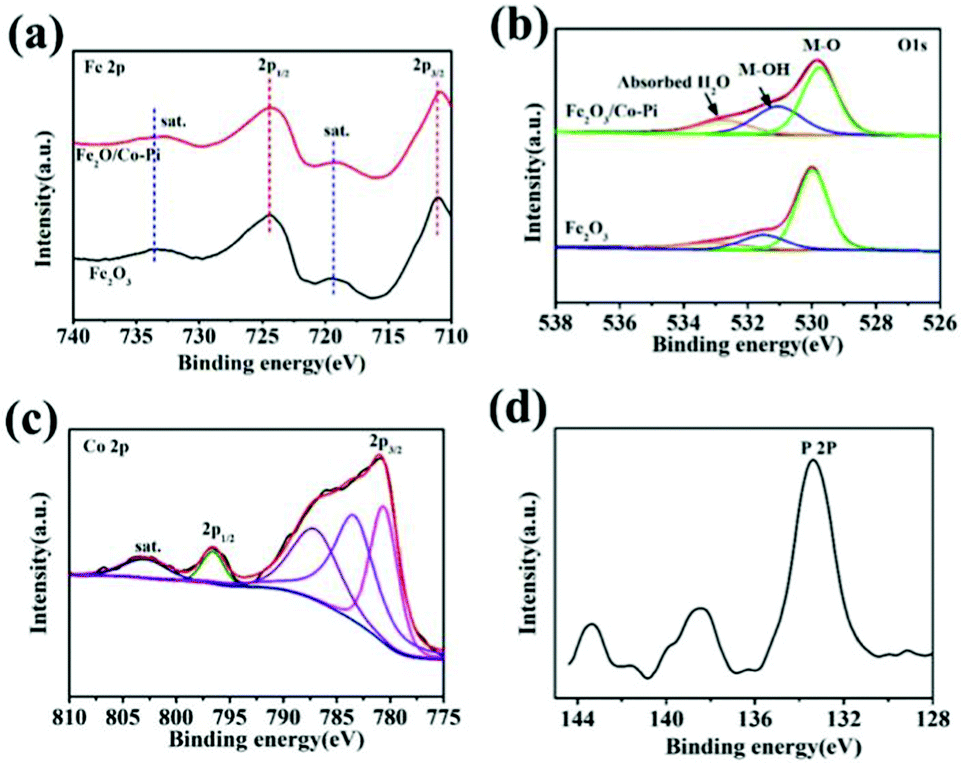
XPS characterization can confirm the element composition and the oxidation state of the element on the surface of samples. The XPS spectra and fitted spectra of the Fe2O3 and Fe2O3/Co–Pi photoanodes are shown in Fig. 5. Among them, Fig. 5a–d are the spectra of Fe 2p, O 1s, Co 2p, and P 2P, respectively. In Fig. 5a, the XPS spectrum of Fe 2p showed that the binding energies of 2p1/2 and 2p3/2 were 724.5 eV and 710.7 eV, respectively. There were two satellite peaks located at 719.3 eV and 733.5 eV. It indicated that there were hematite phase iron oxides in both Fe2O3 and Fe2O3/Co–Pi photoanodes, while there were no Fe(II) and Fe(0) peaks. This analysis was consistent with the XRD and Raman characterization. The peaks at 529.9, 531.5, and 532.7 eV corresponded to the lattice oxygen (M–O), the adsorbed hydroxyl (M–OH lattice) and the hydroxyl in the adsorbed water molecule in iron oxide in Fig. 5b. After the modification of the Co–Pi groups, the binding energy of the M–O bond was negatively shifted by 0.23 eV, which indicated that the electron density near the Fe site increased. It could be observed that after CoPi modification, the peak intensity of M–OH increased. This was because the sample used in the XPS characterization was photoelectrically tested. This made the CoPi group oxidized, and the oxidized CoPi was composed of CoOOH.33 After CoPi deposition, the peak intensity also increased. This was due to the fact that the loading of CoPi would cause more water molecules to be adsorbed on the Fe2O3 surface. It would promote the water oxidation reaction, thereby improving the PEC performance. The spectrum of Co 2p in Fig. 5c showed the peaks at 780.6 eV and 796.6 eV corresponding to the signals of 2p3/2 and 2p3/2, respectively, which were characteristic peaks of Co2+ and Co3+. The peaks at 787.0 eV and 802.8 eV also confirmed the presence of Co2+. In Fig. 5d, the main characteristic peak of P 2p was about 133.4 eV, which corresponded to the phosphate group in PO43−.28 Table S2 (ESI†) presents the contents of each element for Fe2O3 and Fe2O3/Co–Pi photoanodes obtained from XPS characterization. After the modification of the Co–Pi groups, the contents of the Co and P elements in the sample were 1.09 at% and 1.38 at%, respectively. It indicated that the Co–Pi deposited by the immersion process was very small.
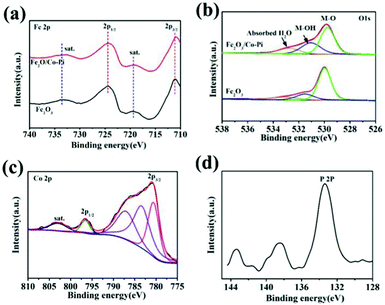 |
| | Fig. 5 XPS spectra and fitted spectra of Fe2O3 and Fe2O3/Co–Pi photoanodes: (a) Fe 2p, (b) O 1s, (c) Co 2p, (d) P 2P. | |
UV-vis absorption spectrum
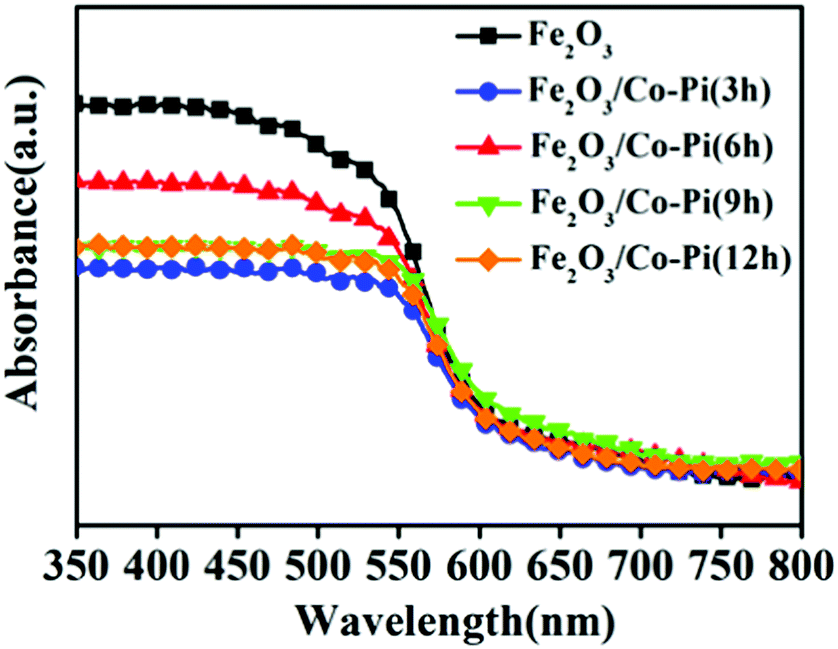
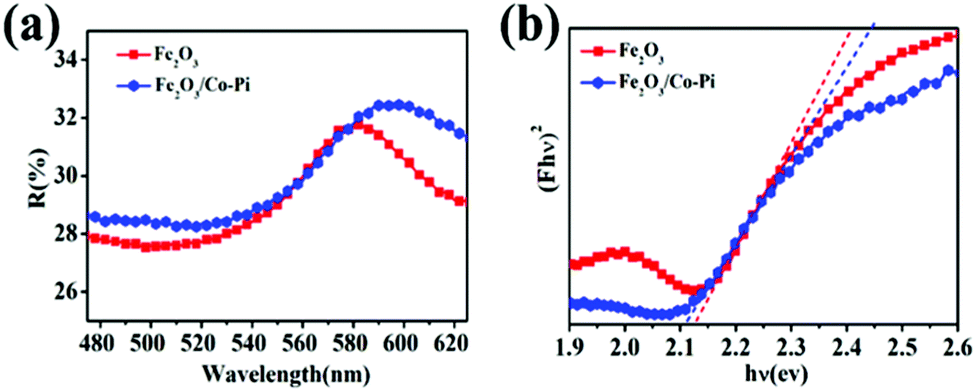
For PEC water splitting, the optical property of the semiconductor materials is one of the important factors. Hence, UV-vis absorption spectra were measured to study the light absorption ability of bare Fe2O3 with various hydrothermal times of 2, 4, 6, 8, and 10 h, various concentrations of the Fe(NO3)3·9H2O precursor of 2, 4, 6, 8, 10, and 12 mM, various calcination temperature of 500, 525, 550, 575, and 600 °C, as well as Fe2O3/Co–Pi photoanodes after immersion in phosphate for various times of 3, 6, 9, and 12 h in Fig. S5–S7 (ESI†) and Fig. 6. Fig. S5 (ESI†) shows that when the hydrothermal time increased from 2 h to 8 h, the light absorption in the 350–800 nm range gradually increased. Fig. S6 (ESI†) indicates that the light absorption in the range of 350–800 nm increased with the increase of the Fe precursor concentration. This was because as the hydrothermal time and concentration of the precursor increased, the thickness of the iron oxide film also increased, thereby increasing the light absorption. Fig. S7 (ESI†) shows that the light absorption of Fe2O3 in the range of 350–525 nm increased with the increasing calcination temperature. This was because the enhanced thickness promotes light absorption ability, which was consistent with SEM analysis. In order to obtain the optimal loading of the Co–Pi groups, we changed the immersion time of the α-Fe2O3 photoanode in 0.1 mol L−1 Na3PO4 solution and 0.01 mol L−1 Co(NO3)2 solution to explore the relationship between the loading and α-Fe2O3 photoelectric performance. The time of the two-step dipping always retained consistent changes throughout the experiment. Fig. 6 is the UV-vis absorption spectra of the Fe2O3/Co–Pi photoanode after immersion in phosphate for various times of 3, 6, 9, and 12 h. It showed that after depositing various amounts of Co–Pi on the surface of α-Fe2O3, the light absorption in the 350–550 nm range decreased. As the immersion time increased, the light absorption decreased slightly, which showed that the modification of the Co–Pi groups did not improve the photoelectric performance by enhancing light absorption. Comparatively speaking, when the two-step immersion time was 6 h, the light absorption of the Fe2O3/Co–Pi photoanode was the best, so we compared the reflection spectrum and band gap of Fe2O3/Co–Pi (6 h) with bare Fe2O3. Fig. 7a is the reflectance curve of Fe2O3 and Fe2O3/Co–Pi photoanodes. Fig. 7b is the (Fhν)2 − (hν) curve, which was calculated by the reflectivity of the Fe2O3 and Fe2O3/Co–Pi photoanodes. The band gaps of bare Fe2O3 and Fe2O3/Co–Pi obtained by extrapolation were 2.11 eV and 2.12 eV, respectively. The data indicated that the modification of Co–Pi groups would not change the internal energy band structure of Fe2O3.
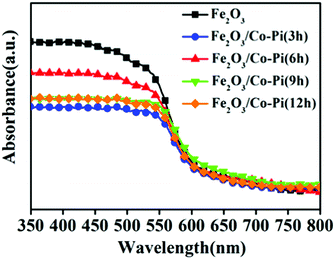 |
| | Fig. 6 UV-vis absorption spectra of the Fe2O3/Co–Pi photoanodes after immersion in phosphate for various time of 3, 6, 9, and 12 h. | |
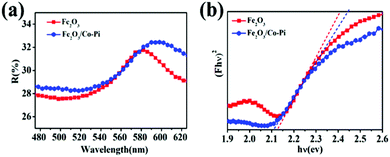 |
| | Fig. 7 (a) Reflectance-wavelength curve of Fe2O3 and Fe2O3/Co–Pi photoanodes, (b) (Fhν)2 − (hν) curves of Fe2O3 and Fe2O3/Co–Pi photoanodes. | |
Photoelectrochemical tests
LSV and chopping I–t curves.
The photocurrent densities under constant and transient condition were measured to study the PEC performance in the solar water splitting. The LSV curves of bare α-Fe2O3 prepared under various hydrothermal times of 2, 4, 6, 8, and 10 h and various precursor concentration conditions of 2, 4, 6, 8, 10, and 12 mM are shown in Fig. S8 and S9 (ESI†). Fig. S8 (ESI†) shows that when the hydrothermal time increased from 2 h to 4 h, the photocurrent density at 1.23 VRHE increased from 0.035 mA cm−2 to 0.084 mA cm−2. However, when the hydrothermal reaction time continued to increase, the photocurrent density decreased instead. This was due to the fact that the extension of the hydrothermal reaction time would increase the thickness of α-Fe2O3, resulting in a longer distance for holes to migrate to the surface. It facilitated the recombination of the holes with the electrons. Fig. S9 (ESI†) shows that in the range of 1.0–1.6 VRHE, as the concentration of Fe(NO3)3·9H2O increased from 2 mM to 12 mM, the photocurrent density of the samples first increased and then decreased. This was because as the concentration of the precursor increases, the thickness of the Fe2O3 film also increased. This increased the light absorption and photocurrent density. However, if the precursor concentration continued to increase, the thickness of the film would be too large. Since the hole diffusion distance of iron oxide is only 2–4 nm, a thickness that is too large could easily recombine electrons and holes.
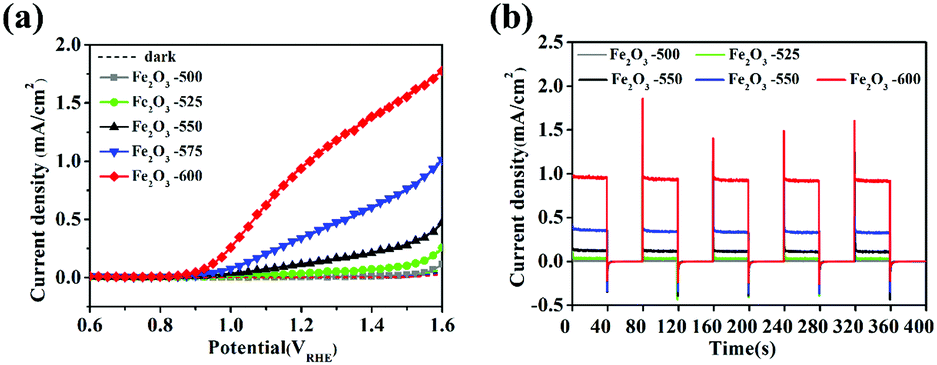
Fig. 8a is the LSV curves of bare Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C. It showed that the photocurrent densities of α-Fe2O3 calcined at 500 °C, 525 °C, 550 °C, 575 °C and 600 °C at 1.23 VRHE were 0.012, 0.047, 0.18, 0.38, and 1.02 mA cm−2, respectively. The current density of all samples in the dark state was almost zero. With the increased calcination temperature, the photocurrent density in the range of 1.1–1.6 VRHE significantly increased. Fig. 8b is the I–t curves of bare Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C under chopped light. It showed that the transient photoelectric response at 1.23 VRHE increased with the increase of the calcination temperature. When the calcination temperature was 600 °C in air condition, α-Fe2O3 had the highest transient photocurrent density. There was still an obvious peak current on transient PEC detection, which indicated that the charge recombination happened at the surface of the bare α-Fe2O3 semiconductor. The results in Fig. S6, S7 (ESI†), and Fig. 8 indicated that the temperature during the annealing had a greater impact on the PEC performance of Fe2O3. The optimum synthesis conditions of bare Fe2O3 were a hydrothermal time of 4 h, iron precursor concentration of 6 mM and calcination temperature of 600 °C for 2 h.
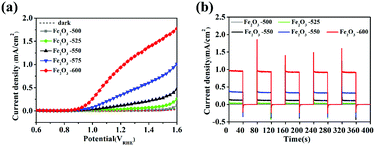 |
| | Fig. 8 (a) LSV curves of Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C; (b) Chopping I–t curves of Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C under chopped light, and the dotted line represents the dark state current density. | |
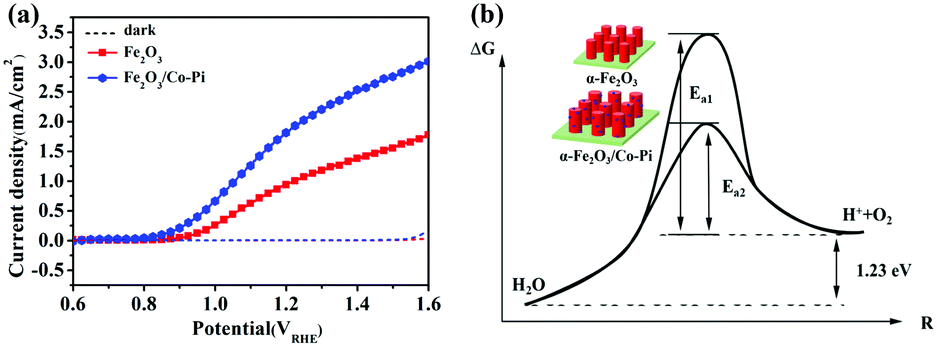
Fig. 9a is the LSV curve of the Fe2O3 and Fe2O3/Co–Pi photoanodes. It showed that the photocurrent density of Co–Pi modified Fe2O3 at 1.23 VRHE increased from 1.02 mA cm−2 to 1.95 mA cm−2. The turn-on potential was reduced from 0.85 VRHE to 0.78 VRHE, which was negatively shifted by 70 mV. Based on the law of the energy conservation
| | | Photocatalysis: Eg ≥ E° + Ea | (9) |
| | | Photoelectrocatalysis: Eg + Ev + E° + EAOP | (10) |
| | 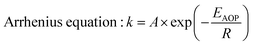 | (11) |
where
E° is the chemical energy at the standard potential for O
2 evolution (1.23 V),
Ea is the activation energy, and
EAOP is the activation energy in PEC water oxidation; the activation overpotential is denoted as AOP;
Eg is the band gap of Fe
2O
3;
EV is the externally applied electric energy at the onset potential.
 |
| | Fig. 9 (a) LSV curve of the Fe2O3 and Fe2O3/Co-Pi photoanodes under dark and light condition. (b) Schematic diagram of Fe2O3/Co–Pi reducing the activation energy of the water oxidation reaction. | |
For the water splitting reaction, the activation energy of the hydrogen evolution reaction (HER) is very low. Thus, here we mainly discussed the activation energy of water oxidation. After Co–Pi was deposited, the initial potential of all samples shifted negatively by about 70 mV. This indicated that less EV was applied during the PEC water oxidation process. Co–Pi deposited on the Fe2O3 electrode could reduce the EAOP of PEC water oxidation, thereby increasing the rate of OER. Fig. 9b is the schematic diagram of Fe2O3/Co–Pi reducing the activation energy of the water oxidation reaction. This result showed that the Co–Pi groups effectively reduced the water oxidation overpotential of Fe2O3 and accelerated the reaction.
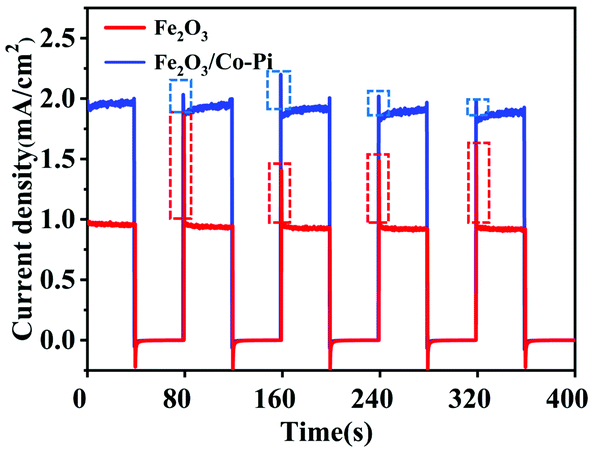
Fig. 10 is the chopping I–t curve of the Fe2O3 and Fe2O3/Co–Pi photoanodes. It showed the transient current–time curve of the samples before and after modification under visible light. During the illumination period, the photocurrent density of Fe2O3 was about 0.9 mA cm−2, and the photocurrent density of Fe2O3 modified with Co–Pi groups was about 1.8 mA cm−2. The dotted frame in Fig. 10 was the peak-shaped current curve at the moment of turning on the light. With illumination, sharp anodic spikes were observed in the case of bare Fe2O3. This was due to the accumulation of photo-generated holes at the photoanode/electrolyte interface and were subsequently used in the recombination process. These transient anode spikes were reduced in the presence of CoPi. This meant that the holes in the Fe2O3 valence band (VB) were rapidly consumed at the semiconductor/electrolyte interface, so that the electron–hole pair recombination process on the electrode surface was reduced.
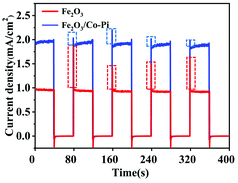 |
| | Fig. 10 Chopping I–t curve of the Fe2O3 and Fe2O3/Co–Pi photoanodes. The dotted line represents the current density in the dark state, and the dotted frame in figure is the peak-shaped current curve at the moment of turning on the light. | |
Applied bias photon-to-current efficiency (ABPE), charge separation efficiency and charge injection efficiency analysis
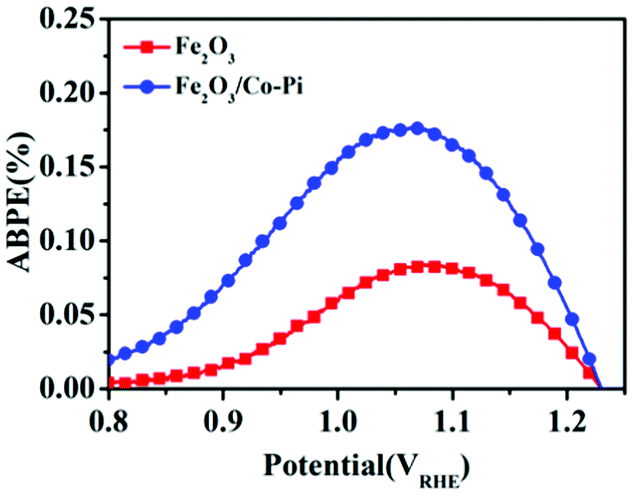
Fig. 11 is the ABPE curves of the Fe2O3 and Fe2O3/Co–Pi photoanodes. It showed that within the voltage range of 0.8–1.23 VRHE, the ABPE of Fe2O3 and Fe2O3/Co–Pi photoanodes both showed a trend of first increasing and then decreasing. The Fe2O3 photoanode had the largest ABPE at 1.05 VRHE with a value of 0.08%. The Fe2O3/Co–Pi photoanode had an ABPE of 0.17% at 1.05 VRHE, which was 2.1 times that of Fe2O3.
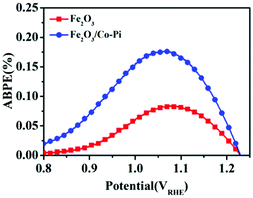 |
| | Fig. 11 Applied bias photon-to-current efficiency (ABPE) curves of Fe2O3 and Fe2O3/Co-Pi photoanodes. | |
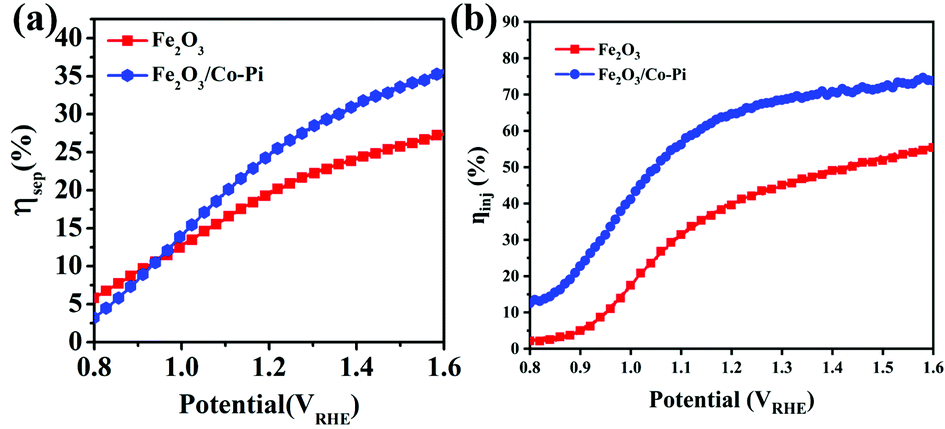
Fig. 12a and b compares the charge separation efficiency (ηsep) and charge injection efficiency (ηinj) of the photoanodes before and after modification. Fig. 12a shows that the ηsep of Fe2O3 at 1.23 VRHE was 20.39%. After Co–Pi modification, Fe2O3 had a similar ηsep with a value of 25.77%. Fig. S11a and b (ESI†) are the LSV curves of bare Fe2O3 and Fe2O3/CoPi without and with 0.5 M Na2SO3. From the curves, the ηinj of Fe2O3 and Fe2O3/Co–Pi could be calculated. The ηinj of Fe2O3 and Fe2O3/Co–Pi at 1.23 VRHE shown in Fig. 12b were 41.76% and 66.67%, respectively. It could be seen that through the modification of Co–Pi, the ηinj of Fe2O3/Co–Pi was 1.6 times that of bare Fe2O3. Combining Fig. 12a and b, it could be obtained that the ηinj of Fe2O3 was improved even more after CoPi modification. This indicated that CoPi groups were more often used as a water oxidation promoter to accelerate the water oxidation reaction (OER).
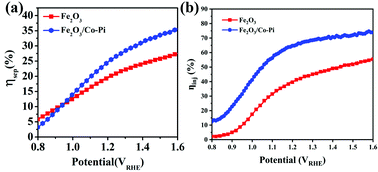 |
| | Fig. 12 (a) Charge separation efficiency and (b) charge injection efficiency of bare Fe2O3 and Fe2O3/Co–Pi photoanodes. | |
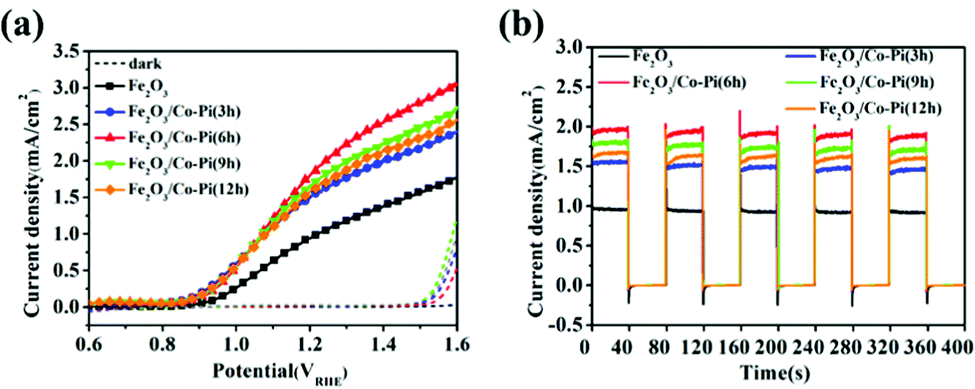
Fig. 13a is the LSV curve of the Fe2O3 and Fe2O3/Co–Pi photoanodes obtained after immersion in phosphate for various times of 3, 6, 9, and 12 h. It showed that after depositing various amounts of Co–Pi groups on the surface of Fe2O3, the photocurrent density in the entire test voltage range was higher than that of the bare Fe2O3. As the immersion time increased from 3 h to 6 h, the photocurrent density gradually increased. However, when the immersion time continued to increase to 12 h, the photocurrent density of Fe2O3 decreased instead. This result was consistent with the phenomenon shown in Fig. 6 that the immersion time was inversely proportional to the light absorption. Fig. 13b is the current–time curve of Fe2O3 and Fe2O3/Co–Pi photoanodes obtained after immersion in phosphate for various times of 3, 6, 9, and 12 h. As shown, the transient photocurrent density at 1.23 VRHE showed a trend of first increasing and then decreasing with the increase of Co–Pi loading. This trend was consistent with the change of the photocurrent density of the LSV curve. When the immersion time in Na3PO4 solution and Co(NO3)2 solution was 6 h, α-Fe2O3/Co–Pi had the best photoelectric performance. Its photocurrent density at 1.23 VRHE was 1.95 mA cm−2, and its on-potential was 0.78 VRHE.
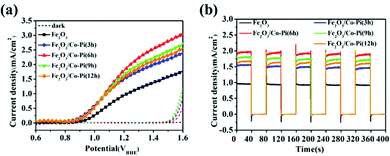 |
| | Fig. 13 (a) LSV curve of the Fe2O3 and Fe2O3/Co–Pi photoanodes obtained after immersion in phosphate for various time of 3, 6, 9, and 12 h and (b) current–time curve under chopped wave, and the dotted line represents the current in the dark state density. | |
Electrochemical results
In order to better understand the nature of the interface between the electrode and the electrolyte, we conducted electrochemical impedance tests on the Fe2O3 and Fe2O3/Co–Pi photoanodes. The EIS Nyquist diagram of Fe2O3 calcined at various temperatures of 500, 525, 550, 575, and 600 °C is shown in Fig. S12 (ESI†). When the calcination temperature increased from 500 °C to 575 °C, the radius of the arc in the high-frequency region of the Nernst diagram gradually decreased. It indicated that the charge transfer resistance decreased. When the calcination temperature continued to rise to 600 °C, the charge transfer resistance increased instead. This was mainly due to the fact that too high temperature would destroy the SnO2 layer on the surface of the conductive substrate, thereby reducing the conductivity of the substrate. Fig. S13 (ESI†) is the digital photo of bare Fe2O3 calcined at 600 °C. Due to the high calcination temperature, the FTO substrate in the picture had been slightly deformed. Combining the SEM analysis, it could be seen that when the calcination temperature increased from 500 °C to 600 °C, the film thickness increased by 110 nm. This increased the thickness of the electric double layer, resulting in an increase in resistance during the charge transfer process. The EIS Nyquist plot for the Fe2O3/Co–Pi photoanode after immersion in phosphate for various time of 3, 6, 9, and 12 h is shown in Fig. S14 (ESI†). The figure showed that the Co–Pi modified sample had a reduced arc radius in the high frequency region of the Nernst plot. It indicated that depositing the right amounts of Co–Pi groups would reduce the charge transfer resistance.
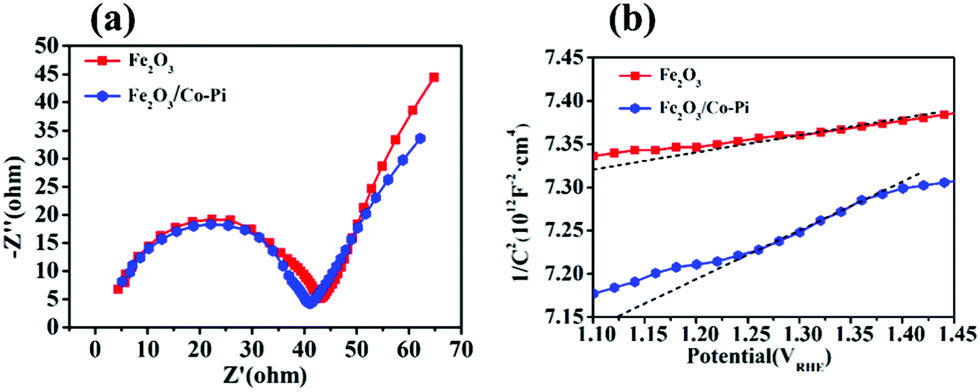
Fig. 14a is the EIS spectrum of the Fe2O3 and Fe2O3/Co–Pi photoanodes. The curve in the figure showed that the radius of the arc of Fe2O3/Co–Pi in the high frequency region was smaller than that of bare Fe2O3. It indicated that the deposition of CoPi changed the electrode/electrolyte interface performance. After the holes in the VB reach the surface of Fe2O3, they were extracted by the CoPi covered on the surface. During the subsequent catalytic water oxidation, CoPi undergoes proton-coupled electron transfers along with cyclic valency changes between Co(II/III) and Co(III/IV). As an effective hole extractor and redox mediator, CoPi could inhibit the recombination of photo-generated carriers at the semiconductor/electrolyte interface and improve the charge transport capability.34Fig. 14b is the Mott–Schottky plots of the bare Fe2O3 and Fe2O3/Co–Pi photoanodes, which showed that the slopes of all samples were positive. It inferred that the Fe2O3 and Fe2O3/Co–Pi photoanodes were both n-type semiconductors and the majority carriers were electrons. The carrier density of the Fe2O3 and Fe2O3/Co–Pi photoanodes could be calculated by the slope of the M–S curve, which were 1.03 × 1017 cm−3 and 3.48 × 1016 cm−3, respectively. The data indicated that the modification of the Co–Pi groups did not increase the carrier density.
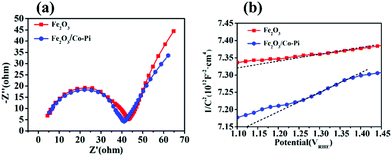 |
| | Fig. 14 (a) EIS Nyquist plots of bare Fe2O3 and Fe2O3/Co–Pi photoanodes measured at a bias of 1.23 VRHE under simulated solar light illumination, (b) Mott–Schottky plots of bare Fe2O3 and Fe2O3/Co–Pi photoanodes. | |
Mechanism analysis
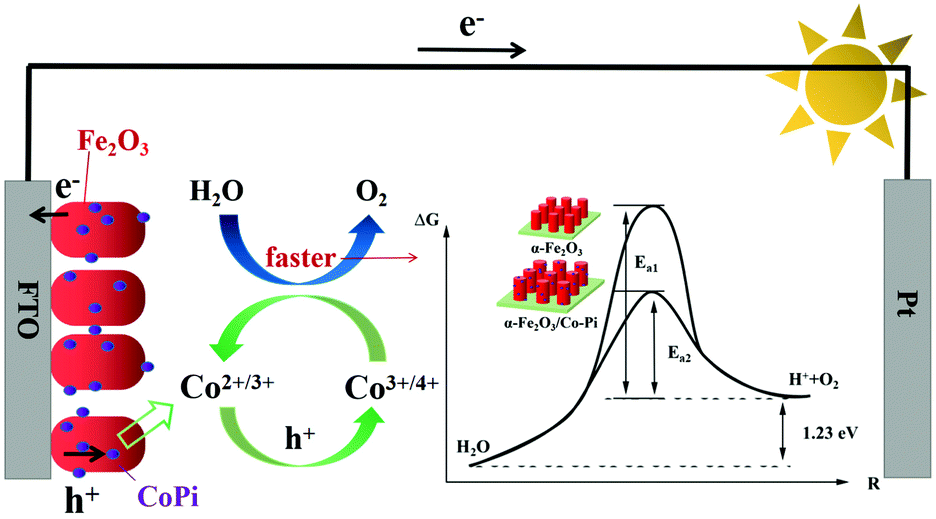
Fig. 15 is the schematic diagram of the mechanism of Co–Pi enhancing the photoelectric performance of the Fe2O3 photoanode. According to the transient I–t curve, the transient anodic spike of Fe2O3 decreased after CoPi deposition. This indicated that CoPi extracts holes on the surface of Fe2O3, thereby inhibiting the accumulation of holes on the electrode surface. The holes transferred to the promoter layer had strong oxidizing ability. This made the Co element in the low-valence state (II/III) oxidized to the high-valence state (III/IV). Then, the high-valence Co got electrons from the water and was re-reduced to the low-valence Co. In the above process, CoPi acted as a redox mediator. According to the LSV curve, after CoPi deposition, the initial potential of Fe2O3 moved negatively from 0.85 VRHE to 0.78 VRHE. According to the law of conservation of energy and the Arrhenius equation, the following conclusions could be derived. CoPi deposited on the Fe2O3 electrode could reduce the EAOP of PEC water oxidation, thereby increasing the rate of OER. CoPi not only acted as a hole extractor and redox mediator, but also a common co-catalyst to promote OER. Through the synergistic effect of CoPi, the PEC performance of Fe2O3 photoanode was improved. The ABPE of Fe2O3/Co–Pi photoanode was 1.51% (4.87 times that of Fe2O3). The ηinj of Fe2O3/Co–Pi was 66.67%, which was 1.6 times that of Fe2O3.
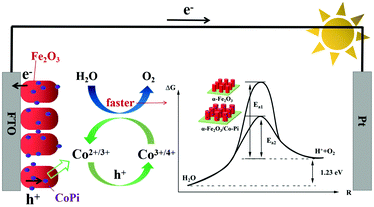 |
| | Fig. 15 Schematic diagram of the mechanism of Co–Pi enhancing the photoelectric performance of the Fe2O3 photoanode. | |
Conclusions
In this paper, a hydrothermal process was used to prepare bare α-Fe2O3, and Co-Pi groups were deposited on the surface of the Fe2O3 photoanode through a two-step dipping process. The effects of the hydrothermal time, concentration of the Fe precursor, annealing temperature, as well as loading of Co–Pi on the performance of the α-Fe2O3 photoanode were studied. SEM showed that Fe2O3 is composed of a short nanorod, and the thickness of the Fe2O3 layer increased with the increase of the calcination temperature. There were no peaks of Co–Pi groups in the XRD of Fe2O3/CoPi, and there were no lattice fringes of Co–Pi groups in the TEM of Fe2O3/CoPi. The Co and P elements signals in XPS indicated that the Co–Pi groups were indeed deposited on the surface of Fe2O3. These measurements showed that the Co–Pi groups on the Fe2O3/CoPi photoanode were amorphous. When the Fe precursor concentration was 6 mM, the hydrothermal reaction time was 4 h, and the calcination temperature was 600 °C, the bare Fe2O3 photoanode had the best photoelectric performance. The photocurrent density of the optimized sample at 1.23 VRHE was 1.02 mA cm−2, which was 29 times that of the sample without conditional exploration. The photocurrent density of Fe2O3/Co–Pi at 1.23 VRHE was 1.95 mA cm−2. CoPi made the initial potential of Fe2O3 negatively shifted by 70 mV. According to the law of conservation of energy, it could be deduced that CoPi acted as an effective co-catalyst to reduce the activation energy of OER. The transient J–t curve showed that the transient anodic spike of Fe2O3 decreased after CoPi deposition. It indicated that the charge accumulation on the surface of Fe2O3/CoPi was reduced. EIS showed that the electrode/electrolyte interface resistance of Fe2O3/CoPi decreased. It showed that CoPi could change the interface performance. Combined with the transient I–t curve and EIS analysis, it could be obtained that CoPi could be used as a hole extractant and a redox mediator to promote charge injection. Through the synergy of CoPi, the ABPE of the Fe2O3/Co–Pi photoanode was 0.17% (2.1 times that of bare Fe2O3). The ηinj of the Fe2O3/Co–Pi photoanode at 1.23 VRHE was 1.6 times that of bare Fe2O3. It was hoped that this work could provide more insights into the synthesis of the Fe2O3 photoanodes, and the role of CoPi on Fe2O3 and other semiconductor photoanodes.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundations of China (No. 21805040/B0605), the Natural Science Foundation of Fujian Province of China (No. 2018J05015), and the Foundation of Fujian Educational Committee (No. JAT170131). We gratefully acknowledge the help of the Dalian Institute of Chemical Physics, Chinese Academy of Sciences.
References
- Y. Zhao, C. Ding, J. Zhu, W. Qin, X. Tao, F. Fan, R. Li and C. Li, Angew. Chem., Int. Ed., 2020, 59, 9653–9658 CrossRef CAS PubMed.
- C. Xu, W. Sun, Y. Dong, C. Dong, Q. Hu, B. Maa and Y. Ding, J. Mater. Chem. A, 2020, 8, 4062–4072 RSC.
- X. Chai, H. Zhang, Q. Pan, J. Bian, Z. Chen and C. Cheng, Appl. Surf. Sci., 2019, 470, 668–676 CrossRef CAS.
- F. Feng, C. Li, J. Jian, X. Qiao, H. Wang and L. Jia, Chem. Eng. J., 2019, 368, 959–967 CrossRef CAS.
- P. S. Basavarajappa, S. B. Patil, N. Ganganagappa, K. R. Reddy, A. V. Raghu and C. V. Reddy, Int. J. Hydrogen Energy, 2020, 45, 7764–7778 CrossRef CAS.
- S. Bai, H. Chu, X. Xiang, R. Luo, J. He and A. Chen, Chem. Eng. J., 2018, 350, 148–156 CrossRef CAS.
- Q. Sun, T. Cheng, Z. Liu and L. Qi, Appl. Catal., B, 2020, 277, 119189 CrossRef CAS.
- M. Zhao, T. Chen, B. He and X. Hu, J. Mater. Chem. A, 2020, 8, 15976–15983 RSC.
- L. Yu, Y. Zhang, J. He, H. Zhu, X. Zhou, M. Li, Q. Yang and F. Xu, J. Alloys Compd., 2018, 753, 601–606 CrossRef CAS.
- R. L. Spray and K. S. Choi, Chem. Mater., 2009, 21, 3701–3709 CrossRef CAS.
- X. Wang, Y. Tuo, Y. Zhou, D. Wang, S. Wang and J. Zhang, Chem. Eng. J., 2021, 403, 126297 CrossRef CAS.
- J. Wang, N. H. Perry, L. Guo, L. Vayssieres and H. L. Tuller, ACS Appl. Mater. Interfaces, 2019, 11, 2031–2041 CrossRef CAS PubMed.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- K. Sivula, F. F. Le and M. Graetzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- J. Li, J. Li, H. Yuan, W. Zhang, Z. Jiao and X. S. Zhao, Chem. Eng. J, 2020, 398, 125662 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Gratzel, Angew. Chem., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS PubMed.
- J. Y. Kim, G. Magesh, D. H. Youn, J. W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681–2688 CrossRef PubMed.
- Y. Hou, Z. H. Wen, S. M. Cui, X. L. Feng and J. H. Chen, Nano Lett., 2016, 16, 2268–2277 CrossRef CAS PubMed.
- Q. J. Bu, S. Li, K. Zhang, Y. H. Lin, D. J. Wang, X. X. Zou and T. F. Xie, ACS Sustainable Chem. Eng., 2019, 7, 10971–10978 CrossRef CAS.
- J. M. Feckl, H. K. Dunn, P. M. Zehetmaier, A. Müller, S. R. Pendlebury, P. Zeller, K. Fominykh, I. Kondofersky, M. Döblinger, J. R. Durrant, C. Scheu, L. Peter, D. Fattakhova-Rohlfing and T. Bein, Adv. Mater. Interfaces, 2015, 2, 1500358 CrossRef.
- F. Li, Z. Zhao, H. Yang, D. Zhou, Y. Zhao, Y. Li, W. Li, X. Wu, P. Zhang and L. Sun, Dalton Trans., 2020, 49, 588–592 RSC.
- C. Du, X. G. Yang, M. T. Mayer, H. Hoyt, J. Xie, G. McMahon, G. Bischoping and D. W. Wang, Angew. Chem., Int. Ed., 2013, 152, 12924–12927 CrossRef.
- Q. Yu, X. Meng, T. Wang, P. Li and J. Ye, Adv. Funct. Mater., 2018, 25, 2686–2692 CrossRef.
- C. H. Liu, Y. Xu, H. Luo, W. C. Wang, Q. Liang and Z. D. Chen, Chem. Eng. J., 2019, 363, 23–32 CrossRef CAS.
- T. Wang, X. Long, S. Wei, P. Wang, C. Wang, J. Jin and G. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 49705–49712 CrossRef CAS PubMed.
- S. N. S. Moraes, D. L. Morgado and M. Nalin, Vib. Spectrosc., 2019, 100, 57–63 CrossRef CAS.
- L. Hu, X. Zeng, X. Wei, H. Wang, Y. Wu, W. Gu, L. Shi and C. Zhu, Appl. Catal., B, 2020, 273, 119014 CrossRef CAS.
- C. G. Shull, W. A. Strauser and E. O. Wollan, Phys. Rev., 1951, 83, 333–345 CrossRef CAS.
- C. H. Li, C. L. Huang, X. F. Chuah, D. S. Raja, C. T. Hsieh and S. Y. Lu, Chem. Eng. J., 2019, 361, 660–670 CrossRef CAS.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- N. M. Randall, J. Qiu and F. Laskowski, ACS Energy Lett., 2018, 2286–2291 Search PubMed.
- S. Alam, T. K. Sahu and M. Qureshi, ACS Sustainable Chem. Eng., 2021, 9, 5155–5165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Including SEM, TEM and XRD images of Fe2O3, the content of each element of Fe2O3 and Fe2O3/CoPi obtained from EDS and XPS, the ultraviolet-visible absorption spectra, stability test, EIS spectra of Fe2O3 and Fe2O3/CoPi, as well as LSV curves of Fe2O3 and Fe2O3/CoPi with Na2SO3, etc. Full experiment details and the additional figures are present See DOI: 10.1039/d1nj04516d |
| ‡ Ai Qin and Wanqing Fang contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*