
Large polarization of push–pull “Cruciforms” via coordination with lanthanide ions†
Sara
Benedini
a,
Yeting
Zheng
b,
Andrea
Nitti
 a,
Mercedes M. A.
Mazza
b,
Daniele
Dondi
a,
Françisco M.
Raymo
b and
Dario
Pasini
*a
a,
Mercedes M. A.
Mazza
b,
Daniele
Dondi
a,
Françisco M.
Raymo
b and
Dario
Pasini
*a
aDepartment of Chemistry and INSTM Research Unit, University of Pavia, Via Taramelli 10 – 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it
bLaboratory for Molecular Photonics, Department of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146-0431, USA
First published on 29th November 2021
Abstract
We report on two different series of conjugated push–pull compounds bearing dimethylamino donor and 1,3-dicarbonyl acceptor functionalities with a “bent” or“cruciform” shape, exhibit striking differences in their supramolecular recognition of lanthanide cations. Both series of compounds are effective to coordinate lanthanide cations (Sc3+ and Eu3+) in MeCN, CHCl3 or MeOH solutions to the 1,3-dicarbonyl acceptor end. The stoichiometry of the complexes are largely dependent on the nature of the lanthanide cation, with Sc3+ forming preferentially 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 metal:ligand complexes, and Eu3+ forming 1:1 metal:ligand complexes. The coordination of the lanthanide cation results in an overall supramolecular polarization of the system and the redshifting of the intramolecular charge transfer band. The polarization of the “cruciform” systems is greatly enhanced with respect to the “bent” chromophores, reaching outstanding values for the red shift (238 nm, corresponding to an energy difference of 1.23 eV) with high binding constants (LogKa >5.5).
:3 metal:ligand complexes, and Eu3+ forming 1:1 metal:ligand complexes. The coordination of the lanthanide cation results in an overall supramolecular polarization of the system and the redshifting of the intramolecular charge transfer band. The polarization of the “cruciform” systems is greatly enhanced with respect to the “bent” chromophores, reaching outstanding values for the red shift (238 nm, corresponding to an energy difference of 1.23 eV) with high binding constants (LogKa >5.5).
Introduction
Conjugated molecules, oligomers and polymers are increasingly used for a variety of applications, which often need a structuring of the organic chromophores in thin films or in the solid state.1 Supramolecular chemistry has a fundamental role in the morphological structuring of the device and can provide, given the reversibility of the noncovalent bonds, rearrangement/reorganization/self-healing properties to the chromophores in the thin film.2The possibility of changing the electronic properties of the π-extended chromophores by a supramolecular coordination event is, instead, relatively less explored. This concept has been developed in the design of molecular sensors, using the changes in the electrochemical/emission properties of the conjugated moiety upon complexation in order to sense the analyte.3 It has been used less frequently, using specific substrates supramolecularly bound to the π-extended backbone, to tune the electrooptic response.4 Bunz and collaborators have explored the supramolecular polarization of two-dimensional, “X-shaped” conjugated chromophores (named “cruciforms”), constructed from two perpendicular π-conjugated linear arms connected through a central aromatic core.5
Lanthanide(III) β-diketonate complexes are of great interest for a wide range of photonic application as they exhibit high quantum efficiency and narrow emission bands.6 Their synthesis is carried out starting from 1,3-dicarbonyl systems through the removal and enolization of one of the acidic hydrogen atoms in position 2 (between the two carbonyls). Several modifications of the core structures have been reported in the literature, to introduce organic chromophores (see example A in Fig. 1), which are attached to one of the carbonyl carbon atoms and function as antennas: they can absorb light in the UV/vis region and then transfer energy to the central lanthanide core for the emission in the desired region. Such complexes can be purified and isolated by conventional methods, for example crystallization. 1,3-Dicarbonyl units are also used as substrates, in combination with a lanthanide or other metal cations behaving as the Lewis acid in organic catalysis: the supramolecular reversibility of the binding to the metal is key for achieving (stereo)selective catalytic properties and high turnover numbers.7
 | ||
| Fig. 1 A prototypical lanthanide(III) β-diketonate complex (left), and the ligands and supramolecular complexes studied in this work (center and right). | ||
Organic π-systems end-capped with an electron donor (D) and an electron acceptor (A) are a class of conjugated molecules widely known as push–pull systems. Donor–Acceptor intramolecular charge-transfer in such D–π–A molecules accounts for distinct optoelectronic properties. Typical electron donors D are substituents with resonance and/or inductive electron donating effects such as OH, NH2, OR and NR2, heterocyclic moieties such as thiophene as well as some metallocenes. The most used electron acceptors A involve substituents featuring resonance and/or inductive electron withdrawing effects such as NO2, CN, CHO and electron deficient heterocyclic compounds.8a We have previously reported on series of “push–pull” chromophores in which the electron-withdrawing molecular fragment is a 1,3-dicarbonyl moiety.8 Two series of compounds have been synthesized and characterized, either “bent” (structure B in Fig. 1), or “cruciform” (structure C in Fig. 1) in shape. We have also previously demonstrated that compounds of class B are able to form supramolecular, reversible complexes with lanthanides which coordinate to the 1,3-dicarbonyl oxygens with variable stoichiometries. When compared with more conventional lanthanide(III) β-diketonate complexes, the absence of acidic protons in the 2 position results in the impossibility to form ligand–metal complexes through enolization.
The peculiar nature of complexes A is testified by a large red shift of the intramolecular charge-transfer absorption band (ICT) upon complexation, resulting in an overall polarization of the supramolecular system, and in potentially interesting and useful properties from the materials science point of view. In addition, ligands B and C, which are formed by a central olefin stator and decorated with either three or four rotors, show in some instances aggregation-induced emissive (AIE) behaviour,9 similarly to the archetypal AIE luminogen tetraphenylethene.10
In this contribution, we report on the comparison and thorough supramolecular characterization in the binding of lanthanides of a series of “bent” and “cruciform” molecular modules. Building on both new and already published contributions, a comprehensive picture of the properties and the potential of these compounds is provided. We show that the polarization of the “cruciform” systems is greatly enhanced with respect to the “bent” chromophores, reaching outstanding values in terms of red shift and of binding ability of the lanthanide ions.
Results and discussion
Complexation response with lanthanides by UV/vis studies
The compounds presented and discussed in this study are shown in Fig. 2. For sake of clarity we have divided them into two different classes (compounds 1 and 2). | ||
| Fig. 2 Chemical structure of compound series 1–2 and labelling of key protons for titration studies. | ||
Simple design considerations prompted us to explore and compare the properties of diketone system 1b with the diester 1a, together with the analogues 2a and 2b. Ketones possess a similar degree of steric hindrance with respect to esters but different electron-withdrawing characteristics, as testified by their σp Hammett parameters (0.45 for COOCH3, 0.50 for COCH3).11
The synthesis of compounds 1 has been carried out through Knoevenagel condensation, starting from the appropriate aldehyde and the 1,3-dicarbonyl compound. The synthesis of compounds 2 has been carried out by silver triflate mediated condensation of commercially available 4,4′-bis(dimethylamino)thiobenzofenone and the appropriate 1,3-dicarbonyl compound. All compounds have been previously reported and characterized.9a We used Eu3+ and Sc3+ as trifluoromethanesulfonate salts, since they are readily dissolved in MeCN. Sc3+ has markedly different ionic radius and polarizability properties with respect to lanthanides, yet it is often used for coordinating 1,3-dicarbonyl moieties as a lanthanide analogue,12 and it can be used freely in 1H NMR titrations as it is diamagnetic.
Chromophores 1a and 1b displayed intense absorption bands in the UV/vis region; in accordance with the previously reported σp Hammett parameters the λmax red shifts passing from 1a to 1b. All relevant parameters are shown in Table 1.
| Entry | Compound | λ max (nm) | Δλmaxb (nm) | LogKac (M−1) |
|---|---|---|---|---|
| a Numbers in parenthesis refer to εcomplex (M−1 cm−1) at the corresponding λmax. b Difference between the λmax of ICT of the free ligand and λmax of complex at saturation. A positive value indicates a red shift of the ICT band upon complexation, a negative value indicates a blue shift. c Numbers in parenthesis refer to the coefficient obtained by the Hill equation. All nonlinear regressions for the best fit reported in Fig. 4 gave high confidence outputs (r2 >0.995). d Data taken from ref. 8a. | ||||
| 1d | 1a | 378 (27500) |
— | — |
| 2d | 1a·Sc | 269 (17800) |
−109 | 4.5 ± 0.1 (2.4) |
| 3d | 1a·Eu | 474 (12080) |
96 | 2.5 ± 0.1 (1) |
| 4 | 1b | 384 (192000) |
— | — |
| 5 | 1b·Sc | 511 (7800) | 127 | 4.5 ± 0.05 (2.2) |
| 6 | 1b·Eu | 494 (6800) | 110 | 4.7 ± 0.1 (1) |
| 7 | 2a | 360 (19500) |
— | — |
| 8 | 2a·Sc | 360 (1500) | — | 5.2 ± 0.1 (1) |
| 9 | 2a·Eu | 565 (3500) | 205 | 4.7 ± 0.05 (1) |
| 10 | 2b | 385 (10900) |
— | — |
| 11 | 2b·Sc | 623 (39100) |
238 | 5.5 ± 0.1 (3.0) |
| 12 | 2b·Eu | 604 (39600) |
211 | 5.7 ± 0.1 (1) |
UV/vis titrations were conducted in dry MeCN using the anhydrous triflate salts of Sc3+, and Eu3+, and representative titrations and binding isotherms of chromophore 1b with Sc(OTf)3 and Eu(OTf)3 are reported in Fig. 3 and Fig. S1–S2 (ESI†). In general, substantial variations of the UV/vis spectra were observed during the titrations, testifying for an effective interaction between the ligands (kept at a constant concentration, see experimental) and the metal salts (which do not absorb in the spectral window examined). In both cases a red shift of the ICT was observed. In the case of Eu3+ (Fig. 3) the binding constant is higher with what reported for 1a (Table 1, entry 3 vs. 6), as well as the shift of the λmax upon complexation, while the stoichiometry of the supramolecular complex was confirmed as 1:1.
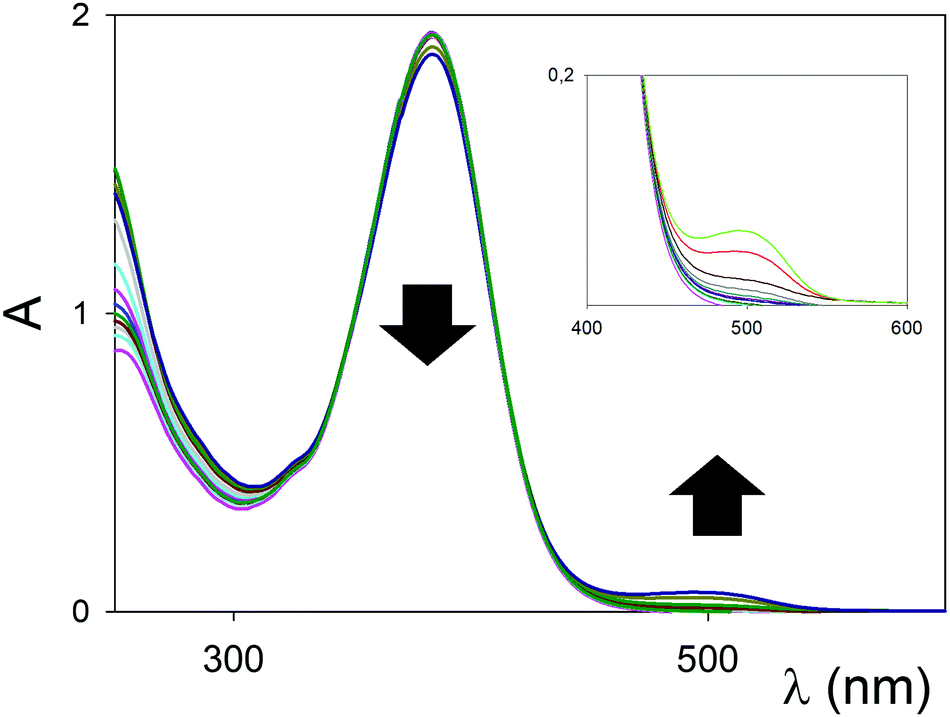 | ||
| Fig. 3 UV/Vis titration (MeCN, 298 K) of 1b (10−5 M) with Eu(OTf)3. | ||
In the case of Sc3+, the change from 1,3-diester in 1a to the 1,3-diketone in 1b (Fig. S1, ESI†) switches the blue shift of the ICT observed with 1a into a red shift (Table 1, entry 2 vs. 5). The previously observed preference of the Sc3+, behaving as a “hard” cation, to the amine moiety of chromophore 1a,8a is therefore switched back to the 1,3-dicarbonyl system in 1b, presumably due to the better coordinating ability of the ketone carbonyls when compared to the ester ones.
The titration profiles at a selected wavelength (Fig. S2, ESI†) clearly indicated that the binding of ligand and metal cation in solution is not just 1:1. The sigmoidal profiles indicated the presence of multiple ligand/metal cation complexes; fitting with the Hill equation gave a value for the apparent average association constant, and an indication of the average stoichiometry of the complex with the Hill number (in parenthesis, column 5 in Table 1).13 The lack of isosbestic points is evidenced in the inset of Fig. S1 (ESI†). The reduction in the intensity of the ICT upon complex formation (ε <8000 M−1 cm−1, Table 1, entries 5 and 6) has been previously observed in other complexes8b and linked to the overall geometry of the complex, which renders in these cases the spectroscopic transition formally forbidden by symmetry consideration, and only weakly allowed by vibronic coupling.
Compounds 2 were examined next. Perhaps surprisingly, other conditions equal, the λmax does not change significantly when passing from the “bent” to the “cruciform” compounds (see, for example, entries 1 vs. 7, or 4 vs. 10 in Table 1). Binding experiments, however, proved to be surprisingly different. The titrations of chromophores 2a–2b with Eu(OTf)3 are shown in Fig. 4. The polarization with the “cruciform” ester-derived 2a is significantly higher than the “bent” one, with a redshift of the λmax upon complexation of 205 nm (Table 1, entry 9). Again, a reduction in the intensity of the ICT upon complex formation and the 1:1 stoichiometry of the supramolecular complex were observed. In the case of Sc3+ (Fig. S3 and S4, ESI†) the titration profile for 2a and the presence of an isosbestic point clearly pointed to a 1:1 binding stoichiometry. An undetectable red shift yet a substantial variation in intensity of the ICT band at 360 nm was also recorded.
 | ||
| Fig. 4 UV/Vis titrations (MeCN, 298 K) of 2a (top, 3 × 10−5 M), 2b (bottom, 3 × 10−5 M) with Eu(OTf)3. | ||
In the case of the diketone “cruciform” 2b with Eu(OTf)3, instead, impressive polarization and enhancement of the molar absorbivity ε upon complexation were detected (Fig. 4, bottom, and entry 12 in Table 1). The shift of 211 nm corresponds to an energy difference of 1.17 eV (see black trace for ligand 2b alone and red trace after the first addition, Fig. 4 bottom). The stoichiometry of the complex was confirmed to be 1:1 (Fig. S4, ESI†), but with a marked enhancement of the binding affinity towards the metal cation (Ka = 513000 M−1).
In the case of Sc3+ (Fig. S3 and S4, ESI†) and 2b, the polarization upon complexation was even more pronounced, with a 238 nm redshift in the ICT band (corresponding to an energy difference of 1.23 eV). The titration profile at a selected wavelength (Fig. S4, ESI†) pointed to a 1:3 metal:ligand stoichiometry, with an apparent average association constant calculated with the Hill equation of 304000 M−1.
1H NMR studies
The complexation properties were further investigated by 1H NMR spectroscopy with Sc(OTf)3. Shifts upon complexation were evident in all tested ligands 1b, 2a and 2b, testifying for the coordination with the metal cation Sc3+ significantly perturbing the π-conjugated system. However, as in the case of UV/vis titrations, differences between the complexes formed could be identified.In the case of the ligand 1b (Fig. 5), the shift of the proton resonances confirmed the complexation to the 1,3-dicarbonyl units and the red shift observed in the UV/vis titration. The singlet at 7.4 ppm, assigned to the vinylidene proton HC (see Fig. 2 for assignments), widens already after the first addition (spectrum B, 0.3 equivalents) of Sc3+, until it becomes very broad under the baseline at the end of the titration (spectrum E, 1.1 equivalents of Sc3+).
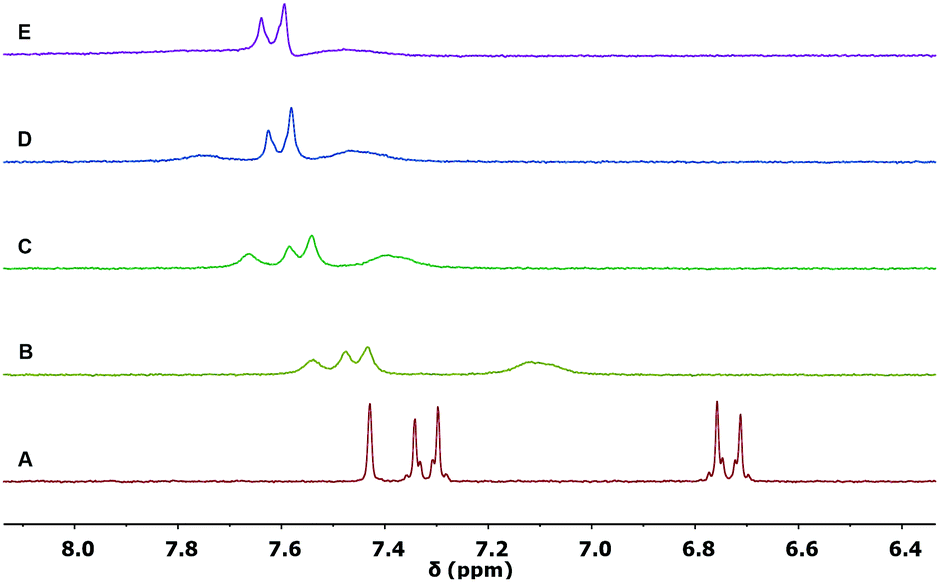 | ||
| Fig. 5 Partial 1H NMR spectra (300 MHz, 298 K) of: (A) ligand 1b (0.005 M in CD3CN); (B) 1b with 0.3 equivalents; (C) 0.6 equivalents; (D) 0.9 equivalents; (E) 1.1 equivalents of Sc(OTf)3. | ||
The HA and HB aromatic proton resonances shift at lower fields (0.36 ppm 0.72 ppm, respectively, from spectrum A to E in Fig. 5), also undergoing a significant broadening. The proton resonances of the methyl groups of the diketone system, which are initially distinguished in the ligand 1b alone because of symmetry, broaden and collapse into a single resonance (Fig. S5, ESI†). The described shifts of the proton resonances appear to be in agreement with the decrease in electron density in the conjugate bridge following the coordination of the cation to the final 1,3-dicarbonyl acceptor. The saturation of the curve is reached at approx. 1 equivalent (Fig. S6, ESI†), confirming the 1:1 stoichiometry of the complex observed in the UV/vis titration, carried out at a much lower concentration.
Since the spectral changes observed in Fig. 5 seem to indicate an intermediate exchange regime with signals broadened by chemical exchange, we performed the titration in the presence of Eu(OTf)3: the paramagnetic shifts could enlarge the chemical shift and slow exchange regime could be reached. However, no slow exchange could be observed in the titration with Eu(OTf)3 (Fig. S7, ESI†).
We also performed the 1H NMR titration using Y(OTf)3, since Yttrium is diamagnetic, but its ionic radius is much closer to the other lanthanides than Scandium. The results, reported in Fig. S8 (ESI†), revealed a very similar trend to that observed with Scandium.
Also in the case of ligand 2a (Fig. S9, ESI†) the shifts of the proton resonances are in agreement with the complexation to the 1,3-dicarbonyl unit: the aromatic protons HA and HB are characterized respectively by a shift at lower fields of 0.64 ppm and 0.84 ppm, respectively. The proton resonances of the dimethylamino groups move to lower fields (0.25 ppm, Fig. S10, ESI†), which is essentially the same shift recorded in the case of 1b. Also in this case, the saturation of the curve is reached at approx. 1 equivalent (Fig. S11, ESI†), confirming the 1:1 stoichiometry of the complex observed in the UV/vis titration at a much lower concentration.
In the case of ligand 2b (Fig. 6), the shifts of the main proton resonances follow the same trends described for 1b and 2a. The saturation of the curve is reached at approx. 0.3 equivalent of Sc(OTf)3 added (Fig. S12, ESI†), confirming the 1:3 stoichiometry of the complex observed in the UV/vis titration at a much lower concentration. Also in this case, titration with Y(OTf)3 revealed a very similar trend to that observed with Sc(OTf)3 (Fig. S13, ESI†).
 | ||
| Fig. 6 Partial 1H NMR spectra (300 MHz, 298 K) of (A) ligand 2b (0.005 M in CD3CN); (B) 2b with 0.3 equivalents; (C) 0.5 equivalents; (D) 0.6 equivalents; (E) 0.9 equivalents; (F) 1.1 equivalents; (G) 1.3 equivalents of Sc(OTf)3. | ||
Solvent-dependent UV/vis studies and emission spectroscopy
The UV/vis absorption spectra of 2b and its complex with Eu3+ are solvent dependent (Fig. 7a). However, the polarity of the solvent and its ability to sustain hydrogen bonds have opposite effects on the electronic transitions of the two systems. A change from polar (MeCN) to non-polar (CHCl3) medium has negligible influence on the band of the ligand, but shifts bathochromically that of the complex by 25 nm. Instead, the spectra in a hydrogen-bond donor solvent (MeOH) show a red shift for the ligand by 19 nm, relative to the other two solvents, but no significant change for the complex. | ||
| Fig. 7 Normalized UV/vis absorption spectra (a) of 2b (30 μM) in MeCN, CHCl3 and MeOH recorded before and after the addition of 1.5 equivalents of Eu(OTf)3. Emission spectrum (b) of an equimolar solution (30 μM) of 2b and Eu(OTf)3 in MeCN with excitation at 605 nm. Bottom: Photograph of a 30 μM solution of Eu(OTf)3 (left), ligand 2b (center), and the 1:1 complex (right). | ||
Excitation of the Eu3+ complex of 2b at wavelengths (<500 nm) where the ligand absorbs does not result in any detectable emission. Either the transfer of excitation energy from ligand to lanthanide is inefficient or nonradiated pathways dominate the deactivation the excited lanthanide. Instead, excitation of the complex at 605 nm results in a broad and weak band with maximum at 663 nm (Fig. 7b), which is presumably a charge-transfer emission.
Molecular modeling
In order to get further insights on the different behaviour of ligands 1 and 2 in their supramolecular recognition of cations, calculations were performed with Gaussian 09 by using DFT-B3LYP method, to obtain the computed conformational minima for free ligand and complexes. In order to verify the method, geometry optimization for ligand 1a was accurately compared with its crystal structure,9f and the results were verified to be superimposable. Fig. 8 shows the minimized structures for ligands 2a and 2b, whereas structures 1a and 1b are reported in Fig. S14 (ESI†).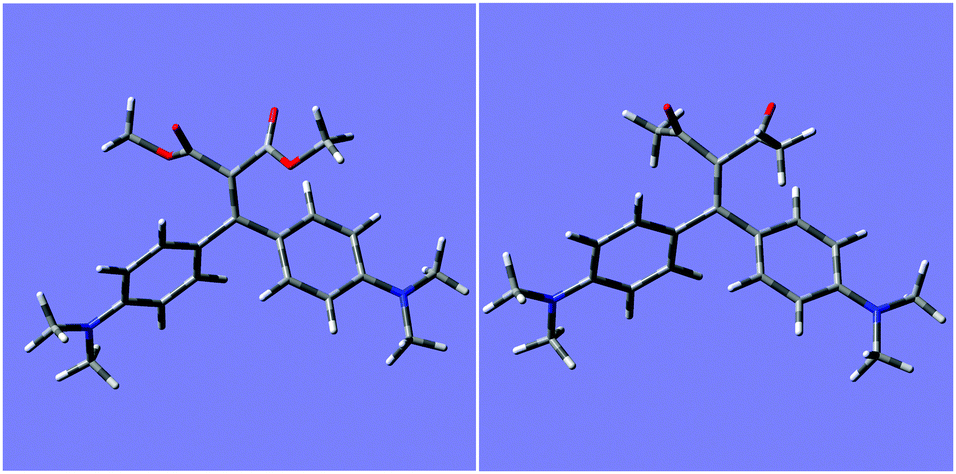 | ||
| Fig. 8 Minimized molecular structures for 2a (left) and 2b (right). | ||
Optimized structures of 1a and 1b show a negligible distortion (<0.4°) of the dihedral angle of the olefin moiety conjugated to phenyl rings (dihedral C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C(Ph ring)), while for 2a and 2b the angle is distorted to −13 and −10°, respectively. This could lead to a reduced conjugation with phenyl rings. The complexation with scandium ion increases the tension of the olefin going to a strained dihedral angle of −64 and −68° for 2a and 2b, respectively. The complexation affects the double bond since the chelating effect tend to form a planar structure. Fig. 9 shows the minimized structures for 1:1 complexes of ligands 2a and 2b, whereas structures for 1a and 1b are reported in Fig. S15 (ESI†).
C–C(Ph ring)), while for 2a and 2b the angle is distorted to −13 and −10°, respectively. This could lead to a reduced conjugation with phenyl rings. The complexation with scandium ion increases the tension of the olefin going to a strained dihedral angle of −64 and −68° for 2a and 2b, respectively. The complexation affects the double bond since the chelating effect tend to form a planar structure. Fig. 9 shows the minimized structures for 1:1 complexes of ligands 2a and 2b, whereas structures for 1a and 1b are reported in Fig. S15 (ESI†).
 | ||
| Fig. 9 Minimized molecular structures for 1:1 complexes with scandium of ligands 2a (left) and 2b (right). | ||
UV-Vis spectra of the ligands and their complexes with scandium ion were calculated by using TD-DFT B3LYP lanl2dz. The solvent effect was evaluated by using the integral equation formalism model (method indicated as default in Gaussian 09) with the internal parameters of acetonitrile. Spectra are reported in Fig. S16–S19 (ESI†) for ligands and S20–S23 (ESI†) for Sc complexes. UV-Vis transitions calculated for the scandium complexes in acetonitrile, considering only the two most intense bands, are reported in Table S1 (ESI†). In all ligands, the lowest energy bands correspond to the π–π* transitions, whereas the other, most intense band at higher energy is related to a ligand-to-metal energy transfer band. Cartesian coordinates for the optimized complexes with Sc(III) are reported in Table S2 (ESI†).
Conclusions
We have reported on the comparison between two different series of conjugated push–pull compounds bearing dimethylamino donor and 1,3-dicarbonyl acceptor functionalities with a “bent” or “cruciform” shape. The two series exhibit striking differences in their supramolecular recognition of lanthanide cations. Both series of compounds are effective to coordinate lanthanide cations (Sc3+ and Eu3+) in MeCN, CHCl3 or MeOH solutions, and the supramolecular coordination occurs unequivocally to the 1,3-dicarbonyl acceptor end. The coordination of the lanthanide cation results in an overall supramolecular polarization of the system and the redshifting of the intramolecular charge transfer band. The polarization of the “cruciform” systems is greatly enhanced with respect to the “bent” chromophores, reaching outstanding values for the red shift (238 nm, corresponding to an energy difference of 1.23 eV) with high binding constants (LogKa >5.5). Excitation of the complex in any of the investigated region did not reveal any efficient energy transfer and emission from the lanthanide core.
This study brings essential insights into simple, tailorable organic chromophores acting very effectively for manipulating the absorbance of lanthanide complexes in the visible region of the solar spectrum. The results will be of great help in designing innovative conjugated sensors, and further efforts into a fine tuning of the molecular structures to match requirements for functional optoelectronic applications can be foreseen.
Experimental
General methods
Compounds 1a, 1b, 2a and 2b were prepared as previously described.9a1H NMR spectra were recorded in CD3CN with a Bruker AMX300 spectrometer, using the solvent residual proton signal as a standard. UV/Vis spectra were recorded with a JASCO V-550 or a Varian Cary 100 Bio spectrometer in aerated quartz cells (path length = 1.0 cm). Emission spectra were recorded with a Shimadzu RF-6000 spectrometer in aerated quartz cells (path length = 1.0 cm).General procedure for the titration experiments
The titration experiments were conducted as follows: to a stock solution of the ligand (solution A) in MeCN (UV/vis spectroscopic grade), were added several aliquots of the guest (solution B). Solution B is formed by the lanthanide triflate at higher concentration dissolved in solution A, in order to maintain the ligand always at the same, constant concentration. 1H NMR titrations were conducted in a single NMR tube, by adding aliquots of solution B to 700 μL of solution A in CD3CN.In the case of a 1:1 binding isotherm, by employing a nonlinear fitting curve program, the plot of absorbance at a selected wavelength against the metal concentration was fitted by eqn (1), thus affording the value of the association constant Ka.
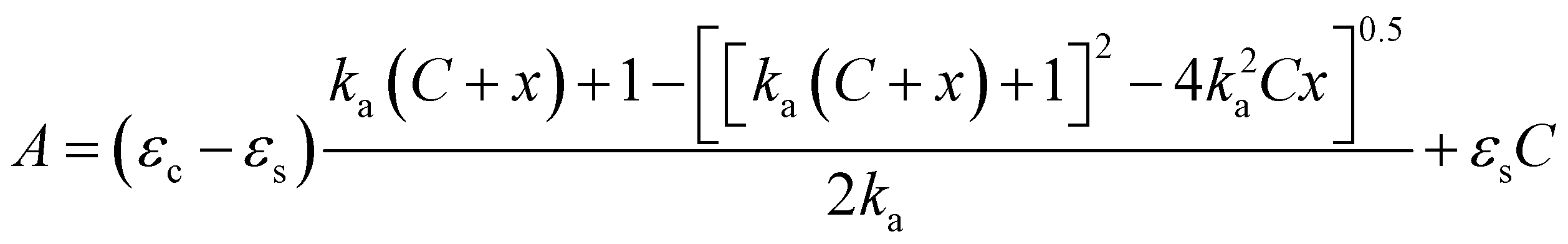 | (1) |
:1 complex.14
The data for other titrations were fitted to a general form of the Hill equation:
| ΔAbs = ΔAbsmax [x]nKa/(1 + [x]nKa) |
Which can be conveniently rewritten in:
| ΔAbs = ΔAbsmax [x]n/[(1/Ka)n + [x]n] | (2) |
Eqn (2) could be fitted employing a nonlinear fitting program according to the general equation: f(x) = a*xb/(cb + xb), obtaining values of a = ΔAbsmax, b = n (the Hill coefficient), c = 1/Ka.13
Molecular modelling
Calculations were performed with Gaussian 09 by using DFT-B3LYP method. The 6-31G(d) basis set was used for light atoms (H, C, N, O), where lanl2dz was used for scandium. No negative frequencies were obtained, demonstrating that structures are energy minimum. Time-dependent density functional theory (TDDFT) was used on the optimized structures to obtain UV-vis spectrum. Spectra are shown uncorrected.Author contributions
Conceptualization D. P.; funding acquisition: F. M. R., D. D. and D. P.; investigation: S. B., Y. Z., A. N., M. M. A. M., D. D., F. M. R., and D. P.; writing – original draft: D. P.; writing – review & editing: F. M. R. and D. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. P. acknowledges grants by MIUR (PRIN 2017 BOOSTER Prot. 2017YXX8AZ), the University of Pavia and INSTM-Regione Lombardia for partial support of this work.Notes and references
- (a) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS; (b) P.-O. Morin, T. Bura and M. Leclerc, Mater. Horiz., 2016, 3, 11–20 RSC; (c) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed; (d) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS; (e) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (f) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- (a) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS; (b) J. G. Mei and Y. Diao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed; (c) C. L. Wang and H. L. Dong, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- (a) A. Zavaleta, A. O. Lykhin, J. H. S. K. Monteiro, S. Uchida, T. W. Bell, A. de Bettencourt-Dias, S. A. Varganov and J. Gallucci, J. Am. Chem. Soc., 2020, 142, 20306–20312 CrossRef CAS; (b) Q. Wang, Q. Zhang, Q.-W. Zhang, X. Li, C.-X. Zhao, T.-Y. Xu, D.-H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef; (c) G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS; (d) F. Wurthner, J. Schmidt, M. Stolte and R. Wortmann, Angew. Chem., Int. Ed., 2006, 45, 3842–3846 CrossRef PubMed; (e) G. Fabbrini, E. Menna, M. Maggini, A. Canazza, G. Marcolongo and M. Meneghetti, J. Am. Chem. Soc., 2004, 126, 6238–6239 CrossRef CAS.
- (a) A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397–408 CrossRef CAS PubMed; (b) A. J. Zucchero, J. N. Wilson and U. H. F. Bunz, J. Am. Chem. Soc., 2006, 128, 11872–11881 CrossRef CAS PubMed.
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; (b) M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496 CrossRef CAS; (c) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS.
- (a) J. Huang, D. Lebœuf and A. J. Frontier, J. Am. Chem. Soc., 2011, 133, 6307–6317 CrossRef CAS PubMed; (b) A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC.
- (a) F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC; (b) M. Caricato, C. Coluccini, D. A. Vander Griend, A. Forni and D. Pasini, New J. Chem., 2013, 37, 2792–2799 RSC; (c) L. Garlaschelli, I. Messina, D. Pasini and P. P. Righetti, Eur. J. Org. Chem., 2002, 3385–3392 CrossRef CAS; (d) D. Pasini, P. P. Righetti and V. Rossi, Org. Lett., 2002, 4, 23–26 CrossRef CAS; (e) D. Pasini, P. P. Righetti and M. Zema, Org. Biomol. Chem., 2004, 2, 1764–1769 RSC; (f) C. Coluccini, P. Metrangolo, M. Parachini, D. Pasini, G. Resnati and P. Righetti, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5202–5213 CrossRef CAS; (g) C. Coluccini, D. Pasini, P. Righetti and D. A. Vander Griend, Tetrahedron, 2009, 65, 10436–10440 CrossRef CAS; (h) C. Coluccini, A. K. Sharma, D. Merli, D. Vander Griend, B. Mannucci and D. Pasini, Dalton Trans., 2011, 40, 11719–11725 RSC.
- (a) A. Nitti, F. Villafiorita-Monteleone, A. Pacini, C. Botta, T. Virgili, A. Forni, E. Cariati, M. Boiocchi and D. Pasini, Faraday Discuss., 2017, 196, 143–161 RSC; (b) M. M. Mróz, S. Benedini, A. Forni, C. Botta, D. Pasini, E. Cariati and T. Virgili, Phys. Chem. Chem. Phys., 2016, 18, 18289–18296 RSC; (c) C. Botta, S. Benedini, L. Carlucci, A. Forni, D. Marinotto, S. Righetto and E. Cariati, J. Mater. Chem. C, 2016, 4, 2979–2989 RSC; (d) C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666–1674 RSC; (e) T. Virgili, A. Forni, E. Cariati, D. Pasini and C. Botta, J. Phys. Chem. C, 2013, 117, 27161–27166 CrossRef CAS; (f) E. Cariati, V. Lanzeni, E. Tordin, R. Ugo, C. Botta, A. Giacometti Schieroni, A. Sironi and D. Pasini, Phys. Chem. Chem. Phys., 2011, 13, 1800518014 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) A. Nitti and D. Pasini, Adv. Mater., 2020, 32, 1908021 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- G. Desimoni, G. Faita, A. Galbiati, D. Pasini, P. Quadrelli and F. Rancati, Tetrahedron: Asymmetry, 2002, 13, 333337 Search PubMed.
- (a) V. Gorteau, G. Bollot, J. Mareda, D. Pasini, D.-H. Tran, A. N. Lazar, A. W. Coleman, N. Sakai and S. Matile, Bioorg. Med. Chem., 2005, 13, 51715180 CrossRef; (b) A. Moletti, D. Pasini and A. Taglietti, New J. Chem., 2007, 31, 352356 Search PubMed; (c) Y. Baudry, G. Bollot, V. Gorteau, S. Litvinchuk, J. Mareda, M. Nishihara, D. Pasini, F. Perret, D. Ronan, N. Sakai, M. R. Shah, A. Som, N. Sorde, P. Talukdar, D.-H. Tran and S. Matile, Adv. Funct. Mater., 2006, 16, 169179 Search PubMed; (d) E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc., 2006, 128, 71727173 CrossRef PubMed; (e) G. Fernández, L. Sánchez, E. M. Pérez and N. Martín, J. Am. Chem. Soc., 2008, 130, 1067410683 Search PubMed; (f) G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097–16103 CrossRef CAS; (g) J. Hamacek and C. Piguet, J. Phys. Chem. B, 2006, 110, 7783–7792 CrossRef CAS.
- (a) J. R. Long and R. S. Drago, J. Chem. Educ., 1982, 59, 1037–1089 CrossRef CAS; (b) M. Asakawa, C. L. Brown, D. Pasini, J. F. Stoddart and P. G. Wyatt, J. Org. Chem., 1996, 61, 7234–7235 CrossRef CAS PubMed; (c) C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 RSC; (d) C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphs and figures. See DOI: 10.1039/d1nj04358g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |