
Synthesis of the new analogs of morpholine and their antiplasmodial evaluation against the human malaria parasite Plasmodium falciparum†
Charu
Upadhyay
a,
Neha
Sharma
b,
Sumit
Kumar
a,
Prem Prakash
Sharma
b,
Diana
Fontinha
c,
Bhupender S.
Chhikara
d,
Budhaditya
Mukherjee
 e,
Dhruv
Kumar
f,
Miguel
Prudencio
c,
Agam P.
Singh
g and
Poonam
*a
e,
Dhruv
Kumar
f,
Miguel
Prudencio
c,
Agam P.
Singh
g and
Poonam
*a
aDepartment of Chemistry, Miranda House, University of Delhi, Delhi, India. E-mail: poonam.chemistry@mirandahouse.ac.in
bLaboratory for Translational Chemistry and Drug Discovery, Department of Chemistry, Hansraj College University Enclave, University of Delhi, Delhi 110007, India
cInstituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Avenida Professor Egas Moniz, 1649-028 Lisboa, Portugal
dDepartment of Chemistry, Aditi Mahavidyalaya, University of Delhi, Delhi, India
eSchool of Medical Science and Technology, IIT Kharagpur, Kharagpur-721302, India
fAmity Institute of Molecular Medicine & Stem Cell Research, Amity University Uttar Pradesh, Noida, India
gInfectious Diseases Laboratory, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110067, India
First published on 3rd December 2021
Abstract
A series of morpholine analogs functionalized with hydroxyethylamine (HEA) pharmacophore was synthesized and assayed for the initial screening against Plasmodium falciparum 3D7 in culture, which suggested that analog 6k is a hit molecule with an inhibitory concentration of 5.059 ± 0.2036 μM. Listed analogs were also assessed for toxicity against liver cells, HepG2, and none of them showed cytotoxicity up to 2 mM. Further, we tested all the compounds against a validated target, plasmepsin X, and the results were supported with molecular docking and molecular dynamic simulation. Overall, these novel compounds demonstrated moderate antiplasmodial activity without any toxic effects, and therefore medicinal chemistry optimization is essential to obtain analogs with improved biological activity.
1. Introduction
Malaria disease, caused by Plasmodium, is one of the most life-threatening diseases affecting 200 million people worldwide, especially in tropical and sub-tropical regions.1,2 Among the Plasmodium species, Plasmodium falciparum (Pf) is the most lethal form of human malaria.3,4 According to a WHO report, a decrement in the mortality rate has been observed since 2011 as the number of deaths reduced from 536![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 40900 in 2019.5 In sub-Saharan countries, the front-line therapeutics, i.e. quinolines, naphthoquinones, antifolates, 8-aminoquinolines, endoperoxides and artemisinin-based combination (ACT) are available to treat the disease.6 Regardless of uninterrupted events towards disease control, inevitable drug resistance has emerged as a major barrier, and thus efforts are necessary to overcome the problem of the decreased efficacy of therapeutics.7,8 However, ACT treatments remain the most effective therapy for malaria disease.9,10 The efficacy of the drugs is compromised at any stage of their life, probably due to their frequent and suboptimal doses,11,12 and ACT resistance is emerging in malaria cases reported along the border of Combodia.3,13,14 Considering the current alarming circumstances, it is necessary to design new, potent, and effective antimalarial drugs, preferably active beyond the asexual blood-stage of malaria.15
000 to 40900 in 2019.5 In sub-Saharan countries, the front-line therapeutics, i.e. quinolines, naphthoquinones, antifolates, 8-aminoquinolines, endoperoxides and artemisinin-based combination (ACT) are available to treat the disease.6 Regardless of uninterrupted events towards disease control, inevitable drug resistance has emerged as a major barrier, and thus efforts are necessary to overcome the problem of the decreased efficacy of therapeutics.7,8 However, ACT treatments remain the most effective therapy for malaria disease.9,10 The efficacy of the drugs is compromised at any stage of their life, probably due to their frequent and suboptimal doses,11,12 and ACT resistance is emerging in malaria cases reported along the border of Combodia.3,13,14 Considering the current alarming circumstances, it is necessary to design new, potent, and effective antimalarial drugs, preferably active beyond the asexual blood-stage of malaria.15
The development of novel chemical libraries based on the validated pharmacophores and their screening against malaria parasites remains one of the potential approaches to finding hit molecules. Looking at the significance of the heterocyclic scaffolds, morpholine analogs have been investigated as high-valued bioactive scaffolds, utilized as potential synthons in medicinal chemistry. Various morpholine analogs have been studied as antioxidants, anti-inflammatory, antimicrobial, analgesics, and anti-cancer agents.16–19 Moreover, morpholine is an integral part of various drug molecules where it acts as an enzyme active-site inhibitor for enzymes such as phosphatidylinositol 3-kinase, squalene synthase, peptidyl transferase, etc., and contributes to favorable pharmacological applications.20 Notably, two well-known antimalarials, namely M5717 and OZ439, containing a morpholine core are under clinical trials,6,21 indicating their suitability as important pharmacophores for the design of novel antimalarial drugs. Hydroxyethylamine (HEA) has also been validated as a potential pharmacophore for antimalarial drug discovery.22,23 Our group has explored many HEA compounds as highly effective against multiple life stages of the malaria parasite (i.e., liver stage, blood stage, and sexual stages), and have ultimately suggested them as multistage antimalarial compounds.24,25 Interestingly, the efficacy of HEA compounds can be easily tuned with the inclusion of high-valued heterocyclic scaffolds.
Subsequently, we designed and synthesized novel morpholine-based HEA compounds anticipating them as potent antiplasmodial agents not only against blood-stage but also liver-stage infection. Being important pharmacophores, the fusion of HEA and morpholine could increase the efficacy parameters and structural diversity. All the fourteen listed compounds were screened against blood-stage and liver-stage activity. Additionally, compounds were evaluated against recombinant Plm X and the results were substantiated with extensive in silico studies.
2. Results and discussion
2.1. Compound synthesis
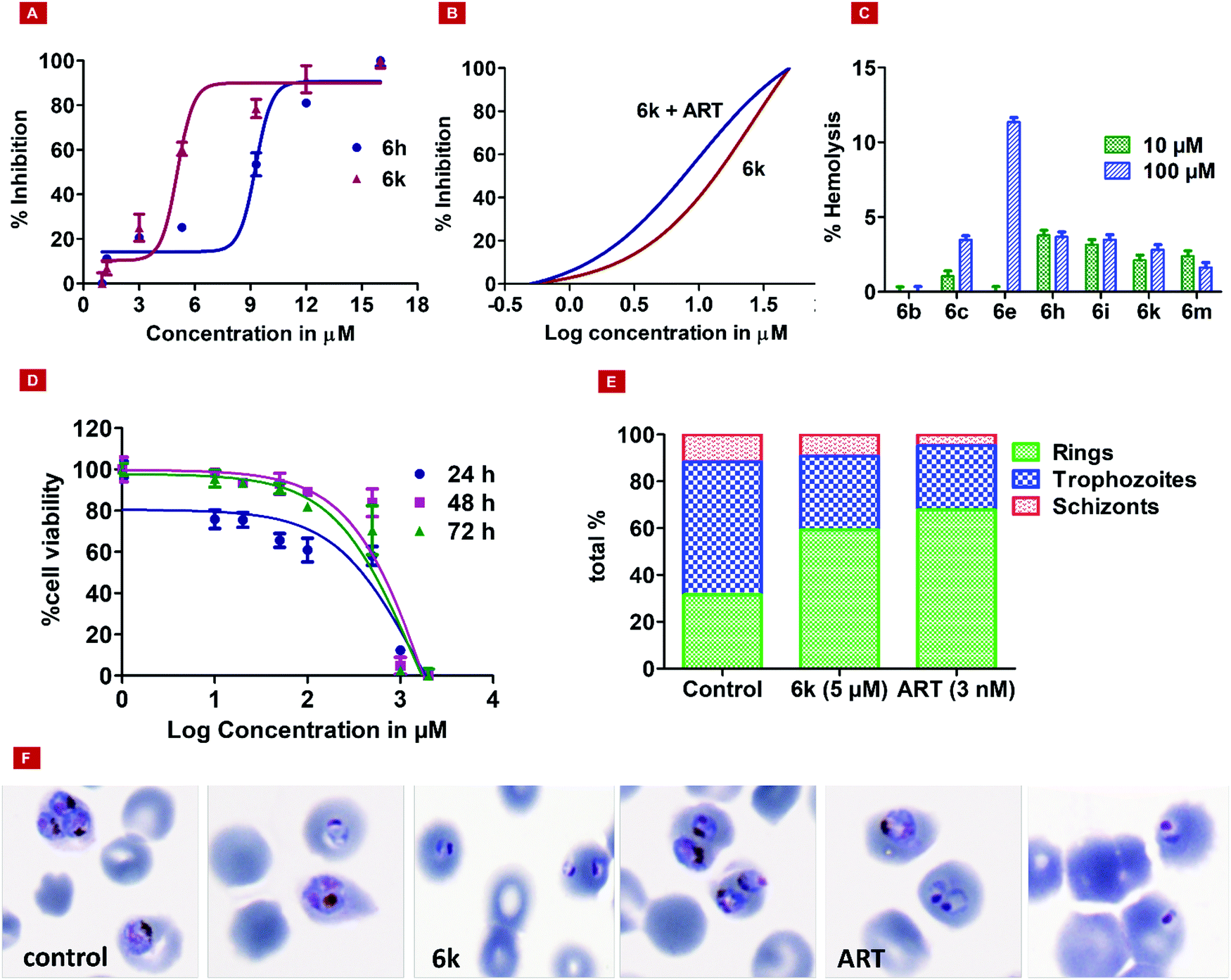
The design of novel compounds based on validated pharmacophores remains one of the important paths to discovering efficacious therapeutics. In this manuscript, we present the fusion of two important and validated pharmacophores, i.e., morpholine and HEA, which have led to fourteen new compounds possessing chemical diversity. In the literature, HEA analogs decorated with various heterocyclic scaffolds have been widely studied as inhibitors of malarial aspartyl proteases, Plms,26 and as potential multistage antimalarials.9,27 Likewise, the morpholine scaffold was explored for medicinal applications including antimalarial drugs.28 Encouraged by this, we designed novel compounds based on morpholine and HEA, which were assessed for ADME profile predictions using the SWISS-ADME server. All molecules strictly obey the Lipinski rule of five, where MW < 500, AccptHB < 10, DonorHB < 5, MlogP < 4.15. There is no violation for any single parameter of Lipinski drug-like characteristics among all synthesized analogs. All the molecules have high GI absorption, while 3, 6b–c, 6e–f, 6i, 6k–m were the only molecules displayed blood–brain barrier (BBB) penetration capabilities. Molecules are moderately soluble having logS of −4.00, soluble (range −4.00 to −2.00), and very soluble (0 to −2.00). Subsequently, the syntheses of fourteen HEA and morpholine-based derivatives were performed by following a simple and convenient method as illustrated in Scheme 1. To obtain structural activity relationship (SAR) insights, chemical diversity was introduced in the desired compounds by embodying aromatic and heterocyclic rings possessing fluorine, bromine, trifluoromethyl, and methyl groups. Our group29–31 has already optimized a facile, rapid, and regioselective32,33 ring-opening of epoxide, (2R,3S)-3-(N-BOC-amino)-1-oxirane-4-phenylbutane with different amines with inversion of the configuration at less hindered carbon atom. The same synthetic route was implemented using morpholine as the amine, which resulted in the Boc-protected intermediate (3). In the next step, deprotection of 3 was carried out, followed by the coupling reaction with functionalized aromatic carboxylic acids, which afforded the final desired compounds (6a–m).34,35
 | ||
| Scheme 1 Synthesis of HEA-morpholine compounds (6a–m). | ||
New compounds were obtained in moderate to good yields. The chemical structure of all the listed compounds was confirmed by NMR (1H & 13C) and mass spectrometry.
2.2. Biological evaluation
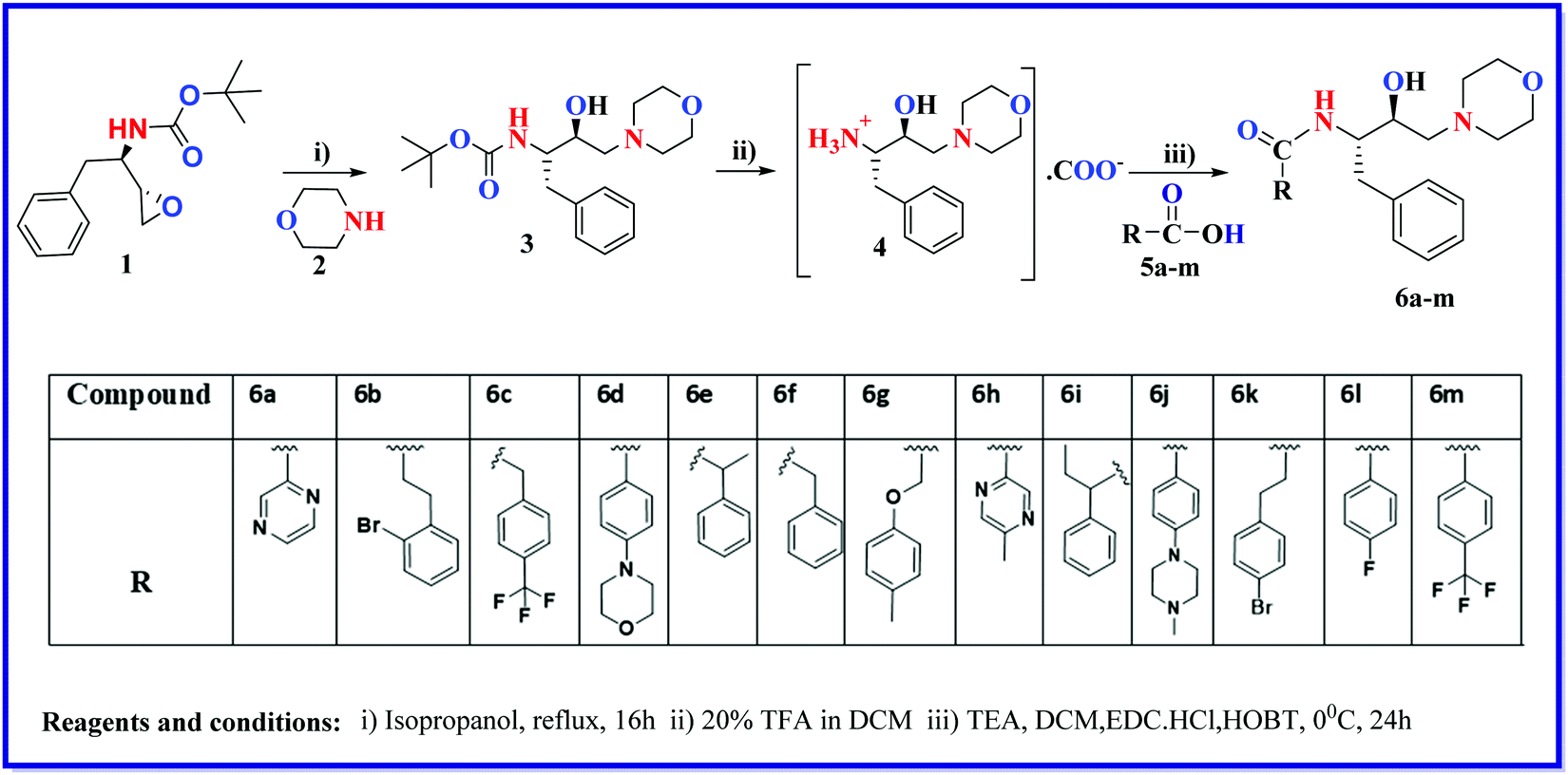
 | ||
| Fig. 1 The efficacy and cytotoxicity effects of compounds. (A) The effects of dose-dependent ABS (asexual blood-stage) of compound 6h and 6k on Pf3D7 strains in culture. An asynchronized parasite population of Pf3D7 infected erythrocytes (4% final hematocrit and 1% parasitemia) was exposed to varying concentrations of test compound 6h and 6k. (B) Two different curves showing the % inhibition of compound 6k alone (red curve) and in combination with ART (blue curve). (C) The effects of the compounds on human RBCs at 10 μM and 100 μM concentrations. (D) Cell viability (%) levels were measured by MTT assay after 24, 48, and 72 h of treatment at different concentrations (1 μM to 2 mM). (E) The percentages of rings, trophozoites, and schizonts in the control, treated (6k), and ART at their IC50 values in the trophozoite stage of the malaria parasite. (F) Photomicrographs of Giemsa-stained blood smears of the control (3.6% parasitemia), 6k, and ART on the trophozoite stage of the parasite morphological appearance. | ||
To evaluate the cytotoxicity of all the listed compounds, an MTT assay was performed against HepG2 (a mammalian liver cell line). The minimal cytotoxic effect was shown by 3, 6a, 6h, and 6j with TC50 > 2 mM. In a separate experiment, all the synthesized compounds were tested on normal human red blood cells (RBCs), to find the hemolysis of RBCs at two different concentrations, i.e., 10 μM and 100 μM. Results are shown in Fig. 1C. Interestingly, no significant lysis of RBCs was found up to the 100 μM concentration. We also performed time course dose-dependent cytotoxicity experiments for hit 6k and evaluated its TC50 at different time intervals, i.e., 24, 48, and 72 hours at different concentrations ranging from 1 μm to 2 mM. The results showed tolerance for hit 6k with TC50 values of 179.9, 496.2, and 383.3 μM at 24, 48, and 72 hours, respectively (Fig. 1D).
| Entry. no. | Code | Structure | TC50 (HepG2) | % reduction of PlmX activity at 1 μM | % inhibition of Pf3D7 at 10 μM |
|---|---|---|---|---|---|
| 1. | 3 |
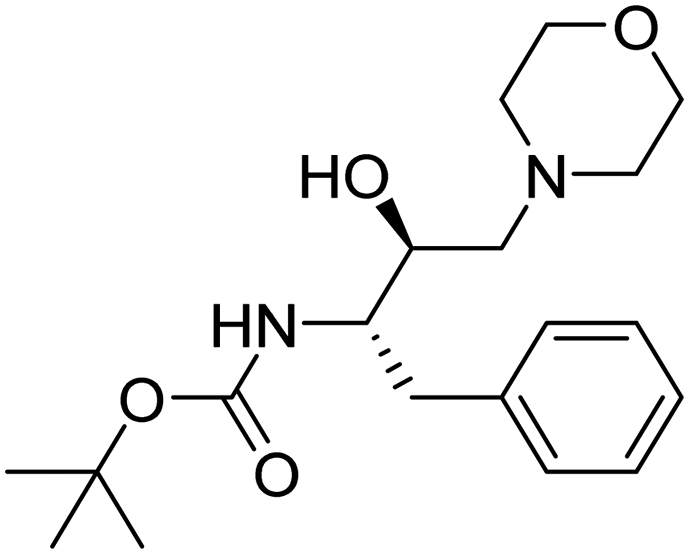
|
>2 mM | 10.9375 | 40.64 |
| 2. | 6a |

|
>2 mM | 11.29167 | 45.74 |
| 3. | 6b |
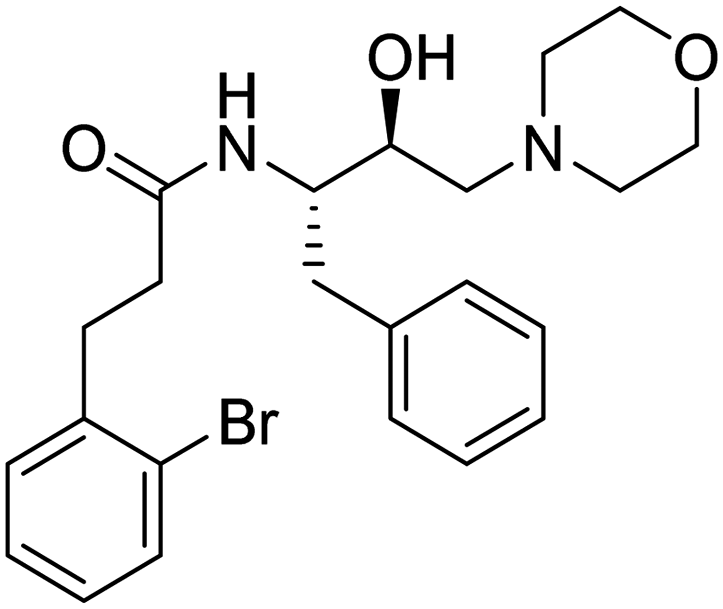
|
486 μM | 10.10417 | 48.61 |
| 4. | 6c |

|
200 μM | 28.89583 | 40.47 |
| 5. | 6d |
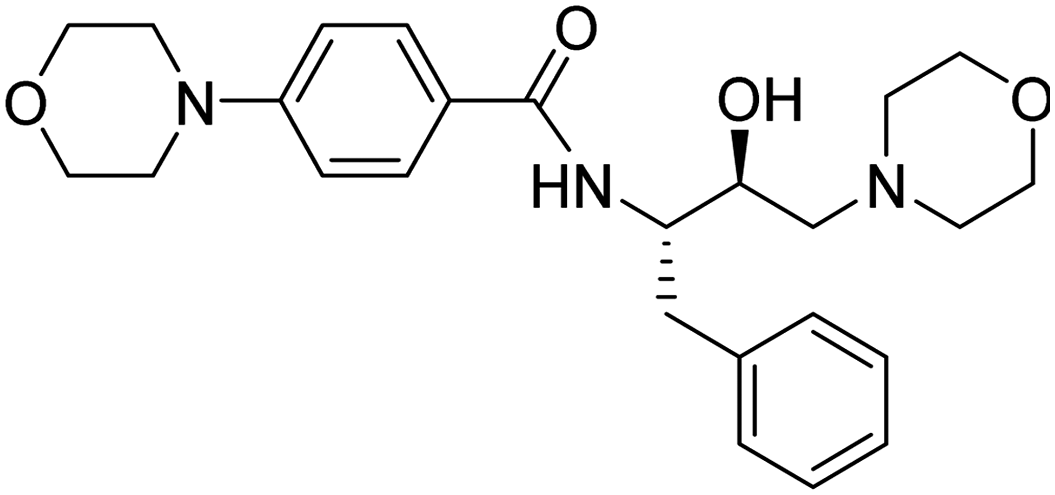
|
>900 μM | 14.1875 | 45.19 |
| 6. | 6e |
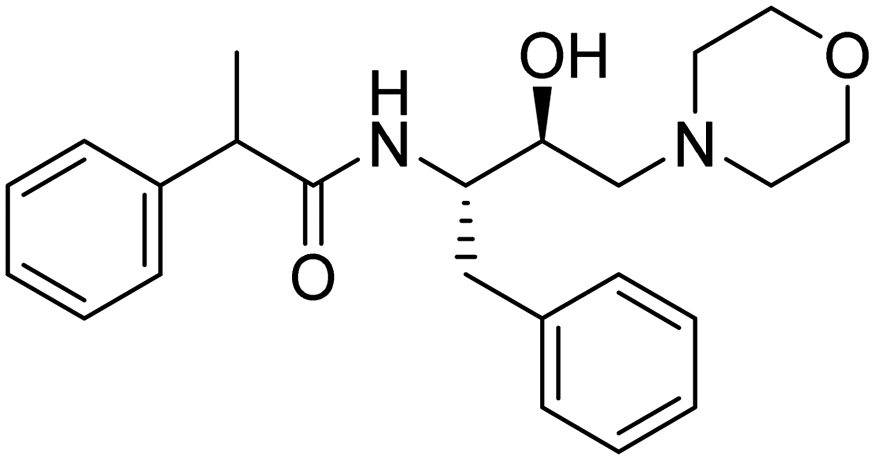
|
>1350 μM | 11.6875 | 51.89 |
| 7. | 6f |
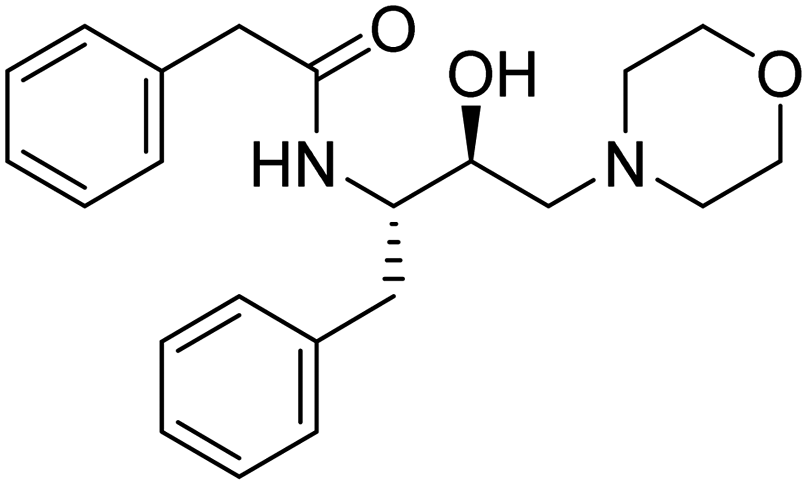
|
>900 μM | 16.875 | 53.26 |
| 8. | 6g |
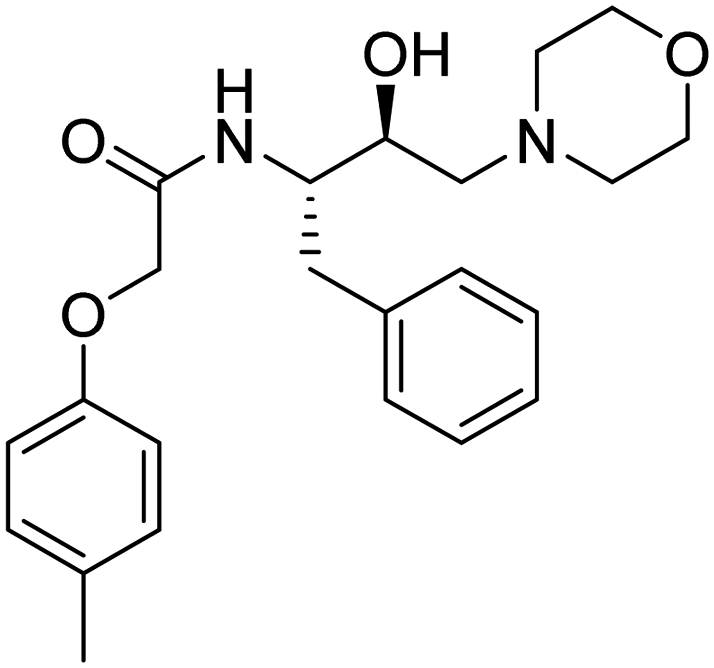
|
1350 μM | 15.52083 | 44.59 |
| 9. | 6h |
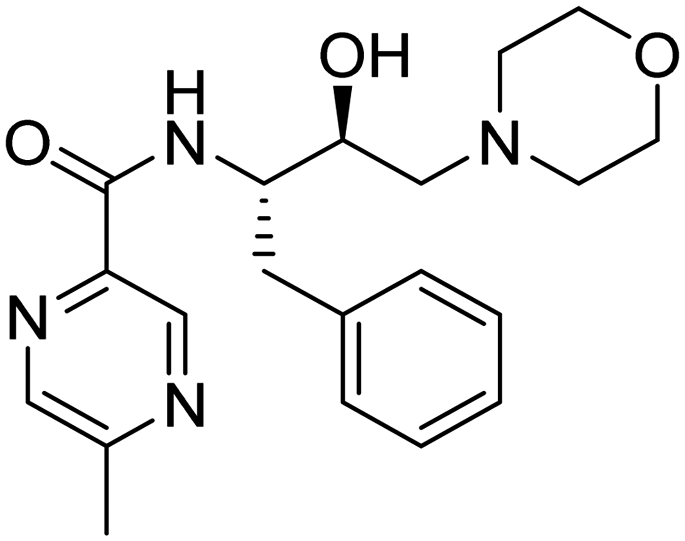
|
>2 mM | 38.41667 | 51.73 |
| 10. | 6i |

|
683 μM | 12.8333 | 34.67 |
| 11. | 6j |
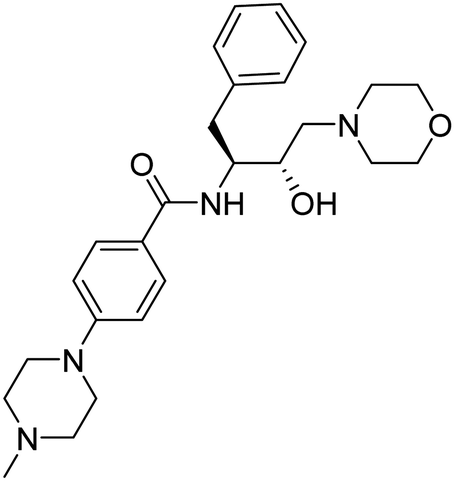
|
>2 mM | 13.95833 | 45.07 |
| 12. | 6k |
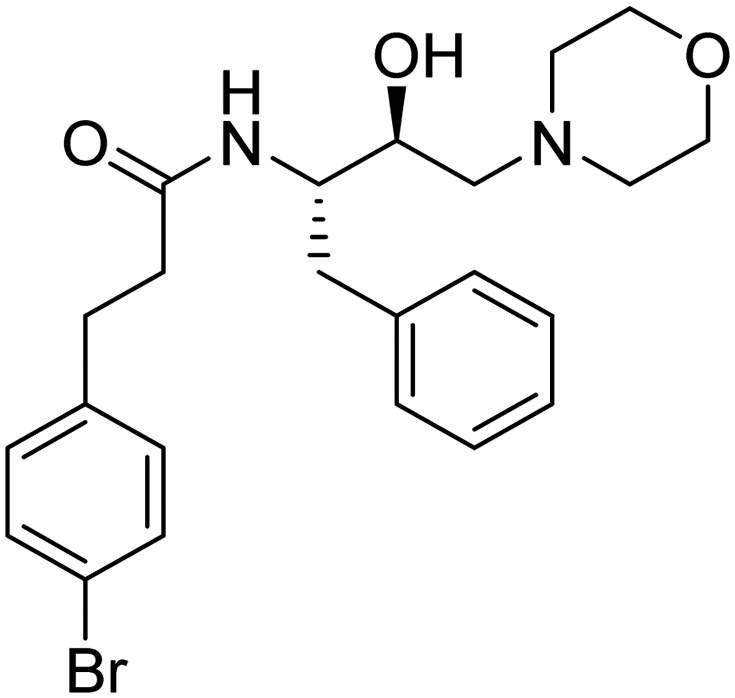
|
233 μM | 30.39583 | 84.17 |
| 13. | 6l |
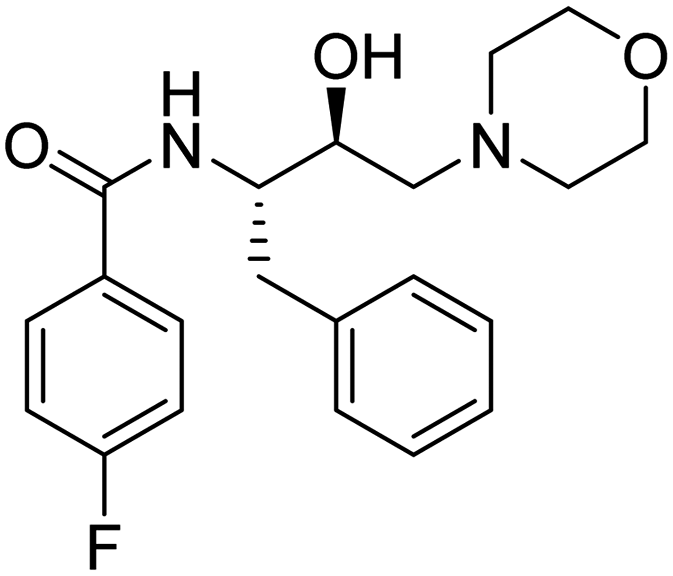
|
>1350 μM | 24.75 | 47.54 |
| 14. | 6m |
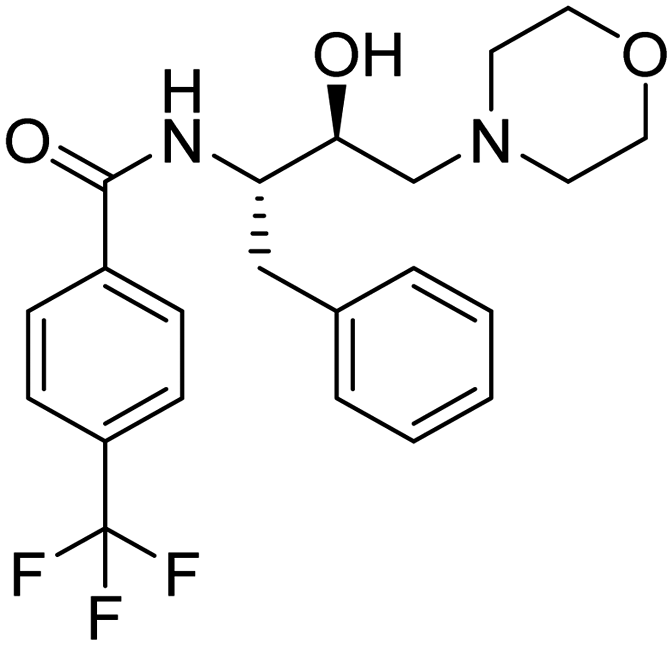
|
1032 μM | 22.72917 | 55.13 |
The developed Plm X model (Fig. S34A, ESI†) was analyzed for its stereochemical geometry by the ProCheck Ramachandran plot. The amino acid residues (Glu_204 and Gln_238), which were found to be in the outlier region, contributed only 0.7% of the total, indicating good structural geometry for the complex. Residues lying in favored, additionally allowed, and generously allowed regions were 87.7%, 11.2%, and 0.4%, respectively (Fig. S34B, ESI†). In silico studies revealed that 6h displayed the best parameters such as docking score, XP Gscore, and binding free energy with the values of −6.12, −6.802, and −79.6 kcal mol−1, respectively (Table 2, entry 1). However, 6k, the most potent molecule in the asexual blood stage, was found to possess a docking score, XP Gscore, and binding free energy of −5.216, −5.891, and −69.26 kcal mol−1, respectively (Table 2, entry 10), in correlation with the anti-proliferative effect against Pf as discussed in the previous sections. The molecular docking analysis indicated that the hydroxyl group and phenyl moiety of HEA interacted with residues Asp_266 and Phe_311 through H-bond and pi–pi interactions, respectively. The nitrogen atom of the morpholine moiety showed interactions with Asp_266 and Phe_311 via the salt bridge and pi cation interactions, respectively (Fig. 2).
| Entry | Compound | Docking score | XP GScore | MMGBSA dG Bind |
|---|---|---|---|---|
| 1 | 6h | −6.12 | −6.802 | −79.6 |
| 2 | 6e | −5.892 | −6.115 | −57.37 |
| 3 | 6j | −5.65 | −6.343 | −64.89 |
| 4 | 6b | −5.492 | −5.721 | −61.69 |
| 5 | 6g | −5.46 | −6.191 | −56.83 |
| 6 | 6m | −5.432 | −5.68 | −50.82 |
| 7 | 6i | −5.425 | −6.11 | −60.88 |
| 8 | 3 | −5.298 | −6.016 | −64.68 |
| 9 | 6f | −5.283 | −5.506 | −45.62 |
| 10 | 6k | −5.216 | −5.891 | −69.26 |
| 11 | 6d | −4.872 | −5.508 | −59.89 |
| 12 | 6c | −4.846 | −5.529 | −62.68 |
| 13 | 6a | −4.771 | −5.453 | −66.15 |
| 14 | 6l | −4.68 | −4.928 | −38.2 |
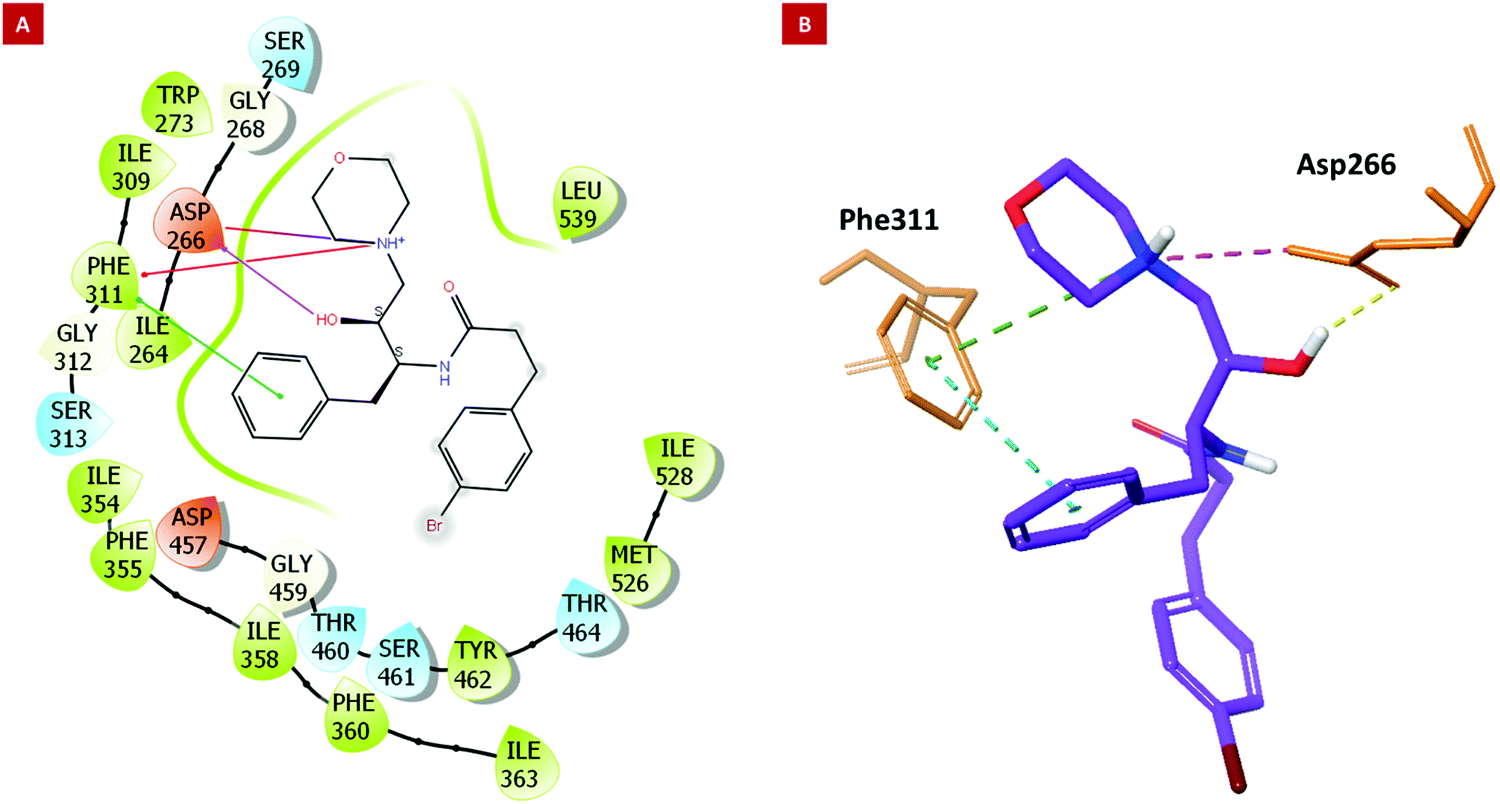 | ||
| Fig. 2 Molecular docking results. (2A) The 2D and (2B) 3D interaction images of 6k with key residues of PlmX. | ||
The amino acid residue Asp_266 is a known catalytic residue of Plm X and interestingly, the hit molecule (6k) showed interaction with the same. Therefore, to further analyze the binding and conformational stability of (6k) with PlmX protein, molecular dynamics (MD) simulation studies were performed.
Firstly, the stereochemical geometry analysis was carried out after performing MD simulation for 50 ns using the Ramachandran plot, which revealed acceptable numbers of residues in the favored, additionally allowed, and generously allowed regions of 81.5%, 17.0%, and 1.1%, respectively. The complex was observed to have only one residue (Asp_518) in the outlier region of the Ramachandran plot (0.4%) (Fig. S34C, ESI†). Overall, the complex showed a sterically acceptable conformation of their receptor Plm X protein molecule after the MD simulation study, indicating the complex to be stable in nature.
Next, the binding behavior of the 6k–Plm X complex was analyzed. The Root Mean Square Deviation (RMSD) of the Cα of the Plm X protein was analysed during the 30 ns simulation of the 6k–PlmX complex. The RMSD plot indicated that the 6k–PlmX complex reached its stable conformation within the first few nanoseconds and maintained its stability till the end of the simulation (Fig. 3). The Cα RMSD of Plm X attained the plateau phase within the first 5 ns of simulation time, where average CαRMSD was observed as 3.45 Å. The stability of ligand RMSD can be viewed in Fig. 3, where an average RMSD of 6k was found as 3.08 Å. The RMSD of both protein Cα and the ligand fluctuated within an acceptable range (3 Å).
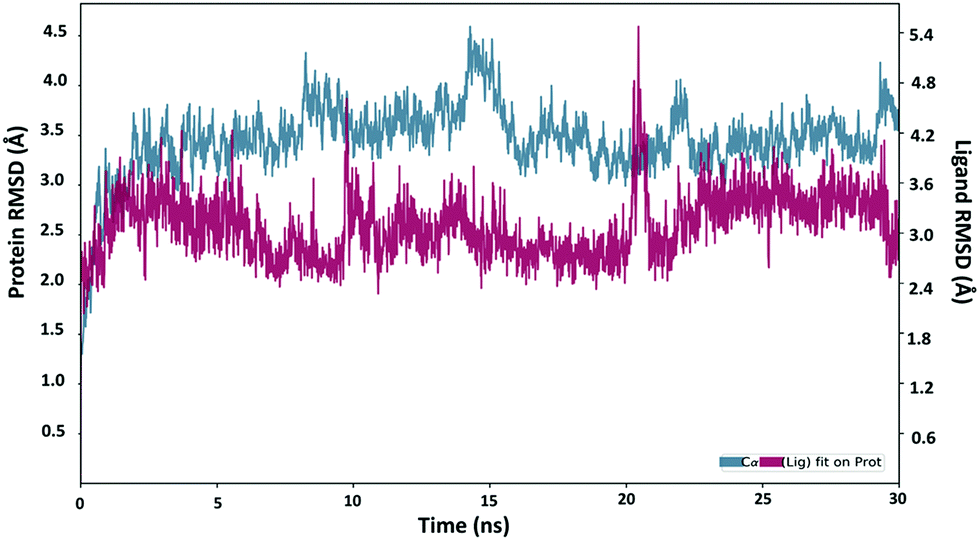 | ||
| Fig. 3 . The RMSD plot obtained for the 6k-PlmX complex. | ||
The Asp266 residue interacted with both the nitrogen atoms of the morpholine ring and the hydroxyl moiety of HEA by 75% and 88% via H-bond, respectively. The 2D interaction poses and the protein–ligand interaction histogram chart are shown in Fig. S35 (ESI†). The histogram showed favoured H-bond interactions with Asp_266; hydrophobic interactions with Ile264, Phe_311, Tyr357, Ile358, Phe360, Ile_363, Tyr_431, Met526, and Ile528; salt bridge with Asp_266, Asp_457, and water bridged interactions with Asp_266, Gly_268, Gly312, Ser313, Tyr357, Tyr_431, Asp_457, Gly459, The_460, and Arg534. Overall, these interactions facilitated 6k structural and conformational stability within the binding pocket. The panels show the total number of specific contacts the PlmX protein made with 6k over the course of the simulation. The PlmX protein made 5–6 contacts with compound 6k during the MD simulation (Fig. S34D, ESI†). The ligand–protein stable association would have contributed to the activity of compound 6k.
 | ||
| Fig. 4 Structure–Activity Landscape Index Similarity Map of the compounds. A scatter plot of the similarity relationships between antiplasmodial scaffolds based on the neighbour similarity FragFp-84% in a layered graph with the root at the top. | ||
3. Conclusion
In this article, we have demonstrated the synthesis, Pf3D7 activity, liver-stage activity, and PlmX activity of novel morpholine analogs. Although the compounds did not exhibit high potency against the malaria parasite, their tolerance up to 2 mM for normal cells combined with negligible lysis in RBCs up to 100 μM concentration were notable observations. Hit analog 6k, possessing the 3,4-bromophenyl substituent also revealed significant activity against malarial aspartyl protease, PlmX, and the results were further supported by in silico studies, supporting the stability of the ligand within the binding pocket. From SAR analysis, it was noted that the presence of a bromine substituent at the para position of the phenyl ring showed improved antiplasmodial activity over the analog 6b that holds bromine at the ortho position of the phenyl ring. Subsequently, medicinal chemistry optimization is essential to enhance the biological efficacy of 6k and obtain lead compounds based on HEA-morpholine.4. Experimental
4.1. General method
The chemicals and solvents used in the syntheses were obtained from commercial sources. Epoxide (2R,3S)-3-(N-BOC-amino)-1-oxirane-4-phenylbutane was purchased from GLR Innovation (New Delhi, India) and the carboxylic acids were purchased from TCI. The completion of the reaction and the purity of the final product were assayed by thin-layer chromatography (TLC). Small amounts of impurities left along with the product were further purified by column chromatography on alumina gel columns (100–200 mesh size, CDH). The melting points of the isolated compound were measured in open glass capillary tubes on a “BUCHI Labortechnik AG CH-9230”. Nuclear magnetic resonance (NMR), 1H, and 13C spectra were recorded using a JEOL ECX-400P NMR Spectrometer using TMS as an internal standard. The splitting patterns were reported as singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Further, a high-resolution Biosystems Q-Star Elite time-of-flight electrospray mass spectrometer was used for the characterization of the compounds. For HPLC analysis, an HPLC system (Gilson, USA) with an analytical column (C18), and a Thermo Separation Spectra SERIES UV100 detector coupled with software were used. The solvent used for the sample preparation for HPLC analysis was a mixture of acetonitrile and water.4.2. General procedure
Epoxide 1 (1 eq., 19 mmol) and morpholine 2 (1 eq., 19 mmol) were dissolved in isopropanol and the reaction was refluxed for 16 h. The completion of the reaction was monitored by TLC. Later, the mixture was allowed to cool and the remaining solvent was evaporated under reduced pressure to obtain compound 3 as a white solid. Compound 3 was dissolved in dichloromethane (DCM), then 20% of trifluoroacetic acid (TFA) was added dropwise at 0 °C and the reaction mixture was stirred on a magnetic stirrer for 2 h to obtain intermediate 4. After monitoring the TLC to ensure the completion of the reaction, DCM was removed under reduced pressure. In order to remove excess TFA, DCM was added and subsequently evaporated three times under vacuum. For the last step, 5a (88 mg, 0.34 mmol) was dissolved in DCM (30 mL) and 1 mL of triethylamine (TEA) was added to it in a round-bottom flask, and the reaction was stirred for 20 minutes. After that, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (97 mg, 0.51 mmol) was added, followed by the addition of anhydrous 1-hydroxybenzotriazole (HOBt) (69 mg, 0.51 mmol) within a time gap of 20 minutes. To the reaction mixture, compound 4 (deprotected HEA) was added at 0 °C and stirred for 24 h at room temperature. The formation of the final product, 6a was confirmed by TLC. The workup was done by removing the excess solvent under reduced pressure. The crude product was extracted using ethyl acetate and water as solvents. Traces of water were removed by treating the organic layer with anhydrous Na2SO4. After evaporation, an oily product was obtained, which was further purified by column chromatography using alumina and ethyl acetate/hexane as eluents (10:90). The final compound 6a was obtained as a white solid.
The above procedure was followed to synthesize compounds 6b-m in fair yields (40–60%).
The structures of the compounds (6a–m) were confirmed by different spectroscopic techniques and the spectra have been provided in the ESI† (Fig. S1–S32).
:9 methanol/chloroform). Yield: 99%. 1H NMR (400 MHz, CDCl3): δ 7.24–7.15 (m, 5H, –C6![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 5), 5.05 (d, J = 9.5 Hz, 1H, –O), 3.69–3.63 (m, 8H, 2x-NC2– + OC2– of morph), 2.90–2.86 (m, 2H, –C2–Ph), 2.53–2.51 (m, 2H, –C2-morph), 2.39–2.37 (m, 1H, –C(OH)CH2–), 2.31 (brs, 1H, –N–), 2.22–2.20 (m, 1H, –NH–C–), 1.35 (s, 9H, –(C3)3C–). 13C NMR (101 MHz, CDCl3): δ 155.91 (s), 138.40 (s), 129.50 (s), 128.44 (s), 126.34 (s), 79.22 (s), 66.99 (s), 65.37 (s), 61.29 (s), 53.62 (s), 53.33 (s), 39.38 (s), 28.44 (s).
:9 methanol/chloroform). Yield: 58%. 1H NMR (400 MHz, CDCl3): δ 9.32 (s, 1H, ortho-NC– of pyrazine), 8.71 (s, 1H, para-NC– of pyrazine), 8.51 (s, 1H, meta-NC– of pyrazine), 8.19–8.17 (d, J = 9.5 Hz, 1H, –N–), 7.26–7.23 (m, 5H, –C65), 4.25–4.23 (d, J = 9.3 Hz, 1H, –OH), 3.83–3.79 (m, 1H, –C(OH)CH2–), 3.65–3.63 (m, 4H, 2x–OC2– of morph), 3.05–3.03 (d, J = 7.7 Hz, 2H, –C2–Ph), 2.97–2.95 (m, 1H, –CH–N–), 2.56–2.53 (m, 2H, –C2-morph), 2.31–2.29 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 162.96 (s), 147.38 (s), 144.53 (s), 144.39 (s), 142.75 (s), 137.80 (s), 129.48 (s), 128.62 (s), 126.67 (s), 66.73 (s), 65.23 (s), 61.48 (s), 52.15 (s), 39.02 (s), 29.78 (s). HRMS m/z calcd [M + H]+ for C19H25N4O3+: 357.1926; found: 357.1916.
:9 methanol/chloroform). Yield: 52%. 1H NMR (400 MHz, CDCl3): δ 7.51–7.49 (d, J = 7.9, 1.0 Hz, 1H, –C (ortho) of Br–C6H4–), 7.29–7.27 (d, J = 6.5 Hz, 1H, –C(meta) of Br–C6H4–), 7.25 (d, J = 1.5 Hz, 1H, –C (para) of Br–C6H4–), 7.22–7.16 (m, 5H, –C65), 7.06–7.02 (m, 1H, –C (meta) of Br–C6H4–), 5.87–5.84 (d, J = 9.4 Hz, 1H, –O), 4.06–4.00 (m, 1H, –C(OH)CH2–), 3.69–3.64 (m, 5H, 2x-OCH2– of morph + –NH–C–), 3.00–2.98 (m, 2H, –C2–(CO)–), 2.86–2.84 (t, J = 8.6 Hz, 2H, –C2–Ph), 2.53–2.43 (m, 4H, 2x-NC2– of morph), 2.30–2.26 (m, 2H, –C2– of morph), 2.19–2.17 (m, 2H, –C2–C6H4–Br). 13C NMR (101 MHz, CDCl3): δ 171.57 (s), 140.05 (s), 138.02 (s), 132.91 (s), 130.83 (s), 129.45 (s), 128.55 (s), 128.17 (s), 127.64 (s), 126.57 (s), 124.35 (s), 66.89 (s), 65.06 (s), 61.26 (s), 53.60 (s), 51.58 (s), 39.02 (s), 36.61 (s), 32.23 (s), 29.80 (s).
:9 methanol/chloroform). Yield: 46%. 1H NMR (400 MHz, CDCl3): δ 7.55–7.52 (d, J = 8.7 Hz, 2x-C– of –C64–CF3), 7.26–7.22 (m, 2H, 2x-C– of –C64–CF3), 7.17–7.15 (m, 5H, –C65), 5.85–5.82 (d, J = 10.0 Hz, 1H, –O), 4.11–4.04 (dd, J = 18.1, 8.7 Hz, 1H, –C(OH)CH2–), 3.70 (brs, 1H, –NH–C–), 3.66–3.62 (m, 4H, 2x –OC2– of morph), 3.52 (s, 2H, –C(CO)–), 2.89–2.87 (d, J = 8.1 Hz, 2H, –C2–Ph), 2.55–2.50 (m, 2H, –C2-morph), 2.23–2.07 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 169.74 (s), 139.17 (s), 137.74 (s), 129.50 (s) 129.34 (s), 128.56 (s), 126.62 (s), 125.82 (s), 125.78 (s), 66.94 (s), 65.51 (s), 61.13 (s), 53.51 (s), 51.52 (s), 43.74 (s), 38.96 (s).
:9 methanol/chloroform). Yield: 51%. 1H NMR (400 MHz, CDCl3): δ 7.69–7.67 (d, J = 8.8 Hz, 2H, ortho, 2x-COC65), 7.29–7.18 (m, 5H, –C65), 6.87–6.85 (d, J = 8.8 Hz, 2H, meta, 2x-COC65), 6.55–6.53 (d, J = 9.2 Hz, 1H, –O), 4.30–4.26 (m, 1H, –C(OH)CH2–), 3.85–3.80 (m, 4H, 2x-OC2 of –C64-morph), 3.79–3.77 (d, J = 3.1 Hz, 1H, –NH–C–), 3.65 (m, 4H, 2x-OC2 of –CH2-morph), 3.23–3.21 (m, 4H, 2x-NC2 of –C6H4-morph), 3.05–2.99 (m, 2H, –C2–Ph), 2.56–2.53 (m, 2H, –C2-morph), 2.35–2.24 (m, 4H, 2x-NCH2– of morph). 13C NMR (101 MHz, CDCl3): δ 166.72 (s), 153.50 (s), 138.23 (s), 129.52 (s), 128.57 (s), 128.47 (s), 126.53 (s), 124.67 (s), 114.13 (s), 67.00 (s), 66.72 (s), 65.35 (s), 61.36 (s), 53.58 (s), 51.85 (s), 48.14 (s), 39.25 (s).
:9 methanol/chloroform). Yield: 55%. 1H NMR (400 MHz, CDCl3): δ 7.29–7.25 (m, 5H, –C65(CH)CH3–), 7.21–7.11 (m, 5H, –C65), 5.85–5.72 (dd, J = 40.1, 9.3 Hz, 1H,–O), 4.06–4.00 (dd, J = 17.1, 8.7 Hz, 1H, –C(OH)CH2–), 3.65–3.59 (d, J = 24.4 Hz, 5H, 2x-OC2– of morph + –C(CO)), 3.52 (m, 1H, –C–NH–), 2.87–2.78 (m, 2H, –C2–Ph), 2.53–2.51 (m, 2H, –C2– of morph), 2.25–2.06 (m, 4H, 2x-NC2- of morph), 1.43–1.41 (d, J = 5.6 Hz, 3H, –C(C3).13C NMR (101 MHz, CDCl3): δ 174.13 (s), 141.18 (s), 137.93 (s), 129.40 (s), 128.95 (s), 128.47 (s), 127.55 (s), 127.26 (s), 126.47 (s), 66.94 (s), 65.59 (s), 61.19 (s), 53.55 (s), 51.48 (s), 47.24 (s), 38.75 (s), 18.41 (s).
:9 methanol/chloroform). Yield: 57%. 1H NMR (400 MHz, CDCl3): δ 7.32–7.23 (m, 5H, –C65CH2CO–), 7.18–7.14 (m, 5H, –CH2C65), 5.85–5.83 (d, J = 9.3 Hz, 1H, –O), 4.08–4.02 (dd, J = 17.0, 7.8 Hz, 1H, –C(OH)CH2–), 3.66 (m, 1H, –CNH–), 3.62 (m, 4H, 2x-OC2– of morph), 3.50–3.48 (d, J = 4.6 Hz, 2H, –C6H5C2CO–), 2.86–2.84 (d, J = 8.2 Hz, 2H, –C2-Ph), 2.54–2.47 (m, 2H, –C2-morph), 2.22–2.09 (m, 4H, 2x-NC2- of morph). 13C NMR (101 MHz, CDCl3): δ 170.88 (s), 137.96 (s), 135.08 (s), 129.42 (s), 129.31 (s), 129.02 (s), 128.53 (s), 127.33 (s), 126.54 (s), 66.99 (s), 65.45 (s), 61.11 (s), 53.53 (s), 51.59 (s), 44.07 (s), 38.86 (s).
:9 methanol/chloroform). Yield: 52%. 1H NMR (400 MHz, CDCl3) δ 7.27–7.22 (m, 4H, ortho, meta 2x –C64–), 7.09–7.02 (d, J = 8.4 Hz, 2H, 2x-meta of methoxy–C64–), 6.97–6.95 (d, J = 9.6 Hz, 1H, para –CH2–C65), 6.80–6.78 (d, J = 8.5 Hz, 2H, 2x-ortho of methoxy-C64–), 4.48–4.37 (qt, J = 21.4 Hz, 2H, –OC2–(CO)–), 4.14–4.08 (m, 1H, –C(OH)CH2–), 3.73–3.69 (m, 1H, –C–NH–), 3.67–3.60 (m, 4H, 2x-OC2– of morph), 2.99–2.94 (d, J = 14.7, 7.7 Hz, 2H, –C2–Ph), 2.53–2.48 (m, 2H, –C2– morph), 2.27 (s, 3H, C3–C6H4–), 2.24–2.16 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 168.45 (s), 155.26 (s), 137.81 (s), 131.46 (s), 130.27 (s), 129.49 (s), 128.57 (s), 126.62 (s), 114.62 (s), 67.54 (s), 66.84 (s), 64.99 (s), 61.15 (s), 53.50 (s), 51.38 (s), 39.00 (s), 20.56 (s).
:9 methanol/chloroform). Yield: 50%. 1H NMR (400 MHz, CDCl3): δ 9.20 (s, 1H, ortho –NC– of pyrazine), 8.38 (s, 1H, meta –NC– of pyrazine), 8.16–8.13 (d, J = 9.7 Hz, 1H, –CH–N–), 7.29–7.24 (m, 4H, ortho, meta 2x –C65), 7.20–7.17 (m, 1H, para –C65), 4.28–4.22 (m, 1H, –C(OH)CH2–), 3.84–3.81 (dd, J = 10.5, 3.7 Hz, 1H, –C–NH–), 3.69–3.61 (m, 5H, –O + 2x –OC2– of morph), 3.06–3.04 (d, J = 7.6 Hz, 2H, –C2–Ph), 2.63 (s, 3H, –pyrazine–C3), 2.59–2.54 (m, 2H, –C2– morph), 2.39–2.28 (m, 4H, 2x-NCH2– of morph). 13C NMR (101 MHz, CDCl3): δ 163.26 (s), 157.16 (s), 143.44 (s), 142.52 (s), 141.70 (s), 137.92 (s), 129.48 (s), 128.59 (s), 126.62 (s), 66.87 (s), 65.32 (s), 61.42 (s), 53.57 (s), 52.00 (s), 39.06 (s), 21.92 (s). HRMS m/z calcd [M + K]+ for C20H26N4O3K+: 409.1648; found: 409.1631.
:9 methanol/chloroform). Yield: 48%. 1H NMR (400 MHz, CDCl3): δ 7.28–7.24 (m, 5H, –(CH)–C65), 7.22–7.10 (m, 5H, –CH2–C65), 5.81–5.79 (dd, J = 9.2 Hz, 1H, –O), 4.06 (m, 1H, –C(OH)CH2–), 3.61–3.59 (m, 4H, 2x –OC2– of morph), 3.21–3.13 (dt, J = 33.4, 7.5 Hz, 1H, –C–(CO)–), 2.90–2.79 (dd, J = 36.7, 7.9 Hz, 2H, –C2–Ph), 2.56–2.39 (m, 2H, –C2-morph), 2.28–2.23 (m, J = 19.4 Hz, 1H, –C–NH–), 2.09–2.06 (m, 4H, 2x –NC2– of morph), 1.83–1.71 (m, 2H, –C2–CH3), 0.86–0.75 (dt, J = 29.6, 7.3 Hz, 3H, –CH2– C3). 13C NMR (101 MHz, CDCl3): δ 173.34 (s), 139.95 (s), 137.91 (s), 129.36 (s), 128.81 (s), 128.44 (s), 127.85 (s), 127.13 (s), 126.43 (s), 66.89 (s), 65.53 (s), 61.20 (s), 55.48 (s), 53.33 (s), 51.44 (s), 38.79 (s), 26.08 (s), 12.46 (s). HRMS m/z calcd [M + H]+ for C24H33N2O3+: 397.2491; found: 397.2489.
:9 methanol/chloroform). Yield: 45%. 1H NMR (400 MHz, CDCl3): δ 7.67–7.65 (d, J = 8.9 Hz, 2H, ortho –C65–CO–), 7.30–7.25 (m, 4H, ortho, meta –C65), 7.20–7.16 (m, 1H, para-C65), 6.88–6.86 (d, J = 8.9 Hz, 2H, meta –C64–CO–), 6.53–6.50 (d, J = 9.3 Hz, 1H, –O), 4.29–4.23 (m, 1H, –C(OH)CH2–), 3.79–3.75 (dd, J = 10.7, 3.5 Hz, 1H, –C–NH–), 3.65–3.62 (m, 4H, 2x –OC2– of morph), 3.30–3.27 (m, 4H, 2x –NC2– of piperazine), 3.08–2.98 (m, 2H, –C2–Ph), 2.56–2.51 (m, 6H, 3x –NC2– of morph), 2.33–2.26 (m, 7H, 2x –NC2 of piperazine + C3– piperazine). 13C NMR (101 MHz, CDCl3): δ 166.76 (s), 153.42 (s), 138.26 (s), 129.52 (s), 128.56 (s), 128.43 (s), 126.50 (s), 124.11 (s), 114.31 (s), 67.02 (s), 65.35 (s), 61.34 (s), 54.87 (s), 53.56 (s), 51.81 (s), 47.87 (s), 46.20 (s), 39.26 (s).
:9 methanol/chloroform). Yield: 60%. 1H NMR (400 MHz, CDCl3): δ 7.52–7.50 (d, J = 7.7 Hz, 1H, –N–), 7.26–7.18 (m, 8H, ortho, meta –C65), 7.07– 7.03 (m, 1H, para –C65), 5.85–5.82 (d, J = 9.3 Hz, 1H, –O), 4.07–4.01 (m, 1H, –C(OH)CH2–), 3.68–3.65 (m, 5H, –C–NH– + –OC2– of morph), 3.03–2.95 (m, 2H, –C2–C6H4–Br), 2.87–2.86 (d, J = 7.6, 2.7 Hz, 2H, –C2–Ph), 2.54–2.50 (dd, J = 10.4, 5.4 Hz, 2H, –C2-morph), 2.47– 2.44 (t, J = 7.6 Hz, 2H, –C2–(CO)–), 2.28–2.15 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 171.32 (s), 139.86 (s), 137.95 (s), 131.58 (s), 130.38 (s), 129.42 (s), 128.55 (s), 126.59 (s), 120.11 (s), 66.95 (s), 64.92 (s), 61.10 (s), 53.59 (s), 51.42 (s), 39.09 (s), 38.20 (s), 30.89 (s). HPLC Purity: 97.6%.
:9 methanol/chloroform). Yield: 40%. 1H NMR (400 MHz, CDCl3): δ 7.75–7.72 (m, 2H, ortho –C64–F), 7.29–7.28 (d, J = 12.4, 3.7 Hz, 4H, ortho, meta –C65), 7.22–7.19 (m, 1H, para –C65), 7.11–7.07 (m, 2H, meta –C64–F), 6.59–6.57 (d, J = 9.2 Hz, 1H, –O), 4.30–4.24 (m, 1H, –C(OH)CH2–), 3.82–3.78 (dd, J = 10.4, 3.8 Hz, 1H, –C–NH–), 3.69–3.62 (m, 4H, 2x –OC2– of morph), 3.09–2.98 (m, 2H, C2–Ph), 2.59–2.54 (m, 2H, –C2-morph), 2.36–2.28 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 166.13 (s), 163.57 (s), 137.96 (s), 129.48 (s), 129.34 (s), 129.25 (s), 128.61 (s), 126.64 (s), 115.80 (s), 66.98 (s), 65.31 (s), 61.38 (s), 53.58 (s), 52.08 (s), 39.19 (s).
:9 methanol/chloroform). Yield: 44%. 1H NMR (400 MHz, CDCl3): δ 7.83–7.81(d, J = 4.3 Hz, 2H, ortho –C64–CF3) 7.69–7.67 (d, J = 8.4 Hz, 2H, meta –C64–CF3), 7.30–7.28 (m, 4H, ortho, meta-C65), 7.25–7.21 (m, 1H, para –C65), 6.68–6.66 (d, J = 9.3 Hz, 1H, –O), 4.32–4.26 (qt, J = 16.4 Hz, 1H, –C(OH)CH2–), 3.68–3.67 (m, 1H, –C–NH–), 3.65–3.64 (m, 4H, 2x –OC2– of morph), 3.09–3.03 (m, 2H, –C2–Ph), 2.60–2.55 (m, 2H, –C2-morph), 2.39–2.30 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 165.91 (s), 137.79 (s), 133.79 (s), 133.49 (s), 133.16 (s), 129.47 (s), 128.65 (s), 127.46 (s), 126.73 (s), 125.75 (s), 125.71 (s) 66.99 (s), 65.28 (s), 61.35 (s), 53.58 (s), 52.16 (s), 39.19 (s).
5), 5.05 (d, J = 9.5 Hz, 1H, –O), 3.69–3.63 (m, 8H, 2x-NC2– + OC2– of morph), 2.90–2.86 (m, 2H, –C2–Ph), 2.53–2.51 (m, 2H, –C2-morph), 2.39–2.37 (m, 1H, –C(OH)CH2–), 2.31 (brs, 1H, –N–), 2.22–2.20 (m, 1H, –NH–C–), 1.35 (s, 9H, –(C3)3C–). 13C NMR (101 MHz, CDCl3): δ 155.91 (s), 138.40 (s), 129.50 (s), 128.44 (s), 126.34 (s), 79.22 (s), 66.99 (s), 65.37 (s), 61.29 (s), 53.62 (s), 53.33 (s), 39.38 (s), 28.44 (s).
:9 methanol/chloroform). Yield: 58%. 1H NMR (400 MHz, CDCl3): δ 9.32 (s, 1H, ortho-NC– of pyrazine), 8.71 (s, 1H, para-NC– of pyrazine), 8.51 (s, 1H, meta-NC– of pyrazine), 8.19–8.17 (d, J = 9.5 Hz, 1H, –N–), 7.26–7.23 (m, 5H, –C65), 4.25–4.23 (d, J = 9.3 Hz, 1H, –OH), 3.83–3.79 (m, 1H, –C(OH)CH2–), 3.65–3.63 (m, 4H, 2x–OC2– of morph), 3.05–3.03 (d, J = 7.7 Hz, 2H, –C2–Ph), 2.97–2.95 (m, 1H, –CH–N–), 2.56–2.53 (m, 2H, –C2-morph), 2.31–2.29 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 162.96 (s), 147.38 (s), 144.53 (s), 144.39 (s), 142.75 (s), 137.80 (s), 129.48 (s), 128.62 (s), 126.67 (s), 66.73 (s), 65.23 (s), 61.48 (s), 52.15 (s), 39.02 (s), 29.78 (s). HRMS m/z calcd [M + H]+ for C19H25N4O3+: 357.1926; found: 357.1916.
:9 methanol/chloroform). Yield: 52%. 1H NMR (400 MHz, CDCl3): δ 7.51–7.49 (d, J = 7.9, 1.0 Hz, 1H, –C (ortho) of Br–C6H4–), 7.29–7.27 (d, J = 6.5 Hz, 1H, –C(meta) of Br–C6H4–), 7.25 (d, J = 1.5 Hz, 1H, –C (para) of Br–C6H4–), 7.22–7.16 (m, 5H, –C65), 7.06–7.02 (m, 1H, –C (meta) of Br–C6H4–), 5.87–5.84 (d, J = 9.4 Hz, 1H, –O), 4.06–4.00 (m, 1H, –C(OH)CH2–), 3.69–3.64 (m, 5H, 2x-OCH2– of morph + –NH–C–), 3.00–2.98 (m, 2H, –C2–(CO)–), 2.86–2.84 (t, J = 8.6 Hz, 2H, –C2–Ph), 2.53–2.43 (m, 4H, 2x-NC2– of morph), 2.30–2.26 (m, 2H, –C2– of morph), 2.19–2.17 (m, 2H, –C2–C6H4–Br). 13C NMR (101 MHz, CDCl3): δ 171.57 (s), 140.05 (s), 138.02 (s), 132.91 (s), 130.83 (s), 129.45 (s), 128.55 (s), 128.17 (s), 127.64 (s), 126.57 (s), 124.35 (s), 66.89 (s), 65.06 (s), 61.26 (s), 53.60 (s), 51.58 (s), 39.02 (s), 36.61 (s), 32.23 (s), 29.80 (s).
:9 methanol/chloroform). Yield: 46%. 1H NMR (400 MHz, CDCl3): δ 7.55–7.52 (d, J = 8.7 Hz, 2x-C– of –C64–CF3), 7.26–7.22 (m, 2H, 2x-C– of –C64–CF3), 7.17–7.15 (m, 5H, –C65), 5.85–5.82 (d, J = 10.0 Hz, 1H, –O), 4.11–4.04 (dd, J = 18.1, 8.7 Hz, 1H, –C(OH)CH2–), 3.70 (brs, 1H, –NH–C–), 3.66–3.62 (m, 4H, 2x –OC2– of morph), 3.52 (s, 2H, –C(CO)–), 2.89–2.87 (d, J = 8.1 Hz, 2H, –C2–Ph), 2.55–2.50 (m, 2H, –C2-morph), 2.23–2.07 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 169.74 (s), 139.17 (s), 137.74 (s), 129.50 (s) 129.34 (s), 128.56 (s), 126.62 (s), 125.82 (s), 125.78 (s), 66.94 (s), 65.51 (s), 61.13 (s), 53.51 (s), 51.52 (s), 43.74 (s), 38.96 (s).
:9 methanol/chloroform). Yield: 51%. 1H NMR (400 MHz, CDCl3): δ 7.69–7.67 (d, J = 8.8 Hz, 2H, ortho, 2x-COC65), 7.29–7.18 (m, 5H, –C65), 6.87–6.85 (d, J = 8.8 Hz, 2H, meta, 2x-COC65), 6.55–6.53 (d, J = 9.2 Hz, 1H, –O), 4.30–4.26 (m, 1H, –C(OH)CH2–), 3.85–3.80 (m, 4H, 2x-OC2 of –C64-morph), 3.79–3.77 (d, J = 3.1 Hz, 1H, –NH–C–), 3.65 (m, 4H, 2x-OC2 of –CH2-morph), 3.23–3.21 (m, 4H, 2x-NC2 of –C6H4-morph), 3.05–2.99 (m, 2H, –C2–Ph), 2.56–2.53 (m, 2H, –C2-morph), 2.35–2.24 (m, 4H, 2x-NCH2– of morph). 13C NMR (101 MHz, CDCl3): δ 166.72 (s), 153.50 (s), 138.23 (s), 129.52 (s), 128.57 (s), 128.47 (s), 126.53 (s), 124.67 (s), 114.13 (s), 67.00 (s), 66.72 (s), 65.35 (s), 61.36 (s), 53.58 (s), 51.85 (s), 48.14 (s), 39.25 (s).
:9 methanol/chloroform). Yield: 55%. 1H NMR (400 MHz, CDCl3): δ 7.29–7.25 (m, 5H, –C65(CH)CH3–), 7.21–7.11 (m, 5H, –C65), 5.85–5.72 (dd, J = 40.1, 9.3 Hz, 1H,–O), 4.06–4.00 (dd, J = 17.1, 8.7 Hz, 1H, –C(OH)CH2–), 3.65–3.59 (d, J = 24.4 Hz, 5H, 2x-OC2– of morph + –C(CO)), 3.52 (m, 1H, –C–NH–), 2.87–2.78 (m, 2H, –C2–Ph), 2.53–2.51 (m, 2H, –C2– of morph), 2.25–2.06 (m, 4H, 2x-NC2- of morph), 1.43–1.41 (d, J = 5.6 Hz, 3H, –C(C3).13C NMR (101 MHz, CDCl3): δ 174.13 (s), 141.18 (s), 137.93 (s), 129.40 (s), 128.95 (s), 128.47 (s), 127.55 (s), 127.26 (s), 126.47 (s), 66.94 (s), 65.59 (s), 61.19 (s), 53.55 (s), 51.48 (s), 47.24 (s), 38.75 (s), 18.41 (s).
:9 methanol/chloroform). Yield: 57%. 1H NMR (400 MHz, CDCl3): δ 7.32–7.23 (m, 5H, –C65CH2CO–), 7.18–7.14 (m, 5H, –CH2C65), 5.85–5.83 (d, J = 9.3 Hz, 1H, –O), 4.08–4.02 (dd, J = 17.0, 7.8 Hz, 1H, –C(OH)CH2–), 3.66 (m, 1H, –CNH–), 3.62 (m, 4H, 2x-OC2– of morph), 3.50–3.48 (d, J = 4.6 Hz, 2H, –C6H5C2CO–), 2.86–2.84 (d, J = 8.2 Hz, 2H, –C2-Ph), 2.54–2.47 (m, 2H, –C2-morph), 2.22–2.09 (m, 4H, 2x-NC2- of morph). 13C NMR (101 MHz, CDCl3): δ 170.88 (s), 137.96 (s), 135.08 (s), 129.42 (s), 129.31 (s), 129.02 (s), 128.53 (s), 127.33 (s), 126.54 (s), 66.99 (s), 65.45 (s), 61.11 (s), 53.53 (s), 51.59 (s), 44.07 (s), 38.86 (s).
:9 methanol/chloroform). Yield: 52%. 1H NMR (400 MHz, CDCl3) δ 7.27–7.22 (m, 4H, ortho, meta 2x –C64–), 7.09–7.02 (d, J = 8.4 Hz, 2H, 2x-meta of methoxy–C64–), 6.97–6.95 (d, J = 9.6 Hz, 1H, para –CH2–C65), 6.80–6.78 (d, J = 8.5 Hz, 2H, 2x-ortho of methoxy-C64–), 4.48–4.37 (qt, J = 21.4 Hz, 2H, –OC2–(CO)–), 4.14–4.08 (m, 1H, –C(OH)CH2–), 3.73–3.69 (m, 1H, –C–NH–), 3.67–3.60 (m, 4H, 2x-OC2– of morph), 2.99–2.94 (d, J = 14.7, 7.7 Hz, 2H, –C2–Ph), 2.53–2.48 (m, 2H, –C2– morph), 2.27 (s, 3H, C3–C6H4–), 2.24–2.16 (m, 4H, 2x-NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 168.45 (s), 155.26 (s), 137.81 (s), 131.46 (s), 130.27 (s), 129.49 (s), 128.57 (s), 126.62 (s), 114.62 (s), 67.54 (s), 66.84 (s), 64.99 (s), 61.15 (s), 53.50 (s), 51.38 (s), 39.00 (s), 20.56 (s).
:9 methanol/chloroform). Yield: 50%. 1H NMR (400 MHz, CDCl3): δ 9.20 (s, 1H, ortho –NC– of pyrazine), 8.38 (s, 1H, meta –NC– of pyrazine), 8.16–8.13 (d, J = 9.7 Hz, 1H, –CH–N–), 7.29–7.24 (m, 4H, ortho, meta 2x –C65), 7.20–7.17 (m, 1H, para –C65), 4.28–4.22 (m, 1H, –C(OH)CH2–), 3.84–3.81 (dd, J = 10.5, 3.7 Hz, 1H, –C–NH–), 3.69–3.61 (m, 5H, –O + 2x –OC2– of morph), 3.06–3.04 (d, J = 7.6 Hz, 2H, –C2–Ph), 2.63 (s, 3H, –pyrazine–C3), 2.59–2.54 (m, 2H, –C2– morph), 2.39–2.28 (m, 4H, 2x-NCH2– of morph). 13C NMR (101 MHz, CDCl3): δ 163.26 (s), 157.16 (s), 143.44 (s), 142.52 (s), 141.70 (s), 137.92 (s), 129.48 (s), 128.59 (s), 126.62 (s), 66.87 (s), 65.32 (s), 61.42 (s), 53.57 (s), 52.00 (s), 39.06 (s), 21.92 (s). HRMS m/z calcd [M + K]+ for C20H26N4O3K+: 409.1648; found: 409.1631.
:9 methanol/chloroform). Yield: 48%. 1H NMR (400 MHz, CDCl3): δ 7.28–7.24 (m, 5H, –(CH)–C65), 7.22–7.10 (m, 5H, –CH2–C65), 5.81–5.79 (dd, J = 9.2 Hz, 1H, –O), 4.06 (m, 1H, –C(OH)CH2–), 3.61–3.59 (m, 4H, 2x –OC2– of morph), 3.21–3.13 (dt, J = 33.4, 7.5 Hz, 1H, –C–(CO)–), 2.90–2.79 (dd, J = 36.7, 7.9 Hz, 2H, –C2–Ph), 2.56–2.39 (m, 2H, –C2-morph), 2.28–2.23 (m, J = 19.4 Hz, 1H, –C–NH–), 2.09–2.06 (m, 4H, 2x –NC2– of morph), 1.83–1.71 (m, 2H, –C2–CH3), 0.86–0.75 (dt, J = 29.6, 7.3 Hz, 3H, –CH2– C3). 13C NMR (101 MHz, CDCl3): δ 173.34 (s), 139.95 (s), 137.91 (s), 129.36 (s), 128.81 (s), 128.44 (s), 127.85 (s), 127.13 (s), 126.43 (s), 66.89 (s), 65.53 (s), 61.20 (s), 55.48 (s), 53.33 (s), 51.44 (s), 38.79 (s), 26.08 (s), 12.46 (s). HRMS m/z calcd [M + H]+ for C24H33N2O3+: 397.2491; found: 397.2489.
:9 methanol/chloroform). Yield: 45%. 1H NMR (400 MHz, CDCl3): δ 7.67–7.65 (d, J = 8.9 Hz, 2H, ortho –C65–CO–), 7.30–7.25 (m, 4H, ortho, meta –C65), 7.20–7.16 (m, 1H, para-C65), 6.88–6.86 (d, J = 8.9 Hz, 2H, meta –C64–CO–), 6.53–6.50 (d, J = 9.3 Hz, 1H, –O), 4.29–4.23 (m, 1H, –C(OH)CH2–), 3.79–3.75 (dd, J = 10.7, 3.5 Hz, 1H, –C–NH–), 3.65–3.62 (m, 4H, 2x –OC2– of morph), 3.30–3.27 (m, 4H, 2x –NC2– of piperazine), 3.08–2.98 (m, 2H, –C2–Ph), 2.56–2.51 (m, 6H, 3x –NC2– of morph), 2.33–2.26 (m, 7H, 2x –NC2 of piperazine + C3– piperazine). 13C NMR (101 MHz, CDCl3): δ 166.76 (s), 153.42 (s), 138.26 (s), 129.52 (s), 128.56 (s), 128.43 (s), 126.50 (s), 124.11 (s), 114.31 (s), 67.02 (s), 65.35 (s), 61.34 (s), 54.87 (s), 53.56 (s), 51.81 (s), 47.87 (s), 46.20 (s), 39.26 (s).
:9 methanol/chloroform). Yield: 60%. 1H NMR (400 MHz, CDCl3): δ 7.52–7.50 (d, J = 7.7 Hz, 1H, –N–), 7.26–7.18 (m, 8H, ortho, meta –C65), 7.07– 7.03 (m, 1H, para –C65), 5.85–5.82 (d, J = 9.3 Hz, 1H, –O), 4.07–4.01 (m, 1H, –C(OH)CH2–), 3.68–3.65 (m, 5H, –C–NH– + –OC2– of morph), 3.03–2.95 (m, 2H, –C2–C6H4–Br), 2.87–2.86 (d, J = 7.6, 2.7 Hz, 2H, –C2–Ph), 2.54–2.50 (dd, J = 10.4, 5.4 Hz, 2H, –C2-morph), 2.47– 2.44 (t, J = 7.6 Hz, 2H, –C2–(CO)–), 2.28–2.15 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 171.32 (s), 139.86 (s), 137.95 (s), 131.58 (s), 130.38 (s), 129.42 (s), 128.55 (s), 126.59 (s), 120.11 (s), 66.95 (s), 64.92 (s), 61.10 (s), 53.59 (s), 51.42 (s), 39.09 (s), 38.20 (s), 30.89 (s). HPLC Purity: 97.6%.
:9 methanol/chloroform). Yield: 40%. 1H NMR (400 MHz, CDCl3): δ 7.75–7.72 (m, 2H, ortho –C64–F), 7.29–7.28 (d, J = 12.4, 3.7 Hz, 4H, ortho, meta –C65), 7.22–7.19 (m, 1H, para –C65), 7.11–7.07 (m, 2H, meta –C64–F), 6.59–6.57 (d, J = 9.2 Hz, 1H, –O), 4.30–4.24 (m, 1H, –C(OH)CH2–), 3.82–3.78 (dd, J = 10.4, 3.8 Hz, 1H, –C–NH–), 3.69–3.62 (m, 4H, 2x –OC2– of morph), 3.09–2.98 (m, 2H, C2–Ph), 2.59–2.54 (m, 2H, –C2-morph), 2.36–2.28 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 166.13 (s), 163.57 (s), 137.96 (s), 129.48 (s), 129.34 (s), 129.25 (s), 128.61 (s), 126.64 (s), 115.80 (s), 66.98 (s), 65.31 (s), 61.38 (s), 53.58 (s), 52.08 (s), 39.19 (s).
:9 methanol/chloroform). Yield: 44%. 1H NMR (400 MHz, CDCl3): δ 7.83–7.81(d, J = 4.3 Hz, 2H, ortho –C64–CF3) 7.69–7.67 (d, J = 8.4 Hz, 2H, meta –C64–CF3), 7.30–7.28 (m, 4H, ortho, meta-C65), 7.25–7.21 (m, 1H, para –C65), 6.68–6.66 (d, J = 9.3 Hz, 1H, –O), 4.32–4.26 (qt, J = 16.4 Hz, 1H, –C(OH)CH2–), 3.68–3.67 (m, 1H, –C–NH–), 3.65–3.64 (m, 4H, 2x –OC2– of morph), 3.09–3.03 (m, 2H, –C2–Ph), 2.60–2.55 (m, 2H, –C2-morph), 2.39–2.30 (m, 4H, 2x –NC2– of morph). 13C NMR (101 MHz, CDCl3): δ 165.91 (s), 137.79 (s), 133.79 (s), 133.49 (s), 133.16 (s), 129.47 (s), 128.65 (s), 127.46 (s), 126.73 (s), 125.75 (s), 125.71 (s) 66.99 (s), 65.28 (s), 61.35 (s), 53.58 (s), 52.16 (s), 39.19 (s).
4.3. Parasite culture
The chloroquine-sensitive (CQS) (3D7) P. falciparum strain was cultured in freshly collected donor erythrocytes in a complete RPMI-based malaria culture medium (CMCM) according to the Malaria Research and Reference Reagent Resource Center (MR4) guidelines.51 Thin blood smears stained with Giemsa were prepared for routine monitoring. Synchronized culture (trophozoites) was taken for the growth progression assay. To establish the morphological changes in the parasite, a blood film must be prepared. A thin film was air-dried and fixed with methanol. Blood films were stained with a 7.5% Giemsa solution (pH of 7.2) for 15 min. Parasitemia was counted using a brightfield microscope, and the morphological changes were noted via imaging.4.4. Cytotoxicity effects of compounds on normal human cells
Cytotoxicity against human cell lines (HepG2) was determined using MTT dye. HepG2 was maintained at 37 °C, 5% CO2 in sterile culture flasks, and complete DMEM medium supplemented with 5% FBS, and gentamicin (40 mg mL−1). Media were refreshed thrice a week. The cells were trypsinized (0.05% trypsin/0.5 mM EDTA), washed, and distributed in 96-well plates (5000 cells per well), followed by incubation for another 24 h at 37 °C. The test compounds and DMSO were added in triplicate. After 3 h of incubation at 37 °C, the supernatant was removed and 100 μL fresh media were added and the kept plates in an incubator at 37 °C, then 100 μL of the MTT solution in complete DMEM was added to each well after 24, 48, and 72 h of incubation. The culture plates were read at 570 nm in a spectrophotometer (Tecan Infinite M200, Nanoquant, UK) with a 490 nm filter at different time points. To determine cytotoxic concentrations, a dose–response curve was prepared.4.5. Hemolysis assay
Hemolysis assay was performed using RBCs suspension 10% (v/v) washed with 1 × PBS (pH 7.4) and resuspended in 1 × PBS. The suspension was treated with compounds at different concentrations for 2 hours at 37 °C. Samples were centrifuged at 800×g for 5 min at room temperature. The supernatant was taken for absorbance at 540 nm to obtain the percentage of RBCs lysis. Triton X-100 with 0.1% (v/v) was used as a positive control. The percentage of RBCs lysis was obtained by using the following formula:| % RBCs lysis = (OD500nm sample − OD500nm PBS)/(OD500nm Triton X-100 0.1% − OD500nm PBS). |
4.6. In vitro drug combination experiment
The in vitro combination assay was done using ART and 6k at different concentrations. A 24-well plate was used to perform the assay and was incubated for 48 hours at 37 °C. After that, thin blood smears were drawn and stained with a buffer solution of the Giemsa stain (9:1). The slides with smears were read with a 100X immersion oil lens of the brightfield microscope.
4.7. Stage-specific study
Parasite cultures with 80% trophozoites, 18% rings, and 2% schizonts were obtained by synchronization with 5% sorbitol. The assay was performed in a 24 well microtiter plate with a 2-fold dilution of culture, and the IC50 drug concentrations of 5 μM (6k) and 3 nM (ART) were used. The plate was incubated for 50 hours at 37 °C. The parasitemia was calculated using Giemsa-stained thin blood smears.524.8. In vitro activity against P. berghei hepatic (liver) stages
In vitro activity against the liver stage of P. berghei infection was assessed as previously described.53,54 Briefly, Huh7 cells were routinely cultured in 1640 Roswell Park Memorial Institute (RPMI) medium supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) glutamine, 1% (v/v) penicillin/streptomycin, 1% non-essential amino acids, and mM 2-(4-(2- hydroxyethyl)piperazin-1-yl)ethanesulfonic acid (HEPES). For drug screening experiments, Huh7 cells were seeded in 96-well plate and incubated overnight at 37 °C with 5% CO2. The stock solutions of the test compounds were prepared in DMSO and were serially diluted in an infection medium, i.e., culture medium supplemented with gentamicin (50 mg mL−1) to get the test concentrations. On the day of the infection, the culture medium was replaced by the serial dilutions of test compounds and incubated for 1 h at 37 °C with 5% CO2. Next, firefly luciferase-expressing P. berghei sporozoites, freshly isolated from the salivary glands of female infected Anopheles mosquitoes, were added to the cultures, and the plates were centrifuged at 1800g for 5 min at room temperature and incubated at 37 °C with 5% CO2. To assess the effect of each compound concentration on the cell viability, at 46 h post-infection (hpi), cultures were incubated with Alamar Blue, according to the manufacturer's recommendations. Parasite load was then assessed by a bioluminescence assay using a multi-plate reader. Nonlinear regression analysis was employed to fit the normalized results of the dose–response curves, and IC50 values were determined using GraphPad Prism 6.0 (GraphPad software).4.9. Recombinant PfPlm X activity assay
Protease cleavage assays were performed with purified recombinant PMX in digestion buffer (25 mM Tris·HCl, 25 mM MES, pH 6.4 and 5.5 for PMX), respectively; 0.2 μM of a synthetic peptide substrate (ThermoFisher scientific, >98% purity). Samples were incubated at 37 °C for 1 hour and the fluorescence was measured using a SynergyH1 multi-well plate reader (BioTek) excited at 340 nm and reading emissions at 490 nm. Samples were gently shaken during incubation. Changes in Relative Fluorescence Units (RFU) were measured for each time point by subtracting the RFU from the blank (without enzyme) for each time point.434.10. Computational
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) LogP), the logarithm of water solubility (ESOL LogS), etc. were calculated. Other predicted properties were GI absorption tendency, BBB permeability behavior, and Lipinski rule.
LogP), the logarithm of water solubility (ESOL LogS), etc. were calculated. Other predicted properties were GI absorption tendency, BBB permeability behavior, and Lipinski rule.
Abbreviations
| ABS | Asexual blood-stage |
| ACT | Artemisinin-based combination therapy |
| ADME | Absorption, distribution, metabolism, excretion |
| BOC | Tert-butyloxycarbonyl |
| DCM | Dichloromethane |
| EDC·HCL | 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| HEA | Hydroxyethylamine |
| HEPES | (4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid) |
| HPLC | High-performance liquid chromatography |
| RBC | Red blood cells |
| HRMS | High-resolution mass spectrometry |
| IC50 | Half-maximal inhibitory concentration; MD, molecular dynamics |
| NMR | Nuclear magnetic resonance |
| P. berghei | Plasmodium berghei |
| PBS | Phosphate-buffered saline |
| Pf | Plasmodium falciparum |
| Plm | Plasmepsin |
| PQ | Primaquine |
| RMSD | Root-mean-squared deviation |
| SAR | Structure–activity relationship |
| TC50 | 50% cytotoxic concentration |
| TFA | Trifluoroacetic acid |
| TLC | Thin-layer chromatography |
| WHO | World Health Organization. |
Author contributions
Conceptualization, C. U., D. K., M. P., A. P. S, Poonam.; methodology, C. U., N. S., S. K, P. P. S., D. F., B. M., B. S. C and D. K.; writing—original draft preparation, C. U., A. P. S. and Poonam; writing—review and editing, C. U., B. S. C, D. K., M. P., A. P. S. and Poonam.; supervision, project administration, and funding acquisition, Poonam. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
Poonam acknowledges Science and Engineering Research Board for financial support under CRG scheme (CRG/2020/005800). CU, NS and SK are highly grateful to CSIR for senior research fellowship, PPS acknowledges DBT, Govt. of India for senior research fellowship.References
- N. Sharma, A. Verma, P. Fnu, P. Kempaiah and B. Rathi, Chem. Biol. Lett., 2019, 6, 14–22 CAS.
- N. M. Pazhayam, J. Chhibber-Goel and A. Sharma, Drug Discovery Today, 2019, 24, 263–271 CrossRef CAS.
- S. Rout and R. K. Mahapatra, Chem. Biol. Drug Des., 2019, 93, 737–759 CrossRef CAS.
- N. D. Pasternak and R. Dzikowski, Int. J. Biochem. Cell Biol., 2009, 41, 1463–1466 CrossRef CAS.
- World Health Organization: World malaria report 2020, https://www.who.int/publications/i/item/9789240015791, accessed march 22, 2021.
- E. G. Tse, M. Korsik and M. H. Todd, Malar. J., 2019, 18, 93 CrossRef PubMed.
- Poonam, Y. Gupta, N. Gupta, S. Singh, L. Wu, B. S. Chhikara, M. Rawat and B. Rathi, Med. Res. Rev., 2018, 38, 1511–1535 CrossRef CAS.
- K. Haldar, S. Bhattacharjee and I. Safeukui, Nat. Rev. Microbiol., 2018, 16, 156–170 CrossRef CAS PubMed.
- N. Sharma, Y. Gupta, M. Bansal, S. Singh, P. Pathak, M. Shahbaaz, R. Mathur, J. Singh, M. Kashif, M. Grishina, V. Potemkin, V. Rajendran, Poonam, P. Kempaiah, A. P. Singh and B. Rathi, RSC Adv., 2020, 10, 35516–35530 RSC.
- M. A. Diallo, M. S. Yade, Y. D. Ndiaye, I. Diallo, K. Diongue, S. A. Sy, M. Sy, M. C. Seck, M. Ndiaye, B. Dieye, J. F. Gomis, D. Sow, A. B. Dème, A. S. Badiane and D. Ndiaye, Sci. Rep., 2020, 10, 8907 CrossRef CAS.
- J. E. Hyde, FEBS J., 2007, 274, 4688–4698 CrossRef CAS.
- K. S. Griffith, L. S. Lewis, S. Mali and M. E. Parise, JAMA, 2007, 297, 2264–2277 CrossRef CAS.
- C. Upadhyay, M. Chaudhary, R. N. De Oliveira, A. Borbas, P. Kempaiah, Poonam and B. Rathi, Expert Opin. Drug Discovery, 2020, 15, 705–718 CrossRef CAS PubMed.
- J. Achan, J. Mwesigwa, C. P. Edwin and U. D’alessandro, Expert Rev. Clin. Pharmacol., 2018, 11, 61–70 CrossRef CAS.
- S. Yahiya, A. Rueda-Zubiaurre, M. J. Delves, M. J. Fuchter and J. Baum, Curr. Opin. Chem. Biol., 2019, 50, 1–9 CrossRef CAS.
- M. Al-Ghorbani, B. A. Bushra, S. Mamatha and S. A. Khanum, Res. J. Pharm. Technol., 2015, 8, 611–628 CrossRef.
- M. Asif and M. Imran, Frontiers in Chemical Research, 2019, 1, 5–12 Search PubMed.
- K. Rupak, S. R. Vulichi and K. Suman, Int. J. Chem. Sci., 2016, 14, 1777–1788 CAS.
- F. Arshad, M. F. Khan, W. Akhtar, M. M. Alam, L. M. Nainwal, S. K. Kaushik, M. Akhter, S. Parvez, S. M. Hasan and M. Shaquiquzzaman, Eur. J. Med. Chem., 2019, 167, 324–356 CrossRef CAS PubMed.
- A. P. Kourounakis, D. Xanthopoulos and A. Tzara, Med. Res. Rev., 2020, 40, 709–752 CrossRef CAS PubMed.
- E. A. Ashley and A. P. Phyo, Drugs, 2018, 78, 861–879 CrossRef CAS PubMed.
- B. Rathi, A. K. Singh, R. Kishan, N. Singh, N. Latha, S. Srinivasan, K. C. Pandey, H. K. Tiwari and B. K. Singh, Bioorg. Med. Chem., 2013, 21, 5503–5509 CrossRef CAS.
- M. C. Souza, T. A. Padua, N. D. Torres, M. F. D. S. Costa, V. Facchinetti, C. R. B. Gomes, M. V. N. Souza and M. D. G. Henriques, Mem. Inst. Oswaldo Cruz, 2015, 110, 560–565 CrossRef CAS.
- P. P. Sharma, S. Kumar, K. Kaushik, A. Singh, I. K. Singh, M. Grishina, K. C. Pandey, P. Singh, V. Potemkin, Poonam, G. Singh and B. Rathi, J. Biomol. Struct. Dyn., 2021, 1–13 Search PubMed.
- N. Sharma, M. Kashif, V. Singh, D. Fontinha, B. Mukherjee, D. Kumar, S. Singh, M. Prudencio, A. P. Singh and B. Rathi, J. Med. Chem., 2021, 64, 8666–8683 CrossRef CAS PubMed.
- K. Jaudzems, K. Tars, G. Maurops, N. Ivdra, M. Otikovs, J. Leitans, I. Kanepe-Lapsa, I. Domraceva, I. Mutule, P. Trapencieris, M. J. Blackman and A. Jirgensons, ACS Med. Chem. Lett., 2014, 5, 373–377 CrossRef CAS.
- S. Singh, V. Rajendran, J. He, A. K. Singh, A. O. Achieng, Vandana, A. Pant, A. S. Nasamu, M. Pandit, J. Singh, A. Quadiri, N. Gupta, Poonam, P. C. Ghosh, B. K. Singh, L. Narayanan, P. Kempaiah, R. Chandra, B. M. Dunn, K. C. Pandey, D. E. Goldberg, A. P. Singh and B. Rathi, ACS Infect. Dis., 2019, 5, 184–198 CrossRef CAS.
- A. Kumari and R. K. Singh, Bioorg. Chem., 2020, 96, 103578 CrossRef CAS PubMed.
- S. Kumar, Y. Gupta, S. E. Zak, C. Upadhyay, N. Sharma, A. S. Herbert, R. Durvasula, V. Potemkin, J. M. Dye, Poonam, P. Kempaiah and B. Rathi, RSC Med. Chem., 2021, 12, 1757–1764 RSC.
- Y. Gupta, S. Kumar, S. E. Zak, K. A. Jones, C. Upadhyay, N. Sharma, S. A. Azizi, R. S. Kathayat, Poonam, A. S. Herbert, R. Durvasula, B. C. Dickinson, J. M. Dye, B. Rathi and P. Kempaiah, Bioorg. Med. Chem., 2021, 47, 116393 CrossRef CAS.
- S. Kumar, C. Upadhyay, M. Bansal, M. Grishina, B. S. Chhikara, V. Potemkin, B. Rathi and Poonam, ACS Omega, 2020, 5, 18746–18757 CrossRef CAS PubMed.
- F. Moschona, I. Savvopoulou, M. Tsitopoulou, D. Tataraki and G. Rassias, Catalysts, 2020, 10, 1117 CrossRef CAS.
- L. Shi, L. Chen, R. Chen and L. Chen, J. Labelled Compd. Radiopharm., 2010, 53, 147–151 CrossRef CAS.
- W. Cunico, C. R. B. Gomes, M. Moreth, D. P. Manhanini, I. H. Figueiredo, C. Penido, M. G. M. O. Henriques, F. P. Varotti and A. U. Krettli, Eur. J. Med. Chem., 2009, 44, 1363–1368 CrossRef CAS PubMed.
- S. Hartwig, M. M. Nguyen and S. Hecht, Polym. Chem., 2010, 1, 69–71 RSC.
- L. Tilley, J. Straimer, N. F. Gnädig, S. A. Ralph and D. A. Fidock, Trends Parasitol., 2016, 32, 682–696 CrossRef CAS PubMed.
- B. Baragaña, I. Hallyburton, M. C. S. Lee, N. R. Norcross, R. Grimaldi, T. D. Otto, W. R. Proto, A. M. Blagborough, S. Meister, G. Wirjanata, A. Ruecker, L. M. Upton, T. S. Abraham, M. J. Almeida, A. Pradhan, A. Porzelle, T. Luksch, M. S. Martínez, T. Luksch, J. M. Bolscher, A. Woodland, S. Norval, F. Zuccotto, J. Thomas, F. Simeons, L. Stojanovski, M. Osuna-Cabello, P. M. Brock, T. S. Churcher, K. A. Sala, S. E. Zakutansky, M. B. Jiménez-Díaz, L. M. Sanz, J. Riley, R. Basak, M. Campbell, V. M. Avery, R. W. Sauerwein, K. J. Dechering, R. Noviyanti, B. Campo, J. A. Frearson, I. Angulo-Barturen, S. Ferrer-Bazaga, F. J. Gamo, P. G. Wyatt, D. Leroy, P. Siegl, M. J. Delves, D. E. Kyle, S. Wittlin, J. Marfurt, R. N. Price, R. E. Sinden, E. A. Winzeler, S. A. Charman, L. Bebrevska, D. W. Gray, S. Campbell, A. H. Fairlamb, P. A. Willis, J. C. Rayner, D. A. Fidock, K. D. Read and I. H. Gilbert, Nature, 2015, 522, 315–320 CrossRef.
- M. Prudêncio, M. M. Mota and A. M. Mendes, Trends Parasitol., 2011, 27, 565–574 CrossRef.
- R. Raphemot, D. Posfai and E. R. Derbyshire, J. Clin. Invest., 2016, 126, 2013–2020 CrossRef.
- I. M. Vera, M. T. G. Ruivo, L. F. L. Rocha, S. Marques, S. N. Bhatia, M. M. Mota and L. Mancio-Silva, JCI Insight, 2019, 4, e127441 CrossRef PubMed.
- A. O. da Silva de Barros, F. L. Portilho, A. P. dos Santos Matos, E. Ricci-Junior, L. M. R. Alencar, C. C. dos Santos, F. J. R. Paumgartten, S. H. Iram, D. Mazier, J.-F. Franetich, F. Alexis and R. Santos-Oliveira, Mater. Sci. Eng., C, 2021, 128, 112275 CrossRef CAS.
- T. Rodrigues, M. Prudêncio, R. Moreira, M. M. Mota and F. Lopes, J. Med. Chem., 2012, 55, 995–1012 CrossRef CAS PubMed.
- P. Pino, R. Caldelari, B. Mukherjee, J. Vahokoski, N. Klages, B. Maco, C. R. Collins, M. J. Blackman, I. Kursula, V. Heussler, M. Brochet and D. Soldati-Favre, Science, 2017, 358, 522–528 CrossRef CAS PubMed.
- G. Munsamy, P. Ramharack and M. E. S. Soliman, RSC Adv., 2018, 8, 21829–21840 RSC.
- J. N. Burrows and D. Soldati-Favre, Cell Host Microbe, 2020, 27, 496–498 CrossRef CAS.
- A. S. Nasamu, S. Glushakova, I. Russo, B. Vaupel, A. Oksman, A. S. Kim, D. H. Fremont, N. Tolia, J. R. Beck, M. J. Meyers, J. C. Niles, J. Zimmerberg and D. E. Goldberg, Science, 2017, 358, 518 CrossRef CAS PubMed.
- A. Kumar Singh, V. Rajendran, S. Singh, P. Kumar, Y. Kumar, A. Singh, W. Miller, V. Potemkin, Poonam, M. Grishina, N. Gupta, P. Kempaiah, R. Durvasula, B. K. Singh, B. M. Dunn and B. Rathi, Bioorg. Med. Chem., 2018, 26, 3837–3844 CrossRef CAS PubMed.
- D. E. Goldberg, Curr. Top. Microbiol. Immunol., 2005, 295, 275–291 CAS.
- B. Baragaña, N. R. Norcross, C. Wilson, A. Porzelle, I. Hallyburton, R. Grimaldi, M. Osuna-Cabello, S. Norval, J. Riley, L. Stojanovski, F. R. Simeons, P. G. Wyatt, M. J. Delves, S. Meister, S. Duffy, V. M. Avery, E. A. Winzeler, R. E. Sinden, S. Wittlin, J. A. Frearson, D. W. Gray, A. H. Fairlamb, D. Waterson, S. F. Campbell, P. Willis, K. D. Read and I. H. Gilbert, J. Med. Chem., 2016, 59, 9672–9685 CrossRef PubMed.
- A. K. Singh, S. Rathore, Y. Tang, N. E. Goldfarb, B. M. Dunn, V. Rajendran, P. C. Ghosh, N. Singh, N. Latha and B. K. Singh, PLoS One, 2015, 10, e0139347 CrossRef.
- F. Mphande, S. Nilsson and A. Bolad, Methods in malaria research, 2008, 1.2008, 1 Search PubMed.
- S. Duffy and V. M. Avery, Int. J. Parasitol.: Drugs Drug Resist., 2017, 7, 295–302 Search PubMed.
- B. Pérez, C. Teixeira, I. S. Albuquerque, J. Gut, P. J. Rosenthal, M. Prudêncio and P. Gomes, MedChemComm, 2012, 3, 1170–1172 RSC.
- F. P. da Cruz, C. Martin, K. Buchholz, M. J. Lafuente-Monasterio, T. Rodrigues, B. Sönnichsen, R. Moreira, F.-J. Gamo, M. Marti, M. M. Mota, M. Hannus and M. Prudêncio, J. Infect. Dis., 2012, 205, 1278–1286 CrossRef CAS.
- S. Kumar, P. P. Sharma, U. Shankar, D. Kumar, S. K. Joshi, L. Pena, R. Durvasula, A. Kumar, P. Kempaiah, Poonam and B. Rathi, J. Chem. Inf. Model., 2020, 60, 5754–5770 CrossRef CAS.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, Nucleic Acids Res., 2018, 46, W363–W367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj04198c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |