
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal control of proton concentration using an electrochemically generated acid compatible with miniaturization†
Janwa El-Maiss ,
Divya Balakrishnan and
César Pascual García*
,
Divya Balakrishnan and
César Pascual García*
Nanomaterials Unit of the Materials Research and Technology Department, Luxembourg Institute of Science and Technology (LIST), Belvaux L-4422, Luxembourg. E-mail: cesar.pascual@list.lu
First published on 21st June 2022
Abstract
Controlling locally produced acidity in miniaturized spaces is of high importance to manage simultaneous chemical reactions. Here, we present a platform that hosts miniaturized micro-reactors each one enabling electrochemical control of the acidity in ∼nL volumes. We demonstrated the local control of chemical reactions with the deprotection of strong acid labile groups in a region of 150 μm of diameter of upstanding glass using high proton concentrations (∼10−1 M) and the acidity contrasts between the cell region and the outside. We demonstrated accurate control of the proton concentration in aqueous and organic solvents and the control of chemical reactions in organic electrolytes achieved with a sulfonated tetrafluoroethylene-based membrane, which isolates the acid generating electrodes from the reagents in the solution. The quantitative control of the acidity by faradaic currents was demonstrated by the calibration of carboxyfluorescein adjusted with external titrations and with a tautomer transition occurring at pH 4.2. To the best of our knowledge, this platform shows the best control of acidity in the smallest volume reported so far.
Introduction
Acidity has a defining role in numerous chemical and biological processes,1 for example, the activity of enzymes,2 the assembly of DNA3 and the solid phase synthesis of biopolymers using acid or base labile protecting groups.4,5 For all these applications, it is important to have a way to predict and control the acidity, which in most of the cases is done by titrating solutions of compounds dissociated in water or organic solvents and limits the miniaturization of chemical reactions. The miniaturization of the control of the acidity has a similar impact on combinatorial chemistry to Moore's law for microelectronics,6 allowing the massive multiplexing of chemical reactions. The electrochemical generation of acidity also allows the regulation of acidity by faradaic currents that can be automatized.7Several solutions for multiplexed combinatorial chemistry are currently available, providing microarrays based on the deprotection of optical labile8 or acid/base labile9 groups achieved by imaging or with the encapsulation of chemical monomers for micro-printing strategies. However, these platforms still lack throughput and versatility to reach widespread use for different applications that would benefit from the control of combinatorial chemistry on a large multiplexing scale. An electrochemically generated acid was also used for the generation of microarrays9,10 using hydroquinone/benzoquinone soluble in the medium, and the protons were electrochemically/photoelectrochemically generated next to the electrodes. Regardless the quasi-reversibility of the processes, these techniques were limited by the fast diffusion of the protons and the limited range of achieved pH. A detailed table of other miniaturized pH-controlled devices is also shown in the ESI.† Electrochemical strategies, in addition to facilitating the electronic management, can ease the miniaturization of the control of chemical reactions using silicon fabrication technologies that would allow a cost-effective production of chips to facilitate access to the technology.11 However, these attempts were not able to provide the necessary specifications to achieve the control of chemistry that would allow them to reach wider use.
A platform for the multiplexed control of chemical reactions with acidity requires reaching high proton concentrations and being able to maintain the contrast between the acid and alkaline regions in miniaturized spaces for times much longer than the characteristic diffusion and reaction times. It also requires an accurate control of the proton concentration in aqueous and organic solvents to allow different chemical strategies including the possibility to control quantitatively the acidity for orthogonal control of chemical reactions at different acidities. Finally, while the quantification of acid in aqueous environments is well predicted by the well-known proton dissociation constants (pKa expressed on a logarithmic scale), in organic solutions, the proton activity is more elusive to determine. In these solvents the pKa of the conjugated acid/base pairs depends also on their interaction with the solvents, and the possible proton carriers that may be added to facilitate the dissociation, or as in the case of electrolytes, to change the properties of the solution.12 Good control in organic solvent for the generalization of chemical reactions requires the generation of a neutral acid from electrodes that would avoid affecting the chemical reactions of the electrolyte while preventing their degradation. Such an acid can be used as a reference for the determination of pKa values in organic solvents.
To control the proton concentration in different solvents, our strategy was to use an electrochemically generated acid using molecules fixed to the electrode surfaces, which was monitored by using a reference fluorescence label. We used a design of an electrochemical reactor where the acidity is generated by proton exchange reactions.13 The platform holds several miniaturized electrochemical reactors, each of which consists of three electrochemical partial cells containing the working, counter, and reference electrodes (WE, CE and RE, respectively) and separated by diffusion barriers. In the cell containing the WE, the acidity is regulated by a voltage bias applied with respect to the reference cell, which controls the release or adsorption of protons by molecules functionalized in the WE. The electrodes are isolated from the electrolyte using a fluorinated proton permeable membrane (Nafion).14 The diffusion barriers ensure that the number of protons in each cell is stable for periods much longer than the dissociation dynamics, while the small dimensions of the cell ensure rapid feedback of the reactions in the cell. The electrochemically generated acid released protons, free of counter ions and free to interact with the elements of the electrolyte. Thanks to these characteristics, the acidity can be controlled and estimated by using faradaic currents.
To monitor the proton activity in our cell, we used the fluorescent dye carboxyfluorescein (FAM). FAM allowed a calibration using an external titration that coincided with the proton concentration calculated using faradaic currents. We also calibrated the system using a special tautomerization process that FAM undergoes at a particular pH, and we used proton quantification to control the acidity in acetonitrile (ACN). We used fluorescence to calibrate the proton activity of acids in an organic electrolyte, observing the formation of super-acids due to the influence of proton carrier salts in the electrolyte. Finally, we used our electrochemical cell for the deprotection of acid labile groups. In our case we proved the deprotection of tert-butyloxycarbonyl (Boc) groups from an APTES monolayer grafted on a glass surface upstanding to the electrodes in the cell of the WE. This kind of reaction added a proof of the activity of the electrochemically generated acid, which in traditional chemistry is obtained after using high acid concentrations. Using a microfluidic platform and cell design similar to the one in ref. 13, we improved and miniaturised the reactor design demonstrating that the production of acidity does not depend on the footprint of the device and that thus, it can be further miniaturised. pH tracking was also improved with online monitoring by faradaic currents calibrated by the FAM transition. Also, the device was firstly used to control chemical reactions other than the pH control with the deprotection of acid labile groups.
Experimental section
Chemicals and instrumentation
4-Aminothiophenol (4-ATP), potassium chloride (KCl), 6-carboxyfluorescein (FAM), tetrabutylammonium hexafluorophosphate (Bu4PF6), hydrochloric acid (HCl), potassium hydroxide (KOH), benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), N,N-diisopropylethylamine (DIPEA), 3-(aminopropyl)triethoxysilane (APTES), trifluoroacetic acid (TFA), rhodamine B isothiocyanate (RITC), absolute ethanol (EtOH), anhydrous acetonitrile (ACN), anhydrous dichloromethane (DCM) and anhydrous dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich and N-tert-butyloxycarbonyl-12-amino-dodecanoic acid (acid-Boc) was purchased from Iris biotech GMBH. All the chemicals were used without further purification. For the preparation of aqueous electrolyte solutions and cleaning purposes Millipore filtered water was used. For the electropolymerization and pH control experiments, a Solartron Modulab XM Pstat 1 mS/s potentiostat (Pstat) was used.Fabrication of microreactors and the microfluidic platform
The electrochemical microreactors were fabricated on silicon chips coated with a dielectric layer of SiO2 using standard techniques of micro-electromechanical systems (MEMS) and chemical and electrochemical surface functionalization methods (Fig. 1a). The chips were integrated with a platform (Fig. 1d) having microfluidic, optical, and electrical access and a pneumatic actuation to open and close the cells. All the details involved in the fabrication process are also reported in the ESI-1.† Briefly, optical lithography was performed to pattern the design of the electrodes. The substrate was spin-coated with an optical resist and the electrodes with contact pads were exposed and developed. Later, 5 and 50 nm layers of Cr and Au were evaporated using an e-beam evaporator. The chip was placed in acetone for lift-off. We then spin coated the chip with SU8 3010 epoxy resist to lithograph the separation barriers. The chip was afterwards developed and hard baked for 60 minutes. The electrodes were electrochemically platinized to increase their surface area. The chip was later activated in a UV ozone cleaner for 30 minutes and functionalized with 0.5 mM of 4-aminothiophenol (4-ATP) in absolute ethanol for 24 hours to form a self-assembled monolayer of the redox active molecules on top of the porous electrodes. This configuration without further modifications was used for experiments with aqueous solutions. The essential difference that we used for the experiments with organic solvents is the isolation of the electrodes from the electrolyte using a sulfonated tetrafluoroethylene (Nafion) based membrane. We incorporated a Nafion film by spin-coating its commercial solution after the functionalization of the chip with the 4-ATP molecules. The Nafion membrane, while avoiding the possibility of cross reactions between 4-ATP molecules and carboxyfluorescein, allows the selective transport of protons from the electrodes to the solution through its sulfonate channels.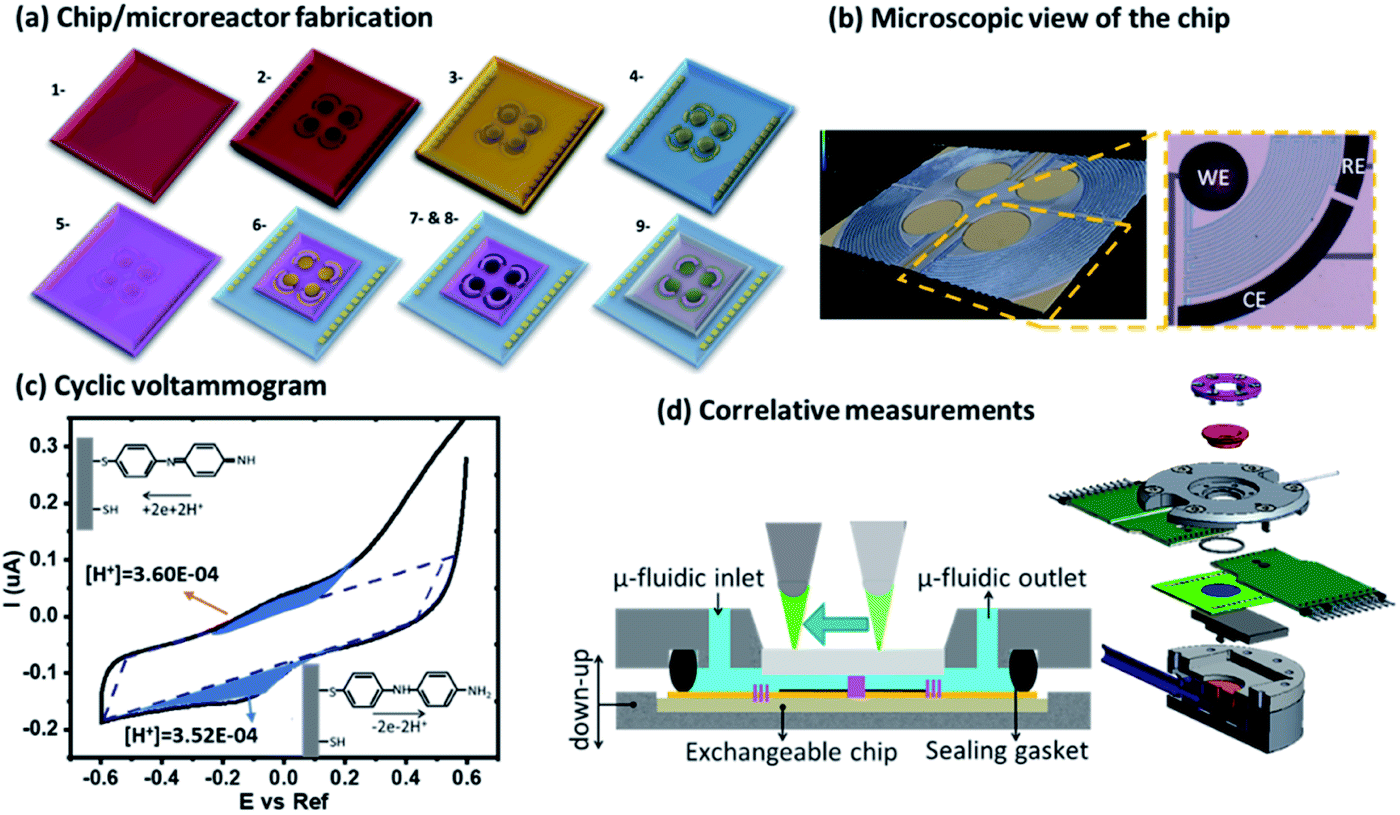 | ||
| Fig. 1 (a) Chip fabrication of the integrated electrochemical cells. (b) 3D microscope picture of the microreactor and a description of the electrode geometry bearing the WE, CE, and RE. (c) Cyclic voltammogram with the extracted faradaic currents represented with blue shadows. (d) Schematic representation of the microfluidic platform with a movable optical setup on top and the exploded view of the platform. | ||
The platform consists of a central liquid reservoir delimited by a circular FFKM gasket. The upper part of the platform allows controling the aperture of the cell between an average of 100 μm between the opened and the closed position, where the barriers delimiting the cells are in contact with the upstanding glass, and the gap in the electrochemical cells of the WE, CE and RE and the diffusion barriers have an average height of ∼5 μm. The open and close positions were actioned by a pneumatic piston. When the cell is in the close position, the total volume of the isolated partial cell of the WE (V) is 0.9 nL. This volume is considered in the article to calculate the concentrations of electrochemically generated acid confined in the volume of the partial cell of the WE. The liquid in the central aperture was managed with syringe pumps through an inlet and evacuated through an outlet in which vacuum could also be applied to achieve good filling. Finally, this cell also allowed optical access into the central aperture through the upstanding glass (this glass will be functionalized in Boc-deprotection experiments). Using an optical microscope, we could follow the fluorescence in the electrolyte, being able to resolve the acidity sequentially between the cells.
Functionalization of the glass slides
For the experiments showing the deprotection of acid labile groups, we functionalized circular microscope slides of 1 cm diameter as shown in Scheme 1. The glass slides were cleaned using isopropanol and acetone and blow dried with nitrogen gas. They were activated using an UV Ozone treatment for 30 min and immersed in a solution of 5% APTES in 40 mL of absolute ethanol at 50° overnight. The samples were cleaned and placed in a solution of PyBOP 742 mg, BOC – 150 mg, DIEA – 248 μL, and DCM 7.5 mL (preactivated for 10 min). The samples were kept in the above solution overnight at room temperature, closed with aluminum foil. The growth of the film was followed by water contact angle measurement and XPS (details in ESI-6†). For each experiment, a functionalized glass slide was mounted on the upstanding lid of the platform, using a dedicated glue. Following the experiments, the glass slides were removed from the platform and stained with rhodamine B isothiocyanate to observe the deprotection fingerprint.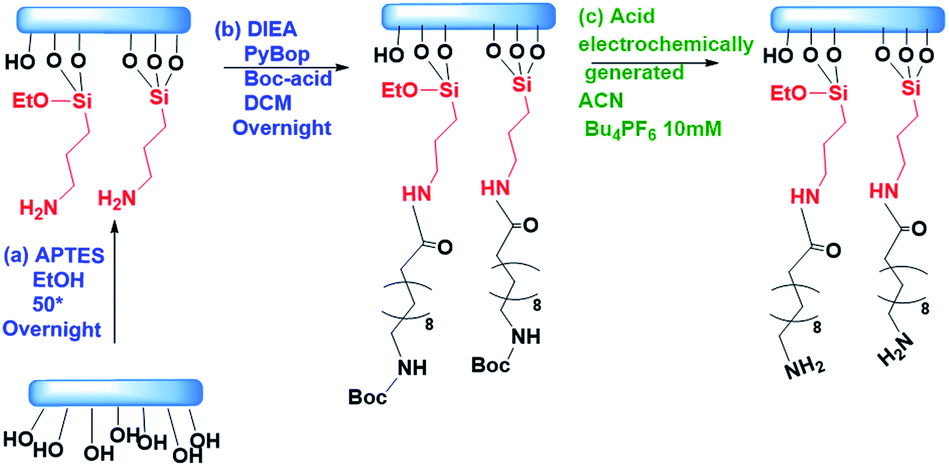 | ||
| Scheme 1 Functionalization of the glass: (a) APTES 5% v/v in absolute EtOH at 50 °C overnight. (b) DIEA 20 eq., PyBOP 20 eq., Boc-acid 20 eq. in DCM overnight. (c) Electrochemical deprotection of Boc in ACN 10 mM Bu4PF6. | ||
Two control samples were prepared for the evaluation of Boc deprotection. A negative control was used with the Boc protected samples and incubated into rhodamine without further treatment. The positive control was prepared after a treatment with 50% TFA acid for one hour, before the same incubation with rhodamine. The fluorescence was evaluated normalizing a spot with a field of view ∼50 μm to the signal from an equivalent glass slide treated with traditional deprotection in TFA acid.
Control of redox reactions in the platform
In our platform, the 4-ATP functionalized electrodes can be biased to exchange protons with the electrolyte. This reaction is more efficient after the dimerization of 4-ATP.15 The dimerization was carried out in water by electro-polymerization with cyclic voltammetry. Before cycling the acid/base control, the cell was placed in the open position and the electrodes were connected to a potentiostat using a three-electrode system. A cyclic voltammetry (CV) program was applied in the range of −0.6 to +0.6 V at a scan rate of 25 mV s−1 for 5 cycles in the presence of the dye (FAM 0.5 μM) in KCl (100 mM) (Fig. 1c). The voltammograms corresponding to the electropolymerisation exhibited two main contributions in the oxidation peaks corresponding to the reversible exchange of protons and the dimerization at ∼0.1 and 0.4 V respectively. As consecutive cycles were performed the main peak corresponding to the dimerization disappeared and the main contribution remained with the peak at 0.2 V corresponding to the two proton exchange reactions. The details of all these reactions were studied in our previous article.15 In some of the chips, other residual contributions that we ascribed to non-dimerized molecules could also be observed between the two peaks of the dimerized molecules and electropolymerisation. The 4-ATP molecules exchanging protons can be assessed by faradaic currents causing proton exchange reactions at low voltages. For the experiments in organic solvents, FAM was used with the same initial concentration 0.5 μM in a solution of 100 mM Bu4PF6 in ACN. In organic medium, CVs were recorded between 0 and 1 V vs. a pseudo-reference with a scan rate of 30 mV s−1. The deprotection of Boc acid labile groups occurs with the application of a linear swipe up from 0 to 0.85 V, a fixed voltage (0.85 V) for 1 min and then a second linear swipe up from 0.85 to 0 V. The electroneutrality is globally maintained between the WE and CE by the difference of the potential. In addition, high ionic strength solution (KCl/Bu4PF6 = 100 mM) was used to avoid increasing the potential difference between both cells and maintain the electroneutrality.Results and discussion
The control of acidity occurs on a customized platform presented in Fig. 1. The electrochemical microelectrodes are fabricated in a multistep process combining spin coating of photoresist, optical lithography, metal evaporation and then functionalization with the redox molecule 4-ATP, as shown in Fig. 1a. The chips bearing four microreactors, each one composed of three electrodes connected through diffusion barriers (WE, CE, and RE), are mounted on the platform (Fig. 1b and d) having a sealable gasket and a microfluidic system for the filling of the system with a pneumatic piston for the opening and closure of the cells. The whole setup holds an optical setup on the top to follow the fluorescence in the cell (Fig. 1d). The fabrication steps of the devices and the platform are shown in ESI-1.† After their polymerization, the 4-ATP molecules dimerize to a redox molecular state that at low applied voltages exchange two protons with the electrolyte for each dimer in the electrode (quasi)-reversibly, and each proton is compensated for by the current of one electron exchanged in the WE.15–17 The CE had the same composition; thus, it balanced the reactions in the WE by reverse proton exchange reactions. However, since the separation by the diffusive barriers keeps both partial cells apart, there are no possibilities for cross proton exchanges. The faradaic currents were fitted considering the contribution of the reversible proton exchange peaks after the subtraction of the contributions from the capacitance, the linear currents attributed to mass transport as well as the non-reversible residual polymerization. The resulting faradaic contributions of the proton exchange reactions were used to calculate the proton concentration generated during the oxidation and re-adsorbed by the electrodes during the reduction cycle (details of the calculations are shown in ESI-3†). The calculations showed similar concentration in the two cases (in the case of Fig. 1c, for instance, 3.6 × 10−4 M and 3.52 × 10−4 M were found for the oxidation and reduction peaks, respectively). This behavior was observed for repetitive cycles (up to hundreds of cycles that we monitored the currents in some of our experiments) validating the quasi-reversibility of the system. During different experiments using different chips we observed a variety of surface functionalization in the electrodes that resulted in different dynamic ranges of pH obtained during the CV cycles, with typical maximum acidities reaching pH between 3 and 1.Monitoring acidity with FAM in aqueous solutions
The changes in FAM fluorescence (pH 7 fixed, 0.5 μM solution in KCl 100 mM) with the applied voltage were tracked focusing a microscope in the WE during the CV measurements and following the peak emission at 525 nm. Fig. 2a reports the time evolution of the CV and fluorescence data corresponding to 4 representative cycles (from cycle 2 to 5, excluding the first oxidation/reduction that usually exhibits spurious contributions from the open circuit state) showing the voltage, the current, and the normalized FAM-fluorescence in black, red, and green, respectively. The current curves reported the typical redox peaks corresponding to the proton exchange reactions, from which for each cycle the faradaic currents could be extracted (quantitative data of the cycles and details of the calculations are shown in ESI-2 and 3†). We attribute the decrease of FAM fluorescence to the increase of acidity in the cell due to the proton exchange reactions. Fig. 2b shows the details in one cycle with the FAM fluorescence (in green) and the calculated charge exchange Q (blue curve) as a function of time extracted from the faradaic currents. Q(t) is expressed on a negative logarithmic scale normalized to the Faraday number and the volume of the cell, to which we added a baseline of Q(0) = 107 (in the units of elementary charge) where the exponent 7 corresponds to a baseline of neutral pH to account for the initial conditions of the electrolyte. In this way the charge is expressed in equivalent units of pH. Fig. 2c shows the fluorescence curve or the FAM vs. pH, calibrated with an ISFET pH meter in a solution that was titrated with KOH. We observed a small delay of the fluorescence with respect to the faradaic currents part of which (∼1 s) can be attributed to the integration time of our spectra, but we did not consider this delay relevant in the overall cycle. We also observed a very good correspondence of the fluorescence obtained in the cell at the equivalent pH calculated from Q and the one at pH produced with the external titration. Due to the cell confinement, this correlation between the acidity and the faradaic currents is a proof of the electrochemical origin of the acid. The maximum fluorescence is obtained when no protons were released in the system or when they are reabsorbed by the 4-ATP molecules after the reduction cycles that correspond to the moment when the normalized faradaic currents indicate pH 7. The intensity decreases when the oxidation of 4-ATP occurs (−0.6 to 0.6 V) and then increases during the reduction fragment of the cycle (0.6 to −0.6 V) where the protons were re-adsorbed. This returning to the normal fluorescence before the protons activity is also consistent with our interpretation that the phenomenon is quasi-reversible, and that the adsorption of the protons released in the system is efficient. In the cycle we detail here, the equivalent pH of the faradaic currents reached values close to 1.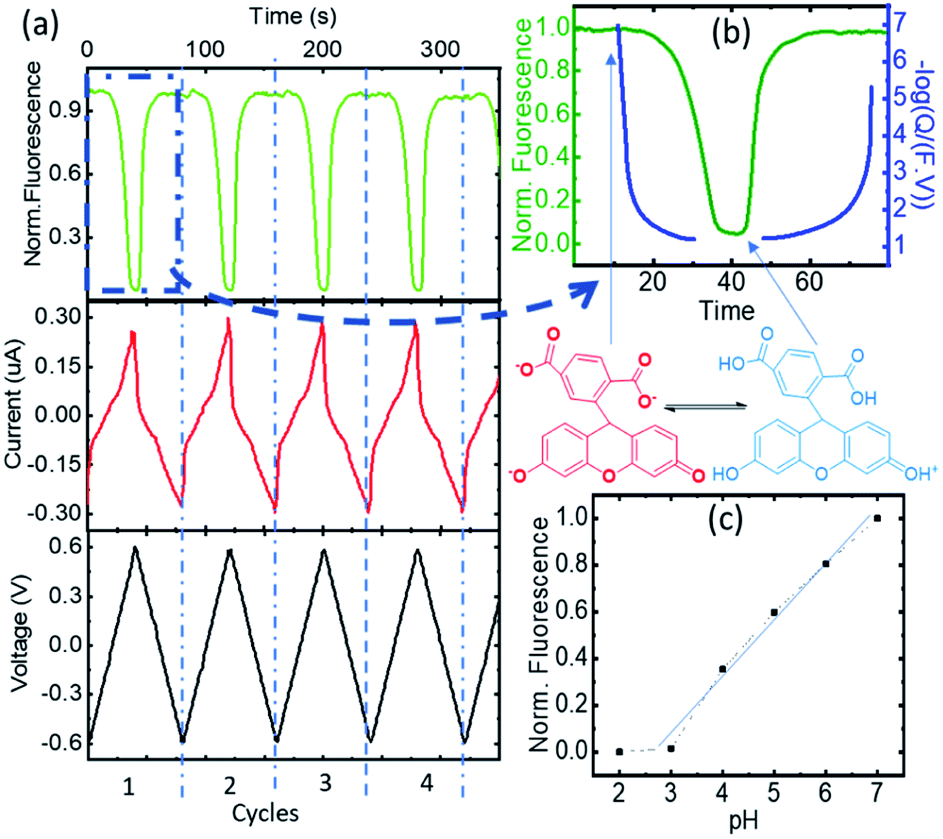 | ||
Fig. 2 (a) The time evolution of the CV and fluorescence data corresponding to cycles 2 to 5, the voltage, the current, and the normalized FAM-fluorescence in black, red, and green, respectively. (b) Normalized fluorescence (green) and faradaic charge expressed as apparent pH = (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Q/(F × V)) (in blue) vs. time. The different active species are indicated. (c) Calibration curve of carboxyfluorescein vs. pH using external titration. Q/(F × V)) (in blue) vs. time. The different active species are indicated. (c) Calibration curve of carboxyfluorescein vs. pH using external titration. | ||
An independent calibration of the relationship of the Q derived from the faradaic currents with the acidity was observed with the tautomerization of FAM. Tautomerism is a solvent-dependent chemical process that commonly results in the relocation of a hydrogen atom and affects the activity of biomolecules and dyes. Either an acid or a base can catalyze the proton transfer.18 FAM has different forms (each one having different tautomers that can coexist in water), and the transition from one to another can be driven by acidity. In aqueous solution, FAM has different acid/base pairs (cation C, neutral N, monoanion M, dianion D, and trianion T) depending on the pH. Aschi et al.18 were the first to present accurate measurements of the pKa values of the different pairs of FAM using spectrophotometric titration, chemometric analysis and quantum mechanical estimations. Based on their paper, we adapted first the nomenclature of the different species by considering the global charge of the tautomer with the groups: (i) the anion family (T (FAM3−), D (FAMH2−) and M (FAMH2−)) that had a similar absorbance at 470 nm (though T and D had their maximum at lower frequencies) denoted as FAM 1, (ii) the neutral tautomer N (FAMH3), which had the lowest absorbance at 470 nm denoted as FAM 2 and (iii) the cation compound C (FAMH4+) that increased the absorbance at 470 nm with respect to FAM 2 but not with respect to FAM 3. FAM 1 to FAM 3 are represented in Fig. 3.
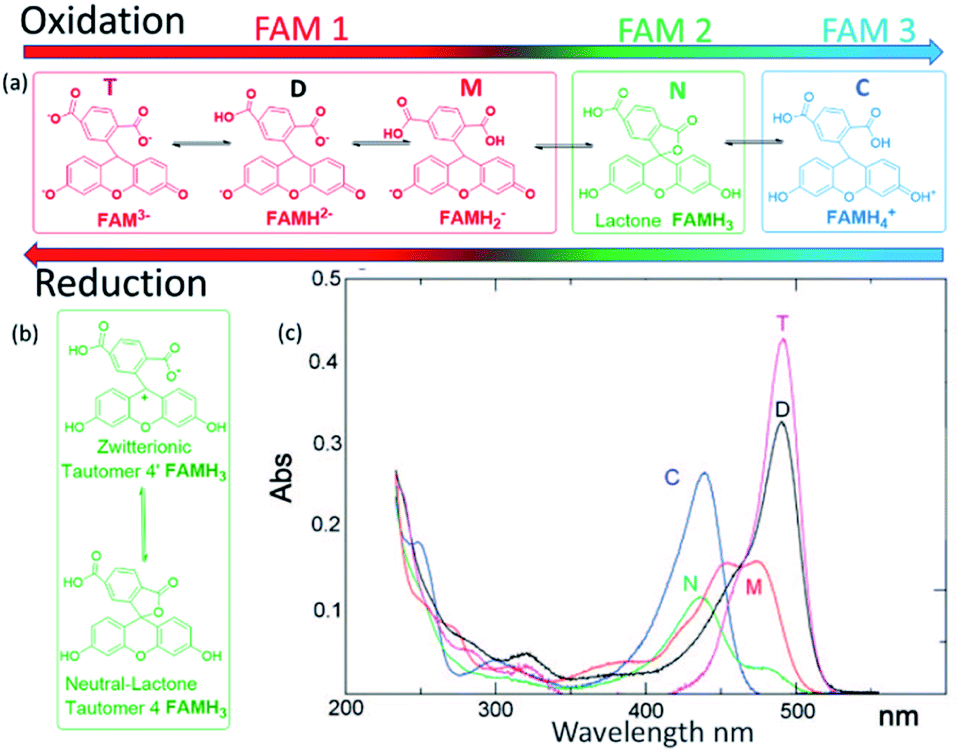 | ||
| Fig. 3 (a) FAM acid/base pairs (FAMH4+ cation C, FAMH3 neutral N, FAMH2− monoanion M, FAMH2− dianion D, and FAM3− trianion T) present during the oxidation and reduction process. FAM 1 is the red shifted, FAM 2 green and FAM 3 blue shifted species. (b) Tautomerization between the lactone FAMH3 and the zwitterionic tautomer, and (c) absorption spectrum of the different species of FAM reported with permission from the literature. | ||
To observe the tautomer transition, we used freshly purchased FAM prepared from a completely dehydroxylated state (FAM 1, shown in red in Fig. 3) without stabilization of the buffer (0.5 μM solution in KCl 100 mM). Fig. 4a shows the time dependent curves of the voltage (black), current (red) and fluorescence (green) from the cell containing the WE. The interpretation of the cyclic voltammograms does not change, as there is no influence of the state of compounds on the electrolyte. All the peaks were fitted to extract Q(t) from the faradaic currents. However, for the preparation of the fluorophore in the electrolyte, we observed a different behavior. Fig. 4b shows a zoomed-in image of the fluorescence of cycle 2 (extended for a time slightly before and after), which is representative of the behavior observed. The most notorious feature is the relative minimum of the fluorescence that can be observed around 150 s occurring at the middle of the oxidation cycle. As the voltammogram continued to complete the oxidation, the descending fluorescence with the oxidation cycle was retrieved to achieve the global minimum at the end of the oxidation cycle and then the maximum of the fluorescence was recovered after the reduction (in this cycle after 210 s). We interpreted the intermediate minimum with the formation of a medium tautomer FAM 2, less fluorescent than the species generated at this instant in the solution with a limited lifetime (few microseconds). This sudden drop in the fluorescence can be explained based on the absorption spectra (Fig. 3c) of the different FAM species. The initial species FAM 1 (shown in red) showed transitions from one acid/base pair to another with a similar absorbance at our excitation wavelength (470 nm). However, the lower absorbance of the neutral lactone N (FAM 2 shown in green in Fig. 3c) at the excitation wavelengths translates into lower emission fluorescence. At this specific moment we recorded the tautomerization of the keto/spiro form (lactone FAMH3) into the zwitterionic equivalent (one of them being less fluorescent than the other). The colored band at the bottom of the fluorescence spectra represents the pH dependence of the different FAM acid/base pairs present in the solution and its evolution with time. Later the transition to cationic FAM 3 recovers the normal behavior. Fig. 4c shows the calculated charge exchanged as a function of time (Q(t)) extracted from the faradaic currents during the oxidation as detailed before. It shows that the pH varied in the solution via the electrochemically generated acid from 7 to 2 passing therefore through all the pKa values of the FAM. Thus, as the generation of protons during the oxidation protonates first the trianionic FAM3− it passes by the neutral (zwitterionic) FAMH3 leading at the end to cationic non-fluorescent FAMH4+. During the reduction of 4-ATP, the protons are re-adsorbed leading to a reverse exchange phenomenon (Fig. 3a), but the tautomerization is no longer observed, showing the instability of the neutral tautomer N.
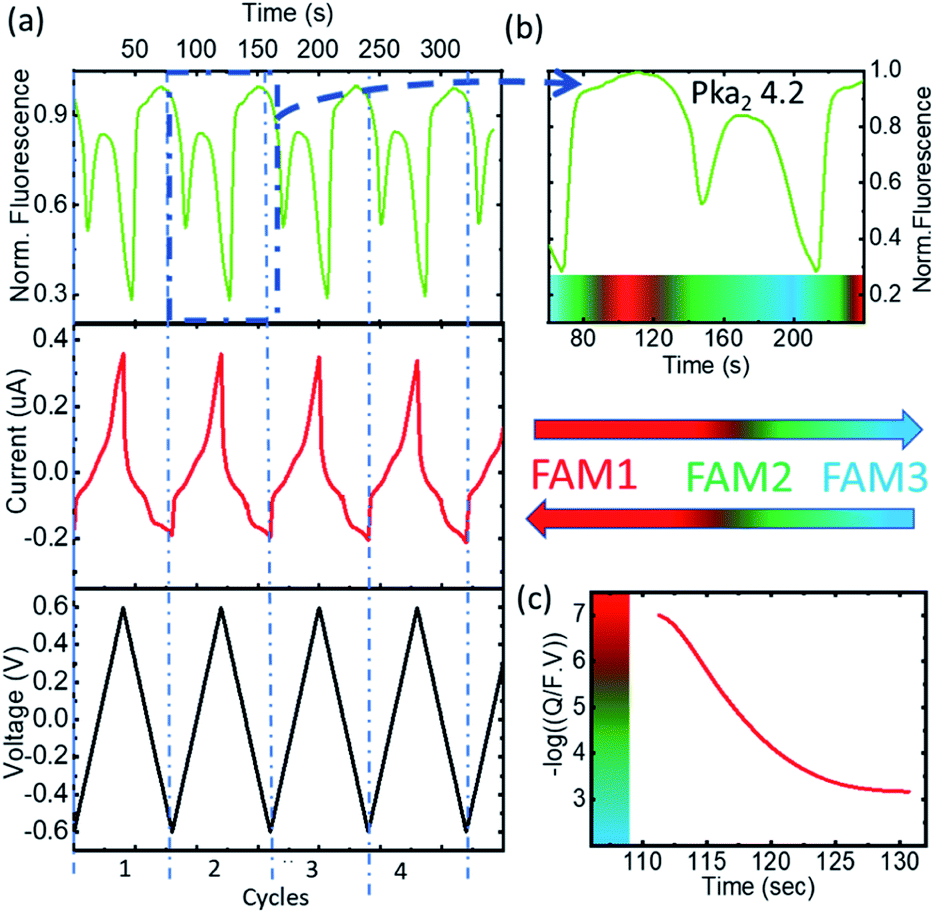 | ||
| Fig. 4 (a) The time evolution of the CV and fluorescence data corresponding to 4 representative cycles, the voltage, the current, and the normalized FAM-fluorescence in black, red, and green, respectively. (b) Normalized fluorescence vs. time and the different species in the medium 1 to FAM 3. (c) Proton concentration (−logQ/(F × V)) vs. time and the different species in the medium. | ||
Fig. 4b and c represent respectively the tautomerization of the neutral tautomer into its zwitterionic state, and the absorbance spectra of all the compounds as reported in the work of Aschi et al.18 This interpretation is consistent with their measurement for the pKa values of the different families:
| At pKa4 = FAM3− + H3O+ ⇆ FAMH2− + H2O, highly fluorescent |
| At pKa3 = FAMH2− + H3O+ ⇆ FAMH2− + H2O |
| At pKa2 = FAMH2− + H3O+ ⇆ FAMH3 + H2O |
| At pKa1 = FAMH3 + H3O+ ⇆ FAMH4+ + H2O, non-fluorescent |
Monitoring acidity with FAM in organic solvents
Following the validation of the method to calculate the proton concentration with the faradaic currents, we deployed the platform for the control of acidity in organic solvents using ACN. Since FAM bares 2 carboxylic radicals, to avoid reactions with the amino groups of 4-ATP in the electrodes we introduced a coating layer with a perfluorinated sulfonate polymer (Nafion 117). Nafion in addition to protecting the 4-ATP groups can transport the electrochemically generated protons from the electrodes to the solution through its sulfonate channels. Fig. 5a shows a few representative cycles (from 2 to 5, also excluding the first cycle to exclude the effects from the open circuit state) from a series of voltammograms obtained in the microreactor, together with the fluorescence obtained from FAM (0.5 μM) dissolved in an acetonitrile-based electrolyte (10 mM Bu4PF6) solution. The CVs vs. time are shown representing the voltage bias in black and the measured current and fluorescence in red and green, respectively. The complete experiment was also carried out for several tens of repetitions without apparent degradation of the faradaic currents of the fluorescence, reflecting the efficiency of Nafion to avoid side reactions between FAM and the electrodes. Fig. 5b shows a representative cycle of CV (cycle 2) with the classical representation of current vs. bias voltage. The faradaic currents show two peaks that we ascribe to the oxidation of the dimerized and monomer forms of 4-ATP at ∼0.3 V and ∼0.55 V, respectively. These peaks were persistent; thus, we believe that the Nafion coating avoids further polymerization. The oxidation peaks are also shifted towards higher bias, as expected due to the increase of the pKa of the electrolyte. We also observe an increase of the bias for the reduction peak, as a consequence of the same effect. The faradaic currents were extracted from each voltammogram cycle to calculate the number of protons produced during each bias ramp (more details of the extraction are shown in ESI-4 Table 2†). The concentration of the protons produced during the oxidation (1.67 × 10−2 M) is almost equivalent to those re-adsorbed for the reduction (1.45 × 10−2 M). The concentration of protons (−log(Q/F × V)) and the relative fluorescence normalized to the maximum value under normal conditions are shown vs. time in the inset of Fig. 5c.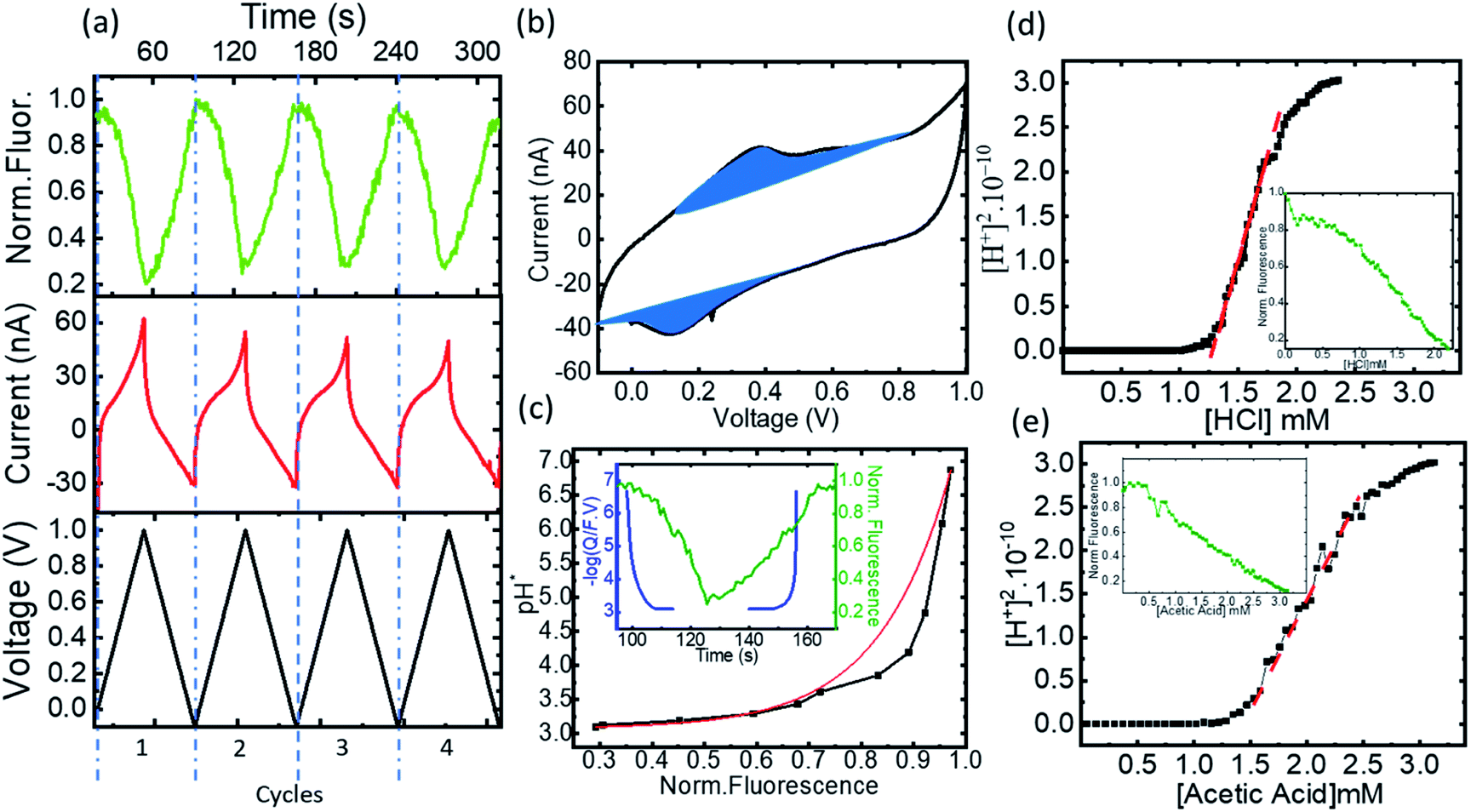 | ||
| Fig. 5 (a) The time evolution of the CV and fluorescence data corresponding to 4 representative cycles, the voltage, the current, and the normalized FAM–fluorescence in black, red and green, respectively, (b) cyclic voltammogram and the extracted faradaic currents, (c) pH*vs. normalized fluorescence and exponential fitting, with the inset showing the normalized fluorescence (green) and proton concentration (−logQ/(F × V)) (in blue) vs. time, (d) transformation of the normalized fluorescence into [H+]2vs. HCl concentration in black, the slope of the corresponding curve in dashed red, inset in green the titration curve of HCl fluorescence vs. the concentration, and (e) transformation of the normalized fluorescence into [H+]2vs. HCl concentration in black, the slope of the corresponding curve in dashed red, inset in green the titration curve of HCl fluorescence vs. the concentration. | ||
In acetonitrile, the electrolyte plays a key role in the mobility of the electrochemically generated protons. Different equilibria are in competition in the solution: the dissociation of the electrolyte, its association with the protons, the protonation of FAM, and the protonation of the solvent itself19 shown as:
| Bu4PF6 ⇆ Bu4+ + PF6− | (1) |
| H+ + PF6− ⇆ HPF6 | (2) |
| H+ + ACN ⇆ ACNH+ | (3) |
| FAM3− + HPF6 ⇆ FAMH2− + PF6− | (4) |
Eqn (1) shows the dissociation of the salt used for the electrolyte conductivity, which acts as the main proton carrier (eqn (2)) as PF6− has a much higher proton affinity than ACN (eqn (3)). In our system, the acidity is generated from the electrodes while the conjugate base remains fixed and isolated from the medium by the Nafion. The proton concentration is similar to that of Bu4PF6, but 100 times higher than the concentration of FAM (FAM 0.5 μM, Bu4PF6 10 mM vs. protons electrogenerated calculated from the faradaic currents ∼14.5 mM). Thus, as the acid generated by the faradaic currents reaches the electrolyte, the protonation of PF6− (eqn (2)) and the migration of protons to FAM (eqn (4)) (faster than the protonation of acetonitrile (eqn (3))) ensure the availability of all the protons to interact with the acid–base pairs of FAM. Also, due to the planar configuration of the electrochemical cell and the electrodes, the typical diffusion length is limited by the height of the cell, which is in the order of a few μm. Thus, the fluorescence in our electrolyte was mainly determined by the equilibrium in eqn (4). Then we can interpret the reversibility of the fluorescence spectra from complete quenching during the oxidation and its total recovery after the reduction as the reporting of FAM fluorescence vs. the proton activity. In the inset of Fig. 5c, we observe a delay of the fluorescence with respect to the faradaic charge, which we attribute to the delay of protons to cross the Nafion membrane. However, as the time interval between the oxidation and reduction is much longer (30 s), the equivalence in Fig. 5 pH* vs. FAM fluorescence is a good approximation over a full cycle. We can define the proton activity of the electrochemically generated acid in the cell as the pH* calculated using the faradaic currents (−log(Q/F × V)). Assuming the delay of the protons to cross the Nafion barriers, we used the correlated temporal dependence of faradaic currents and fluorescence shown in the inset of Fig. 5c to generate FAM fluorescence vs. pH* shown in black in Fig. 5c. We used the relationship of the pH* with FAM fluorescence to calibrate the dissociation of acids in a solvent with the same composition of our electrolyte. We titrated different acids including strong and weak acids (HCl and TFA for the strong and acetic and formic for the weak acids, respectively). We recorded the FAM fluorescence signal corresponding to the titration of the acids using a solvent with the same composition as our medium (insets Fig. 5d and e). To obtain the pKa dependence, we considered the acid dissociation constant:
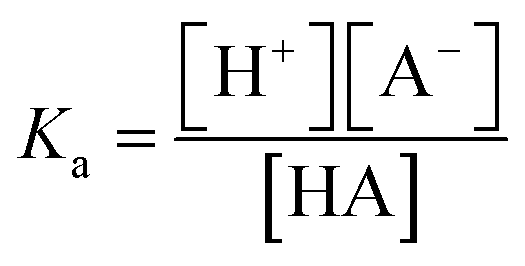
Since the dissociation of ACN is very low (ACN pKa = 40 (ref. 20)), all the protons derive from the acid, and thus the proton concentration in this system can be considered equal to the concentration of the conjugated base:
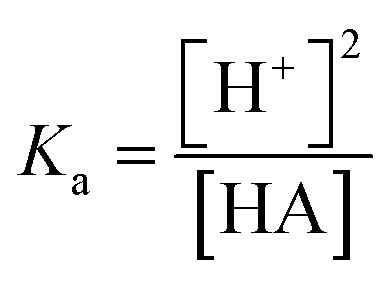
It is noted that pKa from the four acids had similar values in a range from 9 to 10 although some of them are considered weak acids. This similar behavior can already be noticed following the allure of the fluorescence titration curves as the fluorescence vs. acid concentration had a similar behavior in all cases (Table 1).
The strong acidity observed for all the compounds is explained by the influence of the electrolyte Bu4PF6, that creates super-acids due to the ability of PF6− to associate the protons dissociated from the acids already reported in the literature.23 The use of this proton carrier allows increasing the dissociation of the acid, almost similarly to mineral acids dissociated in water. As a result of our calibration of the acidity response of FAM with the faradaic currents, we were able to find the activity of the acids in the complex media that included Bu4PF6.
Deprotection of acid labile groups using the electrochemically generated acid
Following the good control of the acidity reaching very high concentrations of protons, we disposed the generation of acid to deprotect acid labile groups employed in solid phase synthesis. We tested the Boc protecting group, known in peptide and nucleotide synthesis due to the high yield of polymers that it achieves. However, the high yields of purity achieved are also due to its stability that ensures the orthogonality of chemistry during acid/base transitions, which requires a high acid concentration for its removal (∼50% TFA in DCM used in many traditional solid phase syntheses).We proceeded to use glass functionalized with amino groups protected by a Boc terminal function as described in Fig. 6. Fig. 6a represents the deprotection of BOC groups using electrochemically generated protons by chronoamperometry from Nafion protected electrodes in the presence of an ACN based electrolyte (no FAM was included in these experiments). Fig. 6b shows a microscope picture corresponding to one of the electrochemical cells. Black platinized electrodes are seen in the microscope in black. The electrodes are connected by diffusion barriers that are seen in Fig. 6b in grey. The circular electrode is the WE, while the short and long arched electrodes are the RE and CE, respectively. To be sure that we generated enough protons, a pulsed voltage bias to the WE vs. the RE was maintained at 0.85 V for 1 min.
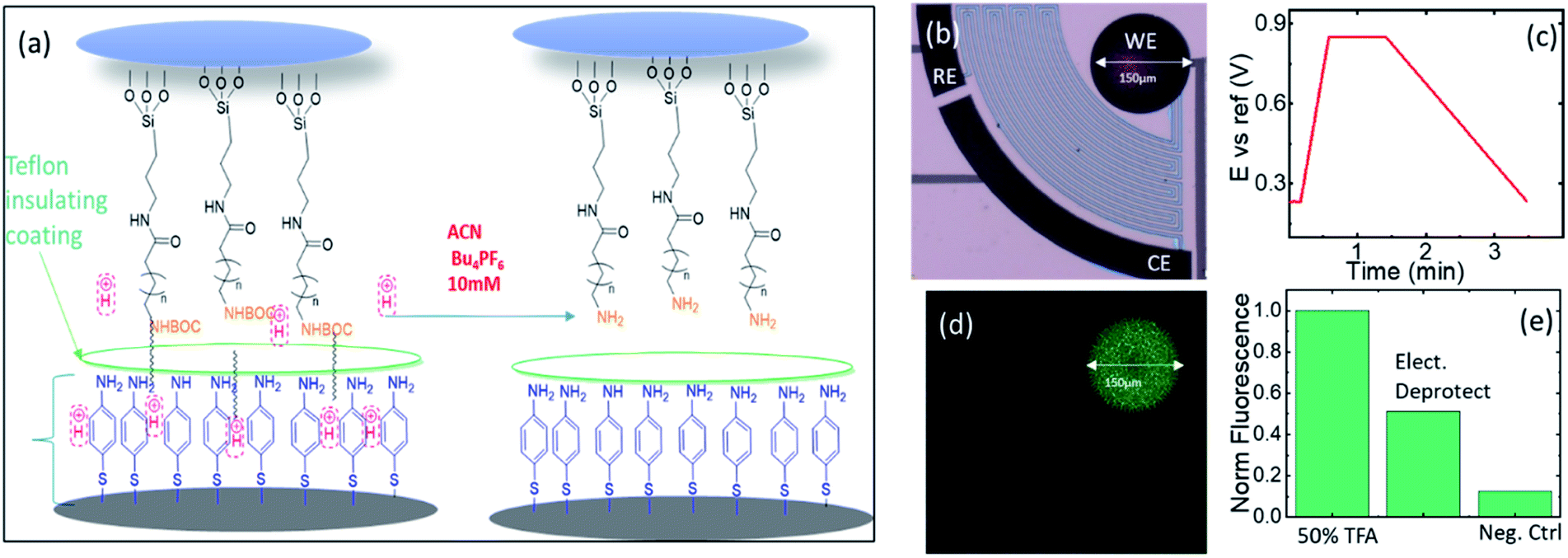 | ||
| Fig. 6 (a) Description of the deprotection of BOC in the electrochemical cell coated with Nafion, (b) full reactor with a circular WE electrode, CE and RE in black and diffusion barriers in grey, (c) electrical bias (pulse) applied on the WE vs. the RE at 0.85 V for 1 min, (d) deprotected cell after incubation in rhodamine B, and (e) comparison of the fluorescence of the electro-deprotected glass with glass treated with TFA 50% and negative control. | ||
Fig. 6c shows the electrical bias used for the generation of acid. After the exposure to this pulse, the glass was removed from the reactor and incubated with rhodamine B isothiocyanate in DMF for an hour to let the isothiocyanate function of the fluorophore react with newly Boc-deprotected amine. Fig. 6d shows the resulting image of the glass after the incubation. The area of the deprotected region is equivalent to the total footprint of our WE both being spots with a radius of ∼150 μm. The fidelity of the fluorescence image in Fig. 6d to the footprint of the WE shows the efficiency of the system to confine the acidity during the chronoamperometry experiments. This also supports the approximation that we have used through the article to consider the protons confined to the partial cell of the WE. We compared the deprotection of the electrochemically generated acid with the positive and negative controls described in the section Materials and methods and ESI-6.† The results are shown in Fig. 6e. The negative control shows that the deprotection was efficient, while the comparison with the positive control yielded lower fluorescence on the glass of the electrochemically generated acid. Nevertheless, since we cannot exclude contributions from fluorescence areas larger than our field of view (due to imperfections of the diaphragm in our microscope system) we believe that the positive control is an upper limit. The deprotection achieved by the electrochemically generated acid was quite significant, considering that the deprotected area had the same footprint of the WE. Since the surface area of the platinized electrodes was ∼10 times the flat area of the glass, we achieved such deprotection with ∼10 molar equivalents of the exposed amino groups.
Conclusion
In this work, we have shown accurate control of acidity by means of an electrochemically generated acid in 150 μm regions. We proved that faradaic currents can accurately control the acidity in the cell both in aqueous and organic solvents. Microreactors provide extremely acidic environments ∼10−1 M, thanks to which, we were able to demonstrate the deprotection of –Boc acid labile groups. The reactor delivers a neutral acid that can be used for the control of many chemical reactions with a Nafion coating that isolated the electrodes.One of the advantages of the electrochemically generated acid in our system is the absence of a conjugate base that could react and interfere in the medium. For organic acidity we used the correspondence of the faradaic currents with the proton concentration in the cell to calibrate the FAM fluorescence. Furthermore, we were able to calculate the pKa values of four different acids in the presence of Bu4PF6, which are different than the values reported in the literature.
To the best of our knowledge, this platform shows the best control of acidity in the smallest volume reported so far. This volume is three orders of magnitude smaller than the last version that we used in our previous publication, as the footprint has decreased to the order of 150 μm. The high fidelity achieved while deprotecting the acid labile groups placed in a glass upstanding the partial cell of our WE shows the high potential of our platform to control addressable chemical reactions. Owing to the high level of miniaturization, it would be possible to highly parallelize such chemical reactions, for example, to achieve high density polymer microarrays using parallelized solid phase synthesis.
Conflicts of interest
The authors declare that they have no competing interests.References
- M. T. Ghoneim, A. Nguyen, N. Dereje, J. Huang, G. C. Moore, P. J. Murzynowski and C. Dagdeviren, Chem. Rev., 2019, 119, 5248–5297 CrossRef CAS PubMed.
- J. Herlet, P. Kornberger, B. Roessler, J. Glanz, W. H. Schwarz, W. Liebl and V. V. Zverlov, Biotechnol. Biofuels, 2017, 10, 234–246 CrossRef CAS PubMed.
- L. Liu, J. W. Liu, Z. M. Huang, H. Wu, N. Li, L. J. Tang and J. H. Jiang, Anal. Chem., 2017, 89, 6944–6947 CrossRef CAS PubMed.
- K. Praveen, S. Das, V. Dhaware, B. Pandey, B. Mondal and S. Sen Gupta, ACS Appl. Bio Mater., 2019, 2, 4162–4172 CrossRef CAS PubMed.
- K. Michigami, T. Sakaguchi and Y. Takemoto, ACS Catal., 2020, 10, 683–688 CrossRef CAS.
- S. E. Thompson and S. Parthasarathy, Mater. Today, 2006, 9, 20–25 CrossRef CAS.
- M. Frasconi, R. Tel-Vered, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2010, 132, 2029–2036 CrossRef CAS PubMed.
- X. Gao, X. Zhou and E. Gulari, Proteomics, 2003, 3, 2135–2141 CrossRef CAS PubMed.
- K. Maurer, J. Cooper, M. Caraballo, J. Crye, D. Suciu, A. Ghindilis, J. A. Leonetti, W. Wang, F. M. Rossi, A. G. Stöver, C. Larson, H. Gao, K. Dill and A. McShea, PLoS One, 2006, 1, 1–7 CrossRef PubMed.
- R. D. Egeland and E. M. Southern, Nucleic Acids Res., 2005, 33, 1–7 CrossRef PubMed.
- E. Chiodi, A. M. Marn, M. T. Geib and M. Selim Ünlü, Polymers, 2021, 13, 1–21 Search PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- D. Balakrishnan, G. Lamblin, J. S. Thomann, A. Van Den Berg, W. Olthuis and C. Pascual-García, Nano Lett., 2018, 18, 2807–2815 CrossRef CAS PubMed.
- P. Umsarika, S. Changkhamchom, N. Paradee, A. Sirivat, P. Supaphol and P. Hormnirun, Mater. Res., 2018, 21, 1–8 Search PubMed.
- D. Balakrishnan, G. Lamblin, J. S. Thomann, J. Guillot, D. Duday, A. Van Den Berg, W. Olthuis and C. Pascual-García, Sci. Rep., 2017, 7, 15401–15412 CrossRef PubMed.
- W. A. Hayes and C. Shannon, Langmuir, 1996, 12, 3688–3694 CrossRef CAS.
- C. R. Raj, F. Kitamura and T. Ohsaka, Langmuir, 2001, 17, 7378–7386 CrossRef CAS.
- M. Aschi, A. A. D'Archivio, A. Fontana and A. Formiglio, J. Org. Chem., 2008, 73, 3411–3417 CrossRef CAS PubMed.
- A. Malloum and J. Conradie, New J. Chem., 2020, 44, 17558–17569 RSC.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Eur. J. Org. Chem., 2021, 2021, 1407–1419 CrossRef.
- F. Eckert, I. Leito, I. Kaljurand, A. Kütt, A. Klamt and M. Diedenhofen, J. Comput. Chem., 2009, 30, 799–810 CrossRef CAS PubMed.
- B. M. Ceballos and J. Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12686–12691 CrossRef CAS PubMed.
- A. K. Srivastava and N. Misra, Polyhedron, 2015, 102, 711–714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00275b |
| This journal is © The Royal Society of Chemistry 2022 |