
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-induced photoluminescence enhancement of CeF3:Tb3+ nanoparticles†
Tingting
Zhao
a,
Xiaoling
Jing
a,
Xueting
Zhang
a,
Chenyi
Li
a,
Ran
Liu
a,
Bo
Liu
a,
Chunxu
Yang
a,
Longhai
Shen
 *b,
Quanjun
Li
*a and
Bingbing
Liu
a
*b,
Quanjun
Li
*a and
Bingbing
Liu
a
aState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, P. R. China. E-mail: liquanjun@jlu.edu.cn
bSchool of Science, Shenyang Ligong University, Shenyang, 110159, China. E-mail: shenlonghai@163.com
First published on 22nd June 2022
Abstract
Rare earth fluorides have been widely used in recent years in the field of solid-state lighting. However, the relationship between the structure and luminescence properties is still unclear. Herein, the photoluminescence and structural transition of CeF3:Tb3+ nanoparticles under high pressure were investigated through in situ photoluminescence and X-ray diffraction measurements. Intriguingly, the photoluminescence of CeF3:Tb3+ nanoparticles displays an enhancement from 18.3 to 33.4 GPa, accompanied by the phase transition from the starting hexagonal phase to the orthorhombic phase. It was found that the distance of luminescent centers increased sharply during the high-pressure phase transition, which weakened the quenching effect and improved transmission efficiency. Our work provides more insight into the optical characteristics and structures of rare earth trifluorides.
Introduction
Rare earth (RE) trifluorides, as one of the famous families of functional materials, have attracted considerable attention because of their unique physical and chemical properties, such as low phonon energy, high resistivity, and high anionic conductivity.1,2 These properties mentioned above are related to the luminescence of RE trifluorides. The light emission originates from the transitions between f–f or d–f electron states which depend on the local symmetry and structure of RE trifluorides.3,4 Researchers had synthesized hexagonal GdF3:Eu3+ nanocrystals that greatly enhanced Eu3+ luminescence as compared to the orthorhombic ones.5 However, it is still a great challenge to explore the influence of structures on the luminescence properties of rare earth fluorides with a large ionic radius (ranging from La to Nd) or small ionic radius (ranging from Tb to Lu, and Sm) due to the difficulty in synthesis of these materials with different crystal structures.6,7It is well known that high pressure is an effective experimental means to change the local symmetry and structures. And many researchers had studied the luminescence properties of RE fluorides with different structures obtained by high pressure means. For example, Zou et al. discovered that the pressure-induced hexagonal phase of EuF3 is beneficial to the enhancement of its photoluminescence (PL) intensity.8 Li et al. observed the pressure enhanced PL intensity in ErF3, which is related to its phase transition process from the orthorhombic structure to a hexagonal structure.9 Gong et al. found that the Eu3+ doped YF3 nanocrystal PL also increased with the phase transition under pressure.10 All the above indicate that high pressure is an effective method to regulate the structures and fluorescence properties of RE fluorides. However, there is no report on the relationship between the structure and luminescence properties of large radius RE fluorides.
As a main fluorescent material, compared with traditional oxide-based ones, the advantage of RE trifluorides lies in that their phonon vibration energy is fairly low, and RE ions as activators can be easily doped into the fluoride matrix.11 Among various large radius rare-earth fluorides, the activator concentration of CeF3 is 100%.12,13 As we all know, CeF3 experiences the phase transformation from the original hexagonal structure into an orthorhombic structure under high pressure.14,15 This laid the foundation for us to further study the fluorescence characteristics of RE trifluorides under high pressure. Lanthanide(III)-doped nanocrystals have attracted great attention as a new kind of bioluminescent label.16 Additionally, doped lanthanide ions can modify the optical properties, and thus different colored fluorescent labels can be generated by changing the dopants.17 Doping Tb3+ in CeF3 brought out a strong green emission from Tb3+ because of the efficient energy transfer from Ce3+ to Tb3+.18–20 As a potential scintillator and tunable laser material, CeF3: Tb3+ nanoparticles also have the conjugate effect with biotin and avidin, which adds to the potential application with biomolecular fluorescent markers.12,13,21 The investigation on the high-pressure behavior of CeF3:Tb3+ nanocrystals would be pretty important for understanding the relationship between the structure and fluorescence characteristics of the REF3 system.
In this work, we explored the PL and the phase transitions of CeF3:Tb3+ nanoparticles under high pressure using in situ PL and X-ray diffraction measurements. The hexagonal to orthorhombic phase transition and enhanced PL properties were discovered in CeF3:Tb3+ nanoparticles under high pressure. These results provide new insight into the relationship between the structure and physical properties of RE trifluorides.
Experimental
Synthesis of CeF3:Tb3+nanoparticles
The experimental method for the synthesis of CeF3 nanoplates is based on a previous report.20 CeF3:Tb3+ (1 mol% Tb3+, 40 nm) nanoparticles were synthesized via a polyol process by employing diethylene glycol (DEG) as solvent. In brief, DEG containing Tb(NO3)3 and Ce(NO3)3·6H2O was stirred and heated in a silicon oil bath in an argon atmosphere. At the same time, NH4F was added to DEG in the oil bath. The obtained suspension was cooled to room temperature and diluted with 50 ml ethanol. The nanoparticles were obtained by centrifugation at a speed of 4500 rpm. Then they were redispersed in ethanol and centrifuged several times to remove the loosely adsorbed solvent molecules on their surfaces. Finally, the as-prepared samples were dried in air.High-pressure measurements
High-pressure experiments were performed by using a diamond anvil cell (DAC) with 400 μm diameter culets. The samples and a ruby ball were loaded into a 120 μm diameter hole drilled in a steel gasket. The pressure was calibrated by the fluorescence emission of ruby.22 Silicone oil was added into the hole as a pressure-transmitting medium. The high-pressure PL measurements were conducted using a home-built optical measurement system with a Horiba Jobin Yvon iHR320 spectrometer and a 360 nm light source. High-pressure X-ray diffraction experiments were performed by using a Rigaku Synergy Custom FR-X (λ = 0.7107 Å). The XRD data were refined using the General Structure Analysis System (GSAS) program to obtain the lattice parameters.Results and discussion
Characterization under ambient conditions
Tb3+ doped hexagonal phase CeF3 with a doping concentration of 1% was synthesized via the polyol method.20 From the low magnification TEM image (Fig. 1a), it can be seen that the CeF3:Tb3+ sample is composed of nanoparticles. The average length of the particles is 40 nm. Fig. 1b clearly displays a well-resolved lattice fringe with a spacing of 0.16 nm, corresponding to the (114) plane of CeF3, which indicates the high crystallinity of the sample. The energy dispersive X-ray analysis images (Fig. S1, ESI†) show that the Tb3+ ions are doped in CeF3, and Fig. 1c (elemental mapping images) indicates that terbium and other elements are distributed uniformly in the CeF3:Tb3+ nanoparticles. According to the previous literature, when Tb(NO3)3·6H2O was added as a raw material, Tb3+ is likely to substitute for Ce3+ in the CeF3 lattice.23 It can be seen from Fig. 1d that Tb3+ ions occupied the Ce3+ sites in CeF3:Tb3+ nanosized particles. The XRD (Fig. 1e) results show that the sample is crystallized well, and all the peaks are consistent with the hexagonal structure of the bulk CeF3.14 The broadened XRD peaks of the CeF3:Tb3+ sample are due to the small size of the nanoparticles. The calculated lattice parameters of the CeF3:Tb3+ nanoparticles (a = b = 0.710 nm; c = 0.726 nm) agree well with the reported values for CeF3 bulk.15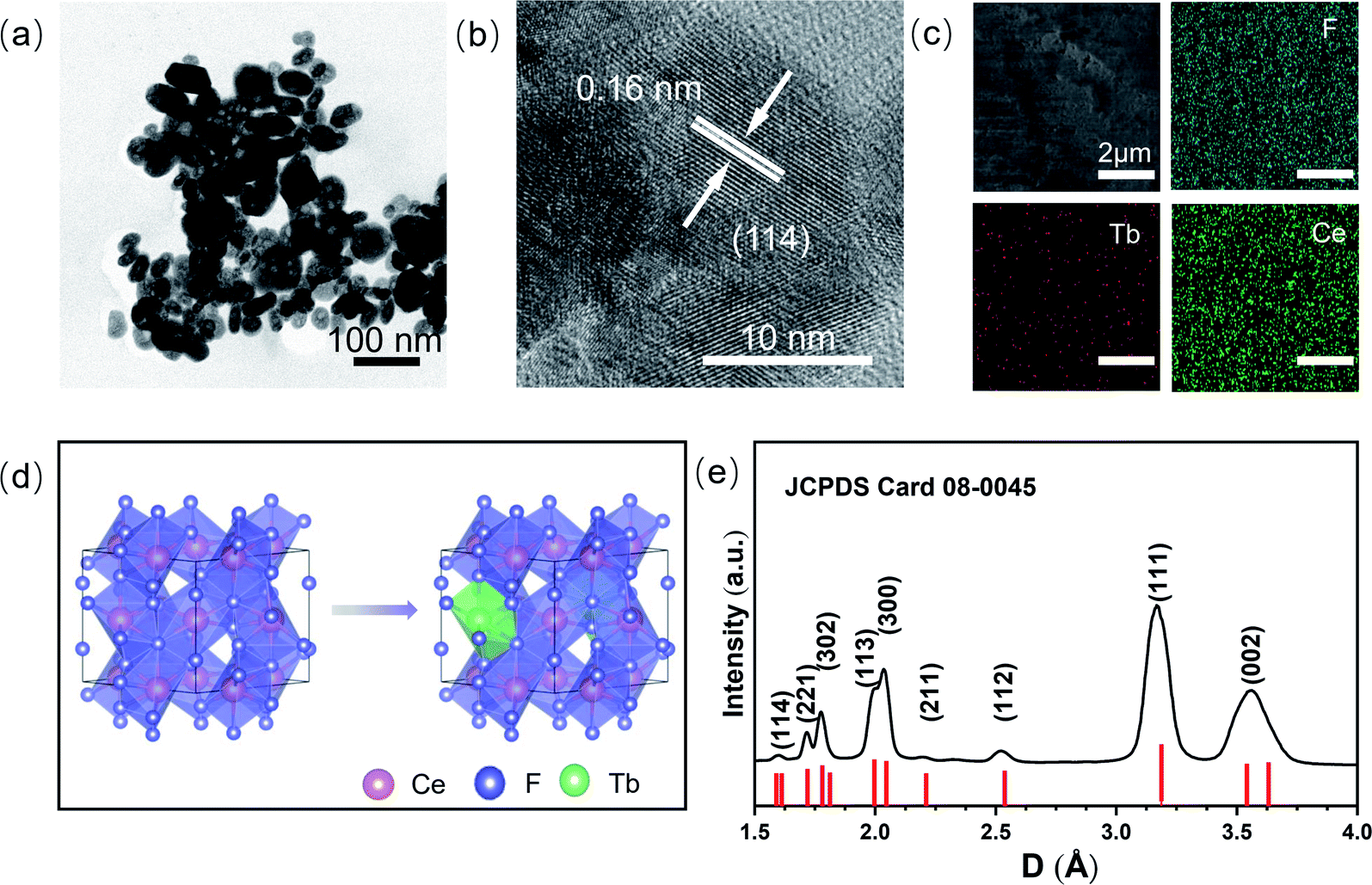 | ||
| Fig. 1 Morphology characteristics of CeF3 nanoparticles doped with Tb3+ ions at ambient pressure. TEM micrographs at (a) low-resolution and (b) high-resolution. (c) Elemental mapping images of Tb3+ ion-doped CeF3 nanoparticles. (d) The polyhedral model before and after doping Tb3+ ions occupying the Ce3+ sites. (e) XRD patterns of the CeF3:Tb3+ nanoparticles, and the standard data for bulk CeF3 (JCPDS Card No. 08-0045). | ||
In situ high-pressure optical and structural measurement
To explore the luminescent properties of CeF3:Tb3+ nanoparticles, we carried out high-pressure PL experiments. As shown in Fig. 2a and b, five distinct peaks in the range of 380–700 nm are observed. The dominant sharp peaks ranging from 450–650 nm are assigned to the 5D4 → 7Fj (j = 6–3) transition of Tb3+ ions; the band ranging from 384–680 nm is attributed to the 5d–4f transition of Ce3+.20 The Ce3+ PL intensities decrease monotonically with increasing the pressure up to 18.3 GPa (Fig. 2a). Upon further compression, the PL intensities of Tb3+ display an increasing trend up to 33.4 GPa (Fig. 2b), which enhanced by about twofold that of the starting pressure. At the same time, we can clearly see that the intensity of the 580 nm luminescence peak is enhanced more than that of the other luminescence peaks, which may be due to the more competitive transition of the 5D4–7F5 energy level. It can be seen that no obvious shift occurred in the PL peak position (Fig. 2d). This is because the RE luminescence originates from the f–f electric dipole transitions of 4f electrons, which are shielded from environmental effects by the outer shell electrons. The pressure dependences of the Ce3+ PL position and intensity in our CeF3:Tb3+ are shown in Fig. 2e and f. The PL of Ce3+ is attributed to the 5d–4f transition, which is not protected like the 4f shell anymore. So Ce3+ PL is vulnerable to external environmental impacts. We can see that the wavelength of Ce3+ shows a blueshift upon increasing the pressure to 20 GPa and then displays a redshift upon increasing the pressure to 33.4 GPa. This intensity change of Ce3+ PL is roughly the same as that of Tb3+, but the final increase is far less. This phenomenon may have originated from the energy transfer between Ce3+ and Tb3+.20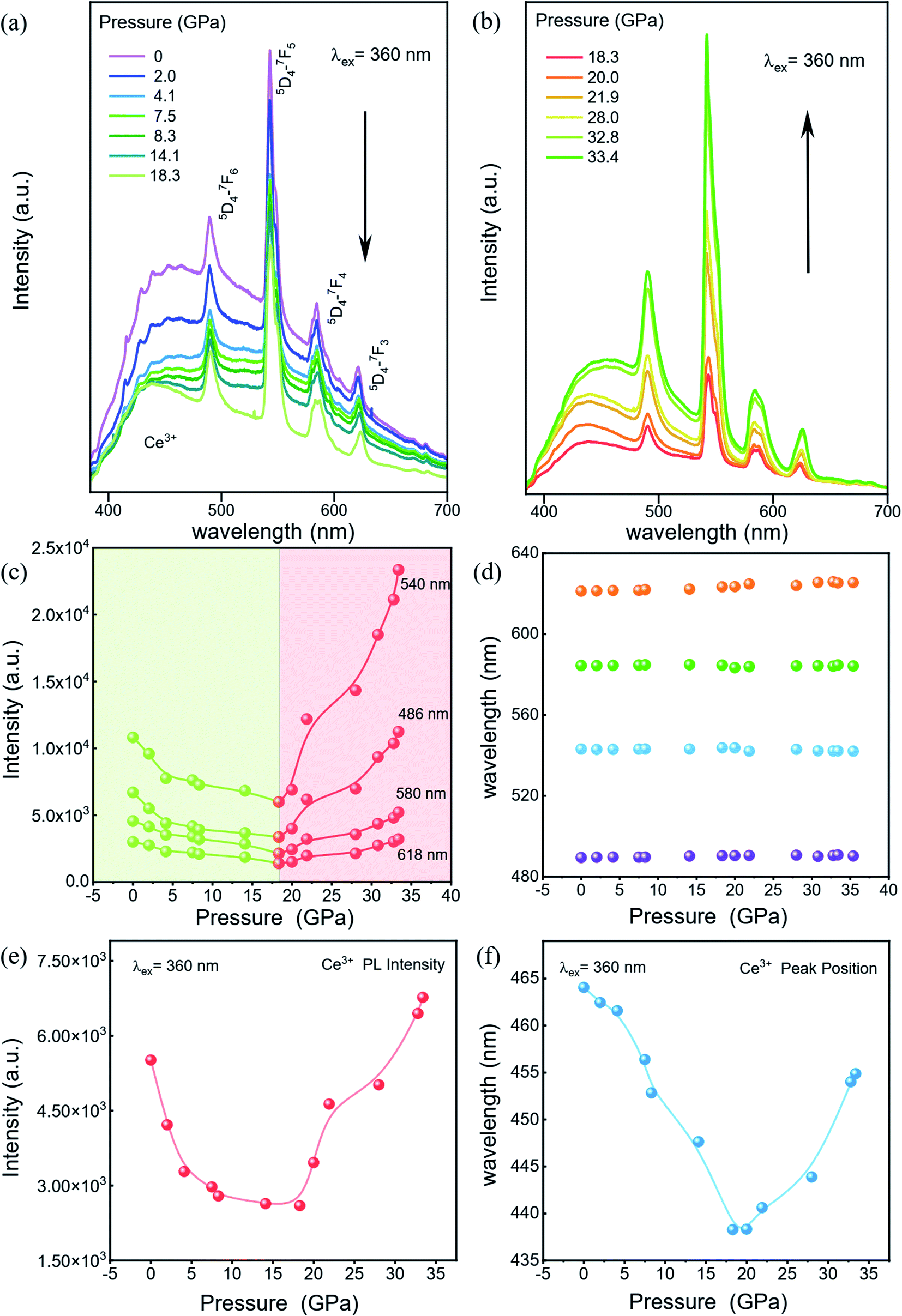 | ||
| Fig. 2 High-pressure PL properties of CeF3:Tb3+. (a) and (b) The PL spectrum changes of CeF3:Tb3+ under pressure. Black arrows indicate the evolution of the PL spectra as a function of pressure. (c) Peak intensity, (d) Peak position evolution of Tb3+ ion-doped CeF3 nanoparticles at various pressures. (e) Peak intensity and (f) Peak position of Ce3+ at various pressures. | ||
We carried out high pressure XRD measurements to reveal the relationship of the structure–optical properties of the CeF3:Tb3+ nanoparticles. The selected XRD patterns are shown in Fig. 3a. With increasing pressure, no apparent change is observed before 16.2 GPa, suggesting that the sample remained in the hexagonal phase. Upon further compression to 18.9 GPa, a new peak gradually appeared at around 14°, manifesting the beginning of a structural phase transition. We can clearly see the new diffraction peaks by peak fitting (Fig. 3b). And the structural phase transition process still progresses until 30.9 GPa. When released under ambient conditions, the structure of the sample returned to its initial phase. According to previous studies, we suggest that the new structural phase is the orthorhombic phase.23,24 In the pressure range 18.9–30.9 GPa, the diffraction peaks of the hexagonal phase gradually weakened. And the intensities of the peaks belonging to the orthorhombic phase get stronger and stronger with increasing pressure, indicating the gradual transition from the hexagonal phase into the orthorhombic phase. Previous studies have shown that the particle size leads to a significant decrease of the pressure in some nanomaterials because of the existence of the unstable cubic phase during the transition period from the hexagonal phase to the orthorhombic phase.15 In addition, a small amount of Tb3+ dopant has a slight increase on the phase transition pressure although it does not affect the order of pressure-induced phase transition.25 But on the whole, the phase transition pressure value of the materials is slightly lower than the bulk CeF3 phase transition value of 20.6 GPa.14
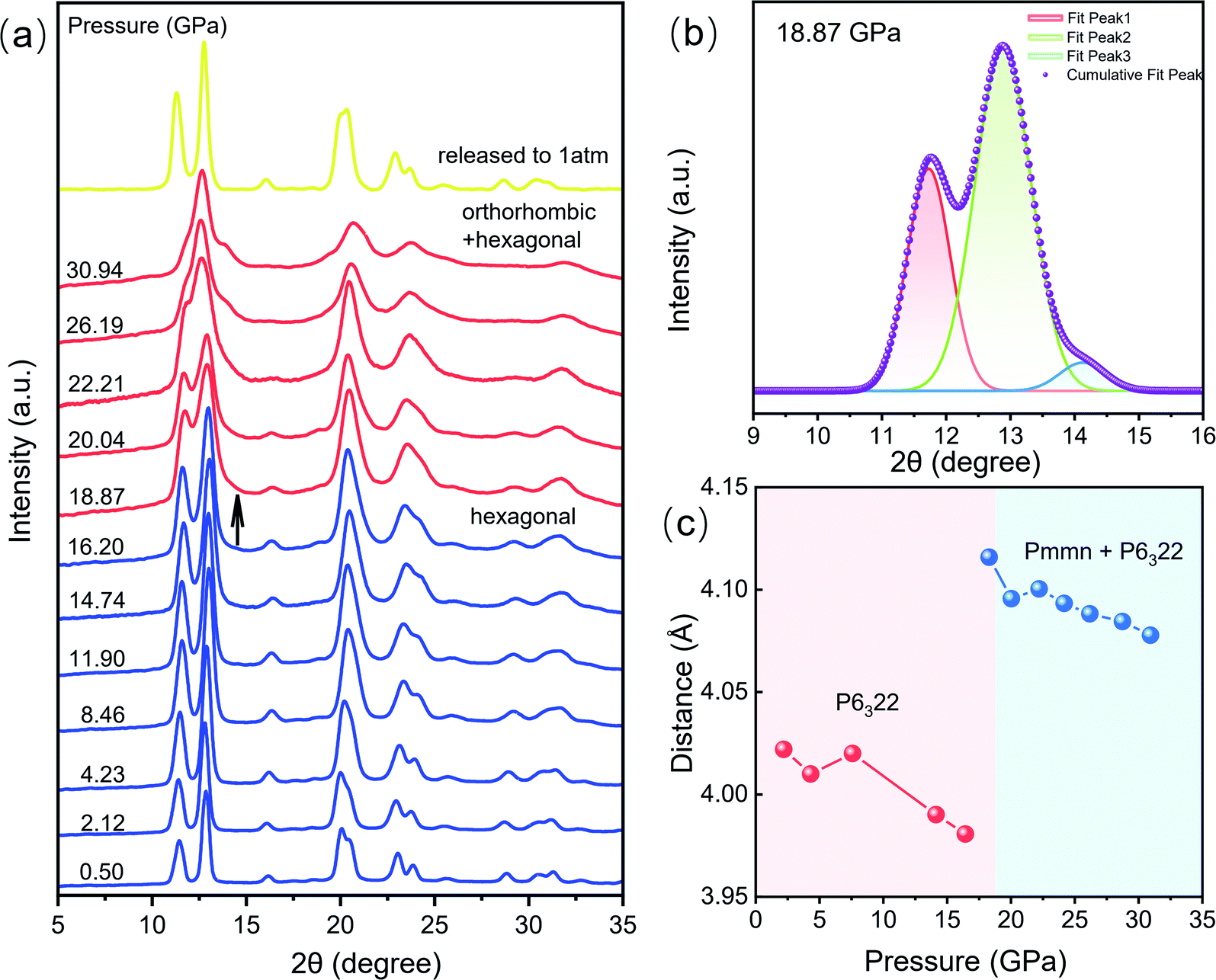 | ||
| Fig. 3 Variation of structural information of CeF3:Tb3+ nanoparticles at different pressures. (a) High-pressure XRD patterns of CeF3:Tb3+ up to 30.9 GPa. (b) Origin peak fitting patterns of 18.9 GPa data. (c) Pressure dependences of the distance of Ce–Tb and Ce–Ce of CeF3:Tb3+ nanoparticles. | ||
The length of Ce–Tb and Ce–Ce distance of CeF3:Tb3+ nanoparticles were obtained by refining the diffraction patterns (Fig. 3c). Fig. S2† shows Rietveld refinement of the diffraction pattern of CeF3:Tb3+ at 18.9 GPa. In the pressure range 2.1–16.2 GPa, the distances between Ce–Tb and Ce–Ce decrease gradually with increasing pressure. But the distances increase significantly above 16.2 GPa accompanied by the structural phase transition. Upon further compression, the Ce–Tb and Ce–Ce distances are reduced steadily up to the highest experimental pressure of 30.9 GPa, but they are still much larger than those of the starting phase in the low-pressure range.
Discussion
Under 360 nm UV light irradiation, CeF3:Tb3+ nanoparticles exhibit bright green emission at atmospheric pressure. Fig. 4a and b show the energy transfer process and the emission spectra of the sample, respectively.20,26,27 The emission spectra consist of a broad band and several peaks. The band located in the range 384–680 nm is ascribed to the 5d–4f transition of Ce3+. The strong sharp peaks between 450 and 650 nm (peaking at 489, 543, 584 and 618 nm), emission of Tb3+, derive from transitions between the excited 5D4 state and the 7Fj (j = 6–3) ground states of Tb3+ ions.26 Energy transfer takes a critical part in improving the emission efficiency of RE materials. The energy transfer process from Ce3+ to Tb3+ takes place by resonance transfer (electric dipole-electric quadrupole interaction) upon excitation with UV light. For the CeF3 system, first, Ce3+ ions are excited by UV light excitation; subsequently, energy transfer occurs from Ce3+ to Tb3+ ions.28–30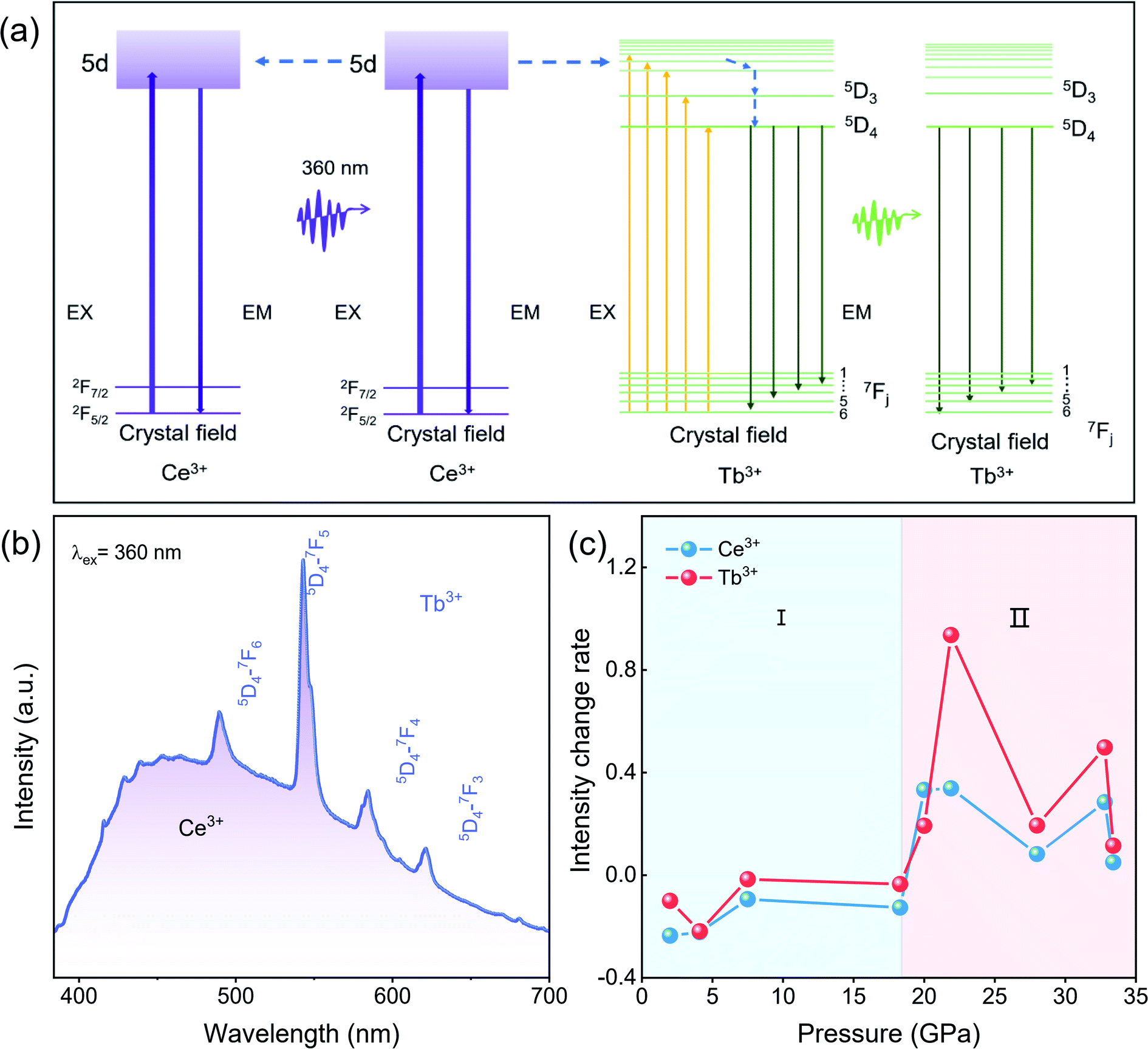 | ||
| Fig. 4 Luminescent properties of CeF3:Tb3+. (a) Energy level scheme of CeF3:Tb3+ with electronic transitions and energy transfer processes. (b) The PL spectra of CeF3:Tb3+ at ambient pressure. (c) The rate of intensity changes for Ce3+ and Tb3+ with pressure. | ||
According to the energy transfer process between Ce3+ and Tb3+ at atmospheric pressure(Fig. 4b), we know that the distance between luminescent centers is crucial to the luminescence of RE fluorides, such as concentration quenching phenomena.31,32 In energy transfer processes of CeF3:Tb3+ under ambient pressure, there is a cascade transfer process between Ce3+ and Ce3+, that is, energy can be transferred from Ce3+ to another Ce3+ or a quenching center except for energy transfer between Ce3+ and Tb3+.32 When the distance between Ce3+ and Ce3+ is close to a certain extent, a cascade transfer process occurs between Ce3+ and Ce3+. And the excitation energy is likely to be transferred to the quenching centers, resulting in energy loss. Considering the decrease of the distance between the luminescence centers from the XRD refinement result (Fig. 3c), it is reasonable to assume that the distance between the luminous centers in the starting hexagonal phase CeF3:Tb3+ is less than the critical value where PL quenching begins. Therefore, the decreased PL intensity in the range of 0–18.3 GPa can be attributed to the decrease in the distance between the luminescence centers, which results in the loss of partially excited light. Similarly, Tb3+ ions gradually approach each other with increasing pressure, and energy migration also takes place between these ions like Ce3+, which adds to the energy loss through easier access to quenching centers.25
Above 18.3 GPa, the PL intensity of the CeF3:Tb3+ nanoparticles display a clear increase with increasing pressure, which is also related to the distance between the luminescence centers. The distance between luminescence centers in the high-pressure orthorhombic phase decreases slowly, but it is larger than that of the starting phase, resulting in the suppression of the quenching phenomenon between the luminescent centers. According to the intensity change rate diagram (Fig. 4c), we can see that the relative intensity changes of Tb3+ are much larger than those of Ce3+ after 20.0 GPa, which means that apart from the fact that partial excited Ce3+ passes energy to Tb3+ instead of conducting the emission process, the nonradiative-relaxation processes down towards the 5D4 state of Tb3+ might probably be simplified as well due to the red shift of Ce3+(Fig. 2f). From another angle, the thus benefited 5D4 → 7Fj (j = 6–3) transition can better compete with other unwanted energy pathways like those from excited Ce3+ or Tb3+ to quenching centers, further promoting the PL. Therefore, the enhanced luminescence intensity in the high-pressure phase is due to the weakened quenching effect and improved transmission efficiency. From the above analysis, we suggest that an ideal distance between RE ions in materials is significantly important for high-quality PL, and high pressure is undoubtedly an effective method to modulate the PL properties through phase engineering.
Conclusions
In summary, we investigated the photoluminescence properties and structural transition of CeF3:Tb3+ nanoparticles by in situ PL and XRD measurements under high pressure. The CeF3:Tb3+ nanoparticles undergo the structural phase transition from the starting hexagonal phase to an orthorhombic phase at 18.9 GPa with the distance between the luminescence centers increasing sharply and then slowly decreasing. The PL intensity of the CeF3:Tb3+ nanoparticles decreases between 0–18.3 GPa, which is due to the fact that the decrease in the distance between the luminescence centers results in the loss of partially excited light. With increasing pressure, the fluorescence intensity increases until 33.4 GPa, which is related to the change of the distance between the luminescence centers, which diminishes the negative quenching effect and improves transmission efficiency. The results provide a deep understanding of the relationship between the structure and fluorescence characteristics of RE fluorides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2018YFA0305900), National Natural Science Foundation of China (11874172, U2032215 and 11634004), and JLU Science and Technology Innovative Research Team (2017TD-01).Notes and references
- L. Wang, P. Li and Y. Li, Adv. Mater., 2007, 19, 3304–3307 CrossRef CAS.
- C. G. Olson, M. Piacentini and D. W. Lynch, Phys. Rev. B, 1978, 18, 5740–5749 CrossRef CAS.
- F. Meiser, C. Cortez and F. Caruso, Angew. Chem., Int. Ed. Engl., 2004, 43, 5954–5957 CrossRef CAS PubMed.
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1–R13 CrossRef CAS.
- X. Zhang, T. Hayakawa, M. Nogami and Y. Ishikawa, J. Alloys Compd., 2011, 509, 2076–2080 CrossRef CAS.
- Z. Allan, T. H. David and H. E. Ted, Inorg. Chem., 1966, 5(8), 1466–1468 CrossRef.
- V. M. Mansnmann, Z. Anorg. Allg. Chem., 1964, 331, 98–101 CrossRef.
- Q. Li, S. Li, K. Wang, J. Liu, B. Liu, K. Yang and B. Zou, J. Phys. Chem. C, 2014, 118, 7562–7568 CrossRef CAS.
- W. Li, X. Ren, Y. Huang, Z. Yu, Z. Mi, N. Tamura, X. Li, F. Peng and L. Wang, Solid State Commun., 2016, 242, 30–35 CrossRef CAS.
- C. Gong, Q. Li, R. Liu, Y. Hou, J. Wang, X. Dong, B. Liu, X. Tan, J. Liu, K. Yang, B. Zou, T. Cui and B. Liu, J. Phys. Chem. C, 2014, 118, 22739–22745 CrossRef CAS.
- C. M. B. Bender and J. M., Chem. Mater., 2000, 12, 1969–1976 CrossRef CAS.
- A. J. Wojtowicz, M. Balcerzyk, E. Berman and A. Lempicki, Phys. Rev. B, 1994, 49, 14880–14895 CrossRef CAS PubMed.
- G. G. Blasse and B. C., Springer:Berlin, 1994, 1–232 Search PubMed.
- T. I. Dyuzheva, L. M. L., G. B. Demishev and N. A. Bendeliani, Inorg. Mater., 2003, 39, 1198–1202 CrossRef CAS.
- P. Wang, Q. J. Li, R. Liu, M. G. Yao, D. M. Li, B. Zou, T. Cui, J. Liu, Z. Q. Chen and B. B. Liu, J. Appl. Phys., 2012, 111, 1–4 Search PubMed.
- F. Wang, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, 1527–1532 CrossRef CAS.
- R. B. Cédric Louis, C. A. Marquette, J.-L. Bridot, S. Roux, G. Ledoux, B. Mercier, L. Blum, P. Pascal and O. Tillement, Chem. Mater., 2005, 17, 1673–1682 CrossRef.
- J. W. Stouwdam and F. van Veggel, Langmuir, 2004, 20, 11763–11771 CrossRef CAS PubMed.
- M. H. Riwotzki K, A. Kornowski and M. Haase, J. Phys. Chem. B, 2000, 104, 2824 CrossRef.
- Z. W. Q. Z. L. Wang and P. Y. Jia, Chem. Mater., 2006, 18, 2030–2037 CrossRef CAS.
- D. Y. Kong, Z. L. Wang, C. K. Lin, Z. W. Quan, Y. Y. Li, C. X. Li and J. Lin, Nanotechnology, 2007, 18, 075601 CrossRef CAS PubMed.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res.: Atmos., 1986, 91, 4673–4676 CrossRef CAS.
- X. Li, W. Zhang, L. Dong, D. Liu and Z. Qi, J. Lumin., 2019, 205, 122–128 CrossRef CAS.
- W. A. Crichton, P. Bouvier, B. Winkler and A. Grzechnik, Dalton Trans., 2010, 39, 4302–4311 RSC.
- C. Gong, Q. Li, R. Liu, Y. Hou, J. Wang, X. Dong, B. Liu, X. Yang, Z. Yao, X. Tan, D. Li, J. Liu, Z. Chen, B. Zou, T. Cui and B. Liu, Phys. Chem. Chem. Phys., 2013, 15, 19925–19931 RSC.
- D. Wang and N. Kodama, J. Solid State Chem., 2009, 182, 2219–2224 CrossRef CAS.
- H. Guo, Appl. Phys. B, 2006, 84, 365–369 CrossRef CAS.
- G. Blasse and A. Bril, J. Chem. Phys., 1969, 51, 3252–3254 CrossRef CAS.
- J. C. Bourcet and F. K. Fong, J. Chem. Phys., 1974, 60, 34–39 CrossRef CAS.
- Z. Wang, Z. Quan, J. Lin and J. Fang, J. Nanosci. Nanotechnol., 2005, 5, 1532–1536 CrossRef CAS PubMed.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063–1070 CrossRef CAS.
- D. J. Shi Chaoshu and W. Yaguang, et al. , Chin. Phys. Lett., 2000, 17(7), 532–533 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00166g |
| This journal is © The Royal Society of Chemistry 2022 |