
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the activity and stability of carbon-supported platinum–gadolinium nanoalloys towards the oxygen reduction reaction†
C. A.
Campos-Roldán
 a,
F.
Pailloux
b,
P.-Y.
Blanchard
a,
D. J.
Jones
a,
J.
Rozière
a and
S.
Cavaliere
*ac
a,
F.
Pailloux
b,
P.-Y.
Blanchard
a,
D. J.
Jones
a,
J.
Rozière
a and
S.
Cavaliere
*ac
aICGM, Université de Montpellier, CNRS, ENSCM, 34095 Montpellier Cédex 5, France. E-mail: sara.cavaliere@umontpellier.fr
bInstitut P′, CNRS, Université de Poitiers, ISAE, ENSMA, UPR 3346, 11 Boulevard Marie et Pierre Curie, Site du Futuroscope, TSA 41123, 86073 Poitiers Cédex 9, France
cInstitut Universitaire de France (IUF), 1 Rue Descartes, 75231 Paris Cedex 05, France
First published on 15th November 2021
Abstract
The activity/stability towards the ORR of PtxGd/C nanoalloys has been enhanced by controlling the atmosphere during the dealloying process. By minimising the formation of porous nanoarchitectures, the ORR activity is increased, and is accompanied by higher activity retention and attenuation of metal dissolution on cycling to high voltage.
Platinum–rare earth metal (Pt–REM) alloys have been reported as being some of the most active and stable electrocatalysts towards the oxygen reduction reaction (ORR), in particular the Pt–Gd system.1,2 From previous studies on Pt–Gd sputter-cleaned polycrystalline surfaces1,3 and non-supported nanoparticles (NPs)2 it can be concluded that the ORR enhancement is related to the formation of a strained Pt overlayer due to the lattice contraction caused by the Pt–Gd alloy core,4 that weakens the binding energy between Pt and oxygen. Moreover, their long-term stability towards the ORR was linked to the Pt–Gd alloy formation energy, as well as the protective Pt overlayer.1 However, the desirable production of actual Pt–Gd NPs dispersed over high surface-area supports is the bottleneck for the implementation of these systems in proton exchange membrane fuel cell (PEMFC) technologies,5–8 since the reduction potential of Gd is far beyond the stability range of water.9 Thus, the Pt–Gd nanoalloy formation represents a big challenge. Recently, Hu et al.9 have developed a solid-state synthesis approach to produce carbon-supported Pt–REM nanostructures directly. Taking the Pt–Gd/C system as model,10 the authors have reported a high active ORR electrocatalyst; however, its stability did not match the expected behaviour. This feature is related to the crystalline phase (cubic Pt2Gd) and the formation of porous nanoarchitectures which are easily degraded under the harsh ORR conditions (nanoalloy segregation, metal dissolution, morphology transitions, etc.),11 resulting in significant performance decay. The formation of such sponge-like structures has been also reported for nanoparticulate systems such as Pt–Ni12 and Pt–Co,13 where they compromise the activity/stability benefits of the alloyed systems. Therefore, the stability enhancement of the novel nanostructured Pt–Gd/C system is of paramount importance for its implementation in PEMFC. Herein, we have produced PtxGd/C nanostructures using a solid-state synthesis method and provide evidence that the formation of the porous structures is minimised (but not suppressed) though a N2-atmosphere dealloying process, leading to ORR activity/stability enhancement.
Following our previous contribution,11 in which we have identified the optimal Pt:Gd composition, PtxGd NPs dispersed over conventional carbon Vulcan XC-72 were prepared. After the synthesis reaction (see details in the ESI†), the XRD pattern of the as-prepared material, Fig. S1,† shows the Pt2Gd and Pt3Gd reflections, along with GdOx and cyanamide crystalline complexes. Such reaction sub-products might be removed via an acidic wash.10 Simultaneously, the dealloying process, i.e. the selective dissolution of Gd, takes place, leading to the formation of a Pt shell around the PtxGd alloy core. The gaseous environment in which the dealloy protocol is performed has a great impact on the free corrosion potential (or open circuit potential, OCP) of a corroding system:14 the presence of oxygen creates a complex mixed OCP, which affects the dealloying kinetics.14 With this knowledge, we have produced and compared the electrocatalysts obtained by the acid wash under air- vs. N2-atmosphere (see detail in the ESI†). Fig. 1 reveals that the remaining sub-products are successfully removed after both leaching protocols, confirming the formation of Pt–Gd alloy (Pt2Gd and Pt3Gd reflections). The XPS spectra, cf.Fig. 1b, support formation of the Pt–Gd alloy with features characteristic of the metallic states of both elements.2 Besides, the binding energy of the Pt signals is downshifted with respect to the Pt/C reference material, suggesting a modification of the Pt electronic structure.15,16
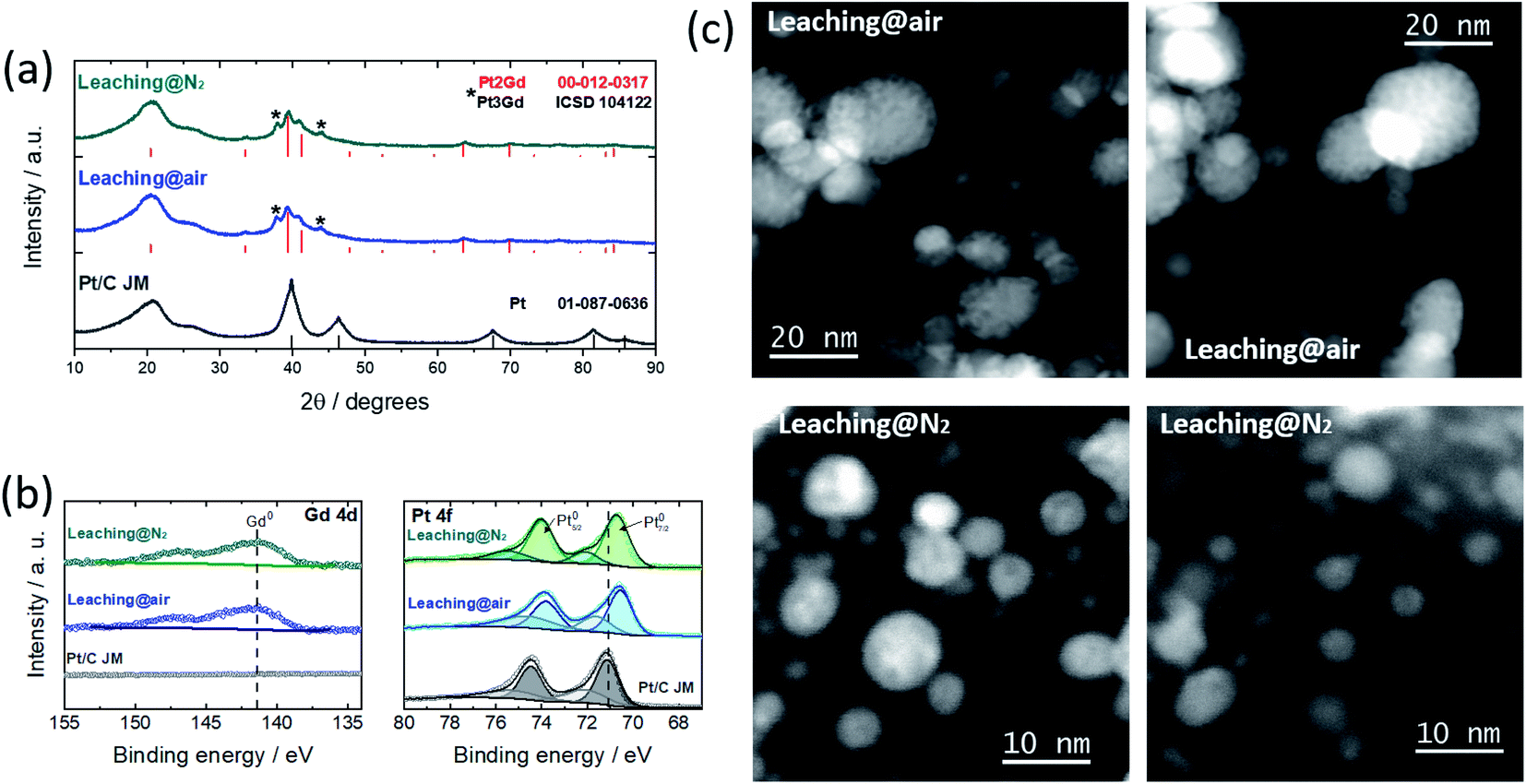 | ||
| Fig. 1 (a) Powder XRD patterns (b) Gd 4d and Pt 4f photoemission lines; and (c) high-angle annular dark field STEM micrographs of the PtxGd/C catalysts washed under air- or N2-atmosphere. 40% Pt/C from Johnson-Matthey is used as reference in (a) and (b). | ||
The metal content and Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Gd ratio, in the bulk and at the near-surface level, were estimated using ICP-MS and XPS, Table 1, and a slightly higher Pt:Gd ratio was observed after the leaching under N2-atmosphere. Moreover, the TEM micrographs shown in Fig. S2† reveal that, regardless of the leaching treatment, a bimodal particle size distribution is observed, mean values of which have the same magnitude (Table 1). The STEM micrographs depicted in Fig. 1c, however, show that porous nanoarchitectures are formed through the leaching under air atmosphere. We have reported that the nanopore formation initiates where Gd is relatively abundant in the particle,11 and its dissolution can proceed until Pt is sufficiently available to form a protective layer and stabilise the structure. This effect is pronounced in NPs with sizes> 10 nm.11 Although the population of these structures is attenuated with the wash under N2-atmosphere, in some regions we have observed porous NPs (Fig. S3†). Therefore, the acid wash under N2-atmosphere substantially modulates (but does not impede) the formation of porous NPs. Gan et al.14 have demonstrated that for dealloyed PtNi3 NPs, the absence of oxygen decreases the dissolution rate of the less noble metal; thus, the Pt surface diffusion is fast enough to stabilise the structure and prevent the formation of nanoporosity.
:Gd ratio, in the bulk and at the near-surface level, were estimated using ICP-MS and XPS, Table 1, and a slightly higher Pt:Gd ratio was observed after the leaching under N2-atmosphere. Moreover, the TEM micrographs shown in Fig. S2† reveal that, regardless of the leaching treatment, a bimodal particle size distribution is observed, mean values of which have the same magnitude (Table 1). The STEM micrographs depicted in Fig. 1c, however, show that porous nanoarchitectures are formed through the leaching under air atmosphere. We have reported that the nanopore formation initiates where Gd is relatively abundant in the particle,11 and its dissolution can proceed until Pt is sufficiently available to form a protective layer and stabilise the structure. This effect is pronounced in NPs with sizes> 10 nm.11 Although the population of these structures is attenuated with the wash under N2-atmosphere, in some regions we have observed porous NPs (Fig. S3†). Therefore, the acid wash under N2-atmosphere substantially modulates (but does not impede) the formation of porous NPs. Gan et al.14 have demonstrated that for dealloyed PtNi3 NPs, the absence of oxygen decreases the dissolution rate of the less noble metal; thus, the Pt surface diffusion is fast enough to stabilise the structure and prevent the formation of nanoporosity.
| Sample | Metal content (% wt) | Pt:Gd ratio (ICP-MS) |
Pt:Gd ratio (XPS) |
Mean particle size (nm) |
|---|---|---|---|---|
| Leaching@air | Pt: 27.2 | Pt4.7Gd | Pt8.3Gd | 7.8 ± 1.2 |
| Gd: 4.2 | 14.1 ± 2.6 | |||
| Leaching@N2 | Pt: 26.5 | Pt4.5Gd | Pt7.8Gd | 7.6 ± 1.4 |
| Gd: 4.6 | 14.9 ± 2.8 |
On the other hand, DFT screenings have suggested that the presence of oxygen induces non-precious metal surface segregation in Pt-based alloys.17 Our results reflect such an effect.
The surface electrochemistry was examined in 0.1 M HClO4 at the beginning of the accelerated degradation test (BoT), cf.Fig. 2a and S4,† when the typical Pt signals in acid electrolyte were acquired. The Hupd region of the PtxGd/C materials present different shapes with respect to Pt/C. This observation supports the change in the Pt–H interactions coming from the alloying effect.18 While such Pt signals seem to be unaltered at the end of the test (EoT) for the PtxGd/C materials, the voltammogram of Pt/C shows that this catalyst underwent evident degradation (Fig. S4†).
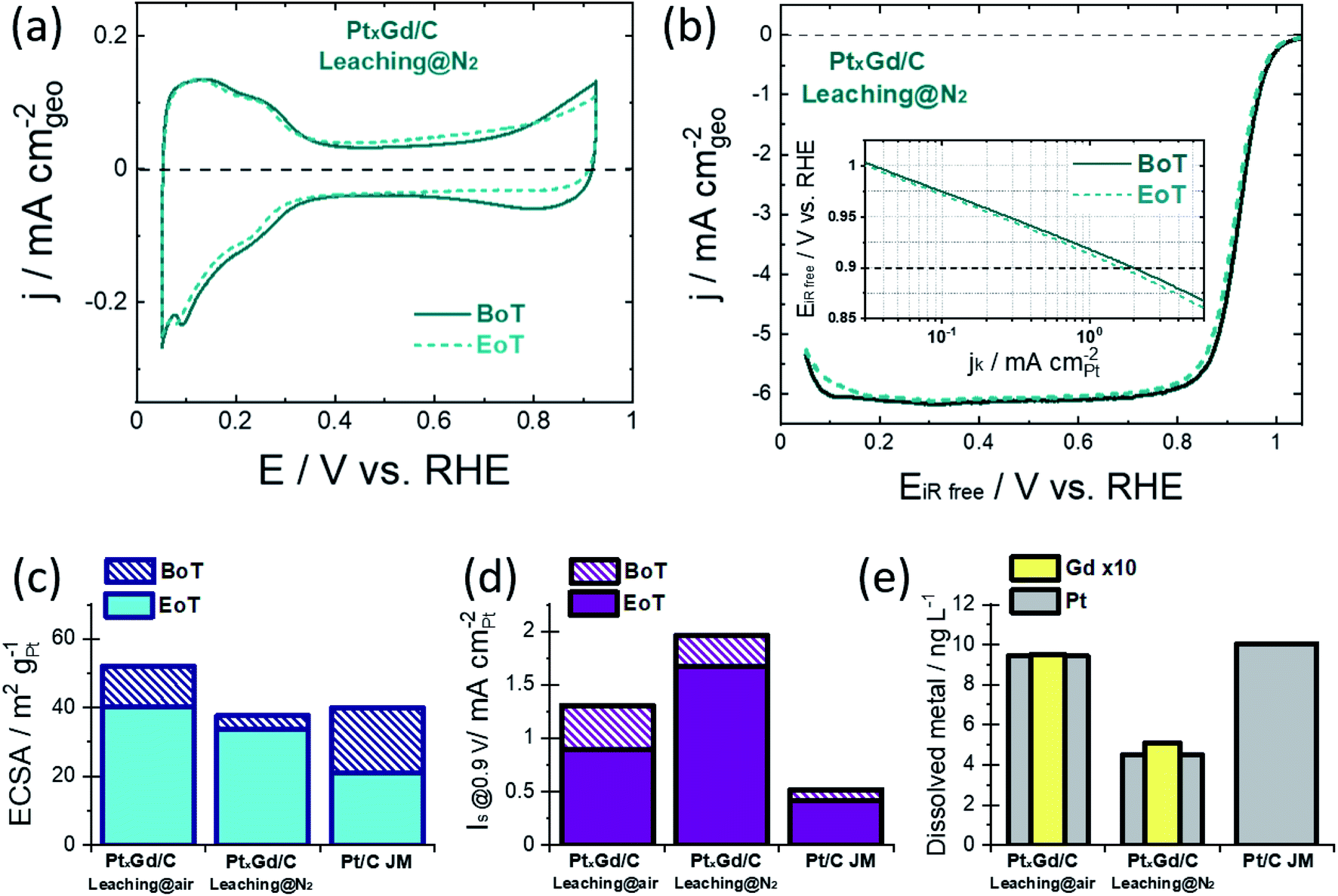 | ||
| Fig. 2 (a) Cyclic voltammograms (N2-saturated 0.1 M HClO4 at 20 mV s−1); (b) ORR polarisation curves (O2-saturated 0.1 M HClO4 at 20 mV s−1 and 1600 rpm) at BoT and EoT (the inset shows the iR-free electrode potential vs. mass-corrected kinetic current normalized by Pt surface); (c) ECSA values at BoT and EoT; (d) specific activity at 0.9 V at BoT and EoT; and (e) dissolved Pt and Gd in the electrolyte of the PtxGd/C catalysts washed under air- or N2-atmosphere. 40% Pt/C from Johnson-Matthey is used as reference in (c)–(e). | ||
The ECSA values were determined by the CO-stripping method (Fig. S5†). One can notice that, for both PtxGd/C materials at the BoT and EoT, there are two CO-oxidation processes, which are related to the mix of agglomerated particles and small NPs.19 The TEM analysis shown in Fig. S2† supports this argument. Moreover, the relationship between the integrated charge (Q) for the Hupd desorption and the CO-stripping process has been considered as an indicator for the surface structural and electronic properties.18Table 2 reports this parameter for the electrocatalysts under comparison. For Pt/C, the QCO/2QHupd is close to 1, in concordance with previous studies.18 In the case of both PtxGd/C materials, the higher QCO value with respect to 2QHupd indicates the well-known changed adsorption properties for Pt alloys.3,18
| Sample | Q CO/2QHupd | ECSACO (m2 gPt−1) | I s @ 0.9 V (mA cmPt−2) | I m @ 0.9 V (A mgPt−1) | Dissolved Pt (ng L−1) | Dissolved Gd (ng L−1) |
|---|---|---|---|---|---|---|
| Leaching@air | 1.09 | 52.01 | 1.31 | 0.68 | 9.45* | 95.24* |
| 1.06* | 40.18* | 0.89* | 0.36* | |||
| Leaching@N2 | 1.12 | 37.70 | 1.96 | 0.72 | 4.51* | 50.07* |
| 1.10* | 33.75* | 1.67* | 0.59* |
Besides, the QCO/2QHupd values for both PtxGd/C materials suggest the formation of the Pt-skeleton structure (0.94 < QCO/2QHupd < 1.5),20 which is maintained at the EoT. We have previously reported the formation of the Pt overlayer in this system, which supports this observation.11
Notably, the ORR polarisation curves reveal a higher activity retention of the PtxGd/C acid-washed under N2-atmosphere, cf.Fig. 2b and S4.† The ECSA and specific-surface activity, at the BoT and at EoT, are shown in Fig. 2c and d. The Pt and Gd dissolution occurring during the degradation protocol was estimated by ex situ ICP-MS measurements of the electrolyte at the EoT, cf.Fig. 2d. Such kinetic parameters are shown in Table 2. In terms of ECSA, the higher value of the air-leached material reflects the higher population of porous nanoarchitectures which, at the EoT, experience a considerable degradation as confirmed by the higher concentration of Gd dissolved into the electrolyte at EoT.
This undesirable degradation is suppressed in the solid NPs produced by the N2-atmosphere leaching. In terms of the ORR intrinsic activity, i.e., the specific activity Is evaluated at 0.9 V, both PtxGd/C electrocatalysts outperform the reference Pt/C material. However, the N2-leached material is characterised by a higher Is, demonstrating that the solid NPs present higher intrinsic activity. Such an effect could be related to the higher Gd concentration at the near-surface region, which induces a stronger strain/ligand effects on the Pt overlayer, boosting the ORR kinetics.
At the EoT, the N2-leached PtxGd/C retained ca. 85% of its initial Is, whereas the air-leached PtxGd/C retained ca. 67%. The higher Gd dissolution could weaken the strain/ligand effect for the air-leached PtxGd/C; thus, the solid NPs confer higher intrinsic ORR activity and stability.
Conclusions
The activity/stability of carbon-supported PtxGd nanoalloys has been improved by minimising the formation of porous nanoarchitectures. This feature was achieved by performing the dealloying process under N2-atmosphere in order to avoid the O2-induced Gd segregation, and favour the stabilisation of solid NPs. Such NPs present higher intrinsic ORR activity and, more important, a notably higher stability. The results in this work might be a baseline to produce high active/stable Pt–REM/C nanoalloys.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. C. A. Campos-Roldán, investigation, methodology, data curation, writing – original draft. F. Pailloux HAADF-STEM experiments, writing – review & editing. D. J. Jones, funding acquisition, conceptualization, writing – review & editing. P.-Y. Blanchard, supervision, writing – review & editing. J. Rozière, conceptualization, writing–review & editing. S. Cavaliere, funding acquisition, conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme/ERC Grant Agreement SPINAM no. 306682 and from the European Union's Horizon 2020 research and innovation programme, Fuel Cells and Hydrogen 2 Joint Undertaking under the GAIA project, Grant Agreement No. 826097. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation programme, Hydrogen Europe and Hydrogen Europe Research. SC acknowledges IUF for financial support.References
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Gronbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 CrossRef CAS.
- A. Velázquez-Palenzuela, F. Masini, A. F. Pedersen, M. Escudero-Escribano, D. Deiana, P. Malacrida, T. W. Hansen, D. Friebel, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2015, 328, 297–307 CrossRef.
- M. Escudero-Escribano, P. Malacrida, M. Hansen, U. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. Stephens and I. Chorkendorff, Science, 2018, 352, 73–76 CrossRef.
- A. F. Pedersen, E. T. Ulrikkeholm, M. Escudero-Escribano, T. P. Johansson, P. Malacrida, C. M. Pedersen, M. H. Hansen, K. D. Jensen, J. Rossmeisl, D. Friebel, A. Nilsson, I. Chorkendorff and I. E. L. Stephens, Nano Energy, 2016, 29, 249–260 CrossRef CAS.
- J. N. Schwämmlein, G. S. Harzer, P. Pfändner, A. Blankenship, H. A. El-Sayed and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, J3173–J3185 CrossRef.
- S. G. Peera, T. G. Lee and A. K. Sahu, Sustainable Energy Fuels, 2019, 3, 1866–1891 RSC.
- T. Chu, M. Xie, D. Yang, P. Ming, B. Li and C. Zhang, Int. J. Hydrogen Energy, 2020, 45, 27291–27298 CrossRef CAS.
- B. Eriksson, G. Montserrat-Sisó, R. Brown, T. Skála, R. Wreland Lindström, G. Lindbergh, B. Wickman and C. Lagergren, Electrochim. Acta, 2021, 387, 138454 CrossRef CAS.
- Y. Hu, J. O. Jensen, L. N. Cleemann, B. A. Brandes and Q. Li, J. Am. Chem. Soc., 2020, 142, 953–961 CrossRef CAS.
- Y. Hu, J. O. Jensen, P. Norby, L. N. Cleemann, F. Yang and Q. Li, Chem. Mater., 2021, 33, 535–546 CrossRef CAS.
- C. A. Campos-Roldán, F. Pailloux, P.-Y. Blanchard, D. J. Jones, J. Rozière and S. Cavaliere, ACS Catal., 2021, 13519–13529, DOI:10.1021/acscatal.1c02449.
- C. Baldizzone, L. Gan, N. Hodnik, G. P. Keeley, A. Kostka, M. Heggen, P. Strasser and K. J. J. Mayrhofer, ACS Catal., 2015, 5, 5000–5007 CrossRef CAS.
- A. K. Schuppert, A. Savan, A. Ludwig and K. J. J. Mayrhofer, Electrochim. Acta, 2014, 144, 332–340 CrossRef CAS.
- L. Gan, M. Heggen, R. O'Malley, B. Theobald and P. Strasser, Nano Lett., 2013, 13, 1131–1138 CrossRef CAS PubMed.
- C. A. Campos-Roldán, G. Ramos-Sanchez, R. G. Gonzalez-Huerta, J. R. Vargas Garcia, P. B. Balbuena and N. Alonso-Vante, ACS Appl. Mater. Interfaces, 2016, 8, 23260–23269 CrossRef.
- S. J. Hwang, S. K. Kim, J. G. Lee, S. C. Lee, J. H. Jang, P. Kim, T. H. Lim, Y. E. Sung and S. J. Yoo, J. Am. Chem. Soc., 2012, 134, 19508–19511 CrossRef CAS.
- M. Sarwar, J. L. Gavartin, A. Martinez Bonastre, S. Garcia Lopez, D. Thompsett, S. C. Ball, A. Krzystala, G. Goldbeck and S. A. French, Phys. Chem. Chem. Phys., 2020, 22, 5902–5914 RSC.
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., Int. Ed. Engl., 2012, 51, 3139–3142 CrossRef CAS.
- C. A. Campos-Roldán and N. Alonso-Vante, J. Solid State Electrochem., 2021, 25, 187–194 CrossRef.
- R. Chattot, I. Martens, M. Scohy, J. Herranz, J. Drnec, F. Maillard and L. Dubau, ACS Energy Lett., 2019, 5, 162–169 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, materials synthesis and characterisation, and complementary XRD, TEM, STEM and electrochemical results. See DOI: 10.1039/d1na00740h |
| This journal is © The Royal Society of Chemistry 2022 |