
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the six stages of the current–time curve and the bilayer nanotubes obtained by one-step anodization of Zr
Pengze
Li†
a,
Heng
Wang†
a,
Yilin
Ni
a,
Ye
Song
 a,
Ming
Sun
*b,
Tianle
Gong
a,
Chengyuan
Li
a and
Xufei
Zhu
*a
a,
Ming
Sun
*b,
Tianle
Gong
a,
Chengyuan
Li
a and
Xufei
Zhu
*a
aKey Laboratory of Soft Chemistry and Functional Materials of Education Ministry, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: zhuxufei.njust@vip.163.com
bSchool of Electronic and Optical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: msun@njust.edu.cn
First published on 6th December 2021
Abstract
The application and growth mechanism of anodic TiO2 nanotubes have been a hot topic in the last ten years, but the formation mechanism of anodic ZrO2 nanotubes has rarely been studied. In one-step constant voltage anodization of Al and Ti, the typical current–time curve has three stages. Moreover, the current–time curves of the three stages can last for 10 min or even 10 hours, resulting in a single layer of nanotubes with the same diameter due to the constant voltage in one-step anodization. However, in this paper, it was found for the first time that the three stages of the current–time curve appeared twice in succession during one-step constant voltage anodization of Zr for only 900 seconds, and bilayer nanotubes with increased diameter were obtained. This six-stage current–time curve cannot be explained by classical field-assisted dissolution and field-assisted flow or stress-driven mechanisms. Here, the formation mechanism and growth kinetics of bilayer ZrO2 nanotubes have been clarified rationally by the theories of ionic current, electronic current and oxygen bubble mold. The interesting results presented in this paper are of great significance for revealing the anodizing process of various metals and the formation mechanism of porous structures.
1. Introduction
Electrochemical anodization and electrochemical synthesis of aluminum, titanium, zirconium and other metals have attracted more and more attention.1–6 Porous anodic alumina (PAA), anodic TiO2 and ZrO2 nanotube structures are widely used in solar cells, supercapacitors and various sensors.7–11 However, the formation mechanism of the porous structure and nanotube structure of this kind of porous anodic oxide (PAO) is very controversial.1,12–18 Both formation mechanisms of PAA and anodic TiO2 nanotubes include the field-assisted dissolution (FAD), dissolution equilibrium, and field-assisted ejection (FAE) theories and field-assisted flow (FAF) model.1,19–24 There are currently two opposite models of pore formation in these PAOs. The first model, digging holes from the oxide surface into the oxide interior, is called the classical FAD theory or dissolution equilibrium theory.19,25 The FAD reaction to form anodic TiO2 nanotubes is TiO2 + 6F− + 4H+ → [TiF6]2− + 2H2O.19–21 It is well known that the anodizing current–time curve shows three stages under constant voltage anodizing conditions in fluoride-containing electrolyte forming TiO2 nanotubes.25–28 The current drops rapidly in the first stage, rises slowly in the second stage, and remains basically stable for a long time in the third stage.25,26 Since anodization is an electrochemical process, there are many factors that determine the final length of nanotubes, including electrolyte composition, temperature, anodizing time, voltage level and anodizing current.25–27 The influence of these factors on the length of nanotubes is reflected in the anodizing current. Therefore, the length of the nanotubes should be related to the size of the anodizing current or the amount of charge in the electrochemical reaction. However, the traditional FAD and FAF theories have nothing to do with the anodizing current or the amount of charge. The greatest limitation of the FAD and FAF theories is that the dissolution reaction and FAF model have nothing to do with the anodizing current and cannot explain the physical and chemical significance of the three stages of the current–time curve.16–18,21 Although dissolution current and hydrolyzing current have been proposed,29,30 in fact, as Thompson et al. pointed out, the above FAD reactions occur only on the oxide surface and do not contribute to the anodizing current across the oxide film.31 The FAD theory proposed by the Manchester group was rejected by themselves in 2013 and 2015.32,33 The growth of the major pores proceeds mainly due to the field-assisted flow of oxide rather than field-assisted dissolution.32,33 They showed that the oxide dissolution rate was in fact very small (less than 1 nm min−1).32,33 Yu et al. showed that the growth rate of porous alumina channels was 150 nm min−1.20 Even though many facts deny the FAD theory and dissolution equilibrium theory,32–43 many researchers still ambiguously explain various experimental phenomena by applying the FAD and dissolution equilibrium theories.44–47The second model is an oxygen bubble model based on the viscous flow model48–50 and the electronic current theory.51–54 The growth manner of the nanotubes is the viscous flow of the barrier oxide layer around the oxygen bubble model to promote the growth of the nanotubes.53–57 Many researchers concluded that the viscous flow is contrary to the expectations of the FAD model of pore formation.1,29,41,48 Many researchers actually accept the above two opposite manners simultaneously. Meanwhile, others accept the viscous flow model, but deny the electronic current and oxygen bubble mold, resulting in the vague dynamics of both the FAD and FAF models and the field-assisted effect.44–51
Compared with the above two models, the growth kinetics of field-assisted dissolution equilibrium theory has been ambiguous for decades.45–51 The physical meanings of FAD and FAF are always unclear.15–20 After discussing the relationship between the volume (or length) of the porous oxide and the quantity of electric charge (the product of electric current and time),24,36,43 the growth kinetics of the oxygen bubble model becomes clearer. That is, the ionic current causes barrier oxide growth, and the electronic current causes oxygen evolution. The barrier oxide grows upward around the oxygen bubble mold to form the nanotubes. Therefore, ionic current and electronic current are the driving forces for the growth of TiO2 nanotubes, rather than the FAD, FAF and stress-driven flow.1,22–24 Pashchanka considered that in the self-organizing systems of PAA, the electroconvective ion transport mechanism prevails and helps overcome the diffusive transport limitations.58 The growth of pores is assisted by the Coulomb forces that are responsible for the motion of electrolyte anions and entrained fluids toward the anode surface.58 The process of self-organization on anodized aluminum is rather complex and multifaceted, and its numerous unique aspects still need to be clarified.58
Compared with anodic TiO2 nanotubes, the formation mechanism of anodic ZrO2 nanotubes is less studied, and most groups directly use the FAD reaction (ZrO2 + 6F− + 4H+ → [ZrF6]2− + 2H2O).59–61 Compared with the anodizing process of Al and Ti, the current–time curve of zirconium under constant voltage is more complicated. There are two-stage and three-stage current–time curves in the anodizing process of zirconium.28,59–61 In this paper, six stages of the current–time curves and bilayer nanotubes were found in a one-step anodizing process of zirconium. To the best of our knowledge, the bilayer nanotube structure and the six stages of the current–time curves are reported for the first time. This paper presents a new interpretation of the original structure and the six stages of the current–time curve.
2. Experimental details
Zirconium foil (0.1 mm thick with a purity of 99.5%) was cut into 1.0 cm × 8.0 cm samples. The samples were polished in a mixed solution (with HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3:H2O = 1:1:2 by volume) for 5 to 10 seconds. The polished samples were immediately rinsed five times in a 2000 mL beaker of water, and then immersed in a 1000 mL beaker full of deionized water for about 30 min. After being taken out of the beaker, the samples were dried with an electric blow dryer. Then insulating tape was affixed to the Zr foil so that the anodized area was 4 cm2 (1 cm × 2.0 cm× 2 sides).
:HNO3:H2O = 1:1:2 by volume) for 5 to 10 seconds. The polished samples were immediately rinsed five times in a 2000 mL beaker of water, and then immersed in a 1000 mL beaker full of deionized water for about 30 min. After being taken out of the beaker, the samples were dried with an electric blow dryer. Then insulating tape was affixed to the Zr foil so that the anodized area was 4 cm2 (1 cm × 2.0 cm× 2 sides).
The electrolyte for zirconium anodization was a glycerol solution containing 1.0 wt% NH4F and 3.5 wt% H2O. The electrolyte was ultrasonically stirred for 60 min to ensure that the NH4F was completely dissolved. A 500 mL plastic beaker filled with approximately 450 mL of electrolyte served as a container for anodization. Zr foil with insulating tape was used as the anode, and a graphite plate about 15 mm wide and 100 mm long was used as the cathode, with a distance of about 2 cm between the anode and the cathode. The anodizing container was placed in a circulating cooling pump to maintain the temperature of the electrolyte at 20 ± 1 °C for the entire anodizing process. The electrolyte was stirred magnetically throughout the anodizing process. Each sample was anodized for 900 seconds at constant voltages of 50 V, 70 V and 80 V. The current–time curve of anodization was recorded automatically by a computer system.57 Each experiment with the same voltage was repeated three times to ensure that the anodizing current curves tested were basically coincident.
After the anodized sample was removed from the electrolyte, it was immediately rinsed five times in a 2000 mL beaker of water, and then immersed in a 1000 mL beaker full of deionized water for about 60 min. Finally, all the samples were dried in a drying oven at 50 °C for 2 hours. When preparing the SEM samples, all zirconium sheets with zirconium oxide film were bent artificially into an Ω shape so that the cross-section morphology of the zirconium oxide film can be observed by FESEM (Zeiss Supra 55).
3. Results and discussion
Fig. 1 shows the SEM images of the morphology of the nanotubes and current–time curve obtained by anodizing zirconium in glycerol electrolyte containing 1.0 wt% NH4F and 3.5 wt% H2O at a constant voltage of 50 V for 900 seconds. The anodic ZrO2 nanotubes and current–time curve shown in Fig. 1 are very similar to those of anodic TiO2 nanotubes.28,57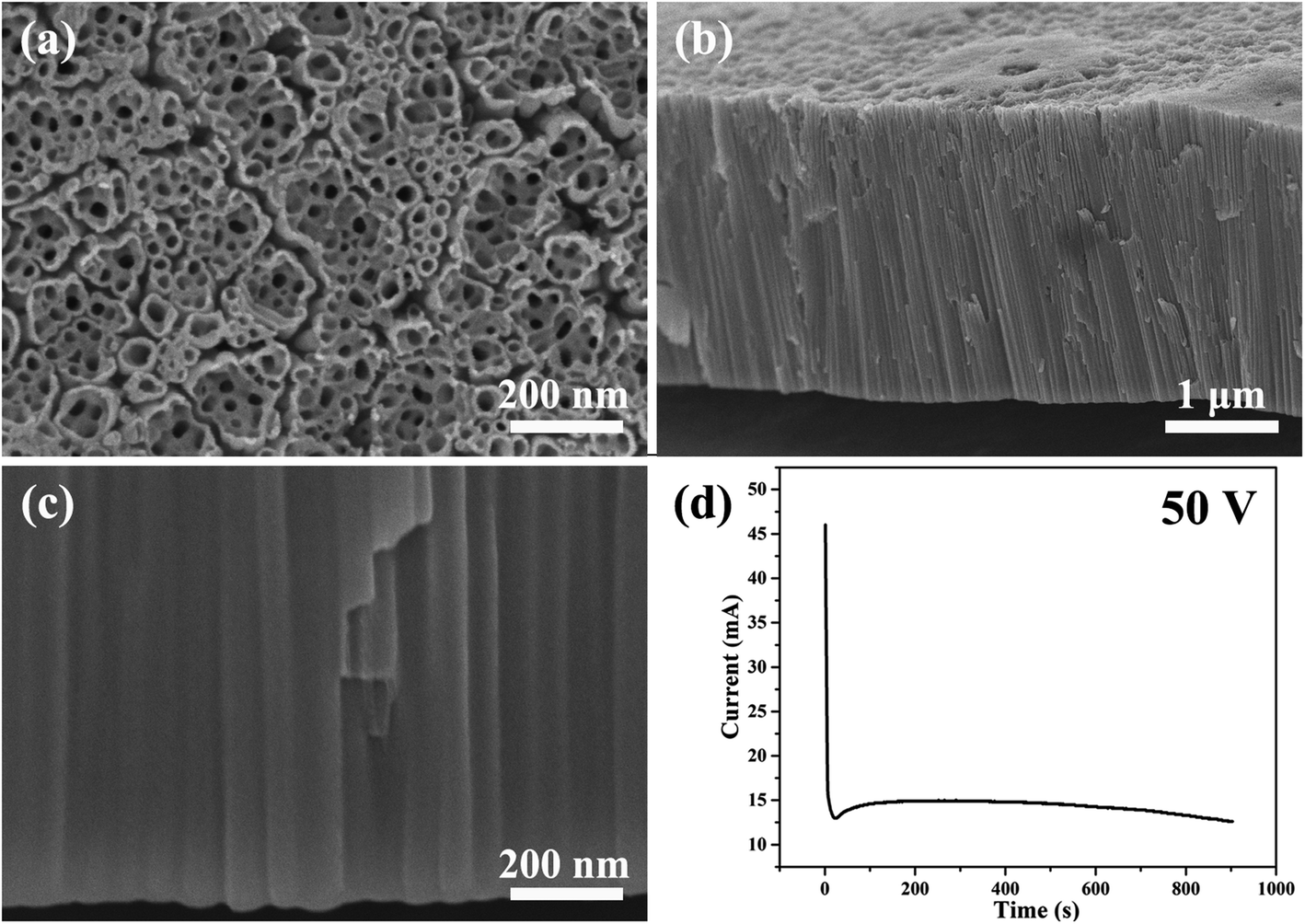 | ||
| Fig. 1 (a–c) The SEM images of anodic ZrO2 nanotubes anodized at a constant voltage of 50 V for 900 s. (d) The corresponding current–time curve over 900 s. | ||
Compared with anodic TiO2 nanotubes, the formation mechanism of anodic ZrO2 nanotubes is also ascribed to the FAD reaction (ZrO2 + 6F− + 4H+ → [ZrF6]2− + 2H2O).59–61Fig. 1d shows the three stages of the current–time curve which are completely consistent with the anodizing process of titanium. In order to explain the current curve in maintaining the basic stability state, Schmuki and Regonini et al. indicated that the steady state appears in the current–time curve because of the dissolution equilibrium model.17,18 That is, the rate of oxide formation at the metal/oxide interface and the rate of field-assisted dissolution at the electrolyte/oxide interface are equal.17–19 The argument that the growth rate of oxide is equal to the dissolution rate has been rejected by Thompson et al.31 The dissolution rate of the oxide is, in fact, several orders of magnitude smaller than the formation rate in the steady-state.31
In fact, this dissolution equilibrium theory was negated by the Skeldon and Hebert groups.32,33 They proved that the field-assisted dissolution rate was very low (less than 1 nm min−1) in PAA.32,33 Thompson et al. indicated that direct measurement of the field-dependent oxide dissolution rate was never achieved.31 Zhang and Yu et al. proved that there is no dissolution equilibrium in the anodizing process of titanium.34,41–43 The growth rate of titanium oxide nanotubes reaches 200 nm min−1, which is much higher than the dissolution rate of titanium oxide in electrolyte (about 1 nm min−1).62 Many references above have negated the FAD dissolution equilibrium, and the three stages of the current–time curve in Fig. 1d could not be explained with FAD and FAF theories; this is the greatest limitation of FAD and FAF theories so far and needs to be solved urgently.
Fig. 2 and 3 show the SEM images of the morphology of the nanotubes and current–time curves obtained by anodizing zirconium in glycerol electrolyte containing 1.0 wt% NH4F and 3.5 wt% H2O at constant voltages of 70 V and 80 V for 900 seconds, respectively. It is interesting to note that as the voltage of one-step anodization increases, the three stages of the current–time curves in Fig. 1d change to six stages of the current–time curves in Fig. 2d and 3e. The same test conditions were applied three times and the same six-stage curves were obtained. To the best of our knowledge, this phenomenon has never been found in the one-step anodization of Al and Ti. In addition, it can be seen that the nanotubes also become bilayer nanotubes, as shown in Fig. 2b, c and 3b–d.
 | ||
| Fig. 2 (a–c) The SEM images of anodic ZrO2 nanotubes anodized at a constant voltage of 70 V for 900 s. (d) The corresponding current–time curve over 900 s. | ||
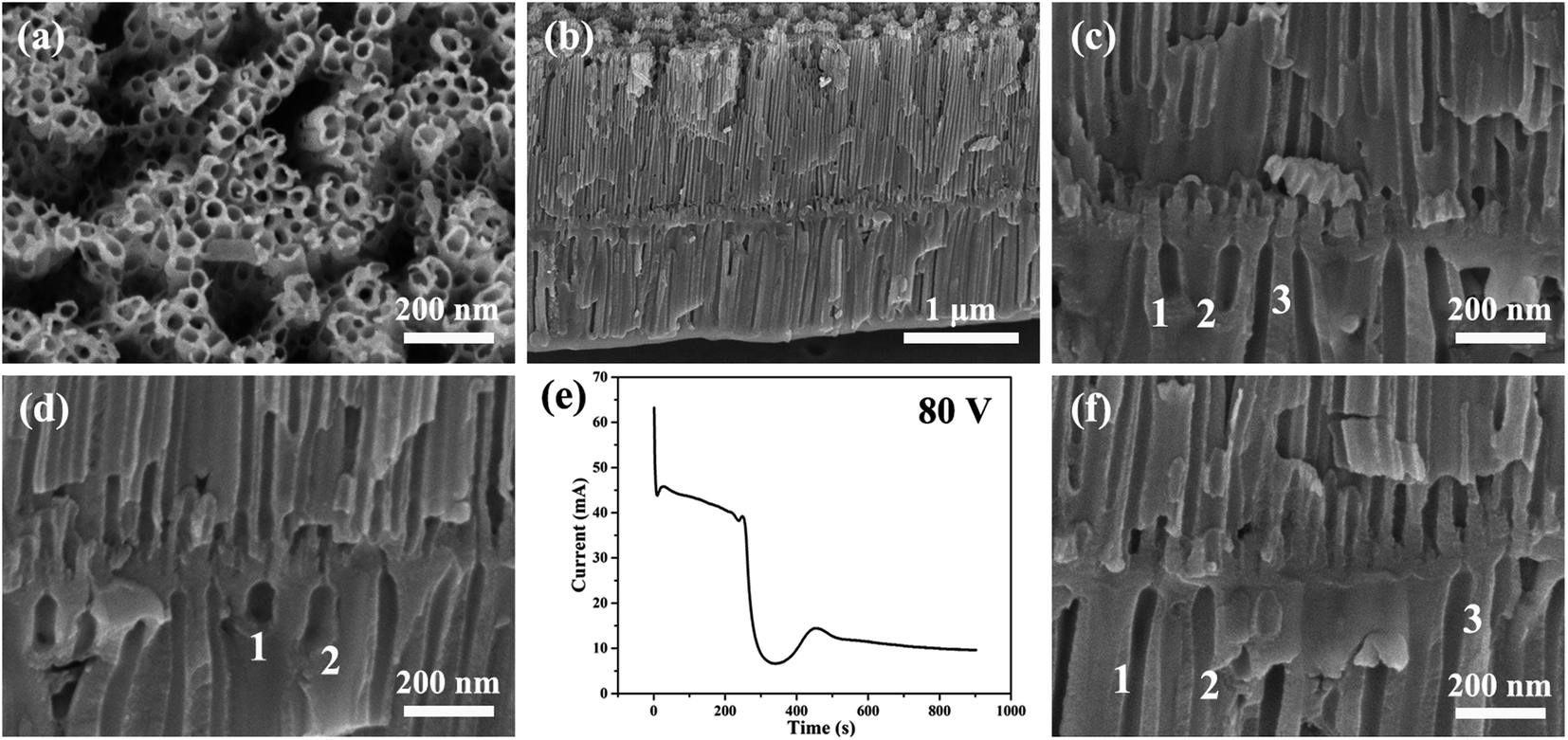 | ||
| Fig. 3 (a–d, f) The SEM images of anodic ZrO2 nanotubes anodized at a constant voltage of 80 V for 900 s. (e) The corresponding current–time curve over 900 s. | ||
As can be seen from Fig. 3c, d and f, the second layer of nanotubes is not fully penetrated by the first layer of nanotubes, and the inner diameter of the second layer of nanotubes is significantly larger than that of the first layer of nanotubes. This means that the number of nanotubes in the second layer is much smaller than the number of nanotubes in the first layer. This proves that the digging manner of the FAD reaction of nanotube formation is not true as shown by the channels of no. 1, 2, and 3 in Fig. 3c.
Obviously, the FAD and FAF theories cannot explain the three-stage curve, let alone the six-stage curve, because there is no connection between the FAD reaction and the anodizing current.31–35 Here, based on the electronic current and the oxygen bubble mold, the six-stage curve and the bilayer nanotube structure discovered for the first time are analyzed in Fig. 4.
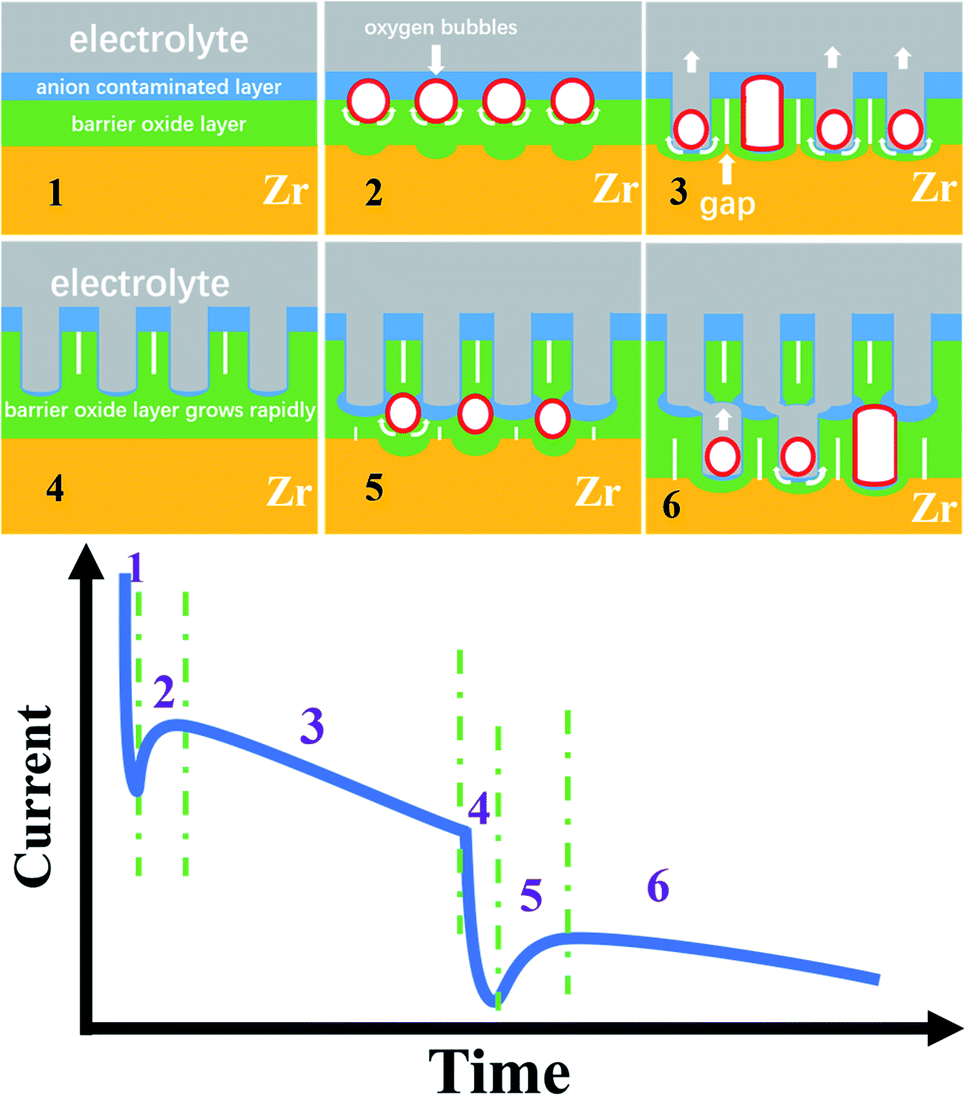 | ||
| Fig. 4 Schematic diagram of the three stages and six stages of growth of anodic ZrO2 nanotubes. | ||
In recent years, many groups have proven the existence of ionic current and electronic current in the metal anodizing process, including the famous Schmuki and Skeldon groups.52–56,63 It is well known that the ionic current Jion may be expressed simply as Jion = Aexp(BUapp/d), where A and B are constants that depend on temperature and electrolyte composition, Uapp is the applied voltage (here, Uapp = 50 V, 70 V, and 80 V), and d is the thickness of the barrier oxide layer as shown in Fig. 4.52–57 Albella et al. proposed that the theoretical expression of electronic current Je is Je = j0exp(αd),52 where α is the impact ionization coefficient of the avalanche, d is the same as above, and j0 is the primary electronic current.52 It is obvious from the above two formulas that the changing trend of ionic current and electronic current with the thickness of the barrier oxide layer is just opposite.54–57 Therefore, the ionic current shows a downward trend in the current–time curve, while the electronic current shows an upward trend in the current–time curve.57
Fig. 4 shows the schematic diagram of the three-stage and six-stage growth of anodic ZrO2 nanotubes. During the normal anodization of Al, Ti, Zr and Ta, the current–time curves all show three stages as shown in Fig. 1d.15–21,64
In stage 1 of Fig. 4, the high ionic current causes ion migration to form a barrier oxide layer (Ti4+ + 2O2− → TiO2), and the rapid growth of the barrier oxide layer leads to a sharp decrease in ionic current. Due to the action of the electric field, the anions in the electrolyte are adsorbed on the oxide surface of the anode, and then the part of the barrier oxide layer near the electrolyte is quickly converted into an anion contaminated layer.52–55 The loss of electrons of anions in the anion contaminated layer is the cause of the generation of electronic current. In fact, the anodizing process is an alternating process dominated by ionic current and electronic current.52–55 The Jion decreases exponentially with the thickness of the barrier oxide layer (Jion = Aexp(BUapp/d)), and the electronic current (Je) increases exponentially with the thickness of the barrier oxide layer (Je = j0exp(αd)). In 1969, Diggle et al. concluded that in the high electric field, the ionic conductance dominates the anodizing process leading to oxide formation; in the low electric field, electronic conductance dominates the anodizing process leading to oxide dielectric breakdown and intense oxygen release on the oxide surface.51 Therefore, Macak et al. suggested that there is an important relationship between the formation of nanotube holes and the oxide breakdown.65 In fact, the breakdown process of oxide is a process in which the increase of electronic current leads to the release of a large amount of oxygen gas.66–68 However, Diggle et al. proposed that the incorporation of anions into the oxide structure is closely associated with the degree of pore formation,51 being the greatest for pore-forming electrolytes, and the least for dense-type electrolytes.51 In other words, the thicker the anion contaminated layer is, the more likely it is to produce pores or nanotubes, and the thinner the anion contaminated layer is, the more likely it is to produce a dense film.25 This theory of nanotube formation beneath the anion contaminated layer,69 which is completely contrary to field-assisted dissolution theory, has been confirmed by gourd-shaped TiO2 nanotube embryos.41
Based on the above discussion, the three conditions for the formation of a nanotube embryo in Fig. 4 are that (1) enough electronic current allows enough oxygen gas to be released; (2) sufficient thickness of the anion contaminated layer ensures that the oxygen bubbles form a bubble mold; and (3) sufficient ionic current causes the newly grown oxide to flow upward around the oxygen bubble mold.
At the end of stage 1 (inflexion), the ionic current decreases to the critical value and the barrier oxide layer increases to a critical thickness. At this critical thickness, a large enough electronic current can maintain the continuous release of oxygen bubbles and have a mold effect. This critical thickness is inversely proportional to the concentration of anions in the electrolyte.57,70
In stage 2 of Fig. 4, enough electronic current causes the anions to lose electrons and transform into oxygen gas more easily (2O2− → O2 (gas) + 4e, or 4OH− → O2 (gas) + 2H2O + 4e).53–55 At the interface of the anion contaminated layer and barrier oxide layer, the oxygen bubble gradually grows into a gas mold. The location of oxygen bubble molds at this interface has been confirmed by double-walled nanotubes.69 Due to the pressure of the anion contaminated layer and external atmospheric pressure, the oxygen bubbles cannot spill over into the electrolyte outside. The oxygen bubble mold under the anion contaminated layer causes the barrier oxide around the bubble mold to grow as shown in stage 2 of Fig. 4. The viscous flow of the barrier oxide grows around the oxygen mold to form the initial nanotube embryo. This gourd-shaped, hollow nanotube embryo formed from the oxygen bubble mold has been discovered in 2021.41 The formation of the hollow nanotube embryos is similar to the hollow molding process of mineral water bottles of PET (polyethylene terephthalate). The plastic or viscous flow of the PET melt, under the action of inner compressed air (inflation pressure 5–8 atm), eventually forms a cylindrical or cube shaped bottle molded by the outer steel cavity mold. The oxygen bubbles in the anodizing process act as gas molds just like the inner compressed air in the molding process of mineral water bottles.
In stage 3 of Fig. 4, the oxygen bubbles escape from the nanotube embryo, and the outer electrolyte enters the bottom of the nanotubes. The barrier oxide at the bottom of the nanotubes partly turns into the anion contaminated layer, creating electronic current and oxygen bubbles at the nanotube bottom. Sufficient ionic current and enough electronic current across the nanotube base are the driving force for nanotube embryo formation and further development.53–55 When the outside electrolyte enters the bottom of the nanotube, the thickness of the barrier oxide layer at the bottom remains roughly the same, and both Je and Jion are at a constant level because both Je and Jion are related to the thickness (Jion = Aexp(BUapp/d), Je = j0exp(αd)). In this stage 3, a steady electronic current easily causes oxygen bubbles to form the mold effect at the nanotube bottom, because the thickness of the anion contaminated layer is thinner than that in stage 1. A stable ionic current causes a new barrier oxide layer to grow upward around the oxygen bubble mold, resulting in upward growth of the nanotube walls.53–57 In general, in one-step constant voltage anodization, the current–time curve of the three stages is maintained continuously as shown in Fig. 1d, and the monolayer nanotube continues to grow under the action of two stable currents, and the diameter of the monolayer nanotube will remain constant as shown in Fig. 1b and c. However, the current–time curve of the six stages shown in Fig. 3e has not been reported. The formation mechanisms of the six-stage curve and the second layer of nanotubes are discussed in detail below.
In stage 4 of Fig. 4, the declining trend of anodizing current is almost exactly the same as that in stage 1, indicating that in stage 4, a new barrier oxide layer is rapidly formed at the base of the first layer nanotubes. In other words, the rapid growth of this barrier oxide layer leads to a rapid decline of ionic current in stage 4. From the morphology, the barrier oxide layer in stage 4 is thicker than that at the bottom of the nanotubes in the first three stages (stages 1, 2, and 3). In fact, it is evident in Fig. 3c and d that there is a thick barrier oxide layer between two layers of nanotubes, and this barrier oxide layer has just been penetrated (a pinhole connecting the upper nanotube and lower channels as shown in Fig. 3c, marked as channels 1, 2, and 3). There is every reason to believe that these channels labeled 1, 2 and 3 in Fig. 3c, d and f are formed by the oxygen bubble mold rather than by the digging manner of FAD reaction. Only the oxygen bubbles in the channels labeled 1, 2 and 3 in Fig. 3c continue to expand, pushing off the top anion contaminated layer as in stage 3. But it is more difficult to push off the thicker layer of anion contaminated above in stage 5 than in stage 2. Therefore, only three small pinholes can be seen above the channels labeled 1, 2 and 3 in Fig. 3c connected to the bottom of the upper nanotubes.
In general, in one-step constant voltage anodization, the current–time curve of three stages is maintained continuously as shown in Fig. 1d, and the monolayer nanotube continues to grow under the action of two stable currents, and the outer diameter of the monolayer nanotube will remain constant as shown in Fig. 1b and c. That is, the outer diameter of the nanotube should remain constant during a one-step anodizing process with a constant voltage of 80 V. However, the diameter of the lower nanotube in Fig. 3c and d is significantly larger than that of the upper nanotube, which also proves that the nanotube is not formed by the digging manner of FAD reaction.
It is clear that the formation of the bilayer nanotubes in Fig. 3 begins at stage 4. Why did the third stage, which was supposed to keep the current constant, suddenly turn into the fourth stage, where the current drops rapidly? Then, what is the intrinsic nature of the sudden drop in ionic current in stage 4? It is helpful to understand the formation mechanism of monolayer and multilayer nanotube structures.
By comparing Fig. 1d and 3e, it can be seen that the current–time curve of anodization at 50 V has three stages, while the current–time curve of anodization at 80 V has six stages. During 900 seconds of anodization at 50 V, the third stage of steady current of 15 mA lasts for about 800 seconds in Fig. 1d. This means that the ionic current and electronic current are at a stable level during the third stage (there is little difference between the two currents), ensuring the stable self-organizing growth of the nanotubes, because the dominant ionic current shows a downward trend in the current–time curve (stage 1 in Fig. 4), and the dominant electronic current shows an upward trend in the current–time curve (stage 2 in Fig. 4).57
However, during 900 seconds of anodization at 80 V in Fig. 3e, the third stage only lasts for about 200 seconds, and the anodizing current in the third stage slowly decreases from 45 mA to 38 mA. The current decrease means that the barrier oxide layer at the bottom of the nanotubes is gradually thickening.34 This indicates that the nanotubes are not undergoing self-organized and stable growth in the third stage of Fig. 3e. Apparently, the rapid drop in the fourth stage current means that a thick barrier oxide layer has formed at the bottom of the nanotube array near the Ti substrate as shown in stage 4 of Fig. 4. The rapid growth of the barrier oxide layer in stage 4 is exactly the same as that in stage 1. After stage 4 is completed, the next stage 5 and stage 6 repeat stage 2 and stage 3 as shown in Fig. 4. The key question in this article is, why is the new barrier oxide layer regenerated in the fourth stage instead of continuing the steady growth of nanotubes as shown in Fig. 1d? This is a complex problem involving ionic conductance of the anodic oxides, electric field level, anion contaminated layer, electrolyte concentration and so on.48–57 The essential reason for the rapid drop in current in the fourth stage to form a new barrier oxide is that the initial ionic current of 38 mA is too large, because in the third stage the ionic current (∼40 mA) is much larger than the electronic current (∼4 mA) in Fig. 3e.28 The two conditions for a steady self-organizing growth of the nanotube are enough electronic current (Je = j0exp(αd)) to form an upward moving oxygen bubble mold, and enough ionic current (Jion = Aexp(BUapp/d)) to form a barrier oxide layer to grow upward around the expansive bubble mold. It can be seen from the above two formulas that only when the thickness of the barrier layer is in a suitable range, the two currents can be in a stable state and the nanotubes can be in a stable self-organizing growth stage.71,72 This is the real nature of self-organized growth, which is still unclear until now. Self-organized and orderly growth means harmonious growth, peaceful growth, and the golden mean (golden section). In contrast, when the ionic current is much larger than the electronic current, in the first and fourth stages in Fig. 4, the ionic current can only increase the thickness of the barrier oxide layer. When the electronic current is far greater than the ionic current, that is, the thickness of the barrier oxide layer reaches the maximum (breakdown thickness), there is oxide breakdown and oxygen gas release but no anodic oxide growth.52,54,68
In stage 5 of Fig. 4, the nanotube embryos of the second layer are formed by oxygen bubble mold, similar to stage 2. After the formation of the new barrier oxide layer in stage 4, the ionic current decreases to a critical value of 6 mA, and the electronic current causes the total current to increase, increasing to a maximum of 13 mA. The oxygen bubble mold is formed at the interface of the anion contaminated layer and barrier oxide layer. It takes more time for the oxygen bubbles in the second layer nanotube to escape, because the oxygen bubbles have to push through the thicker anion contaminated layer on top of them. In Fig. 3e, the fifth stage is about 100 seconds, while the second stage is only about 12 seconds. Due to the larger expansion of oxygen bubbles,24,73 the inner diameter of the second layer of nanotubes in Fig. 3c is much larger than that of the first layer of nanotubes. The pore walls of the second layer of nanotubes are also thicker than those of the first layer.
Stage 6 is similar to the previous stage 3 of Fig. 4, because the lower nanotube has a larger diameter and cannot communicate directly with the bottom of the upper nanotube. Only the oxygen bubbles push up the thicker anion contaminated layer, and the electrolyte can only slowly enter the bottom of the nanotube through the pinholes, so the current in stage 6 gradually decreases. Three pinholes can be seen above the channels labeled 1, 2 and 3 in Fig. 3c connected to the bottom of the upper nanotubes. Two layers of nanotubes are clearly visible in Fig. 3b, and the first layer is longer, because the ionic current is higher when the first layer of nanotubes is growing.
4. Conclusions
Since anodization is an electrochemical process, the growth kinetics of porous anodic oxides should be related to the anodizing current. It is well known that in one-step anodization with constant voltage, the current–time curve usually shows three stages: rapid decrease, slow rise and steady state. However, the classical field-assisted dissolution and flow theories cannot explain the three stages of the current–time curve and the growth kinetics of nanotubes. We conclude that the total anodizing current includes ionic current and electronic current. Ionic current causes barrier oxide growth, and electronic current causes oxygen evolution. In the first stage, ionic current promotes the rapid growth of the barrier oxide, and the ionic current decreases exponentially with the thickness of the barrier oxide. In the second stage, the barrier oxide reaches the critical thickness for generating the electronic current, which causes the total current to rise and forms the oxygen bubble molds. In the third stage, oxygen bubbles overflow from the nanotubes and electrolyte enters the nanotube bottom, where the ionic current and electronic current remain constant and prompt the nanotubes to grow steadily.We have for the first time discovered six stages of the current–time curves in one-step anodization of zirconium at constant voltage in NH4F electrolyte, and have obtained bilayer nanotubes with an increased diameter. None of the above facts can be explained by the dissolution and viscous flow models. Based on the variation of ionic current and electronic current with the barrier oxide thickness, combined with the oxygen bubble model and the viscous flow model, the bilayer nanotubes and the six stages of the current–time curve were reasonably explained. These new ideas presented in this paper improve the growth kinetics of metal anodization and help to regulate the structure of various porous anodic oxides.74–78
Author contributions
Pengze Li: methodology, investigation, and writing. Heng Wang: review & editing, and formal analysis. Yilin Ni: methodology and original draft. Ye Song: supervision. Ming Sun: supervision. Tianle Gong: investigation. Chengyuan Li: formal analysis and validation. Xufei Zhu: conceptualization and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51777097, 51577093) and the National Undergraduate Training Program for Innovation and Entrepreneurship (202110288048).References
- A. Ruiz-Clavijo, O. Caballero-Calero and M. Martín-González, Nanoscale, 2021, 13, 2227–2265 RSC.
- T. Gakhar and A. Hazra, Nanoscale, 2020, 12, 9082–9093 RSC.
- T. T. Wen, H. Y. Tan, S. Chen, P. F. He, S. Yang, C. Deng and S. F. Liu, Electrochem. Commun., 2021, 128, 107073 CrossRef CAS.
- C. P. Yu, Z. L. Zheng, W. C. Zhang, B. Hu, Y. J. Chen, J. H. Chen, K. F. Ma, J. H. Ye and J. W. Zhu, ACS Sustain. Chem. Eng., 2020, 8, 3969–3975 CrossRef CAS.
- L. Shen, Y. Li, W. Zhong, J. C. Wu, J. H. Cheng, L. Jin, X. Hu and Z. Y. Ling, Electrochem. Commun., 2021, 126, 107014 CrossRef CAS.
- L. Vera-Londono, A. Ruiz-Clavijo, O. Caballero-Calero and M. Martín-González, Nanoscale Adv., 2020, 2, 4591–4603 RSC.
- Y. Yu, Y. J. Zhao, K. Li, G. Zhang, K. Y. Yu, Y. H. Ma and Y. Li, Appl. Surf. Sci., 2020, 503, 144316 CrossRef CAS.
- Y. Z. Jiao, S. Y. Cheng, F. Wu, X. H. Pan, A. M. Xie, X. Zhu and W. Dong, Compos. B Eng., 2021, 211, 108643 CrossRef CAS.
- S. Chen, Y. L. Ni, J. P. Zhang, Y. X. Dan, W. C. Zhang, Y. Song and X. F. Zhu, Electrochem. Commun., 2021, 125, 106991 CrossRef CAS.
- S. Y. Cao, D. L. Yu, Y. Lin, C. Zhang, L. F. Lu, M. Yin, X. Zhu, X. Y. Chen and D. Li, ACS Appl. Mater. Interfaces, 2020, 12, 26184–26192 CrossRef CAS PubMed.
- X. Chen, X. R. Xu, J. Cui, C. T. Chen, X. Zhu, D. P. Sun and J. S. Qian, J. Hazard. Mater., 2020, 392, 122331 CrossRef CAS PubMed.
- S. X. Liu, J. L. Tian and W. Zhang, Nanotechnology, 2021, 32, 222001 CrossRef CAS PubMed.
- D. S. Guan and Y. Wang, Nanoscale, 2012, 4, 2968–2977 RSC.
- M. I. Broens, W. R. Cervantes, D. O. Jerez, M. L. Teijelo and O. E. L. Perez, Ceram. Int., 2020, 46, 13599–13606 CrossRef CAS.
- J. T. Domagalski, E. Xifre-Perez and L. F. Marsal, Nanomaterials, 2021, 11, 430 CrossRef CAS PubMed.
- W. Lee and S. J. Park, Chem. Rev., 2014, 114, 7487–7556 CrossRef CAS PubMed.
- F. Riboni, N. T. Nguyen, S. So and P. Schmuki, Nanoscale Horiz., 2016, 1, 445–466 RSC.
- D. Regonini, C. R. Bowen, A. Jaroenworaluck and R. Stevens, Mater. Sci. Eng. R Rep., 2013, 74, 377–406 CrossRef.
- F. J. Q. Cortes, P. J. Arias-Monje, J. Phillips and H. Zea, Mater. Des., 2016, 96, 80–89 CrossRef CAS.
- M. Yu, Y. Chen, C. Li, S. Yan, H. Cui, X. F. Zhu and J. Kong, Electrochem. Commun., 2018, 87, 76–80 CrossRef CAS.
- R. Q. Hang, F. L. Zhao, X. H. Yao, B. Tang and P. K. Chu, Appl. Surf. Sci., 2020, 517, 146118 CrossRef CAS.
- K. R. Hebert, S. Albu, I. Paramasivam and P. Schmuki, Nat. Mater., 2012, 11, 162–166 CrossRef CAS PubMed.
- Q. Dou, P. Shrotriya, W. F. Li and K. R. Hebert, Electrochim. Acta, 2019, 295, 418–426 CrossRef CAS.
- Z. Y. Zhang, Q. Wang, H. Q. Xu, W. C. Zhang, Q. Y. Zhou, H. P. Zeng, J. Yang, J. Zhu and X. Zhu, Electrochem. Commun., 2020, 114, 106717 CrossRef CAS.
- Q. Zhou, M. Tian, Z. Ying, Y. Dan, F. Tang, J. Zhang, J. Zhu and X. F. Zhu, Electrochem. Commun., 2020, 111, 106663 CrossRef CAS.
- J. L. Qin, Z. G. Cao, H. Li and Z. X. Su, Surf. Coat. Technol., 2021, 405, 126661 CrossRef CAS.
- J. E. Yoo and P. Schmuki, J. Electrochem. Soc., 2019, 166, C3389–C3398 CrossRef CAS.
- X. Li, C. Y. Li, T. L. Gong, J. H. Su, W. C. Zhang, Y. Song and X. F. Zhu, Ceram. Int., 2021, 47, 23332–23337 CrossRef CAS.
- Z. X. Su and W. Z. Zhou, J. Mater. Chem., 2011, 21, 8955–8970 RSC.
- C. Cao, J. Li, X. Wang, X. Song and Z. Sun, J. Mater. Res., 2011, 26, 437–442 CrossRef CAS.
- J. Oh and C. V. Thompson, Electrochim. Acta, 2011, 56, 4044–4051 CrossRef CAS.
- A. Baron-Wiechec, M. G. Burke, T. Hashimoto, H. Liu, P. Skeldon, G. E. Thompson, H. Habakaki, J.-J. Ganem and I. C. Vickridge, Electrochim. Acta, 2013, 113, 302–312 CrossRef CAS.
- Ö. Çapraz, P. Shrotriya, P. Skeldon, G. E. Thompson and K. R. Hebert, Electrochim. Acta, 2015, 167, 404–411 CrossRef.
- J. J. Zhang, W. Q. Huang, K. Zhang, D. Z. Li, H. Q. Xu and X. F. Zhu, Electrochem. Commun., 2019, 100, 48–51 CrossRef CAS.
- J. Zhang, Y. T. Yu, P. J. Fang, L. Liu, H. Y. Yue, J. L. Ou and A. J. Han, Electrochem. Commun., 2021, 129, 107086 CrossRef CAS.
- Q. Zhou, D. Niu, X. Feng, A. Wang, Z. R. Ying, J. P. Zhang, N. Lu, J. Zhu and X. Zhu, Electrochem. Commun., 2020, 119, 106815 CrossRef CAS.
- P. Y. Li, D. M. Niu, M. F. He, H. Huang, Z. R. Ying, H. Q. Xu, J. Zhu and X. F. Zhu, J. Phys. Chem. C, 2020, 124, 3050–3058 CrossRef CAS.
- J. W. Cao, Z. Q. Gao, C. Wang, H. M. Muzammal, W. Q. Wang, Q. Gu, C. Dong, H. T. Ma and Y. P. Wang, Surf. Coat. Technol., 2020, 388, 125592 CrossRef CAS.
- N. Lu, J. P. Zhang, Y. X. Dan, M. Sun, T. L. Gong, X. Li and X. F. Zhu, Electrochem. Commun., 2021, 126, 107022 CrossRef CAS.
- R. Z. Zhu, C. Y. Li, P. Li, X. P. Shen, J. Chen, Y. Song and X. F. Zhu, Electrochem. Commun., 2021, 129, 107093 CrossRef CAS.
- T. L. Gong, C. Y. Li, X. Li, H. Y. Yue, X. F. Zhu, Z. Y. Zhao, R. Q. Lv and J. Zhu, Nanoscale Adv., 2021, 3, 4659–4668 RSC.
- M. S. Yu, H. M. Cui, F. P. Ai, L. F. Jiang, J. Kong and X. F. Zhu, Electrochem. Commun., 2018, 86, 80–84 CrossRef CAS.
- Z. Y. Zhang, Q. Q. Liu, M. F. He, F. Tang, Z. R. Ying, H. Q. Xu, Y. Song, J. Zhu and X. F. Zhu, J. Electrochem. Soc., 2020, 167, 113501 CrossRef CAS.
- N. J. Suliali, C. M. Mbulanga, W. E. Goosen, R. Betz and J. R. Botha, Electrochim. Acta, 2020, 337, 135791 CrossRef CAS.
- H. Tsuchiya and P. Schmuki, Nanoscale, 2020, 12, 8119–8132 RSC.
- L. Fialho, C. F. Almeida Alves, L. S. Marques and S. Carvalho, Appl. Surf. Sci., 2020, 511, 145542 CrossRef CAS.
- E. Seçkin and M. Ürgen, Surf. Coat. Technol., 2021, 409, 126840 CrossRef.
- S. J. Garcia-Vergara, P. Skeldon, G. E. Thompson and H. Habazaki, Electrochim. Acta, 2006, 52, 681–687 CrossRef CAS.
- D. J. LeClere, A. Velota, P. Skeldon, G. E. Thompson, S. Berger, J. Kunze, P. Schmuki, H. Habazaki and S. Nagata, J. Electrochem. Soc., 2008, 155, C487–C494 CrossRef CAS.
- J. E. Houser and K. R. Hebert, Nat. Mater., 2009, 8, 415–420 CrossRef CAS PubMed.
- J. W. Diggle, T. C. Downie and C. W. Goulding, Chem. Rev., 1969, 69, 365–405 CrossRef CAS.
- J. M. Albella, I. Montero and J. M. Martinez-Duart, Electrochim. Acta, 1987, 32, 255–258 CrossRef CAS.
- X. F. Zhu, L. Liu, Y. Song, H. B. Jia, H. D. Yu, X. M. Xiao and X. L. Yang, Monatsh. Chem., 2008, 139, 999–1003 CrossRef CAS.
- X. F. Zhu, Y. Song, L. Liu, C. Wang, J. Zheng, H. Jia and X. Wang, Nanotechnology, 2009, 20, 475303 CrossRef PubMed.
- X. F. Zhu, Y. Song, D. Yu, C. Zhang and W. Yao, Electrochem. Commun., 2013, 29, 71–74 CrossRef CAS.
- S. Zhang, D. L. Yu, D. D. Li, Y. Song, J. F. Che, S. Y. You and X. F. Zhu, J. Electrochem. Soc., 2014, 161, E135–E141 CrossRef CAS.
- B. Chong, D. Yu, R. Jin, Y. Wang, D. Li, Y. Song, M. Gao and X. Zhu, Nanotechnology, 2015, 26, 145603 CrossRef PubMed.
- M. Pashchanka, Nanomaterials, 2021, 11, 2271 CrossRef CAS PubMed.
- J. L. Zhao, X. Wang, R. Q. Xu, F. Meng, L. M. Guo and Y. X. Li, Mater. Lett., 2008, 62, 4428–4430 CrossRef CAS.
- S. N. Patel, V. Jayaram and D. Banerjee, Surf. Coat. Technol., 2017, 323, 2–9 CrossRef CAS.
- D. Chopra, K. Gulati and S. Ivanovski, ACS Biomater. Sci. Eng., 2021, 7, 3069–3074 CrossRef CAS PubMed.
- Y. Zhang, W. Cheng, F. Du, S. Y. Zhang, W. Ma, D. Li, Y. Song and X. F. Zhu, Electrochim. Acta, 2015, 180, 147–154 CrossRef CAS.
- A. Mazzarolo, M. Curioni, A. Vicenzo, P. Skeldon and G. E. Thompson, Electrochim. Acta, 2012, 75, 288–295 CrossRef CAS.
- H. A. El-Sayed, C. A. Horwood, A. D. Abhayawardhana and V. I. Birss, Nanoscale, 2013, 5, 1494–1498 RSC.
- M. Alijani, H. Sopha, S. Ng and J. M. Macak, Electrochim. Acta, 2021, 376, 138080 CrossRef CAS.
- L. Li, Z. Zhang, L. L. Bo, Y. J. Cui, Y. M. Xu and Z. F. Zhang, Surf. Coat. Technol., 2020, 404, 126470 CrossRef CAS.
- N. F. Fahim and T. Sekino, Chem. Mater., 2009, 21, 1967–1979 CrossRef CAS.
- Y. Li, H. Shimada, M. Sakairi, K. Shigyo, H. Takahashi and M. Seo, J. Electrochem. Soc., 1997, 144, 866–876 CrossRef CAS.
- M. S. Yu, C. Li, Y. B. Yang, S. K. Xu, K. Zhang, H. M. Cui and X. F. Zhu, Electrochem. Commun., 2018, 90, 34–38 CrossRef CAS.
- Y. L. Zhang, H. W. Fan, X. Ding, Q. H. Yan, L. P. Wang and W. H. Ma, Electrochim. Acta, 2015, 176, 1083–1091 CrossRef CAS.
- B. Chong, D. L. Yu, M. Q. Gao, H. W. Fan, C. Yang, W. H. Ma, S. Y. Zhang and X. F. Zhu, J. Electrochem. Soc., 2015, 162, H244–H250 CrossRef CAS.
- K. Zhang, S. K. Cao, C. Li, J. R. Qi, L. F. Jiang, J. J. Zhang and X. F. Zhu, Electrochem. Commun., 2019, 103, 88–93 CrossRef CAS.
- Y. L. Ni, J. Zhang, T. L. Gong, M. Sun, Z. Y. Zhao, X. Li, H. W. Yu and X. F. Zhu, Surf. Interfaces, 2021, 26, 101419 CrossRef.
- Y. Kim, S. Choi, A. Kim and W. Lee, ACS Nano, 2020, 14, 13727–13738 CrossRef PubMed.
- Y. G. Wang, G. Li, K. Y. Wang and X. Y. Chen, Appl. Surf. Sci., 2020, 505, 144497 CrossRef CAS.
- W. G. Zhang, Y. M. Liu, F. Guo, J. Liu and F. Q. Yang, J. Mater. Chem. C, 2019, 7, 14098–14108 RSC.
- H.-Ch. Shin, J. Dong and M. L. Liu, Adv. Mater., 2004, 16, 237–240 CrossRef CAS.
- M. Martín-Gonzalez, R. Martinez-Moro, M. H. Aguirre, E. Flores and O. Caballero-Calero, Electrochim. Acta, 2020, 330, 135241 CrossRef.
Footnote |
| † Authors Pengze Li and Heng Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |