
Modulating the optical properties and functions of organic molecules through polymerization
Wenbo
Wu
 *a and
Bin
Liu
*bc
*a and
Bin
Liu
*bc
aInstitute of Molecular Aggregation Science, Tianjin University, Tianjin 300072, China. E-mail: wuwb@tju.edu.cn
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheliub@tju.edu.cn
cJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 5th August 2021
Abstract
Organic functional materials with advanced optical properties have attracted much attention due to their broad applications, such as in light-emitting diodes, solar cells, anti-counterfeiting, photocatalysis, and even disease diagnosis and treatment. Recent research has revealed that many optical properties of organic molecules can be improved through simple polymerization. In this review, we discuss the phenomenon, mechanism, and impact of polymerization on the properties of materials, including the polymerization-induced spectral shift, polymerization-enhanced photosensitization, polymerization-enhanced two-photon absorption, polymerization-enhanced photocatalytic efficiency, polymerization-induced room temperature phosphorescence, polymerization-induced thermally activated delayed fluorescence, and polymerization-induced emission using specific examples with different applications. The new opportunities arising from polymerization in designing high performance optical materials are summarized in the future perspective.
 Wenbo Wu | Wenbo Wu obtained his PhD degree in Polymer Chemistry and Physics from Wuhan University in June 2014. After a short stay at Zhengzhou University, he moved to the National University of Singapore as a Research Fellow from 2015 to 2019. In 2020, he joined Tianjin University where he is currently a Professor in the Institute of Molecular Aggregation Science. His current research focuses on the design of high-performance organic/polymeric functional materials for applications in biomedical and optoelectronic fields. |
 Bin Liu | Bin Liu received her PhD degree in Chemistry from the National University of Singapore in 2001. After postdoctoral training at the University of California Santa Barbara, she joined the National University of Singapore where she is currently a Chair Professor in the Department of Chemical and Biomolecular Engineering. Her research focuses on the design and synthesis of organic functional materials and the exploration of their applications in sensing, imaging and optoelectronic devices. |
1. Introduction
Optical materials are a class of functional materials that respond to light, and include fluorescent materials,2 phosphorescent materials,3 photosensitizers (PSs),4 nonlinear optical materials,5 solar materials,6 photocatalytic materials,7etc. Nowadays, they are almost indispensable in our everyday life with broad applications, not only in lighting and displays8 but also in emerging areas, such as phototheranostics,9 photocatalysis,10 anti-counterfeiting,11 solar cells,12etc.Light-responsive pathways for any optically active organic functional materials generally follow the Jablonski diagram (Fig. 1).13 Under suitable light irradiation, molecules can be excited to an excited singlet state (Sn), and the excitons subsequently migrate to the lowest excited singlet state (S1) through internal conversion (IC). According to Kasha's rule, the S1 state is one of the key factors related to the subsequent photophysical processes.14 For fluorescent materials, migration from the S1 state to the ground state (S0) occurs by releasing radiative light energy.15 For photosensitizers (PSs), the excitons can migrate to the lowest triplet excited state (T1) from the S1 state through intersystem crossing (ISC), and with further transfer of energy to normal oxygen gas to form the singlet oxygen state (1O2) for photodynamic therapy (PDT) and photocatalysis.16,17 For room temperature phosphorescent (RTP) materials, the excitons in the T1 state are returned to the S0 state by radiative decay to emit phosphorescence.18,19 The specific process of reverse intersystem crossing (RISC) is the third decay pathway of the excited excitons in the T1 state, through which the excitons can migrate to S1, and subsequently emit delayed fluorescence through radiative decay. This is the mechanism of thermally activated delayed fluorescent (TADF) materials.20,21
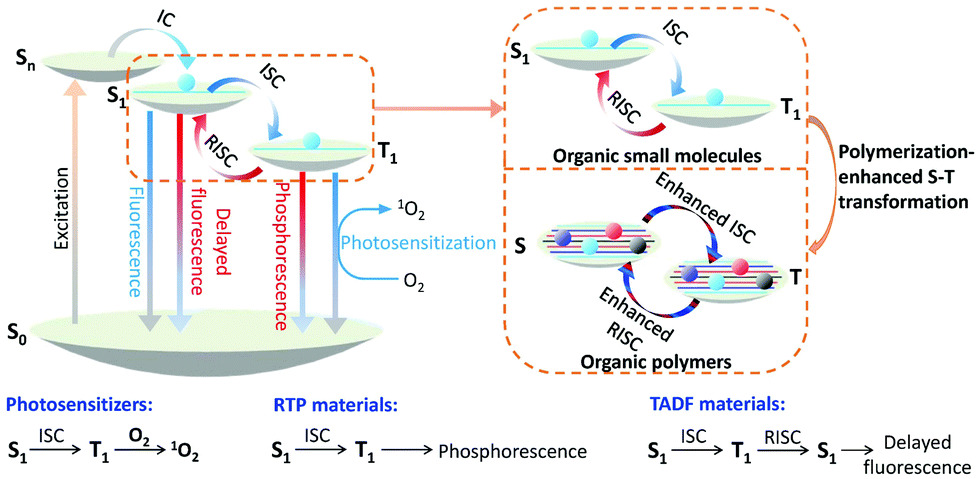 | ||
| Fig. 1 Mechanisms of photosensitizers, RTP materials and TADF materials, which are illustrated by the Jablonski diagram, as well as the general effect of polymerization. | ||
According to the Jablonski diagram above, modulating the communication between the singlet and triplet excitons is one of the key steps in improving the optical performance of many functional materials. Introducing heavy atoms22–25 and decreasing the ΔEST value (the energy gap between the lowest excited singlet state and the triplet state)26–29 are two strategies that have been used widely over the past decade, leading directly to the recent prosperous development of several types of organic functional materials, such as PSs,21–23 RTP materials,24,29 TADF materials,27,28etc. Since both strategies usually require precise molecular design, they are more suitable for small molecules rather than for polymeric materials. So far, most of these organic functional materials have been limited to small molecules.
On the other hand, for polymers, their energy levels are usually very similar. These similar energy levels can subsequently form energy bands (Fig. 1), and their ISC and RISC processes will be different from those of small molecules that have clearly separated energy levels.30,31 In one example, a polymer of (BF2dbm)n (Fig. 2A) was prepared to show an intrinsic long excited-state lifetime of 250 ns, which is more than 100 times longer than that of its corresponding monomer (2.4 ns).30 Density functional theory (DFT) modeling (Fig. 2B) showed that accompanying the increased degree of polymerization, the molecular orbitals (MOs) are split due to interactions between the repeat units, offering more ISC and RISC channels for enhancing both the ISC and RISC processes, which directly enhanced the lifetime of the excitons. This phenomenon was named “polymerization-enhanced intersystem crossing”.30
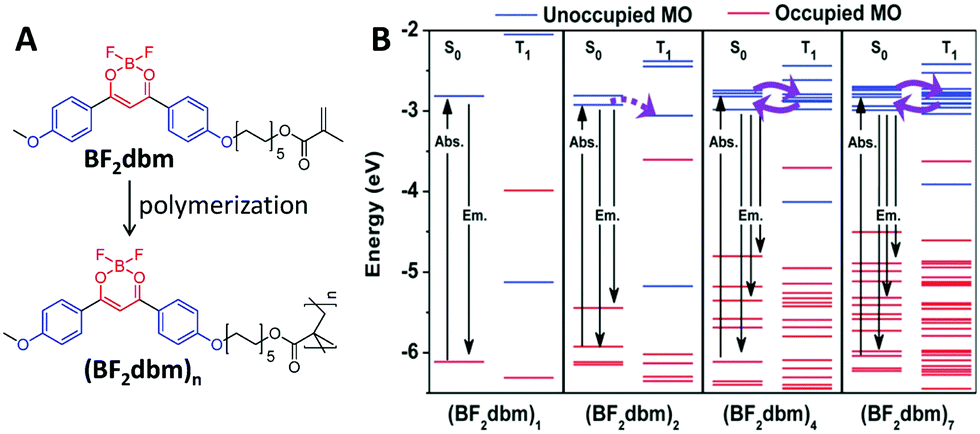 | ||
| Fig. 2 Polymerization-enhanced lifetime of excitons. (A) Chemical structures of BF2dbm and (BF2dbm)n. (B) Calculated selected molecular orbital energy diagrams for the S0 and T1 states of (BF2dbm)n. (B was reprinted with permission from ref. 30, Copyright 2015, Wiley-VCH Verlag GmbH & Co.) | ||
Based on a similar principle, the strategy of polymerization has recently been extended to improve many optical properties, including photosensitization, RTP, TADF, etc. In addition, some other optical properties, such as two-photon absorption, fluorescence etc., can also be modulated by polymerization in some cases. In addition to the improved optical properties, polymeric materials have also shown several other advantages for practical applications, such as high solution processability for preparing devices with low cost, many functional groups for further modification, etc. In this article, we discuss the occurrence, mechanism, and application for some of the materials with optical properties that are significantly improved through polymerization.
2. Polymerization-induced red shift in the absorption and emission wavelengths
Absorption and emission are the basic properties of many optical materials. Conjugated polymers are a special type of polymer with fully conjugated backbones. Compared with small molecules, the counterpart conjugated polymers usually show red-shifted absorption and emission as well as enhanced molar absorptivity due to the extended conjugation length after polymerization.32–34 A typical example of this is shown for BFVBT and PFVBT (Fig. 3A). After polymerization, the absorption peak was red shifted from 475 nm for BFVBT to 523 nm for PFVBT, and the molar absorptivity based on the repeat unit of PFVBT was 1.6-fold that of BFVBT (Fig. 3B). The emission peak of PFVBT was also red shifted by 55 nm compared with that of BFVBT (Fig. 3C).31 Another example involves conjugated polycyanines. As presented in Fig. 3D, both PC1 and PC2 showed a greater than 150 nm red shift compared with the absorption maxima of their corresponding monomeric cyanines of C1 and C2.35,36 The red-shifted absorption with enhanced absorbance endows the conjugated polymers with an improved light-harvesting ability to more effectively facilitate the subsequent processes shown in the Jablonski diagram.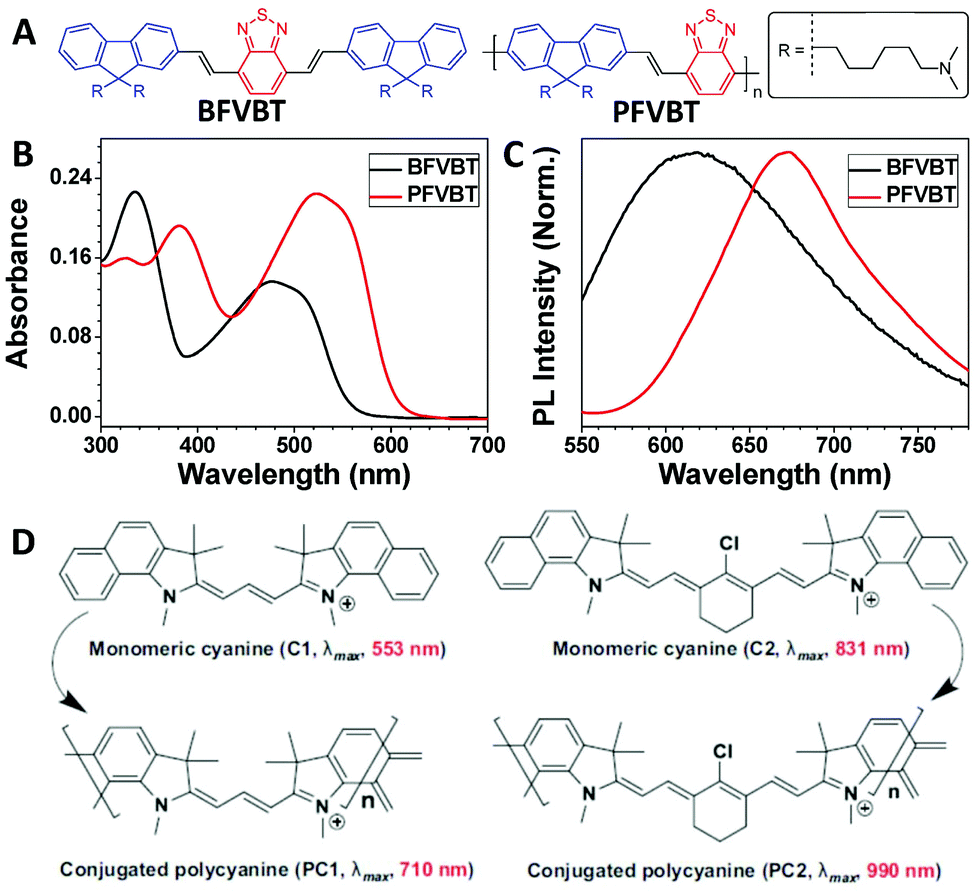 | ||
| Fig. 3 Polymerization-induced red shift in the absorption and emission spectra. (A) Chemical structures of BFVBT and PFVBT. (B) UV-vis spectra of BFVBT and PFVBT in a THF/water mixture (1/99, v/v). (C) Normalized PL spectra (excitation = 480 nm) of BFVBT and PFVBT in a THF/water mixture (1/99, v/v). (D) Chemical structures and maximum absorption wavelengths of monomeric cyanines C1 and C2 and conjugated polycyanines PC1 and PC2 in films. (B and C were reprinted with permission from ref. 31, Copyright 2018, Elsevier; D was reprinted with permission from ref. 36, Copyright 2015, Wiley-VCH Verlag GmbH & Co.) | ||
3. Polymerization-enhanced photosensitization
PDT is a non-invasive treatment method that uses PSs under light irradiation to generate 1O2 or other types of reactive oxygen species (ROS) to kill cancer cells and bacteria.37–39 The 1O2 or ROS generation efficiency of PSs can directly affect their PDT efficacy.40 In 2016, we reported that PTPEAQ41 showed a much higher 1O2 generation efficiency than that of its small-molecule analogue DTPEAQ (Fig. 4A),42 and their 1O2 generation quantum yields were measured as 82% and 38%, respectively. When used for PDT, PTPEAQ also demonstrated a higher phototoxicity to cancer cells than DTPEAQ. However, no detailed understanding of the improvement in 1O2 production for PTPEAQ was then reported.43 At the same time, Schanze's group also reported that the 1O2 generation quantum yield of a series of platinum-containing donor–acceptor–donor-type PSs would be enhanced by extending the effective conjugation length.44 Detailed nanosecond transient absorption spectra revealed that the enhanced conjugation length could increase the contribution of platinum in the frontier orbitals and, therefore, improve the spin–orbit coupling by the heavy-atom effect for the enhanced ISC process.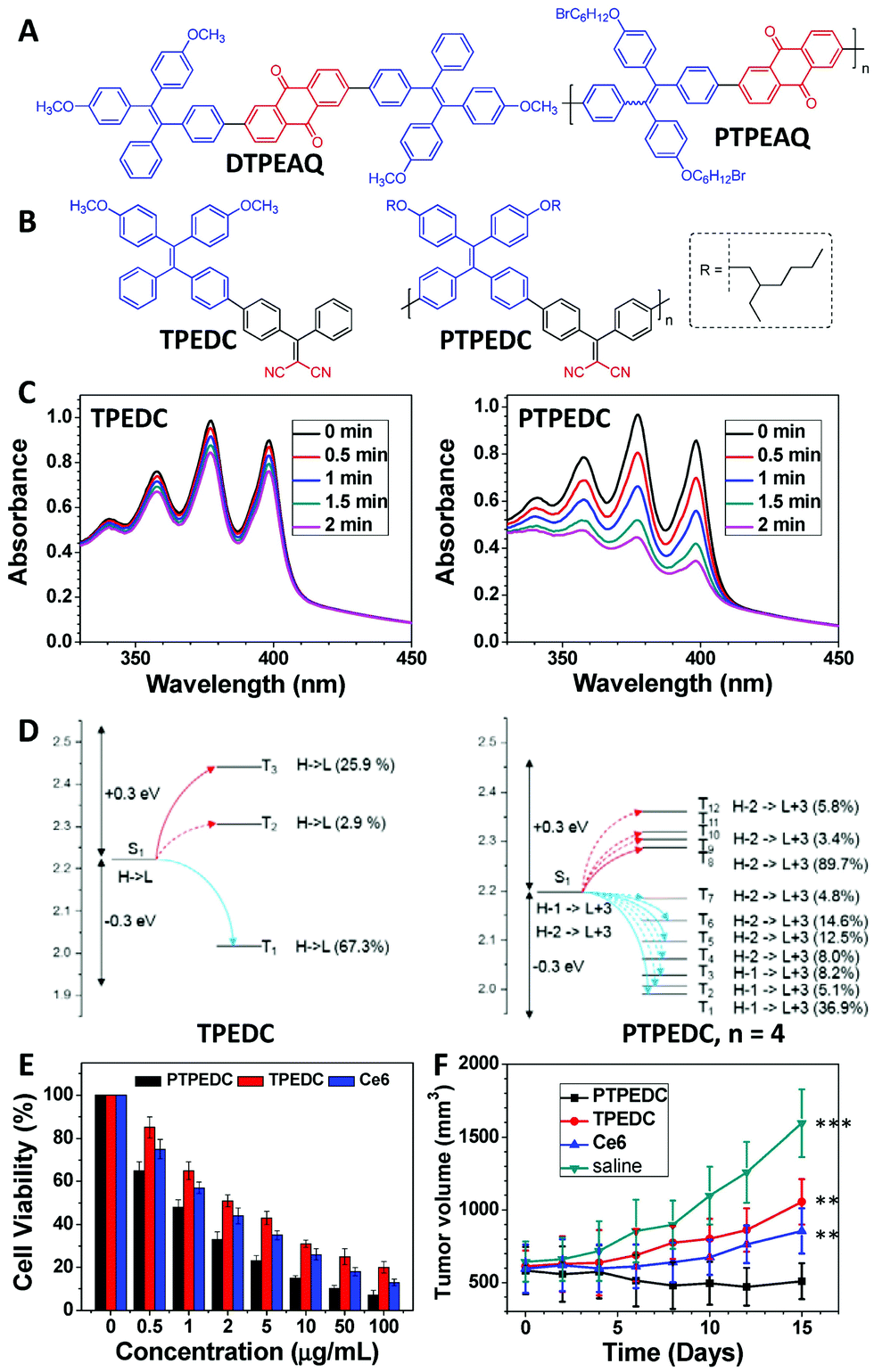 | ||
| Fig. 4 Polymerization-enhanced photosensitization. (A) Chemical structures of DTPEAQ and PTPEAQ. (B) Chemical structures of TPEDC and PTPEDC. (C) The 1O2 generation efficiencies of TPEDC and PTPEDC, measured using ABDA as the indicator. (D) Calculated energy levels and possible ISC channels of TPEDC and PTPEDC. S, T, H, and L stand for singlet, triplet, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO), respectively. The number in parentheses indicates the contribution of the MO to the specific excited state, and the plain and dashed arrows refer to the major and minor ISC channels, respectively. (E) Viability of different NP-treated 4T1 cancer cells under white-light irradiation (60 mW cm−2) for 5 min. (F) Tumor volume measurement of different groups of mice. Error bars indicate the SEM (***, P < 0.001; **, P < 0.01). (C–F were reprinted with permission from ref. 31, Copyright 2018, Elsevier.) | ||
Subsequently, we synthesized several conjugated polymer PSs and their corresponding small-molecule counterparts. These molecules allowed us to observe a universal phenomenon that the conjugated polymer PSs showed a better photosensitization effect than their small-molecule counterparts.31,45 Among these materials, TPEDC and PTPEDC (Fig. 4B) form a good pair of examples. PTPEDC showed a 5.06-fold higher 1O2 generation efficiency than TPEDC (Fig. 4C) due to two main reasons. First, similar to the effect discussed in Fig. 2, DFT modelling indicated that the ISC channels of PTPEDC were much more abundant than those of TPEDC, caused by extremely similar energy levels (Fig. 4D).31 Second, PTPEDC showed an improved light-harvesting ability compared with TPEDC, similar to the previous cases discussed in Fig. 3. Using amphiphilic DSPE-PEG2000 as the polymer matrix, both TPEDC and PTPEDC were encapsulated into nanoparticles (NPs) for further evaluation of their PDT efficacy. As presented in Fig. 4E and F, PTPEDC showed a much better cancer-cell-killing efficiency than that of TPEDC or Ce6 (one of the most commonly used PSs) in both in vitro and in vivo experiments, using 4T1 cancer cells as the model. This phenomenon was named “polymerization-enhanced photosensitization” in 2018.31,45
Subsequent research on PTPEAQ2 by Tang's group (Fig. 5A) further confirmed that the photosensitization effects of conjugated polymers are correlated to the molecular weights (Fig. 5B).46 A series of oligomers and a polymer with the same donor of triphenylamine and acceptor of benzothiadiazole but different molecular weights were prepared to understand the changes in 1O2 generation efficiencies during the polymerization process (Fig. 5C).46 From TB (T is short for triphenylamine in the names of these oligomers, and B is short for benzothiadiazole) to TBTB and then to PTB (Mn = 2000 g mol−1 by gel permeation chromatography, around 5 repeat units), the photosensitization effect was gradually improved with a decreased fluorescence quantum yield (Fig. 5D), confirming the importance of increased conjugation length for improving the photosensitization. In addition, BTB with two acceptors and one donor showed a higher 1O2 production efficiency than that of TBT with two donors and one acceptor (Fig. 5E). The same trend was also observed for BTBTB and TBTBT (Fig. 5E). Time-dependent DFT modeling revealed that the ΔEST values of BTB (0.38 eV) and BTBTB (0.24 eV) were smaller than those of TBT (0.49 eV) and TBTBT (0.41 eV). An even–odd effect in oligomers was proposed: when the number of donors and acceptors is even, the 1O2 generation efficiency is mainly affected by the effective conjugation length; when the number of donors and acceptors is odd, the position and the amount of donor and acceptor could also affect the 1O2 production, in addition to the conjugation length. Following these publications, the polymerization-enhanced photosensitization effect has been used to develop more conjugated polymer PSs for PDT.47–50
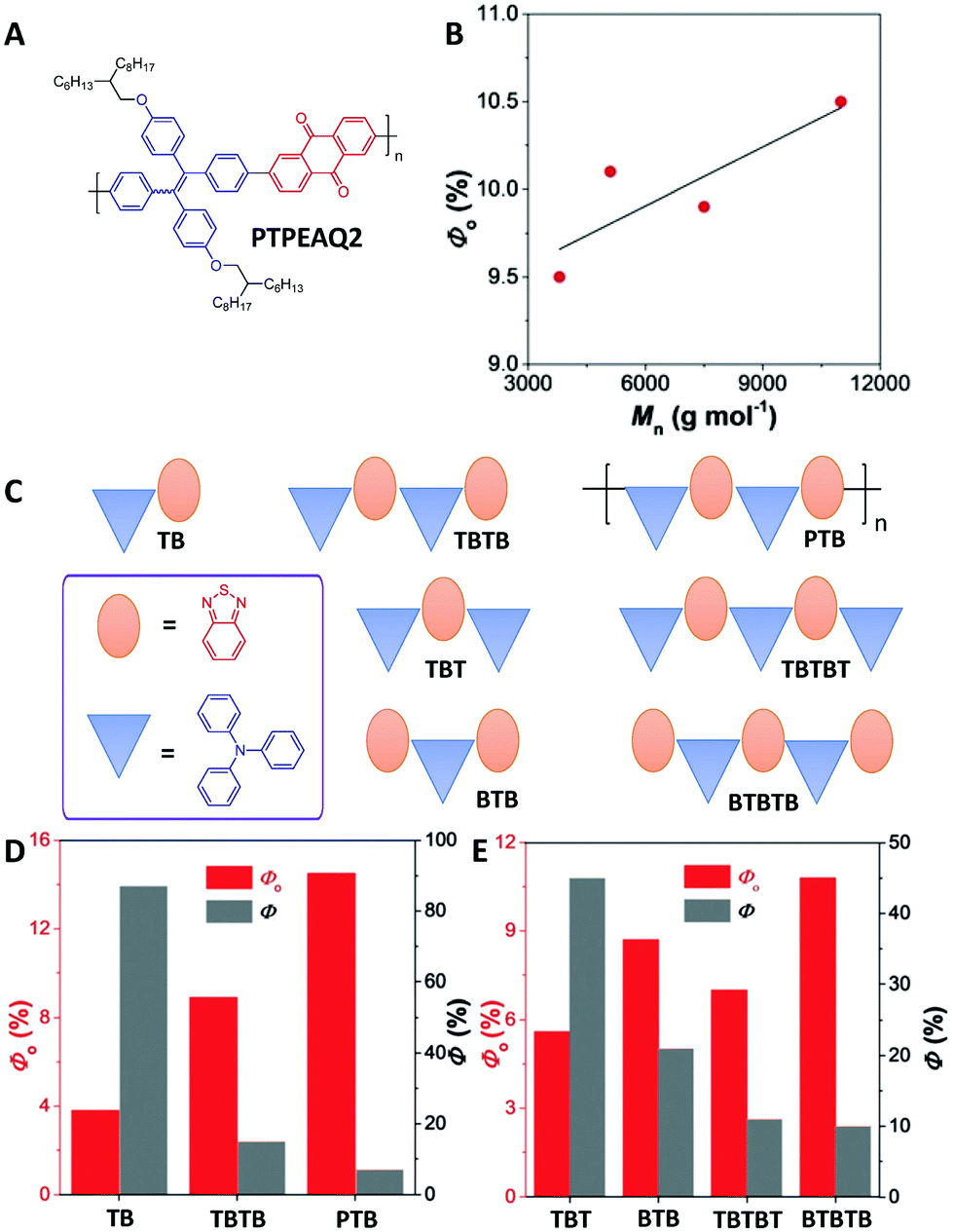 | ||
| Fig. 5 Polymerization-enhanced photosensitization. (A) Chemical structure of PTPEAQ2. (B) 1O2 generation quantum yields of PTPEAQ2 with different number–average molecular weights. (C) Chemical structures of TB, TBTB, PTB, TBT, TBTBT, BTB, and BTBTB. (D) 1O2 generation quantum yield (Φ0) and fluorescence quantum yield (Φ) of TB, TBTB, and PTB. (E) Φ0 and Φf of TBT, BTB, TBTBT, and BTBTB. (B, D and E were reprinted with permission from ref. 46, Copyright 2018, Wiley-VCH Verlag GmbH & Co.) | ||
On the other hand, the penetration depth and accuracy are also important for PDT, and these two parameters can be significantly improved via two-photon technology. In a typical two-photon excited PDT process, two near-infrared photons were used instead of one visible photon to excite the PSs for an improved penetration depth.51 In addition, only PSs at the focus spot of the laser could be excited, which enhances the accuracy in PDT.52
Two-photon excited PDT requires PSs to have a good ROS generation efficiency and two-photon absorption cross-section (2PACS), which are difficult to achieve in one molecule due to their contradictory design principles. For example, a high ROS generation efficiency requires a twisted molecular structure with low conjugation between the donor and the acceptor for a small ΔEST value, while a large two-photon cross-section needs a planar structure with good conjugation.52 In this regard, polymerization not only offers the enhanced photosensitization as discussed above but also allows an extended conjugation length, which is beneficial for two-photon absorption.1,53 Based on the first two-photon excitable PS with aggregation-induced emission (AIE) characteristics (TPEDC, Fig. 6A),54 two conjugated polymers of PTPEDC1 and PTPEDC2 were prepared in 2019.55 From TPEDC to PTPEDC1, the 1O2 generation efficiency and two-photon cross-sections were enhanced 2.27-fold (Fig. 6B) and 3.15-fold (Fig. 6C), respectively. In addition, the 1O2 generation efficiency of PTPEDC1 was already 2.29 times higher than that of the commonly used PS Ce6. With further enhancement of the effective conjugation length by adjusting the linking position of the tetraphenylethene moiety, the performance of PTPEDC2 could be further improved, and it showed 5.48-fold higher 1O2 production under white-light irradiation (Fig. 6B) and a 6.15-fold higher two-photon cross-section than that of TPEDC (Fig. 6C). In two-photon excited photodynamic HeLa cell ablation experiments, PTPEDC2 showed a much better performance than TPEDC and PTPEDC1 within an accurate area of 400 μm × 400 μm (Fig. 6D, red fluorescence indicates dead cells and green fluorescence indicates live cells), and precise PDT on small-sized tumors of around 200 μm diameter in zebrafish was also realized by the two-photon excitation of PTPEDC2.
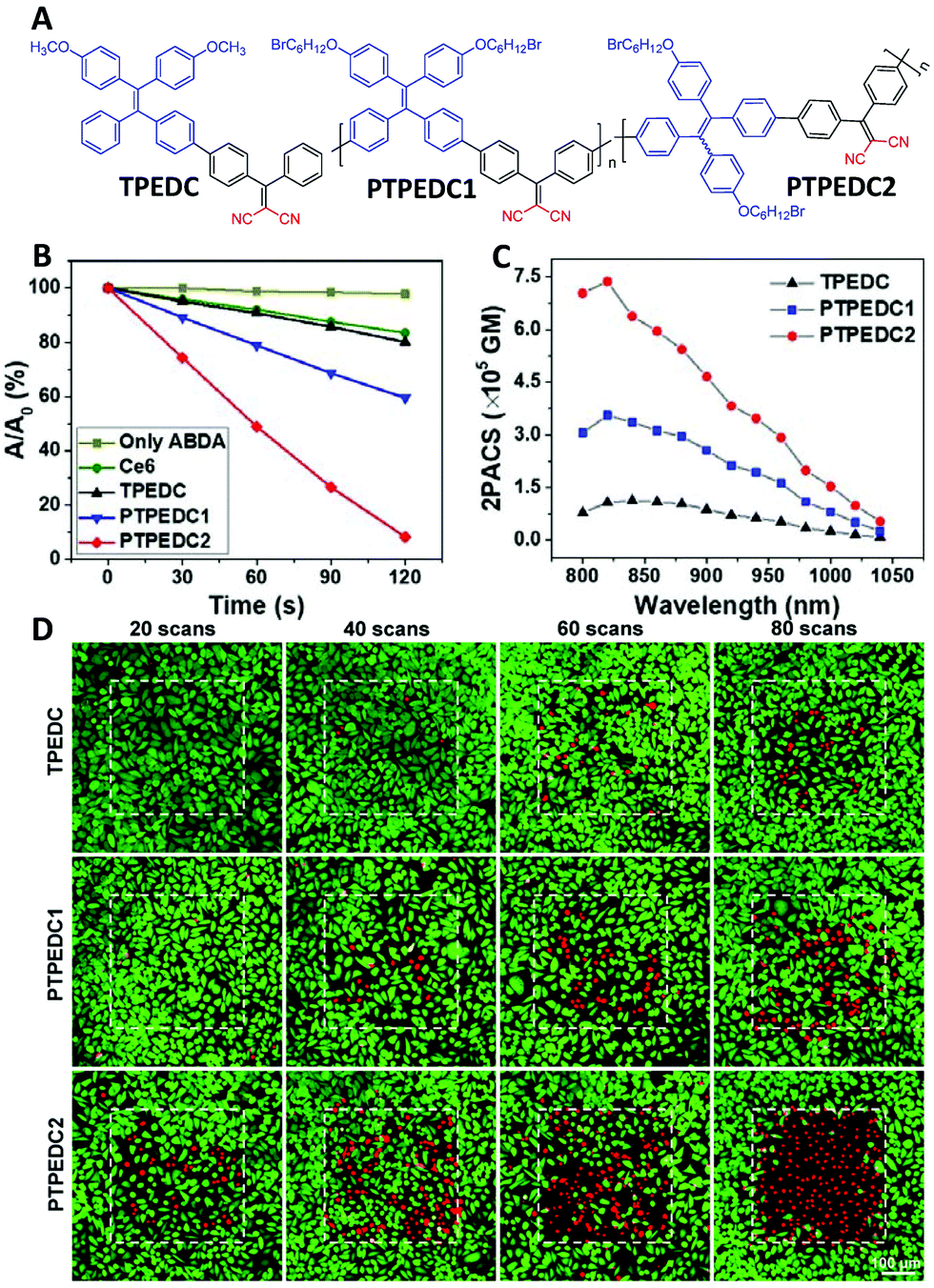 | ||
| Fig. 6 Polymerization-enhanced photosensitization and two-photon absorption cross-section. (A) Chemical structures of TPEDC, PTPEDC1 and PTPEDC2. (B) 1O2 generation efficiencies of TPEDC, PTPEDC1 and PTPEDC2, measured by using ABDA as the indicator. (C) Two-photon absorption cross-sections of TPEDC, PTPEDC1 and PTPEDC2. (D) Photodynamic killing efficiency of TPEDC, PTPEDC1 and PTPEDC2 for HeLa cells under 820 nm two-photon excitation for different times (6 mW, 5.33 s per scan). (B–D were reprinted with permission from ref. 55, Copyright 2019, American Chemical Society.) | ||
Besides PDT, PSs can also be used as photocatalysts as the generated ROS by PSs under light can show a stronger oxidation capacity than oxygen, and many organic reactions, such as Ugi-type oxidation,56 bromination reactions,57 and alcohol oxidation,58etc., have been realized via PS-assisted photoreactions. From this perspective, polymerization can also enhance the photocatalytic efficiency in PS-assisted photoreactions. The PSs of DTF, PTF and CPTF are a group of good examples to confirm this design strategy (Fig. 7A).59 Similar to the above examples, the 1O2 generation efficiency of PTF was 3.63-fold higher than that of DTP after polymerization. Time-dependent DFT modeling clearly indicated that the improved 1O2 generation efficiency was attributed to the increased ISC channels of the conjugated polymers than with the small molecules (Fig. 7B). The cross-linked structure of CPTF was further employed to optimize other properties for photocatalysts, including a good photostability for long-term irradiation, a high specific surface area of 117.2 m3 g−1 for further improving the catalytic efficiency, and a low solubility for easy separation and recyclability after the reaction. Due to the stable and high 1O2 generation efficiency of CPTF, 82% of benzaldehyde could be oxidized to benzoic acid under simulated AM 1.5G irradiation for 4 h when using a very small amount of CPTF as the photocatalyst. The recycling process of this photoreaction was further designed by utilizing the low solubility and high stability of CPTF (Fig. 7C). If 10 g of benzaldehyde was used as the starting material, approximately 6–6.5 g of benzoic acid could be obtained from each cycle, and CPTF retained 85% activity after 10 cycles. CPTF was further used for sunlight-induced wastewater treatment.59 Using CPTF (0.1 mg mL−1) as the photocatalyst, 2 h of simulated AM 1.5G irradiation could totally decompose all Rhodamine 6G and Staphylococcus aureus (Fig. 7D and E). After the wastewater treatment, CPTF could be recovered by filtration for further use. Besides cross-linked conjugated polymers like CPTF, other types of polymers with large specific surface areas, such as conjugated covalent organic frameworks (COFs),60 and amorphous porous organic polymers (POPs),57,61,62etc., have been developed as photocatalysts for oxidation by the same principle. Recently, a microfluidic reactor has been designed by Gao's group to realize the continuous synthesis of chemicals based on photooxidation (Fig. 7F), further improving the synthetic efficiency.63 Using this microfluidic reactor, a famous anti-tumor drug ricobendazole was successfully prepared via the photooxidation of albendazole with a conversion efficiency of 60% using PPET3-N2 as the photocatalyst under white-light irradiation.
 | ||
| Fig. 7 Polymerization-enhanced photocatalytic efficiency. (A) Chemical structures of DTF, PTF and CPTF. (B) Frontier orbital analysis of different model compounds by time-dependent DFT. (C) Sunlight-induced photooxidation of benzaldehyde and the related recycling process. (D) Antibacterial activity of CPTF (0.1 mg mL−1) for S. aureus (1 × 107 counts mL−1) under AM 1.5G irradiation for different time points. (E) Plate images for S. aureus on an LB agar plate supplemented with CPTF and AM 1.5G irradiation for different time points and then grown overnight. (F) Schematic diagram of the microfluidic reactor for photooxidation. (B–E were reprinted with permission from ref. 59, Copyright 2019, Wiley-VCH Verlag GmbH & Co; F was reprinted with permission from ref. 63, Copyright 2019, American Chemical Society.) | ||
4. Polymerization-induced RTP
In addition to producing ROS, triplet excitons can also be used to yield phosphorescence. The radiative transition from triplet states to the ground state is spin forbidden as electric dipole radiation, making the phosphorescence process less effective with a longer life time in comparison with spin-allowed fluorescence.64,65 Therefore, a rigid microenvironment, which can be used for stabilization of the triplet excitons, the prevention of energy dissipation by molecular motions, and the isolation of oxygen and water, is usually necessary for phosphorescence.11 That is why phosphorescence becomes much stronger at low temperature.65 Recent research has indicated that if an extremely rigid microenvironment could be offered to an organic dye with an effective ISC process, strong phosphorescence can be realized at room temperature to facilitate the practical application of phosphorescent materials, and these materials are the RTP materials.66–68Crystallization, including both single-molecule and host–guest systems, is the general strategy to realize a rigid microenvironment, and the overwhelming majority of RTP materials are realized in crystals. On the other hand, the rigid microenvironment can also be achieved via suitable polymerization (for example, cross-linked polymerization), leading to the RTP in an amorphous state. Fig. 8A presents an example of the C1-crosslinked polymer, which was prepared via two-step polymerization.69 The first step of ring-opening polymerization provided the polymer with a hydrophilic backbone, and subsequent reversible addition–fragmentation chain-transfer (RAFT) copolymerization introduced the luminophore into the polymer. The amphiphilic C1-crosslinked polymer can self-assemble into nanoparticles to show bright green phosphorescence with a lifetime of 4 ms in argon-saturated water since the cross-linked structure can restrict the molecular motions, and the hydrophobic polystyrene could isolate the phosphors from water to some extent. As a control, both the monomer C1 and its prototype luminophore Br6A hardly showed any RTP, and nor did the nanoparticles by doping Br6A into the amphiphilic polymer matrix. In addition to RAFT cross-linked polymerization, hydrogen-bond crosslinking70 and Diels–Alder crosslinking71 have also been confirmed to induce the derivatives of Br6A to show RTP after polymerization since these crosslinking methods can also be used to offer a rigid microenvironment to prevent energy dissipation by molecular motions and isolate water and oxygen. The same cross-linked polymerization method has also been used to induce other luminophores to emit RTP.72 These results indicate that suitable cross-linked polymerization is an effective strategy for designing RTP materials.
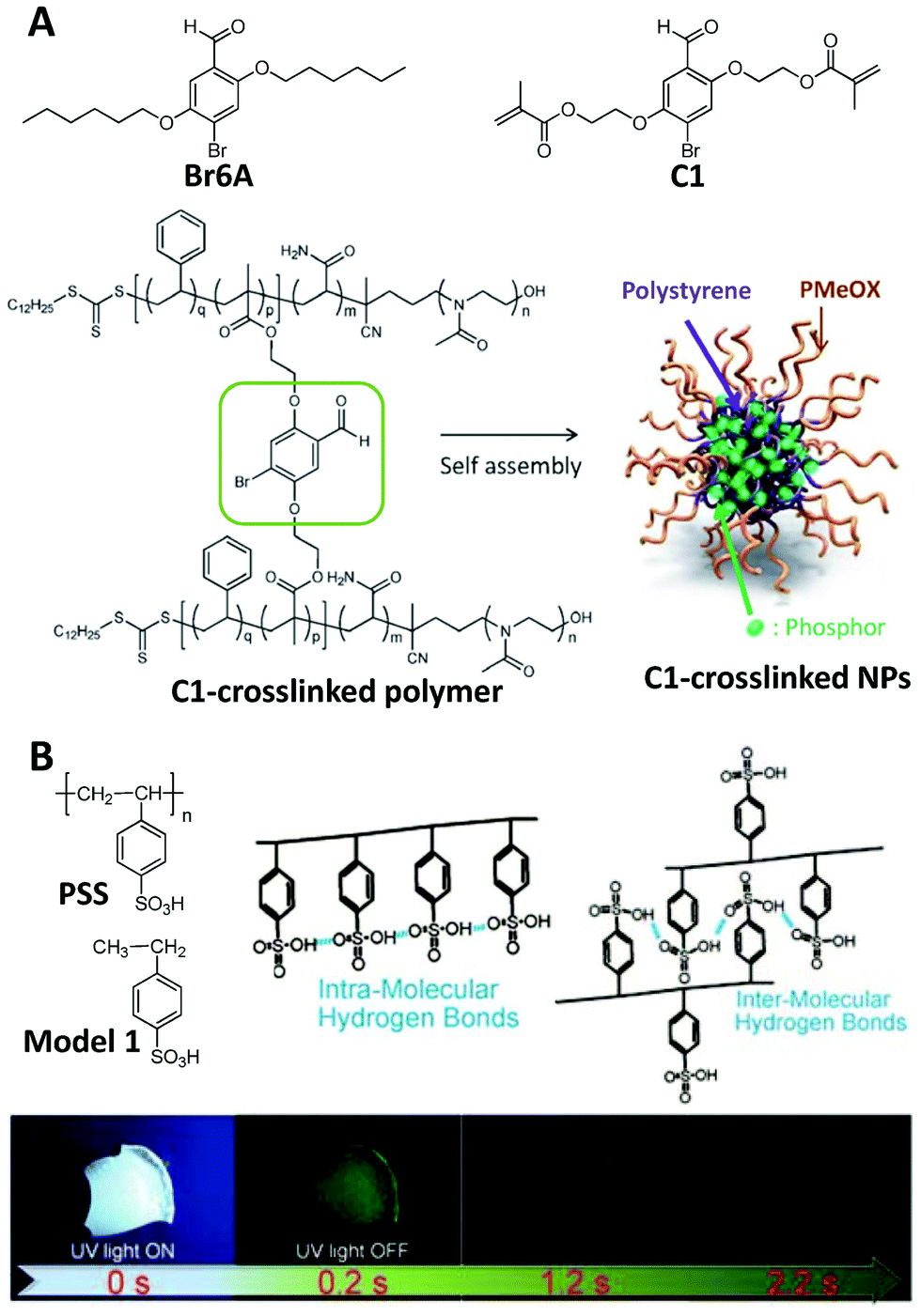 | ||
| Fig. 8 Polymerization-induced RTP. (A) Chemical structures of Br6A, C1, the C1-crosslinked polymer and C1-crosslinked NPs. (B) Chemical structures of PSS and Model 1, the hydrogen bonds in PSS, and luminescence images of a PSS film under 365 nm UV irradiation and after UV irradiation for different time points. (A was reprinted with permission from ref. 69, Copyright 2017, Wiley-VCH Verlag GmbH & Co; B was reprinted with permission from ref. 73, Copyright 2018, Wiley-VCH Verlag GmbH & Co.) | ||
Direct polymerization could also lead to RTP in some special cases. As presented in Fig. 8B, the film of the polymer PSS showed bright green phosphorescence under 365 nm UV irradiation, while the corresponding small molecule of Model 1 was not RTP active at all.73 In addition, the enhanced molecular weight of PSS leads to the increased lifetime of RTP materials. As the weight average molecular weight was increased from 1200 to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 116000 g mol−1, the lifetime of the phosphorescence of PSS was enhanced from 540 ms to 1.13 s. Through polymerization, hydrogen bonding (including both intramolecular and intermolecular types) in PSS can lead to a very rigid microenvironment, to both restrict the motions of the phenyl rings and to isolate oxygen to some extent. After using NaOH or KOH to destroy the hydrogen bonds from the sulfonic acid groups, the RTP lifetime of PSS decreased rapidly.73
116000 g mol−1, the lifetime of the phosphorescence of PSS was enhanced from 540 ms to 1.13 s. Through polymerization, hydrogen bonding (including both intramolecular and intermolecular types) in PSS can lead to a very rigid microenvironment, to both restrict the motions of the phenyl rings and to isolate oxygen to some extent. After using NaOH or KOH to destroy the hydrogen bonds from the sulfonic acid groups, the RTP lifetime of PSS decreased rapidly.73
The polymerization-induced RTP effect prompted the rapid development of RTP polymers besides pure organic crystals and inorganic nanomaterials.74,75 At present, how to control the rigid microenvironment of the phosphorescent centres in polymers remains one of the key concerns for designing high-performance RTP polymers.
5. Polymerization-induced TADF
In the OLED device, the excitons from electrical excitation include both singlet and triplet excitons with an approximate ratio of 1:3. For the TADF emitter-based OLED, the triplet excitons can migrate to singlet states to emit delayed luminescence via the thermally activated RISC process and realize high efficiency. Therefore, pure organic TADF materials have been considered as the third generation of organic light-emitting diode (OLED) materials.20,76 The RISC process is the key pathway of TADF molecules, and decreasing the ΔEST value has therefore become an important principle in designing TADF organic molecules.20,21 However, most of these TADF small molecules have to be fabricated into thin solid films using expensive techniques such as vapor deposition.
To realize solution processable TADF-based OLEDs, a polymeric TADF material was first designed using non-conjugated structures with a high triplet energy level as the spacer to link the TADF moieties together into one polymer chain.77 However, the changes in TADF behaviour during the polymerization process were not discussed in detail. In 2017, Voit's group showed that polymerization could induce a non-TADF unit to show TADF properties.78 They prepared the conjugated polymer of PCZBP (around 14 repeat units calculated via nuclear magnetic resonance) by Yamamoto polymerization of a benzophenone-based donor–acceptor-type monomer, as well as its repeat unit Model 2 as the small-molecule control (Fig. 9A). In polystyrene films (2 wt%), Model 2 had a quantum yield of 3% but did not show any TADF features (Fig. 9B). After polymerization, the photoluminescence quantum yield of PCZBP was improved to 71%, and the intensity-weighted averaged lifetime of the delayed fluorescence was measured as 296 ± 16 μs (Fig. 9B) due to the enhanced RISC process via polymerization. Fig. 9C presents both the fluorescence spectrum at 293 K and the phosphorescence spectrum at 77 K of PCZBP and Model 2 in polystyrene-based solid thin films, which can be used to calculate the exact energy levels of the singlet and triplet states, respectively. Subsequently, the ΔEST value of PCZBP was calculated as 0.023 eV, which was much smaller than that of Model 2 (0.232 eV). To better understand the phenomenon of polymerization-induced TADF, a series of oligomers of CZBP-n with 1–4 repeat units were prepared recently using the same donor and acceptor as PCZBP (Fig. 9D).79 In all these oligomers, the LUMO distributions are localized on the benzophenone side chains, showing very similar LUMO energy levels of around −1.70 eV. However, the HOMOs are distributed in the conjugated backbone of the carbazole donor. Therefore, the extended conjugation from n = 1 to n = 4 could raise the HOMO energy levels, indicating the enhanced electron-donating ability of the donors. Therefore, accompanying the enhanced conjugation length, intramolecular charge transfer was enhanced, leading to smaller ΔEST values (Fig. 9D) for TADF. In fact, when n ≥ 2, CZBP-n became TADF-active.
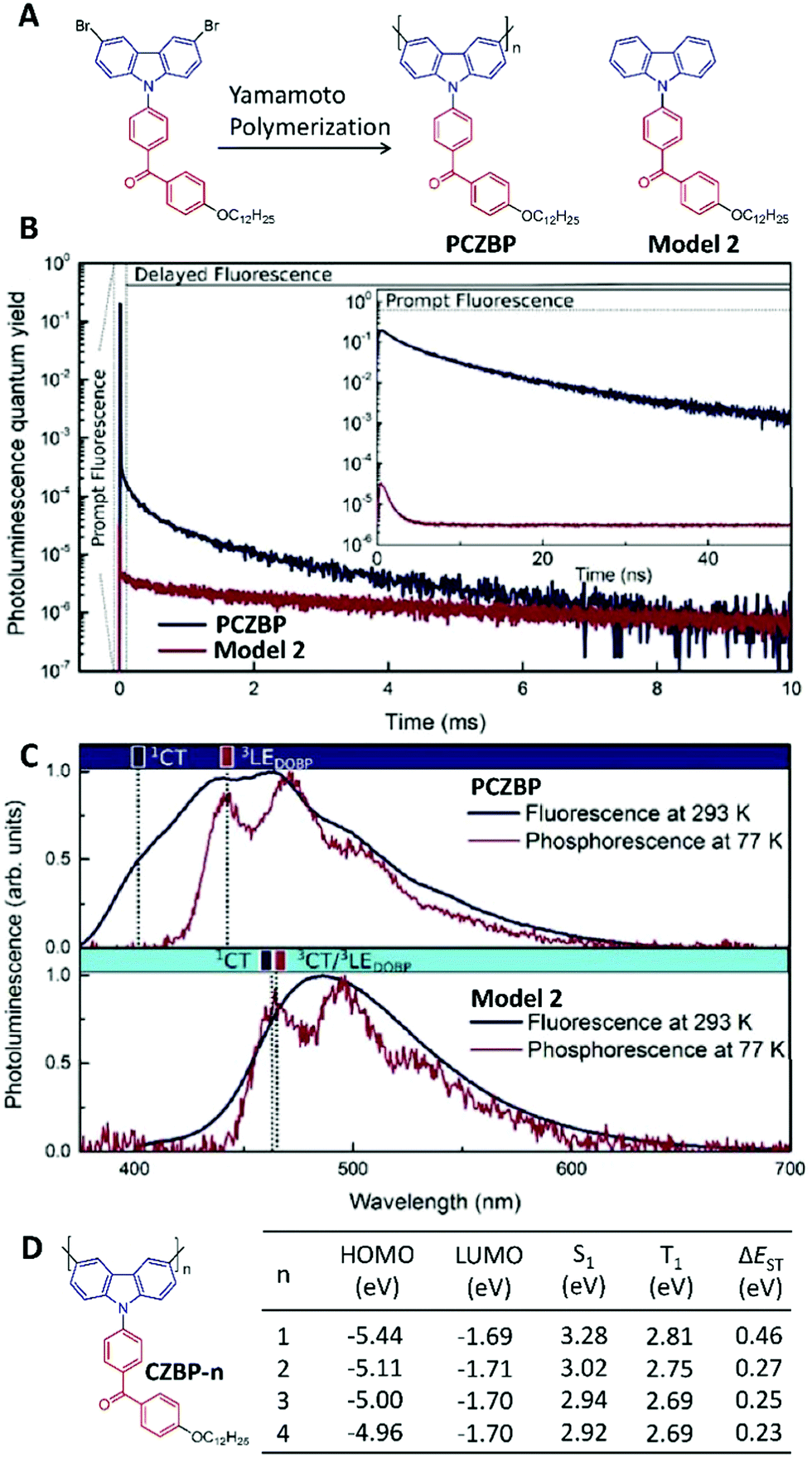 | ||
| Fig. 9 Polymerization-induced TADF. (A) Chemical structures of PCZBP and Model 2. (B) Time-resolved photoluminescence of PCZBP and Model 2 in polystyrene-based solid thin films (2 wt%), where the inset plots their prompt fluorescence characteristics. (C) Steady-state PL spectra at 293 K, and phosphorescence spectra (delayed by 10 ms) at 77 K for PCZBP and Model 2 in polystyrene-based solid thin films (2 wt%). (D) Chemical structures of oligomer molecules CZBP-n as well as their energy levels by time-dependent DFT modelling. (B and C were reprinted with permission from ref. 78, Copyright 2017, Wiley-VCH Verlag GmbH & Co.) | ||
Different from the polymerization-enhanced ISC process, examples of the polymerization-enhanced RISC process are relatively rare, although several TADF polymers have been reported in recent years.21,80 Therefore, more studies are needed to further confirm the improvement of the RISC process by polymerization, which should be beneficial to the development of solution-processable high-performance OLEDs.
6. Polymerization-induced emission
The essential mechanism of nearly all the above effects can be concluded as being that polymerization can enhance the interaction between functional monomers and repeat units and thus change their energy levels for enhanced ISC or RISC processes, which prompt us to check whether or not the enhanced interaction between functional groups could lead to other useful effects. The emission caused by through-space conjugation is a typical example.81 Generally, for some electron-rich groups with separated electrons, such as aromatic rings, ether bonds, hydroxyl groups, ester bonds, amido bonds, nitrile groups, etc., the decreased distance between the groups as a result of polymerization can prompt them to produce clusters with delocalized electrons via enhanced through-space conjugation. This process could induce a polymer with a non-conjugated structure to be emissive (Fig. 10A). The phenomenon was named “polymerization-induced emission” by Tang's group in 2020.81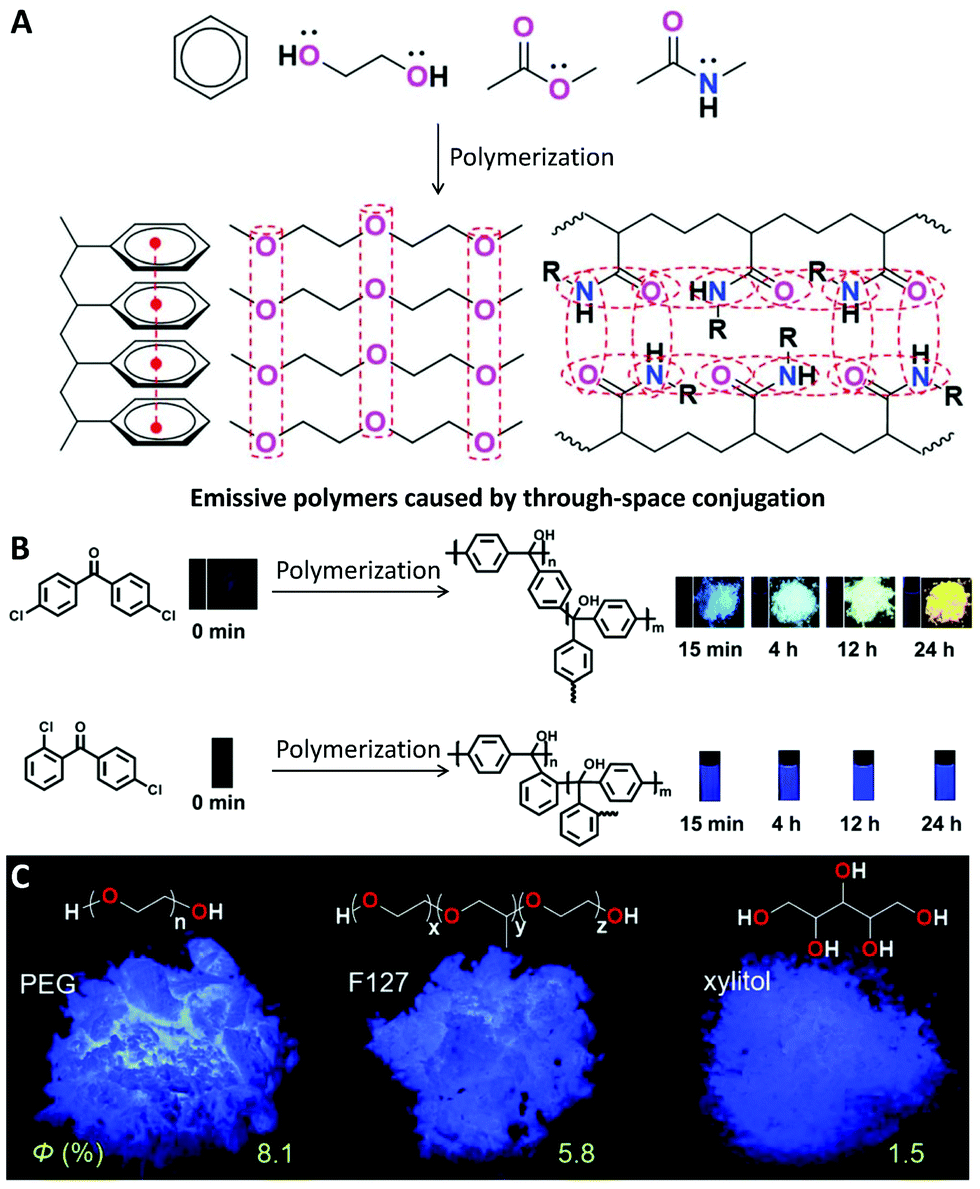 | ||
| Fig. 10 Polymerization-induced emission. (A) General mechanism of polymerization-induced emission by though-space conjugation. (B) Luminescence images of 4,4′-dichlorobenzophenone and 2,4′-dichlorobenzophenone at different polymerization time points under 365 nm UV irradiation. (C) Chemical structures of PEG, F127 and xylitol and their luminescence images in the solid state under 312 nm UV irradiation. (A was reprinted with permission from ref. 80, Copyright 2020, Royal Society of Chemistry; B was reprinted with permission from ref. 81, Copyright 2019, American Chemical Society; C was reprinted with permission from ref. 86, Copyright 2018, Wiley-VCH Verlag GmbH & Co.) | ||
Wan's work clearly demonstrated how the emission changes through the polymerization of dichlorobenzophenone monomers (Fig. 10B).82 Using non-emissive 4,4′-dichlorobenzophenone as the monomer, the obtained polytriphenylmethanols showed AIE properties, and the emission colour was red-shifted from blue to yellow in the solid state by increasing the polymerization time. While using 2,4′-dichlorobenzophenone as the monomer, the obtained polymer showed blue emission in solutions, and the brightness was gradually enhanced by increasing the polymerization time. During polymerization, the rotation of the phenyl groups was limited by the polymer chains, resulting in intramolecular though-space conjugation and charge transfer, further leading to the bright fluorescence.
The polymerization-induced emission effect has also been observed in some other systems without phenyl rings. Tang's group discovered that poly(maleic anhydride) and oligo(maleic anhydride) could emit bright fluorescence, while maleic anhydride itself is not emissive in both solution and the solid state.83,84 The reason is that polymerization could significantly decrease the distance between the carbonyl groups (2.84–3.18 Å) in the polymers to form the carbonyl cluster, and the strong interactions amongst the carbonyl groups induced orbital overlap by though-space conjugation, resulting in a low-lying LUMO state for visible emission. Afterwards, the polymerization of non-emissive N-hydroxysuccinimide methacrylate and poly(N-hydroxysuccinimide methacrylate) confirmed the polymerization-induced emission effect again.85
In fact, the polymerization-induced emission phenomenon was also observed in natural compounds or commercially available compounds. Poly(ethylene glycol) (PEG) and its other macromolecular analogues are typical examples. Although ethylene glycol, ethanol and diethyl ether are not emissive, PEG can show bright blue emission with a quantum yield of 8.1% in the solid state (Fig. 10C).86 Some other macromolecules with similar structures, such as F127,86 xylitol,86 and even hyperbranched polyethers,87 are also emissive in the aggregate state. In addition, the quantum yields of PEG (Mn = 20000) and F127 (Mn = 12000) are much higher than that of xylitol with a molecular weight of 152, indicating that higher molecular weights may also be important for the emission of these non-conjugated systems. The poly(amidoamine)-type dendrimer (PAMAM)88 and hyperbranched polymer89 are other typical commercially available luminescent polymers without traditional conjugated structures. Recently, some polymers have even been reported to show RTP rather than fluorescence via the similar though-space conjugation of clusters, further expanding the concept of polymerization-induced emission.90,91 With these developments, non-conjugated polymers have emerged as a new class of organic luminescent materials.
7. Conclusion and perspectives
In this paper, we summarized the recent progress in how to improve the performance of several types of optically active functional materials through simple polymerization. We discussed the polymerization-induced red shift in absorption and emission, polymerization-enhanced intersystem crossing, polymerization-enhanced photosensitization, the polymerization-enhanced two-photon cross-section, the polymerization-enhanced photocatalytic efficiency, polymerization-induced RTP, polymerization-induced TADF, polymerization-induced emission in non-conjugated systems, etc. In these cases, polymerization played a very important role in enhancing the optical properties through one or more of the following: (1) extending the conjugation of the monomers and subsequently affecting the energy levels of the polymers; (2) decreasing the distance between functional groups and subsequently enhancing intermolecular interactions; and (3) providing a rigid microenvironment to suppress non-radiative transitions, etc. Although many modelling results have been presented to explain most of these phenomena, it is ideal that detailed characterization and fast spectroscopy data are available to reveal all the processes to explain the obtained results more precisely.Based on the successful examples above, we naturally believe that some other properties may also be improved or modulated through polymerization. One example is enhancing the photothermal conversion by polymerization. The polymerization of planar aromatic monomers can enhance the π–π stacking of polymer chains, leading to quenched emission and enhanced non-radiative transitions in the aggregated state. Coupled with the enhanced light-harvesting ability discussed in Fig. 2, we can expect that a higher photothermal conversion can be achieved by conjugated polymers with planar structures. In this regard, the higher the photothermal therapy efficiency, the better the photoacoustic image performance, and an even higher efficiency of sunlight-induced seawater desalination by evaporation can be realized through polymerization of the currently available small molecules.
Despite the success of polymerization in improving the optical performance, several issues or challenges should be studied. First, the polymer type has not yet been carefully screened. Taking polymerization-enhanced photosensitization as an example, nearly all the conjugated polymer PSs reported so far have donors and acceptors in the polymer backbone. Other types of conjugated polymers, such as hyperbranched or even non-conjugated polymers, could be tested to understand how the polymer structures are correlated with the photosensitization functions. The second issue is related to the defects in polymeric materials. So far, there have been only a few studies focusing on understanding how these defects can affect the optical properties. Recent examples have proven that the field effect transistor performance92 and brighter fluorescence93 can be significantly improved by removing the defects of conjugated polymers. A better understanding of these defects will help to further improve the optical properties of polymeric materials, which should also contribute to polymerization-enhanced optical properties. In addition, we should also focus on the precise adjustment of polymeric structures, including the control of both chemical structures and molecular weights, which are directly related to the performance of the obtained polymer. As compared to chemical structures, the effects of molecular weight on the optical properties of materials have been less studied. The examples of both PTPEAQ2 and PSS revealed that different molecular weights could lead to very different optical properties, which prompted us to develop new polymerization methods for more controllable polymerization. It is optimistic that polymerization will continue to be a very effective strategy for yielding new materials with an optimized performance for many innovative high-tech applications.
Abbreviations
| PS | Photosensitizer |
| S | Singlet states |
| T | Triplet states |
| Sn | The nth singlet state |
| Tn | The nth triplet state |
| S0 | The ground state |
| IC | Internal conversion |
| ISC | Intersystem crossing |
| RISC | Reverse intersystem crossing |
| PEX | Polymerization-enhanced intersystem crossing |
| PDT | Photodynamic therapy |
| 1O2 | Singlet oxygen |
| ROS | Reactive oxygen species |
| RTP | Room temperature phosphorescence |
| TADF | Thermally activated delayed fluorescence |
| ΔEST | The energy gap between the excited singlet state and the triplet state |
| DFT | Density functional theory |
| MO | Molecular orbital |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| NPs | Nanoparticles |
| 2PACS | Two-photon absorption cross-section |
| RAFT | Reversible addition–fragmentation chain transfer |
| OLED | Organic light-emitting diode |
| AIE | Aggregation-induced emission |
| ACQ | Aggregation-caused quenching |
| PEG | Poly(ethylene glycol) |
| PAMAM | Poly(amidoamine)-type dendrimer |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Bin Liu is grateful for financial support of the Singapore NRF Competitive Research Program (R279-000-483-281), the NRF Investigatorship (R279-000-444-281), and the National University of Singapore (R279-000-482-133). Wenbo Wu acknowledges the National Natural Science Foundation of China (22075199) for financial support.Notes and references
- W. Wu, R. Tang, Q. Li and Z. Li, Functional hyperbranched polymers with advanced optical, electrical and magnetic properties, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC.
- P. Yang, S. Zhang, X. Chen, X. Liu, Z. Wang and Y. Li, Recent developments in polydopamine fluorescent nanomaterials, Mater. Horiz., 2020, 7, 746–761 RSC.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Near-infrared phosphorescence: materials and applications, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- F. Hu, S. Xu and B. Liu, Photosensitizers with aggregation-induced emission: materials and biomedical applications, Adv. Mater., 2018, 30, 1801350 CrossRef.
- W. Wu, J. Qin and Z. Li, New design strategies for second-order nonlinear optical polymers and dendrimers, Polymer, 2013, 54, 4351–4382 CrossRef CAS.
- G. Chen, J. Seo, C. Yang and P. N. Prasad, Nanochemistry and nanomaterials for photovoltaics, Chem. Soc. Rev., 2013, 42, 8304–8338 RSC.
- L. Yang, Y. Peng, X. Luo, Y. Dan, J. Ye, Y. Zhou and Z. Zou, Beyond C3N4 π-conjugated metal-free polymeric semiconductors for photocatalytic chemical transformations, Chem. Soc. Rev., 2021, 50, 2147–2172 RSC.
- M. Zhang, C.-J. Zheng, H. Lin and S.-L. Tao, Thermally activated delayed fluorescence exciplex emitters for high-performance organic light-emitting diodes, Mater. Horiz., 2021, 8, 401–425 RSC.
- X. Cai and B. Liu, Aggregation-Induced Emission: Recent advances in materials and biomedical applications, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS.
- Z. Cheng, W. Qi, C. H. Pang, T. Thomas, T. Wu, S. Liu and M. Yang, Recent advances in transition metal nitride-based materials for photocatalytic applications, Adv. Funct. Mater., 2021, 31, 2100553 CrossRef CAS.
- J. Yang, M. Fang and Z. Li, Stimulus-responsive room temperature phosphorescence in purely organic luminogens, InfoMater, 2020, 2, 791–806 CrossRef CAS.
- S. Liang, X. Jiang, C. Xiao, C. Li, Q. Chen and W. Li, Double-cable conjugated polymers with pendant rylene diimides for single-component organic solar cells, Acc. Chem. Res., 2021, 54, 2227–2237 CrossRef CAS.
- A. Jabłoński, Efficiency of anti-Stokes fluorescence in dyes, Nature, 1933, 131, 839–840 CrossRef.
- M. Kasha, Energy transfer mechanisms and the molecular exciton model for molecular aggregates, Radiat. Res., 1963, 20, 55–70 CrossRef CAS.
- D. L. Dexter, A theory of sensitized Luminescence in Solids, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Triplet photosensitizers: from molecular design to applications, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- C. A. Robertson, D. H. Evans and H. Abrahamse, Photodynamic therapy (PDT): a short review on cellular mechanisms and cancer research applications for PDT, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS.
- N. A. Kukhta and M. R. Bryce, Dual emission in purely organic materials for optoelectronic applications, Mater. Horiz., 2021, 8, 33–55 RSC.
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, Recent progress in pure organic room temperature phosphorescence of small molecular host–guest systems, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- X.-K. Chen, D. Kim and J.-L. Brédas, Thermally activated delayed fluorescence (TADF) path toward efficient electroluminescence in purely organic materials: molecular level insight, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Recent advances in organic thermally activated delayed fluorescence materials, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, Highly efficient and photostable photosensitizer based on BODIPY chromophore, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS.
- S. Cekli, R. W. Winkel, E. Alarousu, O. F. Mohammed and K. S. Schanze, Triplet excited state properties in variable gap π-conjugated donor–acceptor–donor chromophores, Chem. Sci., 2016, 7, 3621–3631 RSC.
- J. Liu, C. Ji, B. Yuan, X. Liu, Y. Chen, L. Ji and H. Chao, Selectively lighting up two-photon photodynamic activity in mitochondria with AIE-active iridium(III) complexes, Chem. Commun., 2017, 53, 2052–2055 RSC.
- P. She, Y. Yu, Y. Qin, Y. Zhang, F. Li, Y. Ma, S. Liu, W. Huang and Q. Zhao, Controlling organic room temperature phosphorescence through external heavy-atom effect for white light emission and luminescence printing, Adv. Opt. Mater., 2020, 8, 1901437 CrossRef CAS.
- W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C.-J. Zhang, X. Cheng, Y. Yuan, D. Ding, D. Kong and B. Liu, A highly efficient and photostable photosensitizer with near-infrared aggregation-induced emission for image-guided photodynamic anticancer therapy, Adv. Mater., 2017, 29, 1700548 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS.
- P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, Exceptionally fast radiative decay of a dinuclear platinum complex through thermally activated delayed fluorescence, Chem. Sci., 2021, 12, 6172–6180 RSC.
- Y. Lei, J. Yang, W. Dai, Y. Lan, J. Yang, X. Zheng, J. Shi, B. Tong, Z. Cai and Y. Dong, Efficient and organic host–guest room-temperature phosphorescence: tunable triplet–singlet crossing and theoretical calculations for molecular packing, Chem. Sci., 2021, 12, 6518–6525 RSC.
- X. Sun, X. Wang, X. Li, J. Ge, Q. Zhang, J. Jiang and G. Zhang, Polymerization-enhanced intersystem crossing: new strategy to achieve long-lived excitons, Macromol. Rapid Commun., 2015, 36, 298–303 CrossRef CAS.
- W. Wu, D. Mao, S. Xu, Kenry, F. Hu, X. Li, D. Kong and B. Liu, Polymerization-enhanced photosensitization, Chem, 2018, 4, 1937–1951 CAS.
- Z.-P. Yu, K. Yan, W. Ullah, H. Chen and C.-Z. Li, Conjugated polymers for photon-to-electron and photon-to-fuel conversions, ACS Appl. Polym. Mater., 2021, 3, 60–92 CrossRef CAS.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Water-soluble conjugated polymers for imaging, diagnosis, and therapy, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS.
- A. Tsuda and O. Atsuhiro, Fully conjugated porphyrin tapes with electronic absorption bands that reach into infrared, Science, 2001, 293, 79–82 CrossRef CAS.
- T. Geiger, H. Benmansour, B. Fan, R. Hany and F. Nüesch, Low-band gap polymeric cyanine dyes absorbing in the NIR region, Macromol. Rapid Commun., 2008, 29, 651–658 CrossRef CAS.
- Z. Li, T. R. Ensley, H. Hu, Y. Zhang, S.-H. Jang, S. R. Marder, D. J. Hagan, E. W. V. Stryland and A. K.-Y. Jen, Conjugated polycyanines: a new class of materials with large third-order optical nonlinearities, Adv. Opt. Mater., 2015, 3, 900–906 CrossRef CAS.
- M. Champeau, S. Vignoud, L. Mortier and S. Mordon, Photodynamic therapy for skin cancer: how to enhance drug penetration, J. Photochem. Photobiol., B, 2019, 197, 111544 CrossRef CAS.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, The unique features and promises of phthalocyanines as advanced photosensitizers for photodynamic therapy of cancer, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- Q. Jia, Q. Song, P. Li and W. Huang, Rejuvenated photodynamic therapy for bacterial infections, Adv. Healthcare Mater., 2019, 8, 1900608 CrossRef.
- S. S. Lucky, K. C. Soo and Y. Zhang, Nanoparticles in photodynamic therapy, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS.
- W. Wu, G. Feng, S. Xu and B. Liu, A photostable far-red/near-infrared conjugated polymer photosensitizer with aggregation-induced emission for image-guided cancer cell ablation, Macromolecules, 2016, 49, 5017–5025 CrossRef CAS.
- G. Feng, W. Wu, S. Xu and B. Liu, Far red/near-infrared AIE dots for image-guided photodynamic cancer cell ablation, ACS Appl. Mater. Interfaces, 2016, 8, 21193–21200 CrossRef CAS.
- W. Wu, G. C. Bazan and B. Liu, Conjugated-polymer-amplified sensing, imaging, and therapy, Chem, 2017, 2, 760–790 CAS.
- S. Cekli, R. W. Winkel and K. S. Schanze, Effect of Oligomer Length on Photophysical Properties of Platinum Acetylide Donor–Acceptor–Donor Oligomers, J. Phys. Chem. A, 2016, 120, 5512–5521 CrossRef CAS.
- W. Wu, High-performance conjugated polymer photosensitizers, Chem, 2018, 4, 1762–1764 CAS.
- S. Liu, H. Zhang, Y. Li, J. Liu, L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Strategies to enhance the photosensitization: polymerization and the donor–acceptor even-odd effect, Angew. Chem., Int. Ed., 2018, 57, 15189–15193 CrossRef CAS.
- T. Zhou, R. Hu, L. Wang, Y. Qiu, G. Zhang, Q. Deng, H. Zhang, P. Yin, B. Situ, C. Zhan, A. Qin and B. Z. Tang, An AIE-active conjugated polymer with high ROS-generation ability and biocompatibility for efficient photodynamic therapy of bacterial infections, Angew. Chem., Int. Ed., 2020, 59, 9952–9956 CrossRef CAS.
- H. Yao, J. Dai, Z. Zhuang, J. Yao, Z. Wu, S. Wang, F. Xia, J. Zhou, X. Lou and Z. Zhao, Red AIE conjugated polyelectrolytes for long-term tracing and image-guided photodynamic therapy of tumors, Sci. China: Chem., 2020, 63, 1815–1824 CrossRef CAS.
- Q. Xu, F. Lv, L. Liu and S. Wang, Development of a thermo-responsive conjugated polymer with photobleaching-resistance property and tunable photosensitizing performance, Macromol. Rapid Commun., 2020, 41, 2000249 CrossRef CAS.
- L. Hu, Z. Chen, Y. Liu, B. Tian, T. Guo, R. Liu, C. Wang and L. Ying, In vivo bioimaging and photodynamic therapy based on two-photon fluorescent conjugated polymers containing dibenzothiophene-S,S-dioxide derivatives, ACS Appl. Mater. Interfaces, 2020, 12, 57281–57289 CrossRef CAS.
- B. Gu, K.-T. Yong and B. Liu, Strategies to overcome the limitations of AIEgens in biomedical applications, Small Methods, 2018, 2, 1700392 CrossRef.
- W. Wu and B. Liu, Two-photon excitable photosensitizers with aggregation-induced emission and their biomedical applications, Chem. J. Chin. Univ., 2020, 41, 191–203 CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Multiphoton absorbing materials: molecular designs, characterizations, and applications, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS.
- B. Gu, W. Wu, G. Xu, G. Feng, F. Yin, P. H. J. Chong, J. Qu, K.-T. Yong and B. Liu, Precise two-photon photodynamic therapy using efficient photosensitizer with aggregation-induced emission characteristics, Adv. Mater., 2017, 29, 1701076 CrossRef.
- S. Wang, W. Wu, P. N. Manghnani, S. Xu, Y. Wang, C. C. Goh, L. G. Ng and B. Liu, Polymerization-enhanced two-photon photosensitization for precise photodynamic therapy, ACS Nano, 2019, 13, 3095–3105 CrossRef CAS.
- G. Jiang, J. Chen, J.-S. Huang and C.-M. Che, Highly efficient oxidation of amines to imines by singlet oxygen and its application in Ugi-type reactions, Org. Lett., 2009, 11, 4568–4571 CrossRef CAS.
- R. Li, Z. J. Wang, L. Wang, B. C. Ma, S. Ghasimi, H. Lu, K. Landfester and K. A. I. Zhang, Photocatalytic selective bromination of electron-rich aromatic compounds using microporous organic polymers with visible light, ACS Catal., 2016, 6, 1113–1121 CrossRef CAS.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, Singlet oxygen-engaged selective photo-oxidation over Pt nanocrystals/porphyrinic MOF: the roles of photothermal effect and Pt electronic state, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS.
- W. Wu, S. Xu, G. Qi, H. Zhu, F. Hu, Z. Liu, D. Zhang and B. Liu, A cross-linked conjugated polymer photosensitizer enables efficient sunlight-induced photooxidation, Angew. Chem., Int. Ed., 2019, 58, 3062–3066 CrossRef CAS.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F.-S. Xiao, Programming covalent organic frameworks for photocatalysis: investigation of chemical and structural variations, Matter, 2020, 2, 416–427 CrossRef.
- J. Jiang, Z. Liang, X. Xiong, X. Zhou and H. Ji, A carbazolyl porphyrin-based conjugated microporous polymer for metal-free photocatalytic aerobic oxidation reactions, ChemCatChem, 2020, 12, 3523–3529 CrossRef CAS.
- J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Conjugated microporous polymers with rose bengal dye for highly efficient heterogeneous organo-photocatalysis, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- J. Li, Z. An, J. Sun, C. Tan, D. Gao, Y. Tan and Y. Jiang, Highly selective oxidation of organic sulfides by a conjugated polymer as the photosensitizer for singlet oxygen generation, ACS Appl. Mater. Interfaces, 2020, 12, 35475–35481 CrossRef CAS.
- G. Baryshnikov, B. Minaev and H. Ågren, Theory and calculation of the phosphorescence phenomenon, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS.
- T. Itoh, Fluorescence and phosphorescence from higher excited states of organic molecules, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, Crystallization-induced phosphorescence of pure organic luminogens at room temperature, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, The influence of the molecular packing on the room temperature phosphorescence of purely organic luminogens, Nat. Commun., 2018, 9, 840 CrossRef.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Carbazole isomers induce ultralong organic phosphorescence, Nat. Mater., 2021, 20, 175–180 CrossRef CAS.
- Y. Yu, M. S. Kwon, J. Jung, Y. Zeng, M. Kim, K. Chung, J. Gierschner, J. H. Youk, S. M. Borisov and J. Kim, Room-temperature-phosphorescence-based dissolved oxygen detection by core–shell polymer nanoparticles containing metal-free organic phosphors, Angew. Chem., Int. Ed., 2017, 56, 16207–16211 CrossRef CAS.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Tailoring intermolecular interactions for efficient room-temperature phosphorescence from purely organic materials in amorphous polymer matrices, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Suppressing molecular motions for enhanced room-temperature phosphorescence of metal-free organic materials, Nat. Commun., 2015, 6, 8947 CrossRef.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Ultralong room temperature phosphorescence from amorphous organic materials toward confidential information encryption and decryption, Sci. Adv., 2018, 4, eaas9732 CrossRef.
- T. Ogoshi, H. Tsuchida, T. Kakuta, T. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Ultralong room-temperature phosphorescence from amorphous polymer poly(styrene sulfonic acid) in air in the dry solid state, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- M. Fang, J. Yang and Z. Li, Recent advances in purely organic room temperature phosphorescence polymer, Chin. J. Polym. Sci., 2019, 37, 383–393 CrossRef CAS.
- X. Ma, C. Xu, J. Wang and H. Tian, Amorphous pure organic polymers for heavy-atom-free efficient room-temperature phosphorescence emission, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS.
- J.-M. Teng, Y.-F. Wang and C.-F. Chen, Recent progress of narrowband TADF emitters and their applications in OLEDs, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC.
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Thermally activated delayed fluorescence in polymers: a new route toward highly efficient solution processable OLEDs, Adv. Mater., 2015, 27, 7236–7240 CrossRef CAS.
- Q. Wei, P. Kleine, Y. Karpov, X. Qiu, H. Komber, K. Sahre, A. Kiriy, R. Lygaitis, S. Lenk, S. Reineke and B. Voit, Conjugation-induced thermally activated delayed fluorescence (TADF): from conventional non-TADF units to TADF-active polymers, Adv. Funct. Mater., 2017, 27, 1605051 CrossRef.
- Q. Wei, P. Imbrasas, E. Caldera-Cruz, L. Cao, N. Fei, H. Thomas, R. Scholz, S. Lenk, B. Voit, S. Reineke and Z. Ge, Conjugation-induced thermally activated delayed fluorescence: photophysics of a carbazole-benzophenone monomer-to-tetramer molecular series, J. Phys. Chem. A, 2021, 125, 1345–1354 CrossRef CAS.
- T. Jiang, Y. Liu, Z. Ren and S. Yan, The design, synthesis and performance of thermally activated delayed fluorescence macromolecules, Polym. Chem., 2020, 11, 1555–1571 RSC.
- B. Liu, H. Zhang, S. Liu, J. Sun, X. Zhang and B. Z. Tang, Polymerization-induced emission, Mater. Horiz., 2020, 7, 987–998 RSC.
- Y.-N. Jing, S.-S. Li, M. Su, H. Bao and W.-M. Wan, Barbier hyperbranching polymerization-induced emission toward facile fabrication of white light-emitting diode and light-harvesting film, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef CAS.
- E. Zhao, J. W. Y. Lam, L. Meng, Y. Hong, H. Deng, G. Bai, X. Huang, J. Hao and B. Z. Tang, Poly[(maleic anhydride)-alt-(vinyl acetate)]: a pure oxygenic nonconjugated macromolecule with strong light emission and solvatochromic effect, Macromolecules, 2015, 48, 64–71 CrossRef CAS.
- X. Zhou, W. Luo, H. Nie, L. Xu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, Oligo(maleic anhydride)s: a platform for unveiling the mechanism of clusteroluminescence of non-aromatic polymers, J. Mater. Chem. C, 2017, 5, 4775–4779 RSC.
- X. Bin, W. Luo, W. Z. Yuan and Y. Zhang, Clustering-triggered emission of poly(N-hydroxysuccinimide methacrylate), Acta Chim. Sin., 2016, 74, 935–941 CrossRef CAS.
- Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Emission and emissive mechanism of nonaromatic oxygen clusters, Macromol. Rapid Commun., 2018, 39, 1800528 CrossRef.
- X. Miao, T. Liu, C. Zhang, X. Geng, Y. Meng and X. Li, Fluorescent aliphatic hyperbranched polyether: chromophore-free and without any N and P atoms, Phys. Chem. Chem. Phys., 2016, 18, 4295–4299 RSC.
- W. I. Lee, Y. Bae and A. J. Bard, Strong blue photoluminescence and ECL from OH-terminated PAMAM dendrimers in the absence of gold nanoparticles, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS.
- Y.-Z. You, Z.-Q. Yu, M.-M. Cui and C.-Y. Hong, Preparation of photoluminescent nanorings with controllable bioreducibility and stimuli-responsiveness, Angew. Chem., Int. Ed., 2010, 49, 1099–1102 CrossRef CAS.
- D. Wang, X. Wang, C. Xu and X. Ma, A novel metal-free amorphous room-temperature phosphorescent polymer without conjugation, Sci. China: Chem., 2019, 62, 430–433 CrossRef CAS.
- X. Chen, Z. He, F. Kausar, G. Chen, Y. Zhang and W. Z. Yuan, Aggregation-induced dual emission and unusual luminescence beyond excimer emission of poly(ethylene terephthalate), Macromolecules, 2018, 51, 9035–9042 CrossRef CAS.
- Z. Ni, H. Wang, H. Dong, Y. Dang, Q. Zhao, X. Zhang and W. Hu, Mesopolymer synthesis by ligand-modulated direct arylation polycondensation towards n-type and ambipolar conjugated systems, Nat. Chem., 2019, 11, 271–277 CrossRef CAS.
- X. Guo, Y. Zhang, Y. Hu, J. Yang, Y. Li, Z. Ni, H. Dong and W. Hu, Molecular weight engineering in high-performance ambipolar emissive mesopolymers, Angew. Chem., Int. Ed., 2021, 60, 14902–14908 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |