
Modulating the optical properties and functions of organic molecules through polymerization

Abstract
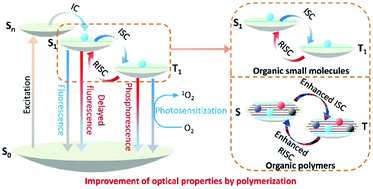
Organic functional materials with advanced optical properties have attracted much attention due to their broad applications, such as in light-emitting diodes, solar cells, anti-counterfeiting, photocatalysis, and even disease diagnosis and treatment. Recent research has revealed that many optical properties of organic molecules can be improved through simple polymerization. In this review, we discuss the phenomenon, mechanism, and impact of polymerization on the properties of materials, including the polymerization-induced spectral shift, polymerization-enhanced photosensitization, polymerization-enhanced two-photon absorption, polymerization-enhanced photocatalytic efficiency, polymerization-induced room temperature phosphorescence, polymerization-induced thermally activated delayed fluorescence, and polymerization-induced emission using specific examples with different applications. The new opportunities arising from polymerization in designing high performance optical materials are summarized in the future perspective.
- This article is part of the themed collection: Special issue in honour of Seth Marder
 Please wait while we load your content...
Please wait while we load your content...

