DOI:
10.1039/D1MH00821H
(Communication)
Mater. Horiz., 2022,
9, 281-293
Design and synthesis of yellow- to red-emitting gold(III) complexes containing isomeric thienopyridine and thienoquinoline moieties and their applications in operationally stable organic light-emitting devices†‡
Received
24th May 2021
, Accepted 12th August 2021
First published on 2nd September 2021
Abstract
A new class of yellow- to red-emitting carbazolylgold(III) complexes containing isomeric thienopyridine or thienoquinoline moieties in the cyclometalating ligand has been designed and synthesized, which showed high photoluminescence quantum yields of over 80% in solid-state thin films. The isomeric effect and extended π-conjugation of the N-heterocycles have been found to remarkably perturb the photophysical, electrochemical and electroluminescence properties of the gold(III) complexes. In particular, the operational lifetimes of organic light-emitting devices based on that incorporated with thieno[2,3-c]pyridine are almost three orders of magnitude longer than that incorporated with thieno[3,2-c]pyridine. This has led to long device operational stability with a LT70 value of up to 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h at a luminance of 100 cd m−2 and a long half-lifetime of 206800 h, as well as maximum external quantum efficiencies of up to 8.6% and 14.5% in the solution-processed and vacuum-deposited devices, respectively. This work provides insights into the development of robust and highly luminescent gold(III) complexes and the identification of stable molecular motifs for designing efficient emitters.
200 h at a luminance of 100 cd m−2 and a long half-lifetime of 206800 h, as well as maximum external quantum efficiencies of up to 8.6% and 14.5% in the solution-processed and vacuum-deposited devices, respectively. This work provides insights into the development of robust and highly luminescent gold(III) complexes and the identification of stable molecular motifs for designing efficient emitters.
New concepts
In this manuscript, we present the design and synthesis of yellow- to red-emitting gold(III) complexes and their applications in operationally stable organic light-emitting devices (OLEDs). Through incorporating six different N-heterocycles into the cyclometalating ligand, these isomeric gold(III) complexes have been found to exhibit distinctly different photophysical, electrochemical and electroluminescence properties. The investigation of the isomeric effect and extended π-conjugation of the N-heterocycles has never been reported in gold(III) systems, and is also scarcely reported in the widely studied iridium(III) and platinum(II) systems. Considering the radical anion stability and the resonance structures of the N-heterocyclic moieties, we believe that a proper choice of the isomer in the emitting material could lead to prolonged device operational lifetimes.
|
Introduction
A great commercial success of active matrix organic light-emitting devices (AMOLEDs) has been demonstrated in the state-of-the-art flat-panel displays, televisions and mobile phones. With continuous research and development, commercial products equipped with a high resolution of 8 K and a high absolute color accuracy for OLED displays are anticipated to be the new standards in the coming years. Since the first report on the utilization of transition metal complexes as dopants in OLEDs,1 tremendous efforts have been made towards the design and synthesis of tailor-made emitters to effectively tune their emission colors to meet the industrial requirements. Meanwhile, comprehensive studies on the device performance based on the structural variations of emitters have also been carried out. In particular, great efforts have been made to realize highly efficient emitters with high photoluminescence quantum yields (PLQYs), short excited-state lifetimes and tunable emission colors ranging from sky blue to saturated red.2–6 To date, with the advancement in sophisticated studies and the development of material synthesis, together with advanced device engineering, high external quantum efficiencies (EQEs) of over 30% have been realized in the OLEDs based on iridium(III)7–10 and platinum(II)11 complexes and thermally activated delayed fluorescent (TADF) compounds12,13 without the use of any light extraction techniques.
In view of the fact that the emission energies of emitters are heavily dependent on the choice of N-heterocyclic moieties and the substituent groups, which could significantly alter the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), structural modifications of the cyclometalating or ancillary ligands are considered as the conventional strategy for color tuning.14 Various types of N-heterocyclic moieties, such as pyridine, quinoline, benzoxazole, benzimidazole, thiophene and carbazole, have been commonly adopted to tune the photoluminescence and electrochemical properties of the metal complexes.15,16 As a result, numerous transition metal complexes incorporated with these N-heterocyclic moieties have been reported and some have been demonstrated to show superior electroluminescence (EL) performances and long device operational lifetimes in the fabricated OLEDs. For example, Tao, Lee and co-workers reported an iridium(III) complex with a benzoxazole-substituted ancillary ligand in 2013,17 in which the bulky ancillary ligand was incorporated to suppress molecular aggregation and to reduce the bimolecular quenching process. The LT50, defined as the operational lifetime at 50% of the initial luminance, of the fabricated device based on this complex was estimated to be over 30000 h at 100 cd m−2.17 Indeed, there have been a number of reports and investigations on the long device operational lifetimes of OLEDs based on transition metal complexes with different host materials and device configurations,18–24 which demonstrated excellent EL performances; however, little is known about the effect of isomeric N-heterocyclic moieties of the metal complexes on the EL properties and the device operational stability. In view of this, it would be motivating to study and investigate the structure–property relationship between the chemical structures of emitters and EL performances of their corresponding devices, which is envisaged to provide important insights into the future development of robust and highly efficient emitters.
Having an isoelectronic d8 configuration to the platinum(II) complexes, gold(III) complexes have been considered as another promising class of emitting materials for OLED applications, as exemplified by their high EL performances25–29 that are comparable with those of the iridium(III) and platinum(II) systems.14,30,31 Interestingly, thermally stimulated delayed phosphorescence (TSDP)32,33 and TADF26,27,34–36 behavior have been recently reported in the gold(III) system. While the design and synthesis of highly efficient gold(III) emitters have been increasingly reported, the investigation of the effect of the isomeric N-heterocyclic moieties on the photophysical, electrochemical and EL properties has never been reported in the gold(III) systems and is also scarcely reported in the widely studied iridium(III) and platinum(II) systems. Capitalizing on our recent results found for the carbazolylgold(III) complexes and their application studies,25,29,37 in which maximum EQEs of up to 21.6% and a LT50 value of over 83000 h at 100 cd m−2 have been achieved,25 structural modifications with the integration of isomeric and π-extended N-heterocycles into the cyclometalating ligand are anticipated to afford a new series of color-tunable, highly efficient and stable gold(III) complexes. On the other hand, metal-containing emitters incorporated with thiophene-fused N-heterocycles, such as thieno[3,2-c]pyridine38–40 and thieno[3,2-c]quinoline41 moieties, which differ from the conventional types of pyridine and isoquinoline moieties, have been one of the attractive emitters for device applications. In particular, it is worth mentioning that the iridium(III) complex, iridium(III) bis(4-phenylthieno[3,2-c]pyridinato-N,C2′) acetylacetonate (PO-01),42,43 has been demonstrated to provide the complementary yellow emission for highly efficient phosphorescent44–46 and TADF47–49 white OLEDs, suggesting the high potential of these emitters incorporated with thiophene-fused N-heterocycles for device applications. However, to the best of our knowledge, the reports on these thiophene-fused N-heterocycle-containing iridium(III) complexes to date are mostly focused on one form of the N-heterocycle, i.e. thieno[3,2-c]pyridine; in addition, the isomeric effect of these N-heterocycles on the properties of the metal complexes has not yet been investigated.
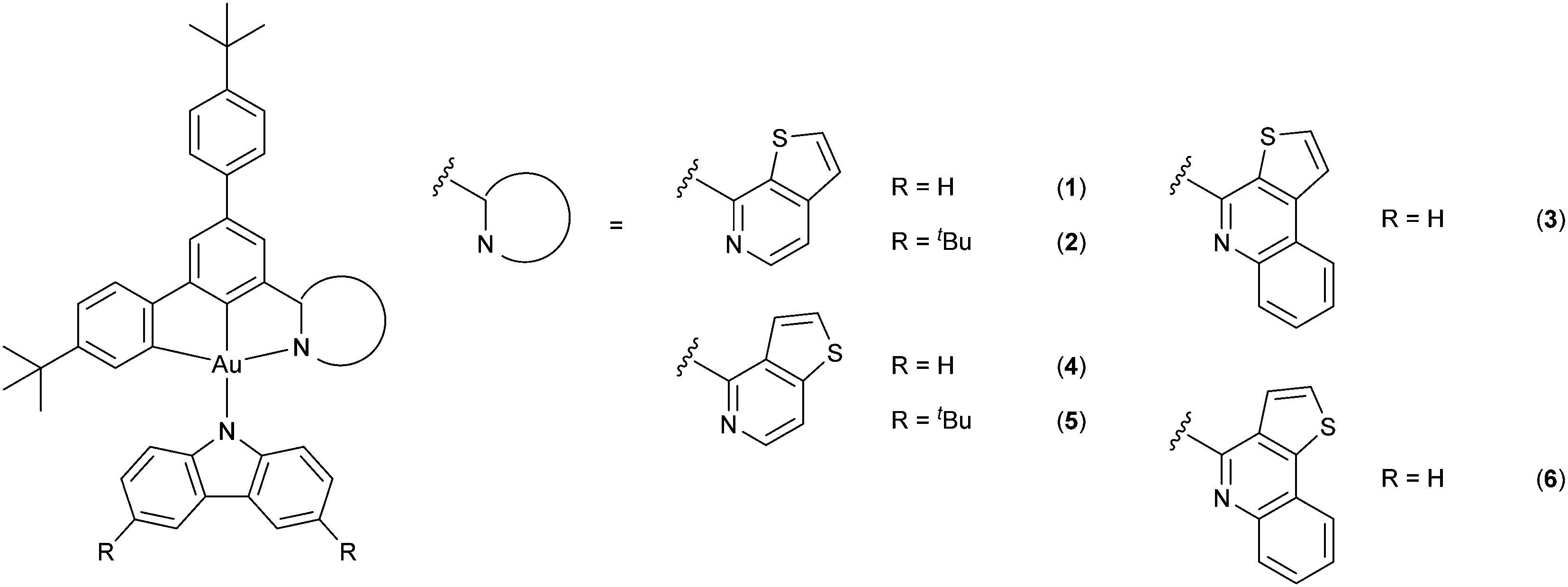
In this contribution, a new class of isomeric thienopyridine- and thienoquinoline-containing carbazolylgold(III) complexes 1–6 has been designed and synthesized, with their molecular structures shown in Scheme 1. Based on the versatile design of the tridentate ligand framework, structurally related N-heterocyclic moieties, namely thieno[2,3-c]pyridine (1-thpy) (1, 2), thieno[2,3-c]quinoline (1-thq) (3), thieno[3,2-c]pyridine (2-thpy) (4, 5) and thieno[3,2-c]quinoline (2-thq) (6), have been incorporated into the cyclometalating ligand. These N-heterocyclic moieties have been found to remarkably perturb the LUMO energy of the complexes, as reflected by the tuning of the emission colors from yellow to red with high PLQYs of over 80% in solid-state thin films. It is worth noting that the isomeric effect and extended π-conjugation of the N-heterocycles have been found to significantly influence the operational stability of the fabricated OLEDs, in addition to the photophysical and electrochemical properties of the complexes. Specifically, the device operational lifetimes based on those incorporated with 1-thpy (i.e.1 and 2) have been found to be almost three orders of magnitude over those incorporated with 2-thpy (i.e.4 and 5), owing to different positions of the sulfur atom and the resonance structures of the thienopyridine moieties. These have led to the long device operational stability with a LT70 value, defined as the operational lifetime at 70% of the initial luminance, of ca. 63200 h at a luminance of 100 cd m−2. Yellow- to red-emitting solution-processed and vacuum-deposited OLEDs based on these complexes have also been realized, exhibiting maximum EQEs of up to 8.6% and 14.5%, respectively. This work is anticipated to provide insights into the development of robust and highly luminescent gold(III) complexes, as well as the identification of stable molecular motifs for designing efficient emitters.
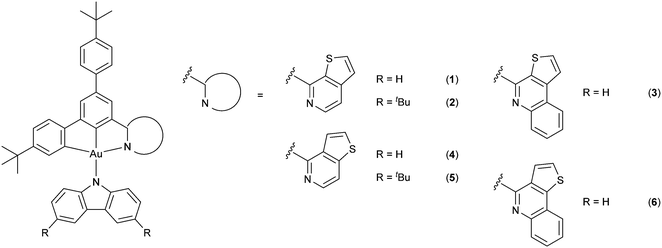 |
| | Scheme 1 Molecular structures of the carbazolylgold(III) complexes. | |
Results and discussion
Synthesis, characterization and thermal stability of the gold(III) complexes
All of the gold(III) complexes were synthesized by reacting carbazole or 3,6-di-tert-butylcarbazole with the corresponding chlorogold(III) precursors in the presence of sodium hydride in degassed tetrahydrofuran (Scheme S1, ESI‡). After isolating the crude product, recrystallization was carried out by diffusion of diethyl ether vapor into a concentrated dichloromethane solution of the complexes to give yellow to orange–red solids. All the carbazolylgold(III) complexes were obtained by vacuum sublimation and characterized by 1H and 13C{1H} nuclear magnetic resonance spectroscopies, high-resolution electrospray ionization mass spectrometry and satisfactory elemental analyses. High-resolution ESI mass spectroscopy with a cryospray source, i.e. cryospary ionization (CSI)-MS, was also employed to provide further support for the high purity of 1 and 4. Thermogravimetric analyses (TGA) were performed to study the thermal stability of these complexes. Fig. S1 (ESI‡) depicts the TGA curves of 1–6 and Table S1 (ESI‡) summarizes their decomposition temperatures (Td). In general, this class of complexes demonstrates a high Td of over 300 °C. Interestingly, the incorporation of thienopyridine (thpy) moieties into the complexes (i.e.1, 2, 4 and 5) results in a similar Td of ca. 330 °C, regardless of the position of the sulfur atom. On the other hand, the positional effect of the sulfur atom in the thienoquinoline (thq) moieties leads to a notable enhancement of Td of almost 60 °C in 3 (Td = 363 °C) relative to 6 (Td = 304 °C). These results suggest the higher robustness of 3 than 6, which is in good agreement with the observation during OLED fabrication (see below).
Electrochemistry
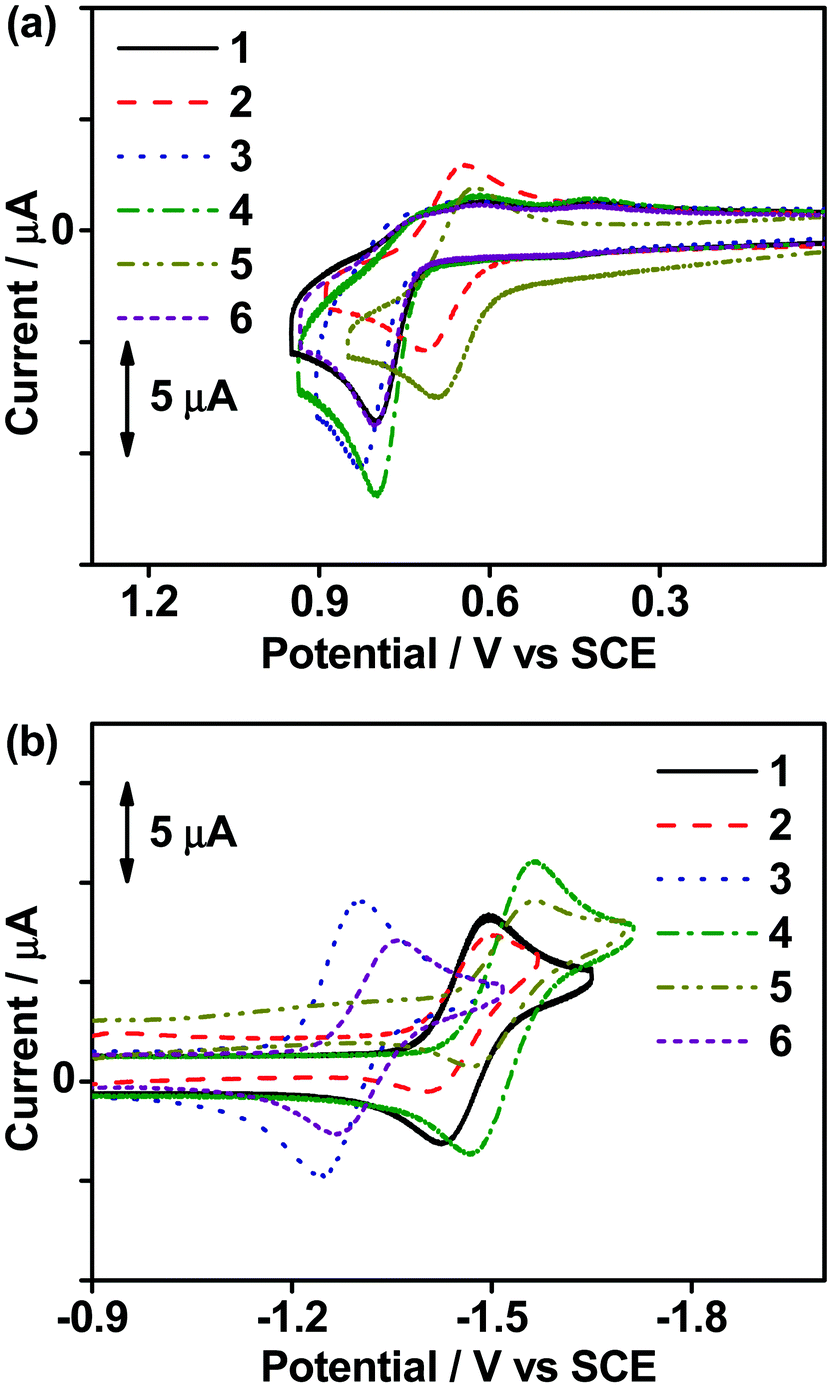
The electrochemical properties of all the complexes in dichloromethane (0.1 M nBu4NPF6) were investigated by cyclic voltammetry at 298 K. In general, except for 2 and 5, all the complexes showed an irreversible oxidation wave at ca. +0.80 V vs. standard calomel electrode (SCE). The cyclic voltammograms of the oxidation and reduction scans of the complexes are shown in Fig. 1 and the data are listed in Table S2 (ESI‡). With the incorporation of σ-donating tert-butyl (tBu) groups into the carbazole, 2 and 5 show a shift of the potential for oxidation to a less positive value and result in a quasi-reversible oxidation couple at ca. +0.66 V vs. SCE. The oxidation waves of all the complexes are assigned as the carbazole ligand-centered oxidation.25,37 On the other hand, all the complexes feature quasi-reversible reduction waves ranging from −1.27 to −1.52 V vs. SCE, which are found to be sensitive to the choice of the cyclometalating ligand. Apparently, the integration of 1-thpy in 1 and 2 results in a reduction potential that is ca. 0.06 V vs. SCE less negative than those of 4 and 5 which bear 2-thpy in the C^C^N ligand, probably due to the better π-accepting ability and thus a more stabilized π* orbital of the C^C^N ligand in 1 and 2. Similar phenomena have also been observed in 3 and 6, which are incorporated with 1-thq and 2-thq moieties, respectively. Apart from these, the extended π-conjugation of the C^C^N ligand in 3 and 6 results in the least negative reduction potential of ca. −1.29 V vs. SCE when compared to the other complexes. Hence, the reduction waves of all the complexes are assigned as the ligand-centered reduction of the C^C^N ligand,25,37 and the experimental trend is found to be in good agreement with the photophysical and computational studies.
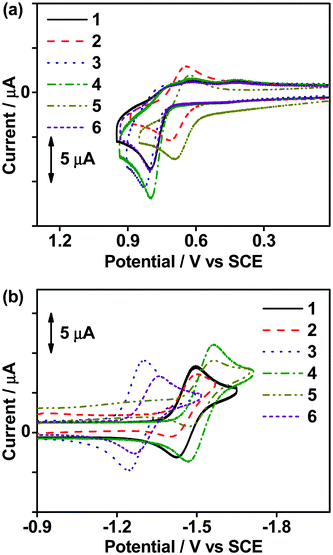 |
| | Fig. 1 Cyclic voltammograms for (a) oxidative and (b) reductive scans of 1–6 in degassed dichloromethane (0.1 M nBu4NPF6) at 298 K. | |
Photophysical properties
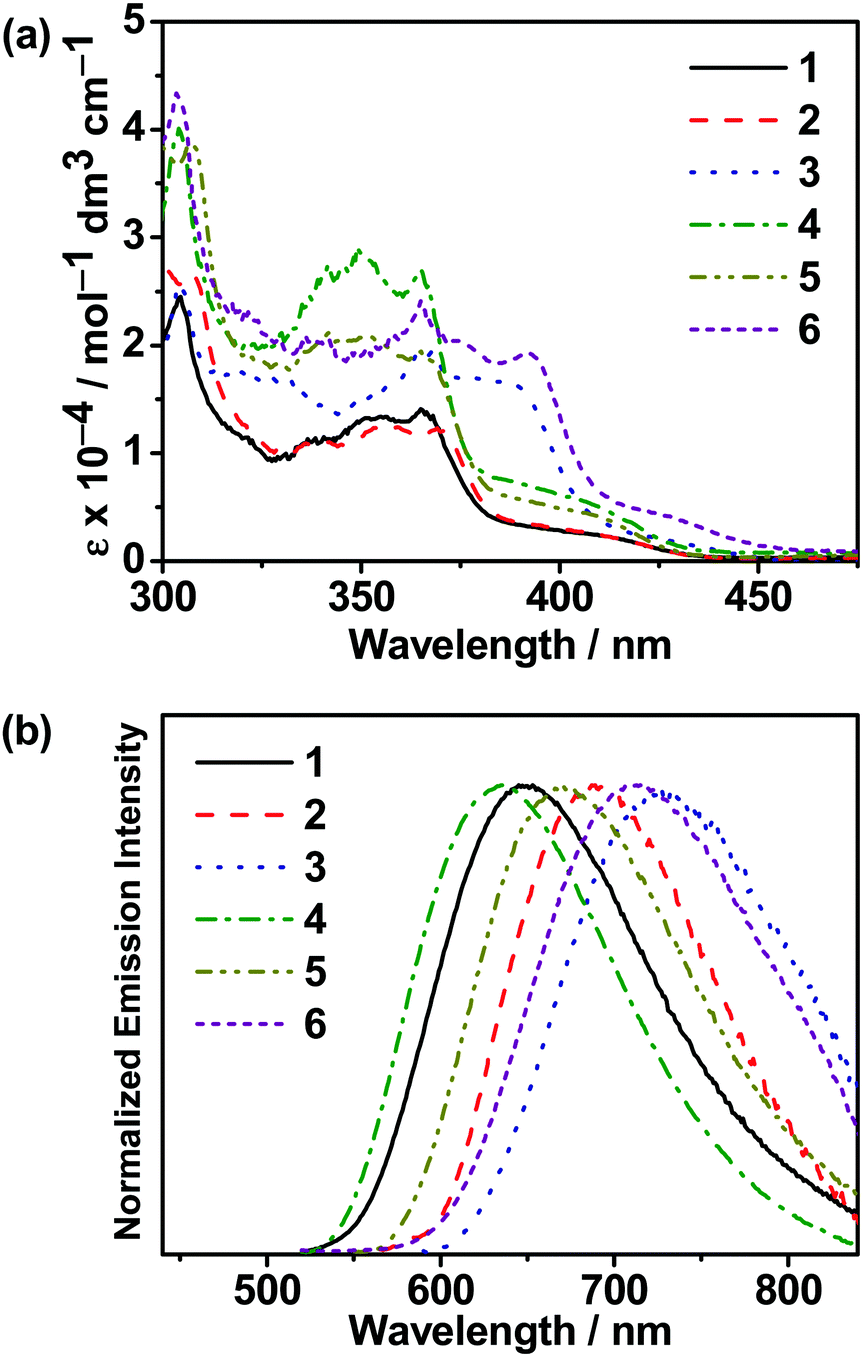
UV–vis absorption and emission studies were conducted on all the carbazolylgold(III) complexes. The intense absorption bands at a wavelength (λ) of ≤ 390 nm are attributed to the spin-allowed intraligand (IL) [π→π*(C^C^N)] transitions with some charge transfer character from the phenyl ring to the N-heterocycles, with an extinction coefficient (ε) on the order of 104 dm3 mol−1 cm−1, while the absorption tails beyond ca. 400 nm are assigned as the ligand-to-ligand charge transfer (LLCT) π[carbazole]→π*[C^C^N] transitions,25,37 which are supported by computational studies. The UV–vis absorption and emission spectra of all the complexes in toluene at 298 K are shown in Fig. 2, while the absorption data are shown in Table S3 (ESI‡) and the luminescence data are summarized in Table 1. Upon excitation at λ ≥ 350 nm in degassed toluene, all the complexes feature intense structureless emission bands peaking at ca. 635–730 nm, with a relatively short excited-state lifetime of 0.1–0.3 μs and a high rate of radiative decay (kr) on the order of 105 s−1. The emissions are assigned to the LLCT π[carbazole]→π*[C^C^N] origin,25,37 as supported by computational studies. With the addition of tBu groups onto the carbazole, 2 displays a bathochromic shift of ca. 960 cm−1 when compared with 1, while extending the π-conjugation in the N-heterocycle in 3 leads to a larger red shift of ca. 1700 cm−1. Similar observations have also been found in 4–6. These results suggest that the increase in aromatic conjugation of the N-heterocycles, rather than the incorporation of tBu groups into the carbazole, is a more effective way to realize red emission. The emission energies are also found to be dependent on the positional effect of the sulfur atom in the N-heterocyclic moieties. For instance, the replacement of 1-thpy in 1 with 2-thpy in 4 results in a slight blue shift of ca. 292 cm−1 owing to their different extents of stabilization of the π* orbital of the C^C^N ligand. Similar observations have been found when comparing 2 with 5 and 3 with 6, which are structural analogues of each other, with 3 featuring the most red-shifted emission band peaking at ca. 730 nm due to the stronger electron-accepting ability of 1-thq to further stabilize the π* orbital of the C^C^N ligand. These results are found to be generally consistent with the electrochemical and computational studies of the complexes. To gain further insights into the photophysical properties of these gold(III) complexes, nanosecond transient absorption (TA) measurements were performed on the isomeric complexes 1 and 4 in degassed toluene at 298 K (Fig. S2 and S3, ESI‡). Both complexes display a very weak ground-state bleaching at ca. 360 nm, a weak absorption band at ca. 450 nm, and a moderately intense, broad absorption band at ca. 530–750 nm. With reference to previous reports on the tridentate ligand-containing gold(III) complexes,50,51 the higher energy TA band at ca. 450 nm can be assigned to the absorption of the radical anion of the C^C^N ligand, while the lower energy broad TA band can be assigned as the absorption of the carbazolyl radical cations. The absorption decay lifetimes are found to be ca. 0.4 μs at 635 and 620 nm for 1 and 4, respectively, which are comparable to their respective emission decay lifetimes in toluene and are supportive to the excited-state origin for their emission bands. It is noteworthy to mention that the radical anion of the cyclometalating ligand could be stabilized by intramolecular hydrogen bonding.52 Given the presence of dipole–dipole interaction between the sulfur and the hydrogen atoms in the cyclometalating ligand of 1 (see below), it is believed that 1 would have a higher stability than 2 in their excited states.
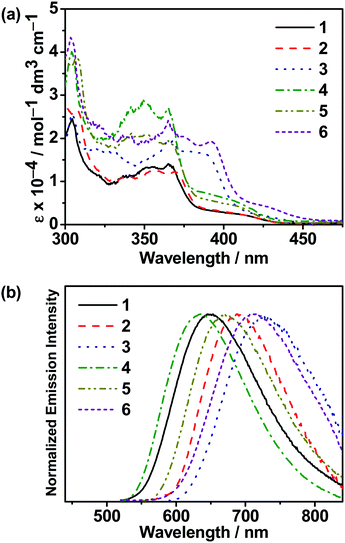 |
| | Fig. 2 (a) UV-vis absorption spectra of 1–6 in toluene at 298 K. (b) Normalized emission spectra of 1–6 in degassed toluene at 298 K. | |
Table 1 Luminescence data of 1–6
| Complex |
Medium (T/K) |
Emission λmax/nm (τo/μs) |
Φ
PL
|
k
r
/s−1 |
k
nr
/s−1 |
|
The relative luminescence quantum yields of the gold(III) complexes in solution were measured at room temperature using quinine sulfate in 0.5 M H2SO4 as the reference (excitation wavelength = 365 nm and Φ = 0.546).
Φ
PL(film) of the gold(III) complexes doped in m-CBP or MCP excited at a wavelength of 310 nm.
Radiative decay rate constant determined from the equation kr = Φem/τ; non-radiative decay rate constant determined from the equation knr = (1 − Φem)/τ.
Measured in EtOH–MeOH–CH2Cl2 (40:10:1, v/v).
Prepared by vacuum deposition.
Prepared by spin coating a blend of complex:host in chloroform.
|
|
1
|
Toluene (298) |
647 (0.3) |
0.20 |
6.7 × 105 |
2.7 × 106 |
|
|
Solid (298) |
603 (0.5) |
|
|
|
|
|
Solid (77) |
562, 588 (3.1) |
|
|
|
|
|
Glass (77)d |
563 (4.1) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
11% v/v in m-CBPe |
562 |
0.82 |
|
|
|
|
5 wt% in MCPf |
554 (3.3) |
0.83 |
2.5 × 105 |
5.2 × 104 |
|
|
10 wt% in MCPf |
563 (2.5) |
0.80 |
3.2 × 105 |
8.0 × 104 |
|
|
15 wt% in MCPf |
570 (2.5) |
0.75 |
3.0 × 105 |
1.0 × 105 |
|
|
20 wt% in MCPf |
578 (1.5) |
0.75 |
5.0 × 105 |
1.7 × 105 |
|
|
10 wt% in CBPf |
564 (2.1) |
0.71 |
3.4 × 105 |
1.4 × 105 |
|
|
10 wt% in TCTAf |
566 (2.1) |
0.64 |
3.0 × 105 |
1.7 × 105 |
|
|
10 wt% in TmPyPBf |
574 (1.5) |
0.58 |
3.9 × 105 |
2.8 × 105 |
|
|
10 wt% in PMMAf |
581 (1.6) |
0.47 |
2.9 × 105 |
3.3 × 105 |
|
2
|
Toluene (298) |
690 (0.1) |
0.04 |
4.0 × 105 |
9.6 × 106 |
|
|
Solid (298) |
630 (0.2) |
|
|
|
|
|
Solid (77) |
614, 666 (0.9) |
|
|
|
|
|
Glass (77)d |
498, 536, 573 (58.4) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
11% v/v in m-CBPe |
602 |
0.43 |
|
|
|
|
5 wt% in MCPf |
592 (1.4) |
0.60 |
4.3 × 105 |
2.9 × 105 |
|
|
10 wt% in MCPf |
594 (1.2) |
0.58 |
4.8 × 105 |
3.5 × 105 |
|
|
15 wt% in MCPf |
596 (1.2) |
0.54 |
4.5 × 105 |
3.8 × 105 |
|
|
20 wt% in MCPf |
604 (0.9) |
0.50 |
5.6 × 105 |
5.6 × 105 |
|
3
|
Toluene (298) |
727 (0.1) |
0.02 |
2.0 × 105 |
9.8 × 106 |
|
|
Solid (298) |
693 (1.2) |
|
|
|
|
|
Solid (77) |
636, 687 (2.7) |
|
|
|
|
|
Glass (77)d |
637 (4.4) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
11% v/v in m-CBPe |
615 |
0.41 |
|
|
|
|
5 wt% in MCPf |
604 (4.5) |
0.57 |
1.3 × 105 |
9.6 × 104 |
|
|
10 wt% in MCPf |
614 (2.7) |
0.52 |
1.9 × 105 |
1.8 × 105 |
|
|
15 wt% in MCPf |
620 (2.6) |
0.47 |
1.8 × 105 |
2.0 × 105 |
|
|
20 wt% in MCPf |
628 (2.3) |
0.43 |
1.9 × 105 |
2.5 × 105 |
|
4
|
Toluene (298) |
635 (0.3) |
0.22 |
7.3 × 105 |
2.6 × 106 |
|
|
Solid (298) |
606 (0.4) |
|
|
|
|
|
Solid (77) |
556, 592 (3.2) |
|
|
|
|
|
Glass (77)d |
512, 553, 599 (3.9) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
11% v/v in m-CBPe |
558 |
0.56 |
|
|
|
|
5 wt% in MCPf |
530 sh, 557 (8.1) |
0.81 |
1.0 × 105 |
2.3 × 104 |
|
|
10 wt% in MCPf |
561 (6.6) |
0.77 |
1.2 × 105 |
3.5 × 104 |
|
|
15 wt% in MCPf |
562 (6.1) |
0.75 |
1.2 × 105 |
4.1 × 104 |
|
|
20 wt% in MCPf |
565 (4.7) |
0.76 |
1.6 × 105 |
5.1 × 104 |
|
|
10 wt% in CBPf |
564 (3.5) |
0.67 |
1.9 × 105 |
9.4 × 104 |
|
|
10 wt% in TCTAf |
566 (2.6) |
0.56 |
2.2 × 105 |
1.7 × 105 |
|
|
10 wt% in TmPyPBf |
572 (2.1) |
0.62 |
3.0 × 105 |
1.8 × 105 |
|
|
10 wt% in PMMAf |
578 (2.0) |
0.43 |
2.2 × 105 |
2.9 × 105 |
|
5
|
Toluene (298) |
671 (0.1) |
0.06 |
6.0 × 105 |
9.4 × 106 |
|
|
Solid (298) |
606 (0.2) |
|
|
|
|
|
Solid (77) |
591, 638 (1.8) |
|
|
|
|
|
Glass (77)d |
514, 558, 601 (2.9) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
5 wt% in MCPf |
580 (1.8) |
0.71 |
3.9 × 105 |
1.6 × 105 |
|
|
10 wt% in MCPf |
589 (1.3) |
0.57 |
4.4 × 105 |
3.3 × 105 |
|
|
15 wt% in MCPf |
593 (1.1) |
0.51 |
4.6 × 105 |
4.5 × 105 |
|
|
20 wt% in MCPf |
599 (1.0) |
0.47 |
4.7 × 105 |
5.3 × 105 |
|
6
|
Toluene (298) |
710 (0.1) |
0.03 |
3.0 × 105 |
9.7 × 106 |
|
|
Solid (298) |
657 (0.6) |
|
|
|
|
|
Solid (77) |
635 (3.1) |
|
|
|
|
|
Glass (77)d |
626 (2.5) |
|
|
|
|
|
Thin film (298) |
|
|
|
|
|
|
5 wt% in MCPf |
594 (4.2) |
0.56 |
1.3 × 105 |
1.0 × 105 |
|
|
10 wt% in MCPf |
600 (3.9) |
0.57 |
1.5 × 105 |
1.1 × 105 |
|
|
15 wt% in MCPf |
609 (3.0) |
0.47 |
1.6 × 105 |
1.8 × 105 |
|
|
20 wt% in MCPf |
619 (2.1) |
0.39 |
1.9 × 105 |
2.9 × 105 |
The emission behaviors of all the complexes in solid-state thin films have been investigated. The normalized photoluminescence (PL) spectra of all the complexes doped at 10 wt% and those of 1 doped at 5–20 wt% in 1,3-bis(N-carbazolyl)benzene (MCP) thin films at 298 K are shown in Fig. S4 (ESI‡), while the normalized PL spectra of 2–6 doped at 5–20 wt% in MCP thin films at 298 K are shown in Fig S5–S9 (ESI‡). The emission behaviors of these complexes in MCP thin films are found to resemble that in the solution state; for instance, a bathochromic shift of the emission maxima from 1 and 4 (ca. 562 nm) to 3 and 6 (ca. 600–614 nm) of the complexes doped at 10 wt% in MCP is observed, which suggests that an interplay of different N-heterocyclic moieties would give different luminescence properties in this class of gold(III) complexes. Similar to the previously reported analogues,25,37 this class of complexes displays slight bathochromic shifts upon increasing the dopant concentration, probably due to the change in the molecular packing and polarity in the host matrix environment.33 It is worth noting that shorter lifetimes in the range of 0.9–4.5 μs are found for all the complexes except 4, suggesting the alleviation of triplet–triplet annihilation in the thin films. Additionally, a thermally enhanced luminescence behavior is found in the emission band upon increasing the temperature of these gold(III) complexes doped in solid-state thin films. The emission spectra of the representative complex 1 doped in MCP at 20 wt% upon increasing the temperature from 77 to 320 K and from 320 to 370 K are shown in Fig. 3 and Fig. S10 (ESI‡), respectively. Apparently, the intensity of the structureless emission band is found to drastically increase by almost 5-fold from 77 to 320 K, while the emission intensity gradually drops upon further increasing the temperature from 320 to 370 K, indicating that the non-radiative decay process becomes more efficient than the thermally enhanced luminescence process when the temperature is higher than 320 K. The excited-state lifetimes of 1 doped in MCP at 20 wt% have also been studied, which are found to be almost unchanged in the range of 200 K (1.5 μs) to 320 K (1.4 μs), suggesting that the thermal upconversion process is almost complete under these temperature conditions.53 These, together with the ΔES1–T1 value of 0.004 eV computed by time-dependent density functional theory (TDDFT) calculations (see below), as well as the high kr on the order of 105 s−1 in both solution and solid-state thin films, are indicative of the TADF behavior in the carbazolylgold(III) complex.36 Attempts to calculate the ΔES1–T1 value by using both the conventional two-level Boltzmann model and the Dias, Penfold and Monkman's fitting have also been performed; however, these methods might not be the most suitable methods due to the involvement of the third state, i.e.3IL, in the present gold(III) complexes, especially at low temperatures.
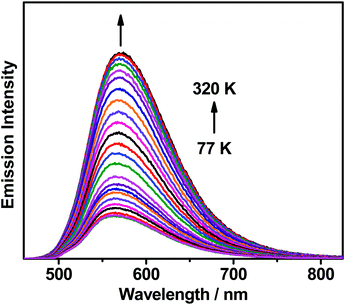 |
| | Fig. 3 Emission spectra of the thin film of 20 wt% 1 doped in MCP upon increasing the temperature from 77 to 320 K. | |
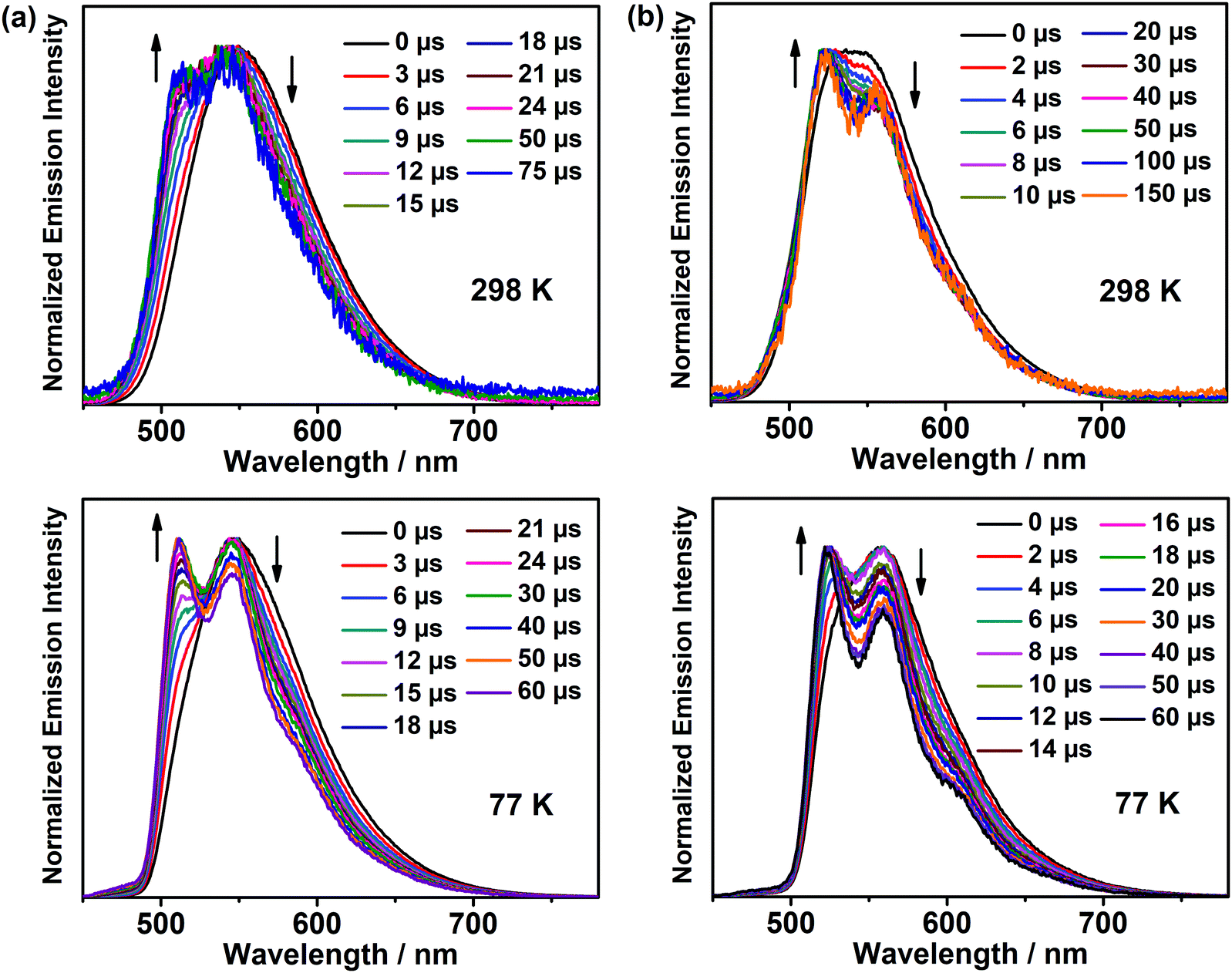
The emission properties of the representative complexes 1 and 4 doped in different host materials in solution-processed thin films were also investigated. Fig. S11 (ESI‡) depicts the normalized emission spectra of 1 and 4 in different host materials at 10 wt% dopant concentration. In general, both complexes display bathochromic shifts upon changing the host matrix from MCP to 4,4′-N,N′-dicarbazole-biphenyl (CBP) to 4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TCTA) to 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) to polymethyl methacrylate (PMMA), which could be ascribed to the charge transfer nature of the excited states and different polarities of the host materials.33 From MCP to PMMA, the bathochromic shifts of ca. 550 and 524 cm−1 were observed for 1 and 4, respectively, accompanied by a drop of almost half of the PLQY. To gain deeper insights into the nature of the excited states of the complexes in solid-state thin films, time-resolved emission studies were performed on 1 and 4 doped in MCP at the 5 wt% dopant concentration, and the corresponding normalized emission spectra at 298 and 77 K are shown in Fig. 4, respectively. In general, both complexes show structureless emission bands at a time delay of 0 μs, but the emission bands gradually change to more vibronic structured when the emission signals are collected with a time delay from 2 to 150 μs. These results indicate the involvement of a mixture of excited states in the solid-state thin films, where these two excited states are close-lying in energy. The longer-lived vibronic-structured emission is found to be in line with its 3IL excited state character, as illustrated in Fig. S12 (ESI‡), showing the emission spectra of the thin films of the corresponding chlorogold(III) precursor complexes doped in MCP. On the other hand, high PLQYs as high as 83% have been realized in these complexes; more importantly, the PLQY remains at ca. 50% for red emission, which is much higher than that reported for the previously reported red-emitting analogues (i.e. ca. 30%).25 These findings demonstrate that through a delicate interplay of the N-heterocycles, the emission energy of the complexes can be readily tuned to span from yellow to red with high PLQYs, representing a promising class of gold(III) emitters for OLED applications.
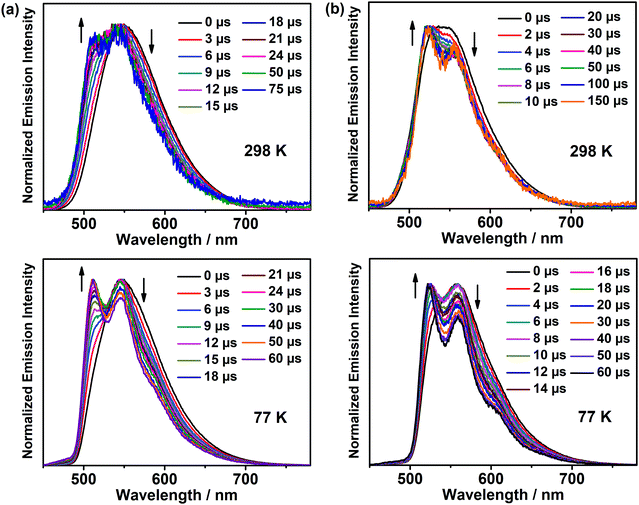 |
| | Fig. 4 Normalized time-resolved emission spectra of (a) 1 and (b) 4 doped at 5 wt% in MCP at 298 K and 77 K. | |
Computational studies
To gain deeper insights into the electronic structures and the nature of the absorption and emission origins of these thpy- and thq-based gold(III) complexes, density functional theory (DFT) and TDDFT calculations are performed on the selected complexes, i.e.1, 3, 4 and 6. The optimized ground-state (S0) geometries (front view) and the selected structural parameters of the complexes are shown in Fig. S13 (ESI‡), while the optimized S0 geometries (side view) of the complexes are shown in Fig. S14 (ESI‡). The Au–N(1) bonds in 3 and 6 are computed to be ca. 2.253 Å, which are significantly longer than those in 1 and 4 (ca. 2.160 Å). In addition, the planes of the carbazole and the C^C^N ligand in 3 and 6 are nearly orthogonal, while those in 1 and 4 have a dihedral angle close to 70° due to the larger steric repulsion between the carbazole and thq moieties in 3 and 6. It is also observed that the positional effect of the sulfur atom can have an effect on the dihedral angle between the planes of the N-heterocyclic moieties and that of the central phenyl ring. As shown in Fig. S14 (ESI‡), the planes of the 1-thpy and 1-thq moieties in 1 and 3, respectively, are inclined at ca. 2–8° to the same side, while those of the 2-thpy and 2-thq moieties in 4 and 6, respectively, are inclined at ca. 10–20° to the other side. Although this phenomenon may lead to a slight perturbation of the molecular packing of the complexes in the solid state, the positional effect of the sulfur atom seems to be insignificant as observed from the molecular orientation studies.
The first fifteen singlet excited states of 1, 3, 4 and 6, computed by the TDDFT/conductor-like continuum model (CPCM) method, are summarized in Table S4 (ESI‡), and the simulated UV–vis spectra, generated by Multiwfn,54 are shown in Fig. S15–S18 (ESI‡). Selected molecular orbitals involved in the transitions are shown in Fig. S19–S22 (ESI‡). The absorption tails computed beyond ca. 400 nm are mainly contributed by the HOMO → LUMO excitation in 1 and 4 and by the HOMO–1 → LUMO excitation in 3 and 6. For all the complexes, the HOMO and HOMO–1 are predominantly the π orbitals localized on the carbazole, while the LUMOs are the π* orbitals predominantly localized on the central phenyl ring and the thpy or thq moieties of the C^C^N ligand. Hence, the absorption tail can be assigned as the LLCT [π(carbazole)→π*(C^C^N)] transition, which supports the experimental energy trend of the absorption tails and their spectral assignments. For all the complexes, the intense absorption bands computed at ca. 340–370 nm are mainly attributed to the HOMO–3 → LUMO excitation, where the HOMO–3 is predominantly the π orbital on the C^C^N ligand. Therefore, these absorption bands can be assigned as the IL [π→π*(C^C^N)] transition with some charge transfer character from the phenyl rings to the thpy or thq moieties, which is in line with the experimental observation.
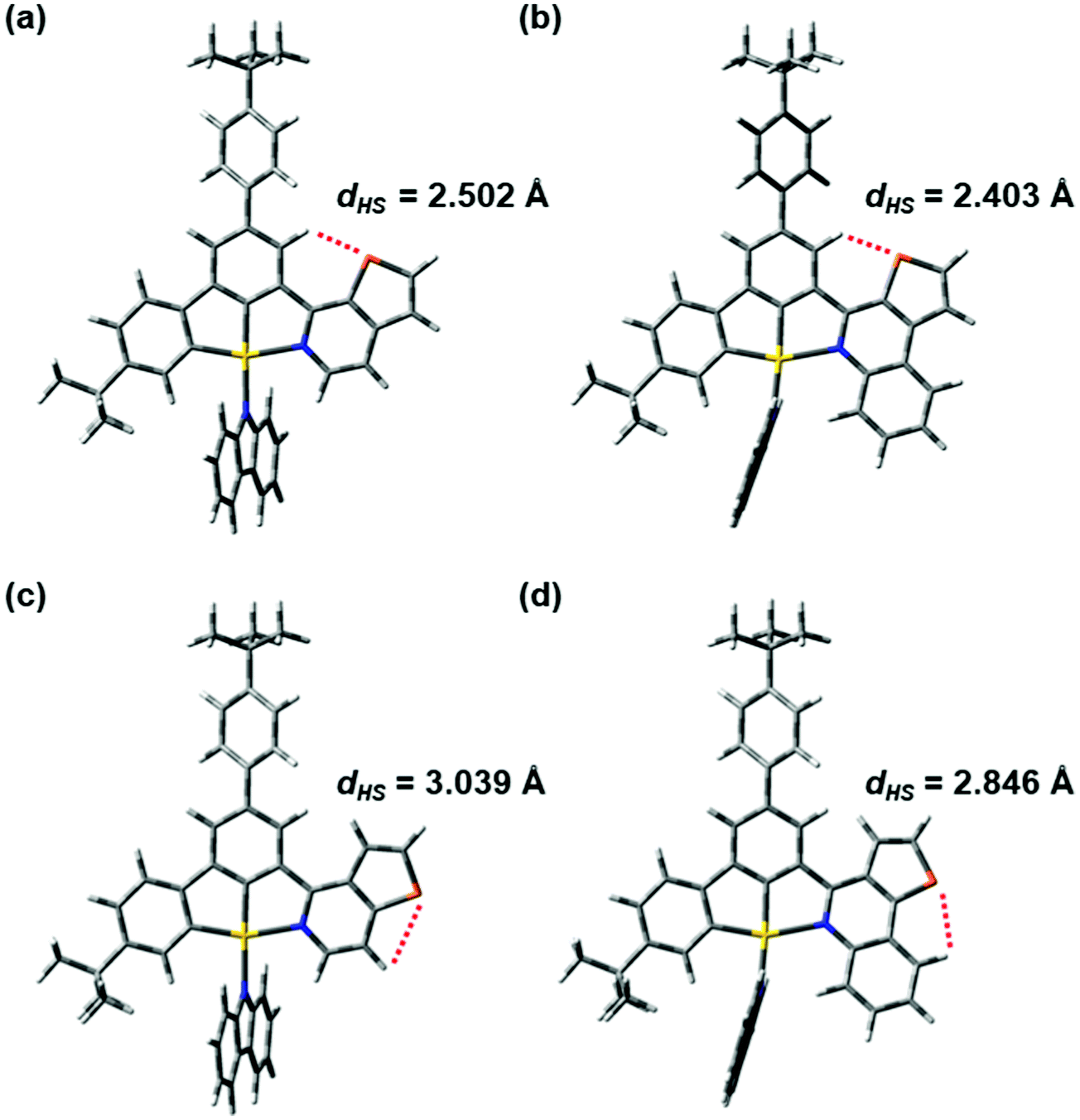
The orbital energy diagram showing the frontier molecular orbitals of 1, 3, 4 and 6 is shown in Fig. S23 (ESI‡). In general, there is no significant difference in the HOMO energies, whereas the LUMO energy decreases from 4 (−2.16 eV) to 1 (−2.26 eV) to 6 (−2.32 eV) to 3 (−2.44 eV). The trend of the HOMO and LUMO energies is in good agreement with the results obtained by cyclic voltammetry, where the potentials for oxidation are more or less the same and the reduction potential is less negative upon going from 4 to 1 to 6 to 3 (Table S2, ESI‡). The LUMO energies of 3 and 6 are in general lower than those of 1 and 4 owing to the stabilization by the more conjugated π–aromatic system in 3 and 6. In addition, it is observed that the LUMO energy of 1 is lower than that of 4, while the LUMO energy of 3 is lower than that of 6, indicating that the LUMO energy is affected by the position of the sulfur atom. In 1 and 3, the sulfur atoms on the 1-thpy and 1-thq moieties, respectively, are pointing towards the central phenyl ring of the C^C^N ligand. The distances between the sulfur atom and the hydrogen atom on the central phenyl ring are 2.50 and 2.40 Å, respectively, in 1 and 3, rendering a dipole–dipole interaction between the sulfur and the hydrogen atoms, which would lead to a higher degree of planarization to stabilize the LUMO energy. On the other hand, in 4 and 6, the sulfur atoms are pointing outward and the dipole–dipole interactions are expected to be insignificant in this case because the distances between the sulfur atom and the hydrogen atom on the 2-thpy or 2-thq moieties are 3.04 and 2.85 Å, respectively. The LUMOs of 4 and 6 may also be destabilized by the steric repulsion between the hydrogen atoms on the central phenyl ring and the 2-thpy or 2-thq moieties. Similar observations have been found at their optimized T1 geometries. In this case, the presence of the dipole–dipole interaction in 1 and 3 would allow them to have a more stable radical anion in their excited states than 4 and 6. Fig. 5 and Fig. S24 (ESI‡) illustrate the distances between the sulfur and hydrogen atoms in the N-heterocyclic moieties of 1, 3, 4 and 6 at the optimized S0 and T1 geometries, respectively. Overall, the HOMO–LUMO energy gap decreases from 4 (2.94 eV) to 1 (2.83 eV) to 6 (2.82 eV) to 3 (2.70 eV), which is in good agreement with the experimental trend. To sum up, the position of the sulfur atom plays an important role in controlling the dipole–dipole interaction and the steric repulsion within the complex, which would influence the HOMO–LUMO energy gap and the molecular packing of the complexes in the solid state.
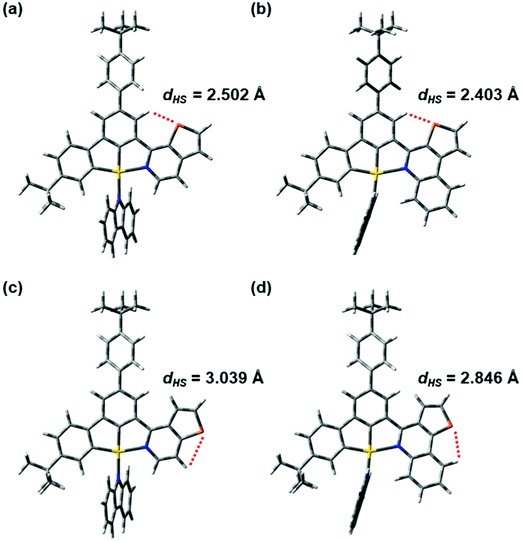 |
| | Fig. 5 The distances between the sulfur and hydrogen atoms in the N-heterocyclic moieties of (a) 1, (b) 3, (c) 4 and (d) 6 at the optimized ground-state geometries. | |
To gain deeper insights into the nature of the emissive states, the geometries of the lowest lying triplet excited state (T1) of 1, 3, 4 and 6 have been optimized with the unrestricted UPBE0 method. Their plots of the spin density are shown in Fig. S25 (ESI‡). In general, the spin density of all the complexes is predominantly localized on the carbazolyl moiety, the central phenyl ring and the thpy or thq moieties of the C^C^N ligand, which supports the LLCT [π(carbazole)→π*(C^C^N)] character in the emissive state. The emission wavelengths of 1, 3, 4 and 6, approximated by the energy difference between the S0 and T1 states at their corresponding optimized geometries, are summarized in Table S5 (ESI‡). The calculated emission wavelength shows a red shift on going from 4 (557 nm) to 1 (572 nm) to 6 (612 nm) to 3 (632 nm), which is in line with the experimental trend observed in the emission spectra. The geometry of the lowest lying singlet excited state (S1) of 1 has also been optimized using TDDFT to gain further insights into the excited states involved in the TADF process. The ΔEST value of 1 is computed to be 0.004 eV. The Cartesian coordinates of the optimized S0 and T1 geometries of the complexes are shown in Tables S6–S15 (ESI‡).
Molecular orientation studies
In light of the recent interest in the horizontal orientation of the complexes for enhancing the device performance,55–58 angular-dependent PL studies have been conducted on the vacuum-deposited thin films of selective complexes 1–4. In this study, 3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl (m-CBP) has been selected as the host material owing to its random orientation in a wide range of deposition temperatures,59 such that any preferential orientation in the thin films would solely be induced by the present gold(III) complexes. Fig. 6 depicts the normalized PL intensities as a function of emission angle for the thin films. In general, the values S = −0.23 to S = −0.27 have been found for 1, 3 and 4. Given that S = 0 indicates a random orientation, while S = −0.50 represents a completely horizontal orientation,55 these results imply that 1, 3 and 4 show a preferential horizontal alignment in the m-CBP host matrix, irrespective of the position of the sulfur atom (i.e.1vs.4) or the extended π-conjugation of the pyridyl moiety (i.e.1vs.3). On the other hand, the integration of −tBu groups into the carbazole in 2 has resulted in S = −0.05, suggesting the nearly random orientation of the complex. This could be due to the perturbation of the molecular orientation in the host matrix by the bulky −tBu groups, which consequently causes a drop of the proportion of horizontally oriented gold(III) complex molecules in the thin film doped with 2. Table 2 summarizes the molecular orientation parameters of 1–4. Given that θ is the angle between the normal of a substrate and the transition dipole moment vector (TDMV),55S = −0.23 to S = −0.27 and S = −0.05 correspond to Θ of 0.82–0.85 and 0.70, respectively. It is worth noting that a high ratio of the horizontal dipole to the total dipole of the emitters (Θ) is advantageous for improving the out-coupling efficiency (ζ) to overcome the theoretical limit of ca. 20–22% for planar OLEDs,55–57 as demonstrated in the previously reported OLEDs based on the horizontally oriented iridium(III) and platinum(II) complexes.7,9,11 More importantly, the value of Θ of up to 0.85 represents one of the highest values in the reported literature among both the phosphorescent and TADF emitters,56,57,60 demonstrating the promising potential of these gold(III) complexes as the emitting material for realizing high-performance OLEDs.
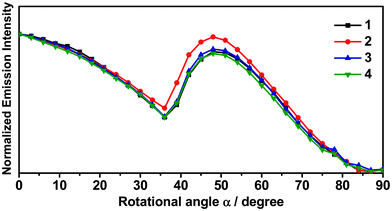 |
| | Fig. 6 Angular-dependent PL intensities of the p-polarized light of 1–4 (11% v/v) in m-CBP thin films with 20 nm thickness. | |
Table 2 Molecular orientation parameters of 1–4
| Complex |
Order parameter (S) |
θ
/° |
Θ
|
(h:v)b |
θ represents the angle between the normal of a substrate and the transition dipole moment vector and is calculated by the equation 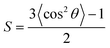 , with the bracketed values 〈…〉 indicating an ensemble average of 〈cos2θ〉. From ref. 55.
Θ represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θ:(1 − Θ) = 〈sin2θ〉 , with the bracketed values 〈…〉 indicating an ensemble average of 〈cos2θ〉. From ref. 55.
Θ represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θ:(1 − Θ) = 〈sin2θ〉![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :〈cos2θ〉 = h:v. From ref. 60 and 61. :〈cos2θ〉 = h:v. From ref. 60 and 61.
|
|
1
|
−0.24 |
65.4 |
0.83 |
0.83:0.16 |
|
2
|
−0.05 |
56.8 |
0.70 |
0.70:0.30 |
|
3
|
−0.23 |
64.9 |
0.82 |
0.82:0.18 |
|
4
|
−0.27 |
67.0 |
0.85 |
0.85:0.15 |
OLED fabrication and characterization
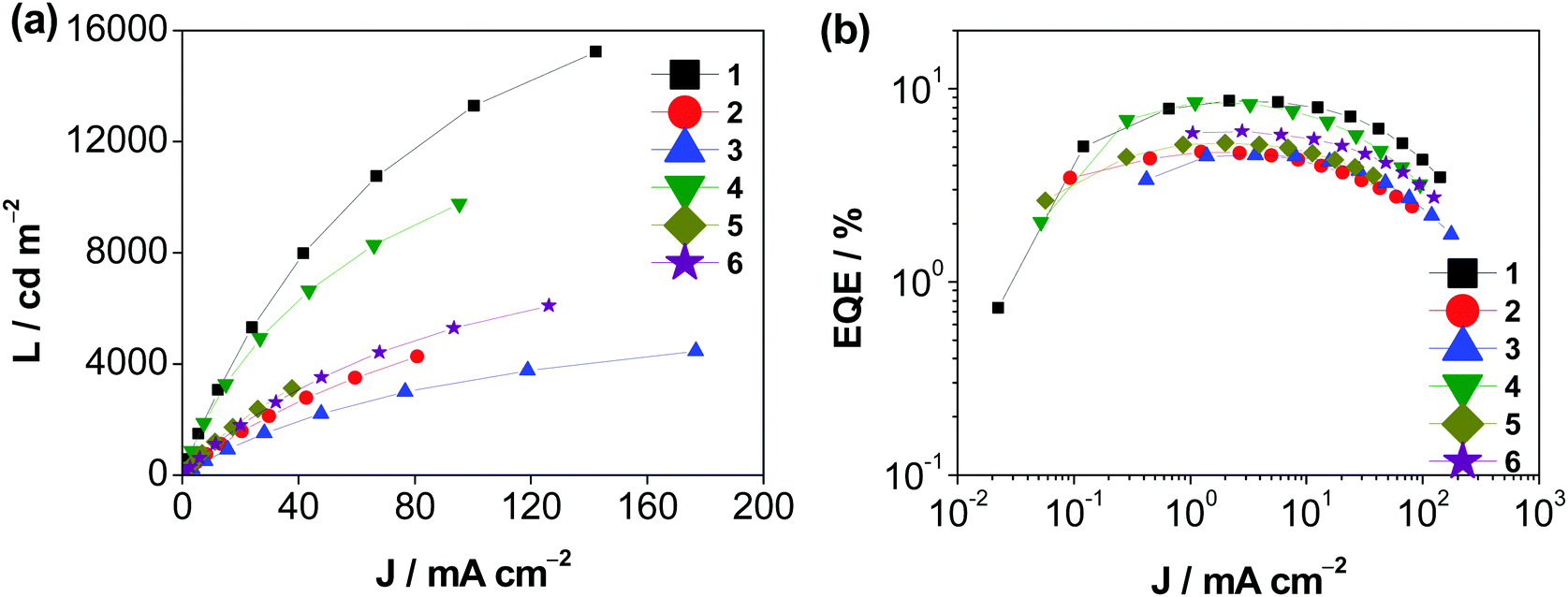
Solution-processed devices with the structure of indium tin oxide (ITO)/poly(ethylene-dioxythioxythiophene):poly(styrene-sulfonic acid) (PEDOT:PSS; 40 nm)/x% Au(III):MCP (30 nm)/tris[2,4,6-trimethyl-3-(pyridin-3-yl)phenyl]borane (3TPYMB; 5 nm)/1,3,5-tri[(3-pyridyl)phen-3-yl]benzene (TmPyPB; 40 nm)/LiF (1 nm)/Al (100 nm) were prepared for the investigation of the EL properties of these isomeric gold(III) complexes, in which the emissive layer was prepared by spin-coating a chloroform solution of Au(III):MCP blend at different concentrations, i.e. 5, 10, 15, and 20 wt%. Fig S26 (ESI‡) depicts the normalized EL spectra of the solution-processed devices based on 15 wt% 1–6. Apparently, all devices exhibit Gaussian-shaped emission bands, in which their EL behavior resembles the PL spectra of the complexes in both solution and solid-state thin films. For instance, the employment of thq moieties in 3 and 6 results in a significant red shift of the emission maxima of the devices. The emission energy is found to be shifted from 564 nm in 1 to 620 nm in 3 when 1-thpy is replaced by the 1-thq moiety. Similarly, a bathochromic shift of ca. 1600 cm−1 is observed upon replacing 2-thpy in 4 (560 nm) by 2-thq in 6 (608 nm). Fig. 7 and Table S16 (ESI‡) show the characteristics of the solution-processed devices. Maximum current efficiencies (CE) of 27.0 cd A−1 together with maximum EQEs of 8.6% has been achieved for the devices made with 1 and 4.
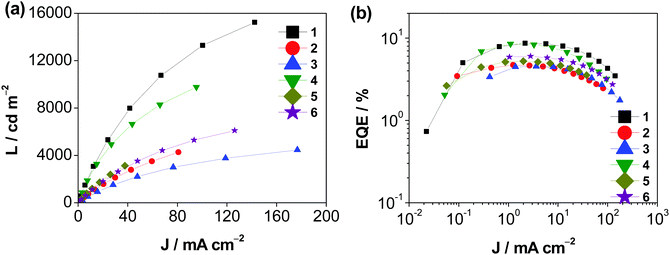 |
| | Fig. 7 (a) J–L curves and (b) EQEs vs. current density of the solution-processed devices based on 1–6. | |
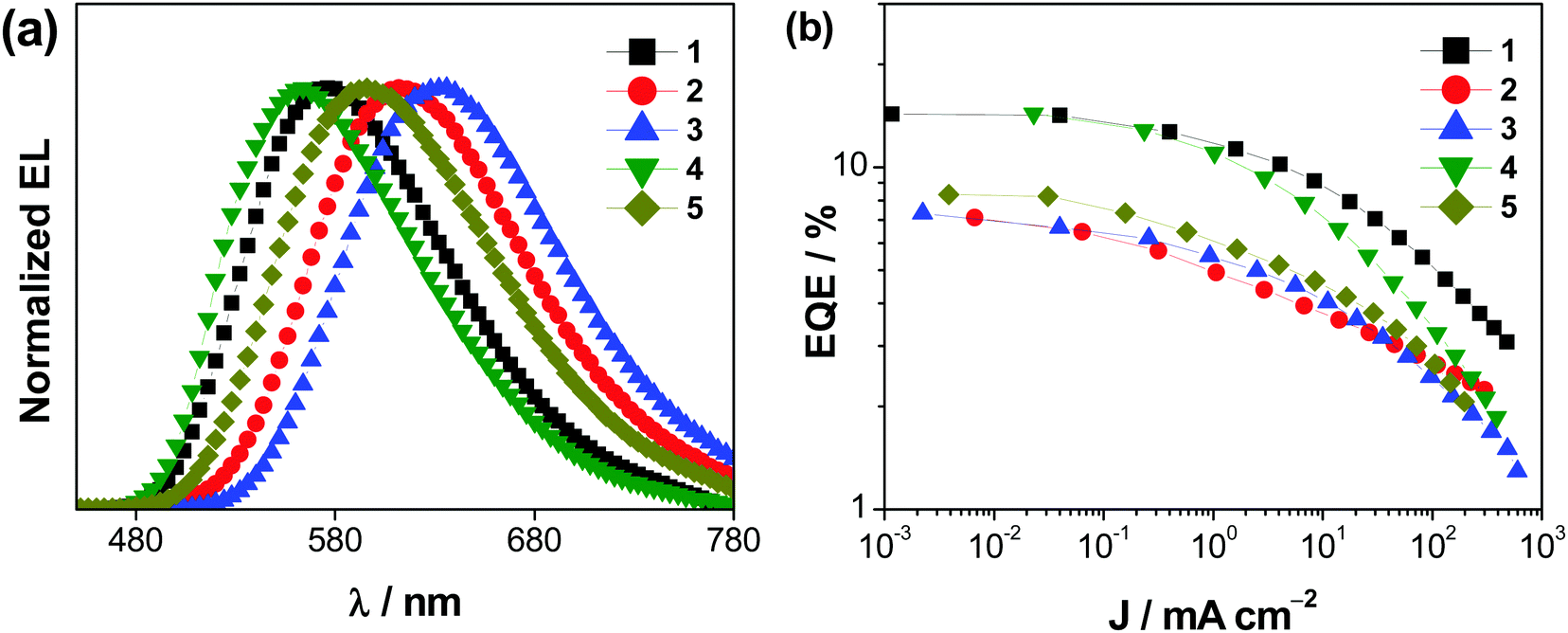
Vacuum-deposited devices based on 1–5 have also been prepared with the configuration of ITO/N,N′-bis(naphthalen-1-yl)-N,N′-bis(phenyl)-2,2′-dimethylbenzidine (α-NPD; 40 nm)/4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TCTA; 5 nm)/x % Au(III):m-CBP (20 nm)/1,3,5-tris(6-(3-(pyridin-3-yl)phenyl)-pyridin-2-yl)benzene (Tm3PyP26PyB; 50 nm)/LiF (1 nm)/Al (150 nm), in which the emissive layer has been prepared by simultaneously co-evaporating the respective gold(III) complex and m-CBP at the desired concentration, i.e. 2, 5, 8, 11 and 14% v/v. Fig. 8 shows the normalized EL spectra and the respective EQEs of the vacuum-deposited devices based on 1–5. These complexes exhibit Gaussian-shaped EL bands, in which the EL behavior shows excellent agreement with the PL spectra of the complexes and the EL spectra of their corresponding solution-processed devices. Yellow- to red-emitting vacuum-deposited gold(III)-based devices have been realized, with maximum CEs reaching 41.6 cd A−1, power efficiencies reaching 37.3 lm W−1, and EQEs reaching 14.5% (see Table S17, ESI‡). Among the complexes that exhibit preferential horizontal alignment (see above), the high Θ is anticipated to improve the out-coupling efficiency of the corresponding devices to give higher EQEs, with 1 and 4 being higher than that of 2. On the other hand, an unusually low EQE of 6.9% was found for the device made with 3. This unusually low EQE may be ascribed to the deep electron trapping in 3 due to its low-lying LUMO level of 3.07 eV. This deep electron trapping inevitably results in a relatively low EQE in the devices based on 3. Devices made with 6 had also been fabricated; however, a poor performance was obtained with an undesired emission band at ca. 454 nm. Fig. S27 (ESI‡) depicts the EL spectra of the vacuum-deposited devices made with 6 at various brightness levels from 0.5 to 90 cd m−2.
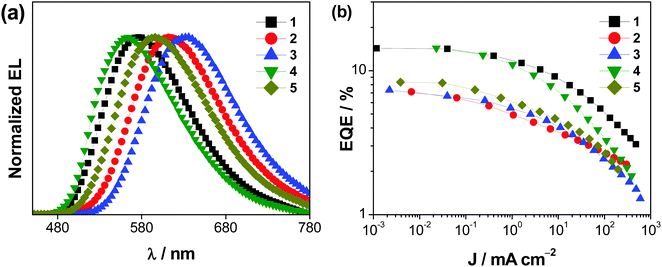 |
| | Fig. 8 (a) Normalized EL spectra and (b) plots of EQEs vs. current density of the vacuum-deposited devices based on 1–5. | |
The operational stability of the vacuum-deposited devices based on 1–5 was measured by accelerated tests at a constant driving current density of 20 mA cm−2. Devices with the configuration of ITO/dipyrazino[2,3-f:2′,3′-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile (HAT-CN; 10 nm)/α-NPD (40 nm)/9,9′,9′′-triphenyl-9H,9′H,9′′H-3,3′:6′,3′′-tercarbazole (TrisPCz; 10 nm)/11% v/v Au(III):m-CBP (25 nm)/2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine (T2T; 10 nm)/2,7-di(2,2′-bipyridin-5-yl)triphenylene (BPyTP2; 40 nm)/LiF (1 nm)/Al (150 nm) were fabricated and encapsulated in a glovebox under nitrogen. Specifically, the operational lifetimes of the devices at which the luminance dropped to 70% of their initial value (LT70) were measured. Fig. 9 shows the relative luminance (L/L0) of the gold(III)-based OLEDs as a function of time, while Table S17 (ESI‡) summarizes the device lifetime data of 1–5. Apparently, the operational stabilities of the vacuum-deposited devices significantly depend on the choice of the N-heterocyclic moieties on the C^C^N ligand. With the incorporation of 2-thpy in 4 and 5, the devices based on these two complexes exhibited LT70 values of 86 and 55 h at 100 cd m−2, respectively. In sharp contrast, devices based on 1 and 2, which are incorporated with 1-thpy, demonstrate long LT70 values of 63258 and 15760 h at 100 cd m−2, respectively. A long operational half-lifetime (LT50), i.e. the time at which the brightness of the device dropped to its half value, of ca. 206800 h has also been determined for the device based on 1. This could be rationalized by the relatively higher radical anion stability of 1–3. As supported by computational studies, the position of the sulfur atom plays an important role in controlling the dipole–dipole interaction and the steric repulsion within the complex. In addition, the higher robustness of 1–3 can be ascribed to the stability provided by the resonance structures of the N-heterocyclic moieties. As shown in Fig. S28 (ESI‡), which depicts the resonance structures of 1-thpy and 2-thpy moieties in the cyclometalating ligands, one more resonance structure is found in 1-thpy when compared with 2-thpy, which leads to an increased overall delocalization of electrons within the C^C^N(1-thpy) (or N(1-thq)) ligand. More importantly, the electronegative nitrogen atom in one of the resonance structures of 1-thpy can best accommodate the negative charge, acquiring an octet of electrons, and thus rendering extra stabilization to the 1-thpy moiety and can possibly account for the higher intrinsic stability of 1–3. It is worth noting that a LT50 value of ca. 206800 h of the device based on 1 is much longer than those based on structurally related pyridine- and isoquinoline-containing gold(III) complexes (i.e. 66000 and 83000 h, respectively),25 which can also be rationalized by the more stable heteroatom-based radical anion of 1 in the excited state.52 It should be highlighted that the high horizontal dipole ratios (Θ) of ca. 0.83–0.85 are found for both 1 and 4, irrespective of the position of the sulfur atom, as confirmed in molecular orientation studies. Given that the LT70 value of the device based on 1 (63258 h) is almost three orders of magnitude longer than that based on 4 (86 h), the isomeric effect is believed to be more significant than the orientation effect of these gold(III) complex molecules in governing the operational stability of the fabricated devices. The device lifetime data of the solution-processed device based on 1 are also recorded. The device shows an estimated LT70 value of 2600 h at 100 cd m−2 (Fig. S29, ESI‡), which is comparable with those of solution-processed devices based on the iridium(III) system.62,63 Although the operational stability of the devices in this work is not as outstanding as those of the best reported and more well-established classes of phosphorescent and TADF emitter-based OLEDs,64 the operational lifetimes of the present devices are one of the longest in this emerging and novel class of gold(III) complexes. In particular, the device based on 1 displays the longest and most impressive operational lifetime as well as the highest EQEs among all the reported orange-emitting gold(III)-based devices.25,26,29,65 These results demonstrate the promising potential of this class of carbazolylgold(III) complexes in both solution-processed and vacuum-deposited OLED applications.
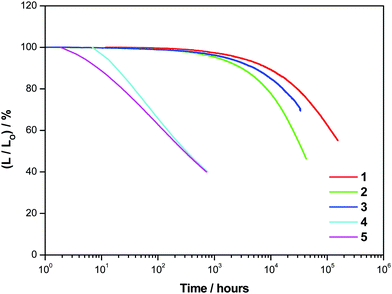 |
| | Fig. 9 Relative luminance, L/L0, of vacuum-deposited devices based on 1–5 as a function of operation time projected at an initial luminance of 100 cd cm−2. | |
Conclusions
To conclude, a new class of carbazolylgold(III) complexes containing isomeric N-heterocyclic moieties has been designed and synthesized, in which their emission energies have been effectively tuned to span from yellow to red. These gold(III) complexes are found to exhibit high thermal stability, high solubility in common organic solvents and high PLQYs of over 80% in solid-state thin films. The isomeric N-heterocyclic moieties have been found to significantly perturb the photophysical, electrochemical and EL properties of the gold(III) complexes. Notably, the use of 1-thpy and 1-thq rather than 2-thpy and 2-thq moieties can render an extra stabilization to the gold(III) complexes, leading to long device operational stability with a LT70 value of ca. 63200 h and a LT50 value of ca. 206800 h at 100 cd m−2 in the gold(III)-based OLEDs. Solution-processed and vacuum-deposited OLEDs based on these complexes have also been realized, with the maximum EQEs of 8.6% and 14.5%, respectively. It is anticipated that the present work provides important insights into the development of robust and highly luminescent gold(III) complexes, and the identification of stable molecular motifs for designing efficient emitters.
Author contributions
V. W.-W. Y. initiated and designed the research. V. W.-W. Y., L.-K. L. and M.-C. T. designed the gold(III) complexes. L.-K. L. and C. C. A.-Y. conducted the synthesis of the gold(III) complexes. L.-K. L. conducted the characterization, photophysical and electrochemical and molecular orientation measurements of the gold(III) complexes. L.-K. L. and M. N. performed and analyzed the computational calculations. S.-L. L., W.-L. C. and M.-Y. C. carried out the OLED fabrication and characterization. V. W.-W. Y. supervised the work. All authors discussed the results and contributed to the manuscript.
Conflicts of interest
V. W.-W. Y., M.-C. T., M.-Y. C and L.-K. L. have filed the following patent applications: The University of Hong Kong, Hong Kong; V. W.-W. Y., M.-C. T., M.-Y. C. and C.-H. L., Molecular design and OLED applications. US provisional patent application no. 16/339,013; The University of Hong Kong, Hong Kong; V. W.-W. Y., M.-C. T., M.-Y. C. and C.-H. L. Molecular design and OLED applications. PCT patent application no. PCT/CN2017/105241.
Acknowledgements
V. W.-W. Y. acknowledges support from The University of Hong Kong (HKU), University Research Committee (URC) Strategically Oriented Research Theme on Functional Materials for Molecular Electronics Towards Materials and Energy Applications and the HKU-TCL Joint Laboratory on New Printable OLED Materials and Technology. The work described in this paper was supported by the Hong Kong Quantum AI Lab Ltd under the AIR@InnoHK administrated by the Innovation and Technology Commission (ITC) and a grant from the General Research Fund (GRF) of the Research Grants Council of the Hong Kong Special Administrative Region, People's Republic of China (HKU17306219). The computations were performed by using the HKU ITS research computing facilities. L.-K. L., W.-L. C. and C. C. A.-Y. acknowledge the receipt of postgraduate studentships from HKU. L.-K. L. acknowledges the receipt of a University Postgraduate Fellowship from HKU. C.-H. Lee is acknowledged for his helpful discussion in this work.
Notes and references
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky, M. E. Thompson and R. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622–1624 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS.
- K.-H. Kim, S. Lee, C.-K. Moon, S.-Y. Kim, Y.-S. Park, J.-H. Lee, J. Woo Lee, J. Huh, Y. You and J.-J. Kim, Nat. Commun., 2014, 5, 4769 CrossRef CAS.
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS.
- K.-H. Kim, E. S. Ahn, J.-S. Huh, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2016, 28, 7505–7510 CrossRef CAS.
- C.-Y. Kuei, W.-L. Tsai, B. Tong, M. Jiao, W.-K. Lee, Y. Chi, C.-C. Wu, S.-H. Liu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2016, 28, 2795–2800 CrossRef CAS.
- K.-H. Kim, J.-L. Liao, S. W. Lee, B. Sim, C.-K. Moon, G.-H. Lee, H. J. Kim, Y. Chi and J.-J. Kim, Adv. Mater., 2016, 28, 2526–2532 CrossRef CAS PubMed.
- T. Komino, Y. Sagara, H. Tanaka, Y. Oki, N. Nakamura, H. Fujimoto and C. Adachi, Appl. Phys. Lett., 2016, 108, 241106 CrossRef.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- G. Zhou, W.-Y. Wong and X. Yang, Chem. – Asian J., 2011, 6, 1706–1727 CrossRef CAS.
- W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709–1758 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- C. Wu, S. Tao, M. Chen, F.-L. Wong, Y. Yuan, H.-W. Mo, W. Zhao and C.-S. Lee, Chem. – Asian
J., 2013, 8, 2575–2578 CrossRef CAS.
- R. C. Kwong, M. R. Nugent, L. Michalski, T. Ngo, K. Rajan, Y.-J. Tung, M. S. Weaver, T. X. Zhou, M. Hack, M. E. Thompson, S. R. Forrest and J. J. Brown, Appl. Phys. Lett., 2002, 81, 162–164 CrossRef CAS.
- C.-H. Chen, L.-C. Hsu, P. Rajamalli, Y.-W. Chang, F.-I. Wu, C.-Y. Liao, M.-J. Chiu, P.-Y. Chou, M.-J. Huang, L.-K. Chu and C.-H. Cheng, J. Mater. Chem. C, 2014, 2, 6183–6191 RSC.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4050–4055 CrossRef CAS.
- H. Fukagawa, T. Shimizu, T. Kamada, Y. Kiribayashi, Y. Osada, M. Hasegawa, K. Morii and T. Yamamoto, Adv. Opt. Mater., 2014, 2, 1070–1075 CrossRef CAS.
- T. Fleetham, G. Li and J. Li, ACS Appl. Mater. Interfaces, 2015, 7, 16240–16246 CrossRef CAS.
- Z.-Q. Zhu, K. Klimes, S. Holloway and J. Li, Adv. Mater., 2017, 29, 1605002 CrossRef.
- G. Li, J. Zheng, K. Klimes, Z.-Q. Zhu, J. Wu, H. Zhu and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 40320–40331 CrossRef CAS PubMed.
- L.-K. Li, M.-C. Tang, S.-L. Lai, M. Ng, W.-K. Kwok, M.-Y. Chan and V. W.-W. Yam, Nat. Photonics, 2019, 13, 185–191 CrossRef CAS.
- D. Zhou, W.-P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong and C.-M. Che, Adv. Sci., 2019, 1802297, DOI:10.1002/advs.201802297.
- D. Zhou, W.-P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS.
- H. Beucher, S. Kumar, E. Merino, W.-H. Hu, G. Stemmler, S. Cuesta-Galisteo, J. A. González, J. Jagielski, C.-J. Shih and C. Nevado, Chem. Mater., 2020, 32, 1605–1611 CrossRef CAS.
- M.-C. Tang, L.-K. Li, S.-L. Lai, W.-L. Cheung, M. Ng, C.-Y. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2020, 59, 21023–21031 CrossRef CAS PubMed.
- T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- M.-C. Tang, M.-Y. Leung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 13115–13124 CrossRef CAS.
- M.-Y. Leung, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 2448–2459 CrossRef CAS.
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629–16632 RSC.
- W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036–14041 CrossRef CAS PubMed.
- C. C. Au-Yeung, L.-K. Li, M.-C. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2021, 12, 9516–9527 RSC.
- W.-K. Kwok, M.-C. Tang, S.-L. Lai, W.-L. Cheung, L.-K. Li, M. Ng, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2020, 59, 9684–9692 CrossRef CAS.
- S.-Y. Huang, H.-F. Meng, H.-L. Huang, T.-C. Chao, M.-R. Tseng, Y.-C. Chao and S.-F. Horng, Synth. Met., 2010, 160, 2393–2396 CrossRef CAS.
- C. Fan, J. Miao, B. Jiang, C. Yang, H. Wu, J. Qin and Y. Cao, Org. Electron., 2013, 14, 3392–3398 CrossRef CAS.
- S. H. Cho and Y. Ha, Mol. Cryst. Liq. Cryst., 2017, 651, 85–90 CrossRef CAS.
- B. Jiang, X. Ning, S. Gong, N. Jiang, C. Zhong, Z.-H. Lu and C. Yang, J. Mater. Chem. C, 2017, 5, 10220–10224 RSC.
-
K. H. Shen, S. T. Yeh, H. L. Huang, I. H. Shen, M. T. Chu and T. S. Shieh, T.W. Pat., I242999, 2004 Search PubMed.
-
K. H. Shen, S. T. Yeh, H. L. Huang, I. H. Shen, M. T. Chu and T. S. Shieh, U.S. Pat., 7445857, 2008 Search PubMed.
- M.-T. L. Lee, M.-T. Chu, J.-S. Lin and M.-R. Tseng, J. Phys. D: Appl. Phys., 2010, 43, 442003 CrossRef.
- Q.-D. Ou, L. Zhou, Y.-Q. Li, S. Shen, J.-D. Chen, C. Li, Q.-K. Wang, S.-T. Lee and J.-X. Tang, Adv. Funct. Mater., 2014, 24, 7249–7256 CrossRef CAS.
- W.-C. Chen, Y. Yuan, Z.-L. Zhu, Z.-Q. Jiang, S.-J. Su, L.-S. Liao and C.-S. Lee, Chem. Sci., 2018, 9, 4062–4070 RSC.
- D. Zhang, L. Duan, Y. Li, D. Zhang and Y. Qiu, J. Mater. Chem. C, 2014, 2, 8191–8197 RSC.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Sci. Rep., 2015, 5, 7859 CrossRef CAS.
- X. Ban, Y. Liu, J. Pan, F. Chen, A. Zhu, W. Jiang, Y. Sun and Y. Dong, ACS Appl. Mater. Interfaces, 2020, 12, 1190–1200 CrossRef CAS.
- V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS.
- C.-H. Lee, M.-C. Tang, F. K.-W. Kong, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 520–529 CrossRef CAS PubMed.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. Sci., 2020, 11, 1192–1204 RSC.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- D. Yokoyama, J. Mater. Chem., 2011, 21, 19187–19202 RSC.
-
T. D. Schmidt, T. Lampe, M. R. Daniel Sylvinson, P. I. Djurovich, M. E. Thompson and W. BrüttingPhys. Rev. Appl., 2017, 8, 037001 Search PubMed.
- K.-H. Kim and J.-J. Kim, Adv. Mater., 2018, 30, 1705600 CrossRef.
- J. Ràfols-Ribé, P.-A. Will, C. Hänisch, M. Gonzalez-Silveira, S. Lenk, J. Rodríguez-Viejo and S. Reineke, Sci. Adv., 2018, 4, eaar8332 CrossRef.
- T. Komino, H. Tanaka and C. Adachi, Chem. Mater., 2014, 26, 3665–3671 CrossRef CAS.
- T. Marcato and C.-J. Shih, Helv. Chim. Acta, 2019, 102, e1900048 CrossRef.
-
C.-K. Moon, Molecular Orientation and Emission Characteristics of Ir Complexes and Exciplex in Organic Thin Films, Springer, 2019 Search PubMed.
- X. Yang, H. Guo, B. Liu, J. Zhao, G. Zhou, Z. Wu and W.-Y. Wong, Adv. Sci., 2018, 5, 1701067 CrossRef PubMed.
- Y. Sun, X. Yang, Z. Feng, B. Liu, D. Zhong, J. Zhang, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 26152–26164 CrossRef CAS.
- S. K. Jeon, H. L. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, 1803524 CrossRef.
- M.-C. Tang, M.-Y. Chan and V. W.-W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Seth Marder in celebration of his contributions to research to the scientific community and on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh00821h |
| § L.-K. L. and C. C. A. Y. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Cathay Chai
Au-Yeung§
,
Cathay Chai
Au-Yeung§


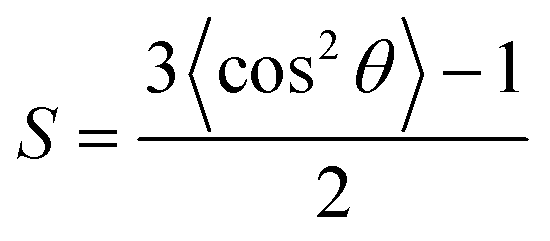 , with the bracketed values 〈…〉 indicating an ensemble average of 〈cos2θ〉. From ref. 55.
b
Θ represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θ:(1 − Θ) = 〈sin2θ〉
, with the bracketed values 〈…〉 indicating an ensemble average of 〈cos2θ〉. From ref. 55.
b
Θ represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θ:(1 − Θ) = 〈sin2θ〉