
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmbient-pressure ozone treatment enables tuning of oxygen vacancy concentration in the La1−xSrxFeO3−δ (0 ≤ x ≤ 1) perovskite oxides†
Geletu
Qing
a,
David
Thompson
 a,
Mourad
Benamara
b,
Clemens
Heske
cd,
Lauren
Greenlee
*e and
Jingyi
Chen
*a
a,
Mourad
Benamara
b,
Clemens
Heske
cd,
Lauren
Greenlee
*e and
Jingyi
Chen
*a
aDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville, USA. E-mail: chenj@uark.edu
bInstitute of Nanoscience and Engineering, University of Arkansas, Fayetteville, USA
cDepartment of Chemistry and Biochemistry, University of Nevada, Las Vegas (UNLV), USA
dInstitute for Photon Science and Synchrotron Radiation (IPS) and Institute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany
eDepartment of Chemical Engineering, Pennsylvania State University, USA. E-mail: greenlee@psu.edu
First published on 8th September 2022
Abstract
Oxygen vacancies in metal oxides can determine their properties. However, it is difficult to reduce the oxygen vacancy concentration in metal oxides without annealing them under high pressure. In this work, we develop a facile approach to control oxygen vacancy content via an ozone treatment under ambient pressure during cooling. This approach is demonstrated for the synthesis of La1−xSrxFeO3−δ (0 ≤ x ≤ 1, 0 ≤ δ ≤ 0.5x) perovskite oxides – an important class of energy-related materials due to their wide range of non-stoichiometry, mixed ionic and electronic conductivity, and the presence of a rare Fe(IV) oxidation state. A series of La1−xSrxFeO3−δ compounds was initially synthesized using a polymerized complex method. The concentration of oxygen vacancies and Fe(IV) were determined by redox titration, and the crystal structures were derived by analyzing X-ray diffraction patterns using Rietveld refinement. Significant amounts of oxygen vacancies were found in the as-synthesized compounds with x ≥ 0.8: La0.2Sr0.8FeO3−δ (δ = 0.066) and SrFeO3−δ (δ = 0.195). The ambient-pressure ozone treatment approach was able to substantially reduce the amount of oxygen vacancies in these compounds to achieve levels near the oxygen stoichiometry of 3 for La0.2Sr0.8FeO3−δ (δ = 0.006) and SrFeO3−δ (δ = 0.021). The oxygenation/deoxygenation kinetics can be tuned by the cooling rate after annealing. As the oxygen vacancy concentration decreases, the structure of SrFeO3−δ evolves from orthorhombic to cubic, demonstrating that the crystal structures in metal oxides can be highly sensitive to the number of oxygen vacancies. The ozone treatment approach developed in this study may thus offer a robust means to tune the properties of a wide variety of metal oxides.
1. Introduction
Oxygen vacancies have a profound effect on both bulk (e.g., ionic/electronic conductivity and optical properties) and surface properties (e.g., catalytic activity) of transition metal oxides.1–3 Changes in oxygen stoichiometry alter the surface electronic structure of metal oxides and thus affect the adsorption strength of reactants and intermediates on the surface, which is critical for surface-catalyzed reactions. A number of studies have demonstrated that catalytic activity varies with the concentration of oxygen vacancies in the transition metal oxides, including materials such as Co3O4, Ca2Mn2O5, SrCoOx, and MnO2 in oxygen evolution/reduction,4–7 SnOx and ZnO in carbon dioxide reduction,8,9 and LaxSrO3−δ and Cr-doped CeO2 in nitrogen reduction,10,11 as well as CeO2, MnO, and Li[Li0.144Ni0.136Co0.136Mn0.544]O2 in battery application.12–14 The oxygen vacancies can also affect other applications of oxides and their hybrids, such as electromagnetic wave absorption.15–18 However, a versatile strategy of controlling the oxygen vacancy in metal oxides remains lacking. Hence, this work focuses on method development of tuning the oxygen vacancy density of metal oxides, in particular perovskite oxides such as La1−xSrxFeO3−δ (0 ≤ x ≤ 1, 0 ≤ δ ≤ 0.5x), which are known to have a wide range of non-stoichiometry.The La1−xSrxFeO3−δ perovskites are also known to be one of the few compound suites that contain Fe(IV), where the substitution of La(III) with Sr(II) results in the oxidation of Fe(III) to Fe(IV); however, oxygen vacancies would also tend to compensate for the decrease in positive electric charge.19–21 The formation of oxygen vacancies is particularly common in compounds that theoretically contain a large amount of Fe(IV). For example, a full substitution of La(III) with Sr(II) to yield SrFeO3−δ is expected to contain only Fe(IV) if it is fully oxygenated (i.e., SrFeO3), but in practice, its oxygen deficiency can vary from δ = 0 to δ = 0.5, depending on the synthesis conditions.22–24 The oxygen stoichiometry of the La1−xSrxFeO3−δ perovskites strongly depends on the synthesis conditions, especially the final annealing temperature and oxygen partial pressure. With the substitution of La(III) with Sr(II), the formation energy of oxygen vacancies in La1−xSrxFeO3−δ decreases significantly and, as a result, the compound tends to be oxygen deficient.25 For example, the La1−xSrxFeO3−δ compounds with x ≤ 2/3 are essentially free of oxygen vacancies (i.e., δ = 0) when they are annealed in air and then cooled down slowly. However, for the compounds with x > 2/3, it is impossible to remove the oxygen vacancies by annealing in air, and the oxygen vacancy concentration increases with the Sr doping level.20,26
Being able to tune or reduce the number of oxygen vacancies in the La1−xSrxFeO3−δ perovskites has drawn considerable attention, because they exhibit a mixed ionic and electronic conductivity,1,27,28 suited for electrocatalysis29,30 and oxygen storage,31 and Fe(IV) is particularly critical for understanding the oxygen evolution reaction (OER) mechanisms in Fe-doped NiOOH electrocatalysts.29 The oxygen deficiency of La1−xSrxFeO3−δ could be finely tuned in the range of δ = 0 to δ = 0.5 by annealing the compounds under a controlled environment. For example, either rapid quenching at high temperatures or annealing under a low oxygen partial pressure, an inert atmosphere (e.g., Ar), or a reducing atmosphere (e.g., 1% H2 in Ar), could introduce more oxygen vacancies into the crystalline structure.20,21,32 However, it is hard to eliminate oxygen vacancies at x ≥ 2/3 even when annealing under pure oxygen is performed.19–21,32 It is even more difficult to yield the fully oxygenated SrFeO3 without extreme conditions, that is, under pure oxygen at extremely high pressure (≥20 MPa).22,33,34
In this work, we have developed an ozone treatment method which can greatly suppress the formation of oxygen vacancies in La1−xSrxFeO3−δ with x ≥ 2/3, including SrFeO3−δ. Ozone is a strong oxidant that has been used to reduce oxygen vacancies; however, these ozone methods were limited to post-treatment, or in some cases coupled with ultraviolet (UV) radiation,35,36 for the removal of the surface oxygen vacancies of metal oxides.37 To our knowledge, ozone has not yet been demonstrated to reduce the oxygen vacancies of the bulk, especially for those cases in which full oxygenation of the bulk is difficult to achieve without a high-pressure oxygen treatment. This study demonstrates a new approach beyond the simple post-treatment in which ozone is applied in situ during the cooling process of the synthesis. A series of powder samples of La1−xSrxFeO3−δ (x = 0.0, 0.2, 0.4, 0.6, 0.8, and 1.0) were first synthesized using a polymerized complex method. The crystal structures of these samples were characterized using X-ray powder diffraction (XRD) and a Rietveld refinement. The content of Fe(IV) and oxygen vacancies in the samples were determined by redox titration. Ozone treatments were then applied to treat those samples with oxygen deficiency (i.e., La0.2Sr0.8FeO3−δ and SrFeO3−δ) while varying temperature, duration, and cooling rate. It was found that a slow cooling rate is the key to achieving high oxygen stoichiometry. As a result, highly oxygenated compounds, including La0.2Sr0.8FeO2.994 and SrFeO2.979, were successfully synthesized via ozone treatment at 225 °C for 6 h with subsequent slow cooling to 25 °C at 0.05 °C min−1. Our findings demonstrate that ozone treatment under atmospheric pressure can replace the high-pressure oxygen annealing in previous studies for the synthesis of highly oxygenated La1−xSrxFeO3−δ perovskites with high Sr/La ratios (x ≥ 2/3).
2. Experimental method
Chemicals and materials
Lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O, 99.9%), strontium nitrate (Sr(NO3)2, ACS grade, 99.0% min), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, ACS, 98.0 + %), ethylenediaminetetraacetic acid (EDTA, 99%), citric acid (anhydrous, ACS grade, 99.5 + %), ammonium hydroxide (NH4OH, 28% NH3), hydrochloric acid (HCl, 99.999% (metals basis), 36.5% min), 1,10-phenanthroline iron(II) sulfate (0.025M aq. soln.), and cerium(IV) sulfate (Ce(SO4)2, 0.1 N standardized solution) were purchased from Alfa Aesar. Ammonium iron(II) sulfate hexahydrate ((NH4)2Fe(SO4)2·6H2O, Mohr's salt, 99.997% trace metals basis), phosphoric acid (H3PO4, 85 wt% in H2O, 99.99% trace metals basis), and iron(III) oxide (Fe2O3, ≥99.995% trace metals basis) were purchased from Sigma-Aldrich. Ultrahigh-purity-grade oxygen (O2, OX UHP300) and ultrahigh-purity-grade argon (Ar, AR UHP300) were purchased from Airgas. All chemicals and materials were used as received, unless specified otherwise. Ultrapure 18 MΩ cm water was used in all experiments.Synthesis of La1−xSrxFeO3−δ (x = 0.0, 0.2, 0.4, 0.6, 0.8, 1.0)
The La1−xSrxFeO3−δ powder samples were synthesized using a polymerized complex method.38 For the synthesis of LaFeO3, 40 mmol of EDTA was mixed with 40 ml of 1 M NH4OH solution in a 500 mL beaker, and then 20 mmol of La(NO3)3·6H2O, 20 mmol of Fe(NO3)3·9H2O, and 60 mmol of citric acid were added to the solution under magnetic stirring. The solution pH was then adjusted to 8.0 by adding 1 M NH4OH solution. The resulting solution was heated on a hot plate with a surface temperature of 135 °C and stirred at 340 rpm for 24 h to form a viscous gel. The obtained gel was then heated in a muffle furnace (Thermolyne 6000, Thermo Scientific) at 200 °C for 6 h to form a powder. [Caution: the reaction mixture expands vigorously and generates a large amount of smoke during the heating; therefore, this process must be carried out in a fume hood with sufficient ventilation. To prevent significant sample loss due to the expansion of the reaction mixture, the beaker containing the gelled sample was placed in a 1-gallon wide-mouth glass jar (Fig. S1, ESI†) that was then placed inside the furnace chamber.] After the heat treatment was completed, the sample powder was collected, ground carefully using a mortar and pestle, and then calcined in the muffle furnace at 900 °C for 5 h in air. The as-obtained power was ground again and then calcined in a high-temperature furnace (KSL-1700X, MTI Corporation) at 1300 °C for 4 h in air, with the heating and cooling rates set to 5 °C min−1. The final product was ground and stored under ambient conditions for future use.For the samples containing Sr, i.e., La1−xSrxFeO3−δ with x = 0.2, 0.4, 0.6, 0.8, and 1.0, the same method was used, but the La precursor was replaced by the Sr precursor in the synthesis, according to the corresponding molar ratio. For the synthesis of La0.2Sr0.8FeO3−δ and SrFeO3−δ, impurities were found in the final product when a large amount of the sample powder was calcined at 1300 °C using a single alumina crucible (cylindrical with covering lid, diameter: 40 mm, height: 40 mm, MTI corporation). To prevent this issue, high-purity samples were synthesized using the following protocols: for La0.2Sr0.8FeO3−δ, the sample powder was equally separated into 8 alumina crucibles, with 4 of them calcined at 1300 °C at a time. For SrFeO3−δ, the sample powder was equally separated into 16 alumina crucibles, with 2 of them calcined at 1300 °C at a time.
Ozone treatment of SrFeO3−δ and La0.2Sr0.8FeO3−δ
The ozone treatment was carried out under atmospheric pressure in a horizontal 1 inch diameter quartz tube furnace (Lindberg/Blue M, Model TF55035A-1), connected to a commercial ozone generator (Enaly, Model 5000BF-1), as shown in Fig. S2 (ESI†). The SrFeO3−δ or La0.2Sr0.8FeO3−δ powder was evenly spread in a quartz boat to maximize the contact between the sample particles and the gas flow. The quartz boat was then placed inside the quartz tube and positioned at the center of the tube furnace. In a typical process, the sample was flushed with oxygen for 30 min to ensure that the sample was under pure oxygen environment. Then, the furnace temperature was increased from room temperature to 650 °C at the rate of 5 °C min−1 and kept at that temperature for 6 h. Immediately after, the furnace temperature was decreased to the ozone treatment temperature (200–400 °C) at the rate of 5 °C min−1 and then kept at that temperature for 2–6 h. The ozone generator was turned on when the furnace temperature reached the ozone treatment temperature. After ozone treatment, the sample was cooled to room temperature under the ozone environment. The ozone generator used in this study can provide ozone at 5% concentration when the oxygen flow rate is ∼750 mL min−1. Given that the ozone treatment was operated at a lower oxygen flow rate, the ozone concentration in all experiments is 5% minimum (min. 5%).X-Ray diffraction and Rietveld refinement
The crystalline structures of the samples were determined by XRD using Cu Kα radiation (Philips PW1830). The diffraction patterns were collected over a 2θ range of 10–90° using a step size of 0.025° at 3 s per step, corresponding to ∼2.7 h of collection time for each measurement to produce an acceptable signal-to-noise ratio. The obtained data were analyzed by the Rietveld method39 using the FullProf Suite program.40 Initial models of the crystal structures, in the format of Crystallographic Information Files (.cif), were obtained from the Crystallography Open Database (COD).41–47 The line shape of the diffraction peaks was modelled using a pseudo-Voigt function, and the background was refined to a linear interpolation between a set of background points with refinable heights. Note that the site occupancy factors for the oxygen ions were not refined since it is difficult to obtain accurate values from the refinement of powder XRD data. Instead, the most abundant phase in each sample was determined by fitting the experimental data with multiple model crystal structures with known site occupancy factors that are available from the COD. The model crystal structure that could best fit the experimental data and result in a reasonable Fe–O bond distance is considered the dominant phase of the sample for refinement.Redox titration
The concentrations of Fe(IV) (y) and oxygen vacancies (δ) in the La1−xSrxFeO3−δ compounds were quantified by redox titration.19,48 All glassware and magnetic stir bars were cleaned with mild detergent, soaked overnight in a 10% nitric acid bath, washed thoroughly with deionized water, and rinsed thoroughly with ultrapure 18 MΩ cm water. The titration procedure starts by adding 50 ml of 6 M HCl to the flask, which is then purged with ultrahigh-purity-grade Ar for 5 min under stirring at 200 rpm to remove atmospheric oxygen; the titration set-up is shown in Fig. S3 (ESI†). The entire titration process was performed under continuous Ar flow. Then, ∼0.1 g of Mohr's salt, containing Fe(II), and ∼0.05 g of the sample powder were added to the solution, followed by rinsing the walls of the flask with 5 ml of 6 M HCl to ensure that all of the added Mohr's salt and the sample powder was rinsed into the solution. After purging with Ar for another 5 min while stirring, the reaction mixture was heated to fully dissolve the sample powder, whose Fe(IV) component is reduced by Fe(II) of the Mohr's salt to yield Fe(III). The temperature and heating time required for the complete dissolution of each sample are listed in Table S1 (ESI†). After complete dissolution, the reaction mixture was cooled down in a water-ice bath for 10 min. Then, 5 mL of 85 wt% H3PO4 was added to the titrate to form a colorless complex with Fe(III), followed by adding one drop of 0.025 M 1,10-phenanthroline iron(II) sulfate as an indicator. Finally, the excess of Fe(II) from the Mohr's salt in the solution was titrated with a 0.015 M Ce(SO4)2 solution. The end point of the titration was indicated by a sudden color change of the solution from red-orange to colorless or light blue. Each sample was titrated three times to report the mean values and standard errors.The Fe(IV) concentration (y) and the oxygen vacancy (δ) in La1−xSrxFe(III)1−yFe(IV)yO3−δ can be calculated using the eqn (1) and (2), based on the charge balance of the chemical formula and the number of moles of Fe(IV) (n, in mol) in a sample with measured mass (m, in g) determined by the titration.
Charge balance:
| 3(1 − x) + 2x + 3(1 − y) + 4y = 2(3 − δ) | (1) |
Fe(IV) determined by titration:
| ym/[MLa(1 − x) + MSrx + MFe + Mo(3 − δ) = n] | (2) |
| δ = (242.747n − 51.285nx − mx)/(15.999n − 2m) | (3) |
With δ known, the Fe(IV) concentration can be calculated using eqn (4).
| y = x − 2δ | (4) |
The stoichiometries of La (1 − x) and Sr (x, 0 ≤ x ≤ 1) are obtained from the molar ratio of the precursors and confirmed by inductively coupled plasma mass spectrometry (ICP-MS) measurement.
3. Results and discussion
3.1. Elemental composition and crystalline structure of La1−xSrxFeO3−δ calcined in air
A series of La1−xSrxFeO3−δ (x = 0.0, 0.2, 0.4, 0.6, 0.8, 1.0) samples were synthesized using a polymerized complex method by first forming the gel from the basic solution of La, Sr, and Fe precursors in the presence of EDTA and citric acid, and then calcining the gel at sequential elevated temperatures to yield the perovskite oxide powders. Fig. 1 illustrates the reaction scheme, along with the crystal structure and the stoichiometry of each resulting compound obtained from XRD Rietveld refinement and redox titration. It was found that the cooling rate of the last calcination step is the key to control the oxygen deficiency in these compounds. The slow cooling rate allows the perovskite oxides to take up oxygen from the atmosphere, leading to a (partial) filling of oxygen vacancies.49 In the synthesis, the cooling rate was kept at 5 °C min−1 for the last calcination step. The elemental composition of the resulting La1−xSrxFeO3−δ compounds was analyzed using ICP-MS. The measured molar ratios of La/Fe and Sr/Fe were very close to the theoretical values of 1 − x and x, respectively, with the differences between the measured and theoretical ratios less than 0.015 (Fig. S4, ESI†). The results suggest that the reaction gives nearly 100 percent yield for the conversion of La, Sr, and Fe precursors to the corresponding perovskite oxide compounds.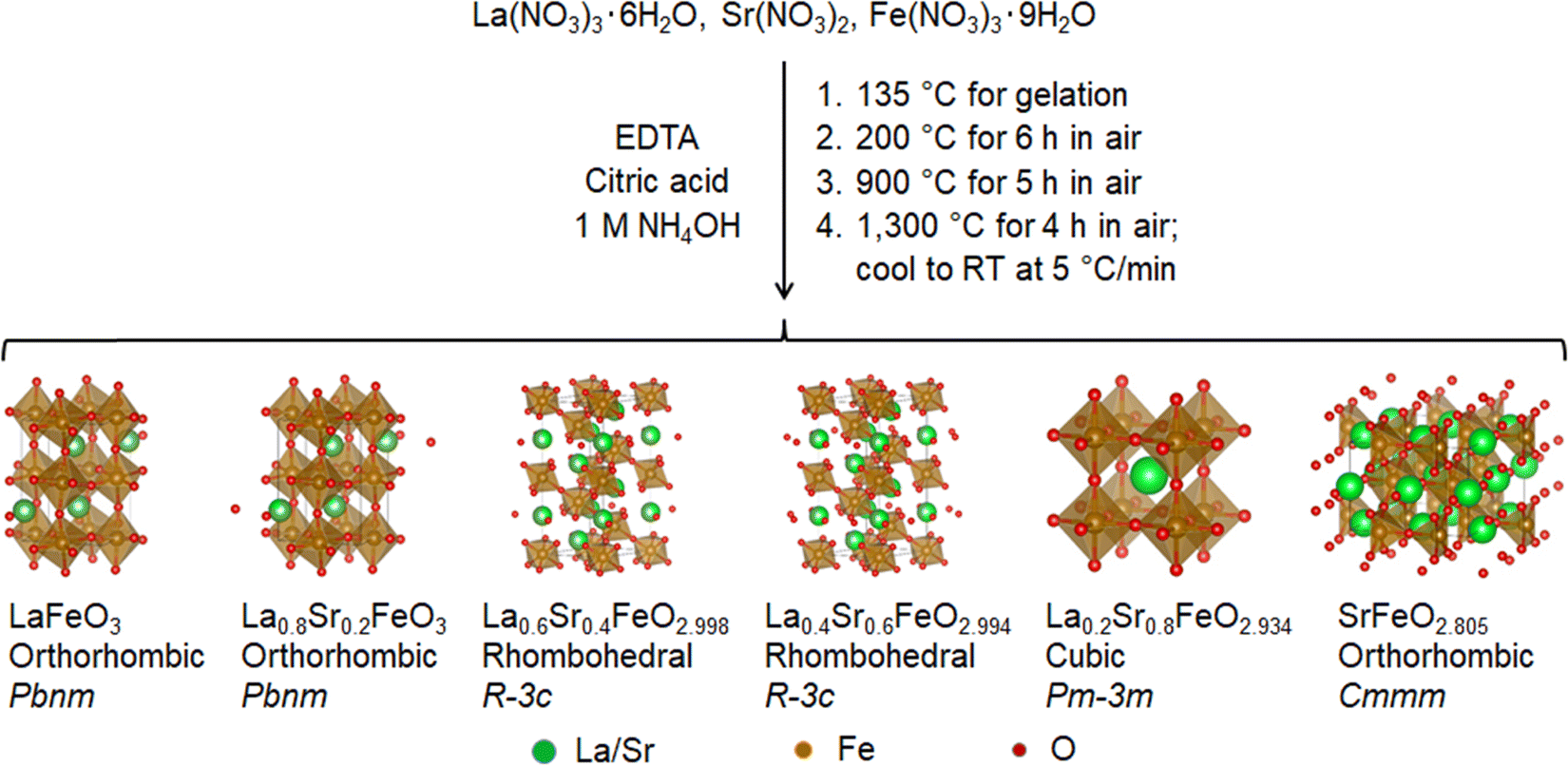 | ||
| Fig. 1 Reaction scheme of La1−xSrxFeO3−δ compounds via a polymerized complex method and calcined in air. The crystal structures were obtained from Rietveld refinements of the XRD patterns and the oxygen stoichiometry was determined by redox titration. | ||
These La1−xSrxFeO3−δ compounds were characterized by XRD to determine their corresponding crystal structures. Fig. 2 displays the XRD patterns of the La1−xSrxFeO3−δ compounds with x = 0, 0.2, 0.4, 0.6, 0.8, and 1.0. The XRD peak positions and their relative intensities are, in general, consistent with previously reported data for these compounds, prepared using a solid-state reaction or the Pechini method.49–51 To further confirm the crystal structure and estimate structural parameters such as lattice constants and atomic distances, we performed a Rietveld refinement on each of these XRD patterns based on known structural information in the literature. For example, it is known that fully oxygenated La1−xSrxFeO3 (i.e., δ = 0) undergoes phase changes as the Sr composition increases, from an orthorhombic phase (Pbnm) for 0 ≤ x ≤ 0.2, to a rhombohedral phase (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) for 0.4 ≤ x ≤ 0.7, and a cubic phase (Pmm) for 0.8 ≤ x ≤ 1.0.19,32,34,50 We also took into consideration the oxygen vacancy effects on the crystal structure in choosing the initial model crystal structures for refinement. This is particularly important for the compounds with Sr composition x > 0.6, where a significant amount of oxygen vacancies were found,20 and where the ordering of oxygen vacancies had a profound effect on the crystal structure.22,32,50 For example, the fully substituted end-member, SrFeO3−δ, was found to have the following four different crystal structures, depending on the oxygen stoichiometry: SrFeO3 (cubic, space group Pmm), SrFeO2.875 (tetragonal, space group I4/mmm), SrFeO2.75 (orthorhombic, space group Cmmm), and SrFeO2.5 (orthorhombic, Ibm2).22 Based on this knowledge, the experimental XRD patterns of La1−xSrxFeO3−δ compounds with 0 ≤ x ≤ 0.6 fit well with the calculated patterns for the corresponding structures of the fully-oxygenated La1−xSrxFeO3 (δ = 0). For La0.2Sr0.8FeO3−δ, a reasonable agreement between the observed diffraction data and calculated profile was achieved using a Pmm cubic structural model with oxygen stoichiometry of 2.96 (i.e., La0.2Sr0.8FeO2.96). This model was chosen based on literature suggestions that La0.2Sr0.8FeO3−δ prepared using a solid-state reaction method with annealing at 1230 °C for 48 h and then furnace cooling to room temperature had an oxygen stoichiometry of 2.96.20 For SrFeO3−δ, the experimental XRD patterns were refined by the four above-mentioned crystal structures (Fig. S5 and S6, ESI†). Based on the results, there could be two phases co-existing in our SrFeO3−δ compound, with a major phase of SrFeO2.75 (orthorhombic, space group Cmmm) and a minor phase of SrFeO2.875 (tetragonal, space group I4/mmm).
c) for 0.4 ≤ x ≤ 0.7, and a cubic phase (Pmm) for 0.8 ≤ x ≤ 1.0.19,32,34,50 We also took into consideration the oxygen vacancy effects on the crystal structure in choosing the initial model crystal structures for refinement. This is particularly important for the compounds with Sr composition x > 0.6, where a significant amount of oxygen vacancies were found,20 and where the ordering of oxygen vacancies had a profound effect on the crystal structure.22,32,50 For example, the fully substituted end-member, SrFeO3−δ, was found to have the following four different crystal structures, depending on the oxygen stoichiometry: SrFeO3 (cubic, space group Pmm), SrFeO2.875 (tetragonal, space group I4/mmm), SrFeO2.75 (orthorhombic, space group Cmmm), and SrFeO2.5 (orthorhombic, Ibm2).22 Based on this knowledge, the experimental XRD patterns of La1−xSrxFeO3−δ compounds with 0 ≤ x ≤ 0.6 fit well with the calculated patterns for the corresponding structures of the fully-oxygenated La1−xSrxFeO3 (δ = 0). For La0.2Sr0.8FeO3−δ, a reasonable agreement between the observed diffraction data and calculated profile was achieved using a Pmm cubic structural model with oxygen stoichiometry of 2.96 (i.e., La0.2Sr0.8FeO2.96). This model was chosen based on literature suggestions that La0.2Sr0.8FeO3−δ prepared using a solid-state reaction method with annealing at 1230 °C for 48 h and then furnace cooling to room temperature had an oxygen stoichiometry of 2.96.20 For SrFeO3−δ, the experimental XRD patterns were refined by the four above-mentioned crystal structures (Fig. S5 and S6, ESI†). Based on the results, there could be two phases co-existing in our SrFeO3−δ compound, with a major phase of SrFeO2.75 (orthorhombic, space group Cmmm) and a minor phase of SrFeO2.875 (tetragonal, space group I4/mmm).
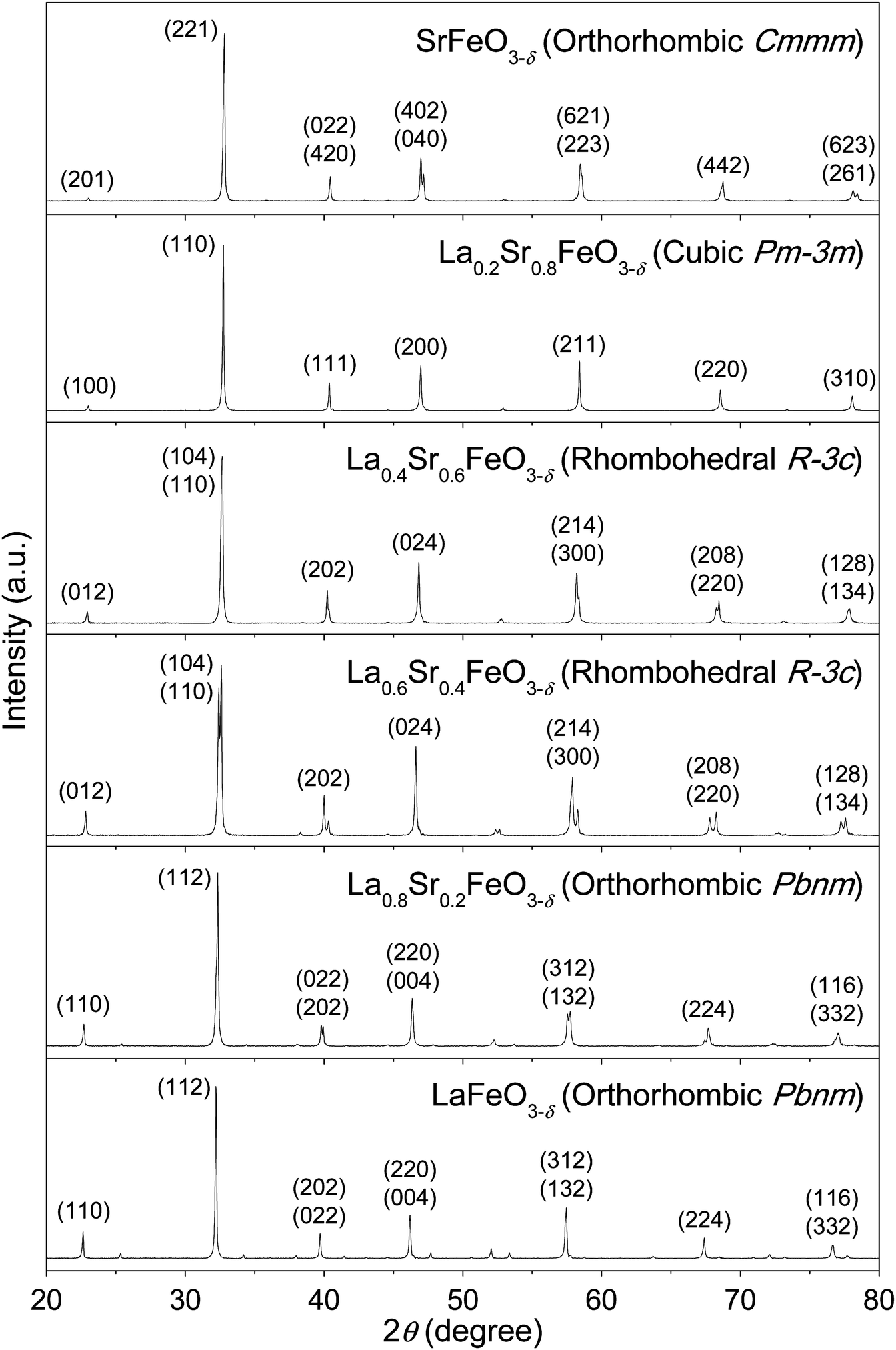 | ||
| Fig. 2 Powder XRD patterns of La1−xSrxFeO3−δ compounds, synthesized using a polymerized complex method and calcined in air, as described in Fig. 1. The patterns were indexed according to the calculated patterns generated using the VESTA software and the crystallographic information files obtained from the Rietveld refinement. | ||
The Rietveld refinement results are plotted in Fig. S7 (ESI†) by overlaying the experimental and calculated patterns for each compound. The structure of each compound refined by the Rietveld method is presented in Fig. 1. The data obtained from the Rietveld method are listed in Table 1. A gradual decrease in the Fe–O bond length with increasing Sr was found, from x = 0.0 to x = 0.8. The lengths of Fe–O bonds in iron oxides are typically 1.91–1.95 Å, 1.990–2.006 Å, and about 2.14 Å for Fe(IV), Fe(III), and Fe(II), respectively.49 These results indicate that an increased percentage of Fe in La1−xSrxFeO3−δ is oxidized from Fe(III) to Fe(IV) with increasing Sr content up to x = 0.8. For the fully-substituted end-member SrFeO3−δ, however, the presence of a large quantity of oxygen vacancies results in the reduction of Fe(IV) to Fe(III). The exact content of Fe(IV) in the La1−xSrxFeO3−δ compounds needs to be determined in order to verify the findings from the XRD analysis, as will be done in the following.
| Sample (x) | 0.0 | 0.2 | 0.4 | 0.6 | 0.8 | 1.0 |
|---|---|---|---|---|---|---|
| Note: a, b, and c are the lattice constants, V is the cell volume, and χ2 is the goodness-of-fit. | ||||||
| Model | LaFeO3 | La0.8Sr0.2FeO3 | La0.6Sr0.4FeO3 | La0.4Sr0.6FeO3 | La0.2Sr0.8FeO2.96 | SrFeO2.75 |
| Space group | Pbnm | Pbnm |
Rc |
Rc |
Pmm |
Cmmm |
| a (Å) | 5.5560 | 5.5521 | 5.5301 | 5.4989 | 3.8728 | 10.9522 |
| b (Å) | 5.5666 | 5.5249 | 7.7098 | |||
| c (Å) | 7.8558 | 7.8196 | 13.4294 | 13.4221 | 5.4707 | |
| V (Å3) | 242.96 | 239.87 | 355.67 | 351.48 | 58.09 | 461.94 |
| χ 2 | 1.41 | 1.29 | 1.56 | 1.64 | 1.77 | 1.52 |
| <Fe–O> (Å) | Fe–O1: 2.0235 | Fe–O1: 1.9986 | Fe–O: 1.9660 | Fe–O: 1.9484 | Fe–O: 1.9364 | Fe1–O1: 1.9244 |
| Fe–O1: 1.9849 | Fe–O1: 1.9641 | Fe1–O3: 1.8806 | ||||
| Fe–O2: 2.0174 | Fe–O2: 1.9915 | Fe2–O2: 1.9356 | ||||
| Fe2–O3: 2.0028 | ||||||
The content of Fe(IV) and the associated oxygen stoichiometry in the La1−xSrxFeO3−δ compounds were determined using redox titration. As illustrated in Fig. 3a, the titration involves a two-step process in which the Fe(IV) in the La1−xSrxFeO3−δ compound is first reacted with an excess amount of Fe(II), and then the remaining Fe(II) is titrated with a standard Ce(IV) solution. As a result, the number of moles of Fe(IV) in the La1−xSrxFeO3−δ compound equals the difference between the numbers of moles of the added Fe(II) and Ce(IV) consumed during the titration. The concentrations of Fe(IV) (y) and oxygen vacancies (δ) in each La1−xSrxFe(III)1−yFe(IV)yO3−δ compound were calculated based on the amounts of the added Mohr's salt or Fe(II), the sample used, and the 0.015 M Ce(SO4)2 solution consumed during titration. The calculation is detailed in the Experimental Method section. The titration results are summarized in Table S2 (ESI†) and are highly reproducible, as indicated by the standard errors of less than 0.004.
 | ||
| Fig. 3 (a) Schematic illustration of the working principle of the redox titration of Fe(IV). (b) Plot of the Fe(IV) (y, left ordinate) and oxygen vacancy (δ, right ordinate) content against Sr content (x) in the as-synthesized La1−xSrxFe(III)1−yFe(IV)yO3−δ compounds calcined in air. The y and δ values were determined from the titration results detailed in the ESI.† The standard error is too small to be seen on the graph, but a full list of the data (including the standard errors) is listed in Table S2 (ESI†). | ||
The stoichiometry of Fe(IV) (y) and oxygen vacancy (δ) are plotted against the Sr content (x) in the La1−xSrxFe(III)1−yFe(IV)yO3−δ compounds in Fig. 3b. A linear increase in y, the stoichiometry of Fe(IV), with increasing Sr composition (x) was observed until x = 0.6 (y = 0.985x with R2 = 0.9989). The slope of the linear fit is 0.985, suggesting that the substitution of La(III) by Sr(II) is nearly completely compensated by the conversion of Fe(III) to Fe(IV) in the La1−xSrxFeO3−δ perovskites (for 0 ≤ x ≤ 0.6). In other words, the number of oxygen vacancies (δ) in these compounds is very small. However, when the Sr composition (x) was further increased from 0.6 to 1.0, the curve deviated from the low-x linear relationship, only reaching y = 0.667 at x = 0.8. At x = 1.0, we even find a slight decrease to y = 0.611. The results suggest that significant amounts of oxygen vacancies are introduced to the La1−xSrxFeO3−δ perovskites with x > 0.6. These observations are consistent with previous studies that show how the content of the oxygen vacancies increases with increased Sr composition and that it is impossible to achieve fully oxygenated compounds with Sr composition x > 0.6 in air.20,26 The results also corroborate the XRD analysis that the SrFeO3−δ compound with oxygen stoichiometry of 2.805 would likely be a mixture of two different phases. This, in turn, agrees well with the previous finding that SrFeO2.75 (orthorhombic, space group Cmmm) and SrFeO2.875 (tetragonal, space group I4/mmm) can coexist in SrFeO2.80.23
3.2. Ozone treatment of highly-oxygen deficient La1−xSrxFeO3−δ compounds
To increase the oxygen stoichiometry of the compounds with high content of Sr (x > 0.6), we applied an ozone treatment at ambient pressure as an alternative to oxygen treatment at high pressure. Ozone is a strong oxidizing agent, known to generate radicals and thus eliminate pathogens and other contaminants in water.52 The radical mechanism makes it possible to fill the oxygen vacancies in highly oxygen-deficient compounds at mild conditions without the use of high pressure. Ozone treatment has been suggested to fully oxygenate the epitaxial La1−xSrxFeO3−δ films with thicknesses varying from 6.9 nm to 52.2 nm;2,53–55 however, so far there has not been a direct evidence of whether full oxygen stoichiometry was achieved in these studies. In this study, we thus further developed the ozone treatment approach and monitored the changes of oxygen stoichiometry by redox titration to demonstrate that the ozone treatment can reverse the loss of oxygen in these high-Sr compounds (i.e., SrFeO3−δ and La0.2Sr0.8FeO3−δ).The ozone treatment was carried out under min. 5% O3/O2 at ambient pressure. We investigated the effects of temperature, duration time, and cooling rate during ozone treatment on the Fe(IV) content and oxygen stoichiometry of the highest Sr containing compound, SrFeO3−δ, which is the most difficult one to be fully oxygenated. Fig. 4a–c, displays the correlation of the ozone treatment conditions and the changes of the Fe(IV) (y) and oxygen vacancy (δ) content of SrFe(III)1−yFe(IV)yO3−δ, monitored by the redox titration. The detailed parameters of each experiment are listed in Table S3 (ESI†). For a fully oxygenated compound, it is expected to have a stoichiometry of SrFe(IV)O3 with y = 1 and δ = 0. We first studied the effect of temperature in the region of 200 to 400 °C (Fig. 4a; E2 to E7 in Table S3, ESI†). It was hypothesized that an increased temperature could lower the activation barrier of the reaction based on the transition state theory (Arrhenius equation). However, the result indicated that with the increase of the ozone treatment temperature, the value of y first increased to reach the highest value of Fe(IV) at 0.887 obtained at 225 °C and then decreased (Fig. 4a). This implies that the temperature effect is not only to increase the rate of the forward reaction (kf) – oxygenation reaction of the ozone treatment, but also to increase the rate of the reverse reaction (kb) – deoxygenation reaction that causes the formation of oxygen vacancies.23 The difference of the temperature effects on kf and kb appear to suggest an optimal temperature for the oxygenation ozone treatment at 225 °C. However, these experiments were done at a similar cooling rate by allowing the furnace to naturally cool to 100 °C, followed by opening the furnace cover for fast cooling to room temperature. In fact, we found that the content of Fe(IV) significantly increased when the cover of the furnace was opened at a lower temperature (Fig. 4b). Varying the temperature at which the furnace cover was opened, the value of y increased from 0.777 to 0.891 for opening at 150 and 80 °C, respectively. This result suggests that the cooling rate plays a significant role in controlling the kf and kb and shifts the oxygenation/deoxygenation reaction equilibrium during ozone treatment.
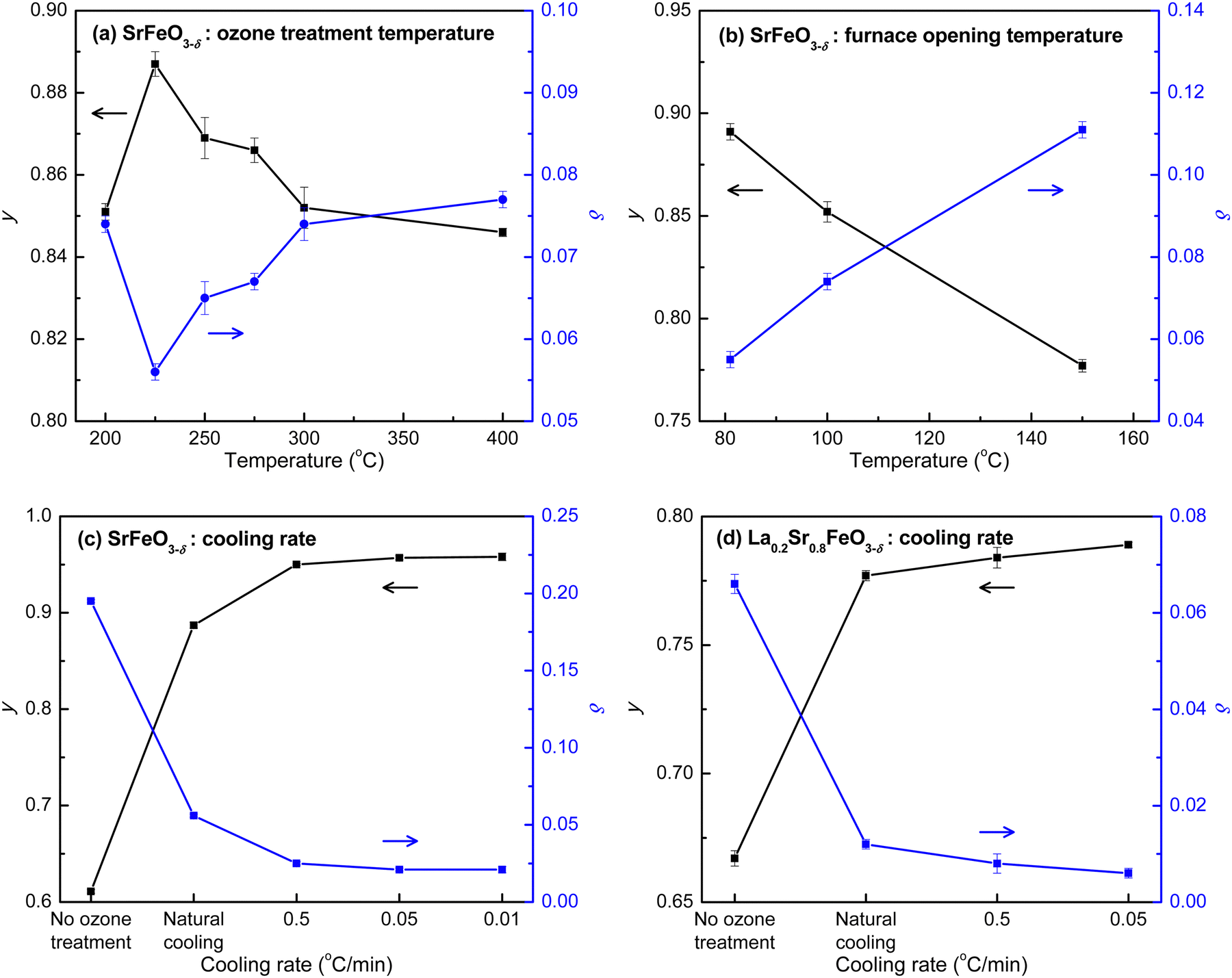 | ||
| Fig. 4 Plots of Fe(IV) (y) and oxygen vacancies (δ) content of SrFeO3−δ (a–c) and La0.2Sr0.8FeO3−δ (d) vs. ozone treatment step at different conditions: (a) SrFeO3−δ samples treated under ozone for 6 h at different temperatures, followed by cooling under ozone to 100 °C, followed by opening the furnace for fast cooling to room temperature; (b) SrFeO3−δ samples treated under ozone for 6 h at 300 °C, followed by cooling under ozone to different furnace-opening temperatures; (c) SrFeO3−δ and (d) La0.2Sr0.8FeO3−δ samples treated under ozone for 6 h at 225 °C with different controlled cooling rates, compared to “natural cooling” and “no ozone treatment”. The y and δ values were determined by the redox titration. | ||
To further examine the effect of the cooling rate, the ozone treatment of SrFeO3−δ was performed under a series of controlled cooling rates (E10 to E12 in Table S3, ESI†). The plot of y and δ against cooling rate in Fig. 4c indicates a significant increase in the content of Fe(IV) with decreased cooling rate. The values of y reached 0.950, 0.957, and 0.958 at cooling rates of 0.5, 0.05, and 0.01 °C min−1, respectively. The value of y increased only slightly when the cooling rate decreased from 0.05 to 0.01 °C min−1, indicating that the maximum value of y that could be obtained using this method is about 0.958. The corresponding highest oxygen stoichiometry can reach 2.979, which is within the range of those obtained via annealing under pure oxygen at high pressure (Table 2). The ozone treatment under controlled cooling rates was also carried out for La0.2Sr0.8FeO3−δ (E13 to E15 in Table S3, ESI†). Similar to the trend of SrFeO3−δ, a significant increase in y was observed with decreasing cooling rate (Fig. 4d). The highest y value, reached at a cooling rate of 0.05 °C min−1, was 0.789, which is only 0.13% different from the theoretical maximum value of 0.8, resulting in a stoichiometry of La0.2Sr0.8FeO2.994 with nearly full oxygen occupation. The results demonstrate that the ozone treatment indeed offers a robust alternative to reverse the oxygen loss and fully oxygenate perovskites at atmospheric pressure.
| Compound | Temperature, time | Oxygen pressure | Cooling condition | Method for δ determination | Ref. |
|---|---|---|---|---|---|
| SrFeO2.979 | 225 °C, 6 h | 101.325 kPa | 0.01 °C min−1 to RT | Redox titration | This work |
| SrFeO2.998 | 400 °C, 24 h | 30 MPa | 1 °C h−1 to 200 °C | Thermogravimetric | 22 |
| SrFeO2.961 | 450 °C, 24 h | 20 MPa | N/A | Redox titration | 19 |
| SrFeO2.98 | 900 °C, 12 h | 20 MPa | 5 °C min−1 to RT | Thermogravimetric | 33 |
| SrFeO2.94 | 900 °C, — | 35.5 MPa | Slowly cooled | Thermogravimetric | 34 |
The corresponding structures of the SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds with different degrees of oxygenation were then characterized by XRD. The XRD patterns of the SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds are shown at the top of Fig. S8 and S9 (ESI†), respectively. The oxygen vacancy concentration has a significant impact on the crystal structures of the SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds, as seen by comparing the peak positions and shapes of the XRD patterns. Detail plots of the major XRD peaks are also shown in Fig. S8 and S9 (ESI†). The peak(s) of each XRD pattern at 2θ = 47° are displayed in Fig. 5 to further elaborate the changes obtained in the XRD patterns for these two sets of compounds, along with the crystal structure indices generated by the Rietveld refinement. This particular 2θ angle is chosen because one peak would arise for the cubic crystal structure, corresponding to the reflection plane (200) that is perpendicular to the c-axis of the cubic structure; however, two peaks would appear for the orthorhombic structure, with one indexed as (402) and the other indexed as (040).
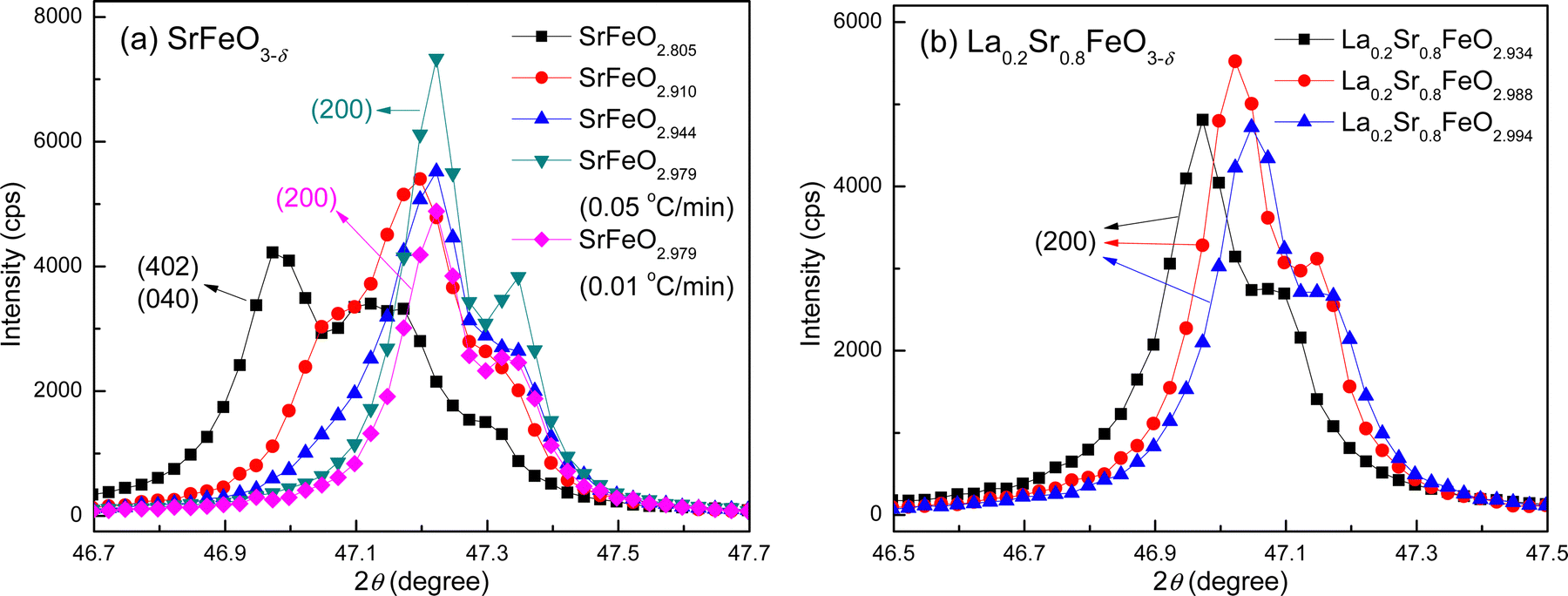 | ||
| Fig. 5 XRD region centered at 2θ = 47° for the ozone treated compounds: (a) SrFeO3−δ, and (b) La0.2Sr0.8FeO3−δ. The full XRD patterns are shown in Fig. S8 and S9 (ESI†). | ||
For SrFeO3−δ, both the peak positions and shapes of the diffraction pattern at 2θ ≈ 47° change significantly with increasing oxygen vacancy content (Fig. 5a). For the two near fully-oxygenated SrFeO2.979 samples, a peak at 47.22° with a shoulder (of about half its intensity) at 47.34° can be assigned to the (200) reflection of the cubic structure. The peak splitting arises from the Cu Kα doublet of the X-ray source. The simulated pattern from Rietveld refinement of the experimental data yields a doublet at 47.18° and 47.30°, in agreement with our experimental observation. For the highly oxygen-deficient SrFeO2.805, three peaks were identified (at 46.97°, 47.13°, and 47.29°), which can be indexed to the two doublet peaks for the (402) and (040) reflections of the orthorhombic structure. The Rietveld refinement of the orthorhombic structure, based on the experimental XRD data, yields simulated (402) reflections at 46.91° and 47.03°, and (040) reflections at 47.11° and 47.23°. The shoulder of the (402) peak at 47.03° overlaps with the major (040) peak at 47.11°, resulting in one broad peak observed in the experimental pattern. A structural transition from a cubic to an orthorhombic structure is observed in SrFeO2.944, with the peak at 47.22° being broadened towards lower angles. The transition is more obvious in SrFeO2.910, with the presence of a shoulder at 47.07°. As a result, the experimental diffraction patterns of SrFeO2.910 and SrFeO2.944 could not be fit using a single structural model in the Rietveld refinement.
For La0.2Sr0.8FeO3−δ, the peak shape remains very similar for all three compounds (La0.2Sr0.8FeO2.994, La0.2Sr0.8FeO2.988, and La0.2Sr0.8FeO2.934), as shown in Fig. 5b. The observed doublet can be assigned to the (200) reflection of the cubic structure based on the Rietveld refinement. As the number of oxygen vacancies increases, the peak shifts to lower angles, suggesting an increase of the lattice constant based on Bragg's Law. This finding is consistent with the Rietveld refinement results.
Table 3 summarizes the results of the Rietveld refinement of the experimental XRD patterns (cubic structures) for the near fully-oxygenated SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds. A comparison of the simulated and experimental XRD patterns is shown in Fig. S10 (ESI†). A decrease in the Fe–O bond distance was observed when decreasing the number of oxygen vacancies in the SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds, which indicates an increase in the oxidation state of Fe. These results are thus consistent with those obtained from the redox titration. For SrFeO3−δ, the Fe–O bond distances estimated from the refinement are 1.9262 Å for SrFeO2.979 (0.05 °C min−1) and 1.9248 Å for SrFeO2.979 (0.01 °C min−1). These values are consistent with the literature reported data of 1.91–1.95 Å for Fe(IV) in iron oxides,49 indicating an oxidation state close to Fe(IV).
| Samples | SrFeO2.979 (0.05 °C min−1) | SrFeO2.979 (0.01 °C min−1) | La0.2Sr0.8FeO2.988 | La0.2Sr0.8FeO2.994 |
|---|---|---|---|---|
| Note: a, b, and c are the lattice constant. V is cell volume. χ2 is goodness-of-fit. | ||||
| Model | SrFeO2.96 | SrFeO2.96 | La0.2Sr0.8FeO2.96 | La0.2Sr0.8FeO2.96 |
| Space group |
Pmm |
Pmm |
Pmm |
Pmm |
| a (Å) | 3.8525 | 3.8496 | 3.8673 | 3.8665 |
| b (Å) | ||||
| c (Å) | ||||
| V (Å3) | 57.18 | 57.05 | 57.84 | 57.80 |
| χ 2 | 1.99 | 2.17 | 1.92 | 1.71 |
| 〈Fe–O〉 (Å) | Fe–O: 1.9262 | Fe–O: 1.9248 | Fe–O: 1.9337 | Fe–O: 1.9333 |
The ozone treatment at a slow cooling rate thus provides a robust approach to reverse the loss of oxygen in the SrFeO3−δ and La0.2Sr0.8FeO3−δ perovskite oxides under ambient pressure, as schematically shown in Fig. 6. The ozone generates radicals that can effectively react with the perovskite lattice and fill the oxygen vacancies in highly oxygen-deficient structures; however, simple ozone applied in post-treatment is not sufficient to fully oxygenate the bulk of these materials. The ozone treatment is required to be applied in situ during the cooling process, starting from an optimized temperature of 225 °C, at which the oxygenation is favorable. More importantly, a slower cooling rate facilitates the complete filling of the oxygen vacancies of the bulk. In this study, the cooling rate of 0.05 °C min−1, starting from 225 °C, resulted in near-zero oxygen vacancies for the perovskites with high Fe(IV) content, quantified by the redox titration as SrFeO2.979 and La0.2Sr0.8FeO2.994. The crystal structures and stoichiometry of SrFeO2.979 and La0.2Sr0.8FeO2.994 compounds are stable in air at room temperature after storage for months, as confirmed by XRD and titration.
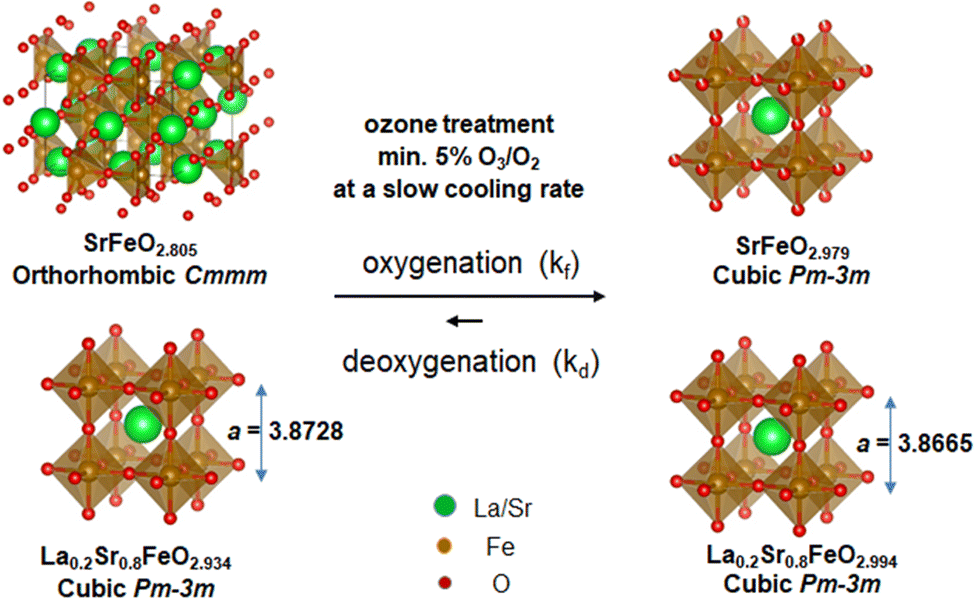 | ||
| Fig. 6 Schematic illustration of the ozone treatment at a slow cooling rate, facilitating the oxygenation reaction to produce samples with near-ideal oxygen stoichiometry (SrFeO2.979 and La0.2Sr0.8FeO2.994). | ||
After ozone treatment, little changes were found in the morphology of the samples for both SrFeO3−δ and La0.2Sr0.8FeO3−δ compounds, as shown in Fig. S11 and S12 (ESI†). They consist of particulates with different sizes ranging from tens of nanometers to tens of micrometers. Based on the EDS analysis, the ratio of Sr to Fe in SrFeO3−δ is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the ratio of La:Sr:Fe is close to the stoichiometry in formula of La0.2Sr0.8FeO3−δ. After storage under ambient conditions for 9 months, the crystal structures of the ozone-treated compounds (i.e., SrFeO2.979 and La0.2Sr0.8FeO2.994) remain the same as the freshly-prepared samples (Fig. S13, ESI†). The titration results indicate that the Fe(IV) stoichiometry of ozone-treated SrFeO3−δ at 0.01 °C min−1 only shows a slight decrease by 0.8%, and the Fe(IV) stoichiometry of ozone-treated SrFeO3−δ and La0.2Sr0.8FeO3−δ at 0.05 °C min−1 decreases by 1.8% and 1.5%, respectively (Table S3, ESI†). This method can couple to the commonly-used calcination method to offer a facile means to reduce oxygen vacancies without the need of high-pressure environments.
:1, and the ratio of La:Sr:Fe is close to the stoichiometry in formula of La0.2Sr0.8FeO3−δ. After storage under ambient conditions for 9 months, the crystal structures of the ozone-treated compounds (i.e., SrFeO2.979 and La0.2Sr0.8FeO2.994) remain the same as the freshly-prepared samples (Fig. S13, ESI†). The titration results indicate that the Fe(IV) stoichiometry of ozone-treated SrFeO3−δ at 0.01 °C min−1 only shows a slight decrease by 0.8%, and the Fe(IV) stoichiometry of ozone-treated SrFeO3−δ and La0.2Sr0.8FeO3−δ at 0.05 °C min−1 decreases by 1.8% and 1.5%, respectively (Table S3, ESI†). This method can couple to the commonly-used calcination method to offer a facile means to reduce oxygen vacancies without the need of high-pressure environments.
4. Conclusions
We have developed an ozone treatment method as a robust alternative to high pressure oxygen treatments to reverse the oxygen loss in the La1−xSrxFeO3−δ perovskites (x > 0.6). Using this method, we have successfully synthesized near fully-oxygenated La0.2Sr0.8FeO2.994 and SrFeO2.979 at ambient pressure. This method complements our synthesis of a series of La1−xSrxFeO3−δ (x = 0.0, 0.2, 0.4, 0.6, 0.8, and 1.0) compounds with near-ideal oxygen stoichiometry, in which annealing in air with slow cooling was sufficient to achieve full oxygen stoichiometry for x ≤ 0.6. Since La0.2Sr0.8FeO3−δ and SrFeO3−δ, however, showed significant amounts of oxygen vacancies when the compounds were calcined in air, we applied the above-mentioned heat-treatment in a mixture of ozone (min. 5%) and oxygen under atmospheric pressure at different conditions by varying temperature, duration, and cooling rate. It was found that the cooling rate is the key to obtaining high oxygen stoichiometry through an optimized balance of the rates of the oxygenation/deoxygenation reactions. In general, a significant increase in the oxygen content of La0.2Sr0.8FeO3−δ and SrFeO3−δ was observed with a decrease of the cooling rate. An ozone treatment at 225 °C for 6 h and subsequent cooling to 25 °C at 0.05 °C min−1 resulted in La0.2Sr0.8FeO2.994 and SrFeO2.979 under atmospheric pressure, achieving an oxygen concentration similar to the compounds obtained via annealing under pure oxygen at high pressure (≥20 MPa). This ozone treatment, using controlled heating and cooling rates, offers an attractive alternative approach to high pressure methods for the synthesis of fully oxygenated perovskites and other complex oxides under atmospheric pressure.Author contributions
Conceptualization, L. G., C. H., J. C.; methodology, G. Q., L. G., C. H., and J. C.; investigation, G. Q., D. T., and M. B.; data curation, G. Q., D. T., M. B., C. H., L. G., J. C.; writing – original draft preparation, G. Q.; writing – review and editing, G. Q., D. T., M. B., C. H., L. G., J. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support for this work from the Department of Energy (Grant Number: DE-SC0021308).References
- T. Maeder and J. G. Bednorz, Influence of Oxygen Stoichiometry on Electrical Transport and Magnetic Properties of Doped Perovskite-Type Ferrate and Manganate Single Crystals, J. Eur. Ceram. Soc., 1999, 19, 1507–1510 CrossRef CAS.
- S. Y. Smolin, M. D. Scafetta, A. K. Choquette, M. Y. Sfeir, J. B. Baxter and S. J. May, Static and Dynamic Optical Properties of La1-xSrxFeO3-δ: The Effects of a-Site and Oxygen Stoichiometry, Chem. Mater., 2016, 28, 97–105 CrossRef CAS.
- K. Y. Zhu, F. Shi, X. F. Zhu and W. S. Yang, The Roles of Oxygen Vacancies in Electrocatalytic Oxygen Evolution Reaction, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Wang and L. M. Dai, Plasma-Engraved Co3O4 Nanosheets with Oxygen Vacancies and High Surface Area for the Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- J. Kim, X. Yin, K. C. Tsao, S. H. Fang and H. Yang, Ca2Mn2O5 as Oxygen-Deficient Perovskite Electrocatalyst for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, Enhancing Perovskite Electrocatalysis through Strain Tuning of the Oxygen Deficiency, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef CAS PubMed.
- F. Y. Cheng, T. R. Zhang, Y. Zhang, J. Du, X. P. Han and J. Chen, Enhancing Electrocatalytic Oxygen Reduction on MnO2 with Vacancies, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- L. L. Li, Z. J. Zhao, C. L. Hu, P. P. Yang, X. T. Yuan, Y. N. Wang, L. Zhang, L. Moskaleva and J. L. Gong, Tuning Oxygen Vacancies of Oxides to Promote Electrocatalytic Reduction of Carbon Dioxide, ACS Energy Lett., 2020, 5, 552–558 CrossRef CAS.
- Z. G. Geng, X. D. Kong, W. W. Chen, H. Y. Su, Y. Liu, F. Cai, G. X. Wang and J. Zeng, Oxygen Vacancies in Zno Nanosheets Enhance CO2 Electrochemical Reduction to CO, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- K. B. Chu, F. Z. Liu, J. W. Zhu, H. Fu, H. Y. Zhu, Y. L. Zhu, Y. Zhang, F. L. Lai and T. X. Liu, A General Strategy to Boost Electrocatalytic Nitrogen Reduction on Perovskite Oxides via the Oxygen Vacancies Derived from a-Site Deficiency, Adv. Energy Mater., 2021, 11, 2003799 CrossRef CAS.
- H. T. Xie, H. B. Wang, Q. Geng, Z. Xing, W. Wang, J. Y. Chen, L. Ji, L. Chang, Z. M. Wang and J. Mao, Oxygen Vacancies of Cr-Doped CeO2 Nanorods That Efficiently Enhance the Performance of Electrocatalytic N2 Fixation to NH3 under Ambient Conditions, Inorg. Chem., 2019, 58, 5423–5427 CrossRef CAS PubMed.
- Y. Hou, J. Wang, C. X. Hou, Y. Q. Fan, Y. J. Zhai, H. Y. Li, F. Dang and S. L. Chou, Oxygen Vacancies Promoting the Electrocatalytic Performance of CeO2 Nanorods as Cathode Materials for Li-O2 Batteries, J. Mater. Chem. A, 2019, 7, 6552–6561 RSC.
- Y. H. Zou, W. Zhang, N. Chen, S. Chen, W. J. Xu, R. S. Cai, C. L. Brown, D. J. Yang and X. D. Yao, Generating Oxygen Vacancies in MnO Hexagonal Sheets for Ultralong Life Lithium Storage with High Capacity, ACS Nano, 2019, 13, 2062–2071 CrossRef CAS PubMed.
- B. Qiu, et al., Gas-Solid Interfacial Modification of Oxygen Activity in Layered Oxide Cathodes for Lithium-Ion Batteries, Nat. Commun., 2016, 7, 12108 CrossRef CAS PubMed.
- S. Zhang, Z. Jia, B. Cheng, Z. Zhao, F. Lu and G. Wu, Recent Progress of Perovskite Oxides and Their Hybrids for Electromagnetic Wave Absorption: A Mini-Review, Adv. Compos. Hybrid Mater., 2022, 1–22 Search PubMed.
- Y. Liu, Z. Jia, Q. Zhan, Y. Dong, Q. Xu and G. Wu, Magnetic Manganese-Based Composites with Multiple Loss Mechanisms Towards Broadband Absorption, Nano Res., 2022, 15, 5590–5600 CrossRef CAS.
- Z. Jia, M. Kong, B. Yu, Y. Ma, J. Pan and G. Wu, Tunable Co/ZnO/C@MWCNTs Based on Carbon Nanotube-Coated Mof with Excellent Microwave Absorption Properties, J. Mater. Sci. Technol., 2022, 127, 153–163 CrossRef.
- Y. Liu, X. Zhou, Z. Jia, H. Wu and G. Wu, Oxygen Vacancy-Induced Dielectric Polarization Prevails in the Electromagnetic Wave-Absorbing Mechanism for Mn-Based MOFs-Derived Composites, Adv. Funct. Mater., 2022, 32, 2204499 CrossRef CAS.
- J. Blasco, B. Aznar, J. Garcia, G. Subias, J. Herrero-Martin and J. Stankiewicz, Charge Disproportionation in La1-xSrxFeO3 Probed by Diffraction and Spectroscopic Experiments, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 054107 CrossRef.
- J. Cheng, A. Navrotsky, X. D. Zhou and H. U. Anderson, Thermochemistry of La1-xSrxFeO3-δ Solid Solutions (0.0 ≤ x≤ 1.0, 0.0 ≤ δ ≤ 0.5), Chem. Mater., 2005, 17, 2197–2207 CrossRef CAS.
- J. B. Yang, W. B. Yelon, W. J. James, Z. Chu, M. Kornecki, Y. X. Xie, X. D. Zhou, H. U. Anderson, A. G. Joshi and S. K. Malik, Crystal Structure, Magnetic Properties, and Mössbauer Studies of La0.6Sr0.4FeO3-δ Prepared by Quenching in Different Atmospheres, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 184415 CrossRef.
- J. P. Hodges, S. Short, J. D. Jorgensen, X. Xiong, B. Dabrowski, S. M. Mini and C. W. Kimball, Evolution of Oxygen-Vacancy Ordered Crystal Structures in the Perovskite Series SrnFenO3n-1 (n = 2, 4, 8, and ∞), and the Relationship to Electronic and Magnetic Properties, J. Solid State Chem., 2000, 151, 190–209 CrossRef CAS.
- Y. Takeda, K. Kanno, T. Takada, O. Yamamoto, M. Takano, N. Nakayama and Y. Bando, Phase Relation in the Oxygen Nonstoichiometric System, SrFeOx (2.5 ≤ x ≤ 3.0), J. Solid State Chem., 1986, 63, 237–249 CrossRef CAS.
- J. B. MacChesney, R. C. Sherwood and J. F. Potter, Electric and Magnetic Properties of the Strontium Ferrates, J. Chem. Phys., 1965, 43, 1907–1913 CrossRef CAS.
- A. M. Ritzmann, A. B. Munoz-Garcia, M. Pavone, J. A. Keith and E. A. Carter, Ab Initio DFT+U Analysis of Oxygen Vacancy Formation and Migration in La1-xSrxFeO3-δ (x = 0, 0.25, 0.50), Chem. Mater., 2013, 25, 3011–3019 CrossRef CAS.
- J. Mizusaki, M. Yoshihiro, S. Yamauchi and K. Fueki, Nonstoichiometry and Defect Structure of the Perovskite-Type Oxides La1-xSrxFeO3-δ, J. Solid State Chem., 1985, 58, 257–266 CrossRef CAS.
- M. Sogaard, P. V. Hendriksen and M. Mogensen, Oxygen Nonstoichiometry and Transport Properties of Strontium Substituted Lanthanum Ferrite, J. Solid State Chem., 2007, 180, 1489–1503 CrossRef CAS.
- J. Mizusaki, T. Sasamoto, W. R. Cannon and H. K. Bowen, Electronic Conductivity, Seebeck Coefficient, and Defect Structure of La1-xSrxFeO3 (x = 0.1, 0.25), J. Am. Ceram. Soc., 1983, 66, 247–252 CrossRef CAS.
- Z. C. Shen, et al., Increased Activity in the Oxygen Evolution Reaction by Fe4+-Induced Hole States in Perovskite La1-xSrxFeO3, J. Mater. Chem. A, 2020, 8, 4407–4415 RSC.
- N. P. Brandon, S. Skinner and B. C. H. Steele, Recent Advances in Materials for Fuel Cells, Annu. Rev. Mater. Res., 2003, 33, 183–213 CrossRef CAS.
- D. D. Taylor, N. J. Schreiber, B. D. Leyitas, W. Q. Xu, P. S. Whitfield and E. E. Rodriguez, Oxygen Storage Properties of La1-xSrxFeO3-δ for Chemical-Looping Reactions-an in Situ Neutron and Synchrotron X-Ray Study, Chem. Mater., 2016, 28, 3951–3960 CrossRef CAS.
- S. E. Dann, D. B. Currie, M. T. Weller, M. F. Thomas and A. D. Alrawwas, The Effect of Oxygen Stoichiometry on Phase Relations and Structure in the System La1-xSrxFeO3-δ (0 ≤ x ≤ 1, 0 ≤ δ ≤ 0.5), J. Solid State Chem., 1994, 109, 134–144 CrossRef CAS.
- H. Falcon, J. A. Barbero, J. A. Alonso, M. J. Martinez-Lope and J. L. G. Fierro, SrFeO3-δ Perovskite Oxides: Chemical Features and Performance for Methane Combustion, Chem. Mater., 2002, 14, 2325–2333 CrossRef CAS.
- A. D. Al-Rawwas, C. E. Johnson, M. F. Thomas, S. E. Dann and M. T. Weiler, Mössbauer Studies on the Series La1-xSrxFeO3, Hyperfine Interact., 1994, 93, 1521–1529 CrossRef CAS.
- H. Y. Chong, S. H. Lee and T. W. Kim, Effect of an Ultraviolet-Ozone Treatment on the Electrical Properties of Titanium-Oxide Thin-Film Transistors Fabricated by Using a Sol-Gel Process, J. Electrochem. Soc., 2012, 159, B771–B774 CrossRef CAS.
- A. Klasen, et al., Removal of Surface Oxygen Vacancies Increases Conductance through TiO2 Thin Films for Perovskite Solar Cells, J. Phys. Chem. C, 2019, 123, 13458–13466 CrossRef CAS PubMed.
- X. Luo, T. Su, X. Xie, Z. Qin and H. Ji, The Adsorption of Ozone on the Solid Catalyst Surface and the Catalytic Reaction Mechanism for Organic Components, ChemistrySelect, 2020, 5, 15092–15116 CrossRef CAS.
- J. I. Jung, M. Risch, S. Park, M. G. Kim, G. Nam, H. Y. Jeong, Y. Shao-Horn and J. Cho, Optimizing Nanoparticle Perovskite for Bifunctional Oxygen Electrocatalysis, Energy Environ. Sci., 2016, 9, 176–183 RSC.
- H. M. Rietveld, A Profile Refinement Method for Nuclear and Magnetic Structures, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- J. Rodriguez-Carvajal, Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- A. Vaitkus, A. Merkys and S. Grazulis, Validation of the Crystallography Open Database Using the Crystallographic Information Framework, J. Appl. Crystallogr., 2021, 54, 661–672 CrossRef CAS PubMed.
- M. Quiros, S. Grazulis, S. Girdzijauskaite, A. Merkys and A. Vaitkus, Using SMILES Strings for the Description of Chemical Connectivity in the Crystallography Open Database, J. Cheminf., 2018, 10, 23 Search PubMed.
- A. Merkys, A. Vaitkus, J. Butkus, M. Okulic-Kazarinas, V. Kairys and S. Grazulis, COD::CIF::Parser: An Error-Correcting CIF Parser for the Perl Language, J. Appl. Crystallogr., 2016, 49, 292–301 CrossRef CAS PubMed.
- S. Grazulis, A. Merkys, A. Vaitkus and M. Okulic-Kazarinas, Computing Stoichiometric Molecular Composition from Crystal Structures, J. Appl. Crystallogr., 2015, 48, 85–91 CrossRef CAS PubMed.
- S. Grazulis, A. Daskevic, A. Merkys, D. Chateigner, L. Lutterotti, M. Quiros, N. R. Serebryanaya, P. Moeck, R. T. Downs and A. Le Bail, Crystallography Open Database (COD): An Open-Access Collection of Crystal Structures and Platform for World-Wide Collaboration, Nucleic Acids Res., 2012, 40, D420–D427 CrossRef CAS PubMed.
- S. Grazulis, D. Chateigner, R. T. Downs, A. F. T. Yokochi, M. Quiros, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, Crystallography Open Database - an Open-Access Collection of Crystal Structures, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef CAS PubMed.
- R. T. Downs and M. Hall-Wallace, The American Mineralogist Crystal Structure Database, Am. Mineral., 2003, 88, 247–250 CrossRef CAS.
- C. Haavik, T. Atake and S. Stolen, On the Enthalpic Contribution to the Redox Energetics of SrFeO3-δ, Phys. Chem. Chem. Phys., 2002, 4, 1082–1087 RSC.
- O. Haas, U. F. Vogt, C. Soltmann, A. Braun, W. S. Yoon, X. Q. Yang and T. Graule, The Fe K-Edge X-Ray Absorption Characteristics of La1-xSrxFeO3-δ Prepared by Solid State Reaction, Mater. Res. Bull., 2009, 44, 1397–1404 CrossRef CAS.
- O. Clemens, M. Kuhn and R. Haberkorn, Synthesis and Characterization of the La1-xSrxFeO3-δ System and the Fluorinated Phases La1-xSrxFeO3-xFx, J. Solid State Chem., 2011, 184, 2870–2876 CrossRef CAS.
- M. Kuhn, S. Hashimoto, K. Sato, K. Yashiro and J. Mizusaki, Oxygen Nonstoichiometry, Thermo-Chemical Stability and Lattice Expansion of La0.6Sr0.4FeO3-δ, Solid State Ionics, 2011, 195, 7–15 CrossRef CAS.
- C. H. Wei, F. Z. Zhang, Y. Hu, C. H. Feng and H. Z. Wu, Ozonation in Water Treatment: The Generation, Basic Properties of Ozone and Its Practical Application, Rev. Chem. Eng., 2017, 33, 49–89 CAS.
- Y. J. Xie, M. D. Scafetta, E. J. Moon, A. L. Krick, R. J. Sichel-Tissot and S. J. May, Electronic Phase Diagram of Epitaxial La1-xSrxFeO3 Films, Appl. Phys. Lett., 2014, 105, 062110 CrossRef.
- Y. J. Xie, M. D. Scafetta, R. J. Sichel-Tissot, E. J. Moon, R. C. Devlin, H. Q. Wu, A. L. Krick and S. J. May, Control of Functional Responses Via Reversible Oxygen Loss in La1-xSrxFeO3-δ Films, Adv. Mater., 2014, 26, 1434–1438 CrossRef CAS PubMed.
- H. Yamada, M. Kawasaki and Y. Tokura, Epitaxial Growth and Valence Control of Strained Perovskite SrFeO3 Films, Appl. Phys. Lett., 2002, 80, 622–624 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00604a |
| This journal is © The Royal Society of Chemistry 2022 |