
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sustainable nano drug delivery system for poorly soluble drug, glipizide: design, in vitro controlled release and kinetics
Debatrayee
Dasgupta
and
Anjali
Patel
 *
*
Department of Chemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara, India. E-mail: anjali.patel-chem@msubaroda.ac.in
First published on 12th September 2022
Abstract
The present work deals with the design of drug delivery systems based on 12-tungstophosphoric acid (TPA)-functionalized MCM-48 nanoparticles (TPA/nMCM-48), for poorly soluble drug GLP. Two DDSs, namely GLP loaded on TPA/nMCM-48 and capped by TPA, were designed and characterized by various techniques. An in vitro release study of GLP was carried out in simulated body fluid (pH-7.4, 37 °C) under stirring conditions followed by the evaluation of the release kinetics and mechanism. The release profiles of the designed DDSs were compared with the release profiles of a commercially available formulation (Glynase) and GLP/nMCM-48. The comparison study indicates that TPA acts better as a functionalizing agent rather than as a capping agent and when compared with the marketed formulation, GLP/TPA/nMCM-48 shows more controlled release of GLP with only 15% cytotoxicity.
Introduction
Diabetes mellitus type 2 (DMT2) is a metabolic disorder, which is characterized by insulin resistance and deficiency, resulting in high blood glucose levels, and is also known as non-insulin-dependent diabetes. DMT2 is a complex disease that is associated with numerous health issues, which include retinopathy, neuropathy, nephropathy, and atherosclerosis, forcing the development of new medications and improved dosage forms to help in combating the disease. As a result, significant research attempts have been aimed towards the implementation of novel pharmacological agents for the treatment of DMT2. In spite of the availability of new agents, many physicians still prefer oral sulfonylurea due to its high tolerance and low cost.1–5Glipizide (GLP), an oral sulfonyl urea hypoglycaemic drug, is prescribed to treat non-insulin-dependent diabetes. GLP reduces blood glucose levels by stimulating insulin release via the SUR1 receptor on K/ATP-sensitive channels found in pancreatic cells.6,7 As GLP is a weak acid (pKa = 5.9), practically insoluble in water under acidic conditions, and highly permeable, the Biopharmaceutics Classification System, BCS, classifies it as a Class II drug. Even though its bioavailability is nearly 100%, its short biological half-life (3.4 ± 0.7 hours) necessitates its administration in two or three doses ranging from 2.5 to 10 mg per day.5,8 Owing to its poor solubility and short half-life, the development of controlled delivery strategies could lead to significant advantages in the clinical use of GLP. In this regard, several drug delivery systems (DDSs) including microspheres,9,10 polymers,11–14 osmotic pump systems,15,16 liposomes,17 hydrogels18–20 and emulsifiers7,21 have been investigated already as drug carriers for GLP.
In this direction, mesoporous silica nanoparticles (MSNs) have emerged as an effective delivery system due to their ability to enhance the bioavailability of lipophilic drugs. MSNs have unique properties such as meso-nanoscale pores (2–50 nm), variable pore sizes, and versatile morphological characteristics (using tunable synthesis techniques). Furthermore, the functionalization of MSNs increases the drug surface interactions and helps in overcoming issues like poor solubility and short bioavailability of poorly soluble drugs.22,23
However, it is observed that irrespective of any drug, MSNs are only functionalized with organic groups such as alkyl, phenyl, alkenyl, carboxyl, amino, hydroxyl, and sulphonyl.24 So, being an inorganic chemist, it was thought of interest to use an inorganic moiety for functionalization and hence, polyoxometalates (POMs) were selected because of their already reported biological activities both in vitro and in vivo, including antitumor, antiviral, antimicrobial, and antidiabetic properties. POMs are negatively charged metal-oxo clusters of high-oxidation-state early transition metal ions [e.g., W(VI), Mo(VI), V(V)]. A literature survey shows that 12-tungstophosphoric acid (TPA), a Keggin type of POM, is mainly used in medicinal chemistry.4,25–28 Tungstate is indeed an oxyanion that has demonstrated insulin-mimetic effects in isolated hepatocytes, raising fructose 2,6-bisphosphate levels, combatting the effects of glucagon on both fructose 2,6-bisphosphate doses and 6-phosphofructo-2-kinase activity, and boosting glycolytic flux. Polyoxotungstates and tungsten metal alone have previously been studied in diabetic mice and rats. In small animals, tungstate has been shown to regenerate pancreatic beta cell function and stimulate insulin secretion.29–31 The aim of the present study is to design a DDS for the controlled release of GLP. It describes the synthesis of TPA-functionalised nMCM-48 and its characterization by using various techniques such as SEM, TGA, FT-IR and TEM. In vitro release studies of unfunctionalized (nMCM-48), functionalized (TPA/nMCM-48) and capped (TPA/GLP/nMCM-48) nanoparticles were carried out in simulated body fluid (SBF, pH-7.4) at 37 °C under stirring conditions to determine whether TPA acts better as a functionalizing agent or as a capping agent for controlled release of GLP. Under the same conditions, the release study of a marketed available formulation of GLP, i.e., Glynase, was carried out and furthermore it was compared with the release studies of the synthesized materials. Different models, such as the zero order, first order and Higuchi models, were also explored to study the kinetics and mechanism. It is observed that the occurrence of cancer in DMT2 patients is relatively higher than in the general population. Recent pharmacoepidemiological studies have revealed that prolonged use of anti-diabetic drugs may be correlated with a lower risk of cancer in DMT2 patients. Long-term use of some diabetes medications, such as gliclazide and glibenclamide alone or in combination with GLP, may result in a dose-dependent reduction in the risk of developing cancer.32,33 Therefore, to check the possibility of the designed DDS for the same, an MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay of our designed DDS was performed using HepG2 cells.
Experimental
Materials
Glipizide was purchased from Sigma Aldrich. Cetyltriethylammoniumbromide (CTAB) was received from Lobachemie, Mumbai. Tetraethylorthosilicate (TEOS), ethanol, 12-tungstophosphoric acid and liquor ammonia (25%) were procured from Merck. The marketed formulation of glipizide, i.e., Glynase (each tablet containing 5 mg of glipizide), was obtained from a pharmaceutical store. The cells for human hepatocellular liver cancer (HepG2) were acquired from the National Centre for Cell Science in Pune, India. All chemicals purchased were of A.R. grade and were utilised with no further purification.Synthesis of nMCM-48
The synthesis of nMCM-48 was carried out by using a previously reported procedure by our group.34 Briefly, 2.4 g CTAB was added to 50 mL of distilled water and allowed to dissolve completely at 35 °C followed by the addition of 50 mL of ethanol and 15.4 mL of 25% liquor ammonia. The solution was stirred for 15–20 minutes. Then, 3.4 g of TEOS was added dropwise and the solution was stirred for 2 h. The resultant white suspension was filtered, washed with distilled water, dried at room temperature and calcined for 6 h at 550 °C. The material thus obtained was designated as nMCM-48.Functionalization of nMCM-48 using 12-tungstophosphoric acid (TPA)
nMCM-48 was functionalized by TPA using an incipient wet impregnation method. 1 g of nMCM-48 was functionalized using 30% (0.3/30 g mL−1 of distilled water) of TPA and then dried at 100 °C for 10 hours. The obtained material was designated as TPA/nMCM-48.Loading of GLP into nMCM-48 and TPA/nMCM-48
To encapsulate GLP, a 1.0 mg mL−1 solution of GLP was obtained by dissolving 20 mg of GLP in a 20 mL solution of water:methanol (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) mixture. In 20 mL of the 1.0 mg mL−1 drug solution, 20.0 mg of the carrier was suspended. This solution was sonicated for 10 minutes to ensure uniform dispersion before being stirred for 24 h at 37 °C to facilitate the effective loading of the drug into the pores. After 24 h, the particles were isolated from the solution using ultracentrifugation and the obtained solid was washed with acetone twice thoroughly to ensure the removal of excess drugs. Following that, the drug-loaded particles were air-dried and stored. The material thus obtained was termed as GLP/nMCM-48. The same procedure was used to load GLP onto TPA/nMCM-48, and the resulting drug-loaded material was termed as GLP/TPA/nMCM-48.
:30) mixture. In 20 mL of the 1.0 mg mL−1 drug solution, 20.0 mg of the carrier was suspended. This solution was sonicated for 10 minutes to ensure uniform dispersion before being stirred for 24 h at 37 °C to facilitate the effective loading of the drug into the pores. After 24 h, the particles were isolated from the solution using ultracentrifugation and the obtained solid was washed with acetone twice thoroughly to ensure the removal of excess drugs. Following that, the drug-loaded particles were air-dried and stored. The material thus obtained was termed as GLP/nMCM-48. The same procedure was used to load GLP onto TPA/nMCM-48, and the resulting drug-loaded material was termed as GLP/TPA/nMCM-48.
The amount of drug entrapped was calculated using the absorbance of the loading media. A calibration plot for GLP at 274 nm was used to measure the concentration of free drug in the supernatant. Using the following equation, the drug's % entrapment efficiency was calculated:

The loading capacity of the drug was calculated using the formula:
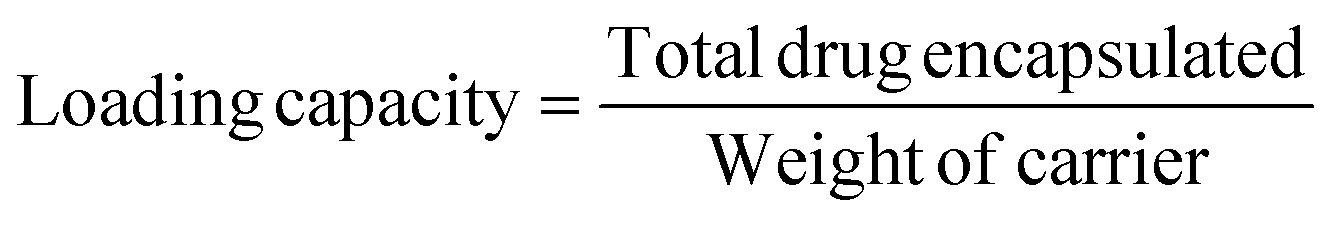
Capping of GLP/nMCM-48 by TPA
To understand the effect of TPA on release rate, the synthesized GLP/nMCM-48 was capped by TPA by an incipient wet impregnation method. Briefly, 30% aqueous solution of TPA (0.3/30 g mL−1 of distilled water) was used for the capping of 1 g of GLP/nMCM-48. The material thus obtained was designated as TPA/GLP/nMCM-48.Characterization
Results and discussion
Solid state characterizations
The TGA curves corresponding to GLP/nMCM-48 and GLP/TPA/nMCM-48 show overall weight loss of 26% and 57% in the temperature range of 30–600 °C, respectively. The greater amount of loss obtained in the case of GLP/TPA/nMCM-48 might be due to the greater amount of drug loaded and loss of TPA. In the case of both nMCM-48 and TPA/nMCM-48, the initial loss up to 100 °C may be attributed to the loss of water of hydration. The remaining decomposition in the temperature range of 100–600 °C could be due to the decomposition of the organic moiety. The curve for TPA/GLP/nMCM-48 is almost similar to that of GLP/nMCM-48 showing the successful capping using TPA. The TG–DTA curves (Fig. 1) give information about the successful loading of the drug inside the nanocarriers. A sharp endothermic peak of the pure drug GLP,7 (Fig. 1(c)), was obtained at 207.5 °C, which corresponds to its melting point. Meanwhile, in the case of the drug loaded nanocarriers, the endothermic peak corresponding to the drug is suppressed, which may be due to the strong bonding of the drug with the carrier after loading resulting in a flattened peak with reduced intensity. Thus, the thermal analysis using TG–DTA implied that the functionalization with TPA and encapsulation of GLP on nMCM-48 was successful.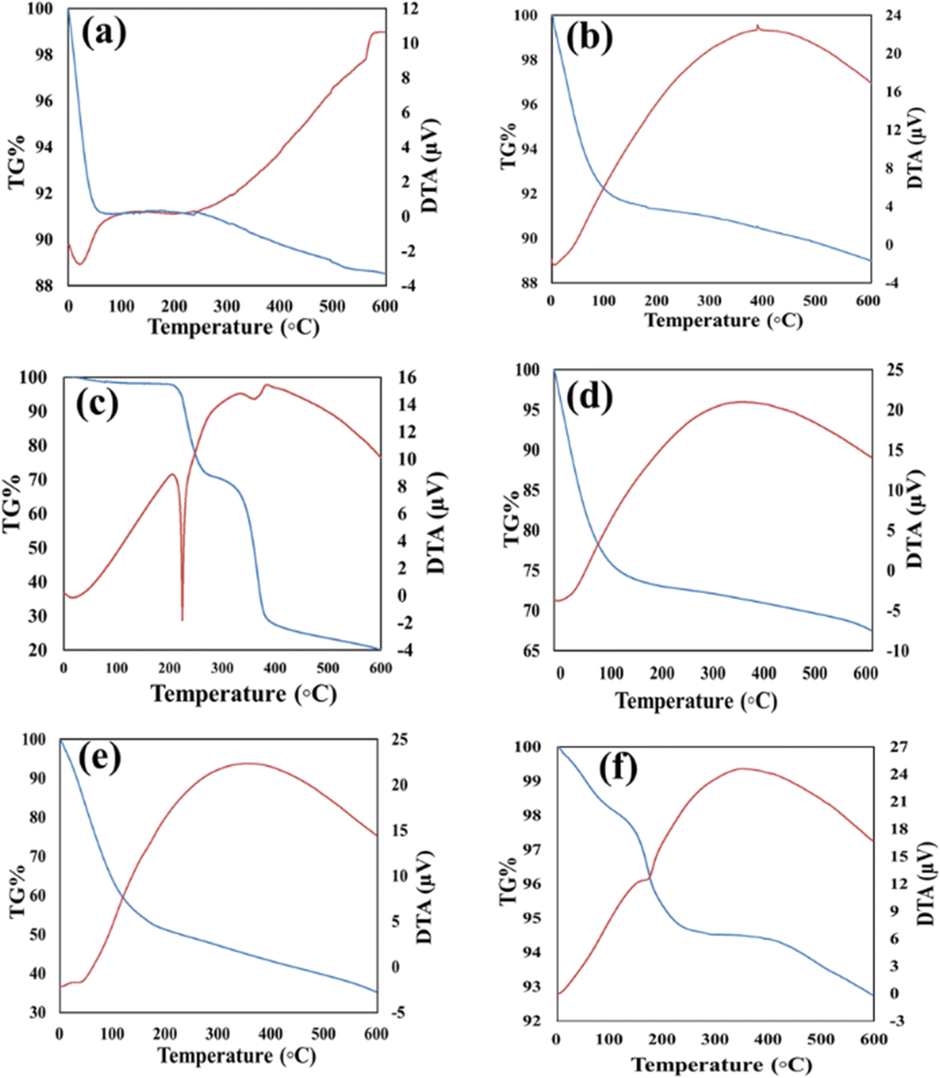 | ||
| Fig. 1 The TG–DTA curves of (a) nMCM-48, (b) TPA/nMCM-48, (c) GLP, (d) GLP/nMCM-48, (e) GLP/TPA/nMCM-48 and (f) TPA/GLP/nMCM-48. Thermal analysis shows the thermal stability of the drug delivery systems and successful loading of TPA and GLP into nMCM-48. | ||
The important assignments of the FT-IR bands with frequency for all the materials and the FT-IR spectra (nMCM-48, TPA, TPA/nMCM-48, GLP, GLP/nMCM-48, GLP/TPA/nMCM-48 and TPA/GLP/nMCM-48) are presented in Table 1 and Fig. 2, respectively. The fingerprint bands demonstrate that the primary structure of TPA remains intact even after impregnation. The FTIR spectra of GLP/nMCM-48 and GLP/TPA/nMCM-48 show bands that match with the frequencies of the corresponding bands present in GLP. Thus, it can be presumed that GLP was loaded into nMCM-48 and TPA/nMCM-48 successfully. The spectra obtained for TPA/GLP/nMCM-48 show all the respective corresponding peaks of TPA, GLP and nMCM-48, indicating successful capping using TPA.
| Compounds | Major peaks (cm−1) | Corresponding bands |
|---|---|---|
| nMCM-48 | 578 | Si–O–Si symmetric stretching |
| 1100, 1250 | Si–O–Si asymmetric stretching | |
| 462 | Si–O bending vibration | |
| TPA | 1088 | P–O symmetric stretching |
| 987 | W–O–W bending | |
| 800 | W–O | |
| TPA/nMCM-48 | 1249 | Si–O–Si asymmetric stretching |
| 956 | W–O–W bending | |
| 810 | W–O | |
| GLP | 3320, 3253 | N–H stretching |
| 1160 | S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
|
| 1690 | CO stretching |
|
| 1525 | Aromatic vibrations | |
| GLP/nMCM-48 | 470 | Si–O bending vibration |
| 1689 | CO stretching |
|
| 1529 | Aromatic vibrations | |
| GLP/TPA/nMCM-48 | 471 | Si–O bending vibration |
| 945 | W–O–W bending | |
| 1529 | Aromatic vibrations | |
| TPA/GLP/nMCM-48 | 595 | Si–O–Si symmetric stretching |
| 1634 | CO stretching |
|
| 1077 | P–O symmetric stretching |
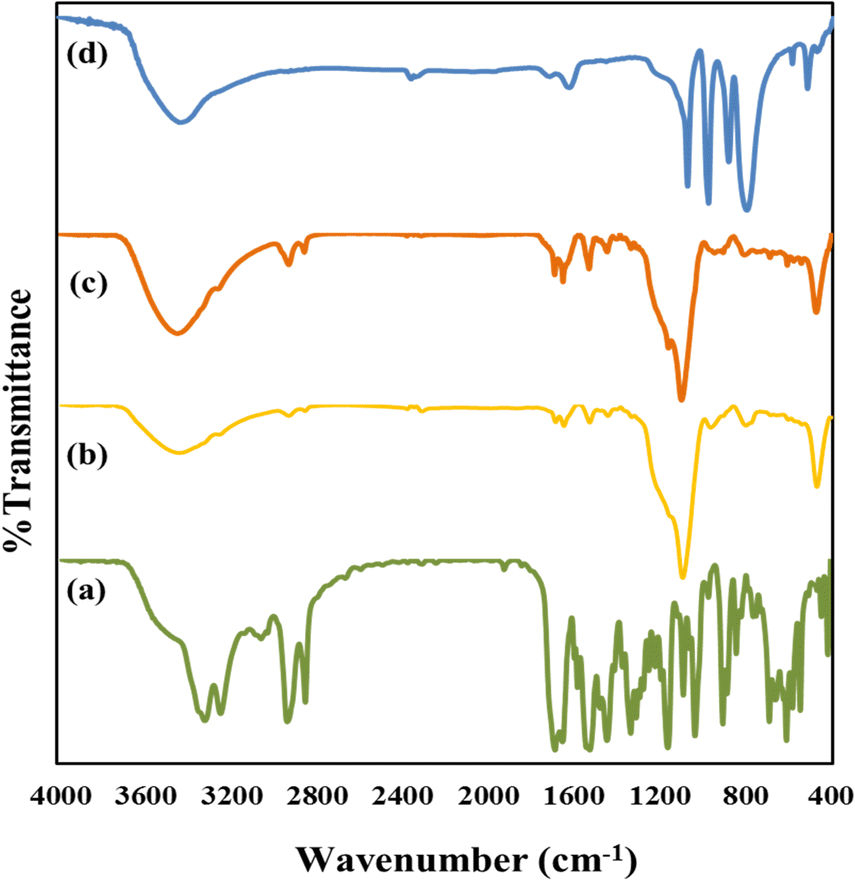 | ||
| Fig. 2 The FT-IR spectra of (a) GLP, (b) GLP/nMCM-48, (c) GLP/TPA/nMCM-48 and (d) TPA/GLP/nMCM-48. The FT-IR spectra show the interaction between the molecules and the successful loading of TPA and GLP into nMCM-48. | ||
The SEM micrographs show the external morphology of the nanocarriers (Fig. 3), which are almost spherical in shape and with uniform particle size overall. The SEM image of TPA/nMCM-48 shows a homogeneous distribution of spheres and no discernible change in the spherical morphology of nMCM-48 even after TPA impregnation. This indicates that TPA has indeed been finely dispersed into the pores. The morphology is retained even after encapsulating and capping with the drug.
 | ||
| Fig. 3 The SEM images of (a) nMCM-48, (b) TPA/nMCM-48, (c) GLP/TPA/nMCM-48, and (d) TPA/GLP/nMCM-48. The SEM micrographs show the spherical morphology of the particles. | ||
The TEM images show well-ordered porous networks with uniform spherical morphology, which supports the nanoporous structure of nMCM-48. The nanocarriers measured between 250 and 300 nm in size. TPA/nMCM-48 exhibited more dark spheres (Fig. 4) than nMCM-48 indicating that TPA was evenly loaded into the maximum number of pores possible. GLP dispersion is also satisfactory in the cases of GLP/nMCM-48 and GLP/TPA/nMCM-48. The TEM images of both the drug-loaded nanoparticles show an ordered porous structure with no agglomeration, indicating that GLP is well dispersed in nMCM-48 and TPA/nMCM-48. Even after functionalization with TPA and encapsulation with GLP, the particles retained their morphology.
 | ||
| Fig. 4 The TEM micrographs of (a) nMCM-48, (b) TPA/nMCM-48, (c) GLP/nMCM-48 and (d) GLP/TPA/nMCM-48. Average particle size of (e) nMCM-48, (f) TPA/nMCM-48, (g) GLP/nMCM-48 and (h) GLP/TPA/nMCM-48. The TEM micrographs show the uniform spherical morphology of the particles. | ||
It is interesting to note that, on the basis of point of zero charge (PZC), for nMCM-48, pH (7.4) is greater than the value of PZC (PZC of silica is in the range of 2–3) and therefore, the surface of nMCM-48 is negatively charged.36,37 As shown in Fig. 5, the negatively charged surface of nMCM-48 will bind with the available counter protons of TPA during functionalization and furthermore, the hydrogen present in GLP will bind with the available terminal oxygen of TPA. Similarly, during capping the negatively charged surface of nMCM-48 will bind with the hydrogen present in GLP and further it will bind with the available terminal oxygen of TPA.
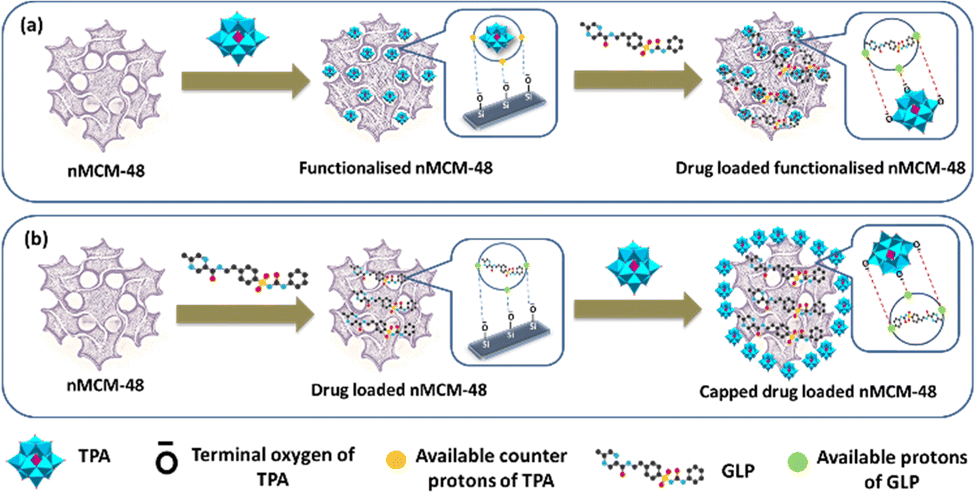 | ||
| Fig. 5 A schematic illustration of the designed DDS and the interaction between the molecules; (a) loading of GLP into TPA-functionalized nMCM-48; (b) capping of TPA into GLP-loaded nMCM-48. | ||
In vitro release study
The amounts of drug loaded per mg of the carriers, nMCM-48 and GLP/TPA/nMCM-48, were found to be 0.4 mg and 0.42 mg, respectively. The % entrapment efficiency of GLP in nMCM-48 was found to be 80%, while for TPA/nMCM-48 it was found to be 84%. The higher percent entrapment efficiency obtained in the case of TPA/nMCM-48 could be explained by the fact that TPA has a free terminal oxygen bond, which enables it to bind to a significantly larger amount of the drug. Since GLP has a pKa value of 5.9 and it is insoluble in acidic media, as stated in the introduction section, the release study was carried out in SBF (at pH-7.4) at 37 °C under stirring conditions.To determine the role of TPA and whether TPA acts as a functionalizing or a capping agent, the following studies were carried out: (i) the release profiles of GLP from GLP/nMCM-48 and GLP/TPA/nMCM-48 were studied, as shown in Fig. 6, to see the role of TPA as a functionalizing agent on release rate of the drug. It shows that at the end of 10 min, nearly 50% of the drug had been released from GLP/nMCM-48. However, only 14% of GLP was released from the GLP/TPA/nMCM-48 system. At 20 min, almost 99% of the drug was released from the GLP/nMCM-48 system, while only 27% of the drug was released from GLP/TPA/nMCM-48. After 110 min, 97% of the drug was released from GLP/TPA/nMCM-48. Thus, more controlled release of GLP was obtained from GLP/TPA/nMCM-48 under the same experimental conditions. (ii) The drug release profiles of GLP from GLP/TPA/nMCM-48 and TPA/GLP/nMCM-48 were compared (Fig. 6). The results show that at the end of 10 min, nearly 50% of the drug had been released from TPA/GLP/nMCM-48, whereas only 14% of GLP was released from the GLP/TPA/nMCM-48 system. After 20 min, almost 99% of the drug was released from the TPA/GLP/nMCM-48 system, while only 27% of the drug was released from GLP/TPA/nMCM-48, and at the end of 110 min, 97% of the drug was released from GLP/TPA/nMCM-48.
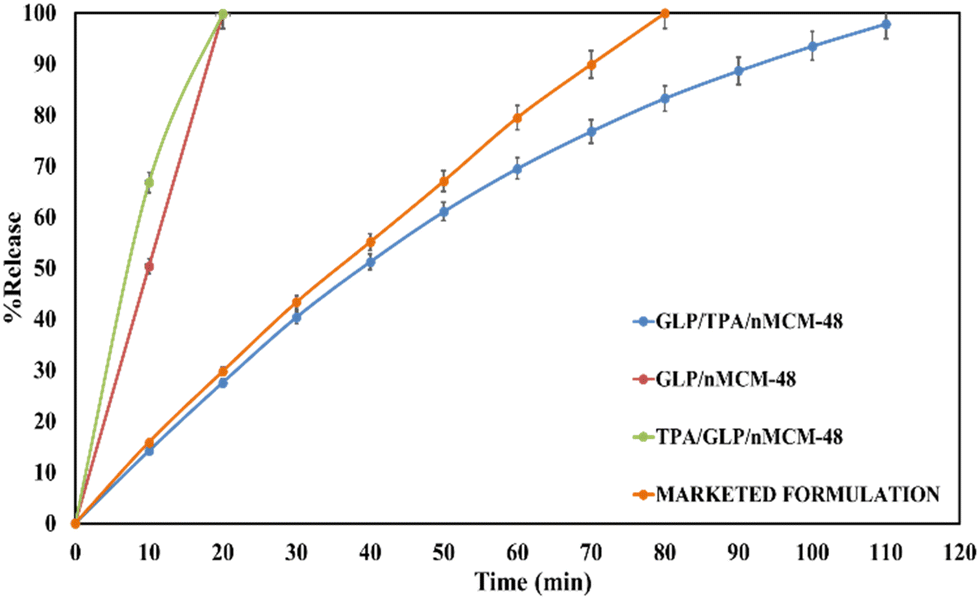 | ||
| Fig. 6 The in vitro release profiles of GLP/nMCM-48, GLP/TPA/nMCM-48, TPA/GLP/nMCM-48 and a marketed formulation at pH-7.4. A controlled release profile of GLP is obtained for GLP/TPA/nMCM-48 amongst all the designed DDSs and when compared with that of the marketed formulation a better release profile is obtained. | ||
From the type of release pattern obtained, it is significant to mention that TPA plays a major role in increasing drug loading capacity as well as drug release rate, as release is slower and better in the GLP/TPA/nMCM-48 system than in the GLP/nMCM-48 or TPA/GLP/nMCM-48 systems. This could be due to blocking of the channels of nMCM-48 by TPA during functionalization and because of possibilities of more favourable interaction between GLP and TPA due to the presence of a free oxygen terminal in TPA. As a result, functionalization with TPA reduces the release rate and results in a more controlled release of GLP. Thus, it can be assumed that TPA acts better as a functionalizing agent rather than as a capping agent. Furthermore, the role of TPA as a functionalizing agent was confirmed by the FT-IR of GLP/TPA/nMCM-48 recorded after the release study (Fig. 7). The spectrum is similar to that of TPA/nMCM-48,35 indicating that TPA acts as a functionalizing agent and that its structure is retained during drug release. Hence, the system GLP/TPA/nMCM-48 is being considered for further detailed studies including kinetics.
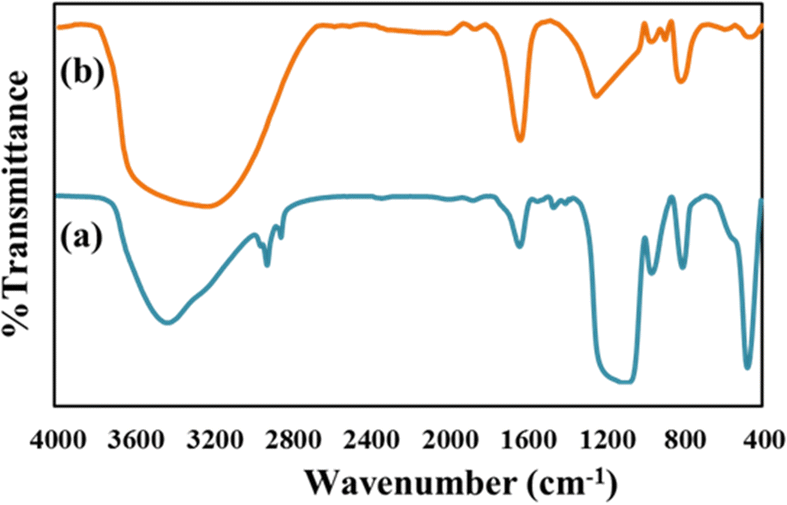 | ||
| Fig. 7 The FT-IR spectra of (a) GLP/TPA/nMCM-48 after the release study and (b) TPA/nMCM-48. These show the retention of the structure even after drug release. | ||
Comparison with a marketed drug and superiority of the designed DDS
Table 2 shows the comparison between the release studies of GLP/TPA/nMCM-48 and a marketed available formulation of GLP, i.e., Glynase. Initially, 29% of GLP is released from Glynase and this increased to 79% after 60 min. GLP/TPA/nMCM-48, on the other hand, releases 27% of the drug initially and can reach 69% after 60 min. Almost 99% of the drug was released by the end of 80 min from Glynase, while only 83% of the drug was released from GLP/TPA/nMCM-48 after 80 min. Thus, it is evident that when compared to Glynase, GLP/TPA/nMCM-48 has a more delayed and controlled release profile.| Sr. No. | Materials | % Release (pH-7.4, 37 °C, and stirring conditions) | ||
|---|---|---|---|---|
| Initial | Up to 80 min | Up to 110 min | ||
| 1. | Marketed formulation | 15.8 | 99.9 | |
| 2. | GLP/TPA/nMCM-48 | 14.2 | 83.0 | 97.8 |
Thus, our designed DDS outperforms the commercially available formulation in terms of more controlled release and increased drug bioavailability. In the coming decades, effective conversion of MSN-based DDSs from laboratory research to clinical implementation will necessitate extensive and systematic research in the field of in vivo nano-biomedical applications. Therefore, it is vital to consider the consequence of silica in the body during and after drug delivery in order to boost the biomedical effectiveness of MSN-based DDSs. It is considered necessary that the carrier material must be chemically resistant enough to secure the loaded drugs during delivery until it is delivered and yet degrade in an appropriate time frame once the drug release is over, to avoid unwanted accumulation and toxicities in the body. But even though the bond of –O–Si– is reasonably strong in silica nanoparticles, with a bond energy of 452 kJ mol−1, it is supposed to break down in aqueous media. In the tetrahedral SiO4 network, the Si centre is particularly prone to nucleophilic attack by the hydroxides in water, resulting in hydrolytic breakdown of the –Si–O–Si– bond. As a matter of fact, silica degrades into biocompatible orthosilicic acid (Si(OH)4), which is excreted in urine. The FDA has accepted silica as “generally recognised as safe” (GRAS). Previous studies on the hydrolytic degradation of silica nanoparticles in vitro and the biocompatibility of MSNs demonstrated by various groups have been published.38,39 Also, literature studies show that oral sodium tungstate administration has also recently emerged as an effective method of treating diabetes. This treatment also has no significant negative side effects and the compound has a low toxicity profile. The impact of tungstate is due in part to the normalisation of sucrase and SGLT1 activity in the brushborder membrane of enterocytes. Furthermore, in animal models of diabetes, some many long-term treatments with tungstate normalise glycaemia by restoring hepatic glucose metabolism.3,29 Since the DDS is a biocompatible material (nMCM-48) and the functionalizing agent (TPA) is proven to have anti-diabetic properties, the system has a better chance of being quickly translated to clinical studies.
Release kinetics and mechanism
To investigate the drug release kinetics and mechanisms,40–43 the data obtained from the release study of GLP from GLP/TPA/nMCM-48 were fitted in different models as detailed in Table 3.| Sr. No. | Model | Equation |
|---|---|---|
| Here, Qt is the amount of drug dissolved in time t and Q0 is the initial amount of drug in the solution (most times, Q0 = 0) and K0, K1 and KH are the release rate constants, respectively. | ||
| (i) | Zero order | Q t = Q0 + K0t |
| (ii) | First order | logQt = logQ0 − K1t/2.303 |
| (iii) | Higuchi model | Q t = KH × t1/2 |
Zero order release kinetic model
For zero-order kinetics, the cumulative drug release is plotted against time to analyse the drug release kinetics. The slope of this plot represents the zero-order rate constant, while the correlation coefficient indicates whether or not the drug release follows zero order kinetics. Fig. 8 depicts a zero order release kinetic model for the GLP/TPA/nMCM-48 system with an R2 = 0.9649 co-relation coefficient.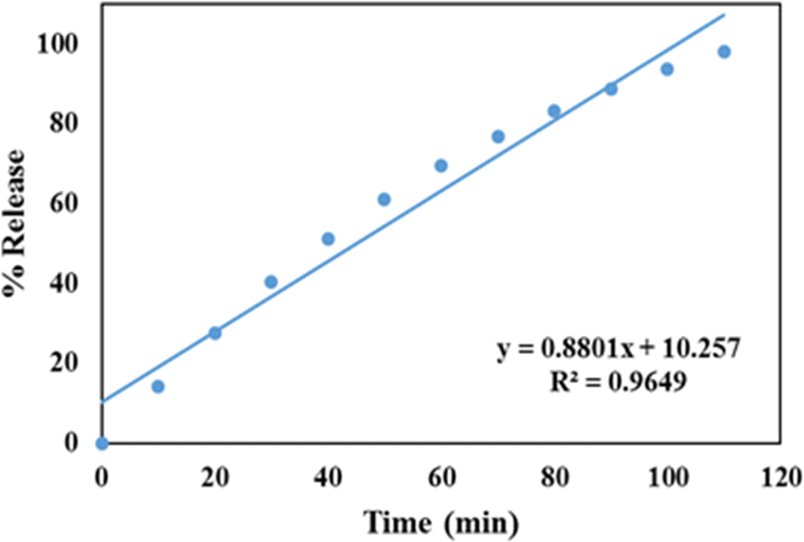 | ||
| Fig. 8 Zero order release kinetic model of GLP/TPA/nMCM-48 showing the plot of cumulative drug release against time. | ||
First order release kinetic model
A first order release kinetic model is used to assess the dissolution of the drug contained in porous matrices. A first-order release kinetic model for GLP/TPA/nMCM-48 is depicted in Fig. 9, with log percent remaining data plotted against time. The value of correlation coefficient of GLP/TPA/nMCM-48 was found to be R2 = 0.9119.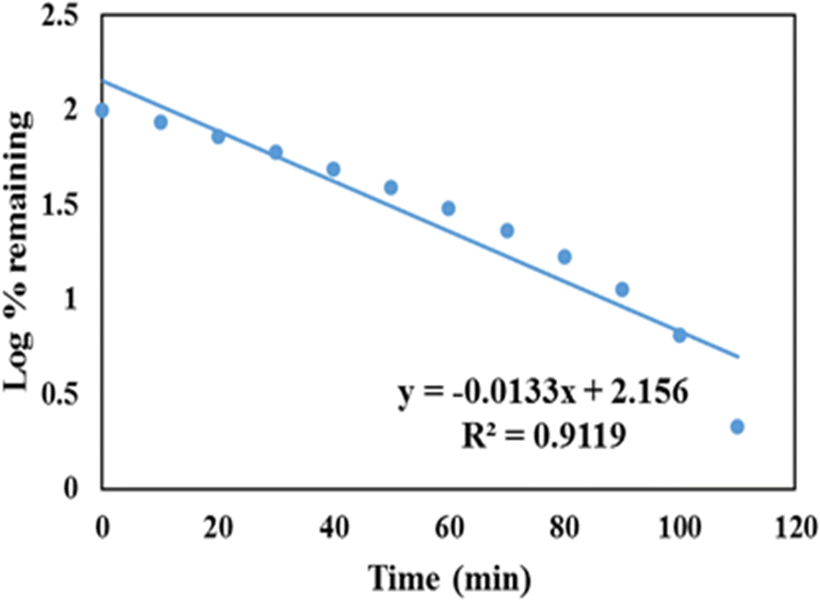 | ||
| Fig. 9 First order release kinetic model of GLP/TPA/nMCM-48 showing the plot of log percent remaining data plotted against time. | ||
Higuchi model
The Higuchi model is involved with the release of drugs from a matrix. According to this model, drug release involves sequential SBF penetration into pores, drug molecule dissolution, and drug molecule diffusion out of the pores. The correlation coefficient for the Higuchi Model (Fig. 10) for the GLP/TPA/nMCM-48 system was found to be R2 = 0.9977. Thus, GLP release from GLP/TPA/nMCM-48 follows Fickian diffusion.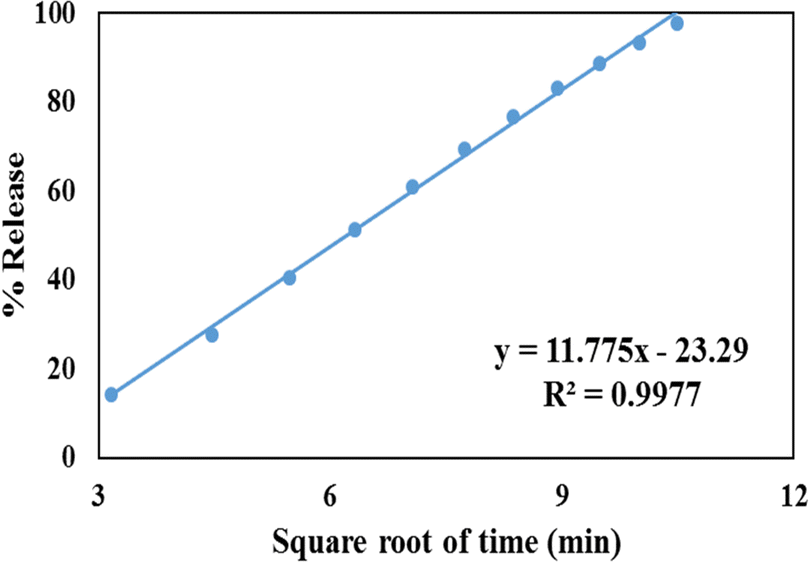 | ||
| Fig. 10 Higuchi model of GLP/TPA/nMCM-48 showing the plot of cumulative drug release against the square of time. | ||
Thus, in terms of kinetics and mechanism, GLP release from the GLP/TPA/nMCM-48 system follows zero order kinetics (Table 4) and the Higuchi model of diffusion.
| Sr. No. | Formulation | Zero order | First order | Higuchi | ||
|---|---|---|---|---|---|---|
| K 0 | R 2 | K 1 | R 2 | R 2 | ||
| 1. | GLP/TPA/nMCM-48 | 0.8801 | 0.9649 | 0.0133 | 0.9119 | 0.9977 |
Evaluation of in vitro cytotoxicity – MTT assay
To check how GLP suppresses cancer, we performed an MTT assay. In this study, GLP/nMCM-48 and GLP/TPA/nMCM-48 were used to treat HepG2 cells. The biocompatibility of the nanocarriers was assessed by measuring the viability of HepG2 cells incubated with different concentrations. At the end of the experiment, metabolically active cells produce a purple formazan and the intensity of the colour indicates the functional state of mitochondria. The designed DDS shows 15% cytotoxicity towards liver cancer cell lines (Fig. 11).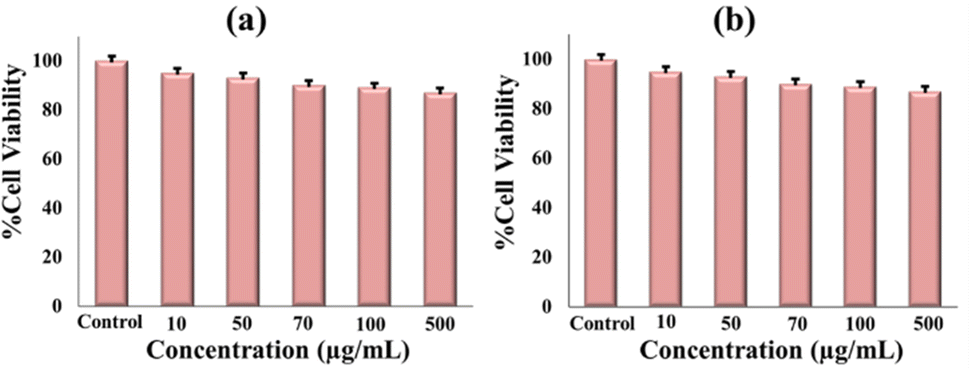 | ||
| Fig. 11 Cytotoxicity assessment of (a) GLP/nMCM-48 and (b) GLP/TPA/nMCM-48 to observe the cell viability at different concentrations. | ||
Novelty and comparison of nMCM-48 as a DDS
The novelty of the present work is that for the first time nMCM-48 was functionalized with an inorganic moiety, TPA, for the controlled release of an antidiabetic drug, GLP. Also, it is observed that a better and controlled release of GLP was obtained compared to the marketed available formulation, Glynase. The release of GLP/TPA/nMCM-48 is nearly 30 min slower than that of the marketed formulation, which makes it more appealing because it is in good agreement with the concept of less frequent dosing in the case of diabetic patients. Furthermore, the designed DDS may be used in the case of DMT2 patients suffering from cancer to suppress the angiogenic mechanism.Conclusions
The current study explored the possibility of controlled GLP delivery from nMCM-48 functionalized with an inorganic moiety, 12-tungstophophoric acid. TPA acts better as a functionalizing agent rather than a capping agent and therefore, GLP/TPA/nMCM-48 had a more controlled release profile. This is due to TPA, which holds the drug molecules for a longer period of time and aids in achieving a controlled and delayed release profile. Furthermore, a direct comparison of the GLP/TPA/nMCM-48 release profile with that of the marketed formulation demonstrates that the current system is better and exhibits more ordered and controlled drug release. GLP release follows the zero order kinetics followed by the Higuchi model with Fickian diffusion. Also taking into consideration the anti-cancer property of GLP, this study shows that GLP is capable of suppressing cancer to some extent with 15% cytotoxicity towards HepG2 cells.Author contributions
A.P conceptualized, supervised, reviewed and edited the final original draft. D.D carried out the data curation, formal analysis, validation and writing of the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to Council of Scientific & Industrial Research, New Delhi, project no. 01 (3002)/19/EMR-II, for financial support. We are thankful to Dr Varsha P. Brahmkhatri, Jain University for SEM Analysis. We are thankful to Dr Sonal Thakore, The Maharaja Sayajirao University of Baroda for the MTT Assay. We are also thankful to the Department of Chemistry, The Maharaja Sayajirao University of Baroda for the TG-DT Analysis.References
- Y. V. Simos, K. Spyrou, M. Patila, N. Karouta, H. Stamatis, D. Gournis, E. Dounousi and D. Peschos, Asian J. Pharm. Sci., 2021, 16, 62–76 CrossRef PubMed.
- Ş. Bâlici, M. Wankeu-Nya, D. Rusu, G. Z. Nicula, M. Rusu, A. Florea and H. Matei, Microsc. Microanal., 2015, 21, 1236–1248 CrossRef.
- M. M. Gómez-Gómez, N. Rodríguez-Fariñas, B. Cañas-Montalvo, J. Domínguez, J. Guinovart and C. Cámara-Rica, Talanta, 2011, 84, 1011–1018 CrossRef PubMed.
- Z. Ilyas, H. S. Shah, R. Al-Oweini, U. Kortz and J. Iqbal, Metallomics, 2014, 6, 1521–1526 CrossRef CAS PubMed.
- K. Bera, J. Khanam, K. P. Mohanraj and B. Mazumder, J. Microencapsulation, 2014, 31, 220–229 CrossRef CAS PubMed.
- H. S. Mali, S. R. Shaikh, S. D. Joshi, V. D. Dhaygude and A. R. Yadav, Int. J. Sci. Res. Sci. Technol., 2021, 741–750 Search PubMed.
- R. N. Dash, H. Mohammed, T. Humaira and D. Ramesh, Saudi Pharm. J., 2015, 23, 528–540 CrossRef.
- V. S. Meka, S. Pillai, S. R. Dharmalingham, R. Sheshala and A. Gorajana, Acta Pol. Pharm. – Drug Res., 2015, 72, 193–204 Search PubMed.
- M. Sharma and P. K. Choudhury, J. Drug Delivery Ther., 2019, 9, 311–315 CrossRef CAS.
- N. Pandya, M. Pandya and V. H. Bhaskar, J. Young Pharm., 2011, 3, 97–104 CrossRef CAS PubMed.
- H. Fael, C. Ràfols and A. L. Demirel, J. Pharm. Sci., 2018, 107, 2428–2438 CrossRef CAS PubMed.
- S. U. Mehsud, G. M. Khan, A. Hussain, M. Akram, M. Akhlaq, K. A. Khan and A. Shakoor, Pak. J. Pharm. Sci., 2016, 29, 779–787 Search PubMed.
- S. Nie, S. Zhang, W. Pan and Y. Liu, Drug Dev. Ind. Pharm., 2011, 37, 606–612 CrossRef CAS PubMed.
- P. C. Naha, H. J. Byrne and A. K. Panda, J. Nanopharmaceutics Drug Deliv., 2013, 1, 74–81 CrossRef.
- H. Pan, H. Jing, X. Yang, W. Pan and T. Chen, Drug Dev. Ind. Pharm., 2017, 43, 780–788 CrossRef CAS PubMed.
- R. K. Verma and S. Garg, Eur. J. Pharm. Biopharm., 2004, 57, 513–525 CrossRef CAS.
- S. Joshi, M. T. Hussain, C. B. Roces, G. Anderluzzi, E. Kastner, S. Salmaso, D. J. Kirby and Y. Perrie, Int. J. Pharm., 2016, 514, 160–168 CrossRef CAS.
- S. Maiti, P. Dey, A. Banik, B. Sa, S. Ray and S. Kaity, Drug Deliv., 2010, 17, 288–300 CrossRef CAS PubMed.
- P. Sun, P. Li, Y. M. Li, Q. Wei and L. H. Tian, J. Biomed. Mater. Res., Part B, 2011, 97B, 175–183 CrossRef CAS PubMed.
- S. Maiti, S. Ranjit, R. Mondol, S. Ray and B. Sa, Carbohydr. Polym., 2011, 85, 164–172 CrossRef CAS.
- A. G. Agrawal, A. Kumar and P. S. Gide, Colloids Surf., B, 2015, 126, 553–560 CrossRef CAS.
- A. Bakhshian Nik, H. Zare, S. Razavi, H. Mohammadi, P. Torab Ahmadi, N. Yazdani, M. Bayandori, N. Rabiee and J. Izadi Mobarakeh, Microporous Mesoporous Mater., 2020, 299, 110115 CrossRef CAS.
- R. Narayan, U. Y. Nayak, A. M. Raichur and S. Garg, Pharmaceutics, 2018, 10, 1–49 CrossRef PubMed.
- Y. Hu, S. Bai, X. Wu, S. Tan and Y. He, Ceram. Int., 2021, 47, 31031–31041 CrossRef CAS.
- M. B. Čolović, M. Lacković, J. Lalatović, A. S. Mougharbel, U. Kortz and D. Z. Krstić, Curr. Med. Chem., 2020, 27, 362–379 CrossRef PubMed.
- T. Yamase, Polyoxometalates active against tumors, viruses, and bacteria, 2013, vol. 54, pp. 65–116 Search PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- D. Li, X. Gao, J. Gu, Y. Tian, Y. Liu, Z. Jin, D. Yan, Y. G. Chen and X. Zhu, Bioinorg. Chem. Appl., 2016, 2016, 3239494 Search PubMed.
- A. Barberà, J. Fernàndez-Alvarez, A. Truc, R. Gomis and J. J. Guinovart, Diabetologia, 1997, 40, 143–149 CrossRef PubMed.
- R. A. Silvestre, E. M. Egido, R. Hernández and J. Marco, Eur. J. Pharmacol., 2005, 519, 127–134 CrossRef CAS PubMed.
- Ş. Bâlici, S. Şuşman, D. Rusu, G. Z. Nicula, O. Soriţău, M. Rusu, A. S. Biris and H. Matei, J. Appl. Toxicol., 2016, 36, 373–384 CrossRef PubMed.
- C. Qi, Q. Zhou, B. Li, Y. Yang, L. Cao, Y. Ye, J. Li, Y. Ding, H. Wang, J. Wang, X. He, Q. Zhang, T. Lan, K. K. H. Lee, W. Li, X. Song, J. Zhou, X. Yang and L. Wang, Oncotarget, 2014, 5, 9966–9979 CrossRef PubMed.
- C. Qi, B. Li, Y. Yang, Y. Yang, J. Li, Q. Zhou, Y. Wen, C. Zeng, L. Zheng, Q. Zhang, J. Li, X. He, J. Zhou, C. Shao and L. Wang, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- D. Pithadia and A. Patel, Appl. Catal., A, 2020, 602, 117729 CrossRef.
- D. Dasgupta, M. Das, S. Thakore, A. Patel and S. Kumar, J. Drug Delivery Sci. Technol., 2022, 72, 103419 CrossRef CAS.
- J. P. Cloarec, C. Chevalier, J. Genest, J. Beauvais, H. Chamas, Y. Chevolot, T. Baron and A. Souifi, Nanotechnol., 2016, 27, 295602 CrossRef PubMed.
- H. P. Boehm, Discuss. Faraday Soc., 1971, 52, 264–275 RSC.
- E. Choi and S. Kim, Langmuir, 2017, 33, 4974–4980 CrossRef CAS PubMed.
- K. Radhakrishnan, S. Gupta, D. P. Gnanadhas, P. C. Ramamurthy, D. Chakravortty and A. M. Raichur, Part. Part. Syst. Charact., 2014, 31, 449–458 CrossRef CAS.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharma., 2010, 67, 217–223 CAS.
- G. P. Panotopoulos nad Z. S. Haidar, Scientifica, 2019, 9153876 Search PubMed.
- H. Baishya, J. Dev. Drugs, 2017, 6, 1–8 Search PubMed.
- E. Ortiz-Islas, A. Sosa-Arróniz, M. E. Manríquez-Ramírez, C. E. Rodríguez-Pérez, F. Tzompantzi and J. M. Padilla, Rev. Adv. Mater. Sci., 2021, 60, 25–37 CAS.
| This journal is © The Royal Society of Chemistry 2022 |