
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pore-confined cobalt sulphide nanoparticles in a metal–organic framework as a catalyst for the colorimetric detection of hydrogen peroxide†
You-Liang
Chen
,
Yi-Ching
Wang
,
Yu-Hsiu
Chen
,
Tzu-En
Chang
,
Cheng-Hui
Shen
,
Chi-Wei
Huang
and
Chung-Wei
Kung
 *
*
Department of Chemical Engineering, National Cheng Kung University, Tainan City, 70101, Taiwan. E-mail: cwkung@mail.ncku.edu.tw
First published on 5th July 2022
Abstract
In this study, nanoparticles of cobalt sulphide solely confined within the nanopores of a water-stable zirconium-based metal–organic framework (MOF), MOF-808, are synthesized by a two-step approach, with the first step being installing spatially dispersed cobalt ions within the entire framework of MOF-808 and the second step being solution-phase sulphurisation. The crystallinity, morphology, porosity, and loading of cobalt sulphide in the obtained nanocomposite are investigated. As a demonstration, the obtained nanocomposite is used as a heterogeneous catalyst for the redox reaction between 3,3′,5,5′-tetramethylbenzidine (TMB) and H2O2 in aqueous acetate buffer solutions. The pore-confined cobalt sulphide nanoparticles show remarkable catalytic activity, and the catalysis has typical Michaelis–Menten kinetics. Relying on the optical change that originated from the generation of oxidized TMB, as a demonstration, the nanocomposite can be utilized in the colorimetric quantification of H2O2.
1. Introduction
Metal–organic frameworks (MOFs) are a relatively new category of nanoporous materials with the characteristics of ultrahigh specific surface area, periodic and interconnected porosity, structural diversity, and tuneable chemical functionality.1–3 These intriguing advantages enable the use of MOF-based materials in numerous potential applications including gas adsorption,4–6 separation,7,8 energy storage,9,10 solar fuel catalysis,11–13 electronic devices,14,15 and sensors.16–18 Notably, MOFs are considered as attractive platforms for heterogeneous catalysis owing to their highly porous nature and intra-framework functionality.19 By the judicious design of the MOF structure and post-synthetic strategies, immobilized catalytically active sites spatially dispersed through the whole framework can be achieved, which should efficiently increase the number of accessible active species to participate in the catalysis.20–24 However, the poor chemical stability of most MOFs in water strongly restricts their applications in aqueous catalysis.1,25,26 Fortunately, the discovery of MOFs constructed from group 4 metal-based nodes, e.g., zirconium-based MOFs (Zr-MOFs),27 which possess remarkable chemical stability in water, has expanded the use of MOFs in various applications in aqueous media and opened up the opportunity for a variety of material designs.18,26,28,29 Therefore, incorporating catalytically active species into water-stable and highly porous Zr-MOFs has been considered as an appealing strategy to design the heterogeneous catalysts utilized in aqueous environments.30–33Transition metal sulphides have garnered great attention as heterogeneous catalysts among various materials.34 However, the in situ synthesis of metal sulphides within the nanopores of MOFs is relatively challenging because most MOFs cannot preserve their structural integrity in environments containing S2− ions, HS− ions, or H2S vapor. Since Zr-MOFs belong to one unique category of MOFs that are chemically stable in H2S vapor,26 we reasoned that Zr-MOFs should be viable scaffolds for the synthesis of pore-confined metal sulphides. In an early study published in 2015, Hupp, Farha and co-workers reported the use of atomic layer deposition (ALD) to deposit spatially separated clusters of cobalt sulphide into a Zr-MOF, with the use of H2S vapor as the precursor;35 similar approaches were also used to synthesize few-atom clusters of metal sulphides installed in Zr-MOFs.36–38 Compared to the few-atom clusters immobilized on the nodes of Zr-MOFs, we reasoned that the nanoparticles of metal sulphide that are physically confined within the MOF pore should possess more similar properties to the bulk metal sulphide, and the particle size could be highly tuneable by simply adjusting the pore size of the selected MOF. The rigid framework of Zr-MOFs is expected to effectively prevent further agglomeration of nanoparticles, which should result in a high external surface area of the metal sulphide that is beneficial for heterogeneous catalysis. However, published examples of metal sulphide nanoparticles confined in the MOF pores are fairly rare. In most reported examples regarding the in situ synthesis of metal sulphides in Zr-MOFs, a one-step method was usually used by mixing the metal precursor, sulphur source, and Zr-MOF crystals together in the solution under solvothermal conditions, which would generate the metal sulphide mainly distributed between the Zr-MOF crystals rather than within the MOF pore.39,40 The only published example to date attempting to synthesize the pore-confined nanoparticles of metal sulphide in Zr-MOFs was demonstrated by Hou, Wu and co-workers in a very recent study; the Zr-MOF crystals were incubated in a solution containing cadmium(II) ions before the addition of the sulphur source during one-pot synthesis, which could result in the coexistence of cadmium sulphide confined within the pore of Zr-MOFs and that located on the external surface of Zr-MOF crystals.41
In this study, we aimed to demonstrate the controlled synthesis of cobalt sulphide nanoparticles that are solely confined within the nanopore of a Zr-MOF, by utilizing a two-step synthetic strategy (see Fig. 1). A self-limiting solution phase process, the solvothermal deposition in MOFs (SIM) technique,42,43 was first used to immobilize the spatially dispersed Co(II) ions on the hexa-zirconium nodes of a six-connected Zr-MOF, MOF-808.6 Thereafter, the cobalt-functionalized MOF-808 (Co-MOF-808) powder was immersed into a solution containing thioacetamide (TAA) as the sulphur source, where the chemical bonds between the cobalt ions and hexa-zirconium nodes were dissociated; the formation of cobalt sulphide nanoparticles that are selectively confined within the pore of MOF-808 could thus occur. Since cobalt sulphide has been reported as a catalyst to catalyse the oxidation of chromogenic substrates, e.g., 3,3′,5,5′-tetramethylbenzidine (TMB), in the presence of H2O2,44–47 as a demonstration here, the obtained pore-confined cobalt sulphide nanoparticles in MOF-808 served as the catalyst for the colorimetric detection of H2O2.
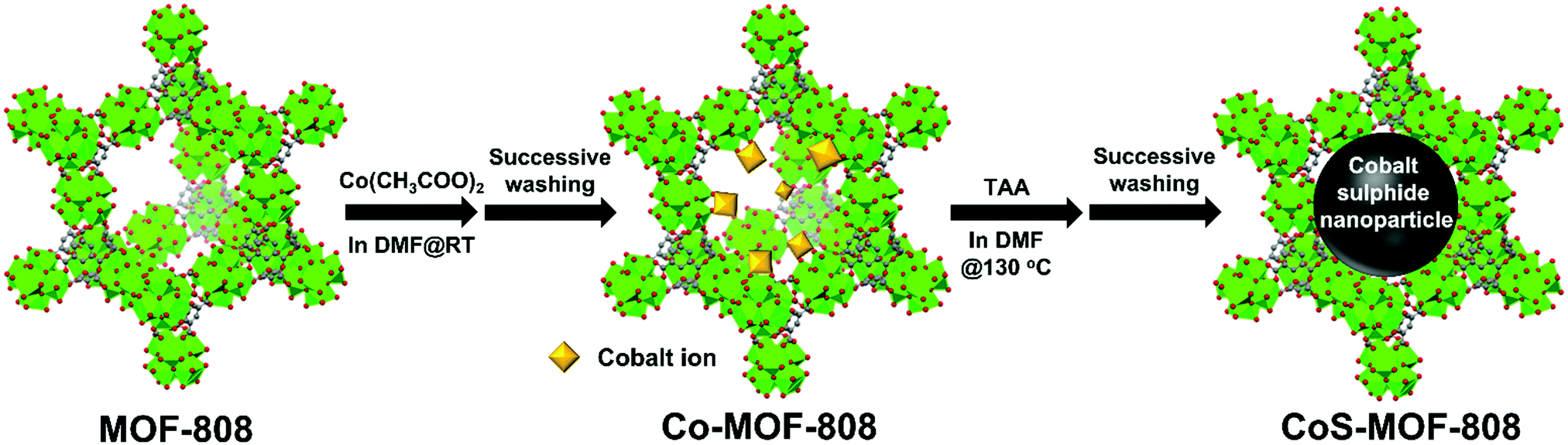 | ||
| Fig. 1 Schematic illustration of the two-step synthetic route for the preparation of CoS-MOF-808. Green, Zr; red, O; and grey, C. For clarity, hydrogen atoms in the structures are not shown. | ||
2. Experimental section
2.1. Chemicals
All chemicals including zirconium(IV) oxychloride octahydrate (ZrOCl2·8H2O, Sigma-Aldrich, 98%), trimesic acid (H3BTC, Sigma-Aldrich, 95%), formic acid (Honeywell Fluka, ≥98%), N,N-dimethylformamide (DMF, ECHO Chemical Co, Ltd, Taiwan, ≥99.8%), hydrochloric acid (HCl, J. T. Baker, 36.5–38.0%), acetone (ECHO Chemical Co, Ltd, Taiwan, 98%), cobalt(II) acetate tetrahydrate (Sigma-Aldrich, reagent grade), thioacetamide (TAA, Alfa Aesar, 99%), acetic acid (Honeywell Fluka, ≥99.8%), sodium acetate trihydrate (Honeywell Fluka, ≥99%), sodium hydroxide (NaOH, Sigma-Aldrich, 99%), dimethyl sulfoxide (DMSO, Duksan Pure Chemicals, 99%), 3,3′,5,5′-tetramethylbenzidine (TMB, Acros Organics, ≥99%), hydrogen peroxide solution (H2O2, Honeywell Fluka, 30–31%), sulfuric acid (H2SO4, Honeywell Fluka, 95.0–98.0%), and nitric acid (HNO3, Honeywell Fluka, ≥65%) were used as received without any further purification. Deionized (DI) water was used in the whole work as the water source.2.2. Synthesis of Co-MOF-808
MOF-808 was synthesized according to the protocol from our previously reported work.48 Thereafter, the self-limiting solvothermal deposition in MOFs (SIM) method was utilized for the installation of cobalt on the hexa-zirconium nodes of MOF-808.42 Initially, 99.0 mg of cobalt(II) acetate tetrahydrate was dissolved in 12 mL of DMF by sonication. Then, 30.0 mg of activated MOF-808 powder was added into the obtained solution, and the mixture was kept at room temperature for 24 h. The resulting solid was washed with 12 mL of fresh DMF three times by centrifugation to completely remove excess cobalt precursor in the MOF crystals. The solid was then washed with 12 mL of fresh acetone three times by centrifugation, with immersing periods of 2 h, overnight, and 2 h in between, respectively. Co(II) decorated MOF-808 was obtained after activating the solid product under vacuum at 80 °C overnight and was named “Co-MOF-808”.2.3. Sulphurisation of Co-MOF-808
For the sulphurisation of Co-MOF-808, 32.4 mg of thioacetamide (TAA) was dissolved in 6 mL of DMF in a microwave vial (2–5 mL, Biotage) by sonication. After adding 30.0 mg of Co-MOF-808 powder into the solution and dispersing the solid by sonication, the vial was crimped and heated at 130 °C for 24 h in an oven. The precipitation of black solid could be observed during the synthetic process. The obtained solid was washed with 6 mL of fresh DMF three times by centrifugation to remove excess reagent, and thereafter washed with 6 mL of fresh acetone three times with immersing periods of 2 h, overnight, and 2 h in between, respectively. A black powder was obtained after drying the solid in a vacuum oven at 80 °C overnight and was designated as “CoS-MOF-808”.2.4. Catalyst tests
To investigate the catalytic activity of each material for the oxidation of TMB in the presence of H2O2, the following experiment was conducted.49 4.0 mg of CoS-MOF-808, Co-MOF-808, or MOF-808 was weighed accurately and first dispersed in 7 mL of acetate buffer solution (0.1 M, pH = 4.03) by sonication. Thereafter, 800 μL of TMB solution (5 mM in DMSO) and 200 μL of H2O2 (40 mM in water) were added into the above-mentioned mixture successively under stirring. It should be noted that the concentrations of TMB and H2O2 in the tested samples here are 0.5 mM and 1.0 mM, respectively. After vigorously stirring at 300 rpm for 10 min, the mixture was immediately filtered by a hydrophilic 200 nm syringe filter to remove the solid, and the resulting solution was subjected to UV-visible (UV-vis) measurement to estimate the content of the oxidized TMB (oxTMB), according to the absorbance change at 652 nm in the UV-vis spectra. Controlled experiments in the absence of a catalyst or H2O2 were also conducted for comparison.2.5. Kinetic measurements
For a catalytic reaction that follows Michaelis–Menten kinetics, the apparent kinetic data could be collected by performing catalyst tests in the presence of various concentrations of H2O2 or TMB while keeping another one constant.49,50 4.0 mg of CoS-MOF-808 was weighed accurately and used here as a catalyst for every sample, and the experimental conditions are as follows. For H2O2 as the substrate (S), the catalyst tests were performed with samples containing 0.5 mM of TMB and various concentrations of H2O2. For TMB as the substrate, the concentration of H2O2 was kept as 35 mM with various concentrations of TMB in the tested samples. By terminating the catalytic reaction through filtration at various reaction times and monitoring the absorbance of each sample at 652 nm, the initial reaction rate (V0) for one certain concentration of the substrate can be obtained according to Lambert–Beer's law. For each substrate, the relationship between the initial reaction rate and substrate concentration ([S]) can be used to gauge the apparent kinetic constants by applying the following equation with Lineweaver–Burk plots,49–51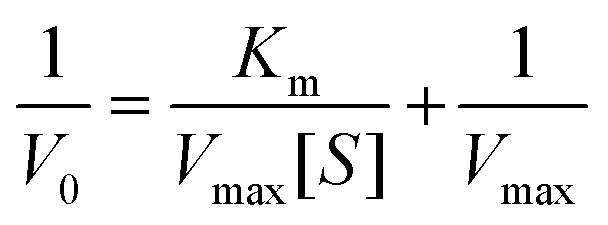 | (1) |
2.6. Turnover number (TON) measurements
The TON is defined as the number of oxTMB produced per active site in this system.52 In order to quantify the TON that can be achieved by the CoS-MOF-808 material here and verify that this material consists of a catalyst for this reaction, the following large-scale experiment was implemented. 2.0 mg of CoS-MOF-808 was weighed accurately and first dispersed in 175 mL of acetate buffer solution (0.1 M, pH = 4.03) by sonication. Thereafter, 20 mL of TMB solution (5 mM in DMSO) and 5 mL of H2O2 (9.8 M) were added into the above-mentioned mixture successively under rapid stirring at 600 rpm. After continuous stirring for 15 min, the catalyst was immediately removed from the mixture using a hydrophilic 200 nm syringe filter. The amount of oxTMB in the resulting solution was quantified by UV-vis spectroscopy.2.7. Detection of H2O2
The chemical sensing experiments for H2O2 were conducted according to the following procedure. 4.0 mg of CoS-MOF-808 was weighed accurately and dispersed in a solution containing 7 mL of acetate buffer solution (0.1 M, pH = 4.03) and 800 μL of TMB solution (5 mM in DMSO). Thereafter, 200 μL of the aqueous solution containing a certain concentration of H2O2 was added into the above-mentioned mixture under vigorous stirring at 300 rpm. After stirring for 10 min, the mixture was immediately filtered using a hydrophilic 200 nm syringe filter to remove the catalyst, and the resulting solution was subjected to UV-vis spectroscopic measurements in order to evaluate the content of oxTMB in the solution.2.8. Instrumentations
The morphologies and energy-dispersive X-ray spectroscopy (EDS) data of MOF-808, Co-MOF-808, and CoS-MOF-808 were investigated using a scanning electron microscope (SEM, Hitachi HR-FESEM SU8010). Pt was sputtered on the samples prior to SEM and EDS measurements. Powder X-ray diffraction (PXRD) patterns were recorded using a RIGAKU Ultima IV. The nitrogen adsorption–desorption isotherms of the samples were obtained using an ASAP 2020 (Micromeritics). All transmission electron microscopy (TEM) images and EDS elemental mapping data were collected using a JEM-2100F (JEOL) at an operating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was performed on a Theta Probe (Thermo Scientific) equipped with a microfocused electron gun, a multi-position aluminium anode, and a monochromated X-ray source. All XPS spectra were corrected by referencing the C 1s peak to 284.8 eV. Inductively coupled plasma-optical emission spectrometry (ICP-OES) and inductively coupled plasma-mass spectrometry (ICP-MS) measurements were performed using a JY 2000-2 ICP-OES spectrometer (Horiba Scientific) and a Thermo-element XR (Thermo Scientific™), respectively. The sample for ICP-OES or ICP-MS was prepared by mixing 0.75 mL of sulfuric acid, 0.25 mL of 30% H2O2 solution, and around 1 mg of MOF-based material in a microwave vial (2–5 mL, Biotage). The vial was crimped and heated at 100 °C in an oil bath for 24 h. The resulting solution was then diluted with 35 mL of 3 wt% HNO3 aqueous solution prior to ICP-OES or ICP-MS measurements. It should be noticed that for ICP-MS samples to quantify the loading of sulphur, the sulfuric acid used for sample preparation was replaced by concentrated nitric acid. Fourier-transform infrared (FTIR) spectroscopy was performed using a Nicolet 6700 (Thermo Fisher Scientific). UV-vis spectra were recorded using a UV-2600 (Shimadzu). The acetate buffer solution was prepared by titrating 0.1 M acetic acid aqueous solution with 0.1 M sodium acetate aqueous solution to reach a pH value of 4.03. A Mettler Toledo T5 autotitrator was used to monitor the pH value. All the washing and solvent-exchange steps throughout the study were conducted using a Heraeus™ Megafuge™ 16 centrifuge (Thermo Fisher Scientific) along with a TX-400 rotor at a rotating speed of 5000 rpm.3. Results and discussion
3.1. Materials characterization
The experimental PXRD patterns of MOF-808, Co-MOF-808, and CoS-MOF-808 with the simulated PXRD pattern of MOF-808 are shown in Fig. 2(a). The diffraction peaks observed in all the PXRD patterns of MOF-808, Co-MOF-808, and CoS-MOF-808 agree well with those in the simulated pattern of MOF-808, indicating that the phase-pure MOF-808 was successfully synthesized and the crystallinity of MOF-808 can remain intact during both the SIM and sulphurisation processes. The simulated PXRD pattern of one commonly seen crystalline phase of cobalt sulphides, Co9S8, is also shown in Fig. 2(a). It should be noticed that no observable diffraction peaks of any cobalt sulphide-based compound, e.g., Co9S8, can be observed in the high-angle region of the PXRD pattern of CoS-MOF-808, which suggests that the cobalt sulphide present in CoS-MOF-808 is amorphous.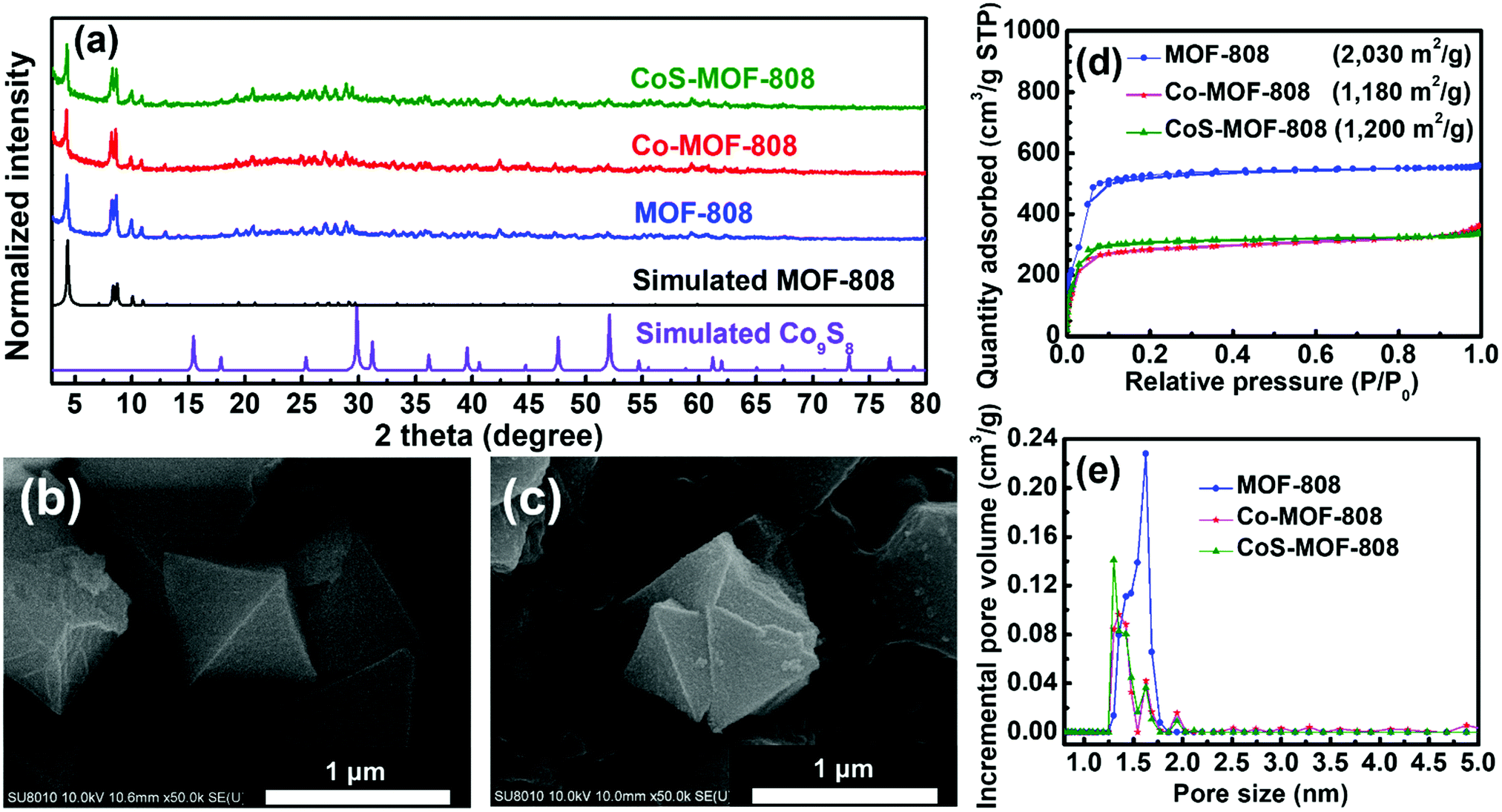 | ||
| Fig. 2 (a) Experimental PXRD patterns of MOF-808, Co-MOF-808 and CoS-MOF-808. The simulated patterns of MOF-808 and Co9S8 are also shown. High-magnification SEM images of (b) MOF-808 and (c) CoS-MOF-808. (d) Nitrogen adsorption–desorption isotherms and (e) DFT pore size distributions of MOF-808, Co-MOF-808, and CoS-MOF-808. BET surface areas are listed in (d). | ||
The surface morphologies of all the materials were examined by SEM, and the SEM images of MOF-808, Co-MOF-808, and CoS-MOF-808 are shown in Fig. S1 (ESI†). It can be observed that the pristine MOF-808 is composed of octahedral crystals with a size of around 1 μm; this morphology is consistent with that of MOF-808 reported previously.48 Both Co-MOF-808 and CoS-MOF-808 reveal the same morphology as that of pristine MOF-808, indicating that there is no obvious change in the surface morphology after both the SIM process and sulphurisation. The high-magnification SEM images of MOF-808 and CoS-MOF-808 are shown in Fig. 2(b) and (c), respectively, which also reveal that the surface morphology of the octahedral MOF crystals is unchanged after the incorporation of cobalt sulphide. The EDS spectra of Co-MOF-808 and CoS-MOF-808 are shown in Fig. S2(a) and (b) (ESI†), respectively, which show that the signal for cobalt can be observed in both spectra and sulphur is present in CoS-MOF-808. This result clearly indicates that the installation of cobalt in MOF-808 by SIM was successful, and both cobalt and sulphur are present in CoS-MOF-808 after sulphurisation. ICP-OES and ICP-MS measurements were further conducted to quantify the loadings of cobalt and sulphur in each material. The ICP-OES result suggests that for Co-MOF-808 and CoS-MOF-808, the average loadings of cobalt are 2.96 Co and 1.32 Co on each hexa-zirconium node, respectively. This finding also implies that there is a partial loss of cobalt during sulphurisation. Furthermore, from the ICP-MS result for CoS-MOF-808, the atomic ratio between cobalt and sulphur was determined as 0.59, which suggests that the amorphous cobalt sulphide present in CoS-MOF-808 has an overall chemical composition of CoS1.69.
The FTIR spectra of MOF-808, Co-MOF-808 and cobalt acetate tetrahydrate, i.e., the precursor used for installing cobalt in MOF-808 during SIM, are shown in Fig. S3(a) (ESI†). Two characteristic peaks originated from the CH3 rocking vibration of acetate ions located at 1059 and 1027 cm−1 can be found in the FTIR spectrum of Co-MOF-808,53 which indicates that some acetate ions were coordinated on either the hexa-zirconium nodes or the installed cobalt ions during the SIM process. In addition, as revealed in Fig. S3(b) (ESI†), one characteristic peak of Co–S stretching located at 610 cm−1 can be found in the FTIR spectrum of CoS-MOF-808,54 which further verifies the presence of cobalt sulphide in CoS-MOF-808.
To investigate the porosity of the materials, the nitrogen adsorption–desorption isotherms of MOF-808, Co-MOF-808, and CoS-MOF-808 were measured, and the results are shown in Fig. 2(d). The pristine MOF-808 shows a Brunauer−Emmett−Teller (BET) surface area of 2030 m2 g−1, which is in good agreement with the reported values for MOF-808.6,48,55 After the installation of cobalt by SIM, the BET surface area decreases to 1180 m2 g−1, but the microporous characteristic of MOF-808 is still present in the isotherm of Co-MOF-808, implying that the major porosity of the MOF is not clogged by the installed cobalt; this observation is quite similar to that of MOF-808 installed with other spatially dispersed metal ions reported in our previous studies.55,56 After sulphurisation, the obtained CoS-MOF-808 reveals almost the same BET surface area (1200 m2 g−1) and porosity as those of Co-MOF-808, which may be attributed to both the increased weight of the material owing to the formation of cobalt sulphide and the decreased weight due to the partial loss of cobalt during sulphurisation; this finding is also in part supported by the ICP-OES data. The DFT pore size distributions of all materials were extracted from their isotherms and are shown in Fig. 2(e). The pristine MOF-808 has the main pore size centred at around 1.6 nm, which is, in general, consistent with the pore size of MOF-808 present in its crystal structure.6 After both the SIM process and sulphurisation, the main pore size of MOF-808 obviously decreases to 1.3 nm with reduced pore volumes, implying that both the installation of cobalt on the nodes during SIM and the formation of cobalt sulphide during sulphurisation uniformly occurred within the micropore of MOF-808. Findings here suggest that after the two-step incorporation of cobalt sulphide, the major microporosity of MOF-808 can still be preserved.
Owing to the amorphous character of the synthesized cobalt sulphide here, XPS analysis was used to further explore the surface chemical state of cobalt sulphide in CoS-MOF-808. The obtained XPS spectra in the regions of Co 2p and S 2p along with the corresponding fitting curves are shown in Fig. S4 (ESI†). One sharp peak located at 778.75 eV along with another broad peak centred at 781.30 eV can be observed in the Co 2p3/2 XPS spectrum of CoS-MOF-808. In addition, another set of dual peaks located at 793.85 and 797.10 eV can also be observed in the corresponding Co 2p1/2 XPS spectrum, as shown in Fig. S4(a) (ESI†). The presence of such four XPS peaks indicates the coexistence of Co2+ and Co3+ in CoS-MOF-808, and the fitting results here agree well with the typical XPS characteristics of Co9S8 reported in previous studies.57–60 In addition, as shown in Fig. S4(b) (ESI†), two XPS peaks located at 162.5 eV and 168.7 eV in the XPS spectrum of CoS-MOF-808 in the S 2p region correspond to the S 2p1/2 peak and satellite peak of the sulphur present in Co9S8, respectively.58,61 The findings from XPS, ICP-OES, ICP-MS and PXRD suggest that the amorphous cobalt sulphide present in CoS-MOF-808 has an overall chemical composition of CoS1.69, and its surface possesses a chemical state similar to that of Co9S8 with the coexistence of Co2+ and Co3+.
Since no obvious difference in the surface morphologies of MOF-808, Co-MOF-808, and CoS-MOF-808 can be observed under SEM, the TEM images of all materials were further obtained in order to probe the morphology within the materials. Fig. S5 (ESI†) shows the TEM images of MOF-808 and Co-MOF-808, which reveal that both materials are composed of octahedral MOF crystals with a smooth surface without any particles formed inside. The EDS elemental mapping data of Co-MOF-808 also show the uniform distributions of Zr and Co within the entire MOF crystal (Fig. S6, ESI†). These findings indicate that after the SIM process, spatially dispersed cobalt sites can be installed on the nodes of the entire MOF structure without generating aggregated cobalt particles; this is consistent with the typical feature of Zr-MOFs after such a self-limiting SIM process.42,43,62 The TEM images of CoS-MOF-808 are shown in Fig. 3(a and b), which reveal a completely distinct morphology compared to those of MOF-808 and Co-MOF-808; several spherical nanoparticles embedded within the MOF crystals can be clearly observed. The EDS elemental mapping data of CoS-MOF-808 are shown in Fig. 3(c–f), which also indicate that both the cobalt and sulphur are uniformly distributed within the entire Zr-MOF crystal. The particle size distribution of these nanoparticles embedded in the MOF estimated from the TEM image of CoS-MOF-808 is shown in Fig. S7 (ESI†). The result indicates that the major particle sizes of these cobalt sulphide nanoparticles range from around 2 nm to 3 nm, which are quite similar to the main cage size present in the crystal structure of MOF-808, ca., 2.2 nm. It should be noted that a considerable number of nanoparticles larger than 3 nm but still less than 7 nm can also be observed in the TEM image, which should be attributed to multiple nanoparticles stacked in the projected direction of the TEM image and some large nanoparticles present on the external surface of the MOF crystal. Findings here suggest that the nanoparticles of cobalt sulphide confined within the nanopore of MOF-808 and uniformly distributed in the MOF crystals can be successfully synthesized.
 | ||
| Fig. 3 TEM images of CoS-MOF-808 at (a) low and (b) high magnifications. EDS elemental mapping signals of (d) zirconium, (e) cobalt, and (f) sulphur, recorded in the rectangular region indicated in the TEM image of CoS-MOF-808 shown in (c). | ||
3.2. Catalysis for TMB/H2O2
In previous studies, a range of inorganic metal oxides and metal sulphides were reported to exhibit enzyme-mimicking activity capable of oxidation of TMB in the presence of H2O2, which can be applied to the colorimetric detection of H2O2 relying on the absorbance change caused by the produced oxTMB.63,64 Since cobalt sulphide was also reported to show such activity,44–46 we reasoned that the pore-confined cobalt sulphide nanoparticles here should be an attractive catalyst for such a reaction. Because such catalytic processes are usually operated in weakly acidic acetate buffer solutions,49 the structural integrities of MOF-808 and CoS-MOF-808 were first tested in such an environment. As shown in Fig. S8 (ESI†), the crystallinity of both MOF-808 and CoS-MOF-808 are well preserved after exposure to the 0.1 M acetate buffer solution with a pH value of 4.03 for 1 h. CoS-MOF-808 was then used as the catalyst to test its catalytic activity for oxidizing TMB in the presence of H2O2. UV-vis absorbance spectra were recorded to examine the formation of oxTMB, which shows strong absorbance at 652 nm. The UV-vis spectra of the solution samples after the catalytic process are shown in Fig. 4. It was observed that the solution containing TMB, H2O2, and the dispersed CoS-MOF-808 catalyst displayed an obvious colour change from colourless to blue after 10 min of reaction (see Fig. 4(b)), and strong absorbance at 652 nm can be found in its UV-vis spectrum. However, when dispersed Co-MOF-808 or MOF-808 was used, only a fairly low absorbance at 652 nm can be observed in the resulting UV-vis spectrum (also see the magnified version of spectra in Fig. S9, ESI†), and this absorbance is almost the same as that of the sample without adding MOF (Fig. 4(a)); this observation indicates that both MOF-808 and cobalt-installed MOF-808 are not active catalysts for the redox reaction between TMB and H2O2. A control experiment with the addition of the CoS-MOF-808 catalyst and TMB without adding H2O2 was also conducted, which also resulted in negligible absorbance at 652 nm after 10 min of reaction (see Fig. 4 and Fig. S9, ESI†). Findings here clearly indicate that pore-confined cobalt sulphide nanoparticles can effectively accelerate the rate of the redox reaction between TMB and H2O2 to generate oxTMB.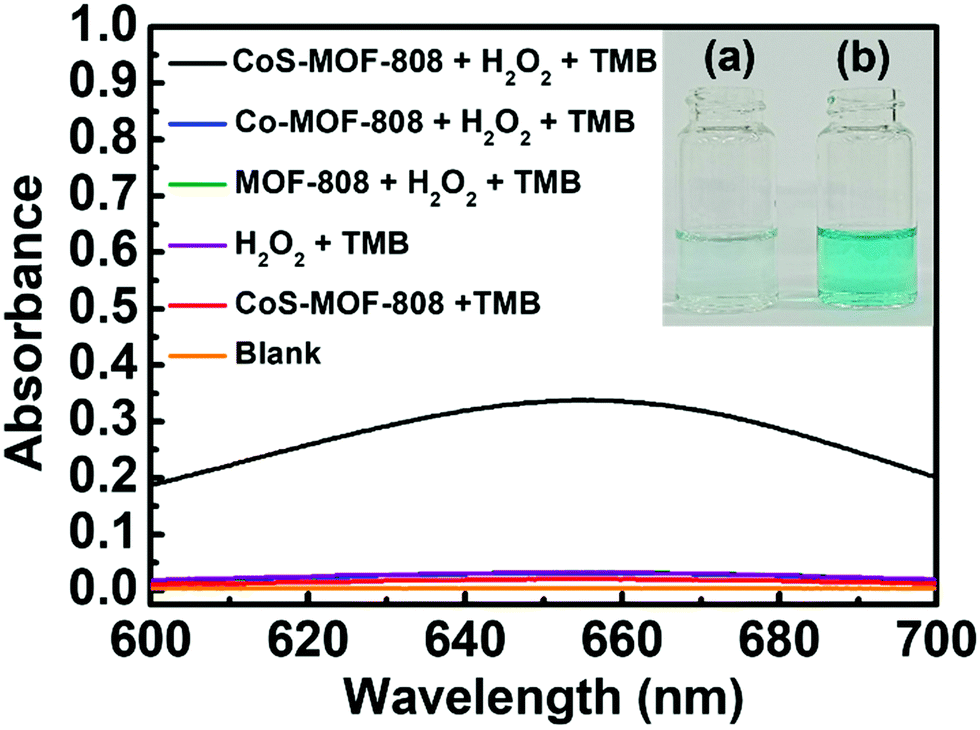 | ||
Fig. 4 UV-vis spectra of the solution samples after 10 min of reaction under stirring at 300 rpm. Each solution is composed of 0.1 M acetate buffer solution (pH = 4.03) and DMSO (v:v = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Concentrations of other species: [H2O2] = 1 mM; [TMB] = 0.5 mM; [MOF-based solid] = 0.5 mg mL−1. It should be noted that “Blank” represents the solution sample containing 0.1 M of acetate buffer solution and DMSO (v:v = 9:1). The inset shows the photograph of the solutions after 10 min of reaction: (a) H2O2 + TMB; (b) CoS-MOF-808 + H2O2 + TMB. :1). Concentrations of other species: [H2O2] = 1 mM; [TMB] = 0.5 mM; [MOF-based solid] = 0.5 mg mL−1. It should be noted that “Blank” represents the solution sample containing 0.1 M of acetate buffer solution and DMSO (v:v = 9:1). The inset shows the photograph of the solutions after 10 min of reaction: (a) H2O2 + TMB; (b) CoS-MOF-808 + H2O2 + TMB. | ||
To define the material as a catalyst for the TMB/H2O2 reaction, it is important to verify the turnover number (TON), which is defined as the number of oxTMB produced per active site of the catalyst, can at least exceed 1. It should be noticed that concerns have been raised in recent studies regarding the TON of the materials for this reaction, and it has been found that some previously reported materials that are capable of accelerating the rate of redox reaction between TMB and H2O2 are in fact the self-limited reagents instead of catalysts.52,65 Thus, to confirm that the pore-confined cobalt sulphide nanoparticles here are the catalyst for a TMB/H2O2 reaction, a 200 mL large-scale catalytic experiment was conducted (also see details in the Experimental Section). As shown in Fig. 5, the amount of oxTMB generated after 15 min of reaction can be calculated from the obtained absorbance value at 652 nm by using Lambert–Beer's law (εTMB = 39000 M−1 cm−1).52 Thereafter, from the difference in the absorbance of samples after the reaction with and without adding 2.0 mg of CoS-MOF-808, the amount of oxTMB generated owing to the presence of CoS-MOF-808 was determined as 4.96 μmol. Since it is not viable to estimate the amount of exposed active sites present in the cobalt sulphide nanoparticles confined in MOF-808 which participate in the reaction, we considered all the atomic cobalt sites present in the material as the active sites in order to obtain the “underestimated” TON (see Section S2 in the ESI,† for details in the calculation). As a result, the obtained TON already achieved 2.83 after 15 min of reaction, which clearly confirms that the pore-confined cobalt sulphide nanoparticles belong to the catalyst for the reaction between TMB and H2O2.
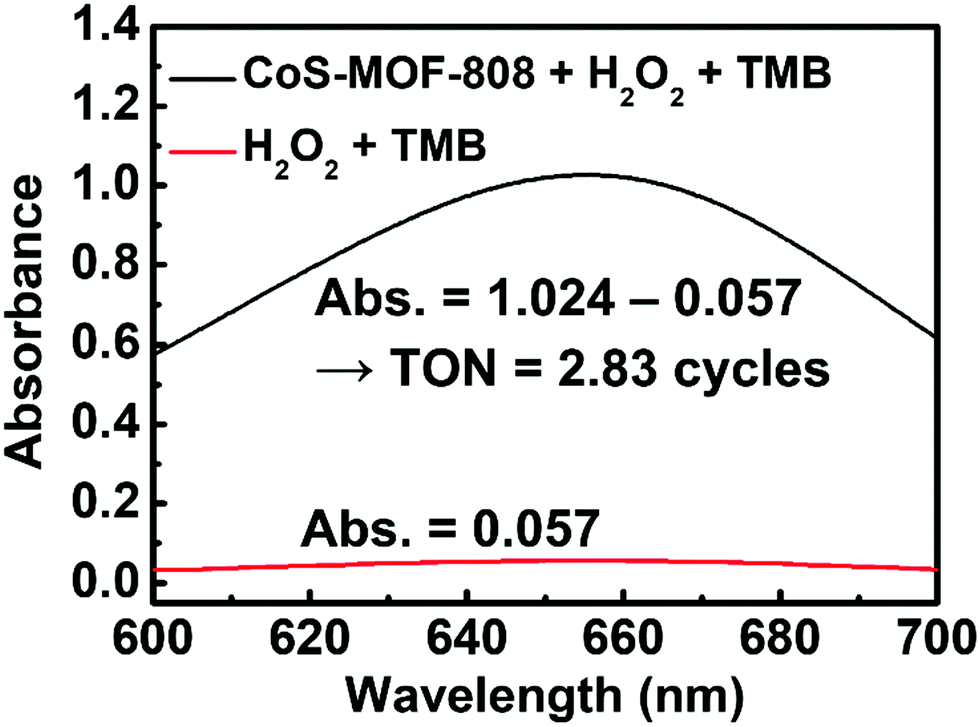 | ||
| Fig. 5 UV-vis spectra of the solution samples after 15 min of reaction under stirring at 600 rpm. Each solution during the reaction is composed of 200 mL of 0.1 M acetate buffer solution (pH = 4.03) and DMSO (v:v = 9:1). Concentrations of other species: [H2O2] = 245 mM; [TMB] = 0.5 mM; [CoS-MOF-808] = 0.01 mg mL−1. | ||
It has been widely reported that the materials such as cobalt sulphide and other transition metal oxides that can exhibit the enzyme-mimicking activity to catalyse the reaction between TMB and H2O2 follow the Michaelis–Menten reaction kinetics.50,63,66 Thus, a series of experiments were carried out to probe the reaction kinetics of this catalytic process occurring on CoS-MOF-808. It should be noticed that since two substrates, i.e., TMB and H2O2, are present here, apparent kinetic parameters of each substrate need to be acquired separately by conducting the experiments with various concentrations of one substrate while keeping another substrate at a fixed concentration.49,50 The experimental details can be found in the Experimental section, and the obtained initial reaction rate (V0) at each concentration of the substrate with H2O2 as the substrate and TMB as the substrate are listed in Tables S1 and S2 (ESI†), respectively. By utilizing the Lineweaver–Burk plot shown in Fig. S10(a) (ESI†), the Km and maximum reaction rate (Vmax) of the CoS-MOF-808 catalyst with H2O2 as the substrate were determined as 35.39 mM and 42.73 × 10−8 M s−1, respectively. On the other hand, according to Fig. S10(b) (ESI†), the Km and Vmax values with TMB as the substrate are 0.24 mM and 37.31 × 10−8 M s−1, respectively (see details in Tables S1 and S2 in the ESI†). According to these kinetic parameters, the Michaelis–Menten models for H2O2 and TMB were further constructed, respectively. As shown in Fig. S10(c) and (d) (ESI†), the experimental data are well consistent with the corresponding model for both TMB and H2O2, which indicates that the reaction between H2O2 and TMB catalysed by CoS-MOF-808 follows the Michaelis–Menten kinetics. It is worth mentioning that ideally, the concentration range used for such kinetic studies should be located between 0.5 Km and 5 Km of the targeted substrate;49 previous studies reporting the use of cobalt sulphide-based materials for the TMB/H2O2 reaction also utilized similar concentration ranges for kinetic studies.46,47 Herein, the use of a H2O2 concentration higher than 45 mM would reduce the reliability of the collected UV-vis data owing to the fast initial reaction rate, and the use of a TMB concentration higher than 0.6 mM exceeds the solubility limit for TMB. Therefore, data of initial reaction rates collected within 25–45 mM and 0.4–0.6 mM were used for the kinetic studies of H2O2 and TMB, respectively. These concentration ranges are well located between 0.5 Km and 5 Km of the corresponding substrates. Table S3 (ESI†) presents the kinetic parameters of some reported materials, including metal oxides, metal sulphides and some MOF-based materials, for the H2O2/TMB reaction. Compared to these reported active materials, CoS-MOF-808 can achieve a medium Km for H2O2, but its Vmax for H2O2 is among the highest in comparison with other reported values. Besides, CoS-MOF-808 also possesses a relatively lower Km value and a significantly higher Vmax value for TMB compared to other materials. This finding indicates that CoS-MOF-808 has strong affinity toward TMB with a high maximum initial rate.
CoS-MOF-808 was further applied for the colorimetric detection of H2O2 by recording the absorbance of the sample at 652 nm after 10 min of reaction between 0.5 mM TMB and a certain concentration of H2O2. As shown in Fig. 6, the absorbance increases obviously with an increase in the concentration of H2O2, and a linear range from 0.05 mM to 0.6 mM can be obtained from the calibration curve. The limit of detection (LOD) was estimated as 7.51 μM based on the signal-to-noise ratio of 3. Table S4 (ESI†) lists the comparison between the sensing performances of the CoS-MOF-808-based H2O2 sensor and those of other reported colorimetric sensors. It is worth mentioning that compared to the nanostructural cobalt sulphide reported previously,44 CoS-MOF-808 can exhibit a similar linear range and a significantly smaller LOD, which implies that the pore-confined cobalt sulphide nanoparticles in MOF-808 can significantly improve the catalytic activity and sensing performance.
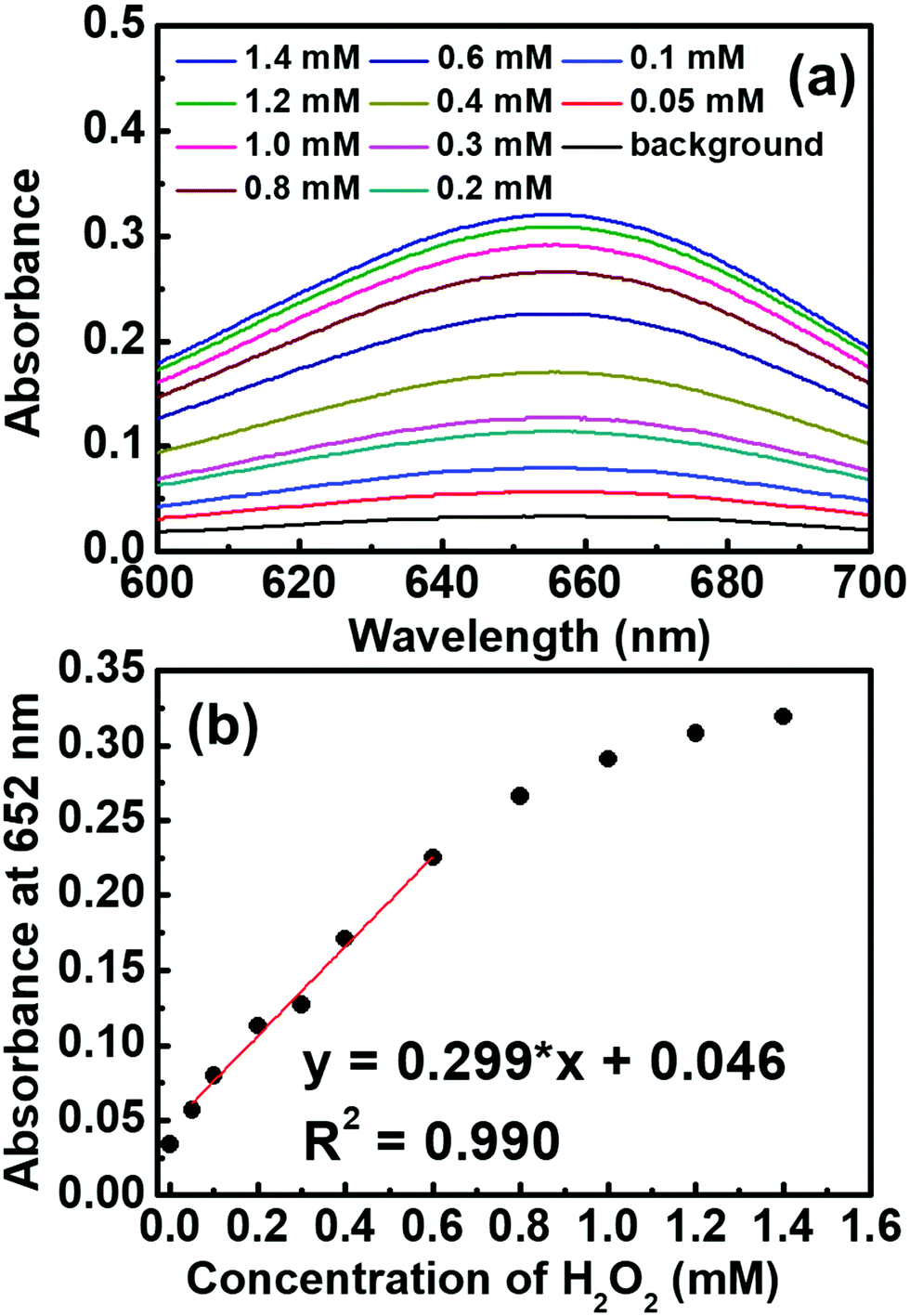 | ||
| Fig. 6 (a) UV-vis spectra of the solution samples after 10 min of reaction under stirring at 300 rpm. Each solution is composed of 0.1 M acetate buffer solution (pH = 4.03) and DMSO (v:v = 9:1) containing a certain concentration of H2O2. Concentrations of other species: [TMB] = 0.5 mM; [CoS-MOF-808] = 0.5 mg mL−1. (b) Absorbance at 652 nm recorded at various concentrations of H2O2 from the data shown in (a), with the corresponding linear calibration line. | ||
4. Conclusions
The controlled synthesis of cobalt sulphide nanoparticles that are confined within the nanopore of a water-stable Zr-MOF, MOF-808, can be successfully achieved by a two-step synthetic strategy, with the self-limiting SIM process to install spatially isolated Co(II) ions on MOF nodes followed by a solution-phase sulphurisation process. The crystallinity, major porosity, and surface morphology of the MOF-808 crystals can still be preserved after the incorporation of pore-confined cobalt sulphide nanoparticles. The obtained cobalt sulphide nanoparticles distribute uniformly within the entire MOF crystal and are mainly composed of amorphous CoS1.69 with a surface chemical state similar to that of Co9S8. The TEM images show that the major particle size of these cobalt sulphide nanoparticles confined in MOF crystals is around 2–3 nm, which is, in general, consistent with the main pore size of MOF-808. The obtained CoS-MOF-808 has been verified as a heterogeneous catalyst for the reaction between H2O2 and TMB. As a result, the CoS-MOF-808 catalyst can be further utilized in the colorimetric detection of H2O2, and a linear range of 0.05 mM to 0.6 mM and a LOD of 7.51 μM can be achieved. Aiming for a range of catalytic applications, ongoing work is focusing on the selective formation of pore-confined metal sulphide nanoparticles in various Zr-MOFs with distinct pore sizes in order to modulate the resulting particle size.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Ministry of Science and Technology (MOST) of Taiwan, under the project 110-2221-E-006-017-MY3. We also acknowledge the financial support from the Yushan Young Scholar Program under the Ministry of Education (MOE), Taiwan. This study was also supported in part by the Higher Education Sprout Project, MOE, Taiwan and the Headquarters of University Advancement at National Cheng Kung University (NCKU). We also thank the Surface Analysis Lab in the Department of Chemical Engineering, National Taiwan University, for recording XPS spectra. The authors also gratefully acknowledge the use of TEM and ICP-MS belonging to the Core Facility Center of NCKU.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J. Dhainaut, M. Bonneau, R. Ueoka, K. Kanamori and S. Furukawa, ACS Appl. Mater. Interfaces, 2020, 12, 10983–10992 CrossRef CAS PubMed.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- A. Cadiau, K. Adil, P. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- K. M. Choi, H. M. Jeong, J. H. Park, Y.-B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- M. B. Majewski, A. W. Peters, M. R. Wasielewski, J. T. Hupp and O. K. Farha, ACS Energy Lett., 2018, 3, 598–611 CrossRef CAS.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- X. Xuan, M. Wang, M. Zhang, Y. V. Kaneti, X. Xu, X. Sun and Y. Yamauchi, J. CO2 Util., 2022, 57, 101883 CrossRef CAS.
- J. J. Calvo, S. M. Angel and M. C. So, APL Mater., 2020, 8, 050901 CrossRef CAS.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- J. O. Kim, J. Y. Kim, J. C. Lee, S. Park, H. R. Moon and D. P. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 4385–4392 CrossRef CAS PubMed.
- S. Pal, S.-S. Yu and C.-W. Kung, Chemosensors, 2021, 9, 306 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- S. Jeoung, S. Kim, M. Kim and H. R. Moon, Coord. Chem. Rev., 2020, 420, 213377 CrossRef CAS.
- U. Ryu, S. Jee, P. C. Rao, J. Shin, C. Ko, M. Yoon, K. S. Park and K. M. Choi, Coord. Chem. Rev., 2021, 426, 213544 CrossRef CAS PubMed.
- M. G. Mohamed, A. F. M. El-Mahdy, M. G. Kotp and S.-W. Kuo, Mater. Adv., 2022, 3, 707–733 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- V. Bon, I. Senkovska, I. A. Baburin and S. Kaskel, Cryst. Growth Des., 2013, 13, 1231–1237 CrossRef CAS.
- S. Yuan, J. S. Qin, C. T. Lollar and H. C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- K. O. Kirlikovali, Z. Chen, T. Islamoglu, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2020, 12, 14702–14720 CrossRef CAS PubMed.
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 11954–11961 CrossRef CAS PubMed.
- B. Gibbons, E. C. Bartlett, M. Cai, X. Yang, E. M. Johnson and A. J. Morris, Inorg. Chem., 2021, 60, 16378–16387 CrossRef CAS PubMed.
- R. R. Chianelli, G. Berhault and B. Torres, Catal. Today, 2009, 147, 275–286 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Nano, 2015, 9, 8484–8490 CrossRef CAS PubMed.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 CrossRef CAS PubMed.
- H. Noh, C.-W. Kung, K.-I. Otake, A. W. Peters, Z. Li, Y. Liao, X. Gong, O. K. Farha and J. T. Hupp, ACS Catal., 2018, 8, 9848–9858 CrossRef CAS.
- L. Yu, W.-G. Cui, Q. Zhang, Z.-F. Li, Y. Shen and T.-L. Hu, Mater. Adv., 2021, 2, 1294–1301 RSC.
- P. Jin, L. Wang, X. Ma, R. Lian, J. Huang, H. She, M. Zhang and Q. Wang, Appl. Catal., B, 2021, 284, 119762 CrossRef CAS.
- Y. Su, Z. Zhang, H. Liu and Y. Wang, Appl. Catal., B, 2017, 200, 448–457 CrossRef CAS.
- K. Gao, H. Li, Q. Meng, J. Wu and H. Hou, ACS Appl. Mater. Interfaces, 2021, 13, 2779–2787 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuno, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, A. E. Platero-Prats, J. Liu, C. W. Kung, H. Noh, M. R. DeStefano, N. M. Schweitzer, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2017, 139, 15251–15258 CrossRef CAS PubMed.
- H. Yang, J. Zha, P. Zhang, Y. Xiong, L. Su and F. Ye, RSC Adv., 2016, 6, 66963–66970 RSC.
- J. Wang, Y. Wang and D. Zhang, J. Colloid Interface Sci., 2020, 561, 327–337 CrossRef CAS PubMed.
- K. Wu, W. Li, S. Zhao, W. Chen, X. Zhu, G. Cui, Z. Liu, Q. Liu, X. Zhang and X. Zhang, Colloids Surf., A, 2020, 602, 125063 CrossRef CAS.
- S. Hashmi, M. Singh, P. Weerathunge, E. L. H. Mayes, P. D. Mariathomas, S. N. Prasad, R. Ramanathan and V. Bansal, ACS Appl. Nano Mater., 2021, 4, 13352–13362 CrossRef CAS.
- W. H. Ho, S. C. Li, Y. C. Wang, T. E. Chang, Y. T. Chiang, Y. P. Li and C. W. Kung, ACS Appl. Mater. Interfaces, 2021, 13, 55358–55366 CrossRef CAS PubMed.
- B. Jiang, D. Duan, L. Gao, M. Zhou, K. Fan, Y. Tang, J. Xi, Y. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tang, G. Nie, M. Liang and X. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS PubMed.
- X. Yan and L. Gao, Nanozymology, Springer, 2020 Search PubMed.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- M. Zandieh and J. Liu, ACS Nano, 2021, 15, 15645–15655 CrossRef CAS PubMed.
- Z. Nickolov, G. Georgiev, D. Stoilova and I. Ivanov, J. Mol. Struct., 1995, 354, 119–125 CrossRef CAS.
- T. Abza, D. G. Dadi, F. G. Hone, T. C. Meharu, G. Tekle, E. B. Abebe and K. S. Ahmed, Adv. Mater. Sci. Eng., 2020, 2020, 2628706 Search PubMed.
- J.-H. Li, Y.-C. Chen, Y.-S. Wang, W. H. Ho, Y.-J. Gu, C.-H. Chuang, Y.-D. Song and C.-W. Kung, ACS Appl. Energy Mater., 2020, 3, 6258–6267 CrossRef CAS.
- C.-H. Shen, Y.-H. Chen, Y.-C. Wang, T.-E. Chang, Y.-L. Chen and C.-W. Kung, Phys. Chem. Chem. Phys., 2022, 24, 9855–9865 RSC.
- P. Tan, Y. Liu, A. Zhu, W. Zeng, H. Cui and J. Pan, ACS Sustainable Chem. Eng., 2018, 6, 10385–10394 CrossRef CAS.
- D. Xiong, Q. Zhang, S. M. Thalluri, J. Xu, W. Li, X. Fu and L. Liu, Chem. – Eur. J., 2017, 23, 8749–8755 CrossRef CAS PubMed.
- S.-H. Chang, M.-D. Lu, Y.-L. Tung and H.-Y. Tuan, ACS Nano, 2013, 7, 9443–9451 CrossRef CAS PubMed.
- J. Mujtaba, L. He, H. Zhu, Z. Xiao, G. Huang, A. A. Solovev and Y. Mei, ACS Appl. Nano Mater., 2021, 4, 1776–1785 CrossRef CAS.
- H. Li, G. Wang, H. Gong and Z. Jin, J. Phys. Chem. C, 2021, 125, 648–659 CrossRef CAS.
- P. Ji, K. Manna, Z. Lin, X. Feng, A. Urban, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 7004–7011 CrossRef CAS PubMed.
- W. Song, B. Zhao, C. Wang, Y. Ozaki and X. Lu, J. Mater. Chem. B, 2019, 7, 850–875 RSC.
- H. Wei and E. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef CAS PubMed.
- S. Scott, H. Zhao, A. Dey and T. B. Gunnoe, ACS Catal., 2020, 10, 14315–14317 CrossRef CAS.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: https://doi.org/10.1039/d2ma00542e |
| This journal is © The Royal Society of Chemistry 2022 |