
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free polypeptide redox flow batteries†‡
Zhiming
Liang§
 a,
Tan P.
Nguyen§
b,
N. Harsha
Attanayake
a,
Alexandra D.
Easley
c,
Jodie L.
Lutkenhaus
*cd,
Karen L.
Wooley
*bcd and
Susan A.
Odom
a
a,
Tan P.
Nguyen§
b,
N. Harsha
Attanayake
a,
Alexandra D.
Easley
c,
Jodie L.
Lutkenhaus
*cd,
Karen L.
Wooley
*bcd and
Susan A.
Odom
a
aDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
bDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: wooley@chem.tamu.edu
cDepartment of Materials Science and Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: jodie.lutkenhaus@tamu.edu
dArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA
First published on 12th July 2022
Abstract
Non-aqueous organic redox flow batteries (NAORFBs) are considered emerging large-scale energy storage systems due to their larger voltage window as compared to aqueous systems and their metal-free nature. However, low solubility, sustainability, and crossover of redox materials remain major challenges for the development of NAORFBs. Here, we report the use of redox active α-helical polypeptides suitable for NAORFBs. The polypeptides exhibit less crossover than small molecule analogs for both Daramic 175 separator and FAPQ 375 PP membrane, with FAPQ 375 PP preventing crossover most effectivley. Polypeptide NAORFBs assembled with a TEMPO-based polypeptide catholyte and viologen-based polypeptide anolyte exhibit low capacity fade (ca. 0.1% per cycle over 500 cycles) and high coulombic efficiency (>99.5%). The polypeptide NAORFBs exhibit an output voltage of 1.1 V with a maximum capacity of 0.53 A h L−1 (39% of the theoretical capacity). After 500 charge–discharge cycles, 60% of the initial capacity was retained. Post cycling analysis using spectral and electrochemical methods demonstrate that the polypeptide backbone and the ester side chain linkages are stable during electrochemical cycling. Taken together, these polypeptides offer naturally-derived, deconstructable platforms for addressing the needs of metal-free energy storage.
Introduction
The production of alternative renewable energy technologies to reduce the carbon footprint of pollution-causing energy sources has become a topic of scientific concern, due to the intermittent nature of energy from renewable resources, such as sun and wind. For load-leveling purposes, redox flow batteries (RFBs) are emerging as large-scale energy storage platforms because of their scalability and safety features.1,2 However, current commercial RFB technologies rely heavily on the use of vanadium and other mineral resources, which are facing a surge in price due to supply scarcity.3,4 As an alternative to vanadium, organic redox-active materials are promising for their general abundance and synthetic versatility.5,6 Organic redox-active species can be used in either aqueous or non-aqueous redox flow batteries (AORFBs or NAORFBs, respectively).5,7,8 However, to further reduce the reliance on mineral resources for both AORFBs and NAORFBs, metal-free electrolytes are of increasing consideration in electrolyte design. AORFBs take advantage of water as an ideal green chemistry solvent, but AORFBs are often faced with adverse effects caused by extreme acidic or alkaline conditions.9,10 The most common redox molecules used in AORFBs are 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO), anthraquinone, viologen-derivatives, and ferrocene derivatives.11 On the other hand, typical small molecule chemistries for NAORFBs are based on TEMPO, phthalimide, anthracene, and quinoxaline derivatives.12 NAORFBs have a larger voltage window and a wider operating temperature range of as compared to AORFBs. Consequently, NAORFBs have emerged as promising options for grid-scale energy storage.In either system, crossover of redox active materials, which reduces the accessible capacity, Coulombic efficiency (CE), and long-term cycle life, remains a major challenge for the widespread application of RFBs.5,7,8 To prevent or reduce crossover, polymeric redox-active materials with large hydrodynamic volumes have been proposed,1 in which size exclusion can be leveraged. More specifically, redox-active polymers including polypyrrole, polythiophene, polyaniline, and polymers with pendant redox-active moieties have attracted scientific interest in the past decade.1,13–19 Other examples include viologen- and TEMPO-containing polymethacrylates for AORFBs and polystyrenes carrying similar redox-active centers for NAORFBs.13,20 An overview of these studies and others1,13–23 reveal several challenges to be addressed. Although redox-active polymers exhibit lower crossover and improved capacity retention, the redox kinetics remain sluggish due to the low diffusion coefficient (<10−7 cm2 s−1) of the polymer in the electrolyte.13,24,25 Further, these systems use non-degradable polymer backbones, which may possess long-term environmental persistence. Accordingly, it is desirable to consider alternative redox-polymer chemistries that exhibit controlled deconstruction and improved diffusion.
Previously, we reported the use of viologen- and TEMPO-containing polypeptides as redox-active materials in solid electrodes for metal-free secondary batteries.26 The polypeptide backbone is comprised of amino acids that link together into α-helical macromolecules. Redox-active pendant groups attached to the polypeptide backbone reversibly stored charge. To demonstrate on-demand deconstruction, the redox-active polypeptides were degraded in the presence of an acid and elevated temperature to yield glutamic acid and byproducts. Although the redox-active polypeptides performed well initially in the solid electrode battery, dissolution of the materials over time limited the functional lifetime. Informed by this prior work, we hypothesize here that these molecules could instead be used in the solution state for metal-free NAORFBs.
In this study, therefore, we explored NAORFBs with redox-active α-helical polypeptides, Fig. 1. Crossover of the polypeptides through different membranes was quantified using an H-cell and then compared to small molecule analogs. Full NAORFBs using metal-free electrolyte were next constructed and assessed using electrochemical impedance spectroscopy and galvanostatic cycling at varying rates. Post-mortem analyses of the anolyte and catholyte were conducted using circular dichroism, Fourier transform infrared (FTIR) spectroscopy, and cyclic voltammetry to examine deconstruction of the polypeptides and evidence of crossover. Overall, this study demonstrates NAORFBs from naturally derived, redox active polypeptide electrolytes with high capacity retention, high Coulombic efficiency, and low crossover. This concept represents an early step toward developing a recyclable RFB, in which the electrolytes could be degraded after operation using acid and heat.
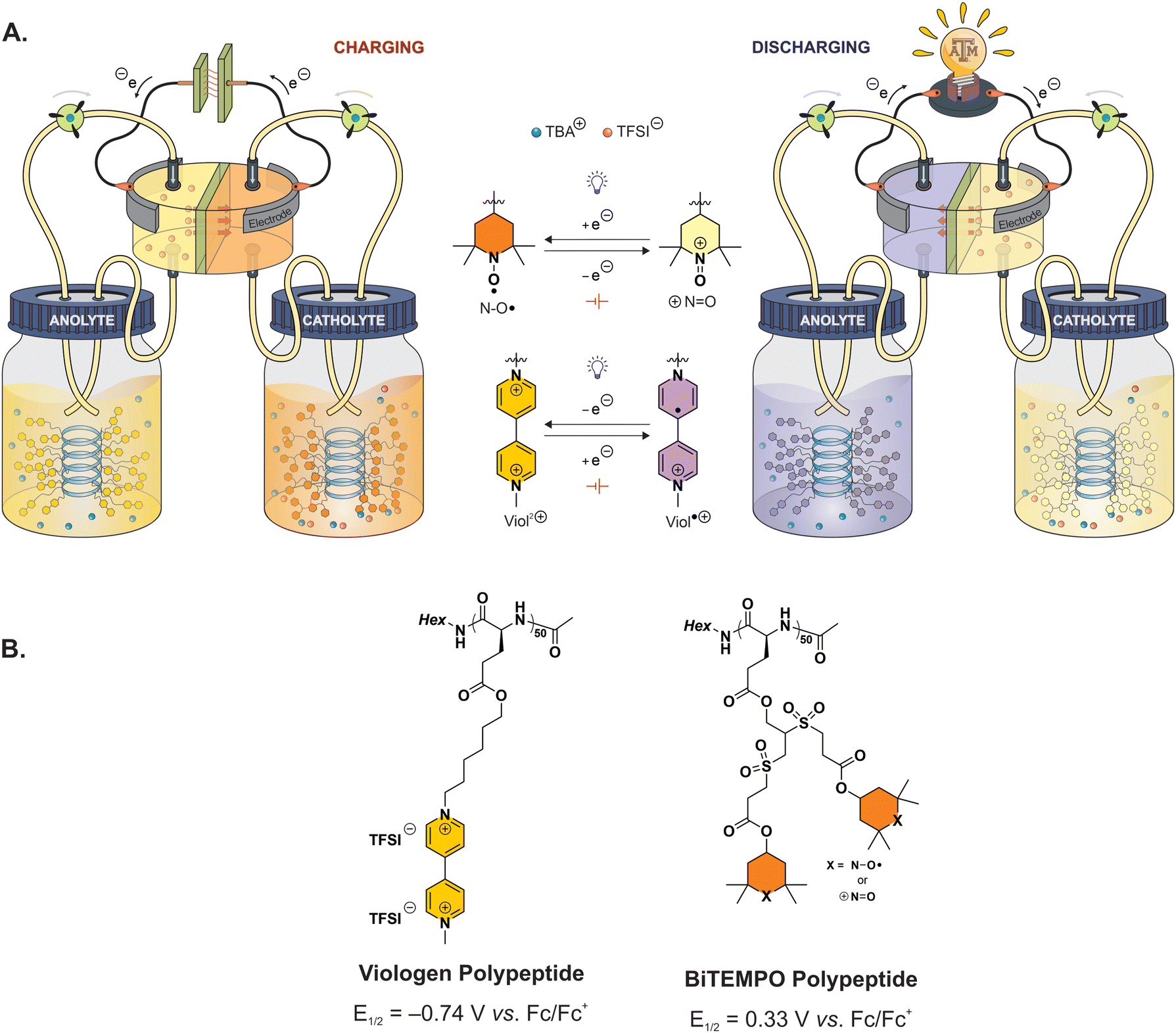 | ||
| Fig. 1 (A) Illustrative design of polypeptide-based redox flow battery during charging and discharging. (B) Molecular structures of the redox-active polypeptides. | ||
Experimental
Materials
3,3-Dithiodipropionic acid, 4,4′-bipyridine, meta-chloroperoxybenzoic acid (m-CPBA, ≤77%), dichloromethane (DCM), N,N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), 2,2-dimethoxy-2-phenylacetophenone (DMPA), N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (4-OH-TEMPO, TEMPOL), diphosgene, hexanes, n-hexylamine, hydrocinnamic acid, iodomethane, iron(II) sulfate heptahydrate (FeSO4·7H2O), L-glutamic acid, methanol, propargyl alcohol, sodium iodide (NaI), triethylamine (TEA), trifluoroethanol (TFE), trimethylsilyl chloride (TMSCl) and triphosgene were purchased from Sigma-Aldrich. DL-Dithiothreitol (DTT) was purchased from Tokyo Chemical Industry (Japan). 6-Chloro-1-hexanol and acetic anhydride were purchased from Alfa-Aesar (USA). All reagents were used as received, except for propargyl alcohol, which was distilled under an atmosphere of N2 prior to use. Ultrapure water was collected from a Milli-Q® integral water purification system (18 MΩ cm). Lithium bis(trifluoromethane)sulfonamide (LiTFSI) was purchased from Alfa Aesar (USA).Electrochemical characterization
Polypeptide cell rate studies and full cell cycling: 7.4 mL (for rate study) or 7.5 mL (for full cell cycling) of 25 mM biTEMPO-polypeptide and 50 mM viologen polypeptide were prepared in 0.5 M TEATFSI/ACN. 5, 10, 15, 20, and 5 mA cm−2 current density was used for the rate studies, and 10 mA cm−2 current density was used for full cell cycling. All electrolytes were pumped into electrolyte reservoirs in an argon-filled glovebox.
The theoretical capacity was calculated by the equation: Qtheoretical = nFC/3600 A h L−1; F is Faraday's constant, n is the number of electrons, and C is the concentration of active species (mol L−1). The concentration of redox active moiety (two TEMPO groups per repeat unit) for the catholyte was 50 mM resulting in Qtheoretical = 1.34 A h L−1.
Syntheses of redox-active polypeptides and their small-molecule analogs
The viologen-iodide and biTEMPO polypeptides were synthesized as previously reported.26 To enhance the solubility of the viologen species in ACN, iodide anions of the viologen-iodide polypeptide and analog were exchanged for TFSI anions prior to testing. The anion exchange was done upon mixing a solution of each viologen species with an equal volume of LiTFSI (6 equiv.), both in H2O. The precipitate was collected via centrifugation, dissolved into ACN, and precipitated into 0.5 M LiTFSI in H2O (×3), followed by precipitation into H2O (×3). The viologen-TFSI polypeptide was obtained as a brown solid and the viologen-TFSI analog as a brown viscous oil.Chemical characterization
Attenuated total reflectance-Fourier-transform infrared (ATR–FTIR) spectra were recorded for powder samples on an IR Prestige 21 system (Shimadzu Corp., Japan) and analysed using IR solution v. 1.40 software. Circular dichroism (CD) spectra of the polymer solutions were recorded on a Chirascan circular dichroism spectrometer from Applied Photophysics (Leatherhead) equipped with a 150-W xenon arc lamp. CD samples were prepared by diluting 25 mM solutions of biTEMPO polypeptide 100× and 50 mM solutions of viologen polypeptide 200× to obtain a final concentration of 0.25 mM. Circular dichroism spectra were acquired between 180 nm and 280 nm, using a wavelength step of 1.0 nm, in a quartz cell with a path length of 1.0 cm, and analysed using Pro-Data version 5 software.Results and discussion
Synthesis of redox-active polypeptides
For the redox flow cell, it is essential to have active materials that are soluble in electrolyte media. We found that viologen polypeptides with small counterions, such as iodide, had limited solubility in organic electrolyte; however, biTEMPO polypeptides were soluble in organic electrolyte. Therefore, the viologen polypeptide's iodide counterions were exchanged for TFSI− to afford a viologen-TFSI polypeptide, hereafter referred to simply as “viologen polypeptide,” that was soluble in the redox flow cell electrolyte. The synthesis and ion-exchange process are described in our previous report26 and the ESI,‡ respectively. With the attainment of soluble viologen and biTEMPO polypeptides, crossover tests and redox flow cell studies were next examined.Crossover tests
Crossover is a serious concern for redox flow cells because it leads to capacity fade during cycling. In this process, active materials undesirably diffuse and transport through the membrane to the other side of the cell. This process can be mitigated by selecting an appropriate membrane and by using larger-sized active materials such as polymers or, as shown here, polypeptides.To examine crossover of the redox active components, an anion exchange membrane (AEM, FAPQ 375 PP) and a separator (Daramic 175) were compared, which were shown to be satisfactory for small-molecule viologen in past work.23 An H-cell consisting of the redox-active species of interest in electrolyte on one side, a blank electrolyte solution of equal volume on the other side, and the membrane or separator between the two was examined.
Cyclic voltammetry (CV) was used to characterize the blank electrolyte side of the H-cell after 6, 12, 24, 48, 72, and 96 h of contact with the anolyte or catholyte via the membrane or separator. As expected, significant crossover was observed for both the redox-active polypeptides and their small-molecule analogs using the Daramic 175 membrane due to its larger pore size of 100 nm (ESI,‡ Fig. S2 and S3).23 The crossover rate of the polypeptides was relatively smaller than the small-molecule analogs (ESI,‡ Fig. S4) due to the larger molecular size. In contrast, the FAPQ 375 PP membrane allowed for considerably lower crossover of the small-molecule analogs over multiple days, and an even smaller amount of crossover was observed for the polypeptides (Fig. 2). The small-molecule viologen analog likely had a higher permeation through the FAPQ 375 PP membrane due to its small size and steric hinderance relative to the biTEMPO analog. Although crossover was small overall for the polypeptides, the biTEMPO polypeptide exhibited slightly more crossover as compared to the viologen polypeptide (inset of Fig. 2). The increased crossover of the biTEMPO polypeptide was likely due to the smaller hydrodynamic size compared to the viologen polypeptide, which had electrostatic repulsion of the side chains due to the 2+ charge, resulting in a larger hydrodynamic size. Taken together, FAPQ 375 PP was chosen as the membrane for flow cell cycling studies because it prevented crossover much more effectively than Daramic 175.
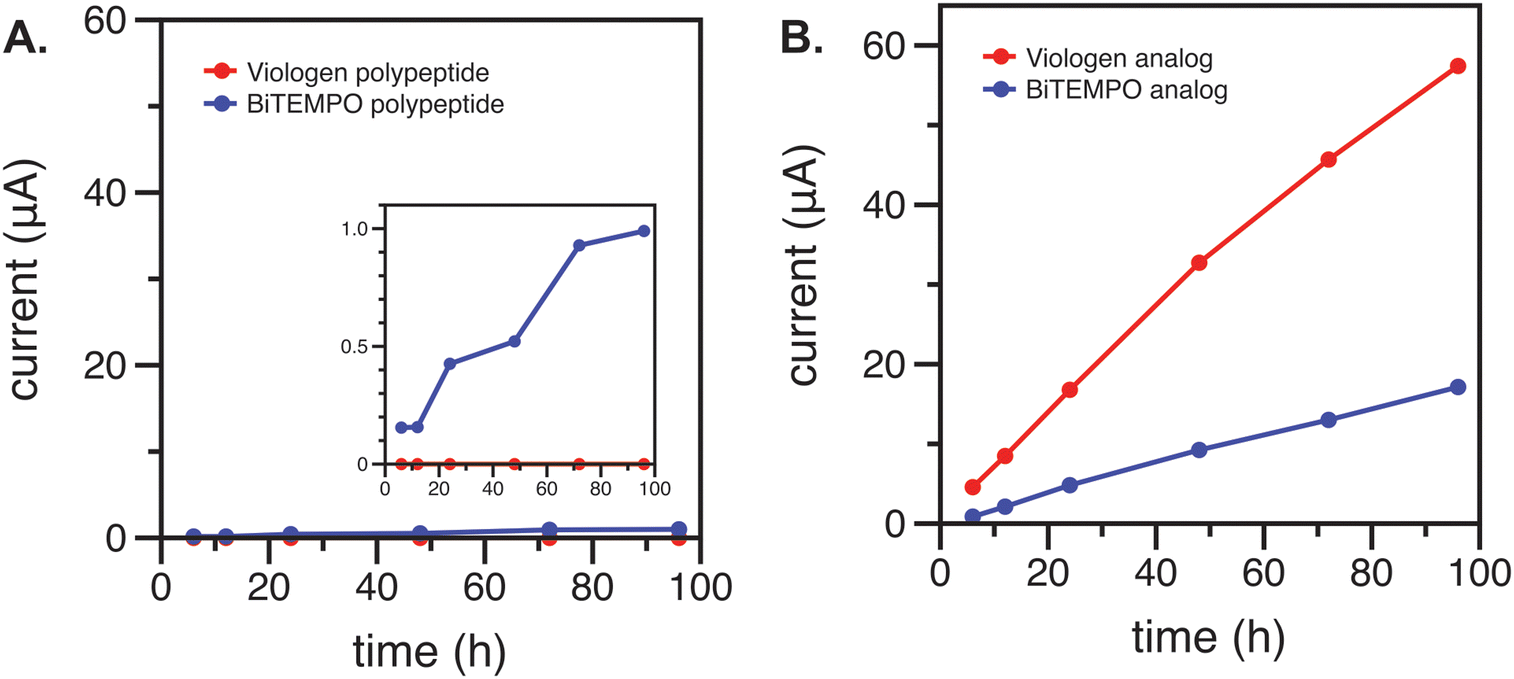 | ||
| Fig. 2 Crossover study of (A) viologen and biTEMPO polypeptides and (B) their small-molecule analogs using an FAPQ 375 PP membrane. The currents were taken as the averages of absolute values of the CV anodic and cathodic peak currents over time for the blank electrolyte side of the H-cell. | ||
Flow cell cycling
The polypeptide redox flow cell was assembled with each reservoir containing 7.5 mL of 50 mM (by repeat unit) viologen polypeptide anolyte and 25 mM (by repeat unit) biTEMPO polypeptide catholyte in 0.5 M TEATFSI in acetonitrile (ACN), yielding a theoretical capacity of 1.34 A h L−1. Prior to cycling, electrochemical impedance spectroscopy (EIS) was performed on circulating electrolytes to assess the flow cell resistance (Fig. 3). The Nyquist plot in Fig. 3 exhibited a high-frequency semicircle, followed by a low frequency tail. From the intercept of the EIS response with the x-axis, the polypeptide flow cell exhibited an Ohmic resistance of ca. 2.5 Ω (or 6.4 Ω cm2). The increased Ohmic resistance could be caused by the relatively low electrolyte salt concentration (0.5 M compared to 1 or 2 M in other reports27), the use of an anion exchange membrane (instead of porous separator), and/or concentration polarization due to the anion shuttling during charge/discharge.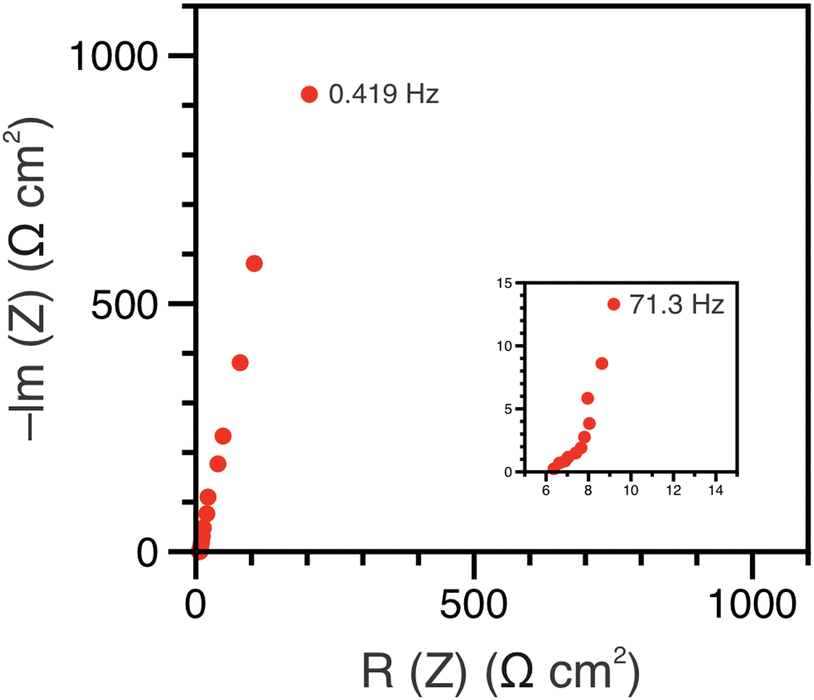 | ||
| Fig. 3 Nyquist plot of the EIS data before galvanostatic cycling for the redox flow cell with a BiTEMPO polypeptide catholyte and viologen polypeptide anolyte. EIS was conducted with circulating electrolytes at open circuit potential with an amplitude of 10 mV in a frequency range of 200 kHz to 0.4 Hz (5 steps per decade). | ||
The flow cell was cycled within a potential window of 0.7 to 1.5 V, and a variable rate experiment was performed to determine trends among applied current density, overpotential, and accessible capacity. The flow cell was cycled at current densities of 5, 10, 15, 20 and 5 mA cm−2 sequentially for five cycles each. The charge and discharge capacities and Coulombic efficiencies at each current density are shown in Fig. 4(A). As expected, the accessible capacity decreased as the current density increased due to increasing cell polarization. At 5 mA cm−2, the polypeptide flow cell presented a capacity utilization of 59% (relative to the theoretical capacity), which decreased to 38% at 10 mA cm−2, 13% at 15 mA cm−2, and complete capacity loss at 20 mA cm−2. Upon returning the current to 5 mA cm−2, the capacity recovered 95% of its initial value, demonstrating that the high current densities did not have adverse impacts on cell stability.
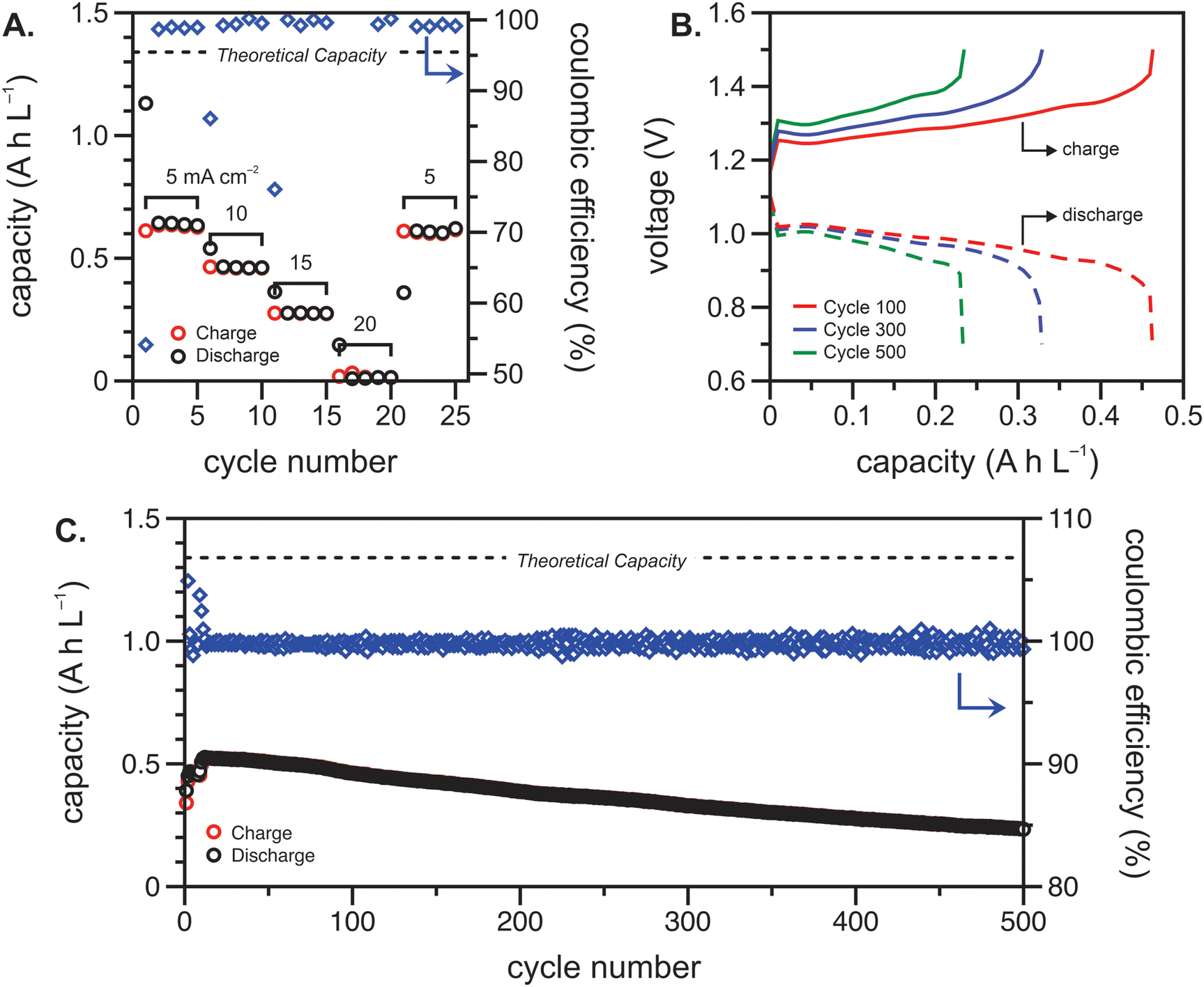 | ||
| Fig. 4 Cycling of the polypeptide flow cell in a potential window of 0.7 to 1.5 V. (A) Capacity and coulombic efficiency vs. cycle number for current densities of 5, 10, 15, 20 and 5 mA cm−2. (B) Galvanostatic charge/discharge curves taken at 100, 300, and 500 cycles. (C) Long-term cycling of the polypeptide flow cell. The current density for (B) and (C) was 10 mA cm−2. The cell consisted of 7.5 mL each of 50 mM (by repeat unit) viologen polypeptide anolyte and 25 mM (by repeat unit) biTEMPO polypeptide catholyte in 0.5 M TEATFSI in ACN. | ||
Constant current cycling was then performed to evaluate the long-term stability of the polypeptide flow cell at 10 mA cm−2 (Fig. 4(B) and (C)). This current density was chosen to balance experimental runtime and the impact of charge/discharge overpotential on the accessed capacity. The high Ohmic resistance and electrolyte concentration polarization led to a sharp change in voltage at low capacities, which could be prevented in future studies through increase active material concentration in the electrolyte (Fig. 4(B)). In this cycling experiment, the polypeptide flow cell had an initial capacity utilization of 0.39 A h L−1 (29% of the theoretical capacity) and reached its maximum of 0.53 A h L−1 (39% of the theoretical capacity) at cycle twelve (Fig. 4(C)). The initial increase in capacity from cycle one to twelve can be attributed to the electrochemical recovery of the nitroxide radical from over-oxidized groups introduced during synthesis.28 From this point on, the capacity faded to 93% and 60% of the original value after 250 and 500 cycles, with the capacity fading ca. 0.1% per cycle. Fig. 4(B) shows selected galvanostatic charge–discharge curves, in which voltage plateaus at about 1.25–1.3 V and 0.95–1.0 V were present for charging and discharging, respectively, showing the nature of the reaction remained essentially the same, albeit with some capacity fade. Nevertheless, Fig. 4(C) shows that the Coulombic efficiencies remained fairly high at >99.5%, which could be due to relatively high current density utilized (compared to the areal resistance).
After cycling, the separate electrolytes were analyzed using CV. If crossover were to occur, one would expect to observe redox peaks attributed to the biTEMPO polypeptide in the viologen polypeptide electrolyte and vice versa. Fig. 5(A) shows a CV of the cycled viologen polypeptide electrolyte, in which the viologen redox peaks decreased after cycling, suggesting some degradation of the redox-active species. Also, minimal crossover of the counter species (biTEMPO polypeptide) was observed, as evidenced by the very small redox peak at 0.4 V vs. ferrocene as an internal standard. Fig. 5(B) shows a CV of the cycled biTEMPO electrolyte, in which the biTEMPO redox peaks decreased after cycling, also suggesting some degradation of the redox-active species; no evidence of viologen crossover was observed in the biTEMPO electrolyte. Of note, Fig. 5(B) shows that the biTEMPO electrolyte exhibited a second redox event at a lower potential attributed to the reduction of the nitroxide radical to form the aminoxy anion, which is consistent with previous literature.28 Taken together, these findings are consistent with the initial crossover study in Fig. 2(A), in which the biTEMPO polypeptide exhibited small – but minimal – crossover relative to the viologen polypeptide. Also, CV indicates that the main mechanism of capacity fade may be degradation of the redox-active moieties, rather than crossover. One possible decomposition mechanism is the irreversible dimerization or disproportionation of the viologen pendant groups, which may be facilitated by helical polypeptide backbone forcing adjacent viologen groups in closer proximity.29,30 This irreversible dimerization has been demonstrated for other extended ring structure organic redox-active molecules.31 As the viologen polypeptide material becomes electrochemically inactive (due to irreversible dimer formation), there are more persistent oxoammonium cations (due to fewer electrons for reduction to the nitroxide radical), which could lead to adsorption (and additional deactivation) in the biTEMPO polypeptide catholyte, thus leading to a reduced peak intensity in CV after cycling.32
 | ||
| Fig. 5 Cyclic voltammetry at 100 mV s−1 of the (A) viologen polypeptide anolyte and (B) biTEMPO polypeptide catholyte before and after cycling of the polypeptide redox flow cell (arrows denote 10 μA). Glassy carbon was used as the working electrode, platinum wire as the counter electrode, un-fritted freshly anodized Ag/AgCl wire as the reference electrode, and ferrocene was used as internal reference. The supporting electrolyte was 0.5 M TEATFSI in ACN. | ||
To examine the nature of the polypeptides before and after cycling, circular dichroism (CD), and attenuated total reflectance-Fourier-transform infrared (ATR–FTIR) spectroscopy were performed on the electrolytes (ESI,‡ Fig. S6–S8). Before cycling, both the viologen and biTEMPO polypeptides exhibited α-helical configurations, as evidenced by minima at 208 and 222 nm in the CD spectra and signatures at 1650 and 1550 cm−1 in the ATR-FTIR spectra.33–35 After cycling, these signatures remained in both the CD and ATR-FTIR spectra, confirming that the α-helical backbone remained intact. However, these methods were not sufficient to directly infer the fate of the redox-active biTEMPO or viologen groups. This is because the signature from the backbone was large relative to the signature from the redox-active groups. However, we note that the ester linkages between the backbone and the redox active group appeared to remain intact, as evidenced by the relatively invariant signature at 1730 cm−1. In our prior work on solid polypeptide batteries, we also observed that the backbone and the ester groups remained intact after cycling.26 Taken together, indirect evidence from ATR-FTIR spectroscopy suggests that the polypeptide and the ester linkages are stable in electrochemical cycling, but the redox active groups themselves may be electrochemically degrading. Therefore, an important future area should be directed at selecting redox active moieties, especially for the anolyte, that are more stable in this flow cell environment.
Conclusions
In summary, we designed and tested an NAORFB with biTEMPO polypeptide catholyte and viologen polypeptide anolyte. The full cell exhibited an Ohmic resistance (ca. 2.5 Ω or 6.4 Ω cm2), low capacity fade (ca. 0.1% per cycle over 500 cycles), and a high Coulombic efficiency (>99.5%). The polypeptide NAORFBs exhibit an output voltage of 1.1 V with a capacity of 0.53 A h L−1 (39% of the theoretical capacity) at 10 mA cm−2. After 500 charge–discharge cycles, 60% of the initial capacity was retained. At a lower discharge current of 5 mA cm−2, the capacity increased to 59% of the theoretical capacity. Post cycling analysis using circular dichroism, ATR-FTIR spectroscopy, and cyclic voltammetry demonstrated that the polypeptides and the ester linkages were stable in electrochemical cycling. The slight, but noticeable, capacity fade was likely due to electrochemical degradation of the redox active groups.The polypeptide-containing NAORFB represents an early step toward developing large-scale energy storage with enhanced sustainability of active materials and alleviating global dependence on strategic metals. More work in the field should focus on the development of additional structured polymers, which could reduce the crossover without significantly increasing the viscosity. Future backbone design should strive to incorporate deconstructable functionalities, as presented here, to further enhance the “greenness” of RFBs, with special attention paid to milder synthetic approaches. As for the electrokinetics, our ongoing work indicates that a 10× improvement in the rate transfer is possible by using the α-helical backbone as opposed to random-coil. Additionally, alternative redox-active moieties with improved electrochemical stability and increased voltage separation should be considered to improve the cycling stability and output cell voltage. Finally, other side group modifications, such as the inclusion of electrochemically stable charged groups, should be considered to improve the solubility of the polypeptides, with the goal of reaching concentrations similar to that of small molecules.
Author contributions
Z. L. and T. P. N. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work by S. A. O., Z. L., and N. A. H. was supported by the National Science Foundation (NSF) under Award # ENG-3200001992. Work by K. L. W. and T. P. N. was supported by NSF under Award # DMR–1507429 and DMR–1905818, and by the Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry (A-0001). Work by J. L. L. and A. D. E. was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES) under Award # DE-SC0014006.References
- Y. Y. Lai, X. Li and Y. Zhu, ACS Appl. Polym. Mater., 2020, 2, 113–128 CrossRef CAS.
- F. Zhong, M. Yang, M. Ding and C. Jia, Front. Chem., 2020, 8, 451 CrossRef CAS PubMed.
- A. Parasuraman, T. M. Lim, C. Menictas and M. Skyllas-Kazacos, Electrochim. Acta, 2013, 101, 27–40 CrossRef CAS.
- G. Kear, A. A. Shah and F. C. Walsh, Int. J. Energy Res., 2012, 36, 1105–1120 CrossRef CAS.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS.
- F. Pan and Q. Wang, Molecules, 2015, 20, 20499–20517 CrossRef CAS PubMed.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 CrossRef CAS.
- W. Liu, W. Lu, H. Zhang and X. Li, Chem. – Eur. J., 2019, 25, 1649–1664 CrossRef CAS PubMed.
- G. C. Y.-C. L. Hongning Chen, J. Energy Chem., 2018, 27, 1304–1325 CrossRef.
- S. Gentil, D. Reynard and H. H. Girault, Curr. Opin. Electrochem., 2020, 21, 7–13 CrossRef CAS.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Energy Environ. Sci., 2015, 8, 3515–3530 RSC.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- E. C. Montoto, G. Nagarjuna, J. S. Moore and J. Rodríguez-López, J. Electrochem. Soc., 2017, 164, A1688–A1694 CrossRef CAS.
- G. Hernández, M. Işik, D. Mantione, A. Pendashteh, P. Navalpotro, D. Shanmukaraj, R. Marcilla and D. Mecerreyes, J. Mater. Chem. A, 2017, 5, 16231–16240 RSC.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J.-F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- A. Vlad, K. Arnould, B. Ernould, L. Sieuw, J. Rolland and J.-F. Gohy, J. Mater. Chem. A, 2015, 3, 11189–11193 RSC.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- C. Wang, W. Zheng, Z. Yue, C. O. Too and G. G. Wallace, Adv. Mater., 2011, 23, 3580–3584 CrossRef CAS PubMed.
- J. Yuan, Z.-Z. Pan, Y. Jin, Q. Qiu, C. Zhang, Y. Zhao and Y. Li, J. Power Sources, 2021, 500, 229983 CrossRef CAS.
- E. C. Montoto, Y. Cao, K. Hernández-Burgos, C. S. Sevov, M. N. Braten, B. A. Helms, J. S. Moore and J. Rodríguez-López, Macromolecules, 2018, 51, 3539–3546 CrossRef CAS.
- S.-H. Shin, S.-H. Yun and S.-H. Moon, RSC Adv., 2013, 3, 9095–9116 RSC.
- Z. Liang, N. H. Attanayake, K. V. Greco, B. J. Neyhouse, J. L. Barton, A. P. Kaur, W. L. Eubanks, F. R. Brushett, J. Landon and S. A. Odom, ACS Appl. Energy Mater., 2021, 4, 5443–5451 CrossRef CAS.
- K. H. Hendriks, S. G. Robinson, M. N. Braten, C. S. Sevov, B. A. Helms, M. S. Sigman, S. D. Minteer and M. S. Sanford, ACS Cent. Sci., 2018, 4, 189–196 CrossRef CAS PubMed.
- G. Nagarjuna, J. Hui, K. J. Cheng, T. Lichtenstein, M. Shen, J. S. Moore and J. Rodríguez-López, J. Am. Chem. Soc., 2014, 136, 16309–16316 CrossRef CAS PubMed.
- T. P. Nguyen, A. D. Easley, N. Kang, S. Khan, S.-M. Lim, Y. H. Rezenom, S. Wang, D. K. Tran, J. Fan, R. A. Letteri, X. He, L. Su, C.-H. Yu, J. L. Lutkenhaus and K. L. Wooley, Nature, 2021, 593, 61–66 CrossRef CAS PubMed.
- J. Yuan, C. Zhang, Y. Zhen, Y. Zhao and Y. Li, J. Power Sources, 2019, 443, 227283 CrossRef CAS.
- Q. Huang, E. D. Walter, L. Cosimbescu, D. Choi and J. P. Lemmon, J. Power Sources, 2016, 306, 812–816 CrossRef CAS.
- V. Singh, S. Kim, J. Kang and H. R. Byon, Nano Res., 2019, 12, 1988–2001 CrossRef CAS.
- C. Kahlfuss, E. Métay, M.-C. Duclos, M. Lemaire, M. Oltean, A. Milet, É. Saint-Aman and C. Bucher, C. R. Chim, 2014, 17, 505–511 CrossRef CAS.
- M.-A. Goulet, L. Tong, D. A. Pollack, D. P. Tabor, S. A. Odom, A. Aspuru-Guzik, E. E. Kwan, R. G. Gordon and M. J. Aziz, J. Am. Chem. Soc., 2019, 141, 8014–8019 CrossRef CAS PubMed.
- H. Qian, M. J. Counihan, H. A. Doan, N. A. Ibrahim, A. S. Danis, W. Setwipatanachai, N. S. Purwanto, J. Rodríguez-López, R. S. Assary and J. S. Moore, J. Mater. Chem. A, 2022, 10, 7739–7753 RSC.
- T. J. Deming, Nat. Mater., 2010, 9, 535–536 CrossRef CAS PubMed.
- A. Lalatsa, A. G. Schätzlein, M. Mazza, T. B. H. Le and I. F. Uchegbu, J. Controlled Release, 2012, 161, 523–536 CrossRef CAS PubMed.
- D. Bairagi, P. Biswas, K. Basu, S. Hazra, D. Hermida-Merino, D. K. Sinha, I. W. Hamley and A. Banerjee, ACS Appl. Bio Mater., 2019, 2, 5235–5244 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated in honour and memory of Professor Susan Odom, our friend and dear colleague. |
| ‡ Electronic supplementary information (ESI) available: Chemical structures, experimental H-cell images, crossover data, CD and IR spectra. See DOI: https://doi.org/10.1039/d2ma00498d |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |