
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-way CO2-responsive dispersions of carbon nanotubes in water†
Ting-Yi
Hsin
,
Vladislav Y.
Shevtsov
and
Yeong-Tarng
Shieh
 *
*
Department of Chemical and Materials Engineering, National University of Kaohsiung, Kaohsiung 81148, Taiwan. E-mail: yts@nuk.edu.tw
First published on 12th July 2022
Abstract
The “grafting to” method was used to functionalize carbon nanotubes (CNTs) with poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) by applying an atom transfer radical reaction. The obtained CNT-g-PDMAEMA was subjected to consecutive quaternization and alkaline hydrolysis to transform PDMAEMA on the surface of CNTs into poly(methacrylic acid) (PMAA). It was found that both CNT-g-PDMAEMA and CNT-g-PMAA could be easily dispersed in an aqueous media and also exhibited the pH- and CO2-responsive behavior. While CNT-g-PDMAEMA remained stably dispersed in an acidic environment and readily precipitated at high pH values, CNT-g-PMAA displayed a reversed direction of responsiveness, staying dispersed in the basic environment and aggregating when the solution's pH was lowered. Most importantly, we demonstrated that CNTs grafted with PDMAEMA could undergo CO2-triggered redispersion, and on the other hand, CNTs grafted with PMAA could be easily switched to the agglomerated state by bubbling CO2 in the basic medium. Elimination of CO2 from the solutions by purging with an inert gas reverted the original appearance of CNTs. Thus, a unique two-way gas responsive system of manipulating CNT dispersibility was created through a sequence of simple chemical modifications. Furthermore, because CNTs acquired the ability to respond to the CO2 trigger, the addition of acids and bases to the solutions to cause coagulation was no longer required, and as a result, the accumulation of salts in the system could be prevented; that altogether establishes a green and environmentally friendly way of manipulating CNT dispersions.
Introduction
Since the early 1990s, carbon nanotubes (CNTs) have become a source of fascination for scientists due to their astonishing toughness and electrical conductivity in combination with the nanoscale size, low density, and high specific surface area.1–5 Achieving stable dispersions of CNTs in a liquid medium is, however, a major challenge that has been addressed by many researchers6–10 and, if accomplished, can facilitate cutting-edge breakthroughs in biochemistry and materials science. Strong π–π interactions between nanotubes caused by van der Waals forces dictate their bundle-like structured appearance, making CNTs practically insoluble in all known common solvents. Therefore, various strategies were developed over the years that aimed to improve the stability and solubility of CNTs. The use of ultrasonic, plasma, and other physical techniques, surfactant-assisted suspension, chemical functionalizations, and many other methods of debundling have already been summarized and extensively discussed by several reviewers.11–13In the last decade, rising environmental awareness in the fields of nanotechnology and polymer chemistry has stimulated investigations related to eco-friendly, “green” ways of dispersing CNTs.14,15 Perhaps the most intriguing innovative approach, which follows this trend, is the utilization of stimuli-responsive polymers. This class of polymers has recently attracted a great deal of interest due to their ability to undergo self-assembly, phase change, or morphological change in response to the stimulations caused by physical or chemical changes in the external environment.
While temperature and pH remain the two most popular stimuli,16 carbon dioxide (CO2), a novel type triggering agent, has received special attention in recent years.17–20 When CO2 dissolves in water, it forms carbonic acid, which has a pKa1 value of 6.33 at 30 °C,21 making it a weak acid. Thus, the addition of CO2 in an aqueous medium lowers its pH, meaning that protonation of amino groups and carboxylate groups can be achieved through a CO2-stimulus avoiding the use of traditional pH-stimulus agents (i.e., hydrochloric acid). Furthermore, if the process is followed by bubbling an inert gas (e.g., N2, Ar), the original functional groups will be recovered through deprotonation due to the removal of CO2 from the aqueous solution. The cycles of introducing and “kicking out” CO2 can be repeated many times resulting in the switchable behavior of water-soluble amine and carboxylate groups-containing polymers. Although the pH-lowering ability of this method is limited due to the weakness of carbonic acid, the use of CO2 is a lot cheaper and safer because it is abundant, nontoxic, and also allows avoiding the accumulation of salts in the system (in comparison to using acids and bases for protonation–deprotonation in conventional pH-triggered manipulations).
Guo et al. reported using a pyrene-labeled amidine-based polymer as a CO2-switchable dispersant that could suspend single-walled CNTs (SWCNTs) in water.22 It was shown that π–π stacking between the CNT surface and the incorporated pyrene allows the aggregation and redispersion of CNTs with the help of CO2 and N2 gas, respectively. Dubois' team adopted a similar approach of de-agglomeration of CNTs, but in their study, instead of the amidine-containing agent, they used another popular multi-stimuli-responsive polymer – poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA).23 It was observed that PDMAEMA was capable of wrapping around multi-walled CNTs (MWCNTs) due to the non-covalent CH-π interactions and thus dispersing MWCNTs in the acidic environment where tertiary amine groups exist in the protonated charged state. Functionalization of CNTs with CO2-responsive polymers is another common solubilization technique. In this scenario, scientists often implement a “grafting from” strategy of attaching polymers to CNTs using surface-initiated atom transfer radical polymerization (SI-ATRP).24 For instance, Kim and coworkers demonstrated the ease of performing the “grafting from” approach by first functionalizing MWCNTs with bromine.25 This method was shown to be an effective tool for enhancing the water solubility of CNTs by their functionalization with PDMAEMA. Additionally, researchers were successful in quaternizing grafted PDMAEMA using bromoethane and obtaining CNT-g-qPDMAEMA, which displayed explicit antibacterial action against Escherichia coli and Staphylococcus aureus. Abraham et al. followed the same synthesis path and grafted poly[2-(diethylamino)ethyl methacrylate] (PDEAEMA) onto MWCNTs.26 The fabricated gas-responsive CNT-g-PDEAEMA membrane showed promising results in oil/water and oil-in-water emulsion separation with a high (>92%) demulsification efficiency. The membrane's reversible surface wettability was achieved by switching the state of PDEAEMA to hydrophobic and back to hydrophilic via treatment with N2 and CO2 gases, which helped to prevent the membrane clogging in the case of a long-term use.
Earlier, in our laboratory, we succeeded in attaching carboxymethyl chitosan (CMC) to COOH-functionalized CNTs by performing a “grafting to” reaction.27 We found that aggregation and redispersion of obtained CNT-g-CMCs in water could be reversibly manipulated by bubbling CO2/N2 and depended on the relative amounts of COOH to NH2 in CMC. In this study, we report that PDMAEMA synthesized via ATRP using a methyl α-bromoisobutyrate (MBIB) initiator can act as a graft-initiating species due to the possession of C–Br chain ends. This feature was used to graft PDMAEMA directly onto the surface of MWCNTs by an atom transfer radical reaction with the carbon–carbon double bonds of CNTs. The prepared CNT-g-PDMAEMA not only could be easily stabilized in water with the help of ultrasonication but also exhibited pH-, temperature-, and CO2-responsive behavior. Afterward, we subjected PDMAEMA grafted to CNTs to quaternization with iodomethane resulting in the preparation of CNT-g-qPDMAEMA, which made the CNT aqueous dispersion highly stable in a wide pH range. Moreover, after quaternization, it became possible to transform qPDMAEMA on the surface of CNTs into poly(methacrylic acid) (PMAA) via an alkaline hydrolysis reaction. Dispersion of CNT-g-PMAA obtained in this manner displayed reversed (compared to CNT-g-PDMAEMA) pH and CO2 sensitivity. The influence of the molecular weight of grafted polymer on the stimuli-responsive properties of the dispersions was also investigated. The developed sequence of functionalization reactions provides a simple alternative way of creating a whole class of “smart” pH- and gas-switchable aqueous CNT dispersions.
Experimental
Materials and characterizations
Chemicals, instruments, and characterizations are described in detail in the ESI.†Synthesis and procedures
ATRP of PDMAEMA-h and PDMAEMA-l. For the preparation of the relatively high molecular weight PDMAEMA (PDMAEMA-h), the CuBr (0.13 g, 9 × 10−4 mol) catalyst and PMDETA (0.31 g, 1.8 × 10−3 mol) ligand were dissolved in 36 mL of EtOH/H2O (1/1 v/v) mixture in a four-neck flask. The vessel was degassed via two freeze–pump–thaw nitrogen cycles, followed by injection of the DMAEMA monomer (14 g, 0.09 mol) and an additional double degassing procedure was done. The MBIB (0.161 g) initiator was added to the mixture via a syringe to initiate ATRP at room temperature (25 °C). The reactor was agitated for 72 h using a magnetic stir bar. The molar ratio of DMAEMA/CuBr/PMDETA/MBIB was 100/1/2/1. The relatively low molecular weight PDMAEMA (PDMAEMA-l) was prepared in an identical manner. The only difference was that the amount of added MBIB increased to 0.483 g, corresponding to the DMAEMA/CuBr/PMDETA/MBIB molar ratio of 100/1/2/3. The reaction products were purified by dialysis in a membrane bag (molecular weight cut off: 6000–8000 g mol−1 for PDMAEMA-h and 3500 g mol−1 for PDMAEMA-l) against deionized water for 3 d to completely remove unreacted DMAEMA, copper ions, and PMDETA. Finally, the solid polymer products were obtained by drying in a vacuum oven for 24 h. The molecular weights of PDMAEMA-h and PDMAEMA-l were determined to be 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 and 5200 g mol−1, respectively, using 1H-NMR spectroscopic analysis. 1H NMR (600 MHz, CDCl3, δ ppm): 0.89–1.05 (C–CH3), 1.25 (C–(CH3)3), 1.82–1.90 (CH2), 2.34 (N–(CH3)2), 2.60 (N–CH2) and 4.06 (O–CH2).
600 and 5200 g mol−1, respectively, using 1H-NMR spectroscopic analysis. 1H NMR (600 MHz, CDCl3, δ ppm): 0.89–1.05 (C–CH3), 1.25 (C–(CH3)3), 1.82–1.90 (CH2), 2.34 (N–(CH3)2), 2.60 (N–CH2) and 4.06 (O–CH2).
Hydrolysis of qPDMAEMA . Conversion of qPDMAEMA-h and qPDMAEMA-l into PMAA-h and PMAA-l respectively was performed by first dissolving the samples (0.1 g) in 1 M NaOH aqueous solutions (10 mL). The hydrolysis reaction was carried out at 90 °C using magnetic stirring. After 24 h, the mixtures were cooled down to room temperature, transferred into the vials, and left alone for 1 h. In order to ensure that the newly formed carboxylic acid groups were protonated, small amounts of 1 N HCl aqueous solution were added carefully to each vial until the pH values of these solutions reached 4. The residual HCl was removed by means of freeze-drying which took approximately 1 d. Then the resulting dry polymer powders were redissolved in DI water and dialyzed for 3 d in the membrane bags (molecular weight cut off: 6000–8000 g mol−1 for PMAA-h and 3500 g mol−1 for PMAA-l). Lastly, purified products were put in a vacuum oven to dry overnight, obtaining pure PMAA end-products. 1H NMR (600 MHz, D2O, δ ppm): 0.95–1.25 (C–CH3) and 1.78–2.05 (CH2).
The procedures of PDMAEMA synthesis via ATRP, quaternization of PDMAEMA to obtain qPDMAEMA, and alkaline hydrolysis of qPDMAEMA resulting in the formation of PMAA, are summarized in Scheme 1.
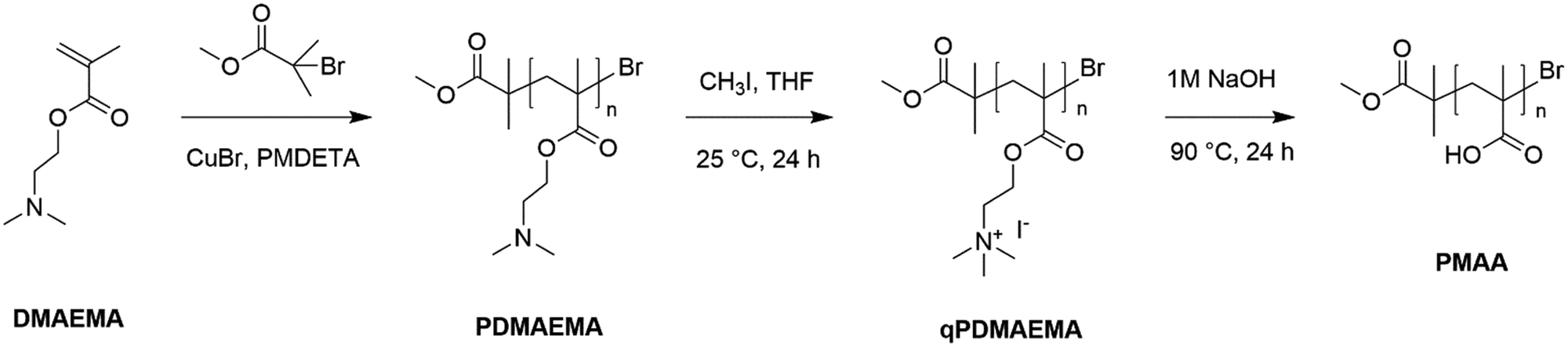 | ||
| Scheme 1 Preparation of PDMAEMA, qPDMAEMA, and PMAA. | ||
Preparation of CNT- g -PDMAEMA, CNT- g -qPDMAEMA, and CNT- g -PMAA . The synthesis was conducted as follows: CuBr (0.13 g, 9 × 10−4 mol), PMDETA (0.31 g, 1.8 × 10−3 mol), and MWCNT (0.3 g) were mixed in 10 mL of EtOH/H2O (1/1 v/v) inside a four-neck round-bottom flask equipped with a magnetic stirring bar, which was then degassed via two freeze–pump–thaw nitrogen cycles. The pre-dissolved PDMAEMA-Br (1 g) in 10 mL of EtOH/H2O (1/1 v/v) was lastly injected into the reactor. Two freeze–pump–thaw nitrogen cycles were repeated to ensure that the oxygen was eliminated entirely from the system. The grafting reaction was initiated by heating the mixture up to 60 °C and left to stir for 2.5 d. The product was separated by filtration, washed with ethanol several times to remove the ungrafted PDMAEMA, and dried in a vacuum oven at 60 °C for 1 d to obtain CNT-g-PDMAEMA powders (CNT-g-PDMAEMA-h and CNT-g-PDMAEMA-l). The yield was 29.5 wt%.
To prepare CNT-g-qPDMAEMA, CNT-g-PDMAEMA (0.1 g) was dispersed in 18 mL of THF. The solution of methyl iodide (1.1 g) in THF (5 mL) was slowly added to the CNT-g-PDMAEMA dispersion while being constantly stirred. Quaternization was conducted at room temperature, and after reacting for 1 d, the mixture was poured into a 10-fold amount of n-hexane to precipitate the product. CNT-g-qPDMAEMA-h and CNT-g-qPDMAEMA-l were separated and dried in a vacuum oven overnight.
The CNT-g-qPDMAEMA (0.05 g) was dispersed in 10 mL of 1 M NaOH aqueous solution, heated to 90 °C, and left to stir for 1 d to hydrolyze it into CNT-g-PMAA. The dispersion was cooled down to room temperature, transferred in a vial, and left alone for 1 h. Small amounts of 1 N HCl aqueous solution were added carefully to the vial to lower the pH to 4. Residual HCl was removed by means of freeze-drying which took approximately 1 d. Then the resulting dry powder was redissolved in DI water and dialyzed for 3 d in the membrane bag (molecular weight cut off: 3500 g mol−1). After drying in a vacuum oven overnight, CNT-g-PMAA-h and CNT-g-PMAA-l products were obtained.
The preparation of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA are summarized in Scheme 2.
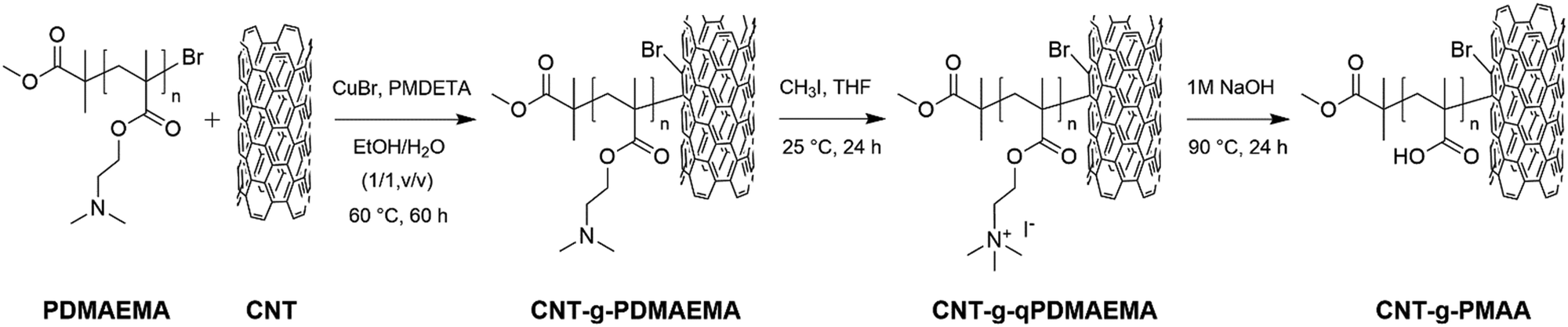 | ||
| Scheme 2 Preparation of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA. | ||
pH-responsiveness of CNT- g -PDMAEMA, CNT- g -qPDMAEMA, and CNT- g -PMAA dispersions . The as-synthesized CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA were ultrasonicated in DI water to prepare dispersions of 0.02 wt% concentrations. The pH of each dispersion was adjusted to pH 4, 7, and 10 at 25 °C using 1 N HCl and 1 N NaOH aqueous solutions to record the phase changes in response to varying pH.
CO 2 -responsiveness of CNT- g -PDMAEMA, CNT- g -qPDMAEMA, and CNT- g -PMAA dispersions . The 0.05 wt% aqueous CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA dispersions were prepared by ultrasonication and adjusted to pH 10 using 1 N NaOH solution. Samples were transferred into the 7 mL vials (5.5 cm height × 1.5 cm diameter) and subjected to bubbling with CO2, followed by bubbling with N2. The temperature of the dispersions was maintained constant (30 °C) throughout all the experiments, and the gase flow rates were set to 100 mL min−1. The phase changes in response to CO2/N2 treatment were recorded 10 min after each CO2 and N2 purging using a digital camera.
Results and discussion
Transformation of PDMAEMA into qPDMAEMA and into PMAA
In our previous report, we comprehensively investigated a sequence of two reactions that allowed the transformation of PDMAEMA into PMAA.28 It was demonstrated that the PDMAEMA, which was highly stable to hydrolysis, if first quaternized into qPDMAEMA using methyl iodide, could be easily converted into PMAA via alkaline hydrolysis. The procedures of PDMAEMA synthesis via ATRP and its consecutive quaternization and hydrolysis were reproduced as previously described. The obtained products were characterized using 1H-NMR (Fig. S1, ESI†) and FT-IR (Fig. S2, ESI†) spectroscopic methods (see the ESI†). Additionally, the temperature-responsive behavior of PDMAEMA and qPDMAEMA were compared in aqueous media. As can be seen in Fig. 1, quaternization of tertiary amine groups in PDMAEMA eliminates its lower critical solution temperature (LCST). PDMAEMA-h reached the cloud point after being heated to 40 °C, whereas qPDMAEMA-h remained completely soluble in water at even higher temperatures (60 °C). The increased water solubility of qPDMAEMA was caused by the formation of highly hydrophilic quaternary ammonium groups that were formed in the polymer sidechains. We reasonably hypothesized that PDMAEMA grafted to CNTs could undergo the same transformations via suggested reactions, thus modifying the functionalization of CNT surface. | ||
| Fig. 1 Temperature-responsive behavior of PDMAEMA-h and qPDMAEMA-h. The 0.05 wt% aqueous solutions of polymers were heated from 25 °C to 40 °C, 50 °C, and 60 °C. | ||
Characterizations of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA
The “grafting to” technique described by Liu et al. was slightly modified and used to graft PDMAEMA directly onto the surface of CNTs.29 The PDMAEMA homopolymer, synthesized by ATRP as described above, carries C–Br chain ends, and therefore is capable of performing the addition and halide atom transfer reaction in the presence of the CuBr catalyst and the PMDETA ligand. Thereby PDMAEMA-Br was covalently incorporated into the CNT surface via the reaction with its –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bonds and bromide atoms transfer, which resulted in formation of CNT-g-PDMAEMA.
C– bonds and bromide atoms transfer, which resulted in formation of CNT-g-PDMAEMA.
Thermal gravimetric analysis (TGA) was used to estimate the amounts of PDMAEMA grafted to CNTs. Fig. 2 shows TGA curves of pristine CNTs, CNT-g-PMAA-l, CNT-g-PDMAEMA-l, CNT-g-PDMAEMA-h, and pristine PDMAEMA-h. The grafting amounts were determined by subtraction of the residual weight of CNT-g-PDMAEMA-h and CNT-g-PDMAEMA-l from that of pure CNTs at 550 °C. The amount of grafted PDMAEMA-h was calculated to be roughly three times greater than the amount of grafted PDMAEMA-l: 36.9 wt% and 10.7 wt%, respectively (Table 1). Taking into account the data obtained by 1H NMR, which showed that the molecular weight of PDMAEMA-h is approximately three times higher than the molecular weight of PDMAEMA-l, it was concluded that the grafting densities of two PDMAEMA samples on CNTs were approximately the same. CNT-g-PMAA-l lost less weight than CNT-g-PDMAENA-l due to the sidechain cleavage caused by hydrolysis. Additionally, Raman spectroscopic analysis was performed to further support the results of TGA. The recorded Raman spectra are displayed in Fig. S3 (ESI†).
 | ||
| Fig. 2 TGA curves of (a) CNTs, (b) CNT-g-PMAA-l, (c) CNT-g-PDMAEMA-l, (d) CNT-g-PDMAEMA-h, and (e) PDMAEMA. | ||
| Samples | 550 °C (wt%) | Grafting amount (wt%) |
|---|---|---|
| CNTs | 98.5 | — |
| CNT-g-PDMAEMA-l | 87.8 | 10.7 |
| CNT-g-PDMAEMA-h | 61.6 | 36.9 |
Electron spectroscopy for chemical analysis (ESCA) was employed to judge whether the conversion of CNT-g-PDMAEMA into CNT-g-PMAA through subsequent quaternization and hydrolysis was achieved. Fig. 3 represents the whole ESCA spectra of CNTs, CNT-g-PDMAEMA, and CNT-g-PMAA. All three samples exhibited C1s peaks at 284 eV and O1s peaks at 533 eV. The N1s signal rose at 402 eV on the CNT-g-PDMAEMA spectrum corresponding to nitrogen atoms of tertiary amine groups. This signal, however, did not appear on the CNT-g-PMAA spectrum, presumably due to the degradation of PDMAEMA. The compositions of C, N, and O on the surface of analyzed samples obtained from Fig. 3 are given in Table 2. An increase in the O content for CNT-g-PMAA indicates the successful transformation of PDMAEMA into PMAA on the surface of CNTs.
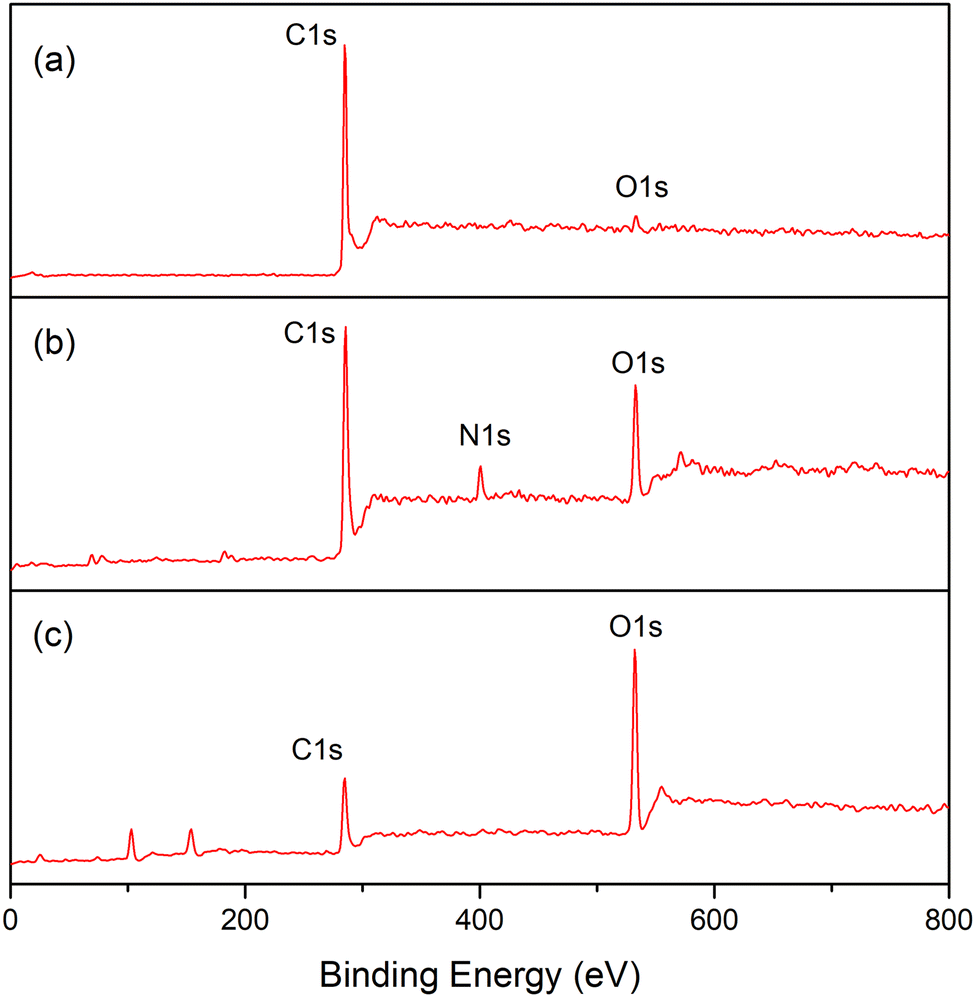 | ||
| Fig. 3 ESCA spectra of (a) CNT, (b) CNT-g-PDMAEMA-h, and (c) CNT-g-PMAA. | ||
| Samples | Content (%) | ||
|---|---|---|---|
| C | N | O | |
| CNT | 98 | 0 | 2 |
| CNT-g-PDMAEMA-h | 78 | 5 | 17 |
| CNT-g-PMAA | 49 | 0 | 51 |
Scanning electron microscopy (SEM) was used to observe the morphology of prepared structures. SEM images of CNTs, CNT-g-PDMAEMA, and CNT-g-PMAA are shown in Fig. 4. The surface of pristine CNTs looked smooth, while the texture of CNT-g-PDMAEMA appeared rougher. The surface of CNT-g-PMAA was coarse, most likely due to the swelling of PMAA.
 | ||
| Fig. 4 SEM images of (a) CNTs, (b) CNT-g-PDMAEMA, and (c) CNT-g-PMAA powders. | ||
Moreover, with the help of SEM, it was found that the morphology of CNT-g-PMAA films cast from the aqueous dispersions varied in response to the changes in dispersions' pH. As shown in Fig. 5, the film cast from CNT-g-PMAA dispersion of pH 4 consisted of nanotubes that looked sticky, glued together. PMAA reportedly has an average pKa of 6.0,30 at pH 4 carboxylic acid groups in PMAA primarily exist in the protonated state. The agglutinated morphology on the left image can thus be attributed to the presence of intermolecular hydrogen bonding between COOH groups in CNT-g-PMAA. This observation correlates well with the visual appearance of the dispersion from which the sample film was cast: precipitation of CNT-g-PMAA occurred at pH 4 in the vial, as can be seen in the photograph displayed at the upper right corner of the SEM image. At pH 7 the concentration of COO− side groups is higher than that of COOH groups, and the excess of hydrophilic COO− groups could lead to expulsion among CNTs, and thus the creation of a uniform layer of discrete nanotubes. At pH 10 the COO− state of carboxylic acid groups in PMAA dominates; we assume that in this case, the tremendous expulsive forces could cause the separated morphology of CNT-g-PMAA particles, as can be seen on the right image in Fig. 5.
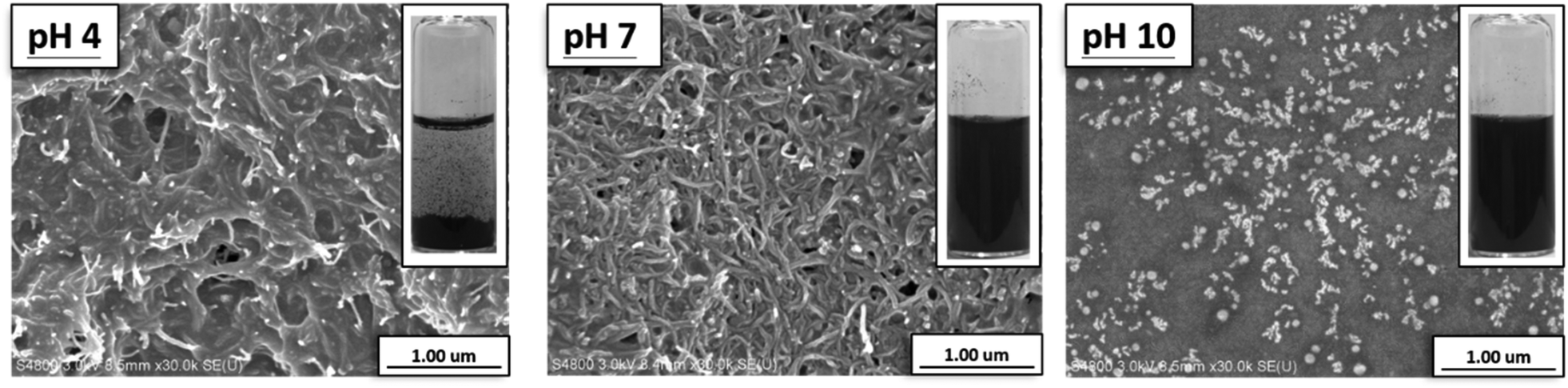 | ||
| Fig. 5 SEM images of CNT-g-PMAA films cast from the 0.01 wt% aqueous dispersions of pH 4, pH 7, and pH 10. A vial at the upper right corner of each image shows the 0.01 wt% aqueous dispersion before the film casting. | ||
pH-Responsiveness of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA dispersions
Since PDMAEMA is a pH- and temperature-responsive polymer, the dispersion of CNT-g-PDMAEMA could be expected to respond to a change in pH and temperature, as shown in Fig. 6. The aqueous solution of pH 4 containing 0.02 wt% CNT-g-PDMAEMA was observed to remain homogeneous after heating up to 60 °C. The 0.02 wt% CNT-g-PDMAEMA dispersion in water of pH 7 started to coagulate at 60 °C. At pH 10, however, the dispersion began to coagulate at temperature as low as 25 °C, clearly aggregated at 50 °C, and turned to completely precipitate at 55 and 60 °C. | ||
| Fig. 6 The dispersions of CNT-g-PDMAEMA (0.02 wt%) in water of pH 4, pH 7, and pH 10 at various temperatures (25, 50, 55, and 60 °C). The samples were dispersed in water by means of 20 min ultrasonic vibration. | ||
Fig. 7 shows aqueous dispersions of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA of different pH values at 25 °C. As can be seen, an increase in the environmental pH led to the destabilization of CNT-g-PDMAEMA. PDMAEMA shows an average pKa of about 7.4.31 At pH 4, most tertiary amine groups in PDMAEMA are protonated, which facilitates its hydrophilic nature and thus the dissolution of the polymer in water. At pH 7, which is very close to pKa of PDMAEMA, the concentrations of the protonated and deprotonated tertiary amine side groups are about the same. The dispersion of CNT-g-PDMAEMA was not very stable at 25 °C, and thus slight precipitation could be seen at the bottom of the vial 24 h after ultrasonication. At pH 10, which is much higher than pKa of PDMAEMA, the concentration of the hydrophilic protonated tertiary amine side groups is much less than that of the hydrophobic deprotonated groups; consequently, significant precipitation of the dispersion occurred immediately after the ultrasonication.
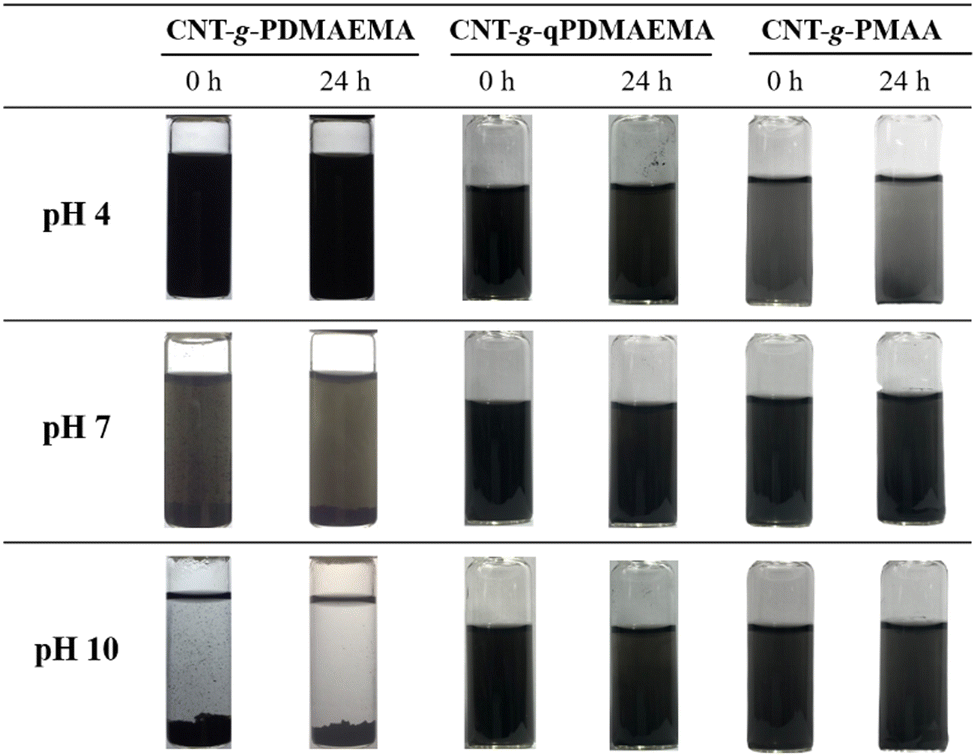 | ||
| Fig. 7 The dispersions of CNT-g-PDMAEMA, CNT-g-qPDMAEMA, and CNT-g-PMAA of 0.02 wt% in water of pH 4, 7, and 10 at 25 °C. These photos were taken immediately or 24 h after 20 min of ultrasonic vibration. | ||
Quaternization of PDMAEMA leads to the formation of positively charged and highly hydrophilic quaternary ammonium groups in polymer sidechains. Therefore, the dispersion of CNT-g-qPDMAEMA in water stayed homogeneous in the pH 4–10 range, not displaying pH-responsive behavior. This highly stable, pH-independent aqueous dispersion can be suggested for use in situations when the antibacterial activity of dispersion is an important criterion because polyquats are known to possess an effective microbe-killing ability.
As previously discussed, the CNT-g-PMAA aqueous dispersion demonstrated pH-responsive behavior (Fig. 5), providing reduced adhesion among CNTs at high pH. From Fig. 7 it is clear that CNT-g-PMAA was not stable in water at 25 °C at pH 4 because this value is less than pKa of PMAA. At pH 7, which is higher than pKa of PMAA, the concentration of the hydrophilic COO− groups is greater than relatively hydrophobic COOH groups, resulting in a stable dispersion of CNTs up to at least 24 h. At pH 10, the concentration of COO− groups is way higher than that of the hydrophobic COOH groups. Predictably, the dispersion of CNT-g-PMAA was very stable in water of pH 10.
As concluded from Fig. 7, the dispersion of 0.02 wt% CNT-g-PDMAEMA was seen to decrease with increasing pH from 4 to 10, whereas that of the CNT-g-PMAA was seen to increase with increasing pH. The change from the acidic-pH-caused dispersion of CNTs to the basic-pH-caused dispersion of the identical CNTs was a consequence of quaternization and followed hydrolysis of the tertiary amine groups in PDMAEMA grafted to CNTs.
CO2-Responsiveness of CNT-g-PDMAEMA and CNT-g-PMAA dispersions
Fig. 8 shows the responses of 0.05 wt% aqueous dispersions of CNT-g-PDMAEMA-h, CNT-g-PDMAEMA-l, CNT-g-PMAA-h, and CNT-g-PMAA-l at pH 10 and 30 °C to 1 min CO2 bubbling followed by N2 bubbling. At pH 10, CNT-g-PDMAEMA-l appeared noticeably more precipitated than CNT-g-PDMAEMA-h, which indicated that given concentration of OH− was able to provide only partial deprotonation of the tertiary amine groups in PDMAEMA-h due to the high molecular weight of this polymer. In contrast, both CNT-g-PMAA-h and CNT-g-PMAA-l at pH 10 looked homogeneously dispersed without a distinctive difference between the two samples; the presence of strongly hydrophilic COO− groups on CNTs in sufficient concentration was evident. | ||
| Fig. 8 Responses of 0.05 wt% aqueous dispersions of CNT-g-PDMAEMA-h, CNT-g-PDMAEMA-l, CNT-g-PMAA-h, and CNT-g-PMAA-l at 30 °C and adjusted to pH 10 to CO2/N2 bubbling for an indicated time. The flow rates of both CO2 and N2 gases were 100 mL min−1 and remained constant throughout the additions. The photos were taken 10 min after each CO2 or N2 bubbling. | ||
Treatment with CO2 for 1 min led to pH reduction from 10 to 4.9 for both CNT-g-PDMAEMA-l and CNT-g-PMAA-l samples, causing redispersion of CNT-g-PDMAEMA-l and aggregation of CNT-g-PMAA-l. Apparently, the dissolution of CO2 in water causes the formation of carbonic acid that subsequently releases protons to convert tertiary amine groups and carboxylate anions into hydrophilic ammonium cations and hydrophobic neutral COOH groups, respectively. Meanwhile, the pH values of CNT-g-PDMAEMA-h and CNT-g-PMAA-h dispersions dropped to 5.1 and 5.3, respectively. Although CO2 bubbling did deagglomerate CNT-g-PDMAEMA-h, it was not able to coagulate CNT-g-PMAA-h due to the lack of protonating capacity of carbonic acid formed in water, considering the high concentration of COO− in PMAA-h. Longer CO2 addition (i.e., 30 min) did not reduce pH any further significantly, and did not cause observable precipitation, meaning that the molecular weight of PMAA needs to be limited to a certain value in order to perform CO2-triggered manipulations with CNTs via the suggested method. Aggregation of this sample could only be achieved by lowering the pH to 4 via the addition of HClaq.
Removal of CO2 from the dispersions by 5 min N2 bubbling showed to be an effective stimulus for CNT-g-PDMAEMA-h and CNT-g-PDMAEMA-l reaggregation, which was accompanied by pH increase to 8.2–8.5. For the CNT-g-PMAA-l sample, however, the N2 trigger worked the other way around, stimulating the redispersion of CNTs by raising the pH of their environment to 8.7. The only downside of this process was that it required longer N2 bubbling (i.e., 30 min).
Conclusions
In summary, a set of two-way CO2-responsive CNTs dispersions was developed based on the chemical functionalization of CNTs with multi-stimuli-responsive polymers. It was shown that PDMAEMA synthesized via ATRP could act as a graft-initiating species, and that allowed synthesis of pH-, temperature-, and CO2-responsive CNT-g-PDMAEMA aqueous dispersions by a simple and effective “grafting to” procedure. Sequential quaternization and alkaline hydrolysis of these products resulted in the transformation of tertiary amine groups-bearing PDMAEMA into carboxylic acid groups-bearing PMAA on the CNT surface. The CNT-g-qPDMAEMA intermediates did not show any stimulus-responsive ability; however, these aqueous dispersions appeared to be highly stable in a wide pH range, and due to the great antimicrobial effect of quaternary ammonium groups, can be suggested for use as a bactericidal media. The opposite response to the pH trigger was observed for obtained CNT-g-PMAA in contrast to CNT-g-PDMAEMA: the sample was found to be stable in alkaline water but destabilized and aggregated in an acidic environment. Most importantly, CNT-g-PDMAEMA and CNT-g-PMAA dispersions displayed the same pattern of responsiveness when the gas trigger was tested. CNT-g-PMAA demonstrated the reversed responsive behavior, precipitating after CO2 bubbling in basic water media, whereas CNT-g-PDMAEMA changed from agglomerated to the dispersed state. The addition of N2 was effective in removing CO2 from the systems reverting the original appearances of CNTs. The high molecular weight (approx. 15 kDa) of grafted polymers can restrict the gas manipulations with these dispersions and needs to be limited. Thus, through the grafting reaction and the series of chemical modifications of PDMAEMA, a class of “smart” multi-stimuli-responsive CNTs was created. Moreover, the suggested way of manipulating the dispersions implies the use of abundant, nontoxic gases, which allows avoiding the addition of any acid/base reagents, providing a more environmentally friendly and green tool for operations with CNTs for researchers in the related fields of science.Author contributions
Ting-Yi Hsin: investigation, methodology, data curation, visualization. Vladislav Y. Shevtsov: validation, methodology, visualization, writing – original draft, writing – review and editing. Yeong-Tarng Shieh: conceptualization, methodology, validation, supervision, project administration, writing – original draft, writing – review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Ministry of Science and Technology, Taiwan, under contract MOST 107-2221-E-390-010-MY3.References
- S. Iijima, Helical microtubules of graphitic carbon, Nature, 1991, 354, 56–58 CrossRef CAS.
- T. W. Odom, J.-L. Huang, P. Kim and C. M. Lieber, Atomic structure and electronic properties of single-walled carbon nanotubes, Nature, 1998, 391, 62–64 CrossRef CAS.
- D. H. S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Chemistry of single-walled carbon nanotubes, Acc. Chem. Res., 2002, 35, 1105–1113 CrossRef PubMed.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Transparent, Conductive Carbon Nanotube Films, Science, 2004, 305, 1273–1276 CrossRef CAS PubMed.
- K. Dirian, M. Á. Herranz, G. Katsukis, J. Malig, L. Rodríguez-Pérez, C. Romero-Nieto, V. Strauss, N. Martín and D. M. Guldi, Low dimensional nanocarbons – chemistry and energy/electron transfer reactions, Chem. Sci., 2013, 4, 4335–4353 RSC.
- S. N. Barman, M. C. LeMieux, J. Baek, R. Rivera and Z. Bao, Effects of Dispersion Conditions of Single-Walled Carbon Nanotubes on the Electrical Characteristics of Thin Film Network Transistors, ACS Appl. Mater. Interfaces, 2010, 2, 2672–2678 CrossRef CAS PubMed.
- P.-C. Ma, N. A. Siddiqui, G. Marom and J.-K. Kim, Dispersion and functionalization of carbon nanotubes for polymer-based nanocomposites: A review, Composites, Part A, 2010, 41, 1345–1367 CrossRef.
- M. Pras, J.-F. Gérard, L. Golanski, G. Quintard and J. Duchet-Rumeau, Key Role of the Dispersion of Carbon Nanotubes (CNTs) within Epoxy Networks on their Ability to Release, Polymers, 2020, 12, 2530 CrossRef CAS PubMed.
- Y. S. Song and J. R. Youn, Influence of dispersion states of carbon nanotubes on physical properties of epoxy nanocomposites, Carbon, 2005, 43, 1378–1385 CrossRef CAS.
- Y.-T. Shieh, G.-L. Liu, H.-H. Wu and C.-C. Lee, Effects of polarity and pH on the solubility of acid-treated carbon nanotubes in different media, Carbon, 2007, 45, 1880–1890 CrossRef CAS.
- O. V. Kharissova, B. I. Kharisov and E. G. de Casas Ortiz, Dispersion of carbon nanotubes in water and non-aqueous solvents, RSC Adv., 2013, 3, 24812–24852 RSC.
- S. Manzetti and J.-C. P. Gabriel, Methods for dispersing carbon nanotubes for nanotechnology applications: liquid nanocrystals, suspensions, polyelectrolytes, colloids and organization control, Int. Nano Lett., 2019, 9, 31–49 CrossRef CAS.
- A. M. Díez-Pascual, Chemical Functionalization of Carbon Nanotubes with Polymers: A Brief Overview, Macromolecules, 2021, 1, 64–83 Search PubMed.
- K. Umemura, R. Hamano, H. Komatsu, T. Ikuno and E. Siswoyo, Dispersion of Carbon Nanotubes with “Green” Detergents, Molecules, 2021, 26, 2908 CrossRef CAS PubMed.
- R. Sadri, M. Hosseini, S. N. Kazi, S. Bagheri, N. Zubir, K. H. Solangi, T. Zaharinie and A. Badarudin, A bio-based, facile approach for the preparation of covalently functionalized carbon nanotubes aqueous suspensions and their potential as heat transfer fluids, J. Colloid Interface Sci., 2017, 504, 115–123 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Tunable pH- and Temperature-Sensitive Copolymer Libraries by Reversible Addition−Fragmentation Chain Transfer Copolymerizations of Methacrylates, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- Q. Zhang, G. Yu, W.-J. Wang, H. Yuan, B.-G. Li and S. Zhu, Preparation of N2/CO2 Triggered Reversibly Coagulatable and Redispersible Latexes by Emulsion Polymerization of Styrene with a Reactive Switchable Surfactant, Langmuir, 2012, 28, 5940–5946 CrossRef CAS PubMed.
- Y. Zhao, K. Landfester and D. Crespy, CO2 responsive reversible aggregation of nanoparticles and formation of nanocapsules with an aqueous core, Soft Matter, 2012, 8, 11687–11696 RSC.
- D. Han, X. Tong, O. Boissière and Y. Zhao, General Strategy for Making CO2-Switchable Polymers, ACS Macro Lett., 2012, 1, 57–61 CrossRef CAS PubMed.
- Y.-T. Shieh, P.-Y. Tai and C.-C. Cheng, Dual CO2/temperature-responsive diblock copolymers confer controlled reversible emulsion behavior, Polym. Chem., 2019, 10, 2641–2646 RSC.
- A. K. Alshamrani, J. R. Vanderveen and P. G. Jessop, A guide to the selection of switchable functional groups for CO2-switchable compounds, Phys. Chem. Chem. Phys., 2016, 18, 19276–19288 RSC.
- Z. Guo, Y. Feng, S. He, M. Qu, H. Chen, H. Liu, Y. Wu and Y. Wang, CO2-Responsive “Smart” Single-Walled Carbon Nanotubes, Adv. Mater., 2013, 25, 584–590 CrossRef CAS PubMed.
- F. Meyer, A. Minoia, J. M. Raquez, M. Spasova, R. Lazzaroni and P. Dubois, Poly(amino-methacrylate) as versatile agent for carbon nanotube dispersion: an experimental, theoretical and application study, J. Mater. Chem., 2010, 20, 6873–6880 RSC.
- C. M. Homenick, G. Lawson and A. Adronov, Polymer Grafting of Carbon Nanotubes Using Living Free-Radical Polymerization, Polym. Rev., 2007, 47, 265–290 CrossRef CAS.
- Y. T. Joo, K. H. Jung, M. J. Kim and Y. Kim, Preparation of antibacterial PDMAEMA-functionalized multiwalled carbon nanotube via atom transfer radical polymerization, J. Appl. Polym. Sci., 2013, 127, 1508–1518 CrossRef CAS.
- S. Abraham, S. K. Kumaran and C. D. Montemagno, Gas-switchable carbon nanotube/polymer hybrid membrane for separation of oil-in-water emulsions, RSC Adv., 2017, 7, 39465–39470 RSC.
- Y.-T. Shieh and Y.-D. Chen, Carboxymethyl-chitosan-modified carbon nanotubes have sensitive CO2-responsive dispersion in water, J. Taiwan Inst. Chem. Eng., 2020, 115, 223–228 CrossRef CAS.
- V. Y. Shevtsov, T.-Y. Hsin and Y.-T. Shieh, Preparation of amphiphilic copolymers via base-catalyzed hydrolysis of quaternized poly[2-(dimethylamino)ethyl methacrylate], Polym. Chem., 2022, 13, 1429–1436 RSC.
- Y.-L. Liu and W.-H. Chen, Modification of Multiwall Carbon Nanotubes with Initiators and Macroinitiators of Atom Transfer Radical Polymerization, Macromolecules, 2007, 40, 8881–8886 CrossRef CAS.
- V. A. Izumrudov, E. Kharlampieva and S. A. Sukhishvili, Multilayers of a globular protein and a weak polyacid: role of polyacid ionization in growth and decomposition in salt solutions, Biomacromolecules, 2005, 6, 1782–1788 CrossRef CAS PubMed.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, A Mechanistic Study of the Hydrolytic Stability of Poly(2-(dimethylamino)ethyl methacrylate, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00305h |
| This journal is © The Royal Society of Chemistry 2022 |