
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of protein nanoparticles for targeted delivery of drugs to cancer cells
Eiry
Kobatake
 *,
Yusuke
Ikeda
and
Masayasu
Mie
*,
Yusuke
Ikeda
and
Masayasu
Mie
School of Life Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan. E-mail: kobatake.e.aa@m.titechac.jp
First published on 24th June 2022
Abstract
Protein nanoparticles (NPs) are potentially valuable carriers for drug delivery, because they are biocompatible, biodegradable, bioabsorbable, and can be easily designed to perform specific functions with high homogeneity when produced by genetic engineering. We constructed protein NPs based on genetically engineered elastin like polypeptides (ELPs) with a fused poly-aspartic acid tail (ELP-D) for use in drug delivery systems (DDSs). To ensure specific delivery of protein NPs, to cancer cells, targeting and internalizing molecules should be present on their surfaces. The iRGD peptide can activate integrin-dependent binding to cancer cells and neuropilin-1 (NRP-1)-dependent internalization into tumor tissues. In the present study, iRGD was genetically fused to the C-terminus of ELP-D to introduce active targeting and cell-penetrating functions to ELP-D NPs. The formation of NPs with iRGD moieties on their surfaces was examined, as well as their targeting ability. Anti-cancer drug paclitaxel (PTX)-loaded protein NPs with iRGD moieties were found to be effective in delivering the drug to cancer cells, becoming internalized into the cells, and inducing cell death.
Introduction
Drug delivery systems (DDSs) help in reducing the side effects of drugs and maximizing their effectiveness by regulating the location and rate of drug release. The design of drug carriers is one of the most important efforts in the development of DDSs, because they require high stability, low antigenicity, biodegradability, accumulation in affected areas, and permeability into cells. To meet these requirements, a wide variety of nanoparticles have been developed as drug carriers, including synthetic polymers, liposomes, virus-based materials, and metals. Synthetic polymers are promising candidates because of their high stability and tunability. Therefore, micelles with various synthetic polymers have been utilized as drug carriers. However, the functionality and heterogeneity of these materials are limited. To overcome these limitations, protein-based nanoparticles are attractive, because they can easily be designed to introduce high functionality and homogeneity using genetic engineering. In addition, biocompatibility and biodegradability are advantages of protein-based nanoparticles.Elastin like polypeptides (ELPs) are often used in the design of drug delivery carriers, owing to their biocompatibility, biodegradability, and drug loading capacities.1–3 Another valuable characteristic of ELPs is the formation of temperature-dependent aggregates, called coacervates, above a specific transition temperature.4,5 However, ELP aggregates are too large to allow for their use as drug carriers.6 Large aggregates increase antigenicity and are trapped by physiological responses. To control the size of aggregates, ELP block copolymers consisting of hydrophobic and hydrophilic ELP blocks were designed by Dreher et al.7 At an appropriate temperature, ELP proteins self-assembled to form spherical micelles of about 30 nm in size. Our ELP-based nanoparticle system was composed of genetically engineered ELPs with fused poly-aspartic acid tails, denoted as ELP-D.8 The formation of the NPs was due to the balance between the aggregation of ELPs and the charge repulsion of the poly-aspartic tails. The genetic design of protein-based NPs offers advantages for the construction of anti-cancer drug carriers, including the ability to display specific tumor targeting proteins and peptides on their surfaces without chemical modification. In our previous study, epidermal growth factor (EGF) was genetically fused to ELP-D to introduce targeting abilities to EGF receptor-overexpressing tumor cells.9 Based on this study, a DNA aptamer which binds to MUC1, a type I transducer glycoprotein overexpressed in various cancer cells, was present on the surface of ELP-NPs.10 Herein, instead of chemical conjugation, DNA aptamers were enzymatically conjugated to protein NPs through the bacteriophage φx174 Gene A* protein. Gene A* protein retains the activity of DNA strand transfer by cleaving its recognition sequence and covalently attaching to the cleavage site by forming a phosphotyrosine diester.11 By exploiting this character, we developed a method for site-specific attachment of single-stranded DNA to a recombinant protein of interest creating a Gene A* protein fusion.12,13 The gene encoding Gene A* protein was fused to the end of the gene encoding the ELP-D fusion protein to conjugate MUC1 aptamers to NPs.10
In addition to targeting specific areas, DDSs must deliver their payload into cells. Various proteins, such as the HIV-1 transactivator TAT protein,14,15 Antennapedia,16 and the herpes simplex virus-1 protein VP22,17 are known to be able to penetrate into the cell cytoplasm. Short cationic cell-penetrating peptides derived from these proteins retain their ability to internalize into cells, and can carry cargoes such as proteins, nucleic acids, and nanoparticles.18,19 Cell-penetrating peptides have no selectivity for cells, and are taken up into almost all types of cells. Therefore, the addition of targeting ability to cell-penetrating peptides in DDS carriers is required to prevent harmful side effects of drugs.
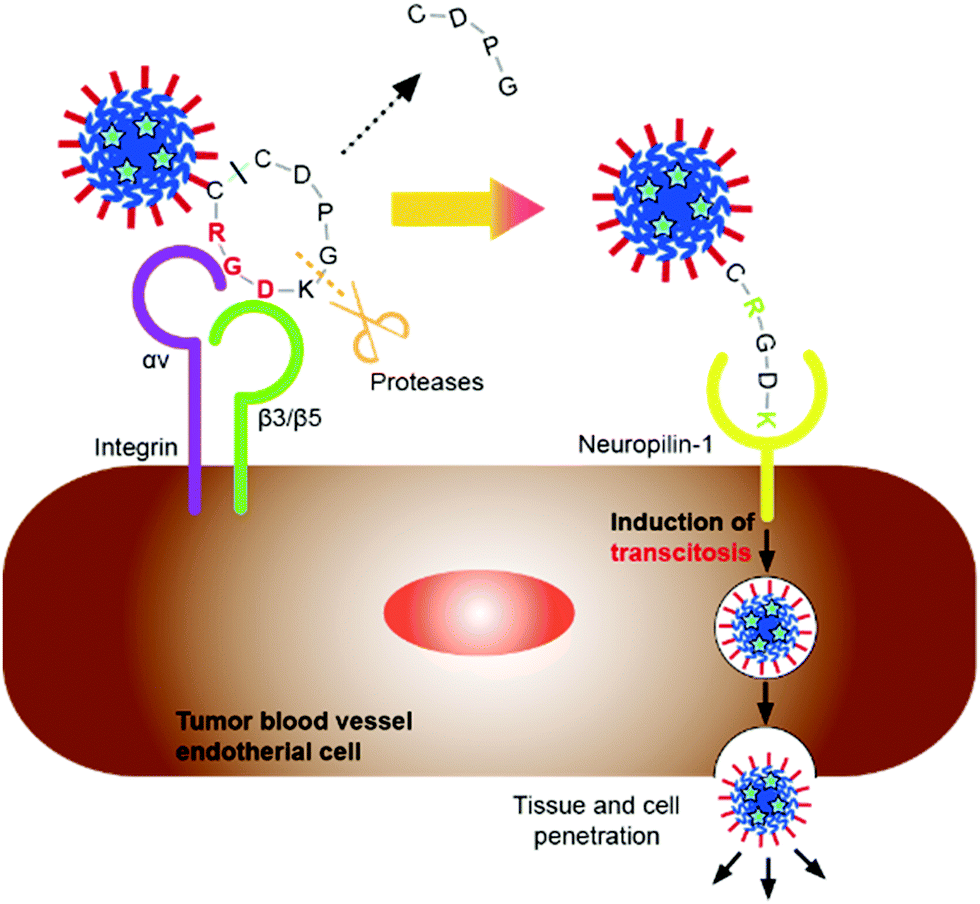
To target delivery into tumors, we paid attention to the internalizing RGD (iRGD). iRGD is a nine-amino acid cyclic peptide which has both cell-penetrating and tumor targeting ability.20,21 iRGD binds to integrin αVβ3 expressed on neovascular endothelial cells, and then the iRGD is cleaved by proteases to expose the C-end rule (CendR) motif. The exposed CendR motif binds to neuropilin (NRP-1), which is highly expressed on various tumors, triggering cell internalization.22 Many studies have shown that iRGD enhances the tumor targeting and penetrating ability of conjugated therapeutic agents or nanovehicles.23–26
In the present study, iRGD was fused to the C-terminus of ELP-D to have iRGD moieties on the surfaces of protein NPs. We evaluated the anti-cancer effects of paclitaxel (PTX)-loaded protein NPs having iRGD on cancer cells. A schematic illustration of the concept of this study is shown in Fig. 1.
 | ||
| Fig. 1 Schematic illustration of this study. | ||
Materials and methods
Materials
Restriction enzymes and ligase were purchased from Takara Bio Inc. (Shiga, Japan). Synthesized DNA oligonucleotides were purchased from FASMAC (Japan). The human lung carcinoma epithelial cell line A549 (RCB0098) was provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. All other chemicals were of analytical grade.Construction of plasmid
The previously constructed plasmid pET28b-(PAVGV)42-D44-CHis9 for the expression of the (PAVGV)42-D44 protein (named AD) was digested with EcoRI and XhoI, and the DNA oligonucleotide encoding the amino acid sequences of EcoRI-SpeI sites, (GGGS)2-iRGD, and HindIII-XhoI sites from its 5′-end was inserted. The resulting plasmid was named pET28b-(PAVGV)42-D44-iRGD-CHis, and expresses (PAVGV)42-D44-iRGD, named ADiR.Expression and purification of proteins
The plasmid pET28b-(PAVGV)42-D44-iRGD-CHis was transfected into E. coli BLR (DE3) competent cells via heat-shock. The transformed cells were inoculated in LB media supplemented with 20 μg mL−1 kanamycin and 20 μg mL−1 tetracycline, and incubated at 37 °C. Protein expression was induced by the addition of isopropyl-β-D-galactopyranoside (IPTG) at a concentration of 1.0 mM when the OD660 reached 0.6. After overnight culture at 25 °C, the cells were harvested by centrifugation, and the cell pellet was resuspended in phosphate buffered saline (PBS) and disrupted by sonication. The cell lysate was centrifuged at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 min at 4 °C to remove insoluble cellular debris. The collected fraction was applied to HIS-Select Nickel Affinity Gels (Sigma-Aldrich, St. Louis, MO, USA) to purify the protein using His-tags. After 1 h of incubation, the column was washed five times with three column volumes of wash buffer (50 mM Na2HPO4, 0.3 mM NaCl, pH 8.0), followed by five washes with three volumes of the same buffer including 20 mM imidazole. The same volume of elution buffer (20 mM Na2HPO4, 0.3 mM NaCl, pH 8.0) including 100 mM imidazole was then added to elute the protein. The purified protein, ADiR, was dialyzed against PBS using slide-A-lizer dialysis cassettes (Pierce, 10000 MW) and analyzed using SDS-PAGE on 12% polyacrylamide gels. The concentration of the purified protein was determined using BCA assay kits (Thermo Fisher Scientific, Waltham, MA).
000g for 15 min at 4 °C to remove insoluble cellular debris. The collected fraction was applied to HIS-Select Nickel Affinity Gels (Sigma-Aldrich, St. Louis, MO, USA) to purify the protein using His-tags. After 1 h of incubation, the column was washed five times with three column volumes of wash buffer (50 mM Na2HPO4, 0.3 mM NaCl, pH 8.0), followed by five washes with three volumes of the same buffer including 20 mM imidazole. The same volume of elution buffer (20 mM Na2HPO4, 0.3 mM NaCl, pH 8.0) including 100 mM imidazole was then added to elute the protein. The purified protein, ADiR, was dialyzed against PBS using slide-A-lizer dialysis cassettes (Pierce, 10000 MW) and analyzed using SDS-PAGE on 12% polyacrylamide gels. The concentration of the purified protein was determined using BCA assay kits (Thermo Fisher Scientific, Waltham, MA).
AD as a control protein was expressed and purified using the same procedure as for ADiR.
Cell adhesion activity of iRGD
Cell adhesion assays were performed in 96-well suspension culture plates (BD Biosciences, San Jose, CA, USA). The surfaces of the wells were coated with 100 μL of various concentrations of ADiR or AD. After incubation for 1 h at 37 °C, each well was washed with PBS, and then blocked with 1% BSA for 1 h at 37 °C. A549 cells were seeded at a density of 4.0 × 104 cells per well in each well. After 2 h of incubation at 37 °C, the numbers of adherent cells were determined using CCK-8 cell counting kits (Dojindo Laboratories, Inc., Kumamoto, Japan).Thermal characterization and dynamic light scattering
The transition temperatures of the purified proteins were evaluated by measuring the turbidity with a spectrometer (DU-7500; Beckman Coulter, Brea, CA, USA) at 350 nm. Purified proteins were dissolved in PBS (pH 7.4) to a final concentration of 500 μg mL−1. The samples were heated from 20 to 50 °C, and subsequently cooled to 20 °C with a temperature gradient of 1 °C min−1.Light scattering data were measured using a Nano-ZS (Malvern Instruments, UK). Samples were equilibrated at the measuring temperature for 10 min prior to data collection. Light scattering data at 90° and 633 nm were collected for at least 10 s x 10 runs. The sizes of the proteins were calculated based on the assumption that the viscosity of PBS was the same as that of water. The particle size, with a single-peaked distribution, was calculated using the cumulant method. Conversion from an intensity distribution to a volume distribution was performed using the software supplied with the Nano-ZS.
Evaluation of drug loading into ADiR NPs
ADiR (0.5 mg mL−1) was mixed with 1-anillinonaphthalene-8-sulfonic acid (1, 8-ANS) in 100 μM PBS, followed by incubation at 25 °C for 10 min. The emission spectrum at 25 °C was recorded at 370 nm excitation using an FP-6500 spectrofluorometer (Jasco, Tokyo, Japan). The samples were incubated for 10 min at 40 °C and the emission spectrum at 40 °C was recorded at 370 nm excitation.Cellular uptake of iRGD nanoparticles
Hydrophobic fluorescent dye coumarin-6 (C-6) was incorporated into ADiR nanoparticles. ADiR (5 μM) and C-6 (50 μM) solutions were mixed, following sonication, then incubated at 42 °C for 15 min to form NPs. The solution was dialyzed against PBS at 37 °C to remove free C-6, and C-6-incorporated NPs (ADiR-C6) were obtained. A549 cells were seeded in 35 mm tissue culture dish plates (BD Bioscience) at a density of 1 × 104 cells per well. After one day of culture, the medium was replaced by a medium without serum. After one day of culture in DMEM medium (1% penicillin/streptomycin, 10% FBS), the medium was changed. After incubation for another day, 60 nM of ADiR-C6 diluted with DMEM containing 50% FBS was added to the same medium. After 1 h, the cells were washed with PBS and observed using fluorescence microscopy. The same experiment was performed using AD-C6 lacking the iRGD sequence as a control.In vitro cytotoxicity
Paclitaxel (PTX) was used as an anti-cancer reagent. ADiR (3 μM) and PTX (30 μM) solutions were mixed, following sonication, then incubated at 42 °C for 15 min to form NPs. The solution was dialyzed against PBS at 37 °C to remove free PTX, and PTX-incorporated NPs (ADiR-PTX) were obtained. A549 cells were seeded in 35 mm tissue culture dish plates (BD Bioscience) at a density of 1 × 104 cells per well. After one day of culture, the medium was replaced with a medium without serum. After incubation for another day, different concentrations of ADiR-PTX were added to the cells. After culturing for three days, the numbers of cells were determined using CCK-8 cell counting kits. Cells were also stained with calcein-AM (Dojindo) and propidium iodide (PI) to assess the cell viability.Results and discussion
Design and expression of proteins
The design of the fusion protein ADiR is shown in Fig. 2. AD was constructed in our previous work, 9 and consists of 42 repeats of the PAVGV sequence, 44 repeats of aspartic acid, and a histidine tag (His-tag). Poly(PAVGV) was employed as the ELP sequence, because it was shown to form stable coacervates at the transition temperature (Tt) due to the rigid packing.27,28 At incubation temperatures above the Tt, the reversible dissociation of the poly(PAVGV) suspension was strongly impeded, even after cooling to a temperature below the Tt, due to the lack of water molecules between the amide groups. The fusion of a poly-aspartic acid chain (D) with ELP allowed the size of the resultant ELP aggregates to be regulated on the nanoscale by charged repulsion of the poly-aspartic acid chains. To add the active targeting property to cancer cells and the cell-permeating property to these protein nanoparticles, iRGD peptides were introduced at the C-terminus of AD. The addition of iRGD peptides at the C-terminus was expected to result in the multivalent display of iRGD on the exterior of the nanoparticles.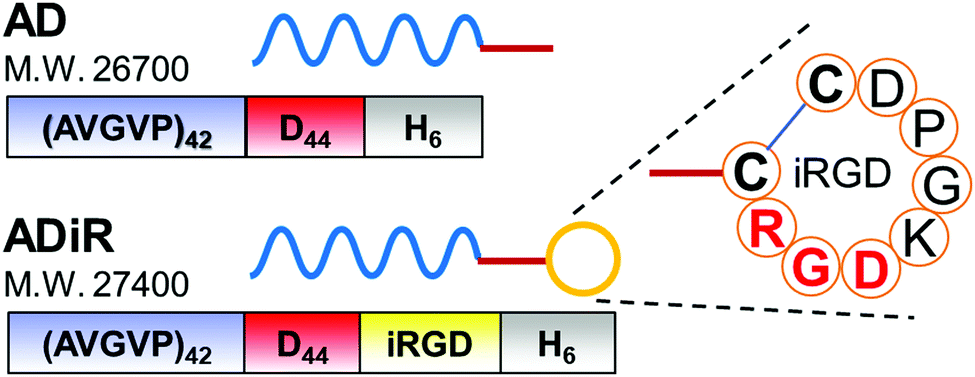 | ||
| Fig. 2 Design of the fusion proteins, AD and ADiR. The amino acid-sequence of the iRGD moiety is indicated. | ||
The constructed plasmids, pET28b-(PAVGV)42-D44-iRGD for ADiR and pET28b-(PAVGV)42-D44 for AD, were expressed in E.coli BLR (DE3) by adding 1 mM IPTG. The expressed proteins were purified from the soluble fraction using His-tag followed dialysis. The expected molecular mass of AD and ADiR is 26.7 kDa and 27.4 kDa, respectively.
Cell adhesion activity of iRGD
To evaluate the cell adhesion activity of the iRGD sequence in ADiR, A549 cells were seeded on a plate whose surface was coated with ADiR or AD. A549 cells express excessive amounts of αVβ3 integrin, which is known to bind with RGD, on their cell surfaces.The well surfaces of 96-well cell culture plates were coated with ADiR or AD of various concentrations. A549 cells were seeded in each well at a density of 4.0 × 104 cells per mL, then after 2 h, the numbers of adhesive cells were determined using CCK-8 assays (Fig. 3A). Few adhesive cells were observed on the AD-coated well surfaces at any concentration of AD. However, on the ADiR-coated well surfaces, the cells adhered in an ADiR concentration dependent manner.
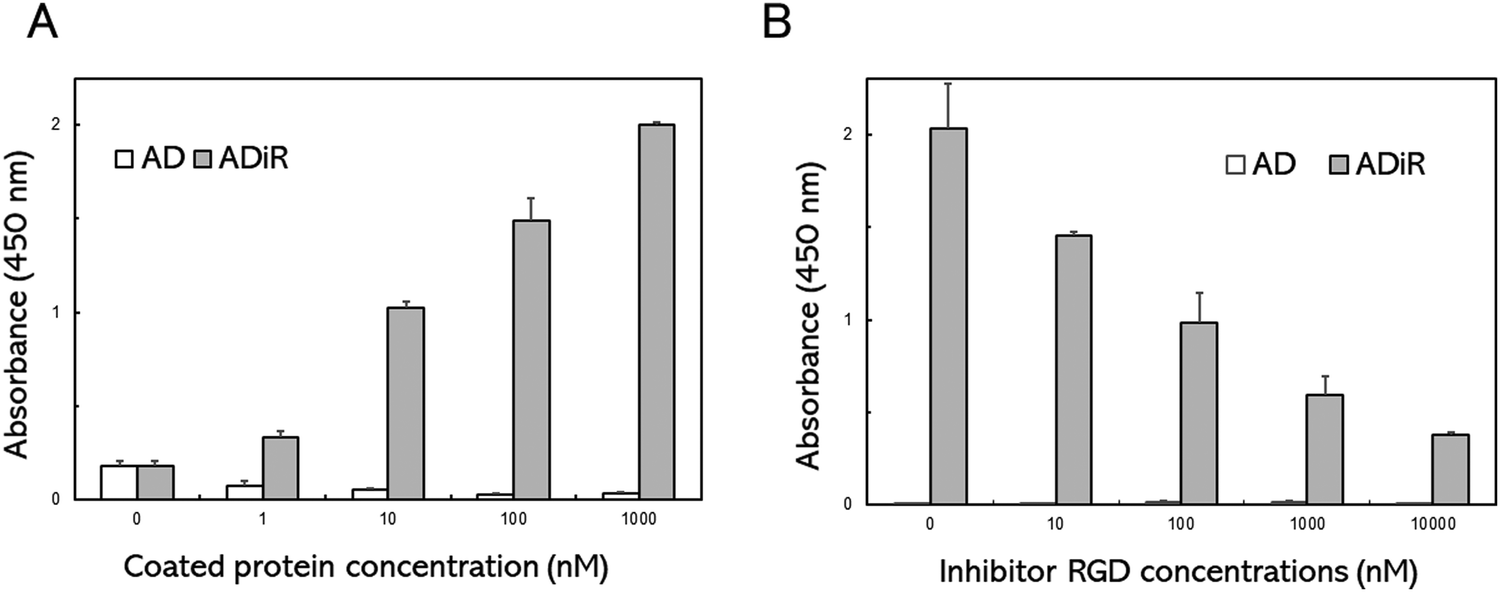 | ||
| Fig. 3 Cell adhesion activity of ADiR. (A) A549 cells seeded on the plate surfaces were coated with ADiR or AD at various concentrations. (B) Various concentrations of RGD peptides were added to cell suspensions before seeding the cells onto the AD- or ADiR-coated plate surfaces. | ||
To confirm that the cell adhesion was caused by specific interactions between RGD and αVβ3 integrin, various concentrations of the RGD peptide were added to cell suspensions before seeding the cells. As shown in Fig. 3B, the adhesion of cells seeded on ADiR-coated well surfaces was inhibited by the presence of the soluble RGD peptide (GRGDSP) in the cell suspension. The number of attached cells decreased with increasing concentrations of the RGD peptide. These results indicate that cells possessing the αVβ3 integrin adhered to ADiR-coated surfaces through the interaction between integrin and the iRGD peptide incorporated into AD.
Formation of thermo-responsive nanoparticles
The thermo-responsive properties of the nanoparticles were evaluated to confirm that ADiR had the ability to form nanoparticles. These properties were determined by continuous monitoring of the optical density at 350 nm while the aqueous solutions were heated from 20 °C to 50 °C, and subsequently cooled (Fig. 4A). In the heating process, both proteins, AD and ADiR, exhibited elevation of the turbidity, involving aggregation. The transition temperatures of AD and ADiR were 34.6 °C and 38.2 °C, respectively. During the process of cooling from 50 °C to 20 °C, no thermal transitions were observed in either protein. Rodorigues-Cabell et al. reported that poly(VPAVG) had a more compact structure than poly(VPGVG) because the alanine residue strongly influences the physical structures of both the folded and unfolded states.27 This causes the irreversibility of ELP consisting of poly(VPAVG). In our previous study,9 it was clear that AD showed irreversibility. The results depicted in Fig. 4A indicate that the addition of the iRGD sequence between D44 and Chis does not affect the irreversibility of AD, although the transition temperature was shifted slightly to a higher temperature.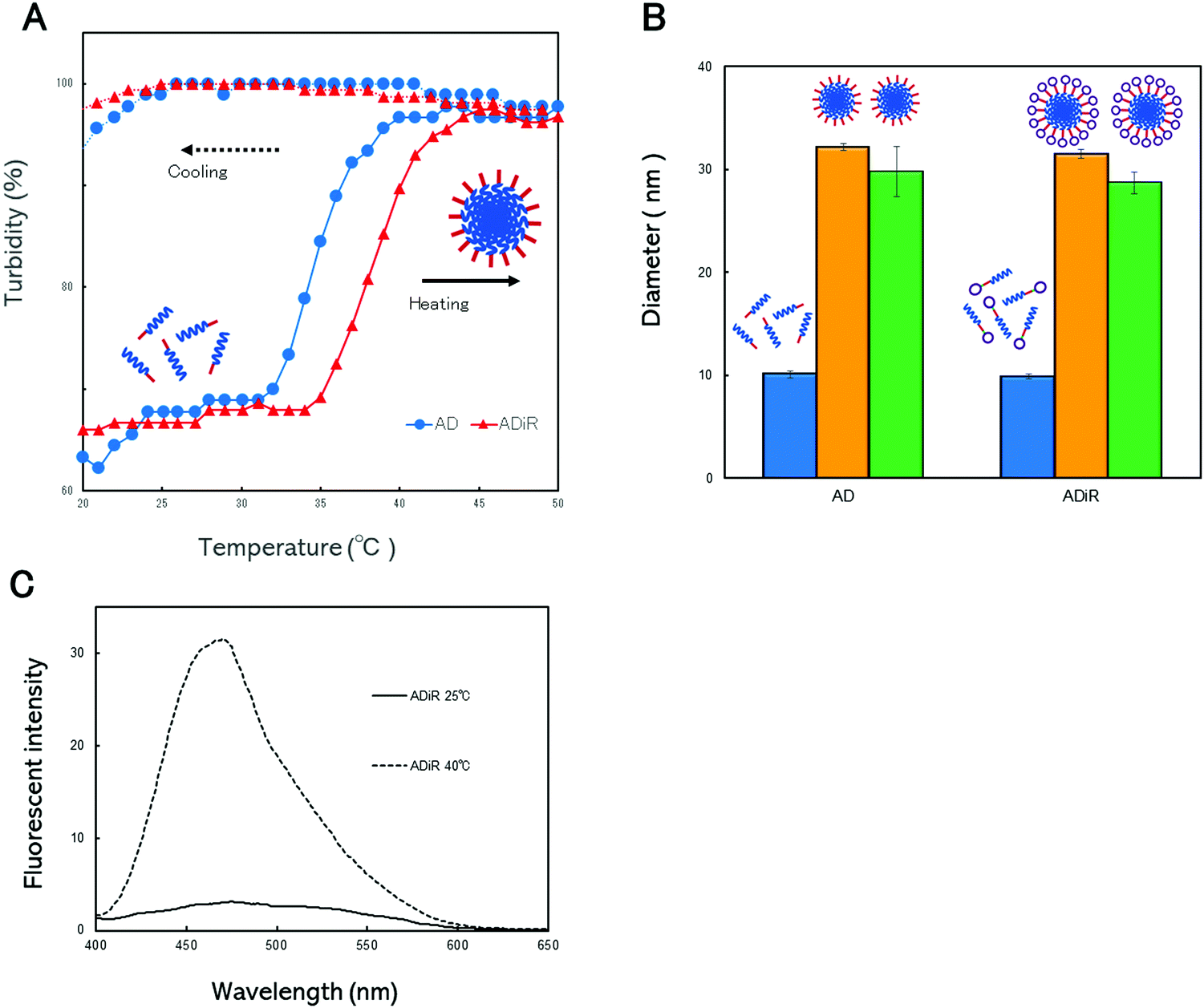 | ||
| Fig. 4 Thermo-responsive nanoparticle formation. (A) Turbidity profile of AD (●) and ADiR (▲). The turbidity profiles of the proteins were obtained by monitoring the optical density at 350 nm. (B) Hydrodynamic size distribution of the nanoparticles was measured by dynamic light scattering (DLS) at 20 °C (blue bar), 37 °C (orange bar), and 20 °C, and cooled after incubation at 37 °C (green bar). (C) Incorporation of hydrophobic molecules into the nanoparticles. The fluorescence spectra of proteins mixed with 1, 8-ANS at 25 °C (solid line) and 37 °C (broken line). | ||
In our previous work,9 we introduced poly-aspartic acid chains to poly(VPAVG) to prevent the formation of aggregation at micro meter sizes. Therefore, the newly designed ADiR based on AD was expected to form nanoparticles following incubation at the transition temperature, due to the presence of poly-aspartic acid chains. The particle sizes were evaluated using dynamic light scattering (DLS) (Fig. 4B). The size distributions of AD and ADiR were first measured at 20 °C, and found to be around 10 nm for both proteins. These results indicated that the proteins existed predominantly as monomers below the transition temperature. Above the transition temperature of 45 °C, the diameters of AD and ADiR were increased to around 30 nm, indicating the formation of nanoparticles. It was supposed that the particle sizes were regulated by charged repulsion of the poly-aspartic acid chains introduced at the 3′-terminus of the proteins. After cooling to 20 °C, the particle sizes of both proteins were maintained at around 30 nm. These results demonstrate that the designed fusion proteins consisting of poly(PAVGV) and a poly-aspartic acid chain, even after the introduction of the iRGD peptide, could form stable nanoparticles via temperature-triggered coacervation.
Evaluation of drug loading into ELP
ADiR formed nanoparticles with a diameter of around 30 nm by heating to above the transition temperature, due to the coacervation character of (PAVGV)n and the charged repulsion of aspartic acid chains. This finding indicates that the nanoparticles have hydrophobic cores with a micelle-like structure. In order to utilize the nanoparticles as drug carriers, the ability of nanoparticles to encapsulate drugs is important. To demonstrate their ability to encapsulate hydrophobic drugs, 1-anilino-8-naphthalene sulfonic acid (1, 8-ANS) was used as a model system of a hydrophobic drug, and was loaded into the nanoparticles. 1, 8-ANS is a well-known fluorescent probe used for the analysis of hydrophobic environments.29 When it binds to the hydrophobic regions of proteins, a blue shift of its emission peak is induced, with an increase of fluorescence intensity. After the addition of 1, 8-ANS to the protein solution, the fluorescence spectra of 1, 8-ANS was evaluated at 25 °C and 40 °C (Fig. 4C). The emission peaks at around 470 nm were slightly increased in the presence of ADiR at 25 °C, compared to 1, 8-ANS in PBS. However, the emission peak at 40 °C was markedly increased for ADiR, while the fluorescence intensity of 1, 8-ANS was not changed at all in PBS. It appeared that 1, 8-ANS was incorporated into the hydrophobic core of the nanoparticles where the ELP was aggregated, resulting in an increased fluorescence intensity. These results indicated that 1, 8-ANS was loaded into nanoparticles consisting of ADiR during its formation by heating. Therefore, the ADiR protein is expected to be useful as a carrier of hydrophobic drugs.iRGD-displayed nanoparticle uptake via the integrin receptor
The nanoparticles consisting of ADiR have an internal hydrophobic ELP sequence, and an external charged D44 sequence. The iRGD sequence is displayed on the surface of the nanoparticles since it is located at the C-terminus of the D44 sequence of ADiR. Therefore, the nanoparticles are expected to be able to target tumor cells expressing high levels of integrin αVβ3 through integrin–RGD interactions.Like conventional RGD peptides, iRGD binds to tumors through integrin αVβ3. The iRGD is then cleaved by protease in the tumor to produce CRGDK/R. The truncated peptide loses its integrin binding activity, but gains an affinity for neuropilin-1 (NRP-1), because of the C-terminal exposure of a CendR sequence (R/KXXR/K).22 Binding of NRP-1 induces the internalization of ADiR nanoparticles into the cells, followed by the release of the contents of the nanoparticles.
To investigate this hypothesis, hydrophobic fluorescent dye C-6 was incorporated into ADiR nanoparticles, which were then added to A549 cells which overexpressed integrin αVβ3. The cells were observed using a confocal fluorescent microscope every 15 min (Fig. 5A). After 15 min, no fluorescence of C6 was observed, indicating that most of the ADiR nanoparticles were in the extracellular field. After 30 min, some fluorescence could be seen in the cells, indicating that some of the nanoparticles had moved into the cells. After 60 min, stronger fluorescence was observed, because most of the ADiR nanoparticles had moved into the cells. After more than 60 min, no further increase of fluorescence was observed. These results suggest that ADiR nanoparticles bind to the cell surface and are internalized into the cells in about 1 h.
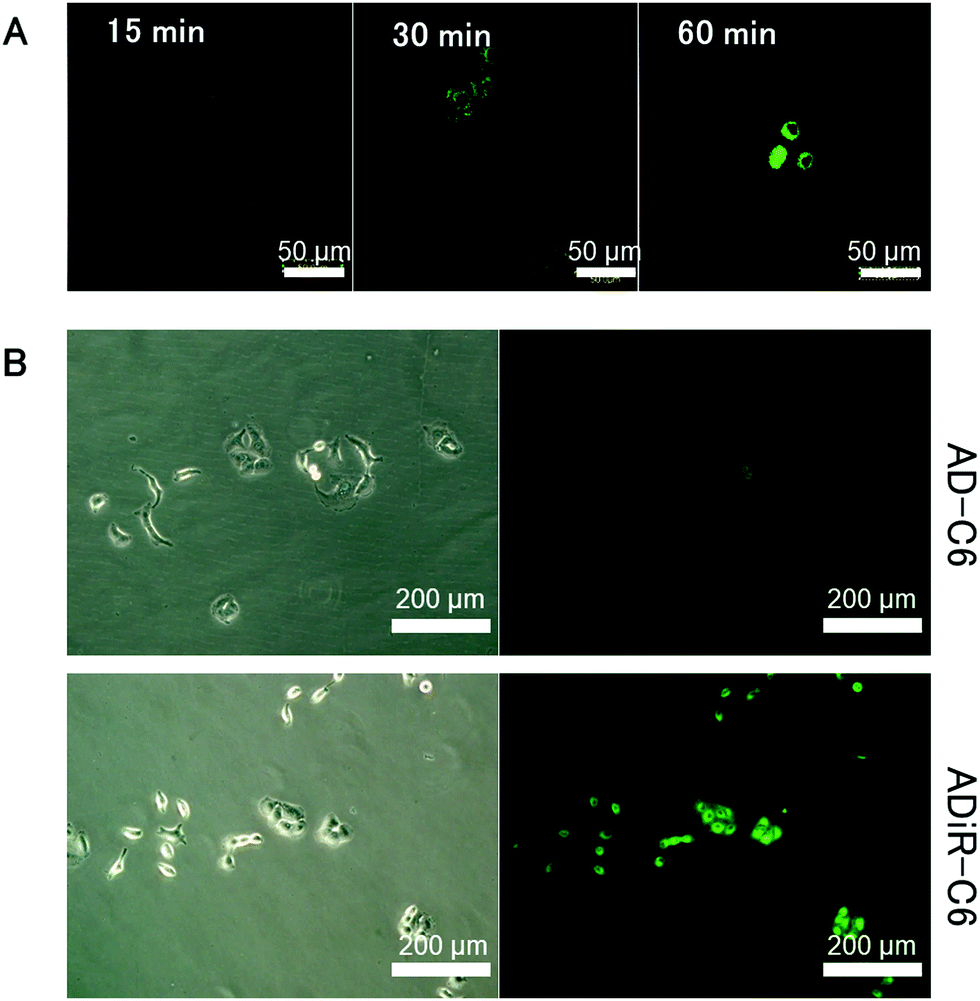 | ||
| Fig. 5 Imaging of the internalization of C-6-incorporated protein nanoparticles into A549 cells. (A) Time course of confocal imaging of ADiR-C6 (B) Imaging of ADiR-C-6 and AD-C-6 at 1 h after addition of nanoparticles. | ||
Based on the above results, the incubation time of ADiR nanoparticles with cells was fixed at 1 h after addition of nanoparticles, and the same experiment was performed on cells cultured in medium containing serum. In serum, proteins tend to adsorb non-specifically to the DDS carrier, which may lead to the disruption of the carrier and interference of targeting. By adding nanoparticles to cells in medium containing serum, it is possible to evaluate the stability of nanoparticles when administered in the blood in the future. Fluorescence from C-6 was observed in cells with ADiR nanoparticles, while no fluorescence was observed in cells with AD nanoparticles lacking the RGD sequence (Fig. 5B). These results also indicate that the iRGD sequences are displayed on the surface of nanoparticles by maintaining their integrin-binding function. Non-specific adsorption of nanoparticles onto the negatively charged surface of cells was suppressed by electric repulsion, since the surfaces of the AD nanoparticles are negatively charged because of the poly-aspartic acid sequence, and internalization of the nanoparticles into the cells by endocytosis was also suppressed.
Some fluorescence could be observed in cells with ADiR-RGD(−) NPs which lacked the RGD sequence, or ADiR-CendR(−) NPs which lacked the CendR sequence. However, this fluorescence was much lower than that of ADiR NPs (data not shown). ADR-RGD(−) NPs were internalized into cells by the NRP-1 pathway, and ADiR-CendR(−) NPs were internalized by endocytosis through the integrin pathway. We concluded that the stronger fluorescence in cells with ADiR NPs was due to the effects of both pathways: NRP-1 and integrin.
Since the non-specific delivery of anti-cancer agents to normal cells may cause some serious side effects, it should be avoided as much as possible. To confirm the specificity of the delivery of ADiR NPs to cancer cells, HEK293 cells were used as normal cells for the same experiments. No fluorescence was observed in cells with ADiR NPs, AD NPs, or C-6 itself (data not shown). These results indicated that ADiR NPs were delivered specifically to cancer cells which overexpressed both integrin αVβ3 and NRP1.
Induction of cell death with PTX-loaded protein nanoparticles
To optimize the concentration of the anti-cancer reagent PTX to produce cell death, ADiR or AD nanoparticles including various concentrations of PTX were prepared, and added to A549 cells. After three days, the cell numbers were determined using CCK-8 assays (Fig. 6A). About 80% of cells were killed by the addition of PTX at concentrations over 250 nM. When ADiR NPs-loaded PTX (ADiR-PTX) were added to cells, the number of living cells decreased with the increase in PTX concentration. It showed almost the same cell death-inducing effect as the positive control (PTX alone) when 1 μM PTX was applied. Therefore, 1 μM of PTX was used for further experiments. AD NPs-loaded with PTX (AD-PTX) also produced cell death in a concentration dependent manner, although their effects were much lower than those of ADiR-PTX. These effects may indicate non-specific delivery of AD-PTX to cells, or leakage of PTX from NPs.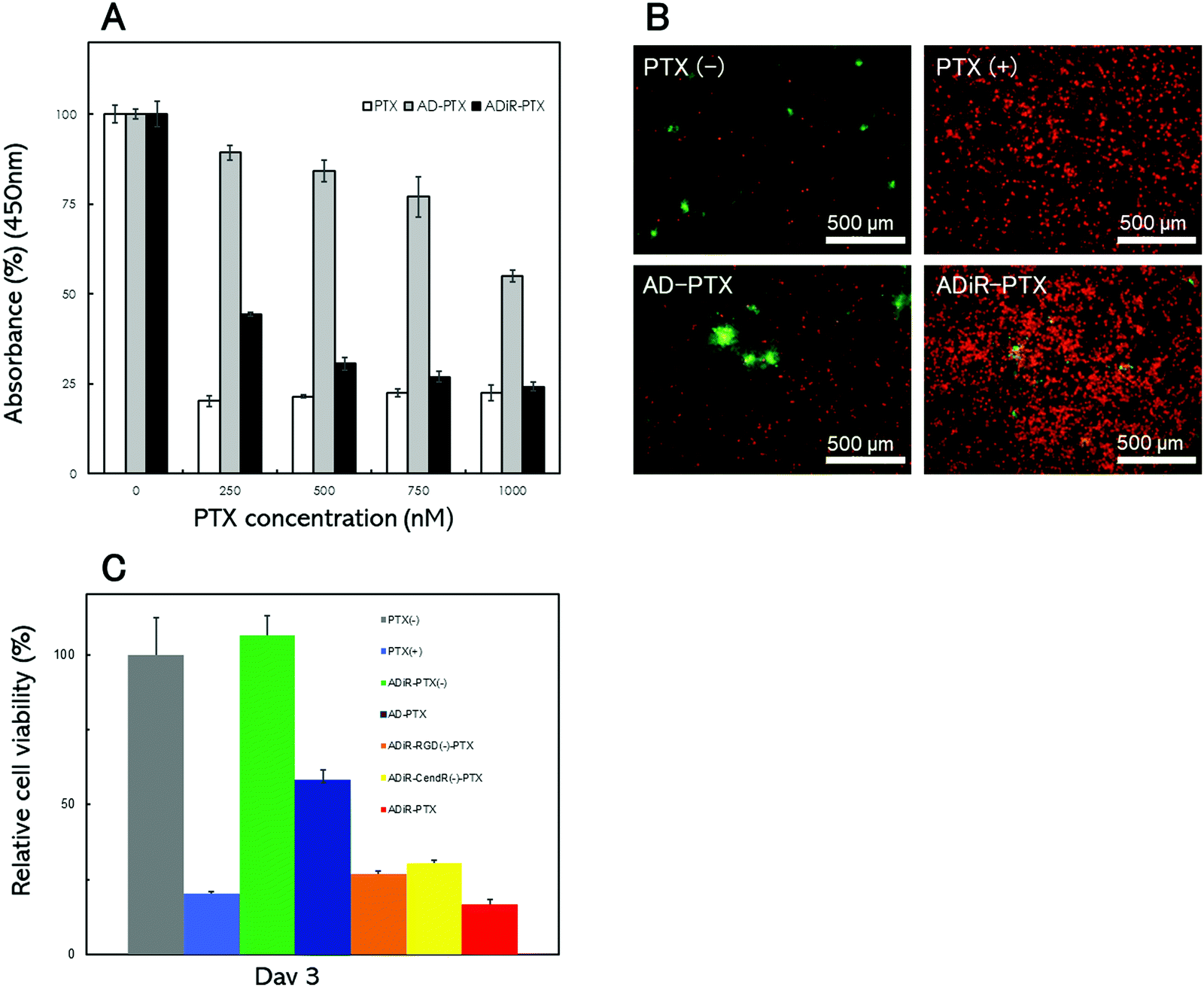 | ||
| Fig. 6 Cell viability after three days of culture. (A) Cell viability with various concentrations of PTX. Cell viability without PTX was considered to be 100%. (B) Fluorescence microscopic images showing cell viability. Living and dead cells were stained with calcein-AM (green) and PI (red), respectively. (C) Cell viability with various protein nanoparticles; gray: PTX (−) as negative control; blue: PTX (+); green: AD-R-PTX (−); dark blue: AD-PTX (+); orange: ADiR-RGD (−)-PTX (+); yellow: ADiR-CendR (−)-PTX (+); red: PTX (+) as positive control. Cell viability without PTX (gray: PTX (−)) was considered to be 100%. | ||
The cells were stained with Calcein-AM and PI, which stain viable and dead cells, respectively (Fig. 6B). PTX-loaded NPs with 200 nM ADiR (ADiR-PTX) were added to A549 cells, and the cells were stained after three days of culture. Few PI-stained dead cells were observed in cells with added PBS (PTX(−)) instead of PTX (PTX(+)), or in NPs with AD (AD-PTX) as a negative control. The number of living cells estimated by CCK-8 assay was set to 100% under this condition, and represented as a relative percentage under other conditions (Fig. 6C). When 1 μM of PTX was added to cells as a positive control (PTX(+)), most of the cells were stained with PI, and about 85% of cells died. The same or higher effect was observed following the addition of ADiR-PTX. Around 40% of the cells were killed by the addition of AD-PTX which did not have the iRGD sequence. It appeared that some PTX molecules leaked gradually from the NPs during incubation for three days. When ADiR NPs without PTX-loading (ADiR-PTX(−)) were added, no cells died, indicating that ADiR NPs alone had no harmful effects.
To confirm that the induction of cell death was due to both the effects of cell targeting by the RGD sequence and cell permeation using the CendR sequence, cell death was examined using PTX-loaded NPs with ADiR deleted. The deleted ADiR sequence –CRGDKGPDC– was substituted with –CRG![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif) KGPDC–, and –CRGDGPDC– in ADiR-RGD(−) and ADiR-CendR(−), respectively (Fig. 6C). Three days after the addition of the NPs, cell viabilities were determined by counting the live cells. The cell viabilities of ADiR-RGD(−)-PTX and ADiR-CendR(−)-PTX were approximately 26% and 30%, respectively. In the case of ADiR-PTX, the cell viability was about 16%, lower than that of deleted ADiR. The effect of both pathways, NRP-1 and integrin, should enhance the internalization of PTX into cells. These results are consistent with the delivery of C6-loaded NPs described in the previous section.
KGPDC–, and –CRGDGPDC– in ADiR-RGD(−) and ADiR-CendR(−), respectively (Fig. 6C). Three days after the addition of the NPs, cell viabilities were determined by counting the live cells. The cell viabilities of ADiR-RGD(−)-PTX and ADiR-CendR(−)-PTX were approximately 26% and 30%, respectively. In the case of ADiR-PTX, the cell viability was about 16%, lower than that of deleted ADiR. The effect of both pathways, NRP-1 and integrin, should enhance the internalization of PTX into cells. These results are consistent with the delivery of C6-loaded NPs described in the previous section.
Conclusions
In this study, protein NPs displaying iRGD were designed for use as a targeted DDS. A fusion protein consisting of an ELP, a poly-aspartic acid chain, and iRGD was genetically constructed. This protein formed stable NPs by heating to above the transition temperature, due to the coacervation of ELPs and the charged repulsion of poly-aspartic acid chains. The hydrophobic anti-cancer drug, PTX, was loaded into these NPs by incubating drugs with the fusion proteins during particle formation. The NPs displayed iRGD on their surfaces, and were delivered to cancer cells via integrin binding, internalized into cells via the NRP-1 pathway, and finally induced cell death. This kind of strategy should contribute to expanding the application of protein NPs as a DDS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Grants-in Aid for Scientific Research from Japan Society for the Promotion of Science (JSPS).References
- I. C. Jenkins, J. J. Milligan and A. Chilkoti, Adv. Healthcare Mater., 2021, 10, 2100209 CrossRef CAS PubMed.
- L. Chambre, Z. Martin-Moldes, R. N. Parker and D. L. Kaplan, Adv. Drug Delivery Rev., 2020, 160, 186–198 CrossRef CAS PubMed.
- J. C. Rodriguez-Cabello, F. J. Arias, M. A. Rodrigo and A. Girotti, Adv. Drug Delivery Rev., 2016, 97, 85–100 CrossRef CAS PubMed.
- D. W. Urry, J. Phys. Chem. B, 1997, 101, 11007–11028 CrossRef CAS.
- A. Chilkoti, T. Christensen and J. A. MacKay, Curr. Opin. Chem. Biol., 2006, 10, 652–657 CrossRef CAS PubMed.
- G. M. Bressan, I. Pasquali-Ronchetti, C. Fornieri, F. Mattioli, I. Castellani and D. Volpin, J. Ultrastruct. Mol. Struct. Res., 1986, 94, 209–216 CrossRef CAS PubMed.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687–694 CrossRef CAS PubMed.
- Y. Fujita, M. Mie and E. Kobatake, Biomaterials, 2009, 30, 3450–3457 CrossRef CAS PubMed.
- R. Matsumoto, R. Hara, T. Andou, M. Mie and E. Kobatake, J. Biomed. Mater. Res., Part B, 2014, 102, 1792–1798 CrossRef PubMed.
- M. Mie, R. Matsumoto, Y. Mashimo, A. E. G. Cass and E. Kobatake, Mol. Biol. Rep., 2019, 46, 261–269 CrossRef CAS PubMed.
- R. Hanai and J. C. Wang, J. Biol. Chem., 1993, 268, 23830–23836 CrossRef CAS PubMed.
- Y. Mashimo, H. Maeda, M. Mie and E. Kobatake, Bioconjugate Chem., 2012, 23, 1349–1355 CrossRef CAS PubMed.
- F. Akter, M. Mie, S. Grimm, P. A. Nygren and E. Kobatake, Anal. Chem., 2012, 84, 5040–5046 CrossRef CAS PubMed.
- S. Fawell, J. Seery, Y. Daikh, C. Moore, L. L. Chen, B. Pepinsky and J. Barsoum, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 664–668 CrossRef CAS PubMed.
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CrossRef CAS PubMed.
- G. Elliott and P. O’Hare, Cell, 1997, 88, 223–233 CrossRef CAS PubMed.
- S. B. Fonseca, M. P. Pereira and S. O. Kelley, Adv. Drug Delivery Rev., 2009, 61, 953–964 CrossRef CAS PubMed.
- J. Stiltner, K. McCandless and M. Zahid, Pharmaceutics, 2021, 13, 890 CrossRef CAS PubMed.
- T. Teesalu, K. N. Sugahara, V. R. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157–16162 CrossRef CAS PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Drug Delivery Rev., 2017, 110–111, 3–12 CrossRef CAS PubMed.
- J. V. Gregory, P. Kadiyala, R. Doherty, M. Cadena, S. Habeel, R. Ruoslahti, P. R. Lowenstein, M. G. Castro and J. Lahann, Nat. Commun., 2020, 11, 5687 CrossRef CAS PubMed.
- H. Hu, B. Wang, C. Lai, X. Xu, Z. Zhen, H. Zhou and D. Xu, Drug Dev. Res., 2019, 80, 1080–1088 CrossRef CAS PubMed.
- J. Wang, H. Wang, J. Li, Z. Liu, H. Xie, X. Wei, D. Lu, R. Zhuang, X. Xu and S. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 19228–19237 CrossRef CAS PubMed.
- K. Wang, X. Zhang, Y. Liu, C. Liu, B. Jiang and Y. Jiang, Biomaterials, 2014, 35, 8735–8747 CrossRef CAS PubMed.
- R. Herrero-Vanrell, A. C. Rincon, R. Alonso, V. Reboto and I. T. Molina-Martinez, J. Controlled Release, 2005, 102, 113–122 CrossRef CAS PubMed.
- P. Schmidt, J. Dybal, J. C. Rodriguez-Cabello and V. Reboto, Biomacromolecules, 2005, 86, 885–891 Search PubMed.
- E. M. Kosower, Acc. Chem. Res., 1982, 15, 259–266 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |