
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
S-Nitroso-N-acetylpenicillamine grafted silicone oil for antibacterial interface applications†
Yun
Qian
 ,
Manjyot Kaur
Chug
,
Hamed
Massoumi
and
Elizabeth J.
Brisbois
*
,
Manjyot Kaur
Chug
,
Hamed
Massoumi
and
Elizabeth J.
Brisbois
*
School of Chemical, Materials and Biomedical Engineering, University of Georgia, 220 Riverbend Road, Athens, GA 30602, USA. E-mail: ejbrisbois@uga.edu; Tel: +1 (706) 542-1243
First published on 16th June 2022
Abstract
Infection remains a significant challenge in healthcare and with medical devices, resulting in two million healthcare-associated infections reported annually in the U.S. alone. Researchers are seeking new antimicrobial materials and therapies to solve the infection challenges associated with biomaterials and devices without bacterial resistance. Nitric oxide (NO) is a new therapy to treat infection, inflammation, and thrombosis, and many materials have been studied to inhibit bacteria by doping NO donor molecules and releasing NO. However, this strategy has been limited by the leaching of the NO donors which delocalizes the NO release. Silicones are widely used in making medical devices and exploring antimicrobial silicones could benefit the existing medical device interfaces. Herein, we report the first NO-releasing silicone oil (SNAP–Si) that exhibits proactive antibacterial effects. Through a two-step reaction, S-nitroso-N-acetylpenicillamine (SNAP) was grafted to poly[dimethylsiloxane-co-(3-aminopropyl)methylsiloxane], and the resulting SNAP–Si oil contained 0.6 mmol g−1 of SNAP and demonstrated storage stability at −20 °C for > 3 weeks. The SNAP–Si oil was infused in medical-grade silicone rubber (SR) surfaces, increasing the hydrophobicity of the SR interface. The SNAP–Si–SR samples released most of the NO payload in 24 h without SNAP leaching, and the highest release flux was around 3.8 × 10−10 mol min−1 cm−2 during the initial 1 h, followed by NO release at a therapeutic level for 6 h as measured using a chemiluminescence NO analyzer. SNAP–Si–SR also exhibited more than 66% and 94% reduction of viable Escherichia coli and Staphylococcus aureus on the surfaces after 3 h, respectively. Due to the easy synthesis, suitable NO release levels without leaching issues, simple infusion on a polymer surface, and antimicrobial effects, the SNAP–Si oil exhibited its potential use to create an antimicrobial medical device surface that can reduce infection challenges.
1. Introduction
Bacterial infection and biofilm formation are prevalent in daily life, and they are critical reasons for many diseases and indwelling medical device failures. Many bacterial strains that can cause infections include Gram-positive Staphylococcus aureus (S. aureus), Staphylococcus epidermidis (S. epidermidis), and Streptococcus pyogenes (S. pyogenes), and Gram-negative Pseudomonas aeruginosa (P. aeruginosa) and Escherichia coli (E. coli).1–3 When these pathogens aggregate and irreversibly attach to surfaces, they form multicellular communities in extracellular polymeric substances known as biofilms.4 Biofilms constitute a protective environment to allow bacteria to grow under hostile conditions. Compared to planktonic cells, bacterial cells in biofilms are much more difficult to kill and can lead to persistent and chronic infections.5 Bacterial infections and biofilms also cause severe diseases with high mortality and morbidity. For example, in 2019, World Health Organization (WHO) reported that infections were associated with more than 30 million deaths worldwide. Five of the top ten causes of these deaths were directly or indirectly associated with bacterial infection or biofilms.6 Additionally, ∼ 80% of the infections in humans are related directly to biofilm formation, and about 26% of all healthcare-associated infections are device-related infections.1 Failure of indwelling medical devices like intravascular catheters,7,8 urinary catheters,3 endotracheal tubes,9 tracheostomies,10 enteral feeding tubes,11 wound drains,12 and others are widely reported1,4,13 These infections and biofilms not only bring risks to the use of these medical devices and implants, but also impact the disease treatments as well as the quality of life of patients. For example, 1.6 million T1D diabetic patients require lifetime exogenous insulin, and continuous subcutaneous insulin infusion (CSII) therapy is vital for these patients.14 However, the infection, inflammation, and encapsulation complications that occur at the insulin cannula infusion site lead to 65% of infusion set failure after 7 days and result in the discontinuation of CSII therapy.15–17 Because of the large number of infection cases, fighting bacterial infection and biofilm formation are critical in both disease treatment and the application of medical devices.Although antibiotics have been used extensively to treat bacterial infections and biofilms, increasing concerns about antibiotic resistance arise, resulting in the growing demands for new therapies that kill bacteria while not inducing resistance.18 Nitric oxide (NO) is a gasotransmitter that is produced endogenously and plays a vital role in regulating various physiological pathways. NO exhibits therapeutic effects such as antithrombosis, antiplatelet, anti-inflammation, angiogenesis, vascular relaxation, as well as antiviral and antibacterial properties.19–24 The antibacterial properties of NO are based on nitrosative and oxidative stress which leads to direct modification of membrane proteins, lipid peroxidation, and DNA cleavage.25 It is noteworthy that NO was reported to eliminate various bacterial strains without an increase in resistance.26 Pathogens like S. aureus, methicillin-resistant S. aureus (MRSA), S. epidermidis, E. coli and P. aeruginosa did not show resistance after NO exposure. Repeated NO exposure of 20 passages of bacteria did not exhibit any increase of minimum inhibitory concentration (MIC).
Despite the excellent antibacterial properties of NO, localized delivery of NO using NO donor molecules is a key method to improve the antimicrobial properties of medical device surfaces. To take advantage of the antibacterial effects of NO and to control NO delivery, polymeric materials such as polysaccharides,27 polyurethane,28 polyvinyl chloride,29 polylactic acid-co-glycolic acid (PLGA),30 polycarbonate–urethane,31 silicone–polyurethane,32 silicone33 have been used. In general, NO donors have been impregnated or blended with the polymers, or covalently linked on the polymer to grant antibacterial effects to the materials.26,28,34 The impregnation, blending, or covalent bonds methods can be used to incorporate NO donors in biomaterials, stabilize the NO donor, and deliver NO at specific sites or interfaces to kill bacteria. However, these impregnation or blending approaches usually show a burst release of NO and NO donor leaching issues. Covalently conjugated NO donor with polymers could avoid the leaching issues by avoiding excessive donor leaching, lowering the initial NO release flux, and tend to have long-term NO release.35 Therefore, covalent linkage of NO to polymer structure can enhance the localized delivery of NO, which is an important concern in medical device field.
As mentioned above, many polymeric materials have been used to deliver NO locally and control the release rate. Among these polymeric materials, silicones (polysiloxane) are widely accepted polymers with a long history in biomedical and bioengineering.36 Due to their immunological inert nature, good mechanical properties, thermal stability, permeability to gases, and biocompatibility, silicone rubbers (solid silicone materials) have been used for contact lenses, cannulas, catheters, grafts and implants, scaffold, wound dressing, while silicone oils (liquid silicones) have been used for cosmetic (e.g. hair care, skin care, etc.) applications and biomedical lubricant applications.37–41 It is noteworthy that liquid silicone oils have been used to create slippery liquid-infused porous surfaces (SLIPs) by soaking materials, such as solid silicone rubbers, with silicone oils to enhance biocompatibility.42–44 To date, silicones have been used to deliver NO for antibacterial purposes, as well as extend the applications of NO in biomedical and bioengineering areas. Several studies have utilized various methods to incorporate NO release properties into silicone materials that exhibited antimicrobial and antithrombotic effects. In these studies reported in the literature related to solid silicone rubbers, SNAP was immobilized to crosslinked silicones by chemical reactions45 as well as impregnated or blended into commercial silicone rubbers.46,47 NO-releasing diazeniumdiolates (NONOates) were also formed in situ in a PDMS-based polyurethane by the reactions between polyethyleneimine and NO gas. Liquid silicone oils were also utilized to fabricate Liquid-infused NO-releasing (LINORel) silicone materials via SLIPs method, where solid silicones were impregnated with SNAP followed by infusing a thin layer of silicone oil on the surface which exhibited reduced SNAP leaching and enhanced antimicrobial properties.33,48 Materials in these studies showed promising antibacterial results, nevertheless, they still have some shortcomings. For example, the reported SNAP–silicone crosslinked polymers went through a complicated synthesis and the crosslinking happened spontaneously, so the manufacturing step (e.g., film casting) must be done immediately at the time of synthesis. Impregnation or blending of SNAP into polymers are more practical as it only requires a simple synthesis of SNAP followed by incorporation in the polymer. However, the approaches that blend or impregnate the small molecule SNAP still exhibit SNAP leaching, similar to other polymeric materials which were impregnated with SNAP. The SNAP leaching delocalizes the NO release from the polymer surface and also result in shorter NO release lifetimes. Therefore, exploring new material solutions that provide NO release without leaching concerns, and making antibacterial surfaces with easy steps could be very beneficial.
Herein, we report the successful development of the first-ever SNAP-grafted silicone oil (SNAP–Si) to deliver NO and to inhibit bacterial infections of biomedical surfaces. SNAP–Si oil provides a simple and feasible way to be infused on medical-grade silicone rubber surfaces and create antibacterial interfaces. Various analytical methods including FT-IR, NMR, and UV-vis were used to confirm the synthesis steps and evaluate the structure and stability of SNAP–Si oil. The NO release and leaching of the NO donor functionalities of SNAP–Si oil-treated silicone samples (SNAP–Si–SR) were also studied. In addition, SNAP–Si–SR surfaces were tested against both Gram-positive S. aureus and Gram-negative E. coli. to evaluate the antimicrobial effects of SNAP–Si.
2. Materials and methods
2.1. Materials
N-Acetyl-D-penicillamine (NAP) was purchased from Sigma Aldrich (USA); S-nitroso-N-acetylpenicillamine (SNAP) was purchased from Pharmablock USA, Inc.; t-butyl nitrite, tetrahydrofuran (THF), dichloromethane (DCM), pyridine, acetic anhydride, ninhydrin, acetic acid, ethanol, ethylenediaminetetraacetic acid (EDTA), and Tween 20 were purchased from Fisher Scientific (USA); phosphate buffer saline solution (10 mM PBS, pH 7.4) was prepared by the protocol from Cold Spring Harbor Laboratory, and all ingredients (sodium phosphate dibasic, potassium phosphate monobasic, sodium chloride, potassium chloride) were purchased from Fisher Scientific. EDTA (100 μM) was added to the PBS to chelate any trace metal in the buffer and prevent catalyzed NO release from the samples. Poly[dimethylsiloxane-co-(3-aminopropyl)methylsiloxane] (NH2–Si, AMS-191) was purchased from Gelest (USA). Medical grade silicone sheet (87315K13) was purchased from McMaster-Carr (USA). All materials were used without purification unless mentioned specifically. Luria Bertani (LB) broth media and agar were purchased from Sigma Aldrich. Bacterial strains Escherichia coli (E. coli, ATCC 25922) and Staphylococcus aureus (S. aureus, ATCC 6538) were obtained from American Type Culture Collection (ATCC).2.2. Synthesis and characterization of S-nitroso-N-acetylpenicillamine grafted silicone oil (SNAP–Si).
The initial amine concentration of aminate silicone oil (NH2–Si) was determined by a modified ninhydrin assay.50 Ninhydrin solution was prepared by dissolving ninhydrin (0.2 g) and acetic acid (0.5 mL) in ethanol (99.5 mL). The sample solution was prepared by dissolving sample (1 mg) in Tween 20 solution (1 mL, 1% w/v in H2O); then, sample solution (0.5 mL), Tween 20 solution (0.5 mL) and ninhydrin solution (0.5 mL) were mixed and heated in boiling water for 10 min. After the addition of ethanol (2.5 mL), the solution was checked by UV-vis at 570 nm and the concentration was obtained using a cysteine calibration curve.
The SNAP concentration of SNAP–Si oil was quantified by a Cary 60 UV-vis spectrophotometer using a SNAP calibration curve in THF. UV-vis spectra were taken within 300–500 nm wavelength at a medium scan speed. Commercial SNAP (Pharmablock) was dissolved in THF at the concentrations of 0.1, 0.25, 0.5, 0.75, and 1 mM, and then the absorbance of each SNAP solution was measured at 340 nm to make a calibration curve. To confirm the SNAP concentration in SNAP–Si oil, SNAP–Si was dissolved in THF (1 mg mL−1 solution), and then its absorbance at 340 nm was measured and used to calculate the concentration of SNAP using the SNAP calibration curve.
2.3. SNAP–Si oil stability
The stability of SNAP–Si oil was monitored by checking the quantity of the SNAP functionality at designed time points using UV-vis spectroscopy. The SNAP–Si oil was placed in amber vials either in −20 °C freezer, rt, or 37 °C incubator to evaluate the storage temperature stability for up to 4 weeks. At the designed timepoint, each sample was removed from storage, dissolved in THF at a 1 mg mL−1 concentration, and measured for absorbance at 340 nm using UV-vis. The percentage of SNAP remaining at each timepoint was quantified with respect to initial absorbance on the first day, and plotted to show the storage stability under different temperature conditions.2.4. Preparation of SNAP–Si–SR and NAP–Si–SR disks
A medical grade silicone sheet was punched into 0.7 cm diameter disks, and then soaked in the SNAP–Si oil or NAP–Si oil in THF (100 mg mL−1) at −20 °C in dark for 12 h. Samples were removed and dried in a fume hood for 12 h before weighing. The soaking process was monitored by swelling ratio, and the mass increase of samples was used to quantify the swelling. Weights of samples after swelling (wt) were compared with their original weights (wi). The swelling ratio of each group was taken with the average of three samples, using eqn (1) below.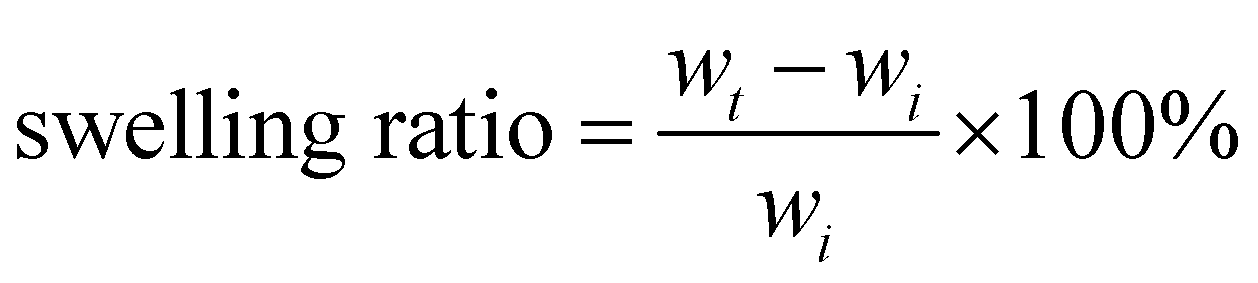 | (1) |
2.5. Water contact angles of disks
Static contact angles of samples were measured by an Ossila Contact Angle Goniometer (Ossila, UK). Sample disks were placed on the sample stage of contact angle goniometer, and 5 μL of deionized water were dropped on the surface. The static contact angles were measured from still frames using the sessile drop approximation, and the results were analyzed by Ossila Contact Angle Software. Three individual water droplets were placed at 3 random locations on the sample surface and then averaged to obtain the average contact angles.2.6. Leaching of the SNAP–Si oil from SNAP–Si–SR disks
In order to evaluate any SNAP–Si leaching from the SR matrix, each SNAP–Si–SR disk was soaked in 1 mL of 10 mM PBS (pH 7.4) containing 100 μM EDTA at 37 °C in an incubator. To determine the amount of SNAP–Si oil leached out, the absorbance at 340 nm of the 1 mL of soaking buffer was measured by UV-vis and the absorption was recorded. At the same time, the sample vial was replenished with 1 mL of fresh 10 mM PBS (pH 7.4, 100 μM EDTA), and incubated at 37 °C until the next reading.2.7. NO release profiles of SNAP–Si–SR disks
The NO released from the SNAP–Si treated disk samples was analyzed by a gold-standard Zysense chemiluminescence Nitric Oxide Analyzer (NOA) 280i as previously reported,29 where the supply nitrogen flow rate and cell pressure were set at 200 mL min−1 and 8.8–9.5 psi, respectively. The sample cell was incubated in a 37 °C water bath to mimic physiological temperature, and 3 mL of PBS buffer (10 mM, pH 7.4, containing 100 μM EDTA) was added to the reaction vessel. Each test started with a short period of baseline measurement, and then SNAP–Si–SR disk was placed in the buffer within the sample cell. The NO released from the sample was purged by continuous N2 flow and was detected in real-time by the chemiluminescence detector at a 1 s interval until it reached steady-state. The NOA data was normalized with the surface area of samples to obtain the flux values with units of mole cm−2 min−1. The NO release was quantified at various timepoints during the experiment to measure the release trends. The samples were incubated in PBS at 37 °C between each measurement.2.8. Evaluating the antibacterial efficacy of SNAP–Si–SR disks
The antibacterial efficacy of SNAP–Si–SR disk samples was evaluated against two strains of bacteria Gram-positive (S. aureus) and Gram-negative (E. coli) in a 3 h antibacterial adhesion assay following previously reported protocol.52 Briefly, an individual bacteria colony was inoculated in LB media and grown to mid-log phase. Cells were extracted from the mid-log phase and the optical density of bacteria suspension was recorded using UV-vis spectroscopy at 600 nm wavelength. Then, the cells were washed and resuspended in fresh media (final bacteria concentration ≈ 107 CFU mL−1). The UV-sterilized SR control, NAP–Si–SR control, and SNAP–Si–SR disks were incubated in the bacterial suspension for 3 h at 120 RPM, 37 °C. After 3 h, samples were briefly rinsed to remove any loosely adhered bacteria on the surface and resuspended in sterile PBS. To detach the adhered bacteria on the samples, bacteria exposed disks were homogenized and vortexed for 60 sec each. The bacteria suspension was then plated on LB agar plates using a bacteria spiral plater (Eddy Jet 2W, IUL instruments). Plates were incubated at 37 °C overnight and the viable colonies on the LB agar plate were enumerated to determine the concentration of bacteria (CFU mL−1) using colony counter (SphereFlash, IUL instruments). The antibacterial activity of SNAP–Si–SR disks compared to controls was determined using eqn (2) and reported as CFU cm−2 of polymer surface area.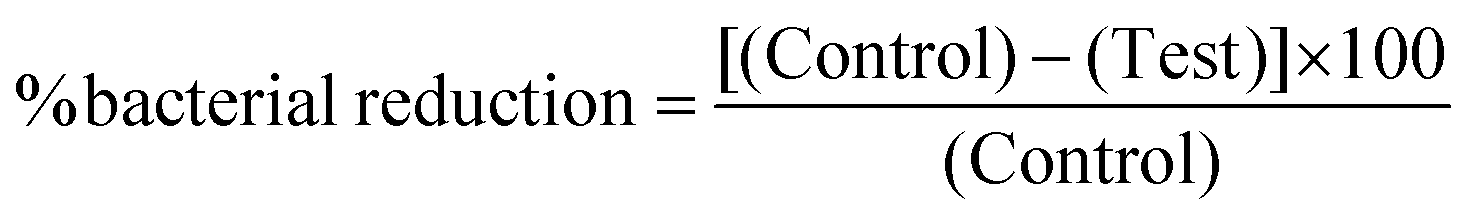 | (2) |
2.9. Statistical analysis
All the data were obtained with sample size ≥ 3, and data are reported as mean ± standard deviation. A two-tailed Student's t-test with a hypothesis of unequal variance and α= 0.05 were used to determine statistical significance.3. Results and discussion
3.1. S-Nitroso-N-acetyl-D-penicillamine grafted silicone oil (SNAP–Si) synthesis
The synthesis of SNAP–Si was achieved via two steps of synthesis: the first step is to graft NAP–thiolactone to the aminated silicone oil (NH2–Si), followed by a second step of nitrosation of the thiol groups (Fig. 1A). This synthesis can be visualized directly due to the color changes. The starting NH2–Si oil was transparent, but it turned green after the nitrosation due to green color of the tertiary S-nitrosothiol SNAP structure, indicating the successful grafting of SNAP moiety on the liquid silicone molecules. The initial amine concentration of NH2–Si was determined by the ninhydrin calibration curve where the amine concentration of the initial NH2–Si was ∼ 1.5 ± 0.41 mmol g−1 (Fig. 1B). After coupling with NAP–thiolactone via a ring-opening reaction, the NH2–Si was converted to NAP–Si which contains the tertiary thiols. The residual amine concentration was ∼ 0.5 ± 0.13 mmol g−1 determined by the ninhydrin test, which indicated a ca. 68% conversion of free amines to immobilized NAP, resulting in ca. 1.0 ± 0.13 mmol g−1 of thiols in the NAP–Si oil. The second step of nitrosation (converting the immobilized R–SH functionalities to R–SNO) resulted in green color silicone oil, and the SNAP concentration was quantified by UV with the calibration curve. The final SNAP concentration was approximately 0.6 mmol g−1 in SNAP–Si, which correlates to a ca. 54% conversion from NAP to SNAP.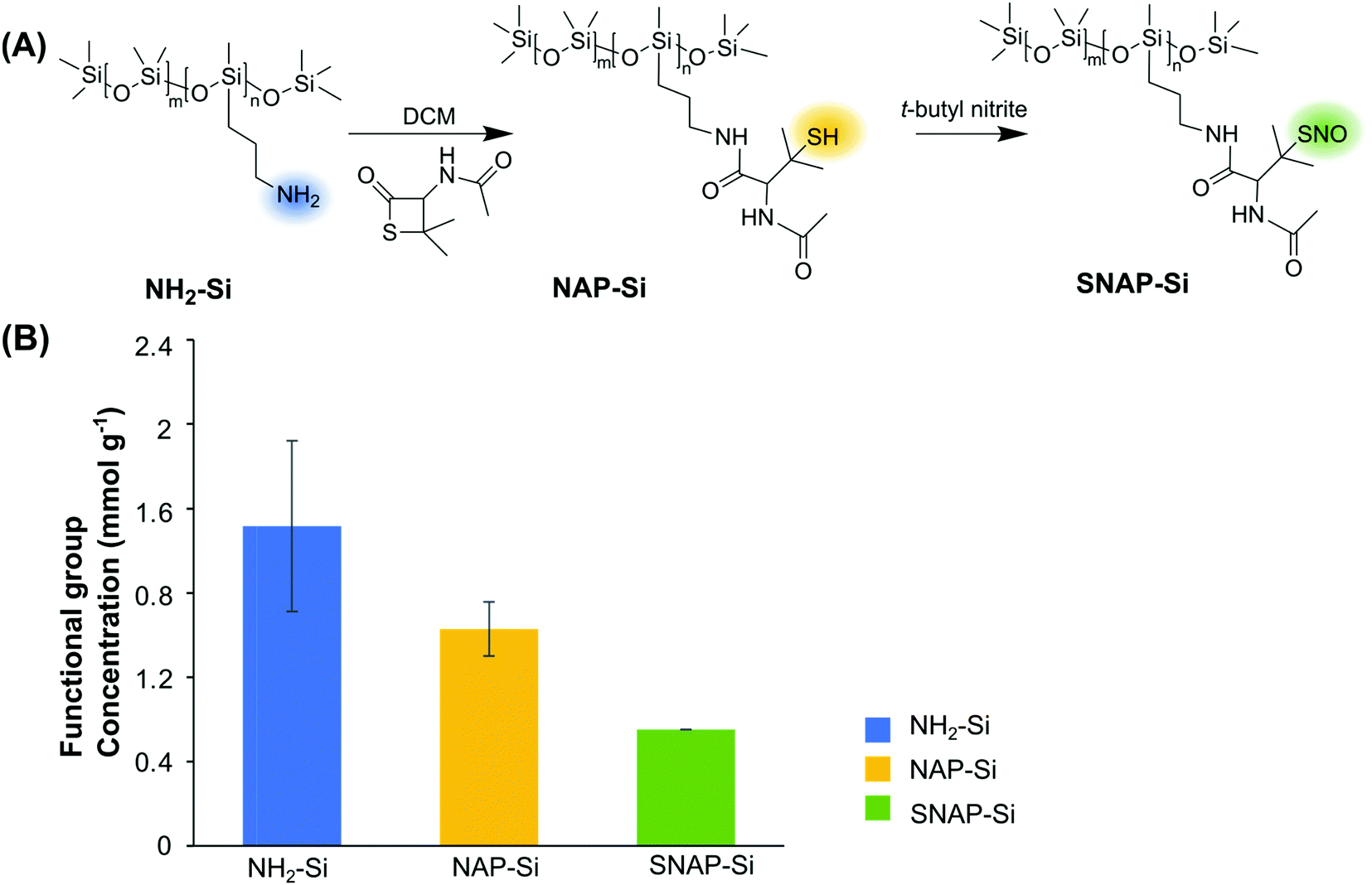 | ||
| Fig. 1 (A) The synthesis scheme of NAP–Si oil and SNAP–Si oil. (B) The concentrations of functional groups on silicone oils as measured by UV-vis spectroscopy assays. The [NH2] of NH2–Si oil was determined by ninhydrin assay, the [HS] of NAP–Si was calculated by subtracting the remaining [NH2] from the initial [NH2] of NH2–Si, and the [SNAP] of SNAP–Si oil was measured by SNAP calibrating curve in THF. (n = 3 and error bars represent standard deviations). | ||
FT-IR and NMR were used to confirm the chemical structures of SNAP–Si. As shown in Fig. 2, NH2–Si oil showed a peak around 1584 cm−1 which was assigned to primary amine N–H bending, and 3424 cm−1 could be either H-bonded silanol or N–H stretching.53,54 For NAP–Si oil, the decrease of amine peak around 1584 cm−1 and the appearance of new secondary amide peaks around 3300 cm−1 suggested that NAP–thiolactone was coupled to the structure. For SNAP–Si oil, the peaks around 1644 cm−1 and 1514 cm−1 appeared to represent the secondary amide C = O stretching and the N–O stretching of the S-nitrosothiol, respectively, demonstrating the SNAP moiety formation.55,56
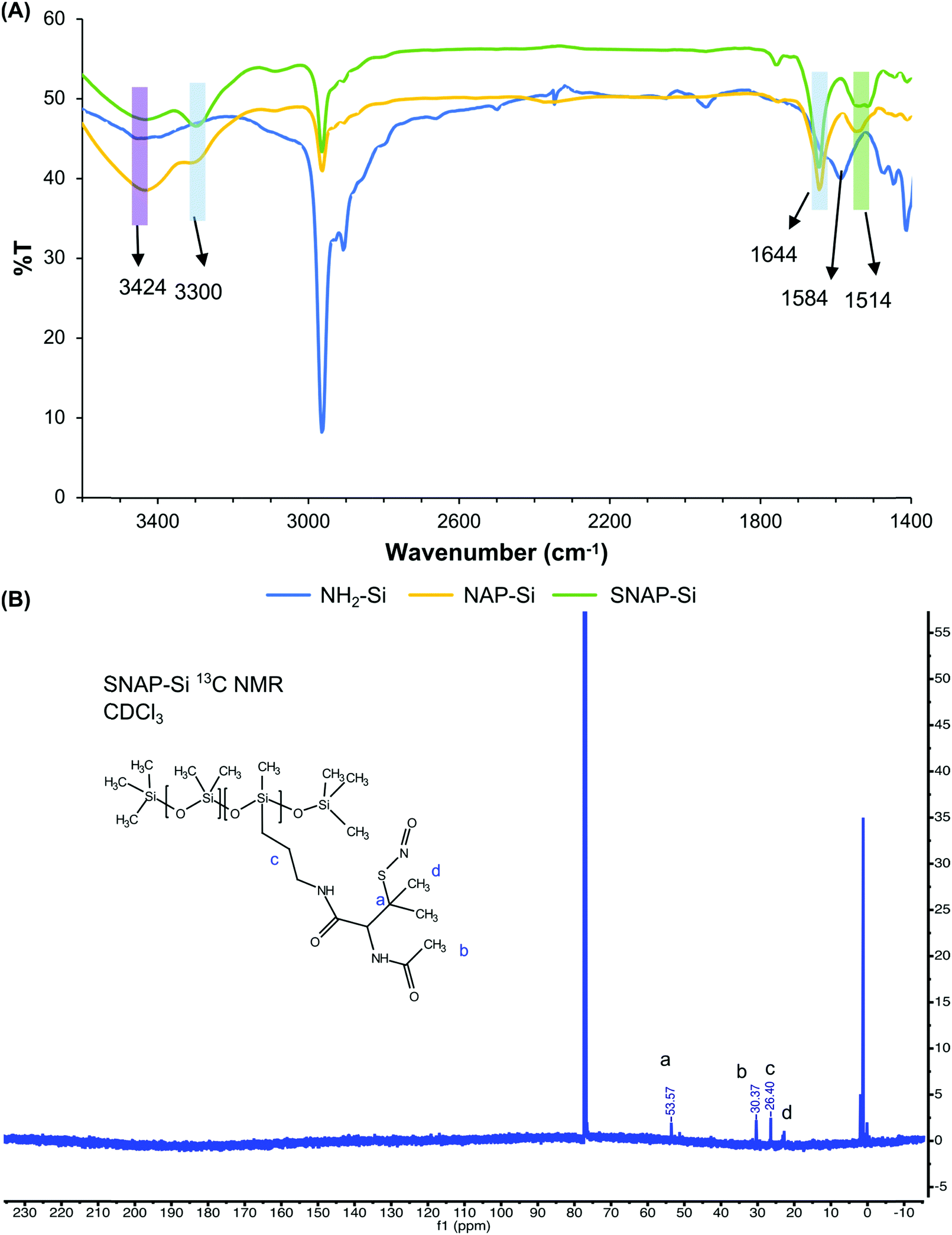 | ||
| Fig. 2 (A) FT-IR of NH2–Si, NAP–Si and SNAP–Si. (B) 13C NMR of SNAP–Si oil using CDCl3. Chemical shift at 53.57 ppm represented the characteristic carbon linked to SNO group. | ||
1H and 13C NMR were performed to confirm the product synthesis using an Agilent/Varian VNMR600 MHz instrument (Fig. 2, Fig. S1–S3, ESI†). Compared with NH2–Si oil, 1H chemical shifts at 1.62, 1.85, and 2.04 ppm appeared in NAP–Si oil, indicating that NAP was tethered to the NH2–Si oil via amide bonds. For NAP–Si oil, 13C chemical shifts were also observed at 22.74, 30.37, 51.29, 60.47, 169.53, and 170.32 ppm (Fig. S2, ESI†), while for NH2–Si oil, 13C chemical shifts were observed at 0.40, 1.18, 1.93, 14.60, 27.61, and 45.46 ppm (Fig. S3, ESI†). The changes of chemical shifts were related to the conjugation of NAP group to NH2–Si side chain. Among these new peaks appeared in NAP–Si, 169.53 and 170.32 ppm were assigned to carbonyls and confirmed the NAP on silicone (Fig. S2, ESI†). Even though the chemical shifts of 1H and 13C NMR for NAP–Si (Fig. S2, ESI†) and SNAP–Si (Fig. S3, ESI†) oils did not show dramatic changes before and after the nitrosation, a small change of 13C NMR in SNAP–Si was observed after the S-nitrosothiol formation. The NAP–Si and SNAP–Si oils had 13C NMR chemical shifts at 51.29 (Fig. S2, ESI†) and 53.57 ppm (Fig. S3, ESI†), respectively, and the 2.28 ppm shift to downfield could be induced by the nitroso group. It is noteworthy that in this study, SNAP moiety was grafted on silicone oil molecules, and the resulting SNAP–Si oil is a homogenous liquid that can be used as a lubricant or to coat polymer surfaces directly and release NO gas at material interfaces.
3.2. SNAP–Si oil stability
NO donors, like SNAP, hold great potential for biocompatibility and antibacterial applications. However, the instability of some NO donor molecules can limit their potential future biomedical applications. For example, SNAP can decompose in the presence of heat or light57,58 which could limit the shelf storage stability. The shelf time of SNAP–Si oil is a critical issue; thus the temperature stability was studied by monitoring the remaining SNAP concentration in SNAP–Si under different temperature conditions (Fig. 3). Three different temperatures (−20 °C, rt, and 37 °C) for up to 4 weeks were used to determine the best thermal conditions for potential storage and transport. The results from the study demonstrated that the SNAP–Si oil samples were much more stable at −20 °C as compared to rt and 37 °C. Data suggests that ca. 89.21% of SNAP was present in the samples after 4 weeks of −20 °C storage relative to initial values obtained from samples on day 0. While the SNAP–Si oil seemed stable at −20 °C, it was found less stable at rt and 37 °C with ca. 0.53 and 0.98% SNAP remaining after only 7 days of storage, respectively. The instability of SNAP–Si oil at these higher temperatures is due to the thermal decomposition mechanism of SNAP.59,60 As the temperature increases, the thermal decomposition of S–NO bond accelerates, generating more NO and sulfonyl radicals from the SNAP–Si, making SNAP–Si oil less stable.60 Prior literature has reported excellent stability of the small molecule SNAP in a range of biomedical polymers, where SNAP is stable in these polymers at room temperature and elevated temperatures of 37 °C for several months (< 10% loss).32,61–63 The stability of SNAP in these materials is due to the polymer–crystal composite that forms, where the SNAP stored in the polymer is in its crystalline form, where intramolecular bonding stabilizes the R–SNO functionality leading to enhanced stability. In contrast, the SNAP–Si oil studied here is in the liquid phase where the SNAP immobilized to the oil is not crystallized, and therefore exhibits the thermal instability observed in this stability study. Overall, the results obtained from the stability study reported here justify that SNAP–Si oil by itself permits long-term storage in colder temperatures (−20 °C) with faster degradation of material at higher temperatures.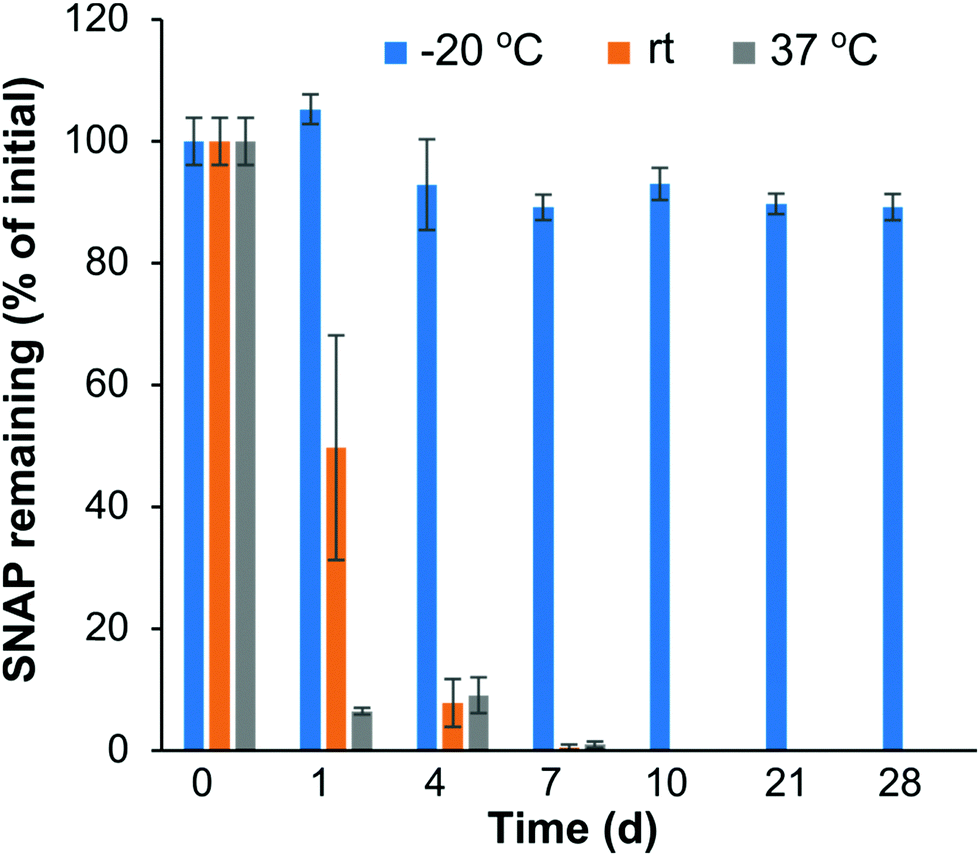 | ||
| Fig. 3 Stability of SNAP–Si was measured at three different storage conditions −20 °C, room temperature (rt), and 37 °C, over 28 d. The absorbance of the SNAP–Si were measured by dissolving the material in THF (1 mg mL−1) at the initial timepoint and after the storage. (n = 3 and error bars represent standard deviations). | ||
3.3. SNAP–Si oil infused silicone disks
The SNAP–Si–SR and NAP–Si–SR disks were prepared by infusing the SR polymer surfaces with either the SNAP–Si oil or NAP–Si oil to create NO-releasing or control surfaces, respectively. The resulting SNAP–Si–SR disks were light green due to the color of SNAP. To infuse the same amount of SNAP–Si and NAP–Si oils into SR disks, the swelling ratio of SR disks was studied. Samples were removed from the oil solutions at designed timepoints during the soaking, gently wiped with Kimwipe to remove excess solution, and weighed. The increased weights represent the infused oil in the disk, where both sample types had increasing weights that reached steady-state infusion after ca. 12 h (Fig. 4). The SNAP–Si–SR and NAP–Si–SR disks gained about 3.7% weight after 12 h of infusion in the respective oils.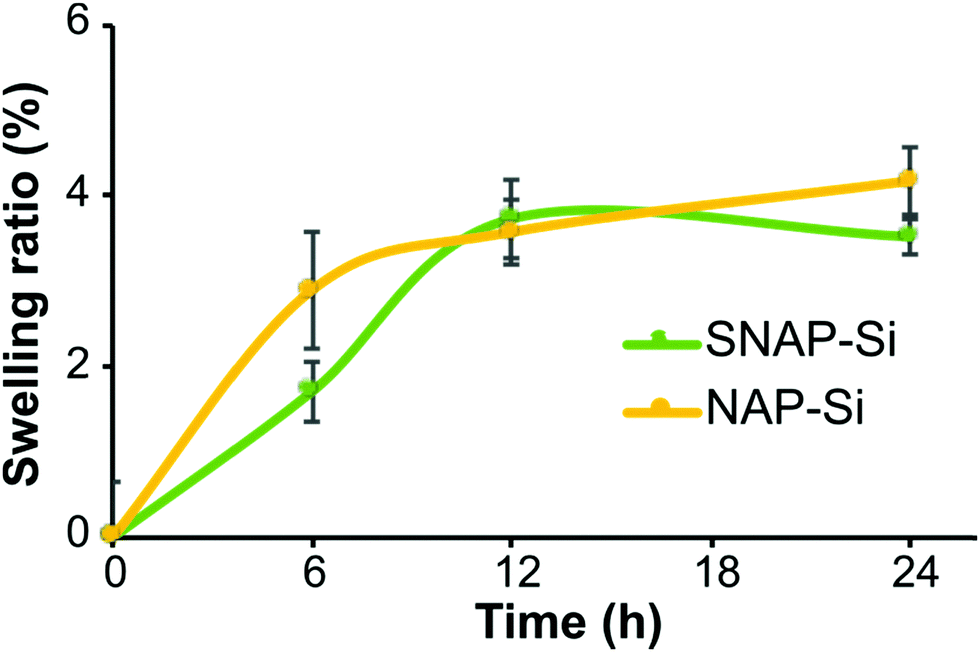 | ||
| Fig. 4 Swelling ratio of silicone disks in different silicone oils. Silicone oils were dissolved in THF at 100 mg mL−1. Disks were soaked in the solutions for the designed time at −20 °C, then taken out and dried in dark overnight before weighing. (n = 3 and error bars represent standard deviations). | ||
3.4. Static contact angles
The static contact angles of disks were determined by an Ossila Contact Angle Goniometer and the static angles results are presented in Fig. 5. The original commercial SR disks had a static contact angle of ∼ 86 ± 4.5°. While the contact angles of silicone materials range from hydrophilic (20–70°) to hydrophobic (95–122°),37,62,64–66 most of the non-hydrogel silicone rubbers are hydrophobic. Despite the experiment errors and other polymeric components which may affect the SR contact angles, a possible reason for the slight reduction in hydrophobicity of the SR material used in this study could be silicone oligomers on the SR surface. Crosslinked silicone rubbers have silicone oligomers on the surface, and these oligomers have been reported to cause contact angle changes due to adaptive wetting behavior. The oligomers could absorb water form a thin oligomer–water lubricant layer, and at the same time the absorbed water could pull more oligomers from the matrix to enhance lubricant layer. Due to the existence of oligomer–water lubricant layer, the contact angle would decrease.67 The NAP–Si–SR had a static contact angle of about 88 ± 1.6°, indicating that surface infusion of NAP–Si oil had almost no changes on SR disk surface wettability properties. However, the static contact angle of SNAP–Si–SR was 103 ± 3.1°, showing that SNAP–Si oil slightly increased the hydrophobicity of the SR surface. These results suggested that the SNAP–Si–SR surface became more hydrophobic than the controls and had similar hydrophobicity like SNAP impregnated silicone materials (104–110°) reported in prior literature.31,68–70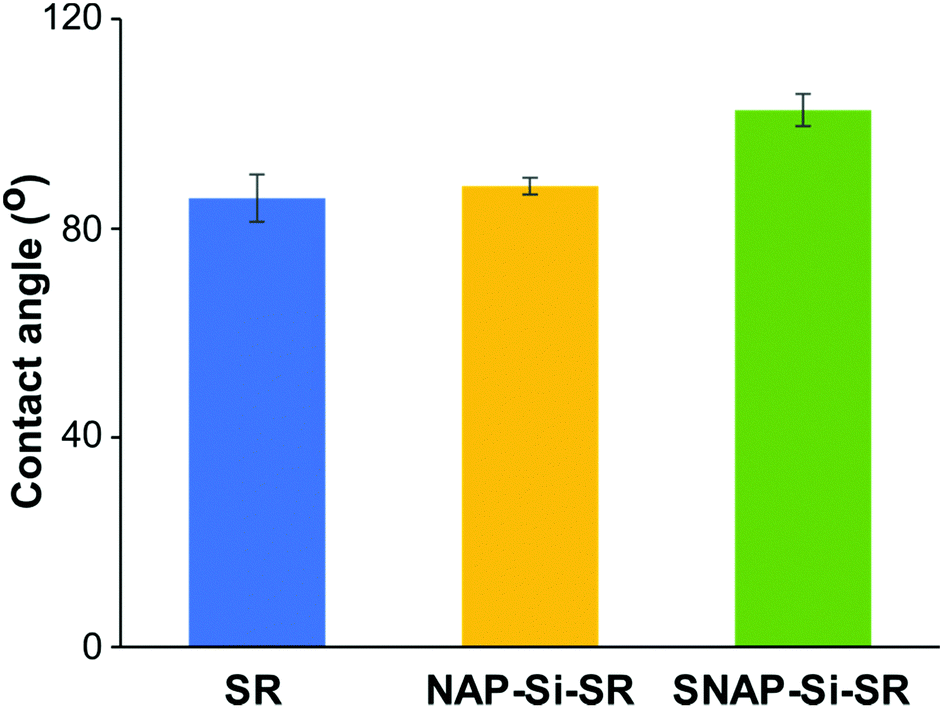 | ||
| Fig. 5 Static contact angles of different disk samples. The contact angles were measured by Ossila Contact Angle Goniometer with 5 μL water droplets on disk surface. (n = 3 and error bars show standard deviations). | ||
3.5. NO release and SNAP leaching of SNAP–Si–SR disks
The NO release profiles of the SNAP–Si–SR disks were measured by a Zysense chemiluminescence NOA 280i at 37 °C while incubated in 3 mL of PBS buffer (10 mM, pH 7.4, with 100 μM EDTA).33 The NO release profiles are shown in Fig. 6. The SNAP–Si–SR was able to release NO at physiologically relevant levels (0.7–3.8 × 10−10 mol min−1 cm−1) for > 6 h, and then continued the low NO flux level (0.2–0.7 × 10−10 mol min−1 cm−1) for up to 2 d until the NO payload was depleted. This high NO flux during the initial period and then gradually decreased NO release trend is commonly seen in other NO-releasing materials.28,32 In addition, the leaching of NO donors is always a safety concern for NO-releasing materials. Thus, the leaching of SNAP–Si oil from SNAP–Si–SR was analyzed using UV-vis spectroscopy (Fig. S4, ESI†). No SNAP–Si oil was detected in incubation PBS buffer, suggesting no SNAP leaching during the NO release period and no inferences of leachates during antibacterial studies.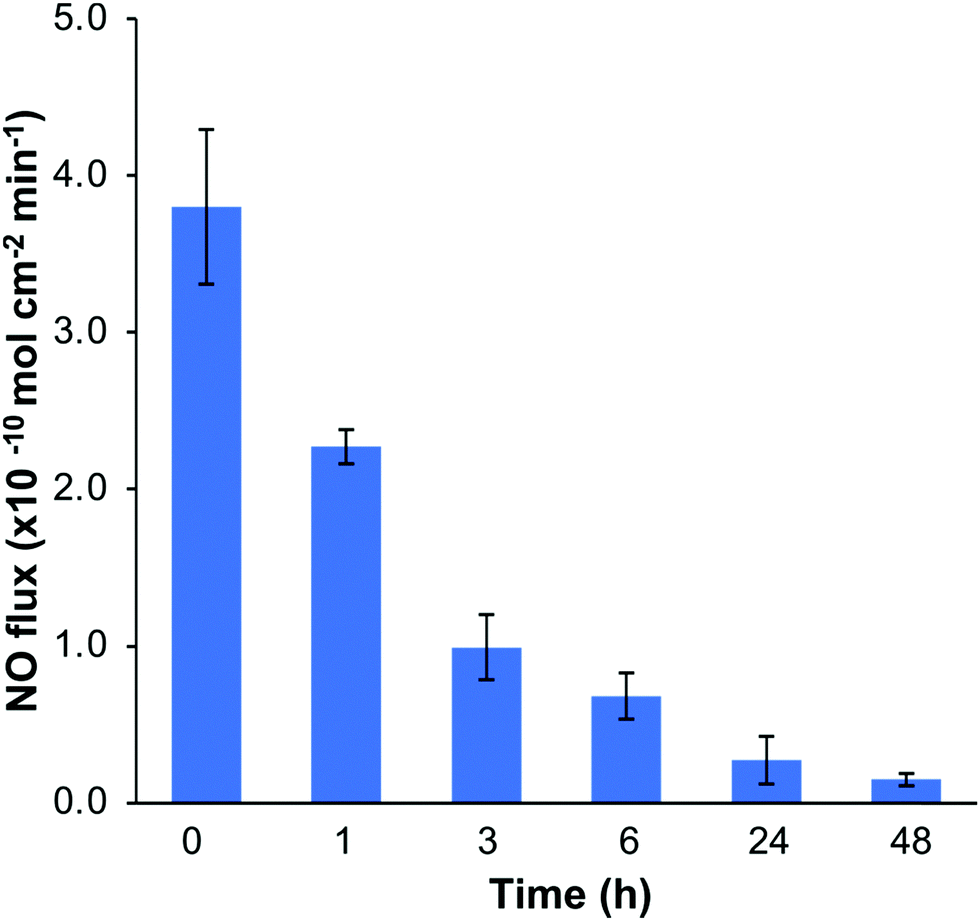 | ||
| Fig. 6 Measurement of real-time NO release using a chemiluminescence nitric oxide analyzer (NOA). The NO flux levels were measured in PBS with 100 μM EDTA at 37 °C. (n = 3, and data represent mean ± standard deviation). | ||
3.6. Bacterial assays of SNAP–Si–SR disks
The antibacterial properties of the SNAP–Si–SR samples and controls were analyzed in a 3 h bacterial adhesion assay against S. aureus and E. coli (n ≥ 3). The bacterial cells adhered to the surface of films were enumerated and normalized to the surface area of the disks (CFU cm−2). The SNAP–Si–SR disks exhibited a 94.1% and 66.1% reduction in viable S. aureus and E. coli, respectively, compared to SR control due to the action of NO (Fig. 7). The SNAP–Si–SR disks killed both S. aureus and E. coli bacteria, and these results corroborate with other NO-releasing surfaces previously reported that exhibit similar broad-spectrum antibacterial activities.29,32,71,72 The NAP–Si–SR control disks did not exhibit significant reductions in bacterial adhesion in comparison to the viable bacteria adhered to the SR controls. Therefore, the antibacterial effects of SNAP–Si–SR were attributed to the active NO release from the polymer surface. NO is known to eradicate bacteria by non-specific processes which include the formation of N2O3 and NO3−1 upon interaction with oxygen, and these oxidative molecules trigger cleavage of bacterial DNA and membranal damage.73 The SNAP–Si–SR samples demonstrated higher antibacterial efficacy against Gram-positive S. aureus as compared to Gram-negative E. coli. This may be attributed to the slight decrease in bactericidal efficiency of NO against Gram-negative bacteria due to the production of flavohemoglobins enzymes that can protect bacteria from reactive nitrogen and oxygen stress generated by NO.74 Moreover, the structural differences between the Gram-positive and Gram-negative bacteria can be another potential reason, where the presence of an additional outer membrane lowers the susceptibility of bacteria against various antibacterial agents.75 Comparable results have been reported in the literature with NO-releasing materials that show decreased bactericidal efficiency against E. coli and other Gram-negative bacteria.68,76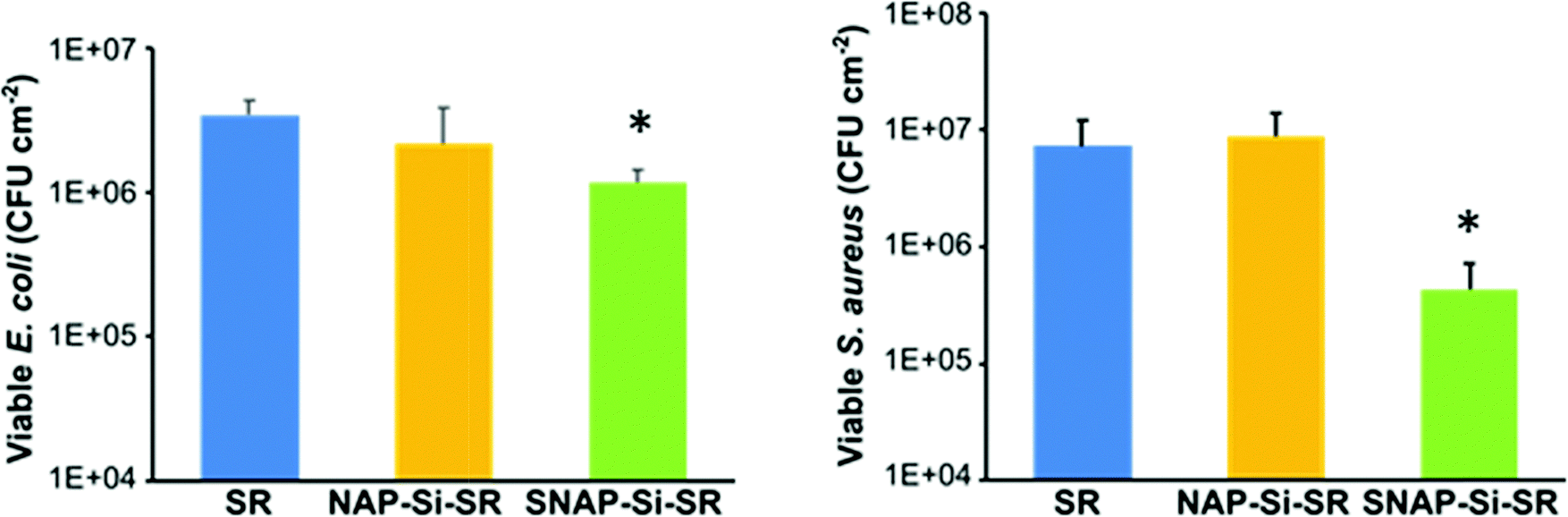 | ||
| Fig. 7 Antimicrobial effects of the SR control, NAP–Si–SR control, and SNAP–Si–SR disks. (n ≥ 3, and error bars represent standard deviation). * = p < 0.05 for SNAP–Si–SR vs. SR control. | ||
Previous reports on SNAP-based biomaterials have demonstrated the antimicrobial effects against various pathogenic bacteria and fungus strains in medical device-related infections such as P. aeruginosa, S. epidermidis, and C. albicans.77–79 Nevertheless, these prior studies involved either blending or impregnating the materials with NO donor SNAP, which are often challenged by leaching of the small molecule NO donors that delocalizes the NO release from the polymer surfaces. In this study, covalently grafting the SNAP moiety to the silicone oil avoided the leaching concerns because the SNAP–Si oil is not miscible with water (as showing in Fig. S4, ESI†). In comparison to traditional antibiotic and metal ion releasing antimicrobial materials, NO-releasing surfaces not only inhibit the bacterial adhesion and kill bacteria without causing resistance and toxicity, but can also provide other biocompatible properties such as reducing surface thrombosis of blood-contacting interfaces.26,31,80,81 Overall, the antibacterial results from this study and the previous success of NO-releasing materials suggests that SNAP–Si oil can be a promising and simple approach to solving bacterial infections for short-term applications. For example, the surfaces of insulin cannulas, intravascular catheters, or other device surfaces could be lubricated with the SNAP–Si oil to infuse a thin layer of the NO-releasing oil on the surface, resulting in the antimicrobial effects observed in this study.
4. Conclusions
In this study, the SNAP–Si oil was successfully synthesized by grafting the NO donor functionality to the liquid silicone oil which was confirmed by UV-vis, FT-IR, and NMR analysis. The SNAP–Si oil was stable in −20 °C freezer storage and retained more than 89% of the initial SNAP after 4 weeks of storage. When SNAP–Si oil was infused and lubricated the SR disk surface, the SNAP–Si–SR samples released physiological NO levels of 3.8 × 10−10 mol min−1 cm−1 during the initial 1 h which continued for up to 2 days without any leaching. The antibacterial effects of the SNAP–Si were tested using a 3 h bacterial adhesion assay against S. aureus and E. coli. The SNAP–Si resulted in a 94.1% and 66.1% reduction in viable S. aureus and E. coli on the SR surfaces, respectively. Results of this study suggest that the SNAP–Si oil is a new NO-releasing silicone-based material that demonstrated antibacterial effects on both Gram-positive and Gram-negative bacteria. Due to the ease of synthesis and excellent broad-spectrum antibacterial effects, the SNAP–Si oil could be potentially used for the development of future antibacterial coating or lubricant for medical devices (e.g. cannulas, catheters, tubing, or topical biomaterial applications).Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Juvenile Diabetes Research Foundation (JDRF) was the funding provider for this project (JDRF 1-SRA-2021-1062-S-B).References
- C. R. Arciola, D. Campoccia and L. Montanaro, Nat. Rev. Microbiol., 2018, 16, 397–409 CrossRef CAS PubMed.
- A. Fusco, L. Coretti, V. Savio, E. Buommino and F. Lembo, et al. , Int. J. Mol. Sci., 2017, 18, 414 CrossRef PubMed.
- F. Rahimi, M. Katouli and S. Karimi, Microb. Pathog., 2016, 98, 69–76 CrossRef CAS PubMed.
- M. Jamal, W. Ahmad, S. Andleeb, F. Jalil and M. Imran, et al. , J. Chin. Med. Assoc., 2018, 81, 7–11 CrossRef PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- The Top 10 Causes of Death, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death, Accessed May 1, 2021.
- L. Zhang, S. Cao, N. Marsh, G. Ray-Barruel and J. Flynn, et al. , J. Infect. Prev., 2016, 17, 207–213 CrossRef PubMed.
- C. Beloin, N. Fernández-Hidalgo and D. Lebeaux, Intensive Care Med., 2017, 43, 443–446 CrossRef PubMed.
- O. Diaconu, I. Siriopol, L. I. Poloşanu and I. Grigoraş, J. Crit. Care Med., 2018, 4, 50–55 CrossRef PubMed.
- C.-Y. Tan, N.-C. Chiu, K.-S. Lee, H. Chi and F.-Y. Huang, et al. , J. Microbiol., Immunol. Infect., 2020, 53, 315–320 CrossRef PubMed.
- N. A. Juma and S. J. Forsythe, in Biofilm-based Healthcare-associated Infections, ed. G. Donelli, Springer International Publishing, 2015, pp. 113–121 Search PubMed.
- P. Barbadoro, C. Marmorale, C. Recanatini, G. Mazzarini and I. Pellegrini, et al. , Am. J. Infect. Control, 2016, 44, 283–288 CrossRef PubMed.
- B. Pittet, D. Montandon and D. Pittet, Lancet Infect. Dis., 2005, 5, 94–106 CrossRef PubMed.
- Type 1 Diabetes Facts, https://www.jdrf.org/t1d-resources/about/facts/, Accessed June 21, 2021.
- T. S. Guinn, G. J. Bailey and R. S. Mecklenburg, Diabetes Care, 1988, 11, 46–51 CrossRef CAS PubMed.
- R. S. Mecklenburg, E. A. Benson, J. W. Benson, Jr., P. N. Fredlund and T. Guinn, et al. , JAMA, 1984, 252, 3265–3269 CrossRef CAS PubMed.
- A. Guenego, G. Bouzillé, S. Breitel, A. Esvant and J.-Y. Poirier, et al. , Diabetes Technol. Ther., 2016, 18, 820–824 CrossRef CAS PubMed.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- P. Vallance, Fundam. Clin. Pharmacol., 2003, 17, 1–10 CrossRef CAS PubMed.
- Y. Qian and J. B. Matson, Adv. Drug Delivery Rev., 2017, 110-111, 137–156 CrossRef CAS PubMed.
- M. R. Garren, M. Ashcraft, Y. Qian, M. Douglass and E. J. Brisbois, et al. , Appl. Mater. Today, 2021, 22, 100887 CrossRef.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- A. Ghaffari, C. C. Miller, B. McMullin and A. Ghahary, Nitric oxide, 2006, 14, 21–29 CrossRef CAS PubMed.
- L. M. Estes, P. Singha, S. Singh, T. S. Sakthivel and M. Garren, et al. , J. Colloid Interface Sci., 2021, 586, 163–177 CrossRef CAS PubMed.
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef PubMed.
- B. J. Privett, A. D. Broadnax, S. J. Bauman, D. A. Riccio and M. H. Schoenfisch, Nitric oxide, 2012, 26, 169–173 CrossRef CAS PubMed.
- M. J. R. Ahonen, J. M. Dorrier and M. H. Schoenfisch, ACS Infect. Dis., 2019, 5, 1327–1335 CrossRef CAS PubMed.
- E. J. Brisbois, J. Bayliss, J. Wu, T. C. Major and C. Xi, et al. , Acta Biomater., 2014, 10, 4136–4142 CrossRef CAS PubMed.
- C. G. Feit, M. K. Chug and E. J. Brisbois, ACS Appl. Bio Mater., 2019, 2, 4335–4345 CrossRef CAS PubMed.
- G. Lautner, M. E. Meyerhoff and S. P. Schwendeman, J. Controlled Release, 2016, 225, 133–139 CrossRef CAS PubMed.
- A. Mondal, M. Douglass, S. P. Hopkins, P. Singha and M. Tran, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 34652–34662 CrossRef CAS PubMed.
- E. J. Brisbois, H. Handa, T. C. Major, R. H. Bartlett and M. E. Meyerhoff, Biomaterials, 2013, 34, 6957–6966 CrossRef CAS PubMed.
- M. K. Chug, C. Feit and E. J. Brisbois, ACS Appl. Bio Mater., 2019, 2, 5965–5975 CrossRef CAS PubMed.
- S. P. Hopkins, J. Pant, M. J. Goudie, C. Schmiedt and H. Handa, ACS Appl. Mater. Interfaces, 2018, 10, 27316–27325 CrossRef CAS PubMed.
- Y. Qian, R. Kumar, M. K. Chug, H. Massoumi and E. J. Brisbois, ACS Appl. Mater. Interfaces, 2021, 13, 52250–52273 CrossRef CAS PubMed.
- J. A. González Calderón, D. Contreras López, E. Pérez and J. Vallejo Montesinos, Polym. Bull., 2020, 77, 2749–2817 CrossRef.
- A. Mata, A. J. Fleischman and S. Roy, Biomed. Microdevices, 2005, 7, 281–293 CrossRef CAS PubMed.
- H. S. El-Zaim and J. P. Heggers, in Polymeric Biomaterials, Revised and Expanded, ed. S. Dumitriu, Taylor & Francis Group, 2001, pp. 79–88 Search PubMed.
- M. N. Dimitrova, J. S. Bee, L. Lu, J. E. Fernandez, Development of Prefilled Syringe Combination Products for Biologics, in Challenges in Protein Product Development, ed. N. W. Warne and H. Mahler, Springer International Publishing, 2018, pp. 203–224 Search PubMed.
- W. Yuan, Z. Hu, M. Liao, Y. Cai and M. Lele, et al. , Int. J. Nanomed., 2012, 5719 CrossRef PubMed.
- T. Aziz, H. Fan, F. U. Khan, M. Haroon and L. Cheng, Polym. Bull., 2019, 76, 2129–2145 CrossRef CAS.
- J. Li, E. Ueda, D. Paulssen and P. A. Levkin, Adv. Funct. Mater., 2019, 29, 1802317 CrossRef.
- N. MacCallum, C. Howell, P. Kim, D. Sun and R. Friedlander, et al. , ACS Biomater. Sci. Eng., 2014, 1, 43–51 CrossRef PubMed.
- C. Wei, G. Zhang, Q. Zhang, X. Zhan and F. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 34810–34819 CrossRef CAS PubMed.
- G. E. Gierke, M. Nielsen and M. C. Frost, Sci. Technol. Adv. Mater., 2011, 12, 055007 CrossRef PubMed.
- E. J. Brisbois, T. C. Major, M. J. Goudie, R. H. Bartlett and M. E. Meyerhoff, et al. , Acta Biomater., 2016, 37, 111–119 CrossRef CAS PubMed.
- G. Lautner, B. Stringer, E. J. Brisbois, M. E. Meyerhoff and S. P. Schwendeman, Nitric oxide, 2019, 86, 31–37 CrossRef CAS PubMed.
- K. H. Homeyer, M. J. Goudie, P. Singha and H. Handa, ACS Biomater. Sci. Eng., 2019, 5, 2021–2029 CrossRef CAS PubMed.
- S. P. Hopkins, J. Pant, M. J. Goudie, C. Schmiedt and H. Handa, ACS Appl. Mater. Interfaces, 2018, 10, 27316–27325 CrossRef CAS PubMed.
- S. Ghalei, S. Hopkins, M. Douglass, M. Garren and A. Mondal, et al. , J. Colloid Interface Sci., 2021, 590, 277–289 CrossRef CAS PubMed.
- M. J. Goudie, P. Singha, S. P. Hopkins, E. J. Brisbois and H. Handa, ACS Appl. Mater. Interfaces, 2019, 11, 4523–4530 CrossRef CAS PubMed.
- M. K. Chug, E. Bachtiar, N. Narwold, K. Gall and E. J. Brisbois, Biomater. Sci., 2021, 9, 3100–3111 RSC.
- D. Chen, X. Hu, H. Zhang, X. Yin and Y. Zhou, Polym. Degrad. Stab., 2015, 111, 124–130 CrossRef CAS.
- S. Ghalei, J. Li, M. Douglass, M. Garren and H. Handa, ACS Biomater. Sci. Eng., 2021, 7, 517–526 CrossRef CAS PubMed.
- S. P. Hopkins and M. C. Frost, Bioengineering, 2020, 7, 9 CrossRef CAS PubMed.
- C. Zhang, T. D. Biggs, N. O. Devarie-Baez, S. Shuang and C. Dong, et al. , Chem. Commun., 2017, 53, 11266–11277 RSC.
- P. G. Wang, M. Xian, X. P. Tang, X. J. Wu and Z. Wen, et al. , Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- N. Hogg, Free Radical Biol. Med., 2000, 28, 1478–1486 CrossRef CAS PubMed.
- D. L. H. Williams, Acc. Chem. Res., 1999, 32, 869–876 CrossRef CAS.
- L. Grossi and P. C. Montevecchi, Chem. – Eur. J., 2002, 8, 380–387 CrossRef CAS PubMed.
- Y. Wo, Z. Li, E. J. Brisbois, A. Colletta and J. Wu, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 22218–22227 CrossRef CAS PubMed.
- M. J. Goudie, E. J. Brisbois, J. Pant, A. Thompson and J. A. Potkay, et al. , Int. J. Polym. Mater., 2016, 65, 769–778 CrossRef CAS PubMed.
- Y. Wo, Z. Li, A. Colletta, J. Wu and C. Xi, et al. , Composites, Part B, 2017, 121, 23–33 CrossRef CAS PubMed.
- M. L. Read, P. B. Morgan, J. M. Kelly and C. Maldonado-Codina, J. Biomater. Appl., 2011, 26, 85–99 CrossRef CAS PubMed.
- S. Vudayagiri, M. D. Junker and A. L. Skov, Polym. J., 2013, 45, 871–878 CrossRef CAS.
- Y. Wo, L.-C. Xu, Z. Li, A. J. Matzger and M. E. Meyerhoff, et al. , Biomater. Sci., 2017, 5, 1265–1278 RSC.
- W. S. Y. Wong, L. Hauer, A. Naga, A. Kaltbeitzel and P. Baumli, et al. , Langmuir, 2020, 36, 7236–7245 CrossRef CAS PubMed.
- M. K. Chug, H. Massoumi, Y. Wu and E. J. Brisbois, J. Biomed. Mater. Res., Part A, 2022, 110, 1263–1277 CrossRef CAS PubMed.
- R. Devine, M. Douglass, M. Ashcraft, N. Tayag and H. Handa, ACS Appl. Mater. Interfaces, 2021, 13, 19613–19624 CrossRef CAS PubMed.
- R. Devine, M. Goudie, P. Singha, M. Douglass and C. W. Schmiedt, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 20158–20171 CrossRef CAS PubMed.
- J. Pant, M. J. Goudie, S. P. Hopkins, E. J. Brisbois and H. Handa, ACS Appl. Mater. Interfaces, 2017, 9, 15254–15264 CrossRef CAS PubMed.
- E. J. Brisbois, R. P. Davis, A. M. Jones, T. C. Major and R. H. Bartlett, et al. , J. Mater. Chem. B, 2015, 3, 1639–1645 RSC.
- F. C. Fang, Nat. Rev. Microbiol., 2004, 2, 820–832 CrossRef CAS PubMed.
- A. Friedman, K. Blecher, D. Sanchez, C. Tuckman-Vernon and P. Gialanella, et al. , Virulence, 2011, 2, 217–221 CrossRef PubMed.
- Z. Breijyeh, B. Jubeh and R. Karaman, Molecules, 2020, 25, 1340 CrossRef CAS PubMed.
- H. Massoumi, R. Kumar, M. K. Chug, Y. Qian and E. J. Brisbois, ACS Appl. Bio Mater., 2022, 5, 2285–2295 CrossRef CAS PubMed.
- M. J. Goudie, J. Pant and H. Handa, Sci. Rep., 2017, 7, 13623 CrossRef PubMed.
- H. Ren, J. Wu, A. Colletta, M. E. Meyerhoff and C. Xi, Front. Microbiol., 2016, 7, 1260 Search PubMed.
- Y. Wo, E. J. Brisbois, J. Wu, Z. Li and T. C. Major, et al. , ACS Biomater. Sci. Eng., 2017, 3, 349–359 CrossRef CAS PubMed.
- Y. S. Kim, M. Y. Song, J. D. Park, K. S. Song and H. R. Ryu, et al. , Part. Fibre Toxicol., 2010, 7, 20 CrossRef PubMed.
- T. Zhang, L. Wang, Q. Chen and C. Chen, Yonsei Med. J., 2014, 55, 283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00414c |
| This journal is © The Royal Society of Chemistry 2022 |