
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of maleimide functionalized poly(3,4-ethylenedioxythiophene) (PEDOT) polymers†
Peter
Sitarik
 ,
Samadhan S.
Nagane
,
Shrirang
Chhatre
,
Yuhang
Wu
,
Quintin
Baugh
and
David C.
Martin
*
,
Samadhan S.
Nagane
,
Shrirang
Chhatre
,
Yuhang
Wu
,
Quintin
Baugh
and
David C.
Martin
*
Materials Science and Engineering, The University of Delaware, Newark, DE 19716, USA. E-mail: milty@udel.edu
First published on 22nd June 2022
Abstract
Here describe the design, synthesis, and characterization of maleimide-functionalized 3,4-ethylenedioxythiophene (EDOT) monomers, and their corresponding polymers (PEDOT) and copolymers. The maleimide functionality can be readily reacted with a number of other moieties and provides access to a wide variety of potentially useful variants of EDOT. Several novel varieties of functionalized 3,4-ethylenedioxythiophene (EDOT+) monomers were developed and investigated, including EDOT-maleimide, EDOT-maleimide-cholesterol, EDOT-maleimide-adamantane, and EDOT-maleimide-cysteine. The corresponding polymers PEDOT+ were prepared by electrochemical deposition onto metallic electrodes. The physical and chemical properties of the resulting polymer films were characterized using optical and electron microscopy, vibrational spectroscopy, cyclic voltammetry, and electrochemical impedance spectroscopy. Post processing of the PEDOT-maleimide films was done using a electrochemical quartz crystal microbalance with dissipation along with X-ray photoelectron spectroscopy. The biocompatibility of the materials was examined using cell viability testing. These materials make it possible to precisely tailor the electrical and biological properties of poly(alkylthiophenes) that are of considerable current interest for a variety of applications including bioelectronic deivces.
Introduction
Materials challenges in biomedical interfaces arise from the inherent mismatch of properties between rigid, solid engineered systems and soft, electrolytic biological systems.1 Nature uses complex organic structures that provide mechanical integrity and durability with a network of communications through ionic pathways. There is intense interest in the development of materials that could directly interface electrical components with specific neurons to mimic the durable, communicative system used by organisms.Conductive polymers (CP) are ideal candidates for biomedical interfaces because they are constructed from organic molecular components and are capable of both electronic and ionic transport. Conductive polymers have also found uses in solar cells, sensors, and mechanical actuators.1,2 One particular advantage of conductive polymers for biomedical device applications is their ability to be electrochemically deposited directly onto electrodes, providing precise spatial and chemical control of the resulting thin film.3,4
The conjugated, doped molecular backbone of the CP allows electrons to flow freely through extended π-orbitals.5 Conductive polymers also facilitate ionic charge transport via the counterions that are used to stabilize the positive charge on the CP backbone. This mix of both ionic and electronic charge carriers make conjugated polymers ideal materials for interfacing solid semiconducting and metallic components with aqueous solutions and biological materials. Furthermore, their rough surface morphology provides a large effective surface area, leading to low impedances and high volumetric capacitance.6,7
Poly(3,4-ethylenedioxythiophene)(PEDOT) is an alkoxy-functionalized thiophene that when chemically polymerized and doped with polystyrenesulfonate (PSS) counterion forms a stable, aqueous dispersion that is available commercially.8 This formulation's success is due to a combination of properties including high conductivity, chemical stability, thermal stability, and mechanical durability. Challenges regarding this material stem from limitations in tunability. Because of their relatively limited solubility, the degree of polymerization in conjugated polymers tends to be low. Improved performance has been seen through the use of solubilizing side groups that make it possible to significantly increase the molecular weight.9
Because the conjugated backbones of CP's are composed of sp2 carbons the polymers tend to be stiff and in the bulk state, brittle. PSS is used as a counterion to stabilize charge and to enhance the mechanical properties, resulting in smooth films that can handle moderate stresses without cracking or delamination. However while PSS is ionically conductive, it is an electrical insulator and so can lower the overall conductivity by keeping the PEDOT-rich domains separated from one another.10 Many post processing techniques attempt to retain interconnectivity of both the PEDOT-rich and PSS-rich domains to maintain high electronic and ionic conductivity.11 Research groups such as that headed by Molly Stevens use this high stability and conductivity to construct mediums for cell stimulation and sensing.12 PEDOT:PSS scaffolds were used along with electrical stimulation to enhance the regeneration of MC3T3-E1 osteogenic precursor cells.13
Though the chemical stability of PEDOT is generally a benefit, it does hinder the ability for functionalization to enhance specificity of the CP to a system.14 Functionality provides a pathway for creating monomers and ensuing polymers with increased solubility, degrees of polymerization, conductivity, surface specific interactions, mechanical durability and tailored growth patterns.
A number of groups have explored the ability to design and synthesize tailored PEDOT's for specific applications. One possibility is to attach a carboxylic acid (EDOT-acid) which provides a monomer with reactive side group on the normally unreactive alkoxy ring of the thiophene.15 The new monomer and polymer had enhanced solubility in aqueous solvents which can help with increasing concentration of polymer solutions and possibly degree of polymerization.16 Similar to most chemical reactions, PEDOT polymerizes mainly in the solution that the monomer is dissolved in. To get useful films it is important to control solutions such that monomer is soluble and polymer precipitates at a polymerization length suitable for the required application.
Other work with EDOT-acid included functionalization through the acid with RGD protein to increase cell adhesion for biomedical interfacing devices.17 EDOT-acid and EDOT-NH2 have both been used to increase adhesion of PEDOT films by using the carboxylic acid and NH2 as a surface binder for further EDOT polymerization.18,19 The use of this technique to form CP films on metal and metal oxide reduced the rate of delamination and improved overall film stability. This shows that the EDOT molecule is still reactive when combined with other functionalities that cause it to act as a pendant group. Formation of an EDOT-acid with an alkyl chain linker has also been done in the work to form bio sensors.20
Recent work from the Berggren group includes the use of functionalized EDOT and thiophene monomers with good solubility and lowered oxidation potentials to form conductive polymers in plants.21,22 Some groups use counterion control to adjust the PEDOT conductive polymers properties using molecules such as hyaluronic acid, heparin, and chondroitin sulfate.23
Since the EDOT monomer is a small molecule (Mw = 142.18 g mol−1), it is important to keep any chemical substituents also small or they may cause the resulting functionalized monomer to lose its ability to polymerize. It has been generally found that adding relatively large substituents such as peptides or fluorescent labels significantly reduces the reactivity of the resulting compound.
Reaction rates should ideally be high for functional EDOT polymerization as it will reduce overall processing time which is important for application both in vitro and in vivo. For industrial applications increased productivity can be achieved if the polymerization is done in the most ideal solvent even if it is not biocompatible. In vivo will have reduced time scales required to produce the polymer films which reduces potential risk to the biological system to exposure of unknown foreign material.
Addition of a maleimide to EDOT provides a thermally crosslinkable chemical structure that can also react through a click reaction with thiol.24 From EDOT-maleimide a number of functional EDOT+'s can be synthesized through click chemistry. EDOT-maleimide has proven to be particularly useful for making functionalized PEDOT as polymerization methods show a high reactivity that is equal to or greater than unfunctionalized PEDOT.
Derivatives that were investigated for this work include cholesterol, cysteine, and adamantane. Cholesterol is a biological precursor for several important bioactive molecules and serves as a membrane stabilizer in animals’ cells by embedding itself in the hydrophobic layer of the plasma membrane. It may also be used by researchers to create more mechanically stable membranes for drug delivery through liposomes.25 Artificial liposomes created with EDOT-cholesterol might allow for the selective introduction of polymerizable EDOT monomer into close association with the plasma membranes of cells. Cysteine is an amino acid that contains a thiol group which allows for molecular coupling using a Michael addition reaction. It has been shown that the coating of otherwise toxic components with cysteine improves water solubility and cell viability.26 Adamantane is a common pharmacological moiety widely used in antiviral drugs. Its incorporation into EDOT might result in a PEDOT variant with enhanced antiviral properties or decreased cytotoxicity.27 We hypothesized that incorporating these biologically important side groups into PEDOT would create hybrid materials of particular interest for biointerfacing.
In this work we describe the design, synthesis, and characterization of EDOT monomers that contain the maleimide functional group. The maleimide provides a convenient handle for the further functionalization of EDOT with a variety of bioinspired groups. These various monomers were polymerized leading to functional PEDOT+ films grown using electrochemical methods. We also demonstrated the retention of the activity of maleimide functional groups on PEDOT-maleimide after electrodeposited on the gold electrode by covalently bonding hydrophobic moieties on the surface through thiol-maleimide click chemistry. The success of the bonding reaction was determined by Electrochemical Quartz Crystal Microbalance with Dissipation (EQCM-D), X-ray photoelectron spectroscopy (XPS), and contact angle measurements. Chemical and physical characterizations of synthesis were done through NMR, FTIR, and biocompatibility with a luminescence assay. The morphologies were examined using optical microscopy and scanning electron microscopy.
Experimental details
Materials
3,4-Dimethoxythiophene, 3-chloro-1,2-propanediol and p-toluenesulfonic acid (p-TSA), 12,12,13,13,14,14,15,15,15-nonafluoropentadecane-1-thiol (fluorinated thiol), maleic anhydride, sodium azide, triphenylphosphine, 1-adamantanethiol, thiolcholesterol, L-cysteine hydrochloride, and glacial acetic acid were purchased from Sigma Aldrich Chemicals (USA) and were used as received. Sodium thiosulfate, sodium sulfate, sodium hydroxide and hydrochloric acid were procured from Fisher Scientific and were used as received. N,N-Dimethylformamide (DMF), tetrahydrofuran (THF), toluene, chloroform, dichloromethane, ethyl acetate, pet ether, phosphate buffered saline 1× (PBS) were procured from Fisher Scientific. DI water was prepared by a Millipore Reference water purification system.Equipment
Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker 400 MHz spectrometer, at resonance frequencies of 400 MHz for 1H NMR and 100 MHz for 13C NMR measurements using CDCl3 and acetone-d6 as solvents. Attenuated total reflectance mode FTIR (ATR-FTIR) spectra of thiophene monomers were recorded on a PerkinElmer Spectrum 100 ATR-FTIR spectrometer. Electrochemical polymerization was carried out using a Metrohm Autolab PGSTAT128N. The commercially available screen printed electrode 223AT from Metrohm Dropsens was used with a gold working electrode, gold counter electrode, and silver pseudoreference electrode with an associated potential of −0.131 V vs. Ag/AgCl.28 Deposition and functionalization characterization for post processing was done using a QCM5140TiAu120-050-Q electrode on a Qsense QEM 401 Electrochemical Quartz Crystal Microbalance-Dissipation (EQCMD). Optical characterization was performed on a Dino-Lite digital camera and Nikon Eclipse LVPOL100. Scanning Electron Microscopy was done with a JEOL JSM-7400F. Luminescent assays were taken on a BMG LABTECH Omega Series Microplate Reader.Synthesis
Scheme 1 depicts the route followed for the synthesis of functionalized EDOT monomers from EDOT-maleimide (EDOT-MA) to EDOT-maleimide-adamantane (EDOT-MA-adamantane), EDOT-maleimide-cholesterol (EDOT-MA-cholesterol) and EDOT-maleimide-cysteine (EDOT-MA-cysteine) starting from 3,4-dimethoxythiophene and 3-chloro-1,2-propanediol.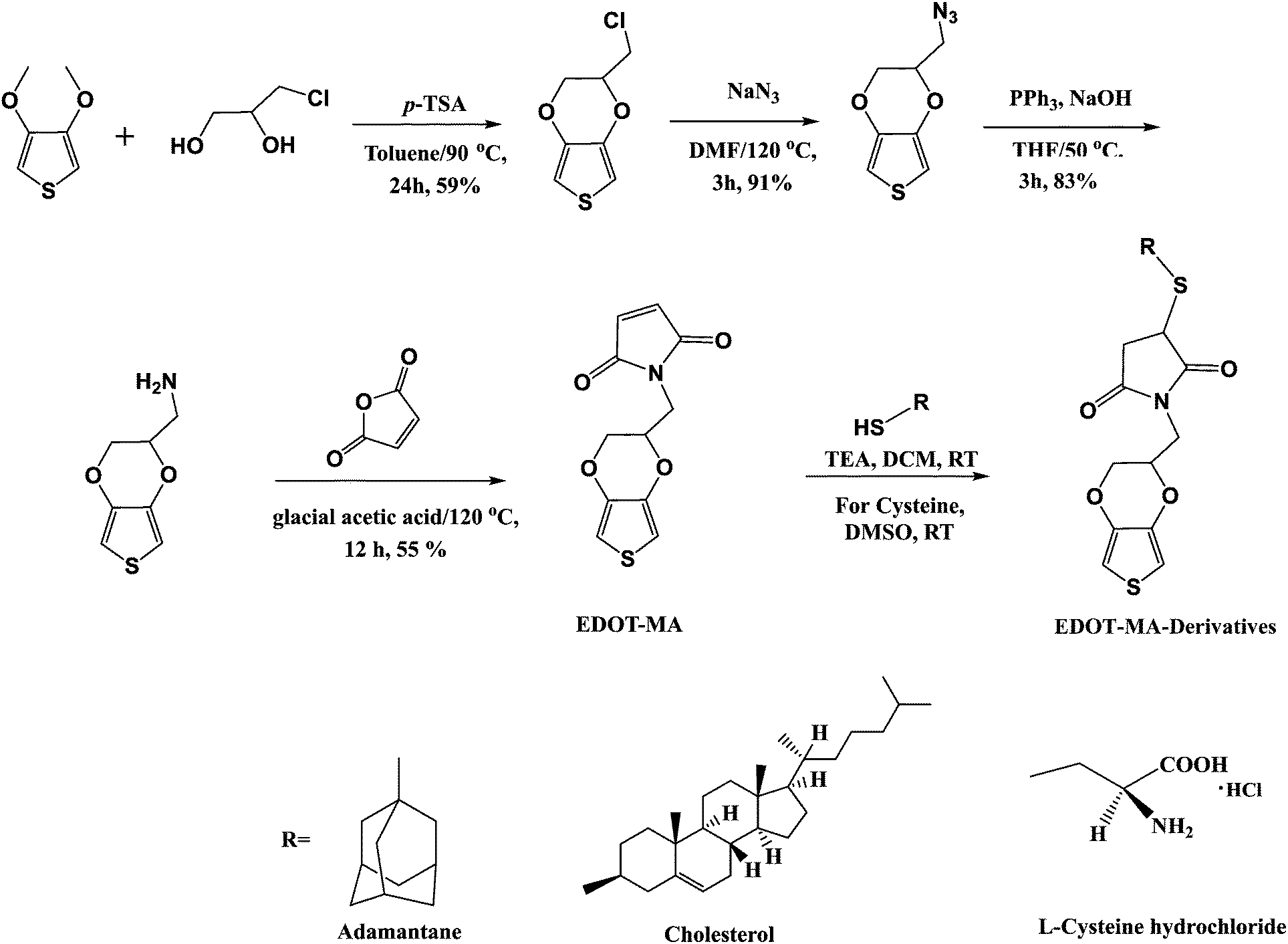 | ||
| Scheme 1 Synthesis of EDOT-MA-derivatives (EDOT-MA-adamantane, EDOT-MA-cholesterol, EDOT-MA-cysteine). | ||
Synthesis of 2′-chloromethyl-3,4-ethylenedioxythiophene (EDOT-Cl)
Into a 500 mL two necked round-bottom flask equipped with a reflux condenser and argon inlet were placed, 3,4-dimethoxythiophene (10.0 g, 69.35 mmol), anhydrous toluene (250 mL) with 3-chloro- 1,2-propanediol (13.16 mL, 157.44 mmol), and p-toluenesulfonic acid (p-TSA) (1.62 g, 6.935 mmol). The reaction mixture was stirred at 90 °C for 24 h. After this time, 3-chloro-1,2-propanediol (13.16 mL) were added to the reaction mixture which was stirred at 90 °C for further 3 h. The reaction mixture was allowed to cool to room temperature and the solvent was evaporated under reduced pressure. The solution of 10% sodium bicarbonate (150 mL) was added to the crude and the product was extracted with dichloromethane (3 × 100 mL). The organic phases were combined, washed with water (2 × 100 mL) and dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The crude product was purified by column chromatography using pet ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane (50:50, v/v) as an eluent to afford EDOT-Cl (7.85 g, 59%). 1H NMR (400 MHz, CDCl3): δ = 6.38 (dd, 2H), 4.40–4.37 (m, 1H), 4.29 (dd, 1H), 4.17 (dd, 1H), 3.76–3.65 (m, 2H), 13C NMR: δ = 141.1, 140.6, 100.1, 72.8, 65.5, 41.3 ppm.
:dichloromethane (50:50, v/v) as an eluent to afford EDOT-Cl (7.85 g, 59%). 1H NMR (400 MHz, CDCl3): δ = 6.38 (dd, 2H), 4.40–4.37 (m, 1H), 4.29 (dd, 1H), 4.17 (dd, 1H), 3.76–3.65 (m, 2H), 13C NMR: δ = 141.1, 140.6, 100.1, 72.8, 65.5, 41.3 ppm.
Synthesis of 2′-azidomethyl-3,4-ethylenedioxythiophene (EDOT-N3)
Into a 100 mL two necked round-bottom flask equipped with argon inlet were added EDOT-Cl (3.0 g, 15.74 mmol) and N,N-dimethylformamide (50 mL). Sodium azide (2.05 g, 31.47 mmol) was added to the solution and the reaction mixture was stirred at 120 °C for 3 h. After cooling, the N,N-dimethylformamide was removed by rotary evaporation under reduced pressure. Then, water (200 mL) was added to the residue and the product was extracted with diethyl ether (2 × 150 mL). The solution dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure to afford EDOT-N3 as a pale yellow oily liquid (2.82 g, 91%).1H NMR (400 MHz, CDCl3): δ = 6.39 (dd, 2H), 4.34–4.30 (m, 1H), 4.20 (dd, 1H), 4.05 (dd, 1H), 3.61–3.48 (m, 2H), 13C NMR: δ = 141.0, 140.6, 100.2, 100.1, 72.4, 65.7, 50.5 ppm.
Synthesis of 2′-aminomethyl-3,4-ethylenedioxythiophene (EDOT–NH2)
Into a 50 mL two necked round-bottom flask equipped with a reflux condenser, a magnetic stirring bar was charged tetrahydrofuran (10 mL), EDOT-N3 (1.0 g, 5.10 mmol), triphenylphosphine (1.6 g, 6.1 mmol) and 2 mol L−1 sodium hydroxide aqueous solution (10 mL). The reaction mixture was stirred at 50 °C for 3 h. The reaction mixture was allowed to cool to room temperature. After evaporation of tetrahydrofuran, 2 M hydrochloric acid solution was used to control the pH below 3. Then the aqueous layer was extracted with dichloromethane (2 × 20 mL) and the combined organic layers were discarded. 1 M sodium hydroxide solution was added to adjust the pH of aqueous layer to 12. The aqueous layer was extracted with dichloromethane (3 × 20 mL) and the combined organic layers were dried with anhydrous sodium sulfate. The solvent was removed under reduced pressure to afford EDOT-NH2 as colorless oil (0.72 g, 83%).1H NMR (400 MHz, CDCl3): δ = 6.33 (dd, 2H), 4.20 (dd, 1H), 4.12 (m, 1H), 4.00 (dd, 1H), 2.97 (m, 2H), 1.32 (s, 2H) 13C NMR: δ = 141.6, 140.5, 99.5, 75.2, 66.6, 42.3 ppm.
Synthesis of 2′-maleimideomethyl-3,4-ethylenedioxythiophene (EDOT-MA)
Into a 50 mL two necked round-bottom flask equipped with a mechanical stirrer, an argon inlet and a reflux condenser were placed EDOT-NH2 (0.20 g, 1.16 mmol), maleic anhydride (0.138 g, 1.40 mmol) and glacial acetic acid (10 mL). The reaction mixture was stirred at 120 °C for 12 h. The reaction mixture was cooled to room temperature and glacial acetic acid removed by evaporation under reduced pressure. The crude product was dissolved in ethyl acetate and the solution was washed with water (3 × 20 mL). The ethyl acetate solution was dried over anhydrous sodium sulfate, filtered and ethyl acetate was removed by evaporation under reduced pressure. The crude product was purified by column chromatography using pet ether:ethyl acetate (70:30, v/v) as an eluent to afford pure EDOT-MA (0.16 g, 55%).
1H NMR (400 MHz, CDCl3): δ = 6.77 (s, 2H), 6.33 (d, 2H), 4.39 (d, 1H), 4.21 (d, 1H), 3.98–3.88 (m, 2H), 3.73 (dd, 1H), 13C NMR: δ = 170.2, 141.0, 134.3, 100.2, 70.9, 66.1, 37.7 ppm.
Synthesis of EDOT-MA-adamantane
To a round bottom flask equipped with a magnetic stirring bar were added EDOT-MA (0.20 g, 0.80 mmol), 1-adamantanethiol (0.135 g, 0.80 mmol), triethylamine (112 μL, 0.80 mmol), and dry dichloromethane (5 mL). The reaction mixture was stirred at room temperature under nitrogen atmosphere for 8 h. The dichloromethane (20 mL) was added to the reaction mixture and the solution was washed with water (3 × 20 mL). The solution was dried over anhydrous sodium sulfate, filtered and dichloromethane was removed by evaporation under reduced pressure. The crude product was purified by column chromatography using pet ether:ethyl acetate (80:20, v/v) as an eluent to afford pure EDOT-MA-adamantane, Fig. S1 and S4 (ESI†).
A similar procedure was followed for synthesis of EDOT-MA-cholesterol, Fig. S2 and S4 (ESI†).
Synthesis of EDOT-MA-cysteine
Into a 25 mL two necked round-bottom flask fitted with a magnetic stirring bar were added EDOT-MA (0.200 g, 0.80 mmol), L-cysteine hydrochloride (0.126 g, 0.80 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at room temperature for 8 h, while consumption of starting material was monitored by TLC. After the reaction was complete DMSO evaporated under reduced pressure to afford EDOT-MA-cysteine, Fig. S3 and S4 (ESI†).Cell culture and cytocompatibility assay
SH-SY5Y neuroblastoma cells were cultured in Dulbecco's Modified-Eagle's Media (DMEM with added glucose and L-glutamine) with additions of 1% Penn-Strep antibiotic solution and 10% fetal bovine serum. A CellTiter-Glo luminescence assay lysed the cells in order to measure the amount of ATP contained within, which was then calibrated to a standard to measure viability. Short time periods, sub-day, were deemed most important as monomer would only be around tissue for a short term polymerization. Concentrations of monomer were 0.01 M in culture media, the standard for polymerizations with an n of 3 for reproducibility.Electrochemical experimentation
:12 by volume water:propylene carbonate cosolvent mixture. Propylene carbonate was used to first dissolve the monomers followed by addition of water for a fully mixed solution. Polymerization was done using galvanostatic methods of current densities 10–100 μA mm−2 for 0.5–5 minutes. Low current densities resulted in controlled polymer film growth. For potentiostatic methods Table 1 lists the polymerization potential peak associated with each EDOT+.
| Monomer | Solvent | Polymerization potential (V) vs. SPE pseudoreference (Ag) |
|---|---|---|
| EDOT | 88:12 water:PPC by volume |
1.1 |
| EDOT-MA | 88:12 water:PPC by volume |
1.0 |
| EDOT-MA-cholesterol | PPC | 1.2–1.4 |
| EDOT-MA-adamantane | Acetonitrile | 1.2 |
| EDOT-MA-cysteine | Acetonitrile | 1.5 |
Polymerization of EDOT-MA-cholesterol
EDOT-MA-cholesterol was electrochemically polymerized in propylene carbonate. Limited film deposition was achieved using a potentiostatic linear sweep from 1.0 to 1.5 V at 5 mV s−1 with a current density of 400 μA cm−2.Polymerization of EDOT-MA-adamantane and EDOT-MA-cysteine
EDOT-MA-cysteine was electrochemically polymerized in acetonitrile using potentiostatic methods. A polymerization potential of 1.5 V was used to create films at current densities of 800 μA cm−2. Long deposition times caused unwanted side reactions on the reference electrode and was accompanied by an increase in impedance.EDOT-MA-adamantane polymerization in acetonitrile required a polymerization potential of 1.18 V to form consistent thin films at a current density of 1.5 mA cm−2.
Stacked Polymerizations of PEDOT and PEDOT+
PEDOT and a PEDOT+ were deposited stepwise with charge densities of 30.8 mC cm−2 onto 223AT screen printed electrodes. In the first case PEDOT was deposited first on the bottom and PEDOT+ deposited second on top of the PEDOT. In the second case PEDOT+ was deposited first on the bottom and PEDOT deposited second on top of the chosen PEDOT +.Electrochemical impedance of PEDOT+ films
Impedance scans were done using the frequency response analyzer in the PGSTAT128N with a typical frequency range of 1 × 10−1 Hz to 1 × 105 Hz. An amplitude of 10 mV was used with two integration cycles to minimize error. 75 μL of 1x phosphate buffered saline solution was used as the conductive medium for impedance scans.Post-processing of PEDOT-maleimide films
For the purpose of examining the availability of maleimide functionalities on the PEDOT-MA surface, a series of Electrochemical Quartz Crystal Microbalance with Dissipation (EQCM-D) explorations were carried out. The acetonitrile solution including 0.01 M EDOT-maleimide and 0.05 M LiClO4 was used in EQCM-D depositions on 1.2 cm diameter Au/Ti QuartzPro QCM-D crystals for preparing PEDOT-maleimide coated crystals, at a constant current of 70 μA for 10 min with a flow rate of 1 mL min−1. Before each deposition, a plasma cleaning for crystals was indispensable for maintaining a contaminant-free surface and stable baseline. In the wake of deposition, those coated crystals were gently rinsed with copious DI water before assessing contact angles. Approximately 4 μL DI water was syringed on coated crystals and contact angles were observed by a Dino-Lite digital camera. 1 mM fluoroalkane–thiol (Fig. S7, ESI†) in dichloromethane with equivalent triethylamine flowed through an EQCM-D cell in the presence of the PEDOT-maleimide coated crystal, which by anticipation should drastically increase the surface hydrophobicity if any trace of thiol-maleimide coupling took place. The flow rate for this trial was 0.34 mL min−1 and the time spanned for 5 hours. The assessment of the contact angle of post-functionalized PEDOT-maleimide followed the same procedure as above. The change of energy dissipation and resonant frequency through all EQCM-D experiments were constantly recorded for further analysis. XPS measurements were done on PEDOT-maleimide samples before and after the flush of the fluoroalkane-thiol solution, which were performed with an Al K alpha gun with a spot size of 400 μm and a pass energy of 20 eV with steps of 0.1 eV. The change of the surface chemistry before and after post-process was revealed by changes in electron peak intensities.Results and discussion
Electrical properties
Many EDOT+s’ like normal EDOT showed limited solubility in aqueous solvents which are the ideal solvents from a green materials perspective. Propylene carbonate (PPC) was tested as a polymerization solvent and in multiple cases was found to solubilize the monomers as well as the polymers which made it inadequate for functionalizing electrode surfaces. An 88:12 by volume mixture of water:PPC was found to be particularly effective for polymerizing some functional EDOT's including EDOT and EDOT-maleimide in this work. PPC is used commercially in cosmetics in various ratios with products containing up to 20% showing limited general biocompatibility issues.29
In addition to having ideal solubility characteristics the use of this solvent mixture lowered the polymerization potential of EDOT monomers when compared to polymerization in PPC, Fig. 1. Lowering the polymerization potential reduces the power required to make CP as well as reducing the chance for side reactions to occur in the solution. In the case of aqueous mixtures, the water splitting potential happens at roughly 1.5 V. By reducing the potential below 1.5 V there will be no bubbling in aqueous mixtures or organic solvents with atmospheric water addition. Bubbling on the electrode surface disrupts the solution for polymerization which affects the kinetics of nucleation and polymerization of monomer. Low voltages are essential for possible polymerization intended for in vivo work to reduce potential damage to the surrounding biological systems.
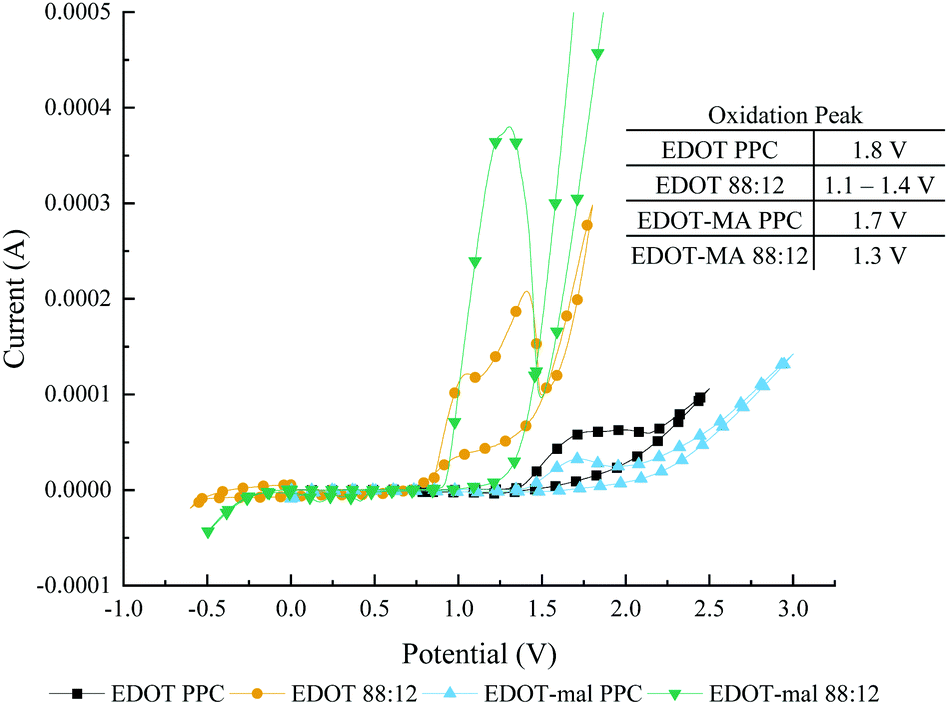 | ||
| Fig. 1 Cyclic voltammetry polymerization by variation in solvent from propylene carbonate to 88:12, H2O:PPC mixed solvent. Peaks determined by local maximums. | ||
Functional PEDOT's growth was observed at different rates and amounts visually and as shown by the electrical spectra, Fig. 2. These films were deposited using the lowest rate (mV s−1) linear sweeps in Fig. S5 (ESI†). EDOT-MA deposited at the highest current densities using the 88:12 by volume H2O:PPC mixed solvent. Lowering the propylene carbonate content even to 10% decreased current densities by a factor of 3 drastically decreasing film formation. Lowering the PPC content decreased monomer solubility and the resulting polymer growth. Fast polymer growth formed large nuclei of PEDOT-MA that grew out of a generally thin film comparatively. In the case of droplet experiments monomer depletion became an issue for a polymerization under 5 minutes. Electrical properties were comparable to those of standard PEDOT electrical polymerization with slightly higher impedances.
 | ||
| Fig. 2 Optical micrographs of polymerization onto gold working electrodes for: (a) EDOT, (b) EDOT-MA, (c) EDOT-MA-cholesterol, (d) EDOT-MA-adamantane, and (e) EDOT-MA-cysteine. The diameter of the central electrode is 1.6 mm. | ||
EDOT-MA-cholesterol was only able to be polymerized in propylene carbonate solvents with limited solubility and polymerization with mixed solvents. The polymerization potentials were higher than that of EDOT and EDOT-MA, but the current densities were the lowest of the functional EDOT's presented in this work. Additionally, minimal polymerization was achieved as evidenced by the cyclic voltammetry where there is a single peak on the first scan with no currents on follow up scans. This would mean that large, thick films would be difficult to achieve of the homopolymer.
EDOT-MA-cysteine and EDOT-MA-adamantane both were only able to be polymerized in acetonitrile to form insoluble PEDOT+ films. Polymerization was possible in other solvent conditions, but for the purpose of forming conductive functional films they were found to be unviable. EDOT-MA -adamantane polymerized into soluble PEDOT+ in propylene carbonate which formed blue PEDOT+ droplets with the addition of small amounts of water to make a mixed solvent. In acetonitrile the polymerization was similar to that of the EDOT-MA-cholesterol electrochemically with polymerization of a thin film on a time scale of a few seconds, though with higher current densities.
In general, the functional groups that were larger, had limited reactive groups, and increased impedance showed decreasing polymerization currents and overall thinner films for a given amount of charge density. The rate of polymerization based on current densities went from EDOT > EDOT-MA > EDOT-MA-cysteine > EDOT-MA-adamantane > EDOT-MA-cholesterol. This correlation can be used to identify suitable functional groups for more diverse polymers in future works.
For this set of EDOT+ EDOT has the highest rate of polymerization as it is small and lowers the impedance when functionalizing a surface with routes for electronic and ionic transport. EDOT-MA increased the molecule size and shows slightly increased impedance, but the maleimide group has reactive functionality in the sp2 carbons. EDOT-MA-cysteine has the lowest molecular weight functional group, an active amine and carboxylic acid, and an impedance that while higher than EDOT is lower than the bare gold electrode. EDOT-MA-adamantane is the next large group with a high impedance polymer form and as an alkyl side group, no easily reactive groups. EDOT-MA-cholesterol like EDOT-MA-adamantane has no easily reactive side groups, increased impedance in the polymer form, and is the largest of the functional groups. These factors combine to decrease the current densities and rate of polymerization for EDOT+.
Determination of the potential for electrochemical polymerization is essential for making consistent, reproducible PEDOT+ films. In this regard linear sweep voltammetry was typically used with a sweeping potential at a set scan rate where it is combined with optical microscopy to determine when polymer growth starts. Decreasing the scan rate has been shown to change the potential of polymerization by lowering it especially when comparing to a standard CV rate of 100 mV s−1. For all EDOT derivatives described in this work other than EDOT-maleimide-cholesterol there was a decrease in both the start and peak in polymerization potential, Table 2 and Fig. S5 (ESI†). Slower scan rates gave the system time to allow monomers to react before the high scan rate increased the potential.
| 1–2 mV s−1 | 20 mV s−1 | 100 mV s−1 | ||||
|---|---|---|---|---|---|---|
| Start (V) | Peak (V) | Start (V) | Peak (V) | Start (V) | Peak (V) | |
| EDOT | 0.74 | 1.14 | 0.82 | 1.39 | 0.82 | — |
| EDOT-MA | 0.9 | 1.02 | 1.02 | 1.33 | ||
| EDOT-MA-cholesterol | 1 | 1.19 | 1 | 1.29 | ||
| EDOT-MA-adamantane | 1.2 | 1.28 | 1.35 | 1.76 | ||
| EDOT-MA-cysteine | 1.57 | 1.66 | — | 1.78 | ||
Electrochemical impedance spectra of PEDOT+ films gave clear changes in the electrical behavior depending on the starting monomer with normal PEDOT performing the best over the frequency range tested of 1 × 10−1 Hz to 1 × 105 Hz, Fig. 3. PEDOT-MA-cholesterol and PEDOT-MA-adamantane performed with the highest overall impedances above 0.6 Hz due to the addition of the large unreactive, hydrophobic groups limiting charge transfer. The PEDOT-MA-cysteine spectrum shows a decreased impedance in between EDOT and gold over the entire spectrum. PEDOT-MA 88:12 H2O:PPC and 86:14 H2O:PPC are both reported with differences due to change in solvent composition. While both were deposited with the same charge density the 86:14 version shows a lower, more uniform impedance, though it still does not match that of PEDOT.
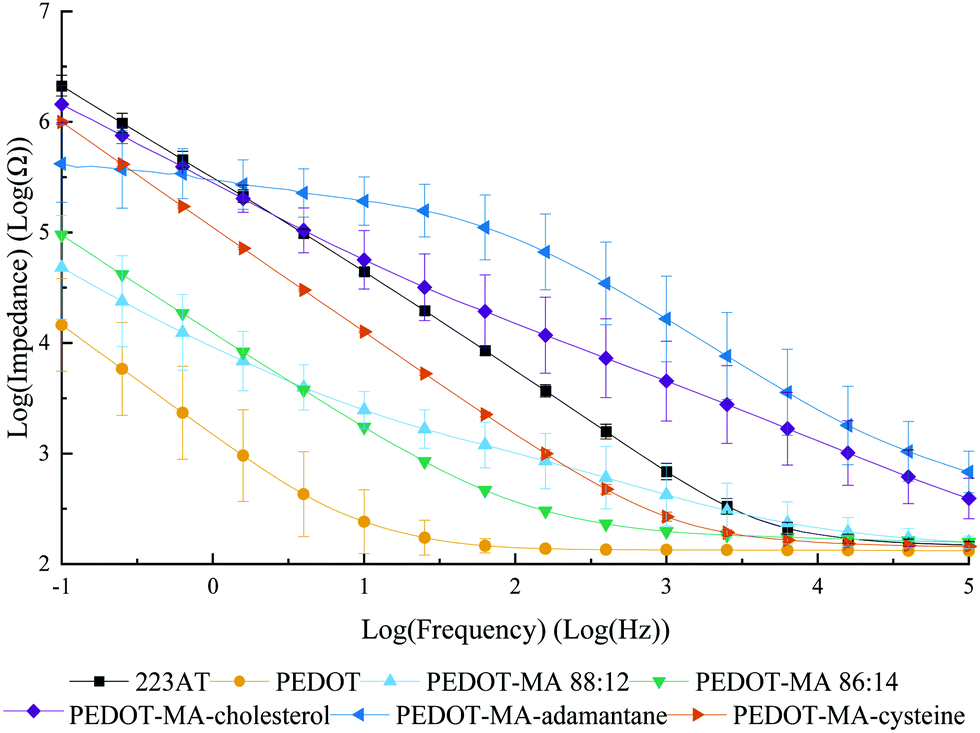 | ||
| Fig. 3 Bode plot of magnitude for PEDOT+ films on 223AT electrodes. | ||
PEDOT Morphology
Morphology of polymers from electrochemical deposition is highly dependent on many factors of which the base electrode material and morphology, the electrical methods for the deposition, and solvent all play an important part.3,30,31 The 223AT gold electrode surface is composed of bumps up to 10 microns in diameter with cracks on the surface, Fig. 4. An uneven surface such as that of the 223AT makes PEDOT+ films grow less even, consistent films. Comparison of the SEM for thick electrochemically grown PEDOT and PEDOT-MA shows differences in the morphology between the two polymers, Fig. 4. The PEDOT-MA film shown here was thick and durable enough to be removed from the electrode surface intact. PEDOT has a typically rough, bumpy surface which helps the conductivity by increasing the available surface area for interaction with the intended system as well as space ion movement. Bumps were on the range of 3 μm to 5 μm with large nodules of size 350 nm to 500 nm distributed evenly along the large bumps of the film. Small 100 nm diameter nodules evenly coat the central surface of the bumps and cluster at the edges that connect to large bumps. PEDOT-MA has a more smooth, flowing surface with underlying bumps and a pleated structure similar to that seen in PEDOT-acid.16 Individual folds are on the scale of 100 μm long with an even distribution of 50 μm to 100 μm between folds and 5–10 μm long cracks along the edges. Along the surface between folds is completely smooth with porosity along the inside edge of the folds with pores that are a few hundred nanometers across. The decrease in roughness and available surface area may play a part into the slightly increased impedance compared to that of PEDOT. | ||
| Fig. 4 SEM of a (left) 223AT gold electrode, (center) electrically grown PEDOT, and (right) electrically grown PEDOT-MA. | ||
Thin PEDOT+ films show pronounced differences between the different functional groups, Fig. 5. PEDOT has its typical rough, bumpy structure coating the entire gold electrode surface, Fig. 5(a). Before reaching a smooth, pleated structure PEDOT-MA looks similar to PEDOT with a bumpy structure, Fig. 5(b). This structure is more rounded and smooth as well as being more connected. Sizing of bumps and nodules for PEDOT-MA is similar to PEDOT except there are no small nodules only large sub-micron nodules that have no discrete boundary and grow into each other.
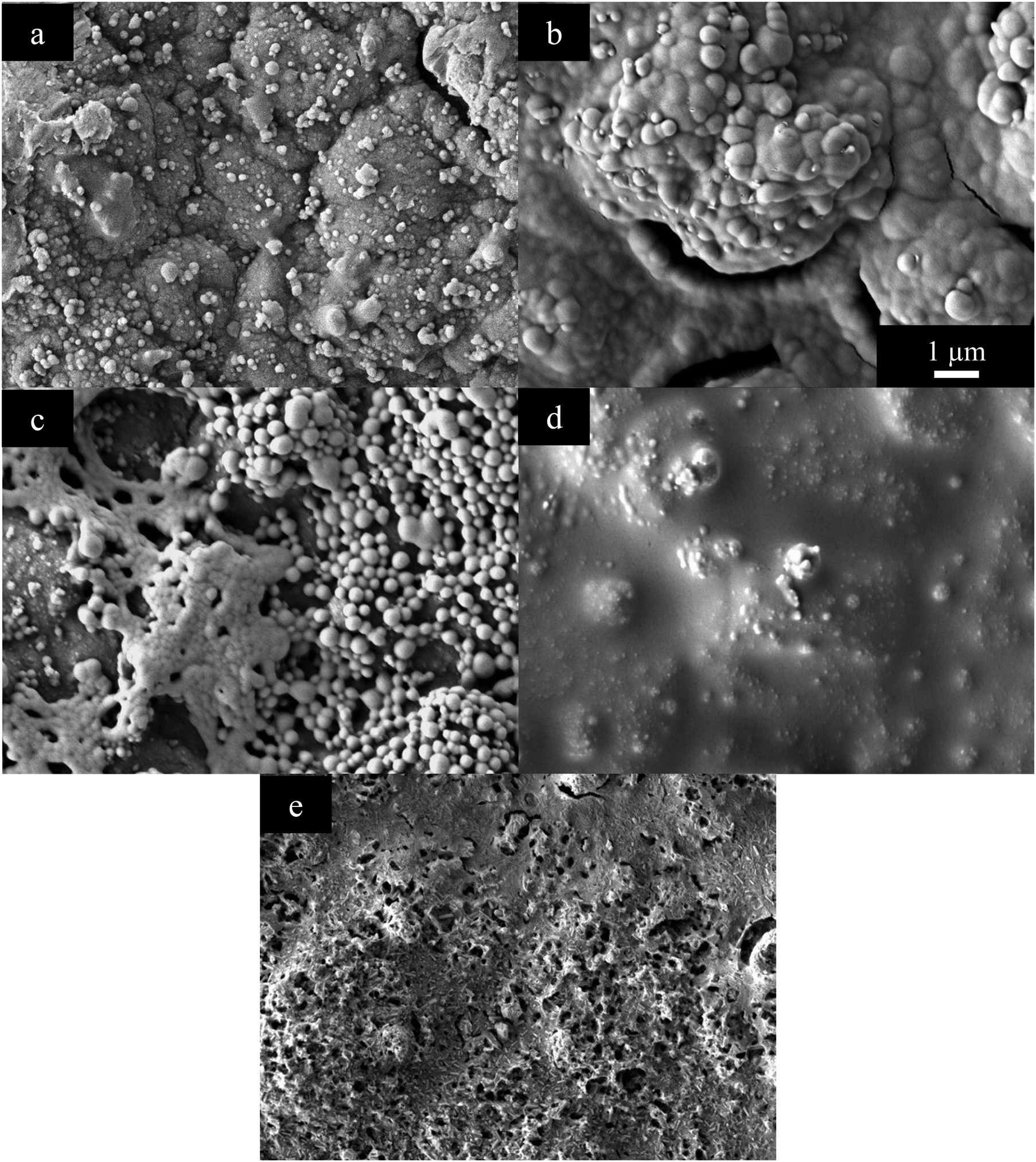 | ||
| Fig. 5 SEM of (a) PEDOT, (b) PEDOT-MA, (c) PEDOT-MA-cholesterol, (d) PEDOT-MA-adamantane, and (e) PEDOT-MA-cysteine electrically grown films. | ||
PEDOT-MA-cholesterol does not form a complete film on the electrode surface and in the SEM shows up as a white interconnected network composed of small spheres with diameters of up to 500 nm, Fig. 5(c). The darker surface underneath these spheres is the gold electrode. The combination of an incomplete film and low surface area due to a smooth, rounded network cause the increased impedance PEDOT-MA-cholesterol. Once the gold surface has been covered there is no more low impedance space for EDOT-MA-cholesterol to grow on which causes a limitation on the thickness. The limited growth is fragile and can be easily scraped away from the gold surface.
PEDOT-MA-adamantane formed a wet looking surface that covers the gold, Fig. 5(d). Unlike the similar hydrophobic polymer, PEDOT-MA-cholesterol, PEDOT-MA-adamantane covers the entire surface of the electrode, but due to decreased surface area to the gold or even PEDOT, there is an increase in impedance. EDOT-MA-cysteine polymerizes to an uneven, jagged surface, Fig. 5(e). 200 nm pores in the film give increased surface area and flow of liquid in and out of the structure. These factors are evidently part of the decreased impedance from PEDOT-MA-cysteine.
Sequential Layered Polymerization of PEDOT and PEDOT+
The idea and use of layering various PEDOT+ functional polymers has been theorized, but previously has been an unexplored area.1 Impedance for layered PEDOT and PEDOT+ had mixed properties of the two polymers used, though the second deposition (top, interfacial layer) has a greater impact than the base layer. This means that bulk PEDOT or PEDOT+ has a lesser effect on the electrochemical properties than the top, surface layer of conducting polymer, Fig. 6. Surface layer morphology and functionalization from altering PEDOT provide the major vectors for controlling the overall solution-electrode interface. | ||
| Fig. 6 Stacked polymerization of PEDOT and PEDOT+ on top of gold in aqueous solution. Layers top to bottom (top) PEDOT, PEDOT+, and gold and (bottom) PEDOT+, PEDOT, and gold with the respective PEDOT and PEDOT+ becoming the dominant top layer. | ||
The EDOT+ variants grew better on PEDOT than on the gold electrode surface. Because of the similarity in structure it is easier for EDOT+ to bond to PEDOT than to the inorganic gold. Increasing surface area from PEDOT also provides more area for EDOT+ to bind and polymerize. Comparing polymerization times of EDOT+ on PEDOT versus on gold shows a decrease in the time to inject the same charge density, polymerizing the same amount of EDOT+, Table S1 (ESI†). Controlling deposition of PEDOT+ on PEDOT allows for the ability to make thin films with tunable properties between that of the two polymer films.
Post processing of PEDOT-maleimide
Controlling the order of functionalization and polymerization allows for making either bulk PEDOT+ (Fig. 7a) or bulk PEDOT-MA with a surface functionalized PEDOT+ (Fig. 7b). Combining the previously discussed sequential layered polymerization of PEDOTs’ with post processing of PEDOT-MA provides alternative methods to make the same PEDOT+ via different routes with more ways to control the overall finished product. A fluorinated thiol component was chosen for post processing to clearly demonstrate the continued activity of the maleimide for a click reaction by characterization of EQCMD, XPS, and contact angle measurements.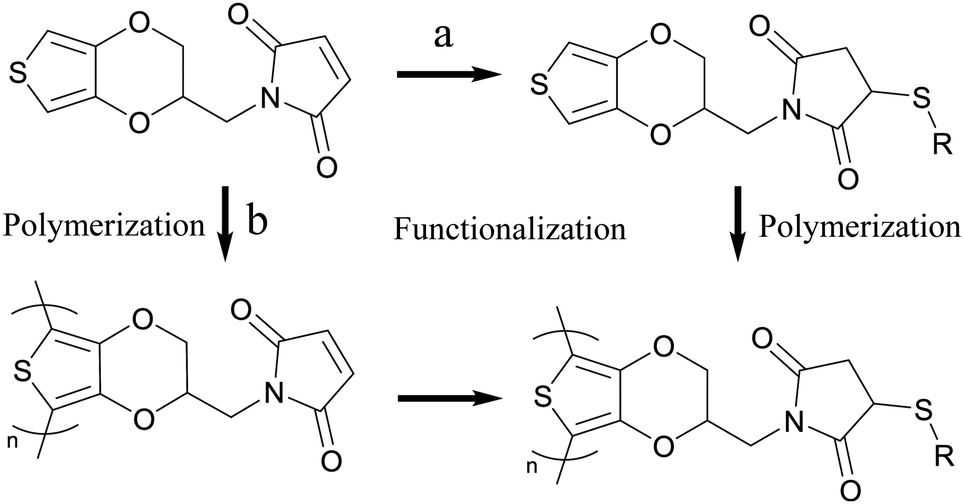 | ||
| Fig. 7 Two routes for making PEDOT+ from PEDOT-MA. Functionalization followed by polymerization (right then down) (a) or polymerization followed by functionalization (down then right) (b). | ||
PEDOT-MA was polymerized in the EQCMD to get values for frequency drop associated with mass deposition and values for dissipation change upon deposition. The ratio of dissipation change to frequency change gives us an idea of film rigidity.32 The Sauerbrey equation can be used to relate change in frequency normalized to the overtone (Δf) to the deposited mass (Δm) for rigid films, eqn (1).33 A film's rigidity is characterized by the change in dissipation (ΔD)/Δf ratio with ratios less than 0.4 ppm Hz−1 considered to be rigid films, Fig. 8.32 For the PEDOT-maleimide films this ratio is 0.036 ppm Hz−1 indicating the application of the Sauebrey equation for deposited mass is valid.
| Δm = −ACfΔf, where Cf = (ρqμ)1/2/(2fo2) | (1) |
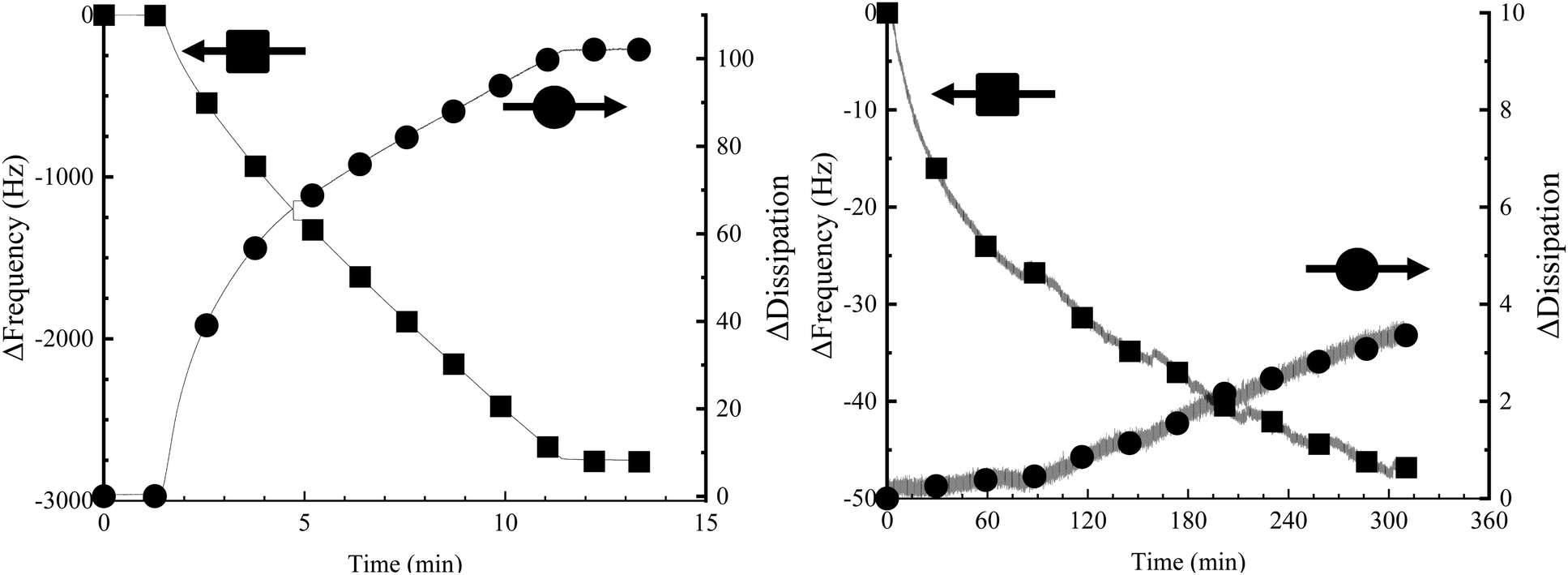 | ||
| Fig. 8 Change in frequency (Δf) and change in dissipation (ΔD) for (left) PEDOT-MA deposition and (right) fluorothiol functionalization. | ||
The Sauerbrey equation consists of: Δf as the observed change in frequency, Δm as the change in mass per cm2, μ is the shear modulus (2.95 × 1010 Pa) of the quartz crystal, ρq is the density of quartz (2.65 g cm−3), f Is the resonant frequency of the unloaded crystal (5 MHz). Combining the constants of fo, μ, and ρq gives a Sauerbrey constant Cf of 17.7 ng (cm−2 Hz−1) for the system used. An observed frequency change of 2750 Hz for the 1.13 cm2 area (A) electrode corresponds to a deposited mass of 55.0 μg cm−2, Fig. 8. Using the charge density of 0.039 C cm−2 with the deposited mass from the Sauerbrey equation gives a charge efficiency for deposition of 1.41 mg C−1 or 5.6 μmol C−1.
Post-functionalization of PEDOT-MA with fluorinated thiol was examined using similar QCM methods. Deposition of fluorinated thiol resulted in a minimal change in frequency of 46.79 Hz which correlates to 0.94 μg cm−2, a significantly lower deposition compared to the changes from EDOT-MA polymerization, Fig. 8. Fluorinated thiol functionalization is expected to be mainly a monolayer on the surface of the bulk PEDOT-MA which corresponds to both less mass deposition and a negligible change in film mechanical properties as shown in the negligible change in dissipation. Repeated experiments show similar results. Control experiments with only triethylamine solution flown over PEDOT-MA gave no changes in both frequency and dissipation, confirming that the deposition was due to the fluorinated thiol Fig. S8 (ESI†).
Examination of the XPS spectra before and after fluorination of PEDOT-MA demonstrates the changes in the fluorine, oxygen, and sulfur atoms. The fluorine spectrum shows the largest change with a large peak after functionalization with the fluorinated thiol, Fig. 9a. Two oxygen peaks are of note for PEDOT-MA at 531.7 eV and 533.2 eV with a slight shift of both peaks by 0.2 eV to a higher energy, Fig. 9b. These peak designations correspond well to other studies on PEDOT where the 531.7 eV peak relates to oxygen double bonded to carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the 533.2 eV peak is associated with an oxygen bonded to two different carbons (C–O–C).34 The single sulfur in PEDOT-MA corresponds to a doublet peak at 163.5 eV for S(2p1/2) and 164.6 eV for S(2p3/2) in the XPS spectra due to spin orbit splitting.35 Functionalization of the polymer lead to an increase in energy of the peak by 0.3 eV and 0.2 eV respectively.
O) and the 533.2 eV peak is associated with an oxygen bonded to two different carbons (C–O–C).34 The single sulfur in PEDOT-MA corresponds to a doublet peak at 163.5 eV for S(2p1/2) and 164.6 eV for S(2p3/2) in the XPS spectra due to spin orbit splitting.35 Functionalization of the polymer lead to an increase in energy of the peak by 0.3 eV and 0.2 eV respectively.
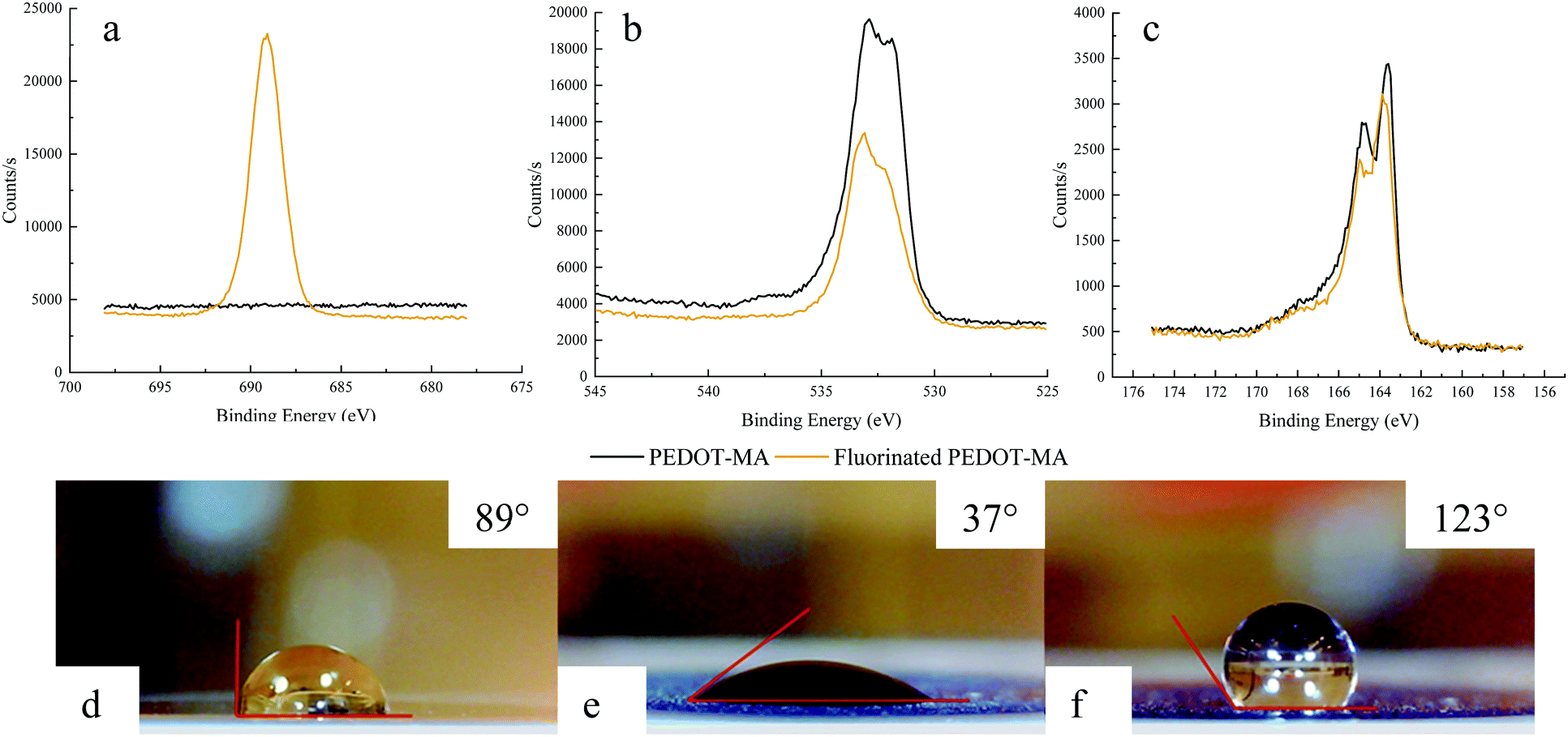 | ||
| Fig. 9 (Top) XPS spectra of (a) fluorine, (b) oxygen, and (c) sulfur for PEDOT-MA films before and after fluorination and (bottom) Contact angle measurements of (d) pristine crystal, (e) PEDOT-MA before post-process, and (f) PEDOT-MA after post-process. Corresponding contact angles are marked in pictures. | ||
The representative results of contact angle measurement are shown in Fig. 9d and e with a clear change on the surface hydrophobicity, contact angles from 37° (before flush) to 123° (after flush), due to the attachment of fluorinated thiol through the thiol-maleimide click reaction. Other characterized samples present the same trend and contact angle changes as shown in Table S2 (ESI†). Excepting that, a blank test with only 1 mM triethylamine dichloromethane solution flowing through the QCM-D cell at the same flow rate resulted in only a 10° change as shown in Table S2 (ESI†).
We also found that green fluorescent protein (GFP) could be attached to the surface using 12 hour exposure times to a 14.1 μg mL−1 GFP solution, details provided in the ESI,† Fig. S9. The above combined results strongly manifested the retention of the activity of maleimide functional groups on PEDOT-MA, which enables possibilities on post-process in contrast to unmodified PEDOT.
The ability to functionalize the monomer or the polymer after electropolymerization provides further avenues for polymer design and functionality. Functionalizing PEDOT-MA after electrochemical polymerization makes it possible to only modify the outermost surface of the sample, not the entire bulk. Characteristics for the polymerization of EDOT+ can vary significantly as shown in this work and post processing allows for the synthesis of PEDOT+'s that might be difficult to deposit. It also significantly reduces the amount of functionalized monomer needed to realize a certain property, which is advantageous since these are typically much more expensive and limited in availability.
Cytocompatibility
A cytocompatibility study of the EDOT+ monomers using SH-SY5Y cells showed that in the case of short time periods, addition of the biofunctional groups in this study improved normalized viability drastically compared to EDOT, Fig. 10. SH-SY5Y are neuroblastoma cancer cells commonly used to simulate neuronal-type cells for in vitro systems.36 Viability can be broken into two groups of high and low viability monomers. At the high viability end there is EDOT-cysteine followed by EDOT-MA-cholesterol and last by EDOT-MA-adamantane. In the low viability end, EDOT-MA shows a minor increase in viability compared to that of the negative control of ethanol at early time points. EDOT showed a marked increase compared to EDOT-MA, but is still an average of less than 30% normalized viability for the entire time period. The side chains improved viability both through the addition of a biological component as well as covering of the toxic maleimide component. Cysteine showing the highest viability corresponds well with its use to increase viability and adhesion of cells to surfaces.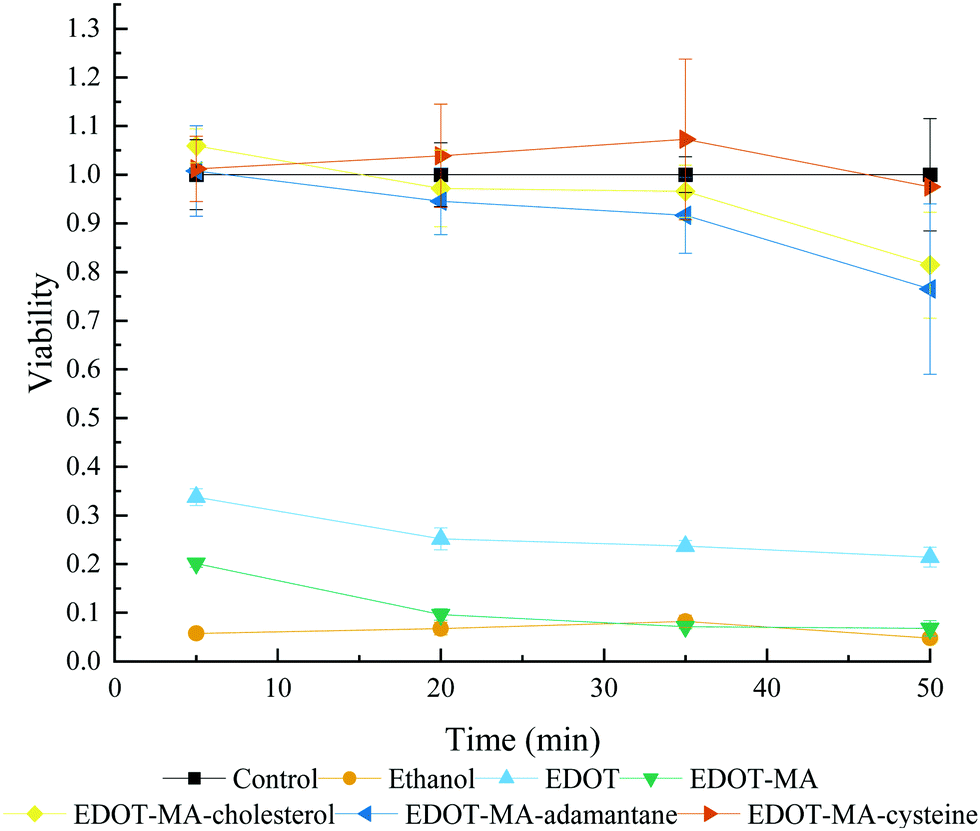 | ||
| Fig. 10 Cytocompatibility assay of variation functional EDOT's at 0.01 M concentration in culture media Polymerization. | ||
Near the end of the studied time frame of 50 minutes there was the beginning of a drop-off in viability for EDOT-MA-cholesterol and EDOT-MA-adamantane. The similarity to the control however shows that for the tens of seconds to minutes required for a polymerization there is a minimal effect on the cell culture. EDOT however has a much lower viability than has been previously reported from our group.37 A different viability assay was used in addition to our tests using only monomers without counterion. From our tests it was found that the common counterion of PSS with and without EDOT in cell cultures showed drastic and unrealistic increases in viability compared to controls, Fig. S10 (ESI†).
Conclusions
A family of novel 3,4-ethylenedioxythiophene monomers was synthesized based on maleimide functional groups. Starting from the base EDOT-maleimide, it was possible to use click chemistry to create a wide variety of different functional EDOT derivatives and their corresponding polymers and copolymers. These derivative polymers and copolymers were characterized with a variety of methods for determination of ideal synthesis methods and future applications. Including this were demonstrations of the electrochemical behavior post processing of PEDOT-MA with thiol bearing reagents, and improvement of the cytocompatibility of monomers. These new materials are of interest for optimizing the performance of PEDOT+ materials for a variety of applications including biomedical devices.Conflicts of interest
There were no conflicts to declare.Acknowledgements
Funding for this work was provided by DARPA HR0011-17–0019, the Air Force Office of Scientific Research MSIT Program for Next Generation Nanosystems, and National Science Foundation DMR-1808048. Support was also provided by the University of Delaware College of Engineering. We would like to thank the faculty and staff here at the University of Delaware including: the Jia group for use of their GFP, Wenbo Wu for help doing XPS, the Advanced Materials Characterization Lab for their characterization equipment, and the Keck Microscopy instate for use of their SEM. Thanks to Andrew White from Nanoscience for consulting on the use and analysis of QCM-D equipment and data. The University of Delaware has a patent filed for the monomers and polymers in this work under patent number: PCT/US2020/048495.Notes and references
- D. C. Martin, MRC, 2015, 5, 131–153 CrossRef CAS.
- D. Mantione, I. del Agua, A. Sanchez-Sanchez and D. Mecerreyes, Polymers, 2017, 9, 354 CrossRef PubMed.
- X. Cui and D. C. Martin, Sens. Actuators, B, 2003, 89, 92–102 CrossRef CAS.
- X. Cui, V. A. Lee, Y. Raphael, J. A. Wiler, J. F. Hetke, D. J. Anderson and D. C. Martin, J. Biomed. Mater. Res., 2001, 56, 261–272 CrossRef CAS PubMed.
- Z. Zhang, Mahmoud Rouabhia and S. E. Moulton, Conductive Polymers Electrical Interactions in Cell Biology and Medicine, CRC Press, 2017 Search PubMed.
- D. A. Koutsouras, P. Gkoupidenis, C. Stolz, V. Subramanian, G. G. Malliaras and D. C. Martin, ChemElectroChem, 2017, 4, 2321–2327 CrossRef.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 CrossRef PubMed.
- S. Kirchmeyer, K. Reuter and J. C. Simpson, in Poly(3,4-ethylene dioxythiophene)-Scientific Importance, Remarkable Properties, and Applications, ed. T. A. Skotheim, Taylor & Francis, Boca Raton, FL, 2007, vol. Conjugated Polymers: Theory, Synthesis, Properties and Characterization Search PubMed.
- H. Yano, K. Kudo, K. Marumo and H. Okuzaki, Sci. Adv., 2019, 5(4) DOI:10.1126/sciadv.aav9492.
- L. Ouyang, C. Musumeci, M. J. Jafari, T. Ederth and O. Inganäs, ACS Appl. Mater. Interfaces, 2015, 7, 19764–19773 CrossRef CAS PubMed.
- L. V. Lingstedt, M. Ghittorelli, H. Lu, D. A. Koutsouras, T. Marszalek, F. Torricelli, N. I. Crăciun, P. Gkoupidenis and P. W. M. Blom, Adv. Electron. Mater., 2019, 5, 1800804 CrossRef.
- D. Mawad, A. Artzy-Schnirman, J. Tonkin, J. Ramos, S. Inal, M. M. Mahat, N. Darwish, L. Zwi-Dantsis, G. G. Malliaras, J. J. Gooding, A. Lauto and M. M. Stevens, Chem. Mater., 2016, 28, 6080–6088 CrossRef CAS PubMed.
- A. G. Guex, J. L. Puetzer, A. Armgarth, E. Littmann, E. Stavrinidou, E. P. Giannelis, G. G. Malliaras and M. M. Stevens, Acta Biomater., 2017, 62, 91–101 CrossRef CAS PubMed.
- X. Strakosas, B. Wei, D. C. Martin and R. M. Owens, J. Mater. Chem. B, 2016, 4, 4952–4968 RSC.
- L. K. Povlich, J. C. Cho, S. Spanninga, D. C. Martin and J. Kim, Polym. Prepr., 2007, 48, 7–8 CAS.
- N. Bhagwat, K. L. Kiick and D. C. Martin, J. Mater. Res., 2014, 29, 2835–2844 CrossRef CAS.
- N. Bhagwat, R. E. Murray, S. I. Shah, K. L. Kiick and D. C. Martin, Acta Biomater., 2016, 41, 235–246 CrossRef CAS PubMed.
- L. Ouyang, B. Wei, C. Kuo, S. Pathak, B. Farrell and D. C. Martin, Sci. Adv., 2017, 3, e1600448 CrossRef PubMed.
- B. Wei, J. Liu, L. Ouyang, C.-C. Kuo and D. C. Martin, ACS Appl. Mater. Interfaces, 2015, 7, 15388–15394 CrossRef CAS PubMed.
- L. Zhang, Y. Wen, Y. Yao, J. Xu, X. Duan and G. Zhang, Electrochim. Acta, 2014, 116, 343–354 CrossRef CAS.
- E. Stavrinidou, R. Gabrielsson, E. Gomez, X. Crispin, O. Nilsson, D. T. Simon and M. Berggren, Sci. Adv., 2015, 1, e1501136 CrossRef PubMed.
- E. Stavrinidou, R. Gabrielsson, K. P. R. Nilsson, S. K. Singh, J. F. Franco-Gonzalez, A. V. Volkov, M. P. Jonsson, A. Grimoldi, M. Elgland, I. V. Zozoulenko, D. T. Simon and M. Berggren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2807–2812 CrossRef CAS PubMed.
- D. Mantione, I. del Agua, W. Schaafsma, J. Diez-Garcia, B. Castro, H. Sardon and D. Mecerreyes, Macromol. Biosci., 2016, 16, 1227–1238 CrossRef CAS PubMed.
- S. Nagane, P. Sitarik, Y. Wu, Q. Baugh, S. Chhatre, J. Lee and D. C. Martin, MRS Adv., 2020, 1–14 Search PubMed.
- S.-C. Hong, K.-M. Park, C. R. Hong, J.-C. Kim, S.-H. Yang, H.-S. Yu, H.-D. Paik, C.-H. Pan and P.-S. Chang, Colloids Surf., A, 2020, 594, 124670 CrossRef CAS.
- X. Wang, W. Dai, X. Li, Z. Chen, Z. Zheng, Z. Chen, G. Zhang, L. Xiong and S. Duo, J. Alloys Compd., 2020, 825, 154052 CrossRef CAS.
- I. Stankova, K. Chuchkov, R. Chayrov, L. Mukova, A. Galabov, D. Marinkova and D. Danalev, Int. J. Pept. Res. Ther., 2019 DOI:10.1007/s10989-019-09983-4.
- Metrohm DropSens, https://www.dropsens.com/en/faqs_dropsens.html, accessed May 19, 2021.
- J. Am. Coll. Toxicol., 1987, 6, 23–51 DOI:10.3109/10915818709095488.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019–4025 CrossRef CAS.
- V. Castagnola, C. Bayon, E. Descamps and C. Bergaud, Synth. Met., 2014, 189, 7–16 CrossRef CAS.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- E. Mitraka, M. J. Jafari, M. Vagin, X. Liu, M. Fahlman, T. Ederth, M. Berggren, M. P. Jonsson and X. Crispin, J. Mater. Chem. A, 2017, 5, 4404–4412 RSC.
- K. Z. Xing, M. Fahlman, X. W. Chen, O. Inganäs and W. R. Salaneck, Synth. Met., 1997, 89, 161–165 CrossRef CAS.
- J. Kovalevich and D. Langford, in Neuronal Cell Culture, ed. S. Amini and M. K. White, Humana Press, Totowa, NJ, 2013, vol. 1078, pp. 9–21 Search PubMed.
- S. M. Richardson-Burns, J. L. Hendricks and D. C. Martin, J. Neural Eng., 2007, 4, L6–L13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00356b |
| This journal is © The Royal Society of Chemistry 2022 |