
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellulose-assisted electrodeposition of zinc for morphological control in battery metal recycling†
B. W.
Hoogendoorn
a,
M.
Parra
a,
A. J.
Capezza
 a,
Y.
Li
a,
K.
Forsberg
b,
X.
Xiao
*a and
R. T.
Olsson
*a
a,
Y.
Li
a,
K.
Forsberg
b,
X.
Xiao
*a and
R. T.
Olsson
*a
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56, 11428 Stockholm, Sweden. E-mail: XionXi@kth.se; rols@kth.se
bDepartment of Chemical Engineering, KTH Royal Institute of Technology, Teknikringen 42, 11428 Stockholm, Sweden
First published on 15th April 2022
Abstract
Cellulose nanofibers (CNFs) are demonstrated as an effective tool for converting electrodeposits into more easily detachable dendritic deposits useful in recycling zinc ion batteries via electrowinning. The incorporation of CNFs at concentrations ranging from 0.01 to 0.5 g L−1 revealed a progressively increasing and more extensive formation of a nacre-like dendritic zinc structure that did not form in its absence. Increasing the CNF concentrations to 0.5 g L−1 resulted in the most extensive formation of dendritic structures. The explanation for the observed phenomenon is the ability of CNFs to strongly interact with metal ions, i.e., restricting the mobility of the ions towards the electrowinning electrode. At the highest concentration of CNFs (0.5 g L−1), in combination with the lowest current density (150 A m−2), electrodeposition was limited to the extent that formed deposits were almost non-existent. The electrodeposition in the presence of CNFs was further evaluated at different temperatures of 20, 40 and 60 °C. The dendritic formation was increasingly suppressed with increasing temperatures, and at a temperature of 60 °C, the electrodeposited morphologies could not be differentiated from the morphologies formed in the absence of the cellulose. The results stemmed from a greater mobility of the metal ions at elevated temperatures, while at the same time suggests an inability of the CNF to strongly associate the metal ions at elevated temperatures. High-pressure blasted titanium electrodes were used as a reference material for accurate comparisons, and electron microscopy (FE-SEM) and X-ray diffraction were used to characterize zinc morphologies and crystallite sizes, respectively. This article reports the first investigation on how dispersions of highly crystalline cellulose nanofibers can be used as a renewable and functional additive during the recycling of battery metal ions. The metal-ion/cellulose interactions may also allow for structural control in electrodeposition of other metal ions.
1. Introduction
Zinc-ion batteries are lower cost alternatives to their lithium-ion counterparts in applications of energy storage.1,2 Zinc is extracted from zinc sulphate-rich ores and refined via electrolysis, accounting for most of the world's zinc production.3 In the battery industry, electrolysis can also be used to recycle and recover metals, similar to lead from the spent lead-acid battery paste, etc., where the electrodeposition is termed as electrowinning.4,5 The possibility to perform the electrowinning continuously would make it an attractive process both from a practical and economical perspective. A continuous electrowinning process would, however, rely on the methods to grow easily removable metal deposits.3,6–8 Efforts to control the nature of the deposits are therefore likely to be in focus in the near future of battery metal recycling. So far, research on the morphology of electrodeposited zinc has mostly focused on suppressing the dendritic zinc growth inside batteries, which is caused by repetitive battery charging that results in limited battery performance.9–11 The formation of dendritic zinc has however also been investigated due to the possibility of taking advantage of superior charge transport properties due to the high specific surface area of a ramified metallic network.12,13 From a recycling perspective, dendritic zinc formation is beneficial due to the facilitated release of the zinc deposited on the cathode (used for electrowinning). A facilitated metal-release from the cathode is relevant for the most electrowinning recycling procedures of leached battery metals.14 In the case of nickel–cadmium batteries, cadmium is deposited on an aluminium surface and scraped off manually before the manufacturing of new electrode materials.15Small organic molecules such as pyridine derivatives, or larger macromolecules like polyethylene glycol (PEG) or polyoxyethylene ether, have historically been used in zinc electrodeposition due to their ability to act as ‘polarizers’, providing more levelled and shiny electroplated surfaces.16–18 Nonetheless, there are shortcomings in using organic additives, including toxicity and pollution, and for an easy release of the electrodeposited material the contrary to shiny and levelled surfaces is preferred.19 With societal developments moving towards the use of abundant environmentally friendly and renewable resources, additives meeting these criteria are of significant interest. Biomacromolecules offer a non-toxic alternative with the ability to bind metal ions in both specific and non-specific ways.20–22 For instance, Xia et al. found that gelatine affected zinc deposits during electrowinning in terms of the crystal orientation and smoothness of the zinc surfaces.23 Another biomacromolecule is cellulose, the most abundant polysaccharide on earth.24 The hydroxyl groups of cellulose are known to bind metal ions, creating hybrid metal oxide/cellulose materials.20,25,26 This ability has, among other things, been used to form nanocomposites via in situ precipitation of metal ions onto cellulose nanofibrils (CNFs).27 Cellulose-based nanocomposites have also been prepared by immersing cellulose fibres in zinc acetate solutions.28 Overall, these abilities make cellulose nanofibers an interesting candidate as an additive within the electrodeposition and recovery of zinc.
In this article, the role of CNFs was studied in the electrodeposition of zinc on titanium substrates used for zinc electrowinning. Different electrochemical conditions were explored, including substrate surface roughness, current densities, and electrodeposition times, before the effect of different CNF concentrations could be accurately determined. The results revealed that dendritic zinc structures are formed due to the presence of CNFs, which facilitate the recovery and detachment of the deposited zinc metal from the electrode. This contrasted with more levelled or randomly organized zinc deposits formed in the absence of CNFs. The dendritic growth phenomenon was dominant at lower temperatures, where the zinc ion/cellulose associations restricted the mobility of metal ions in the electrolyte. Overall, the results represent the first report on how cellulose in its fully defibrillated dispersed state can be used within electrowinning reactions important on larger industrial scales, e.g., in the recycling of battery metals.
2. Experimental
2.1 Materials
Zinc chloride (ZnCl2, ≥98%), potassium chloride (KCl, ≥99.5%), nitric acid (HNO3, ≥65%), sodium hydroxide (NaOH, ≥95%), sodium metasilicate (Na2SiO3, ≥98%), sodium carbonate (Na2CO3, ≥99%) and hydrochloric acid (HCl, 37%) were purchased from Sigma-Aldrich, Sweden. Boric acid (H3BO3, 100%) was purchased from Calbiochem, Merck KGaA, Germany. The zinc standard solution (1000 mg L−1) used for ICP-OES calibration was purchased from Alfa Aesar, Sweden. Titanium- (Grade 1) and zinc-sheets (≥99%) at 0.5 mm thickness were used as electrodes. An aluminium silicate grit (D ∼ 0.20–0.50 mm) and glass beads (D ∼ 0.25–0.42 mm) were purchased from Ahlsell AB, Sweden. The epoxy adhesive for masking the electrodes was produced by Loctite (Power Epoxy, Henkel) and was purchased from Clas Ohlson AB, Sweden. The bacterial cellulose (BC) had been grown from the strains of Acetobacter xylinum and was purchased in the form of cubes with an edge length of ∼1 cm (Chaokoh, Asian Market). Milli-Q water (18.2 MΩ, 25 °C, pH 7.0) was used in the preparation of solutions.2.2 Preparation and characterisation of the cellulose
A mass of ∼2 kg of cellulose was used for the preparation of cellulose nanofibrils (CNFs) via acid hydrolysis extraction and the procedure is illustrated in Fig. S1. BC cubes were rinsed in water (12 h) to remove the remains of preservation and the growth medium. The washed cubes were then treated for 5 min in 1 L of a 10 vol% sodium hydroxide solution (98 °C). Sodium hydroxide was thereafter exchanged with water until a neutral pH (pH ≈ 7) was reached. The cellulose cubes were shredded using a regular kitchen blender before being compressed into a solid to remove excess water in a polyamide mesh (SEFAR PA1000 120/305-35W with 46 μm openings). The CNF content of the semi-dried material (see the appearance of the intermediate material in Fig. S1, ESI†) of the 65 g solid of compressed cellulose was determined to be 12 ± 3 wt% by drying five separate samples in a laboratory oven at 70 °C.Hydrolysis was initiated by adding the shredded cellulose into a 1 L of a 50 vol% H2SO4-solution heated to 60 °C under stirring at 300 rpm. The hydrolysis reaction was allowed to proceed for 4 h before quenching the reaction with 1 L of cold Milli-Q water. The extracted CNF was centrifuged 3 times for 10 min at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × G with the supernatant being exchanged for Milli-Q water between each cycle. The extracted CNF was maintained as an aqueous dispersion with a pH of ∼3 (Fig. S1, ESI†), determined to be 1.55 ± 0.1 wt%. The surface charge of the extracted CNF was determined to be 50 μeq g−1 using polyelectrolyte titration, revealing that the CNF had a negative charge. The nanofibers were measured in length and width from a minimum of 1000 fibres, using the ImageJ software developed by the National Institute of Health, Maryland, USA. The obtained CNF had an average length of 480 ± 30 nm and an average thickness of 25 ± 2 nm, see Fig. 6.
000 × G with the supernatant being exchanged for Milli-Q water between each cycle. The extracted CNF was maintained as an aqueous dispersion with a pH of ∼3 (Fig. S1, ESI†), determined to be 1.55 ± 0.1 wt%. The surface charge of the extracted CNF was determined to be 50 μeq g−1 using polyelectrolyte titration, revealing that the CNF had a negative charge. The nanofibers were measured in length and width from a minimum of 1000 fibres, using the ImageJ software developed by the National Institute of Health, Maryland, USA. The obtained CNF had an average length of 480 ± 30 nm and an average thickness of 25 ± 2 nm, see Fig. 6.
2.3 Zinc electrodeposition
Electrodeposition experiments were carried out in an electrolytic cell with dimensions 170 × 65 × 110 mm. The cell was filled with 1 L of aqueous electrolyte consisting of 60 g L−1 ZnCl2, 160 g L−1 KCl, and 23 g L−1 H3BO3, with or without the CNF added as the wet, never dried, cellulose suspension. The solution was purged with argon for 30 min and the temperature was adjusted for each experiment to 20, 40 or 60 ± 1 °C. Fig. 1a shows the electrolytic cell with the titanium (Ti) electrode, used as the cathode for the electrodeposition, placed in the middle of the cell with a deposition area of 2 cm2. Two 45 × 100 mm zinc sheets were used as anodes, contained in polypropylene mesh bags to avoid the release of debris into the electrolyte solution. The sheets were placed on the sides of the cell as anodes, Fig. 1a, on equal distance from the Ti-electrode. | ||
| Fig. 1 (a) Electrolytic cell setup. (b) Ti-electrodes without abrasive treatment (right), with sandblasting treatment using glass beads (middle) and aluminium silicate treatment (left). (c) Micrograph of the surface of Ti-electrodes with aluminium silicate sandblasted treatment, and the inset shows the magnification of a cavity. SEM images of the surface of substrates (d) after sandblasting treatment with aluminium silicate and (e) without abrasive treatment after electrodeposition. (f) An example of a faceted crystal grown in a cavity. All electrodeposition experiments were carried out at a current density of 150 A m−2 for 20 min at 40 °C. | ||
Ti-electrodes were sandblasted with aluminium silicate (sand) or glass beads (glass), before being cleaned in an alkaline cleaner (40 g L−1 NaOH, 25 g L−1 Na2CO3, and 25 g L−1 Na2SiO3) for 10 min at 90 °C, to remove organic surface impurities. The blasting was carried out to ensure that every Ti-electrode was represented by identical electrode deposition surfaces. After the alkaline treatment, the substrates were rinsed with Milli-Q water and immediately before every electrodeposition experiment, the Ti-electrodes were pickled for 10 min in Aqua regia and rinsed with Milli-Q water.
The pH of the electrolyte was measured before and after each experiment using a pH meter (Seven2Go S8 pH-meter, MettlerToledo, Switzerland) to ensure that the pH of the electrolyte was approximately the same (5.4 ± 0.25). The electrodeposition experiments were carried out for 20 min at current densities of 150 A m−2, 300 A m−2, 500 A m−2, 800 A m−2 and 1000 A m−2. The effect of the electrodeposition time (in absence of the CNF) was further evaluated at current densities of 150 and 1000 A m−2, respectively, by monitoring the crystal growth at time intervals of 1, 5, 30, 60, 300, 600, 1200 and 2400 s.
To investigate the effect of cellulose nanofibers, electrodepositions were performed in aqueous electrolytes containing 0.01 g L−1 and 0.5 g L−1 of the bacterial CNF, in addition to the constituents specified above for the electrolyte. The zinc growth in the presence of the CNF was studied at current densities of 150 A m−2 and 1000 A m−2 at a temperature of 40 °C and the duration of 20 min, which was established as the most representative temperature to elucidate the effect of the CNF (based on the observed depositions at different field strengths). The effect of varying the temperature was further evaluated by performing the electrodeposition at 20, 40 and 60 °C for the electrolyte containing 0.5 g L−1 CNF, for 1, 3 and 7 min. After each experiment, the Zn coated Ti-electrodes were rinsed with Milli-Q water and dried in a vacuum oven at 45 °C for 1 h prior to being characterized.
2.4 Characterisation
ZYGO's 3D Optical Profiler was used to measure the surface roughness of the samples (10× magnification objective, 1× zoom, and 0.83 × 0.83 mm2 field of view). The surface roughness was determined by measuring how much the surface deviated from its average height (Sa). A planar fit was performed to correct for the curvature of the samples.The morphologies of the Ti-electrodes before and after the electrodeposition were observed using a scanning electron microscope (FE-SEM, Hitachi S-4800), and the distributions of elements were measured using energy-dispersive X-ray spectroscopy (EDS). The samples were coated with platinum/palladium (Pt/Pd) in a high-resolution sputter coater (Cressington 208HR) for 20 s at a current density of 80 mA before microscopy imaging.
The samples before and after the electrodeposition were characterised using X-ray diffraction (XRD), and the PANalytical X’pert3 software was used for the peak identification. The diffraction patterns were recorded over a 2θ range from 5° to 80° in steps of 0.008° at 45 kV and 40 mA using Cu Kα radiation (λ = 0.15418 nm).
The concentrations of zinc ions in the electrolyte before and after the electrodeposition were determined using an inductively coupled plasma optical emission spectrometer (ICP-OES, Thermo Fisher iCAP 7400, USA).
3. Results and discussion
3.1 Effect of Ti-electrode surface topography on the electrodeposition of zinc
Fig. 1a and b show the appearances of the titanium electrodes before and after the abrasive blasting treatment. Additionally, Fig. 1c shows a SEM image of the Ti-electrode after the harsher aluminium silicate treatment. The micrograph reveals that cavities with a size of ca. 5–10 μm were formed in the substrate surface, contributing to the roughness of the substrate. As shown in the micrographs and surface texture maps in Fig. S2 (ESI†), the roughness of the surface (deviation from its average height) varied significantly depending on the media used for making the reference electrodes, i.e., aluminium silicate (Sa = 4.20 ± 0.2) > substrates treated with glass beads (Sa = 1.88 ± 0.04) > non-treated substrates (Sa = 0.50 ± 0.02). Fig. S2 (ESI†) also shows how the deposition of zinc occurred on the different substrates. The roughness and texture of the electrode surface was previously demonstrated to significantly affect the nature of the deposition.29,30 It could thus be concluded that the achieved surface roughness values corresponded well with the different hardness values of the blasting media (aluminium silicate grit (7–9 Mohs31) and glass beads (5–6 Mohs32)). Fig. 1d and e, Fig. S2f and S3 (ESI†) show the micrographs of Ti-electrodes after zinc electrodeposition, revealing that the zinc preferred to deposit in the cavities created by sandblasting. Without an abrasive treatment, the Zn instead adhered in a non-preferential manner to the overall smoother surface (Fig. 1e). The EDX images in Fig. S4 (ESI†) confirmed the presence of zinc domains within the cavities of the abraded titanium substrate. Earlier research demonstrated that dislocations and imperfections along the electrode surface are more favourable sites for the initial nucleation.29,30 Aluminium silicate was therefore selected as the blasting medium due to its ability to create the highest surface roughness while at the same time consistently providing identical surfaces for electrodeposition (allowing reproducible electrodeposition experiments with all scratches from the Ti-manufacturing process removed, Fig. 1b). Fig. S5 (ESI†) (XRD) additionally confirmed that the deposited zinc phase was identical regardless of the substrate pre-treatment method.Fig. 1f highlights a representative zinc crystal grown in one of the induced cavities, revealing that the zinc was deposited in the form of flat and smooth crystals with a size of 10–20 μm, always with well-defined facetted edges. These morphologies have been associated with the formation of deposits through the epitaxial growth, which occurs under conditions when the kinetics is controlled by charge transfer and not by mass transfer, or any other phenomena dependent on the ionic diffusion.13,33 As the ions approach the cathode surface, the charge transfer occurs where the zinc ions adsorb along the cathode surface. The atoms can then diffuse along the surface of the electrode and contribute to the crystal growth at the most energetically favourable sites, which explained the very uniform growth of the zinc-metallic structures.10 More facetted crystal structures tend to form when the supply of ionic species is not limited by diffusion in the electrolyte and when the surface diffusivity of the adsorbed atoms is high.13
Fig. S5 (ESI†) shows the XRD patterns of the samples after a 20 min electrodeposition at a current density of 150 A m−2, where the peaks were attributed to Ti-phase (JCPDS-ICDD PDF 44-1294) and Zn-phase (ICDD PDF 04-0831).
Diffractograms confirmed the deposition of Zn crystals onto the Ti-electrode. The presence of the Ti-peaks was consistent with the finding that the zinc did not fully cover the Ti-electrode, as shown in Fig. 1d and e. No differences in the deposited Zn phase could be associated with the diffraction patterns regardless of the nature of the Ti-surface roughness, indicating that the crystal growth was independent of the nucleation on the differently prepared titanium substrates. However, previously it has been shown that increasing current densities can affect the uniformity as well as the morphology of zinc-deposits by promoting the protruding growth.34,35 It was therefore motivated us to investigate an extended range of current densities before evaluating the impact of the CNF on the electrodeposition experiments.
3.2 Effect of the current density in the Zn electrodeposition
Fig. 2 shows micrographs revealing the morphology of the zinc deposits obtained at current densities of 150 A m−2, 300 A m−2, 500 A m−2, 800 A m−2, and 1000 A m−2 after 20 min of deposition. Fig. 2a–c reveal that deposits consisted of solitary facetted hexagonal Zn crystals at current densities lower than 500 A m−2. An increase of the current density to 800 A m−2 resulted in a merging of the hexagonal structures (Fig. 2d), whereas a further increase to 1000 A m−2 allowed a complete coverage of the cathode surface (Fig. 2e). Fig. 2f shows the edges of the Ti-electrodes after 20 min of electrodeposition at current densities of 300 A m−2 and 800 A m−2, revealing that the protuberant zinc deposits significantly increased with an increasing current density. The current crowding effect typically occurs at the corners and edges of the electrode, where a local electric field is intensified, leading to a more extensive zinc growth.36 Furthermore, the protruding structures show that the Zn ions prefer to attach to the already deposited zinc with lower resistance,37 where Ti (39.00 μΩ × cm) has a higher resistivity than Zn (5.45 μΩ × cm).38 | ||
| Fig. 2 Substrate surface morphologies after 20 min of electrodeposition at 40 °C and at different current densities. SEM images of the center of the substrate after electrodeposited at current densities of (a) 150 A m−2, (b) 300 A m−2, (c) 500 A m−2, (d) 800 A m−2, and (e) 1000 A m−2 and (f) the corner of the substrate after electrodeposition at current densities of 300 A m−2 (left) and 800 A m−2 (right). | ||
Fig. 3 shows the X-ray diffractograms after 20 min of electrodeposition at current densities of 150, 500 and 1000 A m−2, respectively. The diffraction peaks related to the Zn phase were the only peaks that could be discerned for current densities of 500 and 1000 A m−2, while the diffraction peaks corresponding to the titanium substrate were only present at 150 A m−2. The lack of titanium peaks was thus synonymous with the fully Zn covered Ti-electrode. The Zn-ion concentrations before and after the deposition were determined through ICP measurements (Table S2, ESI†). The Zn ion concentration of the electrolyte did not decrease throughout the reaction. A more dominant orientational growth of the Zn crystals, with more exposure of the (002) face, always showed at a higher current density of 1000 A m−2 after 20 min deposition, see the inset of Fig. 3. The extended in-plane crystal growth (parallel to the surface) of the (002)-plane could be observed by determining the relative intensities of the (002)/(101) peaks, which were ca. ten times higher after the electrodeposition at 1000 A m−2 compared to the value calculated from ICDD PDF 04-0831. In other studies, an exposure of the (002)-basal planes has been argued to be beneficial due to its high stability, which is likely to hinder corrosion, H2 evolution and by-product reactions, an aspect which in turn significantly improves the electrochemical performance of fuel-cell Zn-ion batteries.39 In this study, the extensive exposure of the (002)-face, associated with a current density of 1000 A m−2 allowed for conditions representing stable Zn–Zn nucleation and growth to show, whereas a current density of 150 A m−2 could be associated with a significantly smaller deposition rate (and total amount) within 20 min.
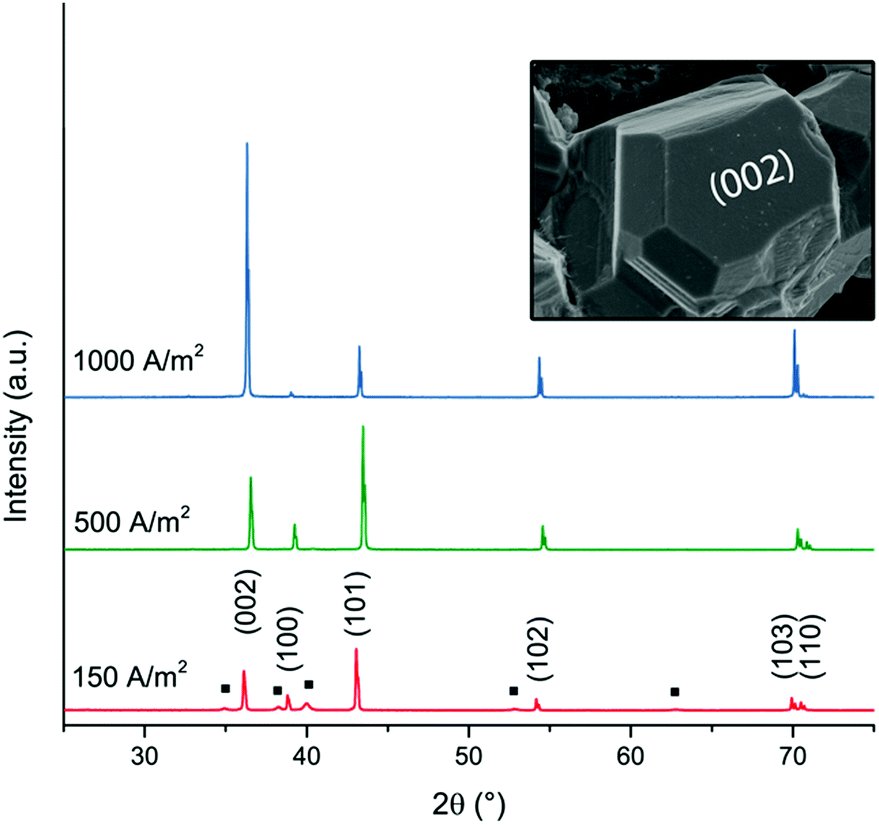 | ||
| Fig. 3 XRD diffractograms of zinc deposited at current densities of 150 A m−2, 500 A m−2 and 1000 A m−2. The peaks corresponding to the titanium phase (JCPDS-ICDD, 44-1294) are labelled with a black square (■). The hexagonal prism analogous to the zinc crystal in Fig. 1f illustrates the corresponding directions of the crystal planes (002) and (100). | ||
3.3 Effect of the electrodeposition time
To understand the growth patterns of the zinc-phase during the electrodeposition reactions, Fig. S6 (ESI†) shows micrographs taken at different deposition times (5 s, 15 s, 30 s, 1 min, 5 min and 10 min). The first formed Zn particles, with facetted structures, showed a diameter of ca. 125 nm already after 5 seconds of deposition (Fig. S6a, ESI†), while smaller nanoflakes (Fig. 4) covered the surface of these particles as a major feature of the earliest growth samples. These smaller particles merged and grew into larger sized particles (with a size of ca. 2–5 μm) within the first minute, while the nanosized flakes completely disappeared. A further increase in the deposition time to 10 min resulted in Zn crystals growing on top of each other as merged crystals, resulting in a transition from having a deposition of singular zinc particles to having a deposited zinc layer. The uniform electrodeposited layer of zinc over the entire electrode could be concluded to have occurred after ten min of deposition, compared Fig. 5a with Fig. 3 (XRD). Overall, the formation and dissolution of the zinc nanoflakes followed by the more extensive growth of the larger Zn-crystals could be concluded to be a typical growth pattern. This trend could be confirmed from observations for the zinc deposited at a current density of 150 A m−2. | ||
| Fig. 4 Micrograph showing zinc electrodeposited after 5 s near one of the edges of the electrode using a current density of 150 A m−2. The hexagonal nanoflakes are visible as merging with its surface. | ||
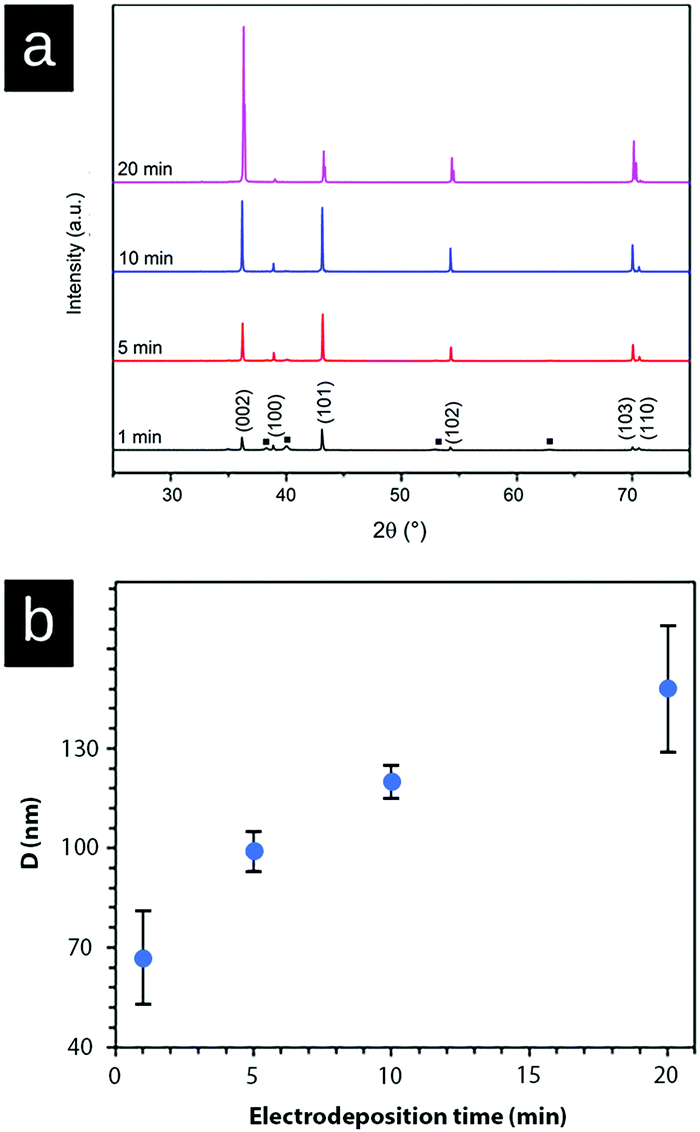 | ||
| Fig. 5 (a) XRD patterns of zinc-deposition on the surface of the Ti substrate at a current density of 1000 A m−2 depending on electrodeposition times. 1 min (black); 6 min (red); 10 min (blue); and 20 min (pink). The peaks corresponding to titanium are labeled with a black square (■). (b) Crystallite size (D) as estimated with the Scherrer equation measured for four different electrodeposition times at a current density of 1000 A m−2. | ||
Fig. 4 and Fig. S7a and b (ESI†) show the nanoflakes with 25–50 nm width as grown on the formed zinc particles at a deposition time of 5 s. The edges of the nanoflakes became more prominent when increasing the deposition time to 30 s and 1 min (Fig. S7c and d, ESI†). Further increases of the deposition time to 5 and 10 min resulted in the disappearance of the hexagonally shaped nanoflake structures on the surface of Zn particles, i.e. similar to the transformation occurring within the first minute at a current density of 1000 A m−2 (Fig. S6a, ESI†). Overall, the deposition at a current density of 150 A m−2 occurred at a slower rate (synonymous with the XRD data in Fig. 3). Fig. 4 shows a ca. 400 nm sized Zn particle observed at one of the edges of the electrode. The entire particle appeared to be composed of an accumulation of the described hexagonal nanoflakes. The observation supported the conclusion that the hexagonal nanoflake particles were always to some extent present in the formation of the larger sized electrodeposited zinc phase.
Fig. 5a shows the X-ray diffraction patterns of the zinc deposited at a current density of 1000 A m−2 measured at 1 min, 5 min, 10 min, and 20 min. The diffractogram shows that the intensity ratio of (002)/(101) diffraction planes changed after 10 min of electrodeposition, demonstrating a stronger growth and representation of the (002)-plane. This intensity ratio transition was never observed at a current density of 150 A m−2 (Fig. 3) within the time frame of 20 min, because of the suppressed and more limited electrodeposition. Fig. 5b shows the average crystallite size of the deposited Zn as calculated from the XRD data (Fig. 5a, peaks (002), (101), (102), (103), and (110)) with the Scherrer equation. The graph demonstrates that the crystallite size of Zn increased (doubled) with the increasing electrodeposition time from ca. 65 nm (1 min) to 145 nm (20 min). The crystallite size was determined as the average from the above peaks because of the anisotropic nature of the crystal growth.40,41 Overall, it was clear from the electrodeposition over time study that the external zinc surface for the fully zinc coated electrodes was dominantly exposing the (002)-plane parallel with the substrate surface.
3.4 Effect of the cellulose nanofiber concentration on the electrodeposition of zinc
Fig. 6a shows the result of the acidic cellulose extraction and the resulting average CNF sizes in terms of the fibre thickness and length of the CNF, whereas Fig. 6b shows the crystalline nature of the CNFs and their X-ray diffraction patterns.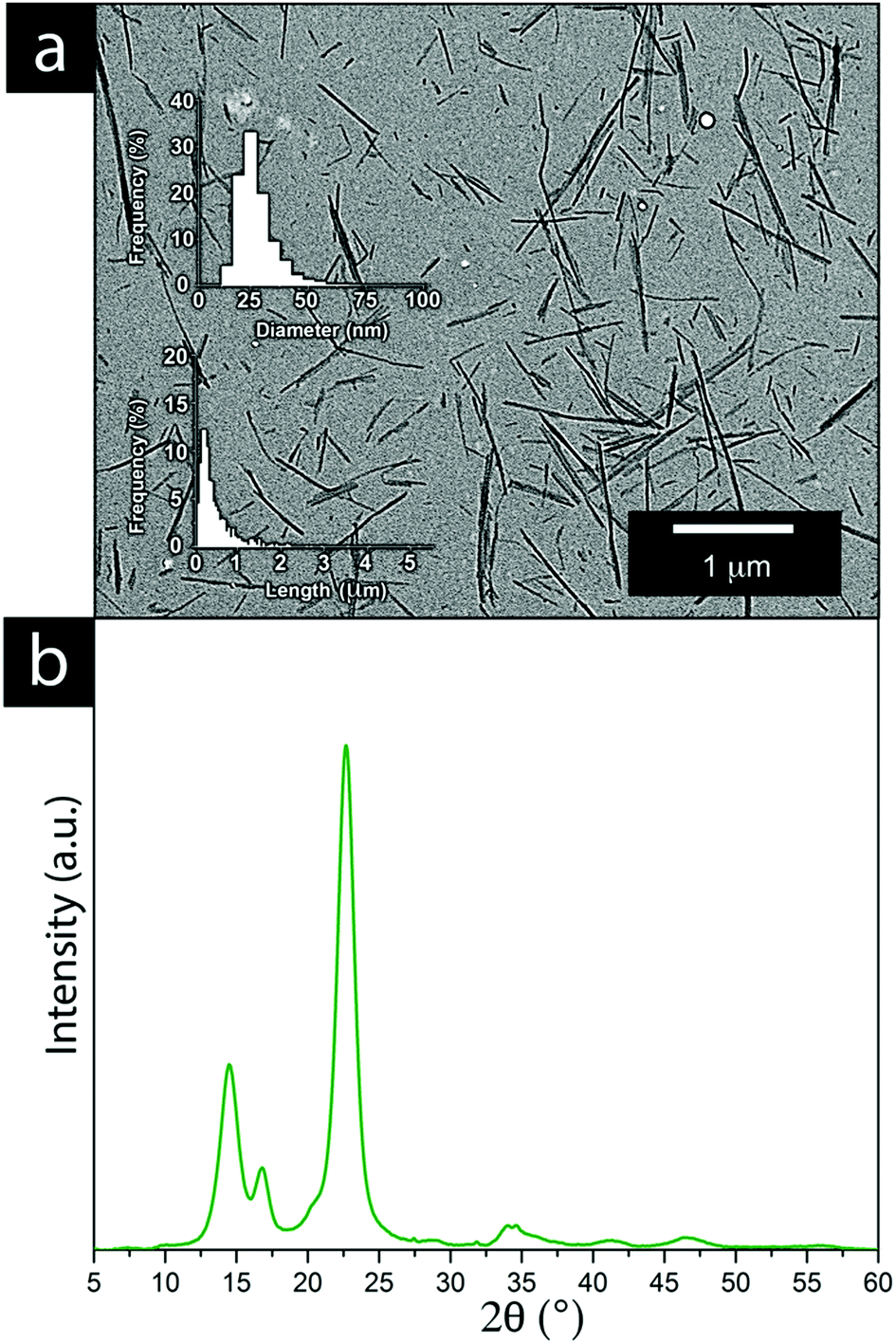 | ||
| Fig. 6 (a) TEM micrographs of the bacterial cellulose obtained after acid hydrolysis, including histograms showing the length and thickness distributions of the bacterial cellulose. (b) XRD diffractogram of the bacterial CNF obtained after acid hydrolysis. | ||
Cellulose nanofibers as an aqueous dispersion allowed for using the CNF in a more defibrillated form, where the fibers had been liberated during hydrolysis. The CNF in a dry state generally comes with limitations from the drying process, which is accompanied by various degrees of irreversible fiber aggregation.42,43 The use of CNF in a dispersed form exposes a larger portion of the nanofiber surfaces to the solution and the ions present in it. The interactions between the metal ions and the nanofiber surface can thereby be maximized to a larger extent, resulting in an overall greater impact on the electrodeposition. It was evident that the extraction resulted in a relatively wide fibre length distribution while their thickness reflected measurements which also included CNFs that remained partially aggregated. The individual CNFs were estimated to be 20–30 nm in thickness, see Fig. 6a. The high crystallinity of the bacterial CNFs is in agreement with previous studies reporting a high crystallinity (often >80%) for the bacterial cellulose (Fig. 6b).44–47
Fig. 8b shows the electrodeposited zinc at a current density of 150 A m−2 when 0.01 g L−1 of the CNF was present in the electrolyte. The deposited zinc morphologies were not significantly altered. The relatively large particles showed almost identical morphologies as for the reference that had been prepared in the absence of the CNF (Fig. 8a).
Occasionally, fibril aggregates with compact spherical zinc deposits along the fibres were visible as a co-deposited bead-shaped features (see Fig. 8b inset). Elemental analysis (EDS) supports that these ca. 200 nm spherical/bead-shaped particles consisted of zinc (Fig. S8, ESI†). However, it was concluded that low concentrations of CNFs did not interfere with the general zinc deposition on the electrode surface. Fig. 8c revealed that the presence of faceted edges had completely disappeared in the samples prepared at a current density of 150 A m−2 and in the presence of 0.5 g L−1 of CNFs.
The results, showing a completely disorganized zinc deposition (Fig. 8c) as compared to the absence of CNFs (Fig. 8a), evidenced that the cellulose strongly interfered with the zinc crystal growth and its resulting morphologies. This has previously been reported as a consequence of the ability of negatively charged CNFs to absorb metal ions.48 Interestingly, as the field strength was increased and a current density of 1000 A m−2 was applied for the electrolyte containing 0.5 g L−1 of the CNF, a marked difference in the characteristics of the electrodeposited material could be observed. Fig. 6d compares the morphology at the edges of two electrodes coated in the presence of 0.5 g L−1 cellulose (right) and its absence (left) at a current density of 1000 A m−2. The formation of pine tree-shaped dendritic structures was only visible when the cellulose was present, with more dendritic splaying occurring in electrode areas where increased field strengths (edges of the electrodes) could be presumed, see Fig. 8d and e, and Fig. S10 (ESI†).
The exact mechanism for dendritic formation is not confirmed, although their formation has been linked to specific effects on the deposition conditions.10,49,50 As the formation of an even deposition relies on a steady supply of ions, the transition to a dendritic structure has been reported as a consequence of a limited mass transfer of metal ions.13 If the mass transfer is limited, a concentration gradient will build up between the liquid/surface interface and the bulk electrolyte.13,51 Within this region, the metallic ion mobility is characterized by the zinc ions taking the shortest and fastest path to already protruding zinc formations, resulting in the build-up of splaying zinc structures. As the CNF was strongly interacting with the metal ions through interactions with the CNF–hydroxyl groups,20,25 which was evidenced for the almost non-existent depositions carried out at a smaller current density (150 A m−2) when the cellulose was present, it is suggested that these interactions limited the diffusion of zinc ions. It has also been reported that dendritic structures may form as a consequence of a low surface diffusivity, which is in agreement with the above reasoning.52 A schematic illustrating the difference between the different scenarios is shown in Fig. 7. Note that the mechanism provided in Fig. 7 (60 °C) essentially describes the same metal formation as discussed in the 3rd paragraph (3.1) where the facetted crystals are grown (in absence of the CNF) due to the undisturbed diffusion path of the metal ions.10
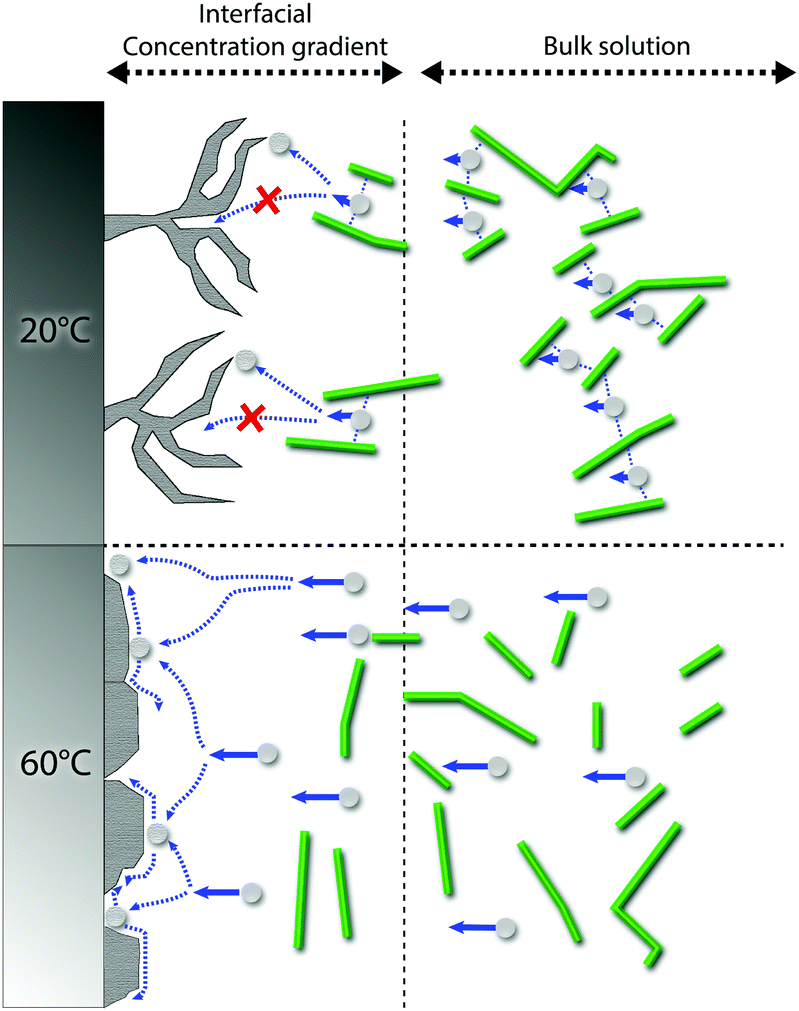 | ||
| Fig. 7 Schematic of the suggested deposition mechanisms for zinc deposition at 20 °C (top) and 60 °C (bottom). The CNF creates a concentration gradient in the interface between the electrode surface and solution, arising from the CNF ability to interact with the zinc ions in the bulk solution at 20 °C, whereas the diffusivity of Zn ions is increased at 60 °C, counteracting the effect of the CNF. At 60 °C, high surface diffusivity and deposition at energetically favourable locations are promoted rather than depositing the ions on the tips of formed dendrites. | ||
The XRD analysis of the samples (Fig. 8f) showed that the orientation in the (002) crystal plane was disfavoured when the CNF concentration was increased to 0.5 g L−1, i.e. the relative peak ratio between the (002) and the (101) lattice planes decreased 10 times for the morphology associated with the pine tree formation. This was in agreement with a transition from a deposited zinc surface dominated by titanium-parallel zinc lattice planes formed during the deposition of the zinc for the same current density (1000 A m−2) in the absence of the CNF, see Section 3.3. It deserves to be mentioned that, at a low current density of 150 A m−2 (0.5 g L−1), the zinc crystals were half as large in size as compared to the absence of the CNF, while for the same concentration of CNF at a current density of 1000 A m−2, the crystallites remained the same in size, as shown in Table S3 (ESI†), which is in agreement with the severely limited deposition and the strong attachment of zinc ions to the cellulose at a lower current density.
 | ||
| Fig. 8 Micrographs of the electrodeposited zinc at a current density of 150 A m−2 and CNF concentrations of 0 g L−1 (a), 0.01 g L−1 (b) and 0.5 g L−1 (c). (d) Micrograph comparing the appearance of the cathode edges after an electrodeposition at 1000 A m−2, without the presence of the CNF (left) and at a CNF concentration of 0.5 g L−1 is shown. (e) Micrograph of dendrite formation after 20 min of electrodeposition at a CNF concentration of 0.5 g L−1 at a current density of 1000 A m−2; Fig. S12 (ESI†) shows the same morphology for 0.1 g L−1. (f) XRD patterns of the zinc-deposited Ti-electrodes after 20 min of depositions at a current density of 1000 A m−2, at different CNF concentrations (0 g L−1, 0.01 g L−1, and 0.5 g L−1). The deposition temperature was always at 40 °C. | ||
3.5 Effect of temperature on the electrodeposition of zinc in the presence of cellulose nanofibers
Fig. 9 shows the results of changing the temperature on the deposited morphologies and how the deposition temperature controls the different ion diffusivities at a current density of 1000 A m−2 in the presence of 0.5 g L−1 of the CNF. A deposition time of 7 min was used in these experiments to capture the growth characteristics before extensive bulk deposition started to occur. Fig. 9a shows that a decrease in temperature resulted in an extensive sheet-like morphology as observed for the 40 °C deposition shown in Fig. 9b. The sheet structures were on average 2–10 μm large in a lateral dimension and stacked in layered structures of 10 sheets or more (see the highlighted area in Fig. 9a). The largest magnification in Fig. 9 reveals that the layered structures, obtained at 20 °C and 40 °C, appeared as an intercalated structure with cellulose nanofibers interspaced between the layered zinc sheets. The cellulose was also bridging the sheets at their corners, interacting more closely with the individual sheets than the sheets themselves (see Fig. 9a and b in the more magnified micrographs). The appearance of the layered deposits thereby resembled a nacre-like structure similar to a brick-and-mortar assembly of the zinc/cellulose-material. In nature, this nacre-like structure provides unique mechanical properties that combines the strength and toughness that, so far, cannot be synthetically produced at a large scale.53 | ||
| Fig. 9 SEM micrographs of the zinc electrodeposited Ti-electrodes after 7 min of electrodeposition at a current density of 1000 A m−2, a CNF concentration of 0.5 g L−1, and temperatures of 20 °C (a), 40 °C (b) and 60 °C (c). A figure serving as the reference, describing the electrodeposition in the absence of cellulose at different temperatures are provided in in Fig. S15 (ESI†). | ||
Fig. 9c shows that, when the temperature was increased to 60 °C, the sheet like morphologies, the dendritic growth of the Zn, and the effect of the CNF cellulose disappeared. In traditional electroplating chemistry, this effect on a macroscopic scale is sometimes referred to as increased throwing power of the system.54 In the case of the electrodeposition of the Zn and its deposition on the electrode, the increased temperature resulted in a deposition of zinc ions similar to the one in Fig. 1f, where no cellulose was present.
This result stems from a strongly favoured diffusion of Zn ions due to the increase in temperature, where the diffusivity of zinc ions was not limited or constrained by the charged cellulose nanofibers (see schematic in Fig. 7). It was also evident from the microscopy that the cellulose in the 60 °C depositions dominantly surrounded the facetted Zn crystals, which appeared to have grown as if no CNF had been present (see Fig. 9c insets and Fig. S14, ESI†).
It has also been shown that the increased temperature can improve the surface diffusion of the metal ions before deposition,52 which would be in agreement with the seemingly similar zinc crystal formation as observed in absence of the CNF (compare crystals in Fig. 9c and 1f).
In summary, the most significant impact of the temperature existed for the lowest temperatures where the zinc ions were most restricted in their mobility. This suggests that the temperature is the external factor that controls the processing window for the controlled growth of dendritic Zn morphologies.
4. Conclusions
The effect of dispersed cellulose nanofibers (CNFs) in zinc recovery during electrodeposition (electrowinning) was investigated. A highly crystalline cellulose nanofiber extracted from the bacterial cellulose was used as the CNF due to its well-defined fiber characteristics. The effect of the CNF concentration (0.01 and 0.5 g L−1) was evaluated for two current densities, 150 and 1000 A m−2, revealing that the cellulose had a marked impact on the electrodeposition of the zinc ions. While CNFs at lower current densities (150 A m−2) led to a suppression of ordered morphologies, the same concentrations at a higher current density (1000 A m−2) resulted in dominant dendritic zinc formation and growth. The dendritic growth was most strongly favored when 0.5 g L−1 and a current density of 1000 A m−2 was used, yielding pine tree-shaped structures on the titanium electrode (cathode). The effect of different temperatures was further investigated, showing that more elevated temperatures (60 °C compared to 20 °C and 40 °C) resulted in an enhanced diffusivity of the Zn ions, essentially diminishing the effect of the CNF on the zinc deposition. In contrast, the lower temperatures resulted in the formation of a nacre-like structure with cellulose inclusions in between the deposited zinc sheets. It could be concluded that cellulose is a promising electrodeposition additive for promoting morphologies that facilitates easy detachment and recovery of the zinc phase when the electrodeposition temperature were low (20–40 °C). Overall, the defibrillated nanosized cellulose is herein reported for the first time as a promising and inexpensive additive of high value in achieving selective and controlled electrodepositions during the recovery of valuable metals (batteries and e-waste).Author contributions
B. W. H. and M. P. performed the experiments. M. P. focused on the experiments concerning the reference zinc electrodeposition, whereas B. W. H. evaluated the reference zinc electrodeposition and carried out most of the CNF study. B. W. H. also carried out most of the isolation and characterization of the bacterial CNF. A. J. C., X. X. and R. T. O. developed analysis methods and analyzed the data. Y. L. and K. F. critically reviewed the data and provided essential feedback to the study. B. W. H., M. P. and R. T. O. wrote most of the manuscript, while A. J. C. and X. X. co-wrote the manuscript, and Y. L. and K. F. discussed the results and commented on the manuscript. Most of the microscopy and X. R. D. were performed by B. W. H. and X. X., R. T. O. identified the concept of cellulose assisted metal ion deposition under electrical fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Knut and Alice Wallenberg Research Foundation for financial support through the Wallenberg Wood Science Center (WWSC). The authors also acknowledge the Swedish Energy Agency's Battery Research Program (PERLI project 48228-1), Vinnova (EcinRaw project 2021-03738) and the Bo Rydin Foundation (Project F30/19) for support.Notes and references
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2021, e2106409, DOI:10.1002/adma.202106409.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- Fundamental Aspects of Electrometallurgy, ed. K. I. Popov, S. S. Djokić, B. N. Grgur, K. I. Popov, S. S. Djokić and B. N. Grgur, Boston, MA, 2002, pp. 175–180 Search PubMed.
- S. Y. Tan, D. J. Payne, J. P. Hallett and G. H. Kelsall, Curr. Opin. Electrochem., 2019, 16, 83–89 CrossRef CAS.
- P. Xing, C. Wang, L. Wang, B. Ma and Y. Chen, Hydrometallurgy, 2019, 189, 105134 CrossRef CAS.
- N. Perez, Electrochemistry and Corrosion Science, Springer, 2004 Search PubMed.
- J. Nettle, D. Baker and F. Wartman, Electrorefining Titanium Metal, US Department of the Interior, Bureau of Mines, 1957 Search PubMed.
- C. A. Young, SME Mineral Processing and Extractive Metallurgy Handbook, Society for Mining, Metallurgy & Exploration, 2019 Search PubMed.
- J. Zhi, S. Li, M. Han and P. Chen, Sci. Adv., 2020, 6, eabb1342 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, P. Pei, M. Wei, X. Liu, Y. Xiao and P. Zhang, Mater. Today Energy, 2021, 100692 CrossRef CAS.
- Y. Yin, S. Wang, Q. Zhang, Y. Song, N. Chang, Y. Pan, H. Zhang and X. Li, Adv. Mater., 2020, 32, 1906803 CrossRef CAS PubMed.
- P.-C. Hsu, S.-K. Seol, T.-N. Lo, C.-J. Liu, C.-L. Wang, C.-S. Lin, Y. Hwu, C. H. Chen, L.-W. Chang, J. H. Je and G. Margaritondo, J. Electrochem. Soc., 2008, 155, D400 CrossRef CAS.
- C. M. Lopez and K. S. Choi, Langmuir, 2006, 22, 10625–10629 CrossRef CAS PubMed.
- X. Xiao, B. W. Hoogendoorn, Y. Ma, S. A. Sahadevan, J. M. Gardner, K. Forsberg and R. T. Olsson, Green Chem., 2021, 23, 8519–8532 RSC.
- A. Biswal, K. Padhy, C. Sarangi, B. Tripathy, I. Bhattacharya and T. Subbaiah, Hydrometallurgy, 2012, 117, 13–17 CrossRef.
- D. Zhao, S. Yang, Y. Chen, Y. Jie, J. He and C. Tang, Hydrometallurgy, 2021, 201, 105567 CrossRef CAS.
- N. Sorour, W. Zhang, E. Ghali and G. Houlachi, Hydrometallurgy, 2017, 171, 320–332 CrossRef CAS.
- S. Das, P. Singh and G. Hefter, J. Appl. Electrochem., 1997, 27, 738–744 CrossRef CAS.
- Q.-B. Zhang and Y.-X. Hua, J. Cent. South Univ., 2012, 19, 2451–2457 CrossRef CAS.
- Q. Xu and L. F. Chen, J. Appl. Polym. Sci., 1999, 71, 1441–1446 CrossRef CAS.
- M. Petukh and E. Alexov, Asian J. Phys., 2014, 23, 735–744 Search PubMed.
- X. Ye, A. J. Capezza, X. Xiao, C. Lendel, M. S. Hedenqvist, V. G. Kessler and R. T. Olsson, ACS Nano, 2021, 15, 5341–5354 CrossRef CAS PubMed.
- Z. Xia, S. Yang and M. Tang, RSC Adv., 2015, 5, 2663–2668 RSC.
- D. Trache, A. F. Tarchoun, M. Derradji, T. S. Hamidon, N. Masruchin, N. Brosse and M. H. Hussin, Front. Chem., 2020, 8, 392 CrossRef CAS PubMed.
- W. Zhou, M. Chen, Q. Tian, J. Chen, X. Xu and C.-P. Wong, Energy Storage Mater., 2022, 44, 57–65 CrossRef.
- R. T. Olsson, M. A. S. Azizi Samir, G. Salazar-Alvarez, L. Belova, V. Ström, L. A. Berglund, O. Ikkala, J. Nogués and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef CAS PubMed.
- S. Galland, R. L. Andersson, M. Salajkova, V. Strom, R. T. Olsson and L. A. Berglund, J. Mater. Chem. C, 2013, 1, 7963–7972 RSC.
- A. Ali, S. Ambreen, Q. Maqbool, S. Naz, M. F. Shams, M. Ahmad, A. R. Phull and M. Zia, J. Phys. Chem. Solids, 2016, 98, 174–182 CrossRef CAS.
- Q. Chen, K. Geng and K. Sieradzki, J. Electrochem. Soc., 2015, 162, A2004 CrossRef CAS.
- L. P. Bicelli, B. Bozzini, C. Mele and L. D'Urzo, Int. J. Electrochem. Sci., 2008, 3, 356–408 CAS.
- Eurogrit, Aluminium Silicate, Technical Data, https://ceindustrie.com/wp-content/uploads/2018/12/Silicate-daluminium.pdf, (accessed 27 April 2022).
- Glass Beads, https://compomat.com/glass-beads/, (accessed 27 April 2022).
- D. A. Cogswell, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 011301 CrossRef PubMed.
- S. Gürmen and M. Emre, Miner. Eng., 2003, 16, 559–562 CrossRef.
- N. Alias and A. A. Mohamad, J. King Saud Univ., Eng. Sci., 2015, 27, 43–48 Search PubMed.
- M. Hughes, C. Bailey and K. McManus, 2007 International Conference on Thermal, Mechanical and Multi-Physics Simulation Experiments in Microelectronics and Micro-Systems, EuroSime 2007, 2007.
- X. Xiao, M. Iwase, G. Yin, M. Nakabayashi, T. Higashi, N. Shibata, K. Domen and T. Watanabe, ACS Sustainable Chem. Eng., 2019, 7, 19407–19414 CrossRef CAS.
- Metals Reference Book, ed. C. J. Smithells and C. J. Smithells, 5th edn, 1976, pp. 1035–1043 Search PubMed.
- M. Zhou, S. Guo, J. Li, X. Luo, Z. Liu, T. Zhang, X. Cao, M. Long, B. Lu, A. Pan, G. Fang, J. Zhou and S. Liang, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed.
- N. M. Pereira, C. M. Pereira, J. P. Araujo and A. F. Silva, J. Electroanal. Chem., 2017, 801, 545–551 CrossRef CAS.
- T. Frade, V. Bouzon, A. Gomes and M. da Silva Pereira, Surf. Coat. Technol., 2010, 204, 3592–3598 CrossRef CAS.
- Y. Peng, D. J. Gardner and Y. Han, Cellulose, 2012, 19, 91–102 CrossRef CAS.
- P. Posada, J. Velásquez-Cock, C. Gómez-Hoyos, A. Serpa Guerra, S. Lyulin, J. Kenny, P. Gañán, C. Castro and R. Zuluaga, Cellulose, 2020, 27, 10649–10670 CrossRef CAS.
- S. M. Santos, J. M. Carbajo, E. Quintana, D. Ibarra, N. Gomez, M. Ladero, M. E. Eugenio and J. C. Villar, Carbohydr. Polym., 2015, 116, 173–181 CrossRef CAS PubMed.
- S. Swingler, A. Gupta, H. Gibson, M. Kowalczuk, W. Heaselgrave and I. Radecka, Polymers, 2021, 13, 412 CrossRef CAS PubMed.
- W. Czaja and D. Romanovicz, Cellulose, 2004, 11, 403–411 CrossRef CAS.
- I. Sacui, R. Nieuwendaal, D. Burnett, S. Stranick, M. Jorfi, C. Weder, E. Foster, R. Olsson and J. Gilman, ACS Appl. Mater. Interfaces, 2014, 6(9), 6127–6138 CrossRef CAS PubMed.
- C. Zhan, P. R. Sharma, H. He, S. K. Sharma, A. McCauley-Pearl, R. Wang and B. S. Hsiao, Environ. Sci.: Water Res. Technol., 2020, 6, 3080–3090 RSC.
- M. Ammam, RSC Adv., 2012, 2, 7633–7646 RSC.
- J. Wang, Y. Yang, Y. Zhang, Y. Li, R. Sun, Z. Wang and H. Wang, Energy Storage Mater., 2021, 35, 19–46 CrossRef.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- J. X. Yu, L. Wang, L. Su, X. P. Ai and H. X. Yang, J. Electrochem. Soc., 2003, 150, C19–C23 CrossRef CAS.
- U. G. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- R. Sekar and S. Jayakrishnan, Plat. Surf. Finish., 2005, 92, 58–68 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00249c |
| This journal is © The Royal Society of Chemistry 2022 |