
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase engineered gallium ferrite: a promising narrow bandgap, room-temperature ferroelectric†
Somdutta
Mukherjee
*a,
Monali
Mishra
ab,
Palash
Swarnakar
c,
Shilpa
Sanwlani
d,
Sukalyan
Dash
b and
Amritendu
Roy
 *c
*c
aMaterials Chemistry Department, CSIR-IMMT Bhubaneswar, Bhubaneswar, 751013, Orissa, India. E-mail: msomdutta@gmail.com
bDepartment of Chemistry, VSSUT Burla, Burla, 768018, India
cSchool of Minerals, Metallurgical and Materials Engineering, Indian Institute of Technology Bhubaneswar, Jatni, 752050, Orissa, India. E-mail: amritendu@iitbbs.ac.in
dDepartment of Physics, Indian Institute of Science, Education and Research Mohali, Punjab, India
First published on 24th March 2022
Abstract
Narrow bandgap oxide ferroelectrics with large polarization are considered promising in novel optoelectronic and photovoltaic devices. In the present work, nanocrystalline gallium ferrite with polar, rhombohedral R3c symmetry is synthesized using the hydrothermal route. Structural characterization using X-ray diffraction, Raman spectroscopy, and electron microscopy confirm the formation of the rhombohedral R3c phase, with two different geometries and narrow size distribution. Electrical characterization corroborated by first-principles density functional theory-based calculations demonstrates room temperature ferroelectricity contributed mainly by triply charged gallium ions. The calculated spontaneous polarization is ∼20 μC cm−2 with a large electronic contribution. Optical characterization predicts a bandgap of ∼2.1 eV. Together with its narrow bandgap and the large contribution of electronic polarization, ferroelectric gallium ferrite could be an interesting system for novel photovoltaic and optoelectronic devices.
1. Introduction
Ferroelectric (FE) materials continue to present challenging physics as well as exciting technological applications in diverse, novel electronic devices including solid-state non-volatile memory elements, field-effect transistors and micro- and nano-electromechanical systems such as sensors, actuators, and transducers.1,2 More recently, FEs have been considered promising for visible light photovoltaic applications wherein spontaneous polarization is presumed to be instrumental for the separation of photogenerated charge carriers. In this regard, halide perovskites3–5 with their favourable optoelectronic characteristics have yielded impressive conversion efficiencies. Perovskite halides, however, suffer from poor stability and potential environmental impacts.6 While improvement towards their stability has been a major research focus,6 the design and development of stable, environment-friendly materials demonstrating an enhanced PV effect could constitute an important avenue in solar cell research.In this context, transition metal oxide (TMOs) based perovskite ferroelectrics render wide opportunities towards tailoring desired functionalities owing to their competing energy scales of various phases and susceptibility to external perturbations.7 However, traditional TMO ferroelectrics demonstrate a large optical bandgap resulting in poor absorption behaviour in the visible part of the solar spectra and therefore yields very small photocurrent vis-à-vis conversion efficiency.8 Thus, tailoring the bandgap is the first step towards designing efficient solar cell materials based on these materials. While selective doping,9–11 solid solution formations8,12 and gap-state engineering13 have been explored to reduce the bandgap of traditional TMO-ferroelectrics, single-phase ferroelectric-multiferroics with a comparatively low bandgap, are superior choices.14
Transition metal oxide ferroelectric-multiferroic bismuth ferrite (BiFeO3 or BFO) is a classic example of a room-temperature multiferroic material with rhombohedral symmetry.15 BFO with a large spontaneous polarization and bandgap of ∼2.7 eV has been shown to possess good PV performance.16,17 However, the bandgap in BFO is far from the ideal value of ∼1.4 eV for efficient absorption of solar radiation. Therefore, it would be desirable to design novel room temperature ferroelectric/multiferroic systems with a narrow bandgap for prospective solar cell applications.
More recently, gallium ferrite (GaFeO3 or GFO) has come up as an alternative room temperature MF with sizable spontaneous polarization and room temperature ferrimagnetism.10,18 Bulk GFO with orthorhombic Pna21 symmetry demonstrates cation site-disorder as well as a wide range of cationic nonstoichiometry which in turn, translates to tunable magnetic vis-à-vis multiferroic transition temperature.19 Nanoscale piezo-response force measurements on epitaxial10 as well as solution deposited18 films demonstrated 180° phase switching upon changing the bias field. Bulk measurement on epitaxially grown films further showed a saturated ferroelectric hysteresis loop.20 The observed poor leakage characteristics could be effectively improved by selective doping.21 As such, GFO is quickly emerging as an effective substitute for BFO. Despite the above attributes, the required electric field for switching the ferroelectric domains in bulk GFO was found to be rather high, ∼1400 kV cm−1.10,20 Thus, to use GFO for commercial device applications, the coercive field of GFO needs to be reduced substantially. In this regard, the possibility of stabilizing a novel ferroelectric phase of the system with a smaller bandgap and coercive field seems exciting from device designing perspectives. GFO has been shown to undergo a series of structural phase transformations across a wide range of hydrostatic pressure.22 Among the observed high-pressure phases,22 a few are polar and keep the promise to demonstrate ferroelectricity. Thus, it would be interesting to explore the feasibility of synthesizing vis-à-vis stabilizing such high-pressure phases at ambient conditions upon applying suitable processing conditions and evaluate their properties for possible room-temperature ferroelectricity/multiferroism. In a recent work, the present authors synthesized GFO nanocrystals with unique truncated hexagonal bi-pyramid (THBP) geometry using a hydrothermal technique.23 Subsequent structural and functional characterization demonstrated an orthorhombic P212121 symmetry with a moderately small bandgap and exciting photocatalytic activity.23
In the present work, we stabilize GFO nanocrystals with rhombohedral R3c symmetry at room-temperature. Electrical, magnetic and optical characterization together with first-principles studies establish the material as a room-temperature ferroelectric with a reduced coercive field and narrow optical band gap (∼2.1 eV). Electrical characterization further demonstrates rectifying characteristics. Phase engineered GFO thus possesses promise for novel optoelectronic and photovoltaic applications and demands further investigations.
2. Experimental and computational details
2.1 Experiments
THBP GFO nanocrystals were synthesized using a hydrothermal method following a protocol as reported earlier by the present authors.23 Gallium nitrate hydrate (Ga(NO3)3·xH2O) (Sigma Aldrich ≥99.9%) and iron nitrate nonahydrate (Fe(NO3)3·9H2O) (Sigma Aldrich ≥98%) were taken in stoichiometric ratio and dissolved in 40 ml deionized water (pH = 6.8). The concentration of precursors in the solution was four times in the case of protocol (ii) in comparison to that of protocol (i). No reducing or shape defining reagent was used. The solution was transferred to a Teflon liner and autoclaved at 523 K for 5 h. In order to obtain the required phase, the as-grown powder was heated for 1 h at 673 K. We would henceforth call this synthesis route protocol (i). However, the yield of the above process is too low to effectively use it for extensive characterization. It was further observed that the required phase could also be stabilized when the amount of precursors was increased to four times keeping other processing parameters identical to our earlier work,23viz., 523 K and 5 h (protocol (ii)).The morphology of the as prepared samples was examined using a field emission scanning electron microscope (FESEM) (Zeiss, Merlin Compact) and a transmission electron microscope. Powder X-ray diffraction patterns of the synthesized crystals were acquired over the 2θ range 10–90° using a Bruker D8 Advance diffractometer with CuKα radiation (λ = 1.5418 Å). Rietveld refinement of the room temperature powder X-ray diffraction data was carried out using FullProf24 with two different space groups, viz., R3c and R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. Thermal analysis (DSC) was carried out using a Differential scanning calorimeter (METTLER TOLEDO). To study the optical properties, ultraviolet-visible (UV-vis) (Shimadzu-UV-2450, Japan) spectra was recorded using a non-absorbing standard BaSO4 powder. An impedance spectroscopy study was conducted in a WayneKerr 6505 B impedance analyser. The bulk ferroelectric hysteresis loop was measured in a Radiant Precision Premier II ferroelectric tester. Piezoresponse force microscopy using the switching spectroscopy mode (Asylum) was performed to study the local response.
c. Thermal analysis (DSC) was carried out using a Differential scanning calorimeter (METTLER TOLEDO). To study the optical properties, ultraviolet-visible (UV-vis) (Shimadzu-UV-2450, Japan) spectra was recorded using a non-absorbing standard BaSO4 powder. An impedance spectroscopy study was conducted in a WayneKerr 6505 B impedance analyser. The bulk ferroelectric hysteresis loop was measured in a Radiant Precision Premier II ferroelectric tester. Piezoresponse force microscopy using the switching spectroscopy mode (Asylum) was performed to study the local response.
2.2 Computation
First-principles calculations were carried out using density functional theory25 as implemented in Vienna Ab initio Simulation Package (VASP)26,27 using the generalized gradient approximation (GGA)28 as well as with suitable application of onsite Coulomb potential (GGA+U with Ueff = 2.0 to 4.0 eV)29,30 to treat the strongly correlated 3d-electrons of Fe ions. We used a rotationally invariant approach31 for Ueff, the magnitude of which was determined based on our previous experience on the material. The Kohn–Sham equation32 was solved using the Perdew and Wang (PW91) functional.33 A large kinetic energy cut-off of the planewaves, 850 eV, was used. Our entire calculations are based on the stoichiometric composition of GaFeO3 with the assumption that no partial interchange of the lattice sites by constituent cations have taken place. We included thirteen valence electrons for Ga (3d104s24p1), eight for Fe (3d74s1) and six for O (2s22p4) ions. We used conjugate gradient algorithm for relaxing the constituent ions within a unit cell constructed using the data obtained from Rietveld refinement of the XRD pattern at room temperature (Table 1). All the calculations were performed at 0 K. Calculation of the electronic structure was carried out using an 8 × 8 × 3 Gamma (Γ-) mesh. Polarization was computed using the Berry phase method within modern theory of polarization.34| Phase: Rc (SG#161) |
||||
|---|---|---|---|---|
| Lattice parameters: a = b = 5.0235 Å; c = 13.6325 Å; α = β =90°; γ = 120° | ||||
| Ion | Wyckoff positions | x | Y | z |
| Ga3+ | 6a | 0.0000 | 0.0000 | 0.3075 |
| Fe3+ | 6a | 0.0000 | 0.0000 | 0.0186 |
| O2− | 18b | −0.3005 | −0.2988 | 0.4062 |
3. Results and discussion
3.1 Structural and microstructural characterization
The room temperature powder XRD pattern of the as-synthesized sample prepared using the modified protocol (protocol – ii) is shown in Fig. 1(a). Rietveld refinement of the powder pattern was attempted using two rhombohedral symmetries, viz., polar R3c (Space Group #161) and non-polar Rc (Space Group # 167). It is not practically possible to differentiate between the above two phases which belong to the same Laue groups, based on powder X-ray diffraction data. However, it is observed that polar R3c (with refined atom positions, 0 0 δ (δ ≠ ¼)) yields a much better goodness of fit in comparison to that of 0 0 ¼ corresponding to non-polar Rc. Accordingly, the symmetry of the phase of the sample is assigned as R3c. Structural parameters obtained from the refinement as well as the refinement parameters are presented in Table 1. Fig. 1(b) schematically depicts the unit cell of R3c-GFO in the hexagonal setting. Our subsequent first-principles study, in section 3.6 and electrical characterization, in section 3.5, further bolster the stability of the R3c symmetry of the material over Rc and thus, corroborate the powder XRD analysis.
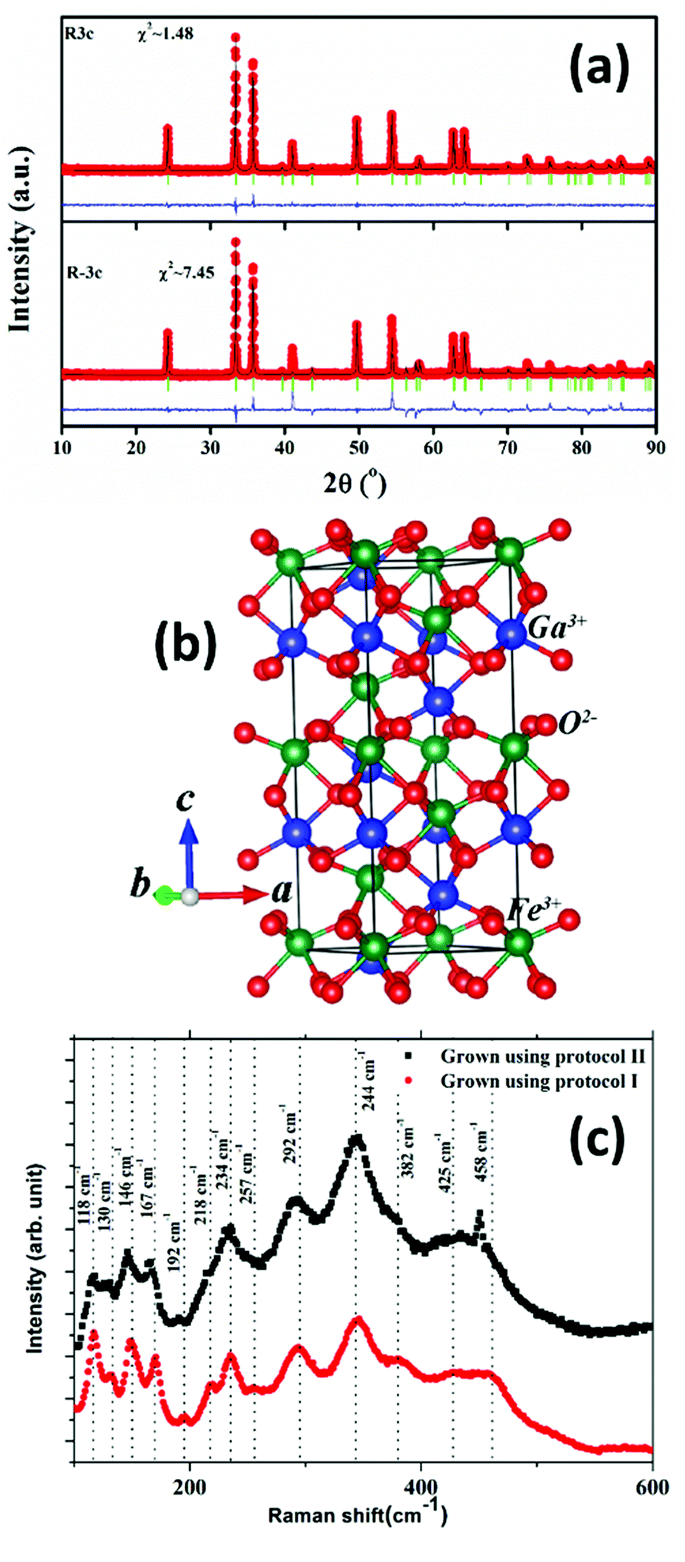 | ||
| Fig. 1 (a) Rietveld refinement of the room temperature XRD pattern of the gallium ferrite (GFO) nanocrystal sample prepared by protocol (ii), using R3c and Rc symmetries. (b) Schematic description of the unit cell of GFO with R3c symmetry. (c) Raman spectra of R3c-GFO synthesized using the two different protocols. | ||
The polar R3c phase with space group #161 is comprised of 30 atoms or 6 formula units within the rhombohedral unit cell in the hexagonal setting. Following group theory, the irreducible representation that presents the Raman active modes at the zone-center is Γ = 4A1 + 9E i.e. a total of 13 modes are expected. As can be seen from Fig. 1(c), 13 distinct Raman peaks are identified over the frequency range 100–600 cm−1 for both the R3c GFO nanocrystals grown using protocol (i) as well as protocol (ii). A comparison of the peak positions with those of R3c-BFO suggests modes between 152–261 cm−1 involve the Fe–O bending and stretching of bonds within FeO6 octahedra, while oxygen motions dominate above 261 cm−1.35 Raman modes related to Ga–O bonds are expected to appear below ∼170 cm−1 though a blue shift of mode positions is expected due to the lower reduced mass of Ga–O compared to Bi–O.35
 | ||
| Fig. 2 Field emission scanning electron micrographs (FESEM) of the GFO nanocrystals synthesized using (a) protocol (i), and (b) protocol ii. The insets show the high magnification images. (c) High resolution transmission electron micrograph a GFO nanocrystal produced using protocol (i). The inset shows a single GFO nanocrystal. (d) TEM micrograph of the GFO sample synthesized using protocol (ii). The inset shows the diffraction pattern obtained from a single nanocrystal. | ||
Transmission electron microscopy studies conducted on both the samples are shown in Fig. 2(c and d). Fig. 2(c) inset shows a single GFO nanocrystal prepared using protocol (i) demonstrating THBP geometry. High resolution TEM imaging of the nanocrystals (Fig. 2(c)) demonstrates atomic arrangements within parallel lattice planes without significant disruption in the periodic arrangement suggesting formation of defect free nanocrystals. Using the Rietveld data, we calculated interplaner spacing of several high index planes within R3c symmetry. The measured interplaner spacing from the HRTEM images substantiates the presence of (104) planes. Fig. 2(d) presents the TEM image of samples prepared using protocol (ii). While the edges of the samples were too thick to produce any lattice images, the diffraction pattern (inset, Fig. 2(d)) predicts rhombohedral symmetry with lattice parameters similar to those predicted by Rietveld refinement, discussed above.
3.2 Mechanism of formation of GFO nanocrystals with varied crystal symmetry and shape
The mechanism of oxide nanostructure evolution during hydrothermal synthesis is explained in light of the kinetics and thermodynamics of the processing, that provide complementary descriptions. The critical balance between the two, determines the crystal symmetry, size and shape of the synthesized nano-crystalline phase. In the present work, we observe that GFO nanocrystals are formed with two different crystal symmetries, viz., P212121 and R3c and geometries, i.e., truncated hexagonal bipyramid (THBP) and cubic geometries under protocol (i) (without annealing)23 and (ii), respectively, described in section 2. In either of the two processes, no capping agent was used and the synthesized nanoparticles are of fairly uniform size.The evolution of the GFO nanostructure begins with the progress of the monomer forming reaction under thermal activation:23 Ga(NO3)3 + Fe(NO3)3 + 3H2O = GaFeO3 + 6HNO3 With time elapsed, monomer concentration in the solution increases rapidly. Beyond the critical supersaturation level, nucleation (self-nucleation) starts. The process of nucleation involves crossing the activation energy barrier by a monomer from the bulk of the solution to a growing nucleus. A smaller activation barrier would favour nucleation of a particular crystalline phase. The driving force for crystallization is the change in chemical potential (Δμ) while the monomer travels from the dissolved phase to the solid crystalline phase:
Δμ = μS − μL + −kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnS lnS | (1) |
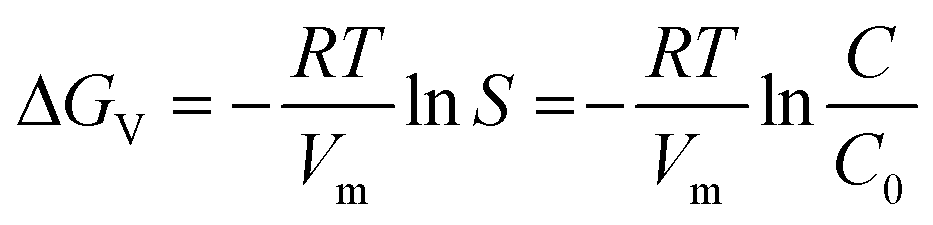 | (2) |
Assuming homogenous nucleation, for a critical sized, spherical nucleus of radius r, the net change in free energy can be expressed as the sum of bulk and surface contributions:39
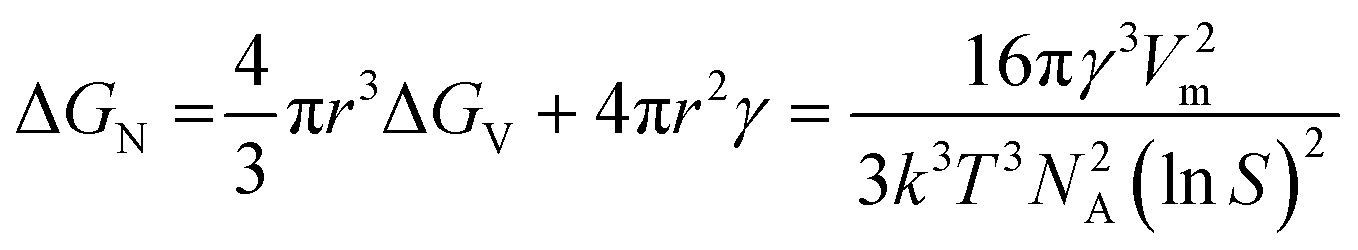 | (3) |
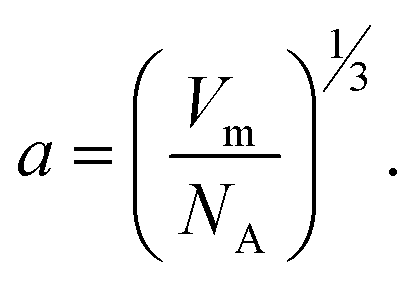 40 Thus, for a qualitative study, γ could be described as:
40 Thus, for a qualitative study, γ could be described as: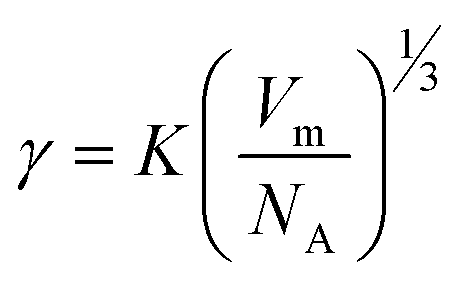 | (4) |
Here, NA is Avogadro's number. With the above substitution, the expression for ΔGN becomes:
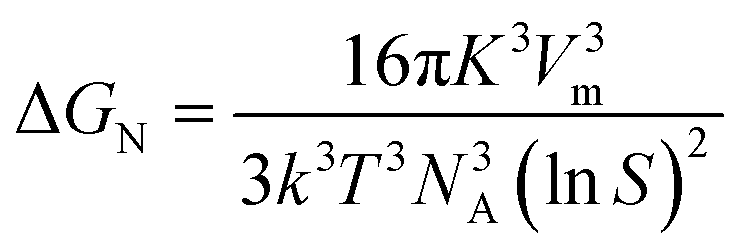 | (5) |
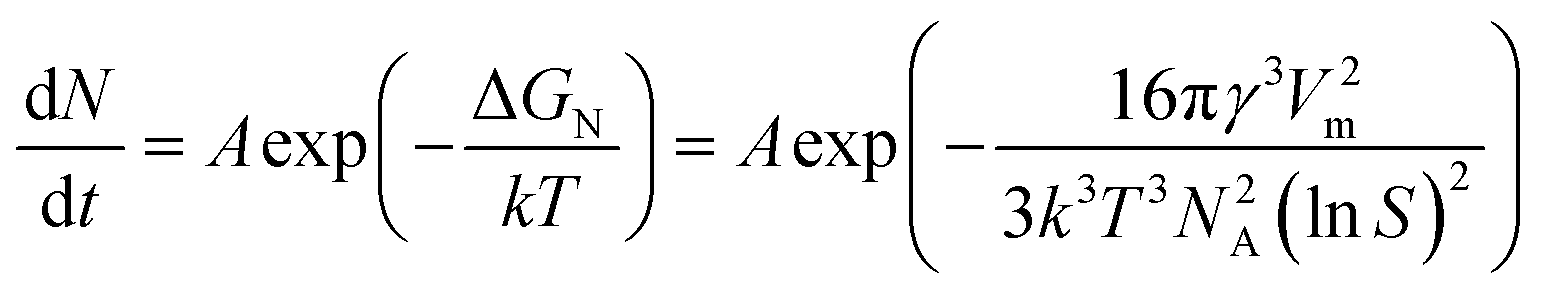 | (6) |
Our earlier study suggests that GFO nuclei formed within the solution demonstrate a shift from isotropic growth at lower temperature to THBP geometry at 250 °C suggesting a kinetics driven growth mechanism.23 For achieving uniform size distribution of the particles, the growth should be diffusion-limited. However, it should be noted that there are no straightforward guidelines to predict the morphology of the particle since there are a number of factors that affect the morphology of the growing particle. The equilibrium shape (Gibbs–Wulff theory) of a particle is one that minimizes the surface energy for a given enclosed volume.41 The isotropic surface energy would therefore result in spherical particles. A low monomer concentration in the solution further promotes spherical particle formation. A high monomer concentration on the other hand, favours anisotropic growth along directions with a lower activation energy barrier. The diffusion controlled growth model proposed by Peng42 has been highly successful in explaining the shape variation of inorganic nanoparticles with varied monomer concentrations in the solution and can be used here. In the present work, under an identical temperature, the shape of the crystal changes to cubic when the precursor concentration in the solution is changed to four times. Apparently, the above modification is due to a change in supersaturation level during the synthesis process. It is believed that the supersaturation level influences the chemical potential in the bulk solution as well as the oriented attachment of the monomers.38 In dilute solution, the system can reach a dynamic equilibrium by dissolution and regrowth of various facets with varied energies, resulting in the formation of nanospheres. A moderate supersaturation, on the other hand, results in directional growth. Variation in the chemical potential among the facets with different crystallographic orientation favours preferential migration of the monomers resulting in shapes such as THBP in intermediate monomer concentration and cubic in high monomer concentration. Similar supersaturation-driven shape evolution has been previously reported in TiO237 and SmVO438 where spherical shaped nanospheres are formed at a low monomer concentration while very high concentration promotes nanowire formation.
3.3 Optical property
Fig. 3(a) inset shows the optical absorption spectra of the R3c-GFO nanocrystal over the range 400–700 nm. It shows strong absorption in the wavelength range of 550–700 nm suggesting that the GFO nanocrystal can absorb a considerable amount of visible light. The absorption edge at ∼600 nm can be attributed to 6A1g → 4T2g (∼2.0 ev) transition originating from the crystal field splitting of FeO6 octahedra.43 The optical band gap of the THBP-GFO nanocrystals was estimated using Tauc relation:44 (hνα)1/n = B(hν − Eg), where ‘h’ is Planck's constant (6.626 × 10−34 m2 kg s−1), ν, represents the frequency of incident light, α, absorption coefficient, B, proportionality constant and Eg is the bandgap of the material. The value of the exponent n denotes the nature of the optical transition. For direct optical transition, n = ½ while n = 2 represents indirect transition from the valence to conduction bands. Since the material was found to behave as a direct bandgap semiconductor, as shown in the following DFT study, a straight line fit on the linear part of (αhυ)2vs. hυ plot (Fig. 3(a)) was made to determine the bandgap of the material. The bandgap, estimated from the x-axis intercept is found to be ∼2.1 eV. The observed bandgap is smaller than that in the bulk phase as well as significantly lower than that of BFO. This narrow bandgap renders the material to absorb a significantly large part of the solar spectrum and thereby promises an improved photocurrent response.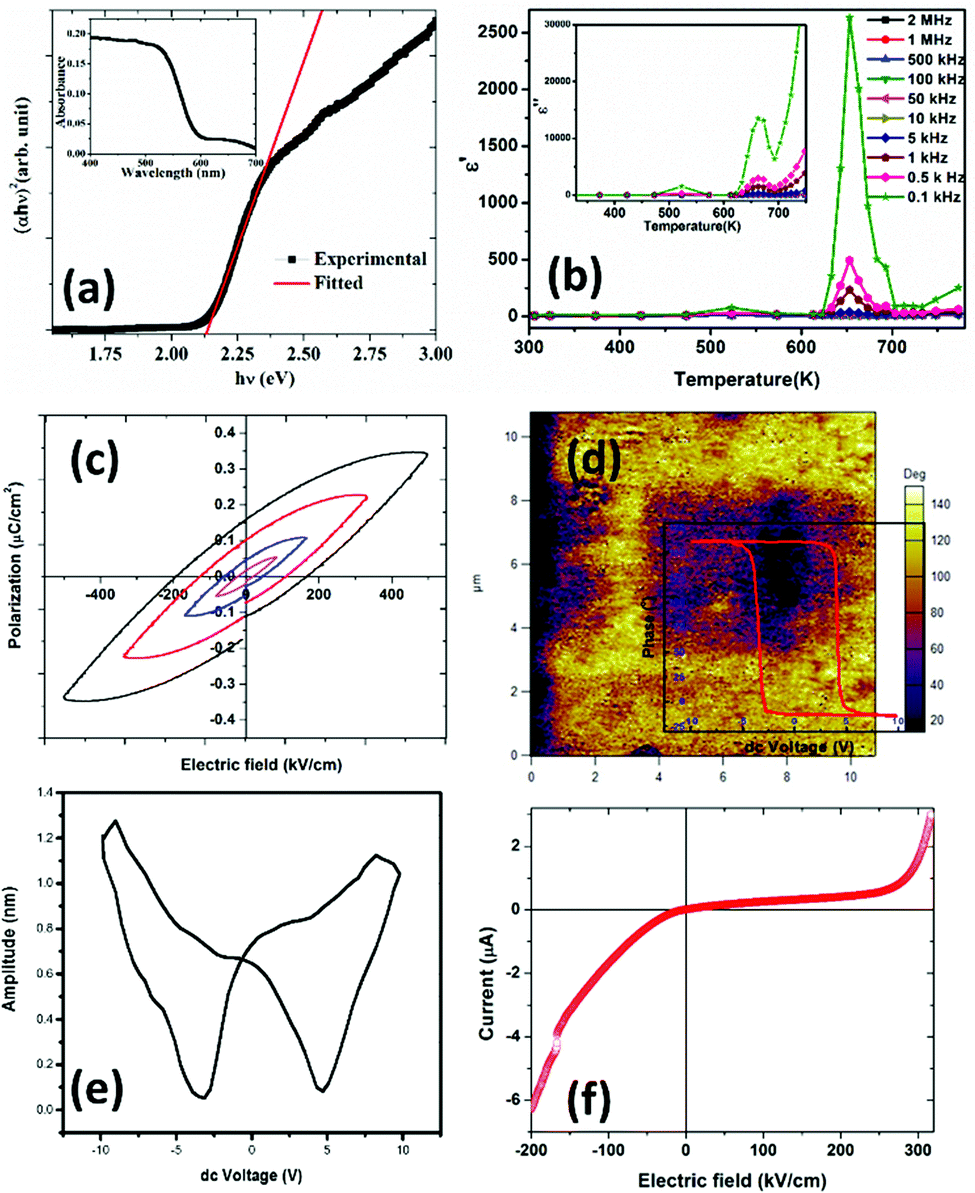 | ||
| Fig. 3 (a) Tauc plot of optical absorption data demonstrating an optical band gap. The inset shows the absorption spectra of the R3c phase of GFO nanocrystals. (b) Real part of the dielectric constant (ε′) versus temperature at certain frequencies; the inset plots the imaginary component dielectric constant (ε′′) versus temperature at different frequencies within R3c-GFO. (c) Bulk polarization hysteresis data plotted as a function of applied voltage on a ∼600 μm thick film of GFO on an ITO coated glass substrate. (d) PFM phase and (e) butterfly loop of GFO on ITO at room temperature. (f) Room temperature current–voltage plot on GFO thick-film on an ITO coated glass substrate demonstrating rectifying characteristics. | ||
3.4 Electrical characterization
Temperature dependent impedance measurement of the sample, prepared using protocol (ii), was performed over a frequency range, 100 Hz–2 MHz. A similar exercise was conducted on the sample synthesized using protocol (i) and subsequent analysis is presented in the ESI.† The two data are identical. Using the impedance data, we calculated the real and imaginary part of the dielectric constant by using the following equation: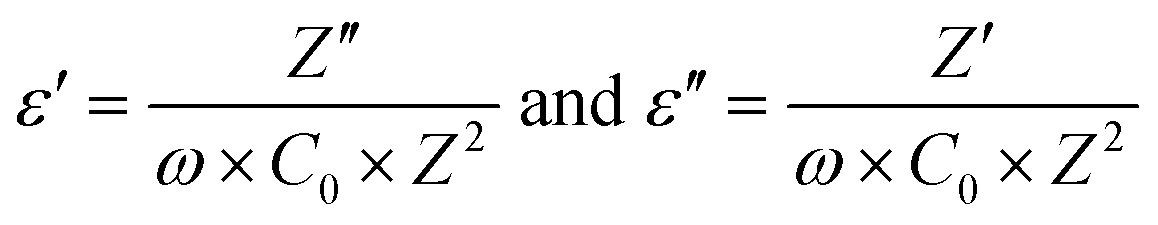 | (7) |
The real part of the dielectric constant (ε′) for different frequency values are plotted as a function of temperature, as shown in Fig. 3(b). Z′ and Z′′ are real and imaginary parts of complex impedance Z; ω = 2πf where f is the measured frequency and C0 is free space capacitance. It is observed that the present R3c phase possesses a high frequency dielectric constant, ε ∼7 which is significantly smaller than traditional ferroelectrics such as BaTiO3 over the temperature range, 300–600 K. The magnitude of the high frequency dielectric constant increases to maximum ∼49 at the highest temperature used, 750 K. The dielectric constant depends upon different types of polarizations, i.e., interfacial, dipolar, ionic and electronic polarization with increasing frequencies. While interfacial polarizations operate at lower frequencies, at high frequency, only dipolar (orientational), ionic and electronic polarizations are responsible. The constant magnitude over the entire range of temperature and frequency indicates that the observed dielectric constant is intrinsic in nature without interfacial contributions. As the temperature increased both ε′ as well as ε′′ exhibit anomalies at ∼523 K and 653 K. The anomalies are prominent at lower frequencies as the ε′ reaches maximum values 78 and 2633 at 523 and 653 K, respectively at 100 Hz. Temperature anomalies of ε′ and ε′′ are suggestive of structural phase transitions over the temperature window. This structural phase transformation is characteristic of ferroelectric materials where symmetry increasing structural transition translates to dielectric anomaly.
P vs. E plots demonstrate hysteresis behaviour. With increasing applied field (E), the size of the loop increases. The shape of the loop is typical of lossy ferroelectrics.45 Early works on BFO46 and GFO47 report similar ferroelectric hysteresis loops. In fact, our DC current–voltage data on the sample demonstrates the presence of a large leakage current in the sample possibly hindering the true ferroelectric nature of the sample. Inappropriate sample preparation (sintering at high temperature could not be performed to avoid possible irreversible phase transformation) could be one of the origins of poor data.
The presence of multivalent Fe-ions could also contribute to increased leakage. However, with this nature of unsaturated ferroelectric hysteresis characteristics, one cannot conclusively confirm the presence of ferroelectricity in the sample. In order to further probe the possible ferroelectric character in the material and to minimize the effect of sample quality on the prospective ferroelectric property, local measurement using piezo-response force microscopy was conducted on the sample. In piezoresponse force microscopy, a 10 μm × 10 μm area of the sample was subjected to a positive bias, allowing all the domains to align unidirectionally. After that a 2 μm × 2 μm area within the poled sample was applied to a reverse bias, making the domains align in opposite directions. The phase image together with switching spectroscopy is presented in Fig. 3(d). The switching spectroscopy data and the phase image of the PFM study demonstrate 180° phase switching upon swapping the electric field direction. The corresponding amplitude plot (Fig. 3(e)) allows the piezoelectric co-efficient to be determined. The piezoelectric coefficient is derived using 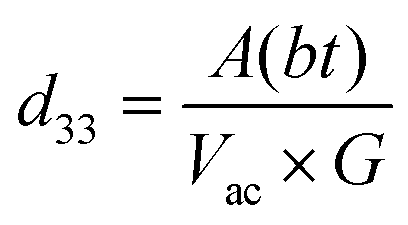 , where A(bt) is the change in thickness or deformation (amplitude of butterfly loop) and Vac is the AC voltage given to the cantilever to apply an electric field. G is directly related to the quality factor of tuning of the cantilever,
, where A(bt) is the change in thickness or deformation (amplitude of butterfly loop) and Vac is the AC voltage given to the cantilever to apply an electric field. G is directly related to the quality factor of tuning of the cantilever, 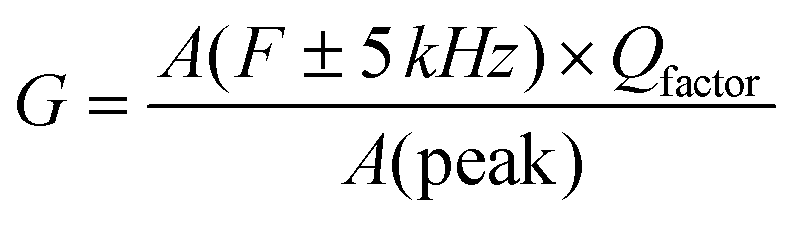 . The calculated piezoelectric coefficient was found to be d33 ∼11 pm V−1. The d33 value is comparable to the value reported for the epitaxial thin film of BFO48 and other similar piezoelectric materials. For prospective photovoltaic applications, it is important that GFO demonstrates rectifying characteristics, since this behaviour allows charge carriers of opposite polarities to separate out. In order to characterize the current–voltage behaviour of the material, a thick film (thickness, ∼600 μm) of GFO on an ITO coated glass substrate was prepared. As shown in Fig. 3(f), with forward bias the current is very small and remains almost parallel to the voltage axis before, and it suddenly shoots up at a voltage of ∼160 V. In the reverse bias, however, the current demonstrates a gradual increase with field. The above behaviour is similar to that of a diode which is required for separation of the photoexcited charge carriers. It is therefore observed that phase engineered GFO demonstrates a rectifying character, important for its potential PV application.
. The calculated piezoelectric coefficient was found to be d33 ∼11 pm V−1. The d33 value is comparable to the value reported for the epitaxial thin film of BFO48 and other similar piezoelectric materials. For prospective photovoltaic applications, it is important that GFO demonstrates rectifying characteristics, since this behaviour allows charge carriers of opposite polarities to separate out. In order to characterize the current–voltage behaviour of the material, a thick film (thickness, ∼600 μm) of GFO on an ITO coated glass substrate was prepared. As shown in Fig. 3(f), with forward bias the current is very small and remains almost parallel to the voltage axis before, and it suddenly shoots up at a voltage of ∼160 V. In the reverse bias, however, the current demonstrates a gradual increase with field. The above behaviour is similar to that of a diode which is required for separation of the photoexcited charge carriers. It is therefore observed that phase engineered GFO demonstrates a rectifying character, important for its potential PV application.
3.5 First-principles studies
First-principles calculations were performed within the framework of density functional theory.49 We started with the structural information obtained from the Rietveld refinement of the room temperature X-ray diffraction pattern of the sample prepared using protocol (ii). The ionic positions were relaxed at the experimental cell volume until the force on individual ions became less than 0.0005 eV Å−1. To determine the possible magnetic order, spin-polarized calculations within ferro- and antiferro-magnetic spin configurations of the Fe3+ ions were performed. It was found that anti-ferromagnetic orderings of the Fe-spins corresponds to 71 meV per atom lower in energy than ferromagnetic ordering within R3c symmetry of GFO. To probe further and therefore to determine possible antiferromagnetic ordering, a 2 × 2 × 1 supercell was constructed. The total energy of the 2 × 2 × 1 supercell was computed by enforcing different antiferromagnetic orders of the Fe3+-ions, viz., A-, C- and G-type antiferromagnetic orders. It was observed that the G-type antiferromagnetic order of the Fe3+ ions corresponds to the lowest energy, 23 meV per atom and 44 meV per atom lower with reference to C-AFM and A-AFM orderings, respectively. Thus, one can predict that stoichiometric GFO within R3c symmetry possesses G-type antiferromagnetic order. Our calculations however did not consider possible site disorder among the Fe3+ and Ga3+ ions, commonly observed in the bulk gallium ferrite.50 Experimental magnetization versus temperature measurement with a small probing field shows a weak magnetic response possibly suggesting AFM-order (please see the ESI†). The stability of the R3c phase was further evaluated by calculation of the phonon spectra, where no negative frequencies were observed bolstering the stability of the current phase under experimental conditions (please refer to the ESI†). Further electronic structure calculations were carried out within the antiferromagnetic configurations of the Fe-spins without further consideration of specific antiferromagnetic order. Fig. 4(a) plots the electronic band structure, total and site projected density of states computed using GGA. It is observed that the material demonstrates direct bandgap characteristics albeit a much smaller bandgap (Eg ∼0.5 eV) than that observed in our experimental study reported above. Such underestimation of the electronic bandgap within DFT studies using GGA is very common due to localized electronic states.51,52 However, the band structure and site projected density of states computed here are good to describe the electronic structure in a qualitative manner. It is observed that the top of the valence band is predominantly made up of O 2p states while the bottom of the conduction band is composed of Fe 3d electronic states. To further treat the localized states of the Fe ion, on-site Coulomb (Hubbard) potential (U = 2–4 eV) was applied, within the GGA+U technique. The optimization of the Hubbard potential was accomplished by computing the electronic bandgap and comparing the bandgap with that obtained from the experiment. It is found that Ueff = 4.0 eV results in an Eg ∼2 eV, closely corresponding to the experimental observation. Furthermore, the qualitative description of the band structure and DOS remained similar with and without application of Ueff.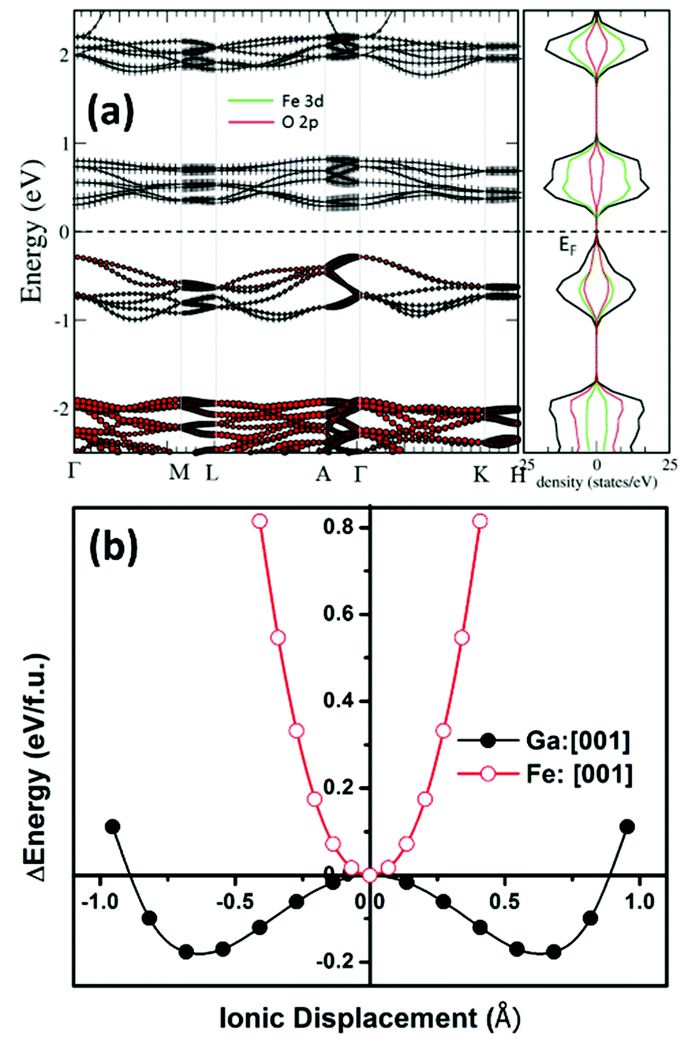 | ||
| Fig. 4 (a) Electronic structure of R3c-GFO calculated using GGA. The figure in the left plots the band structure along the high symmetry directions. The right panel plots the total and angular momentum resolved density of states (DOS). (b) Energy landscape of R3c-GFO while it switches between the polar states via centrosymmetric phase demonstrating double-well, characteristic of ferroelectrics when Ga-ions are moved with respect to the surrounding O-ions, along the polar axis. | ||
To unravel the possible ferroelectric character of the material within R3c symmetry, DFT calculations were performed towards finding possible equivalent energy states which is the signature of ferroelectricity. In typical ferroelectric materials, the equivalent low energy polar states are separated by a high energy state with higher symmetry and the transition path follows an energy double-well. The immediate higher symmetry super-group is Rc which has a close symmetry relation with the present R3c phase. In temperature-dependent dielectric measurements, we observed anomalies around ∼525 K and 650 K, respectively, possibly indicating symmetry-increasing phase transformation. At this moment, we are not very sure whether the dielectric anomaly is due to R3c → Rc transformation though this transformation is quite likely. The direction of spontaneous polarization of the R3c structure within the hexagonal setting is [0001] i.e., along the c-axis of the unit cell. The transition between the polar R3c states through the centrosymmetric Rc state takes place by means of coordinated displacements of the ions. Therefore, to compute a possible ferroelectric energy double-well, we transformed the structure with R3c symmetry to Rc using the utility program PSEUDO53 within the Bilbao crystallographic server. In the process, the unit cell parameters were not symmetrized which may possibly contain some symmetry-breaking strain. With reference to the Rc symmetry, Ga3+ and Fe3+ ions were moved along the [0001] direction in both positive and negative senses and the corresponding total energy of the system was computed. Fig. 4(b) plots the total energy of the system with reference to the Rc phase, as it transforms to R3c. It is observed that while shifting of the Fe3+-ion along the c-axis increases the total energy of the system monotonically, shifting the Ga3+-ion along [0001] lowers the total energy of the system up to a minimum which corresponds to the R3c phase. Further shifting of the Ga3+-ion, however, resulted in the increase of the total energy. Thus, R3c-GFO demonstrates an energy double-well when Ga3+-ions are shifted along the direction of spontaneous polarization, [0001]. As mentioned earlier, this double-well formation of the polar crystal is the signature of the ferroelectricity in the system. Similar double-well formation was also demonstrated in the bulk orthorhombic Pna21 phase of GFO,10 multiferroic bismuth ferrite (BFO)54 with R3c symmetry and other prototype ferroelectrics.55 While bulk GFO demonstrates requirement of large energy10 vis-à-vis a large voltage for polarization switching20 the present nanostructure with R3c symmetry requires much smaller energy, even smaller than that required by BFO with R3c symmetry54 suggesting requirement of a small switching voltage. Furthermore, the contribution to the ferroelectricity in R3c-GFO is mainly from the Ga3+-ions in contrast to the bulk phase where a systematic movement of the Fe3+-ions had been demonstrated to result in the ferroelectric instability within the Pna21 phase. However, BFO with R3c symmetry demonstrates ferroelectricity originating from the Bi3+-ions.
The magnitude of the spontaneous polarization was computed using the Berry phase method as described in the Modern theory of polarization.34 In this method, the electronic contribution to the total polarization is described by the Berry phase. For GFO with R3c symmetry, Berry phase computation yields a polarization lattice, 6.05 + n × 73.31 μC cm−2 along [0001] direction, where n is an integer. In order to estimate the spontaneous polarization of the system, Berry phase calculation was further computed on the high symmetry Rc phase, −15.34 + n × 73.31 μC cm−2 along the [0001] direction. The difference between the polarization of the above two phases yields the spontaneous polarization, Ps of the R3c phase, 21.39 μC cm−2. The entire calculation was carried out within GGA. Application of U, within GGA+U with different Ueff values, resulted in Ps of 25.08, 23.12, and 24.01 μC cm−2 for Ueff = 2, 3, and 4 eV, respectively. Estimated spontaneous polarization is markedly higher than the one obtained from the P–E loop in the previous section. This could be attributed to poor sample quality and defects in the sample. It is further observed that the electronic contribution to the total polarization in the system is comparable with the ionic contribution which potentially suggests a large shift current (related to the bulk photovoltaic effect) and thus promises the material's potential in novel photovoltaic applications. However, this aspect will be taken up in our follow-up work.
4. Conclusion
Hydrothermal synthesis under controlled processing conditions yields a novel, polar phase of gallium ferrite with rhombohedral R3c symmetry with unique geometry. Powder X-ray diffraction, Raman spectroscopy, and electron microscopy were employed to understand the phase evolution. It was argued that burst nucleation followed by diffusion controlled growth under high supersaturation resulted in the formation of the present phase with two different morphologies driven by the surface energies of different crystal surfaces. The optical absorption study reveals a moderate bandgap of ∼2.1 eV. Electrical measurement and scanning probe microscopy in conjunction with first-principles density functional theory based calculations demonstrated the signature of ferroelectricity. Berry phase calculation under modern theory of polarization showed a large contribution of electronic polarization to the spontaneous polarization of the material. This large electronic polarization together with a narrow bandgap would allow for novel optoelectronic and photovoltaic device designing.Author contributions
M. M., P. S. S. S., and S. M. prepared samples and/or conducted characterization. A. R. did DFT calculations. S. M. and A. R. were responsible for data analyses, preparation, and revision of the manuscript. S. M., S. D., and A. R. were responsible for the overall supervision of the entire work.Conflicts of interest
All authors have read and agreed to the published version of the manuscript.Acknowledgements
A. R. thanks J. Bhagyaraj (IIT Kanpur) and Loknath Sathua (IIT Bhubaneswar), and D. Maurya (Virginia Tech.) for their help in electron microscopy. S. Mukherjee acknowledges DST for financial assistance (IFA13-MS-03) under the INSPIRE faculty award program to undertake this work. The work was supported by SERB, Govt. of India through project no. CRG/2019/003828.References
- J. F. Scott, Science, 2007, 315, 954–959 CrossRef CAS PubMed.
- C. A. F. Vaz, Y. J. Shin, M. Bibes, K. M. Rabe, F. J. Walker and C. H. Ahn, Appl. Phys. Rev., 2021, 8, 41308 CAS.
- G. Hodes, Science, 2013, 342, 317–318 CrossRef CAS PubMed.
- M. D. McGehee, Nat. Mater., 2014, 13, 845–846 CrossRef CAS PubMed.
- M. Antonietta Loi and J. C. Hummelen, Nat. Mater., 2013, 12, 1087 CrossRef PubMed.
- Q. Wang and A. Abate, Adv. Mater. Interfaces, 2018, 5, 1800264 CrossRef.
- C. N. R. Rao, Annu. Rev. Phys. Chem., 1989, 40, 291–326 CrossRef CAS.
- R. Nechache, C. Harnagea, S. Li, L. Cardenas, W. Huang, J. Chakrabartty and F. Rosei, Nat. Photonics, 2014, 9, 61 CrossRef.
- W. S. Choi, M. F. Chisholm, D. J. Singh, T. Choi, G. E. Jellison Jr. and H. N. Lee, Nat. Commun., 2012, 3, 689 CrossRef PubMed.
- S. Mukherjee, A. Roy, S. Auluck, R. Prasad, R. Gupta and A. Garg, Phys. Rev. Lett., 2013, 111, 087601 CrossRef PubMed.
- H. An, J. Y. Han, B. Kim, J. Song, S. Y. Jeong, C. Franchini, C. W. Bark and S. Lee, Sci. Rep., 2016, 6, 28313 CrossRef CAS PubMed.
- I. Grinberg, D. V. West, M. Torres, G. Gou, D. M. Stein, L. Wu, G. Chen, E. M. Gallo, A. R. Akbashev, P. K. Davies, J. E. Spanier and A. M. Rappe, Nature, 2013, 503, 509 CrossRef CAS PubMed.
- H. Matsuo, Y. Noguchi and M. Miyayama, Nat. Commun., 2017, 8, 207 CrossRef PubMed.
- J. He, C. Franchini and J. M. Rondinelli, Chem. Mater., 2017, 29, 2445–2451 CrossRef CAS.
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463–2485 CrossRef CAS.
- M. M. Seyfouri and D. Wang, Crit. Rev. Solid State Mater. Sci., 2020, 1–26 Search PubMed.
- S. M. Young, F. Zheng and A. M. Rappe, Phys. Rev. Lett., 2012, 109, 1–5 Search PubMed.
- M. Mishra, A. Roy, A. Garg, R. Gupta and S. Mukherjee, J. Alloys Compd., 2017, 721, 593–599 CrossRef CAS.
- S. Mukherjee, V. Ranjan, R. Gupta and A. Garg, Solid State Commun., 2012, 152, 1181–1185 CrossRef CAS.
- S. Song, H. M. Jang, N. S. Lee, J. Y. Son, R. Gupta, A. Garg, J. Ratanapreechachai and J. F. Scott, NPG Asia Mater., 2016, 8, e242–9 CrossRef CAS.
- V. Singh, K. Brajesh, S. Sahu, A. Garg and R. Gupta, J. Am. Ceram. Soc., 2019, 102, 7414–7427 CrossRef CAS.
- R. Arielly, W. M. Xu, E. Greenberg, G. K. Rozenberg, M. P. Pasternak, G. Garbarino, S. Clark and R. Jeanloz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1–8 CrossRef.
- M. Mishra, I. Mukherjee, A. K. Mall, A. Mitra, S. Dash, S. Chatterjee, S. Mukherjee and A. Roy, J. Mater. Chem. A, 2018, 6, 13031–13040 RSC.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 192, 55–69 CrossRef.
- R. O. Jones and O. Gunnarsson, Rev. Mod. Phys., 1989, 61, 689–746 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- B. Himmetoglu, A. Floris, S. de Gironcoli and M. Cococcioni, Int. J. Quantum Chem., 2014, 114, 14–49 CrossRef CAS.
- S. A. Tolba, K. M. Gameel, B. A. Ali, H. A. Almossalami and N. K. Allam, The DFT+U: Approaches, Accuracy, and Applications, ed. G. Yang, IntechOpen, Rijeka, 2018, vol. 1 Search PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 1651–1654 CrossRef PubMed.
- P. Hermet, M. Goffinet, J. Kreisel and P. Ghosez, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 220102 CrossRef.
- K. P. Rice, A. E. Saunders and M. P. Stoykovich, J. Am. Chem. Soc., 2013, 135, 6669–6676 CrossRef CAS PubMed.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, T. Kipp, R. Cingolani and P. D. Cozzoli, J. Am. Chem. Soc., 2008, 130, 11223–11233 CrossRef CAS PubMed.
- T.-D. Nguyen, C.-T. Dinh, D.-T. Nguyen and T.-O. Do, J. Phys. Chem. C, 2009, 113, 18584–18595 CrossRef CAS.
- T.-D. Nguyen, Nanoscale, 2013, 5, 9455–9482 RSC.
- L. Z. Mezey, D. Marton and J. Giber, Period. Polytech., Mech. Eng., 1977, 21, 189–216 Search PubMed.
- C. Rottman and M. Wortis, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 6274–6277 CrossRef CAS.
- X. Peng, Adv. Mater., 2003, 15, 459–463 CrossRef CAS.
- Y. Ogawa, Y. Kaneko, J. P. He, X. Z. Yu, T. Arima and Y. Tokura, Phys. Rev. Lett., 2004, 92, 47401 CrossRef CAS PubMed.
- J. Tauc and A. Menth, J. Non-Cryst. Solids, 1972, 8–10, 569–585 CrossRef CAS.
- J.-H. Park, B.-K. Kim, J.-G. Park, I.-T. Kim, H.-J. Je, Y. Klm and S. J. Park, Ferroelectrics, 1999, 230, 151–156 CrossRef.
- M. M. Kumar, V. R. Palkar, K. Srinivas and S. V. Suryanarayana, Appl. Phys. Lett., 2000, 76, 2764–2766 CrossRef CAS.
- Z. H. Sun, Y. L. Zhou, S. Y. Dai, L. Z. Cao and Z. H. Chen, Appl. Phys. A: Mater. Sci. Process., 2008, 91, 97–100 CrossRef CAS.
- J. M. Vila-Fungueiriño, A. Gómez, J. Antoja-Lleonart, J. Gázquez, C. Magén, B. Noheda and A. Carretero-Genevrier, Nanoscale, 2018, 10, 20155–20161 RSC.
- R. O. Jones and O. Gunnarsson, Rev. Mod. Phys., 1989, 61, 689–746 CrossRef CAS.
- T. Arima, D. Higashiyama, Y. Kaneko, J. P. He, T. Goto, S. Miyasaka, T. Kimura, K. Oikawa, T. Kamiyama, R. Kumai and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 64426 CrossRef.
- J. P. Perdew, W. Yang, K. Burke, Z. Yang, E. K. U. Gross, M. Scheffler, G. E. Scuseria, T. M. Henderson, I. Y. Zhang, A. Ruzsinszky, H. Peng, J. Sun, E. Trushin and A. Görling, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2801–2806 CrossRef CAS PubMed.
- H. Baltache, R. Khenata, M. Sahnoun, M. Driz, B. Abbar and B. Bouhafs, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 344, 334–342 CrossRef CAS.
- C. Capillas, E. S. Tasci, G. de la Flor, D. Orobengoa, J. M. Perez-Mato and M. I. Aroyo, Z. Kristallogr., 2011, 226, 186–196 CrossRef CAS.
- P. Singh, A. Roy, A. Garg and R. Prasad, J. Appl. Phys., 2015, 117, 184104 CrossRef.
- R. E. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ma00089j |
| This journal is © The Royal Society of Chemistry 2022 |