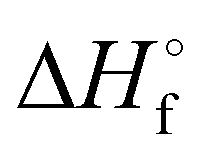DOI:
10.1039/D2MA00088A
(Paper)
Mater. Adv., 2022,
3, 3479-3489
3-(Nitromethylene)oxetane: a very versatile and promising building block for energetic oxetane based monomers†
Received
25th January 2022
, Accepted 3rd March 2022
First published on 4th March 2022
Abstract
In the field of energetic materials, older developments (e.g., RDX, ONC, CL20) are increasingly replaced by more environmentally benign, less expensive and likewise or more powerful compounds. This is mainly achieved through nitrogen-rich motifs like tetrazoles. However, such materials are mostly used as formulations containing polymeric energetic binders. Unfortunately, prior art binders show very poor performances and therefore reduce the overall performance. To address this problem, new monomers with enhanced performance are a prerequisite. Since the majority of energetic binders is oxetane-based, we chose 3-(nitromethylene)oxetane as a promising building block. It exhibits an explosophoric group, has recently become commercially available and provides suitable monomers by elegant and cost-efficient one-pot syntheses via conjugate addition. Herein, we report derivatives based on 1H-tetrazole, 1H-tetrazole-5-amine and the rather exotic but extremely powerful primary explosives 5-azido-1H-tetrazole (5AzT) and 5-nitro-2H-tetrazole (5NT). The sensitivities toward external stimuli like impact, friction, and electrostatic discharge were assessed by BAM standard procedures. As all molecular structures were elucidated by X-ray diffraction, Hirshfeld analysis was applied to explain the surprisingly low sensitivities found for the 5AzT- and 5NT-derivatives. Further, the compounds were studied by vibrational- and multinuclear NMR spectroscopy (1H, 13C, 14N), differential scanning calorimetry, and elemental analysis. Their performance was calculated using the EXPLO5 V6.04 thermochemical code. Based on obtained values, the 5AzT- and 5NT-derivatives outperform prior art energetic oxetanes and TNT. Therefore, their performance was additionally demonstrated and evaluated by a small-scale shock reactivity test (SSRT).
Introduction
Since the first synthesis of 5-cyano-2-phenyltetrazole by Emil Fischer in 1878,1 tetrazoles have been an important structural unit widely used in chemistry. Their applications cover a broad spectrum from dyes and agrochemicals2,3 to pharmaceuticals due to their fungicidal, antibacterial, antiallergic4 and as later discovered, cytostatic effects.5 In addition, they play a key role in the field of energetic materials due to their high nitrogen content and the associated, positive enthalpy of formation.6 The aromatic system also often provides a high thermostability.3,6 Particularly interesting are 5-substituted derivatives featuring nitrogen- and oxygen-rich groups. Due to the high nitrogen content, they mainly form molecular nitrogen upon decomposition and serve as motifs for more environmentally benign, “greener” energetic materials.7 Next to their application in propellant systems, they are suitable for the synthesis of powerful, heavy metal-free primary explosives such as K2DNABT8 or high-performance secondary explosives such as TKX-50 which competes with benchmark compounds such as CL20 or octanitrocubane (ONC) regarding performance while being less expensive.9 However, the performance of such energetic materials seems to have reached a maximum for now. As these materials are often used as formulation containing high proportions of binders, improved energetic polymers offer additional room for enhanced performance. Unfortunately, all prior art energetic monomers and their corresponding polymers show performances inferior to TNT which was already prepared in 1863 by Wilbrand. Hence, new monomers with higher performances are very desirable. Since energetic binder research is mainly focused on polyoxetanes (e.g., poly(AMMO), poly(BAMO), poly(NIMMO))10 while tetrazoles represent a key motif in the development of new energetic materials, tetrazole-based oxetanes appear as a promising approach. In this context, we identified 3-(nitromethylene)oxetane as suitable starting material. It was first prepared in 2008 by Wuitschik via condensation of nitromethane and oxetan-3-one.11 Since then, it has proven to be an excellent Michael acceptor and reacts readily with a wide variety of heteroatom- and carbon nucleophiles under 1,4-selectivity and has been used in drug discovery to obtain 3,3-disubstituted oxetanes.12 For instance, it was employed by Carreira et al. to prepare oxetane peptidomimetics.13 It is of interest to the field of energetic materials as it already features an explosophoric nitro group while offering the possibility to prepare monomers in one-pot syntheses via conjugate addition. To provide monomers based on inexpensive and commercially available materials, we selected 1H-tetrazole and 1H-tetrazol-5-amine, while 5-azido-1H-tetrazole (5AzT) and 5-nitro-2H-tetrazole (5NT) were selected to gain especially high performance. The latter represent primary explosives2,14,15 with a performance comparable to high-explosives such as RDX.15,16 Among monocyclic tetrazoles, 5AzT features the highest nitrogen content (88.3%), a detonation velocity (Vdet.) of 8986 ms−1 and a detonation pressure (pC–J) of 32.7 GPa.14 5NT (Vdet. 9457 ms−1; pC–J 39.0 GPa) outperforms both 5AzT and RDX (Vdet. 8750 ms−1; pC–J 34.6 GPa).15 Accordingly, the addition products show calculated performances (EXPLO5 V6.04) superior to prior art energetic oxetanes and TNT as secondary explosive. For verification, a small-scale shock reactivity test (SSRT) was performed.16 As the molecular structure of all target compounds was elucidated by single-crystal X-ray diffraction, Hirshfeld analysis was applied which made the surprisingly low sensitivity of the 5AzT- and 5NT-derivative comprehensible based on interactions in the crystal. Overall, we obtained satisfying to high yields in each case – even if poor nucleophiles have been used indicating the excellent acceptor properties of the nitroalkene. In general, various known energetic compounds can be directly added to 3-(nitromethylene)oxetane to provide new oxetane monomers in an almost assembly line manner. Therefore, we expect the nitroalkene to contribute to a faster progress in the field of energetic oxetane-based monomers and polymers.
Results and discussion
Synthesis
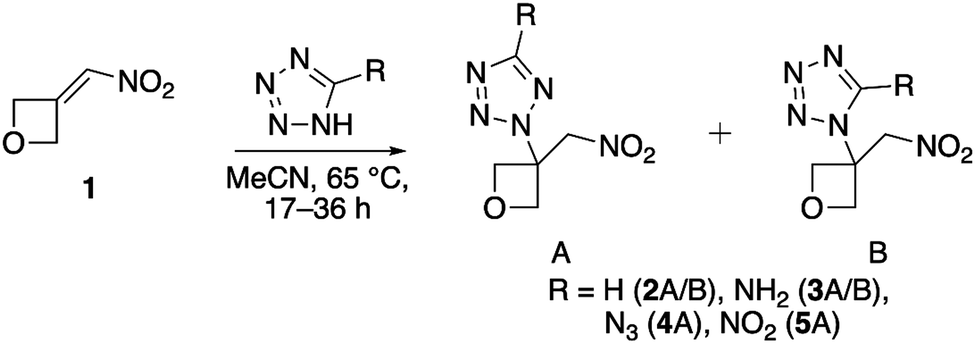
The precursor to all target compounds, 3-(nitromethylene)oxetane, was used as provided by Spirochem AG. 1H-Tetrazole and 1H-tetrazol-5-amine were used as provided by ABCR. 5-Azido-1H-tetrazole was prepared according to the literature by 1,3-dipolar cycloaddition using cyanogen bromide and two equivalents of sodium azide in aqueous methanol solution.14 Protonation of the intermediate (sodium salt) using 2 M hydrochloric acid gave the neutral compound which was extracted with diethyl ether. The organic phase was split into several plastic vials for evaporation to ensure quantities no larger than 100 mg for safety reasons. 5-Nitro-2H-tetrazole was prepared accordingly by acidifying an aqueous solution of sodium 5-nitrotetrazolate dihydrate followed by extraction of the neutral compound with ether and splitting into batches as described. The salt was prepared according to literature methods.17 All aza-Michael additions were first carried out analogously to literature by equimolar conversion of the respective tetrazole derivatives with nitroalkene 1 in anhydrous DMSO.18 Unfortunately, subsequent liquid–liquid extraction (brine, EA/Et2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) caused a significant loss of material leading to yields in a range of 30–40% and impurities caused by the addition of water to unreacted nitroalkene during work-up. Therefore, the solvent was changed to dry acetonitrile (argon atmosphere) which allowed direct monitoring by thin-layer chromatography (TLC). In addition, the need for liquid–liquid extraction was hereby eliminated, as the solvent can be removed by rotary evaporation. For all reactions, a quantitative conversion was observed (TLC) using a reaction temperature of 65 °C and reaction times between 17 and 36 h (Scheme 1).
:2) caused a significant loss of material leading to yields in a range of 30–40% and impurities caused by the addition of water to unreacted nitroalkene during work-up. Therefore, the solvent was changed to dry acetonitrile (argon atmosphere) which allowed direct monitoring by thin-layer chromatography (TLC). In addition, the need for liquid–liquid extraction was hereby eliminated, as the solvent can be removed by rotary evaporation. For all reactions, a quantitative conversion was observed (TLC) using a reaction temperature of 65 °C and reaction times between 17 and 36 h (Scheme 1).
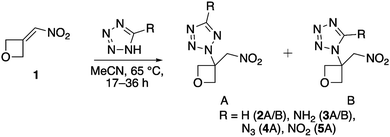 |
| | Scheme 1 General method for the preparation of compounds 2–5via an aza-Michael reaction. | |
This was confirmed by 1H NMR spectroscopy after solvent removal by rotary evaporation. In the case of 1H-tetrazole derivative 2 (A/B) an isomeric ratio (N2/N1) of 2:1 was determined by 1H NMR spectroscopy after 17 h and separation was achieved by column chromatography (SiO2, EA/petroleum ether/toluene 3:1:1) to give 86% yield of compound 2A (with regard to the theoretical amount of N2-isomer) and 71% yield of 2B, respectively. Despite that 1H-tetrazol-5-amine is only sparingly soluble in acetonitrile, full conversion was obtained in case of compound 3 (A/B) after 24 h. Hereby, the initial suspension turned into a solution upon first product formation and became a suspension again as the less polar N2-isomer precipitated. An isomeric ratio of roughly 9:1 (N2/N1) was assessed by 1H NMR spectroscopy. The solubility behavior was utilized to isolate 3A in high purity and yield (77%) by evaporating two thirds of the solvent volume, cooling (fridge, 5 °C), and removal of the liquid phase. The remaining solid was washed with a small volume of cold acetonitrile and the supernatant was again removed to give 3A as colorless solid. As impurities concentrate in the collected liquid phase, the N1-isomer (3B) was isolated by filtration through a silica plug using aforementioned solvent mixture to remove residual N2-isomer. After rotary evaporation, ethyl acetate was added to dissolve crude 3B. The solvent was then allowed to evaporate slowly at ambient conditions. Upon formation of colorless crystals, the supernatant was removed by means of a syringe to afford pure material with a yield of 35% due to work-up and purification losses. Furthermore, negligible trace amounts of compound 3C were obtained. In this case, the conjugated addition took place at both the endo- and exo-cyclic NH-position of 1H-tetrazol-5-amine. Unfortunately, the amount was insufficient for analytic purposes, but a few single-crystals formed and the structure was elucidated by X-ray diffraction. As 5-azido-1H-tetrazole is a weaker nucleophile by comparison, quantitative conversion toward compound 4 required a slightly prolonged reaction time (36 h) and raised equivalents with respect to the employed tetrazole (1.3 equiv.). According to 1H NMR spectroscopy, the formation of the N2-isomer was found to be strongly favored resulting in an isomer ratio (N2/N1) of roughly 10:1. Due to this excess, we renounced the isolation of the N1-isomer in favor of avoiding column chromatography as its necessity often excludes compounds from application in the field of energetic materials due to increased costs and a lack of scalability. After rotary evaporation, the obtained colorless oil was dissolved in dichloromethane (DCM) and filtration through a silica plug removed surplus 5-azido-1H-tetrazole. After rotary evaporation, overlaying of the obtained oil with a small amount of toluene and deep-freezing (−30 °C) resulted in crystals of the N2-isomer in form of large, colorless rods. The toluene layer was removed to afford 4A in high yield (78%). In case of the addition of 5-nitro-2H-tetrazole to nitroalkene 1, an almost quantitative conversion was observed after 17 h. Unfortunately, higher reaction times led to a dark brown color of the reaction solution indicating partial decomposition. Correspondingly, a significant impurity of the crude product was encountered. This can be attributed to undesired side reactions due to the high acidity of 5-nitro-2H-tetrazole. An isomeric ratio (N2/N1) of 12:1 was obtained according to 1H NMR spectroscopy indicating a strong favoring of the N2-isomer. As in case of 4, the isomeric mixture was dissolved in DCM after rotary evaporation and filtered through a silica plug to remove excess tetrazole and impurities to give 5 as off-white solid slightly contaminated with N1-isomer according to 1H NMR spectroscopy. Due to the extremely similar polarity, separation of the isomers by column chromatography failed using various solvent mixtures. Recrystallization was largely complicated by the extremely high solubility of compound 5 in nearly all laboratory solvents. However, recrystallization from hot toluene helped to reduce the fraction of N1-isomer (4.8%) in the target product and compound 5 was isolated with a still satisfying yield of 51%.
Crystallography
Single crystals of compound 1 were obtained by slow evaporation of a saturated chloroform solution. The same method was applied to compounds 2 (A, B), 3 (A, B, C), 4 and 5 using ethyl acetate as solvent. Detailed crystallographic data and the discussion of the N1-isomers (2B, 3B) as well as compound 3C can be found in the ESI.†
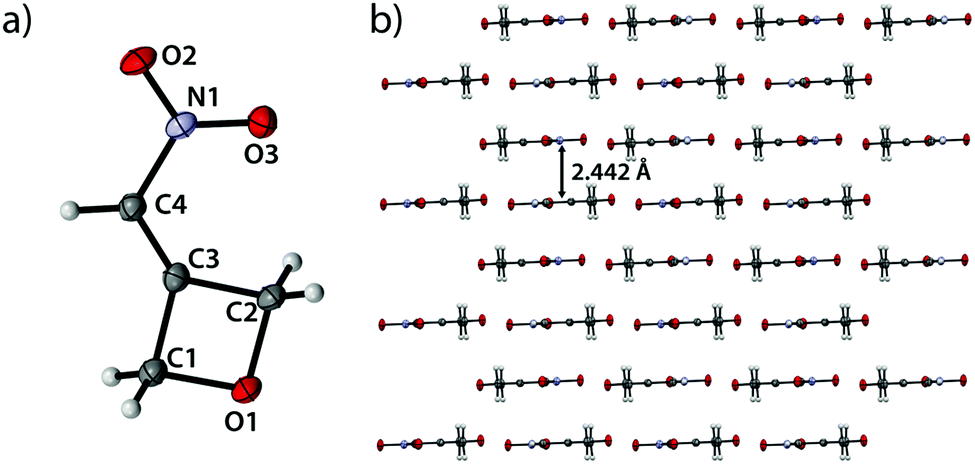
3-(Nitromethylene)oxetane (1) crystallizes in the monoclinic space group C2/c with eight formula units per cell and a density of 1.589 g cm−3 (143 K) corresponding to a room temperature density of 1.550 g cm−3 (Fig. 1).
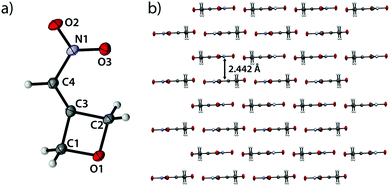 |
| | Fig. 1 (a) Molecular structure of compound 1 in the crystal. Thermal ellipsoids drawn at 50% probability level. (b) View along b axis. | |
Bond angles of the oxetane moiety range from 90.16(9)° (O1–C1–C3) over 90.37(9)° (O1–C2–C3) to considerably small angles of only 88.32(9)° (C1–C3–C2). The largest angle is found at the oxetane oxygen atom (C1–O1–C2) with an angle of 91.13(9)°. The large deviations from the ideal tetrahedral angle of sp3-hybridized carbon atoms (109.5°) give insight to the increased p-character of the endocyclic bonds and the prevailing ring tension. The shortest endocyclic bond is observed between C1 and O1 (1.460(2) Å) while the longest is found between C3 and C1 with a length of 1.503(2) Å. The ring itself is essentially planar with a puckering angle of only 1.23° which is considerably smaller than the puckering angle of unsubstituted oxetane with 8.7(2)° at 140 K.19 The double bond between C3 and C4 shows a typical value of 1.327(2) Å as well as the C4–N1 bond with a length of 1.442(2) Å.20 The nitro group itself is twisted by only 0.4° against the oxetane ring plane (C1–C2–C3). Therefore, the entire molecule is essentially planar. The view along the b axis (Fig. 1b) shows compound 2 forming closely adjacent, step-like and pairwise parallel layers with step heights of only 0.226 Å and 0.285 Å. These parallel segments in turn form layers with a minimum spacing of 2.442 Å and are rotated 180° relative to each other.
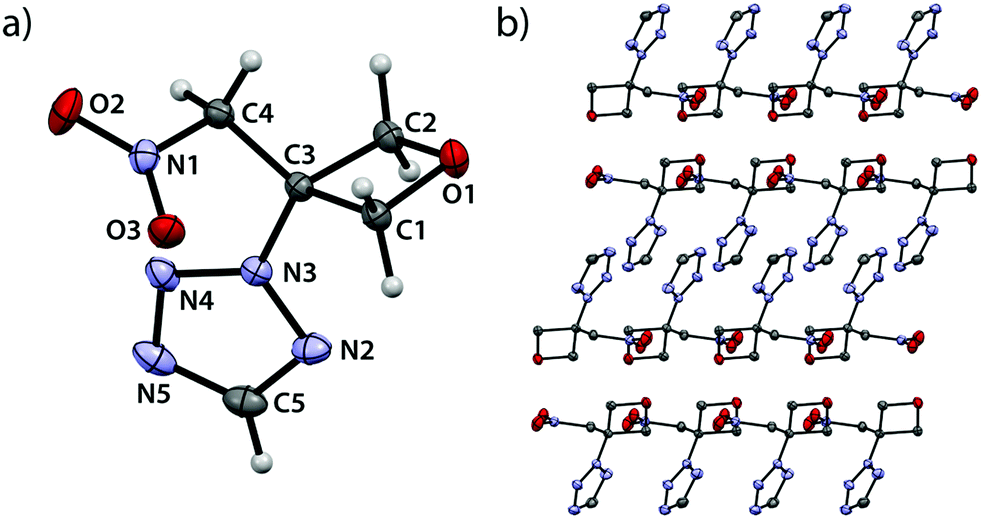
2-(3-(Nitromethyl)oxetan-3-yl)-2H-tetrazole (2A) crystallizes in the monoclinic space group P21/c with four formula units per unit cell and a density of 1.609 g cm−3 (102 K) which correlates to a room temperature density of 1.560 g cm−3 (Fig. 2).
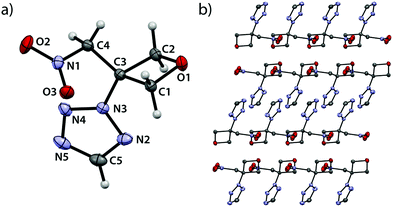 |
| | Fig. 2 (a) Molecular structure of compound 2A in the crystal. Thermal ellipsoids drawn at 50% probability level. (b) View along b axis. | |
As expected, all oxetane bond angles show large deviations from the ideal tetrahedral angle ranging from 91.5(1)° (O1–C1–C3) over 91.1(1)° (O1–C2–C3) to only 85.1(1)° (C1–C3–C2). An angle of 91.9(1)° is found at the oxetane oxygen atom (C1–O1–C2). The shortest bond is observed between O1 and C2 (1.449(2) Å) and the longest between C2 and C3 (1.548(2) Å). A rather small puckering angle of 6.95° (102 K) is found in comparison to the unsubstituted parent compound (10.7(1)°, 90 K).19 The single bond between C3 and the tetrazole moiety features a length of 1.464(2) Å which is shorter than in similar tetrazole-derivatives.21–23 The bond between C3 and C4 is shorter (1.506(2) Å) than the average bond length between sp3-hybridized carbons atoms.20 A deviation from typical values is also found for the bond between C4 and N1 (1.500(2) Å) which is longer compared to similar nitroaliphatic compounds (1.490(3) Å) and therefore expected to be weaker.24 Furthermore, the view along the b axis (Fig. 2b) shows alternating layers with one layer being made up of two bands, each containing the oxetane rings and the nitromethylene groups. The other layer comprises the tetrazole moieties which interlock emanating from aforementioned bands. Within this layer, the tetrazolyl-substituents themselves align in parallel layers.
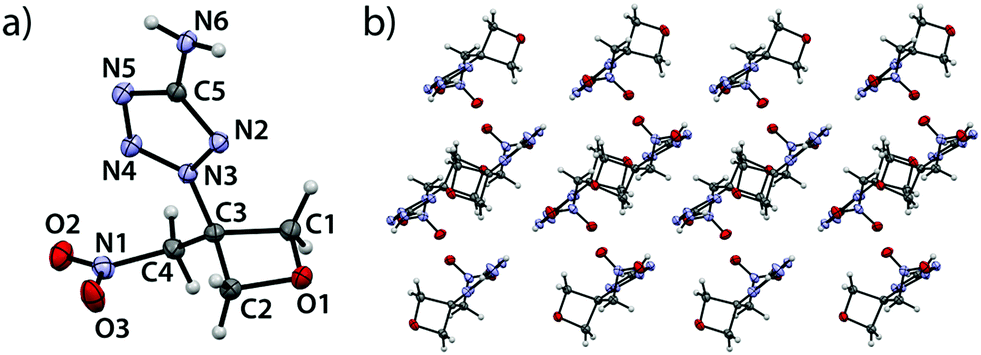
2-(3-(Nitromethyl)oxetan-3-yl)-2H-tetrazol-5-amine (3A) crystallizes in the triclinic space group P–1 with four entities per unit cell and a density of 1.584 g cm−3 (100 K) corresponding to 1.535 g cm−3 at room temperature (Fig. 3).
 |
| | Fig. 3 (a) Molecular structure of compound 3A in the crystal. Thermal ellipsoids drawn at the 50% probability level. (b) View along b axis. | |
In addition, the unit cell is made up of two pairs of crystallographically independent entities. The oxetane ring shows bond angles between 91.5(1)° (O1–C1–C3), 91.3(1)° (O1–C2–C3) down to 85.1(1)° (C1–C3–C2) and 91.5(1)° at the oxetane oxygen atom (C1–O1–C2). The longest bond within the oxetane ring is observed between C1–C3 (1.544(3) Å) and the shortest between O1 and C1 (1.452(2) Å). The oxetane ring is folded with a puckering angle of only 8.82° (100 K) which is surprisingly close to unsubstituted oxetane (10.7(1)°, 90 K).19 The bond between C3 and N3 is found to be considerably shorter (1.463(2) Å) than a typical bond between a tetrazole nitrogen atom and a sp3-hybridized carbon atom.21–23 The bond between the amino group (N6) and carbon atom C5 features a length of 1.363(3) Å and is therefore slightly longer than in the parent compound 1H-tetrazol-5-amine (1.333(1) Å).25 Both amino group and tetrazole ring lie in the same plane. The C3–C4 bond is longer (1.514(3) Å) than typical bonds between sp3-hybridized carbons,20 while the C4–N1 bond is longer (1.499(2) Å) than in similar nitroalkanes (1.485 Å).24 The view along the b axis (Fig. 3b) shows oxetane rings forming tunnel-like corridors in which the rings are twisted against each other in a pairwise manner so that the endocyclic oxygen atoms point away from each other. The tetrazole substituents are arranged in two different types of adjacent, pairwise parallel layers which intersect at an angle of 11.8°. These types of layers alternate within the crystallographic framework. Similar geometries and bond length were found for the entity not depicted in Fig. 3.
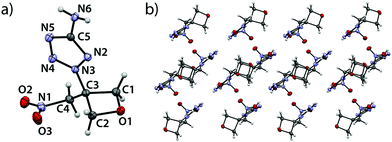
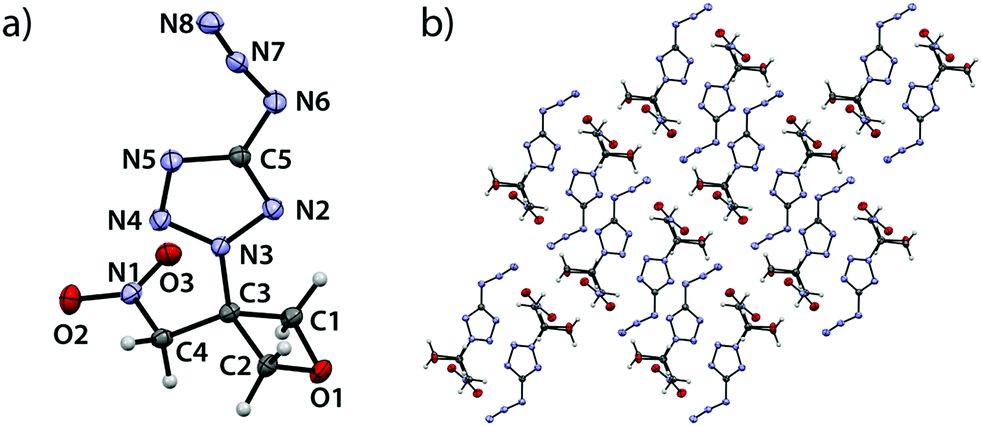
5-Azido-2-(3-(nitromethyl)oxetan-3-yl)-2H-tetrazole (4) crystallizes in the monoclinic space group P21/n with four formula units per cell and a density of 1.646 g cm−3 (123 K) corresponding to a room temperature density of 1.601 g cm−3 (Fig. 4). As in aforementioned compounds, extreme bond angles are found within the ring ranging from 91.49(8)° (O1–C1–C3) over 91.42(7)° (O1–C2–C3) to values of only 84.94(7)° (C1–C3–C2). The largest angle is found at the oxetane oxygen atom (C1–O1–C2, 91.73(7)°). The shortest bond is observed between O1 and C1 (1.451(1) Å) while the longest is again found between C2 and C3 (1.543(1) Å). A puckering angle of 6.85° (123 K) is found which is rather small for a substituted oxetane species taking the corresponding angle in the parent compound oxetane into account (8.7(2)° at 140 K).19 The bond between oxetane and tetrazole ring (C3–N3) shows a length of 1.463(1) Å and is thereby shorter than in comparable N2-alkylsubstituted tetrazoles.21–23 The azido group is twisted against the tetrazole ring plane by an angle of 12.41° and angulated (N8–N7–N6, 172.0(1)°) which can be explained by hyperconjugation effects.26 The single bond between C4 and N1 is significantly longer (1.498(1) Å) than a typical bond between a sp3-hybridized carbon atom and a nitro group (1.485 Å).24 Therefore, it is assumed to be rather weak. The view along b axis (Fig. 4b) shows an arrangement of the formula units in alternating layers – one layer consisting of two parallelly arranged bands containing both the oxetane- and nitromethylene moiety while the other is made up by interlocking tetrazole moieties originating from aforementioned bands. In this arrangement, two formula units oppose each other rotated by 180°, causing the opposing oxetane rings to point in opposite directions. Correspondingly, the tetrazole moieties originate from these bands in opposite spatial directions and are rotated by 180° with respect to each other. They thus form pairwise parallel layers with a spacing of 0.483 Å. In the crystal, these paired layers occur in two spatial orientations and intersect at an angle of 70.9°.
 |
| | Fig. 4 (a) Molecular structure of compound 4 in the crystal. Thermal ellipsoids drawn at the 50% probability level. (b) View along b axis. | |
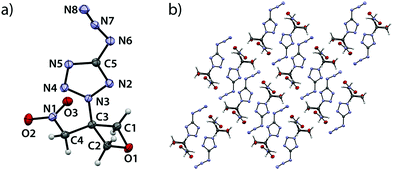
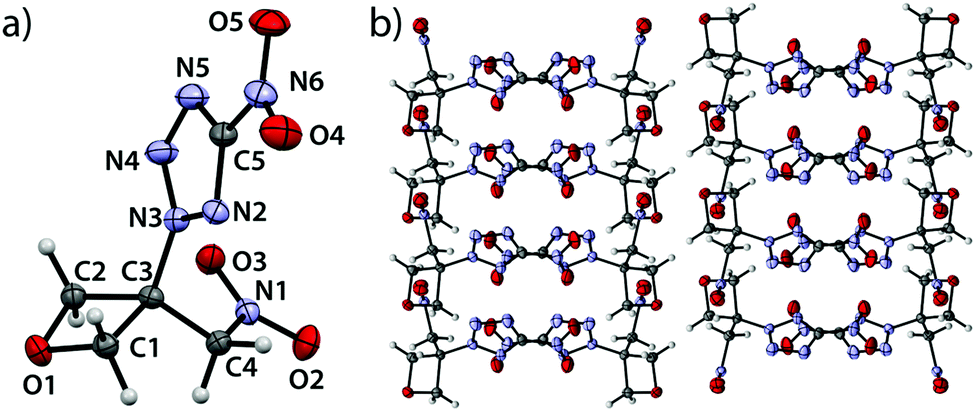
5-Nitro-2-(3-(nitromethyl)oxetan-3-yl)-2H-tetrazole (5) crystallizes in the orthorhombic space group Pbca with eight formula units per cell at a density of 1.729 g cm−3 (123 K) corresponding to 1.682 g cm−3 at ambient conditions (Fig. 5).
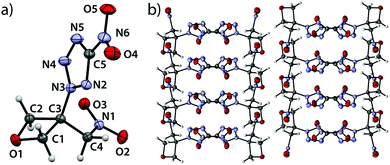 |
| | Fig. 5 (a) Molecular structure of compound 5 in the crystal. Thermal ellipsoids drawn at the 50% probability level. (b) View along a axis. | |
The largest bond angle (91.9(1)°) of the oxetane motif is found at the oxygen atom (C1–O1–C2) while all other endocyclic angles range from 91.1(1)° (C3–C2–O1) over 91.0(1)° (O1–C1–C3) down to 85.2(1)° (C2–C3–C1). The shortest bond is observed between O1 and C1 with a length of 1.446(2) Å while the longest (1.544(2) Å) is found between C1 and C3. The ring puckering angle was determined to be 10.1° (123 K) and is thus again close to the puckering angle of the unsubstituted parent compound at a comparable temperature, as in the compounds described above.19 Since the substitution of the ring often leads to an increase of disadvantageous eclipsing interactions and correspondingly larger puckering angles, the influences of the substituents in compounds 2–5 seem to compensate each other to a certain extent. The nitro group is slightly rotated with respect to the tetrazole plane by an angle of 6.38°. The C3–N3 bond shows a slightly increased length of 1.469(2) Å which is longer and thereby weaker than in compounds 2, 3 and 4 and may correlate with the lower thermal stability of compound 5. The C5–N6 bond features a length according to expectation (1.450(2) Å).20 The C3–C4 shows a typical bond length of (1.509(2) Å).20 The C4–N1 bond is slightly longer (1.496(2) Å) than in comparable compounds.24 The view along a axis (Fig. 5b) shows the same layered structure as described for compound 4. One layer contains interlocked tetrazole moieties, the other two parallel bands contain the oxetane scaffold. Also, two formula units oppose each other being rotated by 180°. Thereby, the tetrazole moieties form two types of pairwise parallel layers (distance: 1.227 Å) which intersect at an angle of 76.9°.
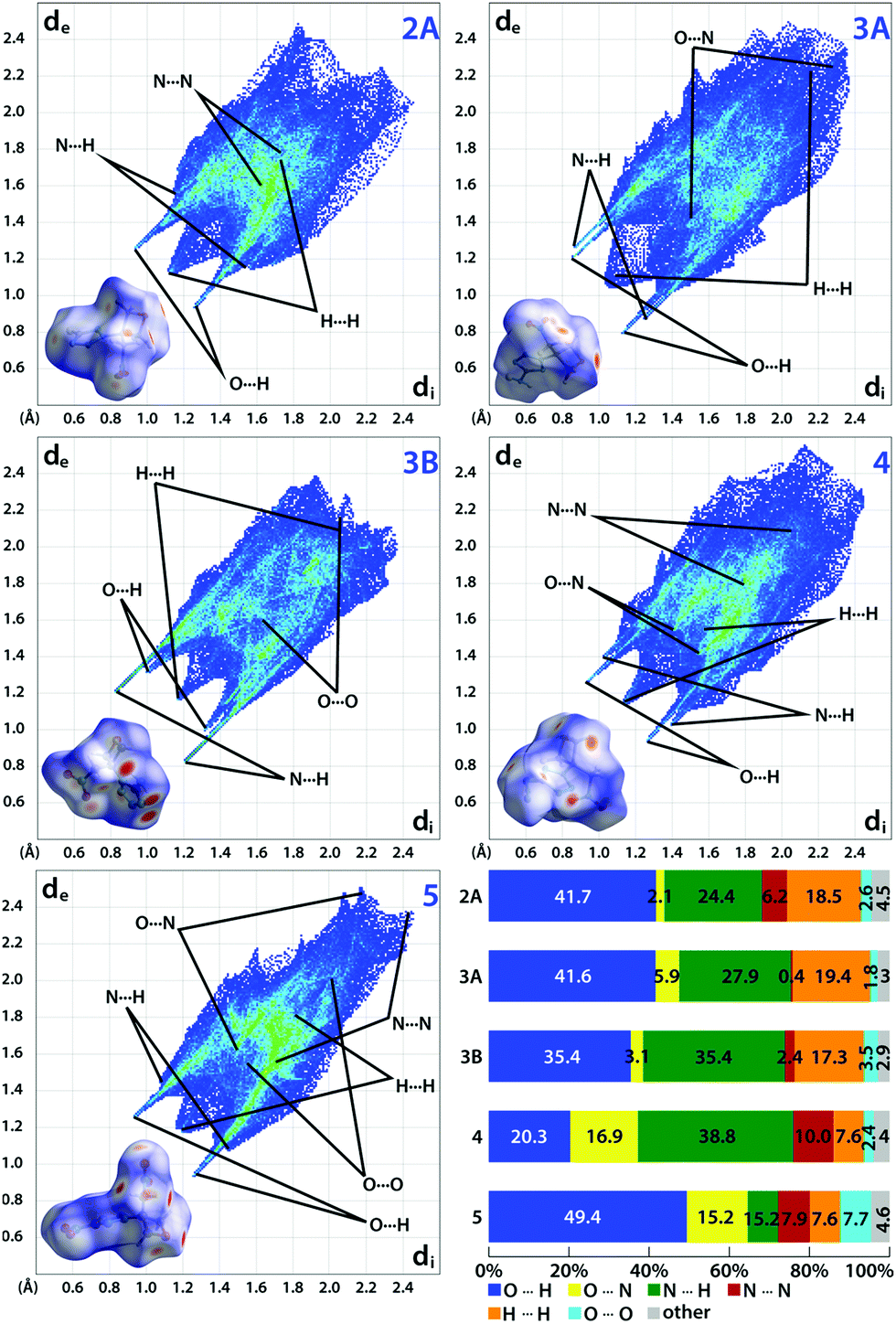
Hirshfeld analysis
When an energetic material is exposed to a mechanical stimulus, a vertical compression and/or a horizontal sliding of layers in the crystal leads to internal strains.27 If the associated strain energy is high enough, bonds may break and trigger the material's decomposition.28 Here, intermolecular interactions can impose a destabilizing or stabilizing effect depending on their nature and strength. For this reason, the sensitivity of an energetic material is linked to close contact interactions in the crystal.28,29 A powerful tool to analyze these interactions is Hirshfeld analysis. The method's fundamentals were already developed in 1977.30 Since then, the method was continuously improved – especially by Spackman and McKinnon.31,32 Ultimately, the development of the CrystalExplorer software in 2004 allowed a user-friendly application.33 However, the method was not applied to energetic materials until 2014.34 Since then, Hirshfeld analysis is used to estimate the sensitivity of energetic materials and to render experimentally assessed values plausible based on the crystal structure. As suitable crystallographic data was obtained for compounds 2A, 3A, 3B, 4 and 5, we calculated their Hirshfeld surfaces (HFS) using CrystalExplorer V17.5.35 On this surface, close contacts are indicated by red dots. To summarize all interactions and their distances di + de (di, distance from HFS to the closest atom interior; de, distance from HFS to closest atom exterior), we used a 2D fingerprint plot (Fig. 6).32,35 Distances larger than 3 Å correspond to weak interactions, distances below 2.4 Å are linked to strong interactions.28 The analysis of the interaction populations and their distance allows a weighting of their influence. In general, insensitive compounds are often designated by high populations of strong, stabilizing O⋯H and N⋯H interactions which form a rather rigid intermolecular 3D network which counteracts a detrimental sliding of layers to a certain extent.28 In addition, correspondingly low percentages of destabilizing H⋯H, O⋯N and O⋯O interactions are found among insensitive compounds. For example, compound 2A exhibits a very high fraction of strong (di + de < 2.4 Å), stabilizing O⋯H (41.7%) and N⋯H interactions (24.4%). These are contrasted by repulsive, destabilizing H⋯H interactions (18.5%) and N⋯N interactions (6.2%). However, the 2D Fingerprint plot shows that only a small proportion of the H⋯H interactions are strong (di + de < 2.4 Å). Therefore, they only cause minor destabilization. The same is true for the N⋯N interactions which are also weak according to their distance (di + de > 3.2 Å). All remaining interactions have a negligible contribution below 5%. Hence, a very insensitive material can be expected which correlates well to obtained values for its impact (IS) and friction sensitivity (FS) values of 40 J and 360 N, respectively. Very similar proportions for strong, stabilizing O⋯H (41.6%) and N⋯H interactions (27.9%) were found for compound 3A. These are counterbalanced by weak O⋯N interactions (5.9%) and H⋯H interactions (19.4%) which are also mostly weak. Compared to 2A, a similar sensitivity can be assumed which was confirmed in practice (IS: 35 J, FS: 360 N). The difference of 5 J is likely to be linked to the higher melting temperature of 3A, which allows less energy to be dissipated by partial melting upon impact. As the ratio of stabilizing to destabilizing interactions barely changes for 3B, a similar sensitivity was expected and found (IS: 40 J, FS: 360 N). Since 5-azido-1H-tetrazole is very sensitive (IS: < 1 J, FS: < 5 N),14 compound 4 was expected to show a slightly lower but still comparable sensitivity. However, the high proportion of strong and stabilizing O⋯H (20.3%) and N⋯H (38.8%) interactions is only counteracted by O⋯N (16.9%), N⋯N (10%) and H⋯H interactions (7.6%) which are mostly weak giving the impression of a rather low sensitivity. Indeed, surprisingly low values were assessed (IS: 2–3 J, FS: 120 N). Accordingly, the estimation based on Hirshfeld analysis turned out to be correct. Compound 5, which is a derivative of the also very sensitive 5-nitro-2H-tetrazole (IS: < 1 J, FS: < 5 N),15 was found to be dominated by strong, stabilizing O⋯H (49.4%) and N⋯H (7.6%) interactions. These are contrasted by a small fraction of destabilizing N⋯N (7.9%), H⋯H (7.6%), O⋯N (15.2%) and O⋯O (7.7%) interactions. As these are largely very weak, a sensitivity comparable to compound 4 was assumed. Against expectation, practical tests revealed an impressively insensitive material (IS: 40 J, FS: 360 N). Although similar sensitivities have been expected for compounds 4 and 5, Hirshfeld analysis estimated correctly that both compounds are significantly less sensitive than the parent tetrazoles and can be handled more safely (Table 1).
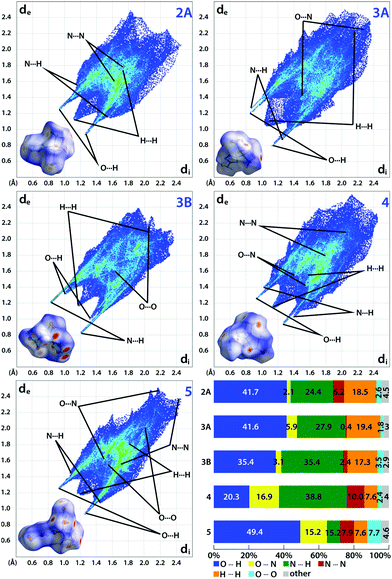 |
| | Fig. 6 Calculated Hirshfeld surfaces and 2D fingerprint plots of compounds 2A, 3 (A, B), 4 and 5 showing important close contacts in the crystal. The bar chart summarizes the interaction percentages. | |
Table 1 Physicochemical and energetic properties of compounds 2–5 together with TNT and the prior art monomer NIMMO for comparative purposes
|
|
2A
|
2B
|
3A
|
3B
|
4
|
5
|
TNT
16,36
|
NIMMO
|
|
Impact sensitivity (BAM drop hammer, method 1 of 6).
Friction sensitivity (BAM friction apparatus, method 1 of 6).
Oxygen balance regarding carbon monoxide (ΩCO = (nO – xC – yH/2)(1600/FW)).
Melting point (DSC, β = 5 °C min−1).
Temperature of decomposition (DSC, β = 5 °C min−1).
Density at 298 K (ρX-ray/1.028).
Standard molar enthalpy of formation.
Detonation energy.
Detonation temperature.
Detonation velocity.
Detonation pressure at C–J-point.
Volume of detonation products.
|
| Formula |
C5H7N5O3 |
|
C5H8N6O3 |
|
C5H6N8O3 |
C5H6N6O5 |
C7H5N3O6 |
C5H9NO4 |
| FW [g mol−1] |
185.14 |
|
200.16 |
|
226.16 |
230.14 |
227.12 |
147.13 |
| ISa [J] |
40 |
40 |
35 |
40 |
2(3) |
40 |
15 |
> 40 |
| FSb [N] |
360 |
360 |
360 |
360 |
120 |
360 |
> 353 |
360 |
| N + O [%] |
63.75 |
|
65.97 |
|
70.77 |
71.28 |
60.76 |
53.02 |
| ΩCOc [%] |
−47.53 |
|
−47.96 |
|
−35.37 |
−20.86 |
−25.3 |
−59.81 |
|
T
m
/Tdec.e [°C] |
94/182 |
145/169 |
161/183 |
129/177 |
56/185 |
138/160 |
81/290 |
−14/170 |
|
ρ
[g cm−3] |
1.56 |
1.59 |
1.54 |
1.56 |
1.60 |
1.69 |
1.64 |
1.19 |
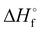 [kJ mol−1]
[kJ mol−1] |
125.9 |
154.3 |
106.4 |
151.0 |
487.9 |
148.1 |
−219.0 |
−268.9 |
|
EXPLO5 V6.04
|
| −ΔEU°h [kJ kg−1] |
3888 |
4041 |
3627 |
3842 |
4629 |
4761 |
4380 |
3949 |
|
T
C–J
[K] |
2707 |
2770 |
2564 |
2657 |
3271 |
3336 |
3190 |
2507 |
|
D
C–J
[m s−1] |
6924 |
7078 |
6950 |
7121 |
7452 |
7686 |
6809 |
5906 |
|
p
C–J
[GPa] |
17.2 |
18.2 |
16.9 |
17.9 |
20.8 |
23.5 |
18.7 |
10.6 |
|
V
0
[dm3 kg−1] |
760 |
758 |
789 |
789 |
766 |
742 |
639 |
827 |
Physicochemical and energetic properties
The thermal behavior of compound 4 was assessed by differential thermal analysis (DTA) at a heating rate of 5 °C min−1. The thermal stability of all other compounds was assessed by differential scanning calorimetry (DSC) at the same heating rate. The precursor to all compounds, 3-(nitromethylene)oxetane (1) melts at 52 °C and decomposes rather early at 165 °C. Regarding compounds 2–5, the lowest thermal stability is observed for compound 5 which decomposes at 160 °C while all other compounds (2–4) decompose in a narrow interval of 182–185 °C. All compounds melt before they decompose. Here, compound 2A is especially interesting as it melts at 94 °C potentially allowing melt-cast applications. The highest melting point is found for compound 3A (161 °C). The heats of formation of compounds 2–5 were calculated using the Gaussian16 program package on the CBS-4M level of theory using the atomization method.37,38 All compounds were found to be endothermic with high positive heats of formation between 106.4 and 154.3 kJ mol−1 exceeding common secondary explosives like TNT (−75.3 kJ mol−1)36 and RDX (79.1 kJ mol−1)39 while compound 4 shows an outstanding heat of formation of 487.9 kJ mol−1. The sensitivities toward external stimuli like shock and friction were assessed using a BAM (Bundesanstalt für Materialforschung) drop hammer apparatus and a BAM friction tester according to STANAG 4489 and STANAG 4487 modified instruction, respectively.40,41 Here, compounds 2 (A, B), 3B and 5 were found to be insensitive according to the UN classification for the transport of dangerous goods with an impact sensitivity of 40 J and a friction sensitivity of 360 N.42 The low sensitivity of 5 can be partially explained by stabilizing intermolecular interactions (see Hirshfeld discussion). A marginally lower sensitivity was found for compound 3A (IS: 35 J, FS: 360 N) which can be classified as “less sensitive”.42 Generally speaking, the impact sensitivity of compound 4 is high (2–3 J), but at the same time surprisingly low taking the very sensitive parent compound 5AzT into account. Furthermore, the IS is only marginally below the value (3 J) of PETN (pentaerythritol tetranitrate) as widely used booster explosive.16,36 The friction sensitivity is even the same as for RDX (120 N) and half of PETN (60 N).36 However, compound 4 needs to be classified as “sensitive” to “very sensitive”.42 The energetic performance of compounds 2–5 was calculated using the EXPLO5 V6.04 thermochemical code.43 Compounds 2A and 3A show detonation pressures of 17.2 GPa and 16.9 GPa and detonation velocities of 6924 ms−1 and 6950 ms−1, respectively. A slightly higher performance was calculated for their corresponding N1-isomers (2B, 3B) as they show a higher density and heat of formation (Table 1). The performance of compound 2 (A, B) and 3 (A, B) is therefore notably higher than in case of prior art oxetane monomers like NIMMO (10.6 GPa, 5906 ms−1) and comparable to TNT. Especially high performance was found in case of 4 (20.8 GPa, 7452 ms−1) and 5 (23.5 GPa, 7686 ms−1). Therefore, both compounds are not only superior to prior art energetic oxetanes, but even outperform TNT.36 To the best of our knowledge, they are currently the most powerful energetic oxetanes known next to 3,3-dinitratooxetane (DNO) that we described recently.44 However, since DNO decomposes at 93 °C, they offer a better balance between performance, sensitivity, and thermal stability, rendering them the more promising candidates for the preparation of performant energetic polymers.
Table 2 CBS-QB3 enthalpies (Hartree) and gas-phase enthalpies of formation (kJ mol−1)
| Fragment |
Formula |
−H(CBS-QB3) [H] |
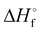
(g) [kJ mol−1] |
|
2A
|
C5H7N5O3 |
692.997417 |
+209.0 |
|
1
|
C4H6NO3 |
435.621358 |
+67.3 |
|
2
|
CHN4 |
257.208636 |
+581.0 |
|
3
|
C4H5N4O |
448.828069 |
+453.7 |
|
4
|
CH2NO2 |
244.028508 |
+149.8 |
|
5
|
C5H7N4O |
488.049907 |
+441.7 |
|
6
|
NO2 |
204.848641 |
+26.7 |
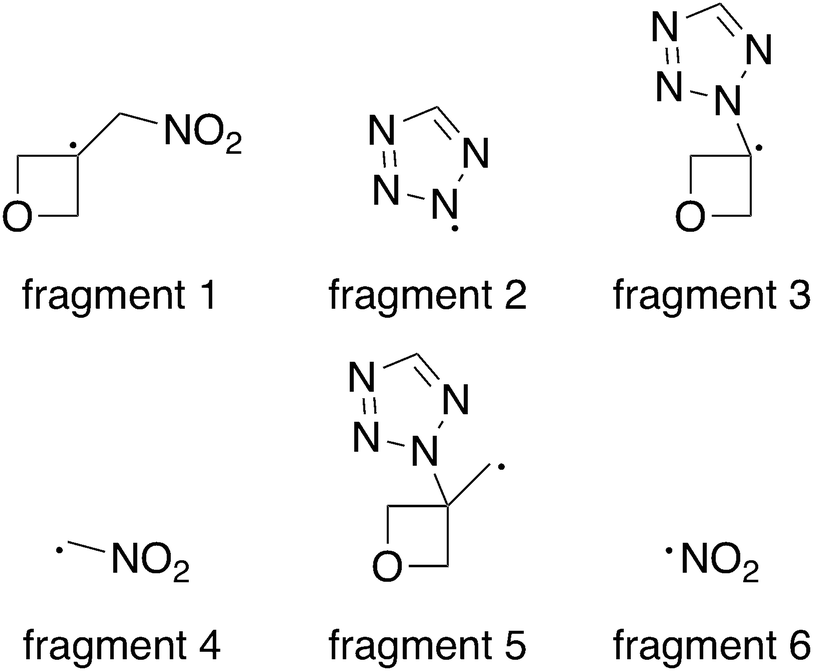
Bond dissociation energy calculation
Since all compounds with exception of 5 decompose in a narrow temperature interval (182–185 °C), a trigger bond inherent to all compounds was anticipated. When this bond breaks under thermal stress, it initiates the exothermic decomposition.28 As both the oxetane and the 1H-tetrazole motif are usually quite thermostable, three bonds were under particular suspicion – the C–N bond between both ring structures, the C–N bond between the exocyclic methylene group and the nitro group as well as the C–C bond between oxetane ring and the nitromethylene group. Therefore, the respective bond dissociation energy (BDE) was calculated on the CBS-QB3 level of theory using 2A as model compound and its crystal structure as input file for the calculation using Gaussian 16.38 For this purpose, the molecule was formally split into the radical fragments of interest (Fig. 7) and their gas phase enthalpies of formation were calculated. The obtained values for compound 2A and each fragment are summarized in Table 2. The BDE is obtained as the enthalpy difference between the molecule and the combined enthalpy of the respective fragments. Hereby, a BDE of 439.3 kJ mol−1 was assessed for the C–N bond between tetrazole and oxetane ring while the C–C bond shows a similar BDE of 394.5 kJ mol−1. As a considerably lower value of 259.4 kJ mol−1 was found for the C–NO2 bond, we identified it as trigger bond. Since compound 5 decomposes even earlier, the C–NO2 bond of the tetrazole motif can be assumed as trigger bond in this case. To increase the thermal stability of the investigated compounds, the nitro group should be reduced to provide an amino group. This would also allow further functionalization of the compounds.
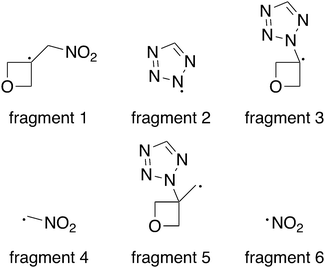 |
| | Fig. 7 Radical fragments used for the BDE calculation. | |
Hot-plate test
The N2-isomers of compounds 2–5 were subjected to a hot-plate test. In each case, 50 mg were placed on a copper witness plate which was subsequently heated by means of a Bunsen burner (see ESI†). Compounds 2A, 3A and 5 showed a violent deflagration with flame heights of up to 70 cm. Contrary to this, azidotetrazole-derivative 4 detonated violently upon heating leaving a dent on the witness plate. Instantaneous detonation of compound 4 was also observed when small crumbs were exposed to a flame on a steel syringe needle further proving its very fast deflagration-to-detonation transition (DDT) even in small, unconfined amounts. To the best of our knowledge, 4 is the first oxetane-based compound to exhibit this behavior otherwise characteristic of primary explosives. Therefore, the compound may represent an important leap towards yet unknown oxetane-based and potentially polymerizable primary explosives.
Hot-needle test
The hot-plate test of compound 4 did not only show its astonishing explosive power, but also its capability to undergo DDT like an primary explosives.16 However, to be classified as such, additional criteria need to be fulfilled. One is a positive response (detonation) upon contact of a needle heated to red heat with a Bunsen burner (Fig. 8). In case of compound 4, crackling noises indicated partial DDT, but the material mainly deflagrates while spreading on the copper plate due to gas generation. This behavior can be explained by the low melting point of compound 4 (55.8 °C) which allows the material to dissipate external, thermal energy. As a consequence, only material with direct contact to the needle is initiated while surrounding material melts, deflagrates or spreads. A more positive result is obtained when the material is prevented from spreading by confining it under a Tesafilm® strip (Fig. 9).
 |
| | Fig. 8 Partial deflagration of unconfined compound 4 upon contact to a hot needle. | |
 |
| | Fig. 9 Compound 4 confined under Tesafilm (left) and scattered material after partial detonation upon contact to a hot needle. | |
In this case, partial detonation is observed. As neither complete detonation nor full deflagration occurred, the test is negative. However, compound 4 is very close to the typical behavior of a primary explosive.
Initiation test
Another criterion for the classification as primary explosive is a positive initiation test. In this test, a small sample (50–100 mg) is placed on top of a booster explosive of rather high sensitivity (PETN) within a copper shell. The shell itself is placed on top of a copper witness plate. Then, the sample is exposed to a spark provided by an electrical igniter. In the case of a positive result, the sample will detonate and thereby trigger detonation of the adjacent booster charge. This causes the copper shell to burst and the witness plate to be perforated (Fig. 10). The test was performed using 50 mg of loosely packed compound 4 and 200 mg of PETN as booster charge. However, the test was negative three times in a row. Analysis of the charge showed that compound 4 had only partially melted upon exposure to sparks provided by the electrical ignitor. However, different results may be obtained using additives that improve its spark-response.
 |
| | Fig. 10 Schematic setup of the initiation test (left) and actual test in a sandbox (right). | |
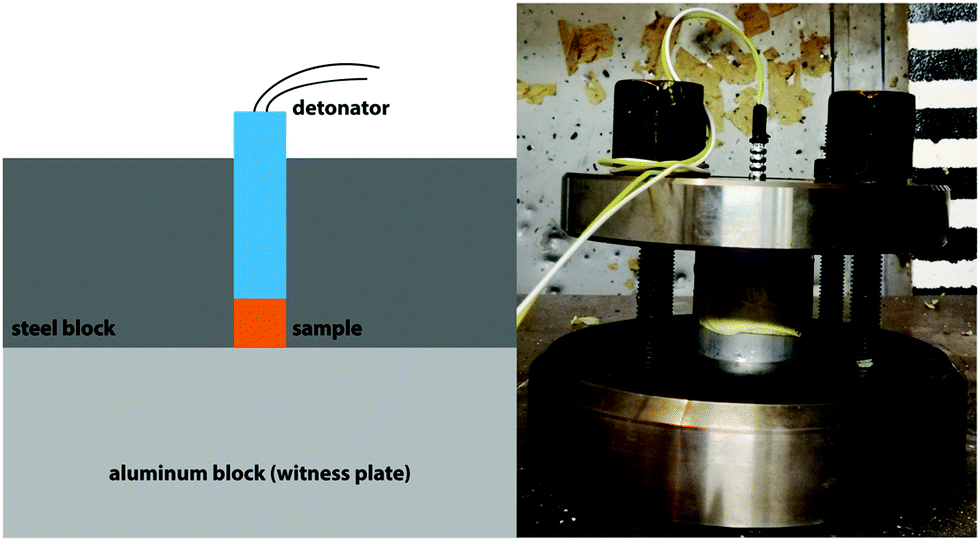
Small-scale shock reactivity test (SSRT)
As their calculated performance is superior to TNT, a SSRT was performed with compounds 4 and 5. In this test, the shock reactivity (explosiveness) can be determined even below a material's critical diameter and without requiring transition to detonation.45 Therefore, it combines the advantages of both lead block and gap test46 requiring amounts of roughly 500 mg. In each test, the same sample Volume VS is used (284 mm3). Therefore, the required mass of explosive me was calculated by the formula me = Vs·ρe·0.95 where ρe is the density of the explosive. The sample is filled into a perforated steel block with an aluminum witness block underneath and pressed (3 tons, 5 seconds). Subsequently, the charge is fired using a commercial Orica Dynadet C2 detonator (Fig. 11). The obtained witness plate dents were measured using a profilometer to compare the relative performance. As compounds 4 and 5 are superior to TNT according to EXPLO5 calculations, it was used as reference. Table 3 summarizes the employed amounts and the corresponding dent volumes.
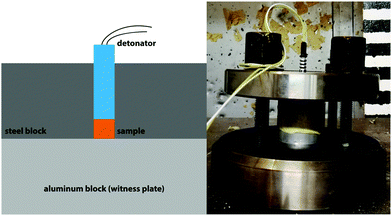 |
| | Fig. 11 Schematic (left) and actual SSRT setup (right). | |
Table 3 Results of the SSRT. Mass of explosive versus dent volume
|
|
4
|
5
|
TNT |
|
m
e (mg) |
411 |
454 |
464 |
|
D
V (mm3) |
917 |
715 |
971 |
|
D
V/me (mm3 mg−1) |
2.23 |
1.57 |
2.09 |
As volume-based method (284 mm3 per sample), the SSRT shows the largest indentation for TNT, followed by compound 4 and 5. Here, the result for compound 5 seems to contradict the calculated performance. However, the low dent volume may be attributed in part to the fact that 5 partially liquefied during pressing causing a reduced density. This in turn negatively affects the performance.16 We assume the impurification with the N1-isomer as cause of this behavior. Compound 4 actually shows a performance comparable to TNT as expected. Further, when the dent volume is correlated with the employed mass (mm3 mg−1), compound 4 indeed outperforms TNT. This agrees well with the EXPLO5 calculation and a higher performance can be anticipated for the pure (solid) N2-isomer of compound 5.
Conclusions
By aza-Michael addition between the commercially available 3-(nitromethylene)oxetane and selected tetrazoles, four new energetic oxetane derivatives were prepared by simple, but elegant one-step syntheses. Each product (isomeric mixture) was obtained in moderate to high yield proving the excellent acceptor properties of nitroalkene 1, especially with respect to the low nucleophilicity of 5-azido-1H-tetrazole and 5-nitro-2H-tetrazole. The products form two groups – compounds 2 and 3 are fully based on commercially available materials and inexpensive tetrazoles while compounds 4 and 5 have been prepared from tetrazoles that belong to the most nitrogen-rich and powerful of their kind. These have been rarely used as building blocks for energetic materials due to their high sensitivity and low thermostability. However, compounds 4 and 5 successfully demonstrate that 5-azido-1H-tetrazole and 5-nitro-2H-tetrazole are tamable and can provide derivatives with moderate (4) or even surprisingly low sensitivity (5). Despite of their non-elaborate synthesis and simple structure, the former group (2, 3) exhibits a performance notably higher than state-of-the-art energetic oxetanes. Binders prepared thereof may find application as insensitive, high-performing and rather nitrogen-rich fuels. The other group (4, 5) shows calculated performances higher than in the case of TNT as common secondary explosive. Hence, they are not only superior to prior art monomers (e.g., AMMO, BAMO, NIMMO), but are probably among the most powerful known oxetane derivatives, along with DNO. They are therefore suitable to prepare binders for energetic formulations where performance is of utmost importance. In addition, compound 4 is a first leap towards oxetane-based and potentially polymerizable primary explosive which are yet unknown. As the C–NO2 bond of the target compounds acts as trigger bond, a reduction of the nitro group is likely to provide higher thermostabilities next to the possibility of further functionalization. Since 3-(nitromethylene)oxetane readily reacts with a broad variety of carbon- and heteroatom nucleophiles, a wide variety of known energetic compounds can be directly employed as Michael donors (e.g., azoles). We therefore conclude that it represents a very potent scaffold for the rapid synthesis of a large number of chemically diverse energetic oxetane monomers. We further anticipate that it will thereby facilitate the preparation of energetic binders with enhanced performance to mitigate the existing performance gap between binders and highly performant fillers in energetic formulations.
Author contributions
The manuscript was written through contributions of all authors.
Abbreviations
| AMMO | 3-Azidomethyl-3-methyloxetane |
| BAMO | 3,3-Bis(azidomethyl)oxetane |
| CL20 | 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane |
| EA | Ethyl acetate |
| K2DNABT | Potassium 1,1′-dinitramino-5,5′-bistetrazolate |
| NIMMO | 3-Nitratomethyl-3-methyloxetane |
| RDX | Hexahydro-1,3,5-trinitro-1,3,5-triazin |
| TNT | 2,4,6-Trinitrotoluene. |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support by the Ludwig-Maximilian-University (LMU), the Office of Naval Research (ONR) under grant no. ONR.N00014-16-1-2062 and the Strategic Environmental Research and Development Program (SERDP) under contract no. WP19-1287 is gratefully acknowledged. The authors also thank Stefan Huber for his help regarding the determination of sensitivities. Furthermore, we thank Marcus Lommel for providing generous amounts of sodium 5-nitrotetrazolate.
References
- E. Fischer, Liebigs Ann. Chem, 1878, 190, 67–183 CrossRef.
-
J. Stierstorfer, PhD thesis, Ludwig-Maximilians-University Munich, 2009.
- G. I. Koldobskii, V. A. Ostrovskii and V. S. Popavskii, Chem. Heterocycl. Compd., 1981, 17, 965–988 CrossRef.
-
F. R. Benson, The Tetrazoles, Wiley, New York, 1967 Search PubMed.
- M. Uchida, M. Komatsu, S. Morita, T. Kanbe and K. Nakagawa, Chem. Pharm. Bull., 1989, 37, 322–326 CrossRef CAS PubMed.
- N. Fischer, T. M. Klapötke, J. Stierstorfer and C. Wiedemann, Polyhedron, 2011, 30, 2374–2386 CrossRef CAS.
- T. M. Klapötke, M. Stein and J. Stierstorfer, Z. Anorg. Allg. Chem., 2008, 634, 1711–1723 CrossRef.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Angew. Chem., Int. Ed., 2014, 53, 8172–8175 CrossRef CAS PubMed.
- N. Fischer, D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Mater. Chem., 2012, 22, 20418–20422 RSC.
-
H. G. Ang and S. Pisharath, Energetic Polymers: Binders and Plasticizers for Enhancing Performance, Wiley, Weinheim, 2012 Search PubMed.
-
G. Wuitschik, PhD thesis, ETH Zürich, 2008.
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS PubMed.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed.
- J. Stierstorfer, T. M. Klapötke, A. Hammerl and R. D. Chapman, Z. Anorg. Allg. Chem., 2008, 634, 1051–1057 CrossRef CAS.
-
M. C. H. Sabaté, PhD thesis, Ludwig-Maximilians-University, 2008 Search PubMed.
-
T. M. Klapötke, Chemistry of High-Energy Materials, DeGruyter, Berlin, 2019 Search PubMed.
- T. M. Klapötke, D. G. Piercey, N. Mehta, K. D. Oyler, M. Jorgensen, S. Lenahan, J. S. Salan, J. W. Fronabarger and M. D. Williams, Z. Anorg. Allg. Chem., 2013, 639, 681–688 CrossRef.
- M. McLaughlin, R. Yazaki, T. C. Fessard and E. M. Carreira, Org. Lett., 2014, 16, 4070–4073 CrossRef CAS PubMed.
- P. Luger and J. Buschmann, J. Am. Chem. Soc., 1984, 106, 7118–7121 CrossRef CAS.
-
A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, Appendix A: Typical Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals, In Struct. Correl., 1994, DOI:10.1002/9783527616091.app1, pp. 752–858.
- A. O. Martirosyan, M. V. Aleksanyan, S. S. Terzyan, Y. Z. Ter-Zakharyan, R. V. Agababyan, A. A. Karapetyan, S. L. Mndzhoyan and R. A. Tamazyan, Pharm. Chem. J., 2001, 35, 169–171 CrossRef CAS.
- I. N. Polyakova, V. V. Saraev, A. S. Gavrilov and E. L. Golod, Crystallogr. Rep., 2009, 54, 441–448 CrossRef CAS.
- F. Han, L. Yang, Z. Li and C. Xia, Org. Biomol. Chem., 2012, 10, 346–354 RSC.
- Y.-L. Lam, L. L. Koh and H. H. Huang, J. Chem. Soc., Perkin Trans. 2, 1993, 175–180, 10.1039/P29930000175.
- H. Fujihisa, K. Honda, S. Obata, H. Yamawaki, S. Takeya, Y. Gotoh and T. Matsunaga, CrystEngComm, 2011, 13, 99–102 RSC.
-
T. M. Klapötke, in Moderne Anorganische Chemie, ed. E. Riedel, DeGruyter, Berlin, 3rd edn, 2008, ch. 1, pp. 1–168 Search PubMed.
- J. Zhang, Q. Zhang, T. T. Vo, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 1697–1704 CrossRef CAS PubMed.
- D. E. Dosch, M. Reichel, M. Born, T. M. Klapötke and K. Karaghiosoff, Cryst. Growth Des., 2021, 21, 243–248 CrossRef CAS.
- M. Reichel, D. Dosch, T. Klapötke and K. Karaghiosoff, J. Am. Chem. Soc., 2019, 141, 19911–19916 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem. – Eur. J., 1998, 4, 2136–2141 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- Y. Ma, A. Zhang, X. Xue, D. Jiang, Y. Zhu and C. Zhang, Cryst. Growth Des., 2014, 14, 6101–6114 CrossRef CAS.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17 (2017), J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef PubMed.
-
T. M. Klapötke, Energetic Materials Encyclopedia, DeGruyter, Berlin, Boston, 2021 Search PubMed.
- T. Altenburg, Thomas M. Klapötke, A. Penger and J. Stierstorfer, Z. Anorg. Allg. Chem., 2010, 636, 463–471 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Rev. C.01), Gaussian Inc., Pittburgh, PA, 2016 Search PubMed.
- G. Krien, H. H. Licht and J. Zierath, Thermochim. Acta, 1973, 6, 465–472 CrossRef CAS.
- NATO Standardization Office, NATO STANAG 4489: Explosives. Impact Sensitivity Tests, 1999.
- NATO Standardization Office, NATO STANAG 4487: Explosives. Friction Sensitivity Tests, 2002.
- UN Recommendations on the Transport of Dangerous Goods. In Manual of Tests and Criteria, 2009.
-
M. Sućeska, EXPLO5 Version 6.04, Zagreb, 2017.
- T. M. Klapoetke, M. Born, T. Fessard, L. Göttermann, J. Stierstorfer and M. Voggenreiter, Chem. Commun., 2021, 57, 2804–2807 RSC.
-
J. E. Felts, H. W. Sandusky and R. H. Granholm, AIP Conference Proceedings 2009, American Institute of Physics, College Park, MD, Vol. 1195, pp. 233–236.
-
A. Homburg, J. Köhler and R. Meyer, Explosives, Wiley-VCH, Weinheim, 6th edn, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra (1H, 13C, 14N), Crystallographic information, DTA and DSC graphs, Hot-plate test of compounds 2–5 (video, hot plate test.mp4), Hot-needle test of compound 4 (video, hot needle test.mp4), Flame test of compound 4 (video, flame test.mp4). X-ray data for compounds 1 (CCDC: 2119618), 2A (CCDC: 2119623), 2B (CCDC: 2119795), 3A (CCDC: 2119619), 3B (CCDC: 2119620), 3C (CCDC: 2119621), 4 (CCDC: 2119622), 5 (CCDC: 2119624). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ma00088a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jakob
Plank
a and
Thomas M.
Klapötke
a,
Jakob
Plank
a and
Thomas M.
Klapötke
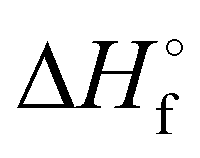 [kJ mol−1]
[kJ mol−1]