
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylthio-based asymmetric liquid crystals: unravelling the substituent effects and intercalated cybotactic nematic and smectic phases†
Yuki
Arakawa
 *a,
Yuko
Ishida
a,
Yukito
Sasaki
a,
Shunsuke
Sasaki
b,
Masatoshi
Tokita
c and
Hideto
Tsuji
a
*a,
Yuko
Ishida
a,
Yukito
Sasaki
a,
Shunsuke
Sasaki
b,
Masatoshi
Tokita
c and
Hideto
Tsuji
a
aDepartment of Applied Chemistry and Life Science, Graduate School of Engineering, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi, Aichi 441-8580, Japan. E-mail: arakawa@tut.jp
bUniversité de Nantes, CNRS, Institut des Matériaux Jean Rouxel, IMN, F-44000 Nantes, France
cDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1, Ookayama, Meguro-ku, Tokyo 152-8550, Japan
First published on 4th March 2022
Abstract
To reveal not only the effects of alkylthio groups but also an unknown layered smectic (Sm) phase formed by short-chain-based liquid crystals (LCs), a novel series of asymmetric 1,4-phenylene bis(benzoate)s possessing 4,4′-alkylthio and alkoxy groups with various chain lengths (SOn, n = 1–8) was established. All members formed well-defined enantiotropic LC phases. The SOn homologs with n = 2–7 exhibited only a nematic (N) phase, and the longest SO8 showed an inclined smectic C (SmC) phase in addition to the N phase. Notably, the shortest SO1 formed an enantiotropic orthogonal smectic A (SmA) phase as well as the N phase. The primary effects of the alkylthio and alkoxy groups were identified for each phase transition over varied chain lengths and through comparison with those of their symmetric alkoxy- and alkylthio-based counterparts (OOn and SSn, respectively). In addition, X-ray diffractometry unraveled surprising results, wherein the N and SmA phases of SO1 were found to possess a layer period that was half of the molecular length, thereby suggesting half-intercalation of the intermolecular cores. These phases are distinct from the conventional N and Sm phases without such core intercalation. The remarkable formation of half-intercalated LC phase was discussed based on plausible intermolecular interactions (e.g., multiple hydrogen bonds due to the presence of esters) implied by single-crystal structural analyses. Furthermore, the birefringence increased when replacing the alkoxy group with the more polarizable alkylthio group.
1. Introduction
Liquid crystals (LCs), which are anisotropic and fluidic mesophases with properties between three-dimensionally ordered solid crystals and isotropic liquids, have enabled the widespread development of LC displays (LCDs) in the last few decades. Moreover, the self-assembly and stimuli-responsive properties of LCs have attracted much attention from various interdisciplinary fields beyond those involving LCDs. The fundamental properties of LCs, including their phase transition behaviors as well as their optical and electronic properties, primarily depend on their mesogenic core structures. Typically, chain-containing substituents such as alkyl and alkoxy groups are appended to the mesogenic core structures to effectively induce LC phases and confer flexibility onto the LC molecules, thereby improving their molecular affinity and miscibility while also lowering their phase transition temperatures. In addition, the elements present in these substituents and their bonds connecting to the mesogenic cores (such as methylene and oxygen for alkyl and ether, respectively), as well as their alkyl chain lengths, also significantly affect the properties of LCs and are therefore crucial in related molecular design.In addition to conventional alkyl and alkoxy groups, alkylthio (i.e., alkylsulfanyl or thioether: R–S–R) groups are also useful in LC materials. Because the sulfur atom is heavier and more electron rich than the commonly used carbon and oxygen atoms, it endows LC materials with a higher polarizability, larger dispersion forces, and elevated highest occupied molecular orbitals (HOMO). Thus, alkylthio-based LCs are used as high-birefringence materials1–8 and organic LC semiconductors with high charge mobilities.9–14 In particular, the alkylthio group is often employed in the molecular design of discotic LCs,9,12–26 since the intermolecular interactions of the more polarizable sulfur atoms help induce and stabilize the columnar (Col) phases in discotic molecules.22,23 In addition, the alkylthio group has greater flexibility and bulkiness, in addition to a lower structural anisotropy, compared with the alkyl and alkoxy groups. These properties arise from the longer C–S bond lengths (∼1.7 Å) and the smaller C–S–C bond angles (∼100°) than those of alkyl and alkoxy groups (e.g., ∼1.5 and ∼1.4 Å for the C–C and C–O bond lengths; and ∼110° and 118° for the C–CH2–C and C–O–C bond angles, respectively). Such steric effects of the alkylthio groups have benefits in inducing not only the above-mentioned Col phase,22,23 but also the cubic phase,27 which could effectively fill large spaces around the columns and gyroids in terms of free volume effects.28
In contrast, the greater flexibility and steric bulkiness imparted by the C–S–C bonds prevent rod-like molecules from rearranging into LC phases and reduce the LC temperature ranges. As a result, compared to their alkyl- and alkoxy-based counterparts, alkylthio-containing rod-like molecules usually exhibit LC phases monotropically, only across a narrow temperature range, or not at all.1–5,29,34–36 On the other hand, enantiotropic LC phases were observed from some types of alkylthio-based calamitic molecules.4–8,29–40 However, there has been no report of covalent-bonded homologous series in which all members exhibit enantiotropic LC phases, while only one such hydrogen-bonded homologous series is known.35 The lack of dataset of more versatile covalent-bonded homologous series showing the enantiotropic LC phases allowed us only to infer the effects of alkylthio groups on LC behaviours based on few of its homologous series. For example, as typical LC motifs, 1,4-phenylene bis(benzoate) (PBB) homologs41–48 possessing symmetric alkoxy-groups (OOn, Fig. 1) are known to display well-defined enantiotropic LC phases.41–46 In addition, we reported the phase transition behavior of PBB homologs with symmetric alkylthio groups (SSn, Fig. 1).34 SSn with n = 1, 2, or 6 exhibit enantiotropic LC phases with narrower temperature ranges, whereas those with n = 3–5, 7, or 8 show monotropic N phases. Thus, SSn typically exhibits poor mesogenic ability compared to OOn. In the paper,34 we noted that the shortest chain SS1 homolog forms layered Sm phase, which is unusual and counter to the general trend where longer chains facilitate the formation of layered Sm phases due to enhanced microphase separation. However, because of its elusive monotropic nature with a narrow temperature range, the structure of this abnormal Sm phase for SS1 could not be further investigated. Moreover, although such Sm phases were also reported for other molecules with short chains,44,45,49 their structures have also remained unknown to date.
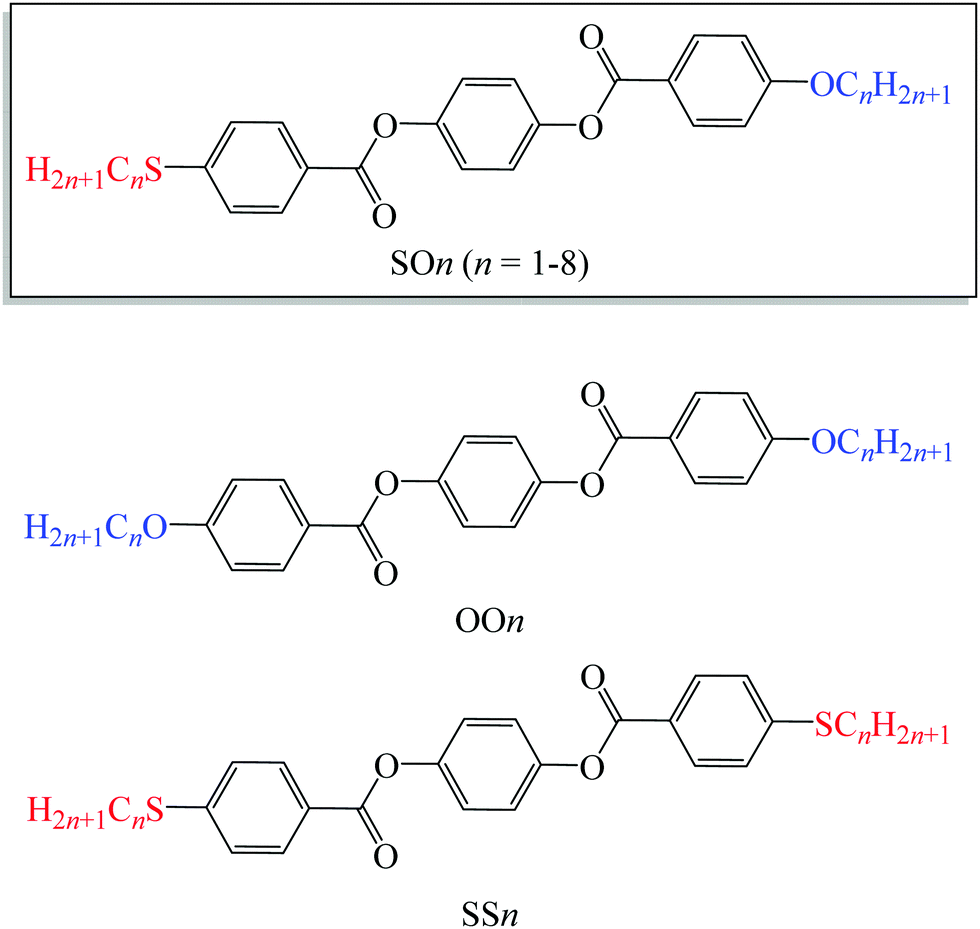 | ||
| Fig. 1 Molecular structures of LC homologs bearing alkylthio and alkoxy groups (SOn) and previously reported alkoxy- (OOn) and alkylthio-based (SSn) LC homologs. | ||
To realize a homologous series in which all members exhibit well-defined enantiotropic LC phases, an alkylthio-appended highly anisotropic structure such as PBB should be combined with an asymmetric substituent such as an alkoxy and alkyl group at the other end. The asymmetric substitution of alkylthio and alkoxy groups is expected to expand the LC temperature ranges beyond those found in the symmetric SSn homologs, enabling us to investigate not only the properties of alkylthio-based LCs but also the enigmatic Sm phases formed by the short-chain molecules. In addition, this raises the questions how the alkylthio and alkoxy (or alkyl) groups affect the overall phase transition temperature, and which structure has a dominant effect when multiple structures coexist in molecular homologs. Currently, there is only one published report on alkylthio- and alkyl-substituted asymmetric diphenyl-acetylene-based LC homologs, some of which show only monotropic LC phases.33 To the best of our knowledge, however, no asymmetric LC homologous series bearing alkylthio and alkoxy (or alkyl) groups in which all members exhibit LC phases have been established. Therefore, the competing substituent effects have yet to be revealed for such homologous series.
In these contexts, we herein report a novel homologous series of asymmetric PBB homologs possessing alkylthio and alkoxy groups with various alkyl chain lengths (CnH2n+1) at the 4,4′-positions (SOn, n = 1–8, Fig. 1). Their phase transition behavior, mesophase structures, and birefringence properties are investigated50 and compared with those of previously reported symmetric alkoxy- and alkylthio-based homologs (OOn and SSn, respectively, Fig. 1). In addition, competing substituent effects on the LC properties between the alkylthio and alkoxy groups are discussed in detail upon varying the chain lengths. Furthermore, the structure of the unknown Sm phase is revealed for short-chain molecules.
2. Experimental
2.1. Synthesis
All SOn homologs were prepared as outlined in Scheme S1, using the general procedures described in the ESI.† The molecular structures were determined using 1H and 13C nuclear magnetic resonance (NMR) spectroscopy on a JNM-ECX500 (500 MHz for 1H and 126 MHz for 13C) or a JNM-ECS400 (400 MHz for 1H and 100 MHz for 13C) spectrometer (JEOL Ltd., Tokyo, Japan). All NMR spectra were obtained using solutions of the samples in deuterated chloroform with a tetramethylsilane internal standard (Kanto Chemical Co., Tokyo, Japan). High-resolution mass spectrometry (HRMS) was also conducted using high-performance liquid chromatography (HPLC) (Agilent 1200 HPLC-Chip and 6520 Accurate-Mass Q-TOF). The molecular characterization data are provided in the ESI.†2.2. Phase identification and thermal properties
Phase identification and analysis of the phase transition behavior were performed using polarized-light optical microscopy (POM; Olympus BX50) with a Linkam LK-600PM hot stage. The phase transition temperatures and the associated enthalpy changes were determined using differential scanning calorimetry (DSC, Shimadzu DSC-60 instrument). The heating and cooling cycles were performed at a rate of 3 °C min−1 under an N2 flow (50 mL min−1). In addition, thermal stability of SO1 was evaluated by thermogravimetry analysis (TGA, Shimadzu, TGA-50 instrument) at a rate of 3 °C min−1 under an N2 flow (50 mL min−1). All POM images and DSC and TGA curves, except those presented in the main text, are shown in the ESI.†2.3. X-ray diffraction measurements
The mesophase structures of SOn (n = 1, 6, and 8) were investigated using X-ray diffractometry (XRD, Bruker D8 DISCOVER diffractometer) with a Cu Kα X-ray source and a Vantec-500 detector. The specimens were stored in glass capillary tubes (diameter: 1.5 mm; WJM-Glass Müller GmbH) and sandwiched between a pair of permanent magnets in a folder. After heating to the isotropic (Iso) phase temperature to erase the thermal histories of the samples, the measurements were carried out at different mesophase temperatures upon cooling.2.4. Birefringence measurements
The birefringence (Δn) of SO6 was measured according to our previous study51 for comparison with those of OO6 and SS6.34 SO6 was inserted into a uniaxially rubbed planer alignment cell (with a polyimide surface and a thickness of 5 μm, EHC Co., Tokyo, Japan) by capillary action at its Iso temperature (controlled by a Mettler FP82HT hot stage). After filling the cell with SO6, the uniaxial alignment state of the N phase was confirmed by POM using a Nikon LV100 Pol optical microscope (Tokyo, Japan). Transmittance through the nematic SO6 medium with the N director at 45° under the cross-Nicol condition was measured using a USB4000 spectrometer (Ocean Insight, Orlando, FL, USA) through the Nikon LV100 Pol optical microscope. The transmittance was obtained upon cooling from the Iso phase temperature, while the temperature was controlled using a Mettler FP82HT hot stage. The transmittance (T) was fitted to eqn (1) along with Cauchy's equation, eqn (2):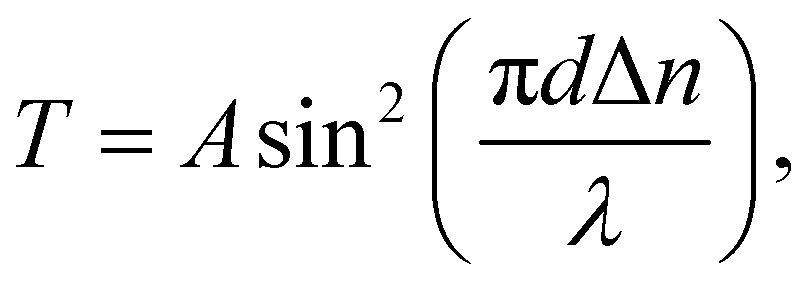 | (1) |
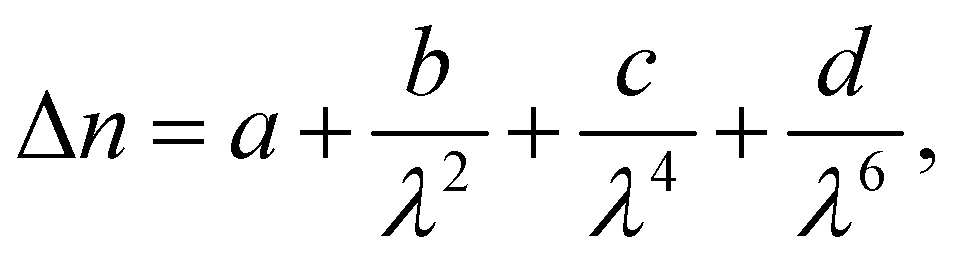 | (2) |
3. Results and discussion
3.1. Phase transition behavior
| n | Phase | T/°C | ΔH/kJ mol−1 | Phase | T/°C | ΔH/kJ mol−1 | Phase | T/°C | ΔH/kJ mol−1 | Phase |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cr | 170.3 | 18.1 | SmA | 194.6 | 4.2 | N | 283.0 | 1.5 | Iso |
| 153.7 | 12.7 | 193.6 | 4.2 | 281.5 | 1.6 | |||||
| 2 | Cr | 216.8 | 40.2 | — | N | 259.3 | 1.7 | Iso | ||
| 207.2 | 40.7 | 257.0 | 1.8 | |||||||
| 3 | Cr | 181.9 | 31.2 | — | N | 214.6 | 1.4 | Iso | ||
| 177.3 | 30.6 | 213.9 | 1.4 | |||||||
| 4 | Cr | 158.4 | 32.9 | — | N | 210.0 | 1.9 | Iso | ||
| 153.9 | 33.0 | 209.0 | 1.9 | |||||||
| 5 | Cr | 146.6 | 33.6 | — | N | 189.0 | 1.3 | Iso | ||
| 140.2 | 33.7 | 188.2 | 1.3 | |||||||
| 6 | Cr | 130.3 | 30.2 | — | N | 185.1 | 1.6 | Iso | ||
| 124.2 | 29.4 | 184.1 | 1.6 | |||||||
| 7 | Cr | 129.0 | 34.6 | — | N | 174.2 | 1.3 | Iso | ||
| 123.3 | 34.2 | 173.4 | 1.4 | |||||||
| 8 | Cr | 122.9 | 31.3 | SmC | 127.5 | 2.4 | N | 171.6 | 1.7 | Iso |
| 113.5 | 30.0 | 126.9 | 2.4 | 170.6 | 1.7 |
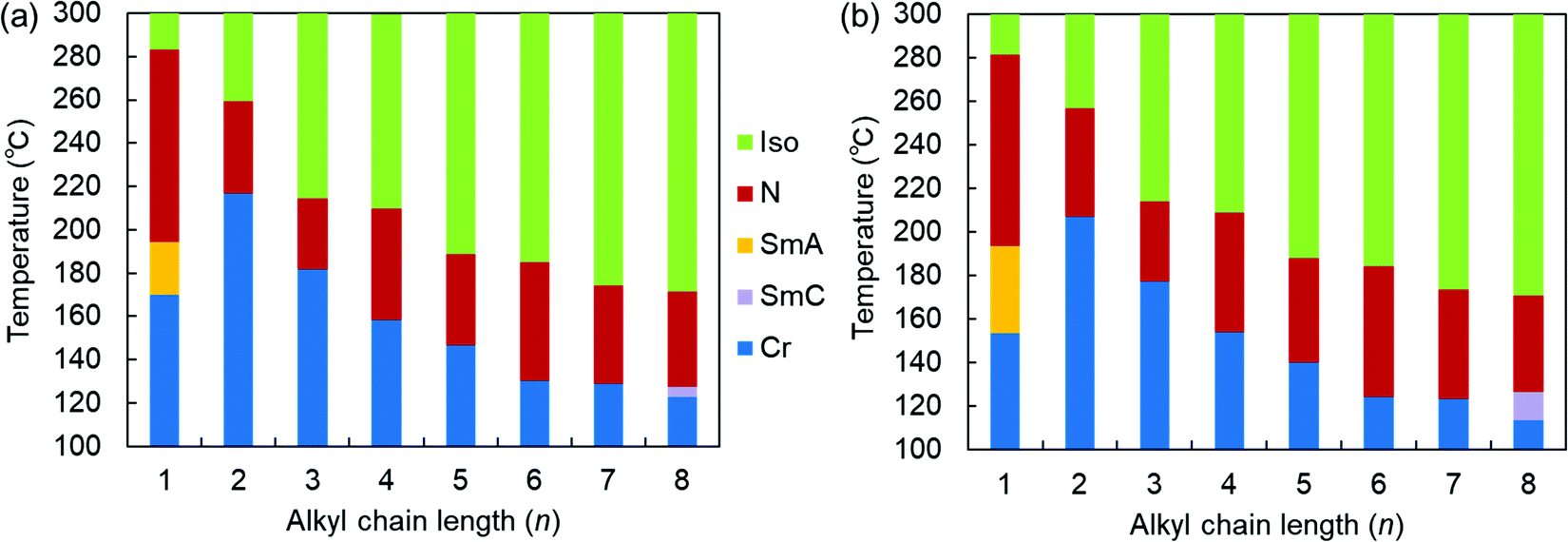 | ||
| Fig. 2 Phase sequences of the SOn homologs as a function of n upon (a) heating and (b) cooling at a rate of 3 °C min−1. | ||
More specifically, SOn with n = 2–7 exhibited only enantiotropic N phases, whereas the longest-chain homolog, SO8, exhibited an enantiotropic tilted layered smectic C (SmC) phase as well as the enantiotropic N phase. The formation of SmC phase is a consequence of microphase separation between the aliphatic chains and the aromatic cores, which is facilitated by longer alkyl chains. POM observations of SO8 upon cooling revealed a streak-like second schlieren texture in the N phase [Fig. S8(b) in the ESI†], which is typical of an NCyb phase containing SmC-like clusters (i.e., a NCybC phase).52–54 These phases were also found for the SS6 and OO6 homologs.34 During the transition from the N to the SmC phase upon cooling, a transition bar was observed and a less fluid schlieren texture with four-brush singularity appeared, indicating a homeotropic alignment in the tilted SmC phase [Fig. S8(c) and (d), ESI†]. This schlieren texture in the SmC phase partly appeared along the streak lines in the NCybC phase. The enantiotropic SmC–N phase transition of SO8 is considered a first-order transition, as shown in the DSC curves (Fig. S15, ESI†).
Remarkably, the shortest-chain homolog (SO1) enantiotropically exhibited not only an N phase but also a layered orthogonal SmA phase [Fig. 3(a) and (b), respectively]. Fig. 3(b) shows a fan-shaped texture typical to Sm phases, and this texture grew from a bâtonnet texture. The formation of a layered SmA phase in SO1 is unusual, because the formation of Sm layered structures is typically facilitated by long alkyl chains due to enhanced microphase separation, as exemplified by SO8. Nevertheless, similar abnormal Sm phase formation was previously reported for other PBB analogs bearing short methyl and ethyl chains (abbreviated as PBB-1 and PBB-2 in the present study)44,45 and PBB-S1 (namely SS1 in the present paper).34 These phases were reported to be monotropic SmA phases in very narrow temperature ranges according to POM observations. In contrast, the present SO1 homolog exhibited an enantiotropic SmA phase within a practical temperature range, and so it was further investigated by XRD (see Section 3.2.2). The DSC curves [Fig. 3(c)] show that the SmA–N transition of SO1 is strongly first order, with an enthalpy change of ΔH = 4.2 kJ mol−1, which is similar to those of previously reported analogues PBB-1 and SS1.34,44 This first-order transition likely differs from the typical second-order nature of SmA–N transitions. SO1 exhibited different melting temperature (Tm) values during the 1st and 2nd heating [184.1 and 170.3 °C, respectively, Fig. 3(c)] due to the presence of crystalline polymorphs; the latter is listed in Table 1. Furthermore, because of the high-temperature LC phase over 250 °C, thermal stability of SO1 was examined by TGA, and the obtained TGA curve is shown in Fig. S16 (ESI†). Slight weight loss or thermal decomposition started over 250 °C and the weight loss at 290 °C was within 5%. Slight thermal decomposition of the sample must have influenced the phase transition behavior and related analyses but these could not be critical.
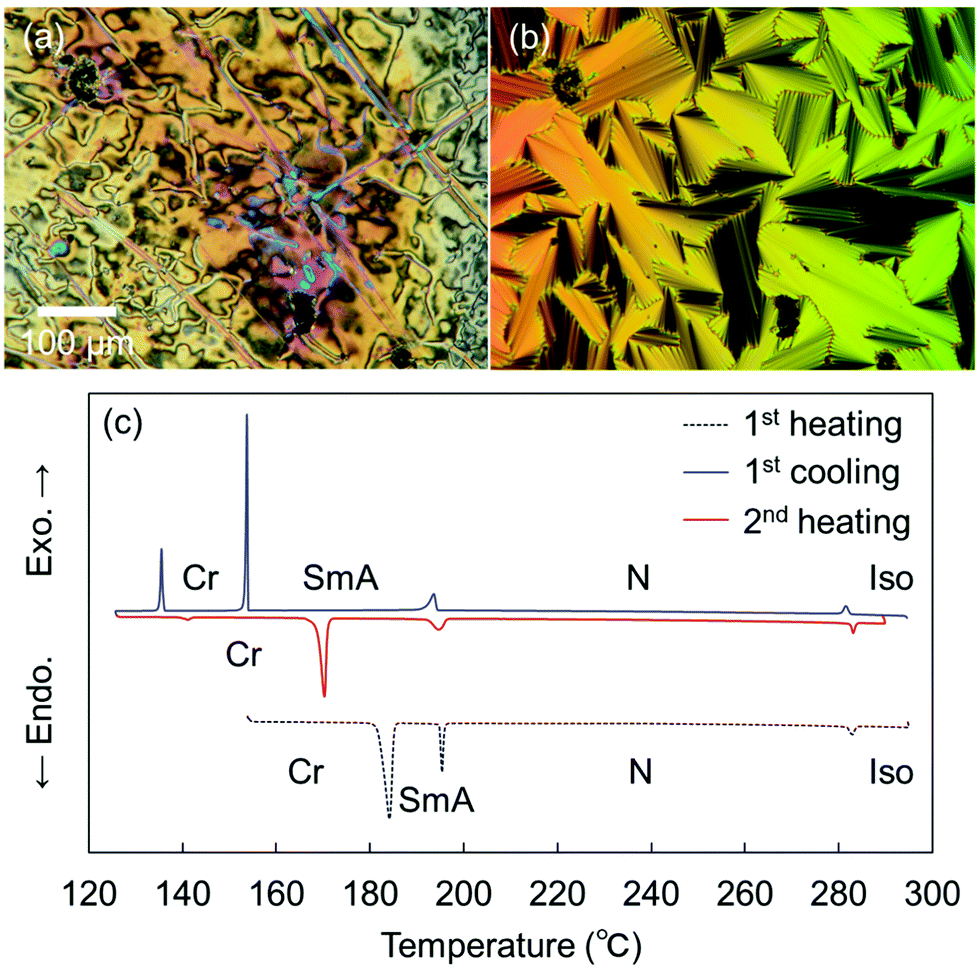 | ||
| Fig. 3 POM images of SO1 (a) in the nematic (N) phase at 195 °C and (b) in the SmA phase at 190 °C, upon cooling, and (c) DSC curves of SO1 collected at a rate of 3 °C min−1. | ||
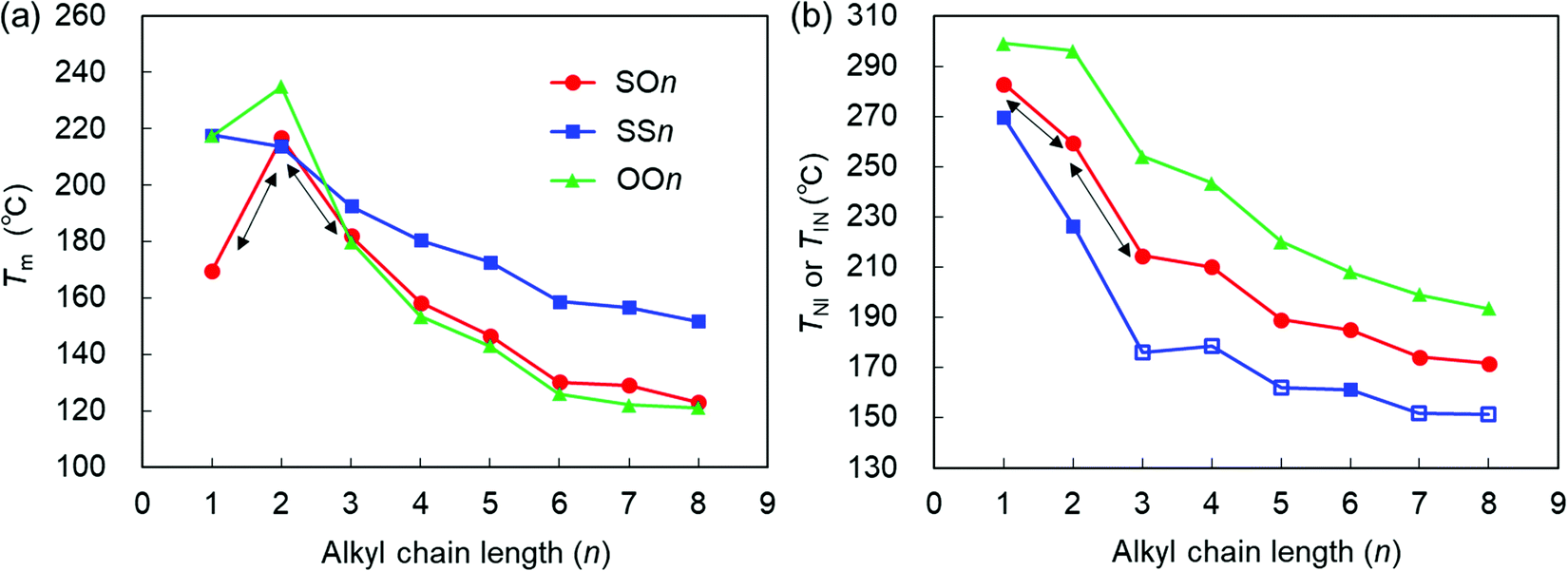 | ||
| Fig. 4 (a) Tm and (b) TNI (filled symbols) or TIN (open symbols) of the SOn, SSn, and OOn homologs (n = 1–8) upon heating (10 °C min−1) as a function of n. The data for OOn45 and SSn34 are drawn from the literature. | ||
Here, it should be emphasized that the Tm values of the asymmetric SOn homologs (n = 1–3) exhibit a typical odd–even oscillation, as represented by the black arrows in Fig. 4(a). Interestingly, this odd–even oscillation of SOn is ascribed to the parity of the alkoxy chains at the other end. This provides an insight into the competing effects between the alkylthio and alkoxy groups on the value of Tm. In short-chain homologs (n = 1–3), the conformational flexibility and steric effects of the alkylthio group at one end did not outweigh the effect of the alkoxy group at the other end of the molecule, which indicates that molecular shape anisotropy also significantly affected the Tm and elevated that of SO2. The Tm values of the homologs with n > 3 decreased in the order SSn > SOn > OOn. In other words, Tm increases upon replacing the alkoxy group with an alkylthio group. More specifically, the symmetric alkylthio-based SSn homologs possess significantly higher Tm values than both the SOn and OOn homologs, which is ascribed to the stronger dispersion forces brought by the two symmetric, highly polarizable alkylthio groups. This interaction effect becomes more pronounced for longer alkyl chains, which could be assisted by enhanced microphase separation or molecular affinities [Fig. 4(a)],36,40 resulting in significantly higher Tm values for molecules containing alkylthio groups than their alkoxy counterparts. Nonetheless, the Tm values of SOn are only marginally higher than those of OOn, which is ascribable to two competing effects. Although the dispersion forces provided by the alkylthio group elevates Tm, this effect is canceled out by asymmetric substitution or molecular inhomogeneity that tends to suppress it.
The TNI or TIN values of the three homologous series decrease in the order OOn > SOn > SSn, which implies that they decrease upon substituting the alkoxy group with an alkylthio group [Fig. 4(b)]. This trend seems contradictory to the stronger dispersion force expected for alkylthio groups. The stronger dispersion force of the more polarizable sulfur atom makes a larger contribution to increase the phase transition temperatures than that from the oxygen atom, as observed in the Tm values. Therefore, there must be other factors that explain lowering of TNI or TIN upon replacement the alkoxy with alkylthio groups. One of the possible rationales is the steric effects of alkylthio groups such as their greater conformational flexibility, bulkiness, and lower molecular shape anisotropy.
Upon increasing n from 1 to 2, the TNI values of SSn, SOn, and OOn decrease by approximately 43, 22, and 3 °C, respectively, indicating that the magnitude of reduction in TNI increases upon replacing the alkoxy group with the alkylthio group. This is due to the greater influence from the increased intrinsic conformational and steric effects of the alkylthio groups when lengthening the alkyl chains by one carbon atom, compared to the case of alkoxy groups. Overall, the TNI or TIN values of all three series of homologs decreased with increasing n, and their LC phase temperature ranges decreased in the order OOn > SOn > SSn upon substituting the alkoxy group with the alkylthio group.
Interestingly, the trend of TNI or TIN (OOn > SOn > SSn) is the reverse of that of their Tm values (SSn > SOn > OOn for n ≥ 3), as shown in Fig. 4. This result supports the view that the melting point of the solids is more likely to be influenced by the relative polarizability or attractive interactions imparted by the substituents, whereas the TNI or TIN values are primarily determined by the conformational and steric effects of the substituents rather than their attractive interactions. In addition, it should be emphasized that for the short chains (n = 1–3), in contrast to the case where the Tm of the asymmetric SOn homologs exhibit an odd–even oscillation, their TNI values show monotonous changes (i.e., a decrease or increase), as represented by the black arrows in Fig. 4. This suggests that the conformational flexibility and bulkiness effects greatly impact the value of TNI. For example, when alkylthio bonds are under steric constraints (e.g., by incorporation into ring structures), sulfur-based rings lead to higher Tm and TNI or TIN values, in addition to enhanced LC phase stability compared with the oxygen-based counterparts, although they likely exhibit a lesser degree of molecular anisotropy than their ether-based counterparts.57 On the other hand, in the case of side-chain LC polymers, the presence of a longer alkylthio spacer renders an alkoxy-based smectogenic azobenzene polymer exclusively nematogenic and drastically decreases its phase transition temperatures.40 These facts can also be interpreted by the significant roles of the flexibility and bulkiness imparted by the C–S–C bonds.
Among the homologs bearing longer alkyl chains, the SmC phase was observed in OO742,45,46 and SO8, but not in SS8,34 thereby indicating that the alkylthio group hindered the formation of an Sm phase in SSn. The alkylthio group can either induce the formation of a highly ordered phase due to the enhanced intermolecular interactions,4,27,40 or hinder the Sm phase formation through the greater conformational and steric effects.30,34 These trends are counterbalanced by the anisotropy of the mesogenic core structures and the alkyl chains. For larger anisotropic mesogenic cores such as the present PBBs that tend to form N phases, formation of the Sm phase was found to be inhibited at the tested chain lengths (n = 1–8). For relatively small mesogenic core structures, the formation of a highly ordered Sm phase is facilitated by the alkylthio groups.4,30,33,40,58,59
3.2. Liquid crystal phase structures
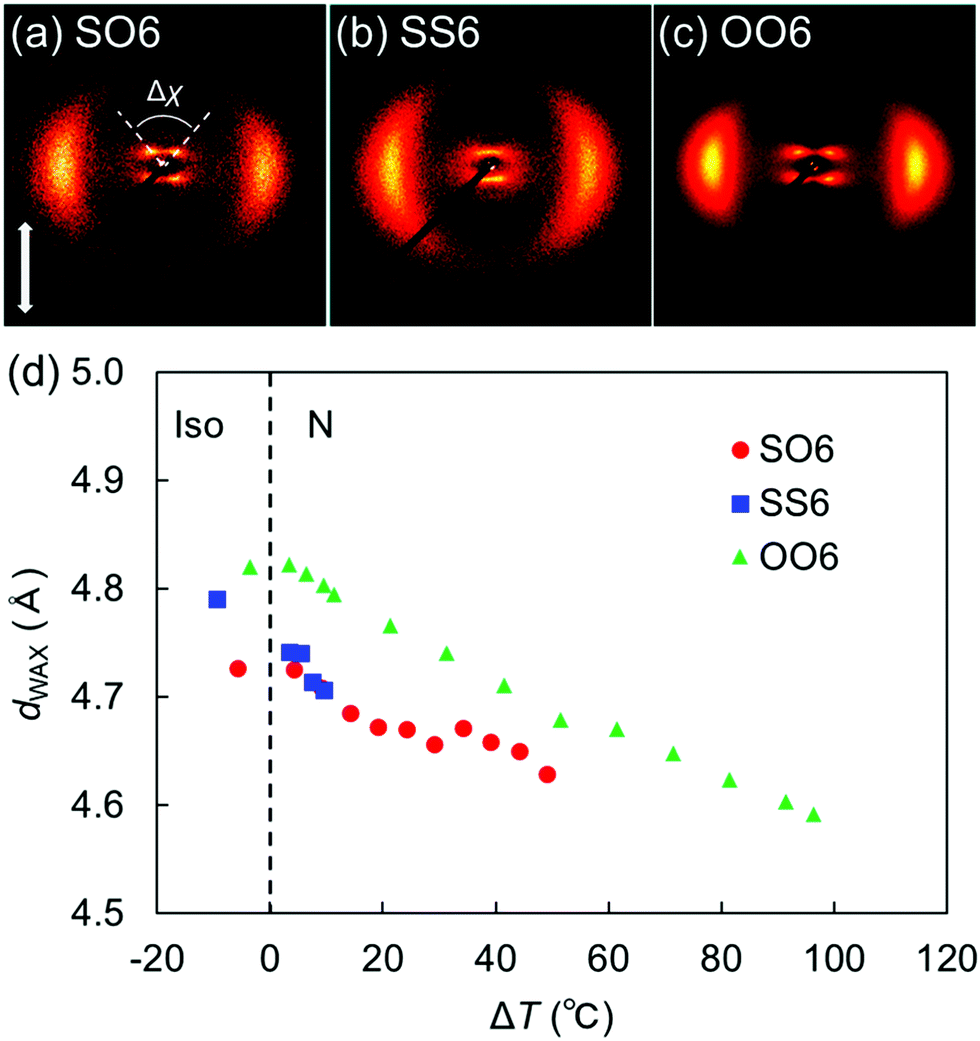 | ||
| Fig. 5 2D XRD patterns of the N phases of (a) SO6, (b) SS6, and (c) OO6. Panels (b) and (c) were adapted from our previous work.34 (d) Lateral intermolecular distances (dWAX) in SO6, SS6, and OO6 as a function of the shifted temperature (ΔT = TIN − T). The data for SS6 and OO6 were taken from our previous work.34 Plots for ΔT < 0 denote temperatures in each Iso phase. | ||
Fig. 5(d) plots the d-spacing values from the wide-angle diffractions (dWAX, representing the average lateral intermolecular distance) of SO6, SS6, and OO6 as a function of the shifted temperature, ΔT = TIN −T, where T is the measurement temperature upon cooling from the Iso phase. The dWAX values of all homologs monotonically decreased with decreasing temperature. In both their Iso and N phases, SO6 and SS6 exhibited similar dWAX values, which were smaller than those of OO6. This is consistent with the typical trend that alkylthio-based molecules in the LC and Iso phases usually possess smaller dWAX values than their alkoxy counterparts.4,30,34 This trend is ascribable to two factors, namely the larger dispersion forces of the alkylthio groups and the stronger electron-donating properties of the alkoxy groups. The former could lead to smaller lateral molecular distances, whereas the latter might generate repulsive forces between the mesogenic cores, which might in turn lead to larger distances between the alkoxy-based molecules (i.e., OO6) than between the alkylthio-containing molecules (i.e., SO6 and SS6). The dispersion forces from the sulfur atoms in SO6 contribute to its intermolecular packing in random head-to-tail and tail-to-head orientations.36 In addition, the orientational order parameters (S) were estimated for SO6, OO6, and SS6 from their wide-angle diffractions (Fig. S17, ESI†) according to the literature.60 When compared for a similar value of ΔT, the S values of SO6 were estimated to be higher than those of SS6 and OO6, and that of OO6 was found to be the lowest. The lower S values for SS6 than for SO6 could be due to its significantly narrower N phase temperature range, which is close to its Iso phase region.
The split peak maxima of the small-angle diffraction in the NCybC phase of SO6 were located at 3.7–4.0°, and the corresponding d-spacing (dSAX) was ∼23 Å. In addition, the half values of the angular splitting of the small-angle scattering maxima (Δχ/2) were estimated as shown in Fig. 5(a). The Δχ/2 value of SO6 in the NCybC phase was estimated to be ∼46–48°. The apparent molecular length (L) in the NCyb layers of SO6 was found to be ∼33–34 Å by d = L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosβ, assuming d (layer spacing) = dSAX and β (tilt angle) = Δχ/2. The estimated L values are similar to the molecular length of SO6 (i.e., ∼32 Å). This suggests that the Δχ/2 of SO6 may reflect the tilt angle in the SmC-like layer clusters, as well as that SO6 molecules would not intercalate among the neighboring SmC-like layer clusters [Fig. 6(d)]. The Δχ/2 values in the NCybC phase of SO6 (∼46–48°) are between those of SS6 and OO6 (∼40° and ∼53°, respectively).34 This means that the Δχ/2 values decrease upon displacing the alkoxy groups with alkylthio groups. This trend can be interpreted by molecular tilting in the SmC-like layer clusters. The larger dispersion forces of the sulfur atoms at the edge of mesogenic cores enhance lateral packing of the mesogenic core moieties, in combination with the dominant CH⋯π and π⋯π interactions among the cores to decrease the tilt angles and be toward non-tilted SmA-like clusters.4,30,34,36,40
cosβ, assuming d (layer spacing) = dSAX and β (tilt angle) = Δχ/2. The estimated L values are similar to the molecular length of SO6 (i.e., ∼32 Å). This suggests that the Δχ/2 of SO6 may reflect the tilt angle in the SmC-like layer clusters, as well as that SO6 molecules would not intercalate among the neighboring SmC-like layer clusters [Fig. 6(d)]. The Δχ/2 values in the NCybC phase of SO6 (∼46–48°) are between those of SS6 and OO6 (∼40° and ∼53°, respectively).34 This means that the Δχ/2 values decrease upon displacing the alkoxy groups with alkylthio groups. This trend can be interpreted by molecular tilting in the SmC-like layer clusters. The larger dispersion forces of the sulfur atoms at the edge of mesogenic cores enhance lateral packing of the mesogenic core moieties, in combination with the dominant CH⋯π and π⋯π interactions among the cores to decrease the tilt angles and be toward non-tilted SmA-like clusters.4,30,34,36,40
Subsequently, the 2D XRD patterns of the N and SmC phases of SO8 were obtained (Fig. 6). In this case, broad wide-angle peaks at 2θ = 19.2° (dWAX = 4.6 Å) and dumbbell-like split small-angle diffractions at 3.3° (dSAX = 26.6 Å) were observed at 140 °C [Fig. 6(a)], indicating the tilted NCybC phase. The Δχ/2 value or tilt angle was estimated to be 43°, similar to that of SO6. The NCybC–SmC phase transition occurred during X-ray irradiation of the NCybC phase at 130 °C [Fig. 6(b)]. As a result, two small-angle diffractions from the SmC layers appeared on the meridional line, in addition to the dumbbell-shaped diffractions from the NCybC layers. The small-angle diffraction of the SmC layers at 2θ = 3.3° corresponds to dSAX = 26.7 Å, which equals that of the pure SmC phase at 120 °C [Fig. 6(c)]. The SmC layer was mostly parallel to the equatorial line, and the molecules inclined from the layer normal or the meridional line. The tilt angle of the SmC phase at 120 °C was ∼20°, almost half the value of the SmC-like cybotactic clusters in the NCybC phase at 140 °C (43°). In addition, the dSAX of the SmC phase (∼27 Å) was less than the molecular length of 35 Å. Therefore, it was assumed that the alkyl chains intercalated into neighboring layers of the SmC phase [Fig. 6(d)]. Although the detailed layer structures of the NCybC and SmC phases differ somewhat from each other in terms of the tilt angles and intercalation, the structural similarities of their inclined LC phases indicate that Sm-like clusters in the upper NCybC phases likely influence the development of subsequent SmC phase structures.4,61
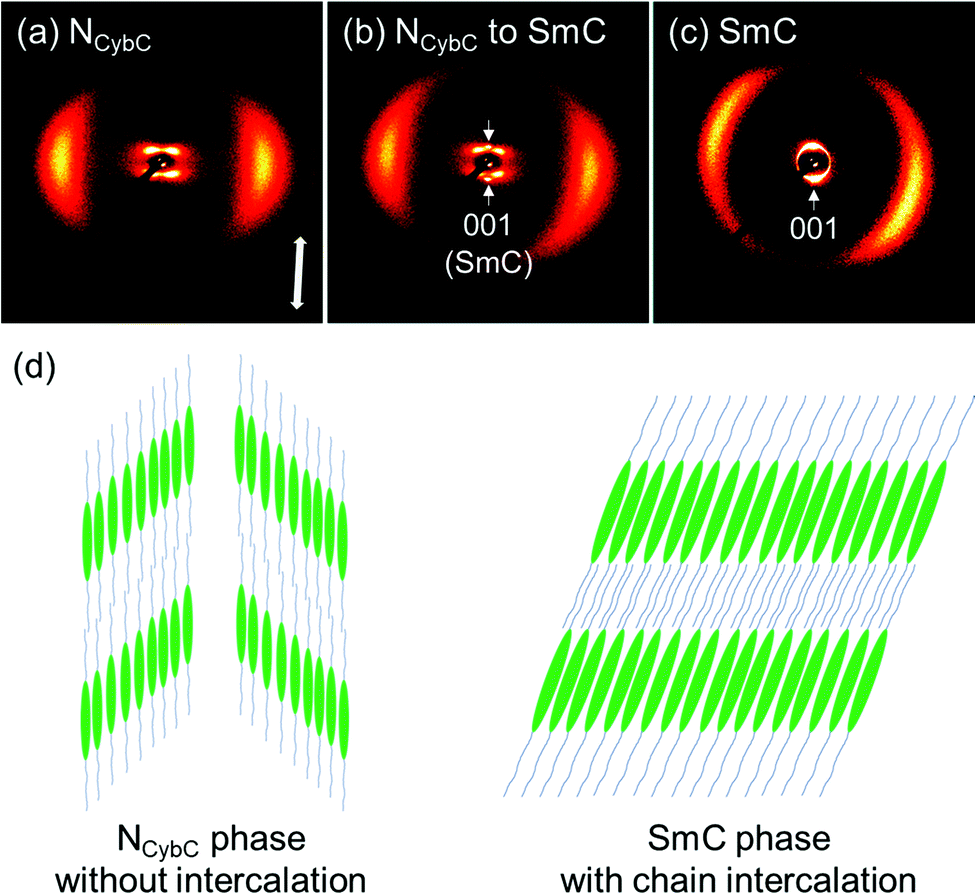 | ||
| Fig. 6 2D XRD patterns of SO8 (a) in the cybotactic N (Ncyb) phase with a tilted SmC cluster (NCybC phase) at 140 °C, (b) during the N–SmC phase transition at 130 °C, and (c) in the SmC phase at 120 °C. (d) Schematic packing models of the NCybC and SmC phases. | ||
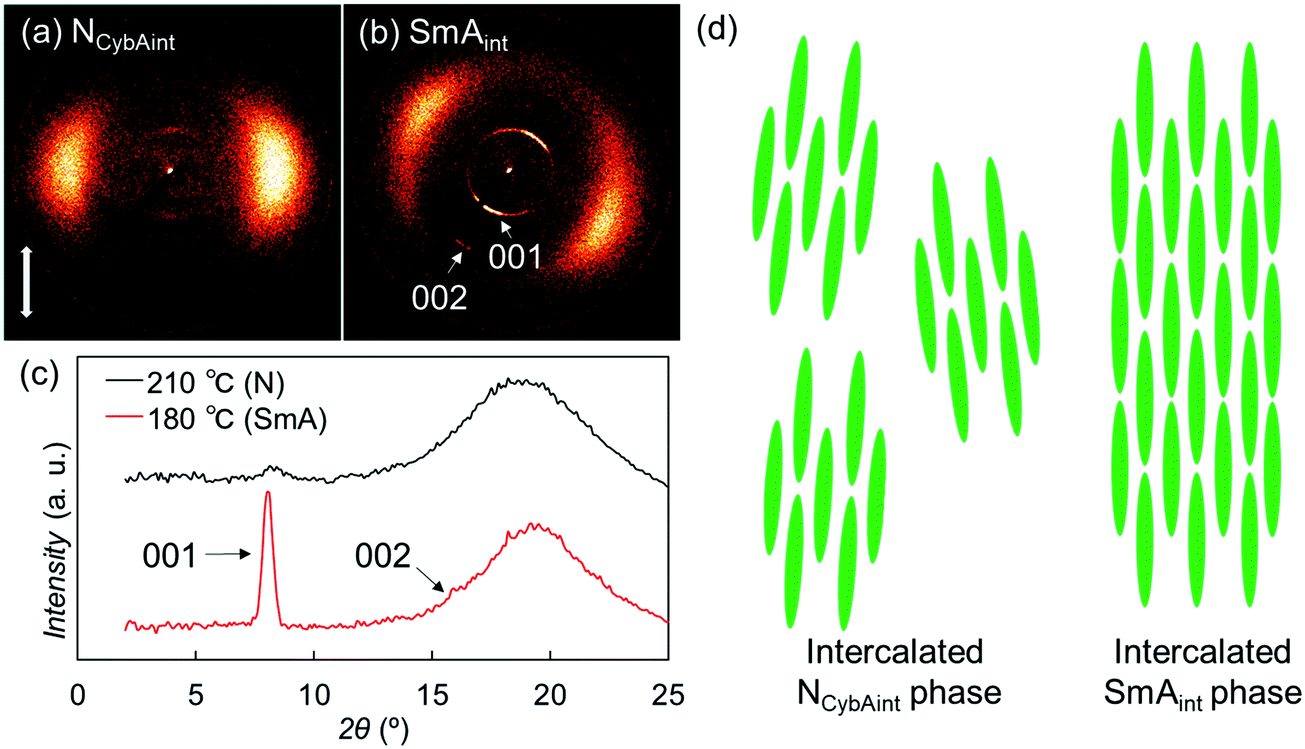 | ||
| Fig. 7 2D XRD patterns of SO1 (a) in the N phase at 210 °C and (b) in the SmA phase at 180 °C. (c) The corresponding 1D XRD profiles. (d) Schematic representation of the possible packing models of the NCyb phase with an SmA-like cluster (NCybAint phase) and the intercalated SmA (SmAint) phase of SO1. | ||
For the Sm phase, broadened wide-angle and sharp small-angle diffractions were detected at 2θ = 19.3° (dWAX = 4.6 Å) and 8.0° (dSAX = 11 Å) [Fig. 7(b) and (c)]. Surprisingly, the value dSAX = 11 Å is also almost half the molecular length, suggesting the presence of intercalated layer structures similar to those of the upper NCybAint phase. In addition, the second-order diffraction peak of the layer structure was observed at 2θ = 16.0°. The relatively broad wide-angle diffractions indicate the fluidity of the Sm phase and the lateral liquid-like correlations of molecules in the Sm layers, which identify this phase of SO1 as an intercalated SmA (SmAint) phase [Fig. 7(d)].62 The small-angle diffractions at 2θ = 8.0° and 16.0° were indexed as 001 and 002 reflections [Fig. 7(b) and (c)]. Considering the similarities in the molecular structures and the phase transition behaviors among OO1, SS1, and the other analogues, their monotropic SmA phases34,44,45 could also be classified as the same SmAint phase with half-intercalated layer structures. The methyl chain is generally too short to facilitate microphase separation to form layered Sm phases. Therefore, the intercalated cybotactic clusters and Sm layers in the NCybAint and SmAint phases are likely ascribable to the strong core–core interactions arising from the highly anisotropic molecular structures that do not depend on the microphase separation driven by long alkyl chains. In addition, as described earlier in Section 3.2.1, PBB homologs with longer alkyl chains exhibited the NCybC phase with tilted SmC-like clusters and SmC phase.34 Lengthening the alkyl chains led to a change from the non-tilted and intercalated phases of SO1 to the non-intercalated and tilted ones, which are ascribed to the enhanced microphase separation and free volume effect to cause the molecular tilts.28
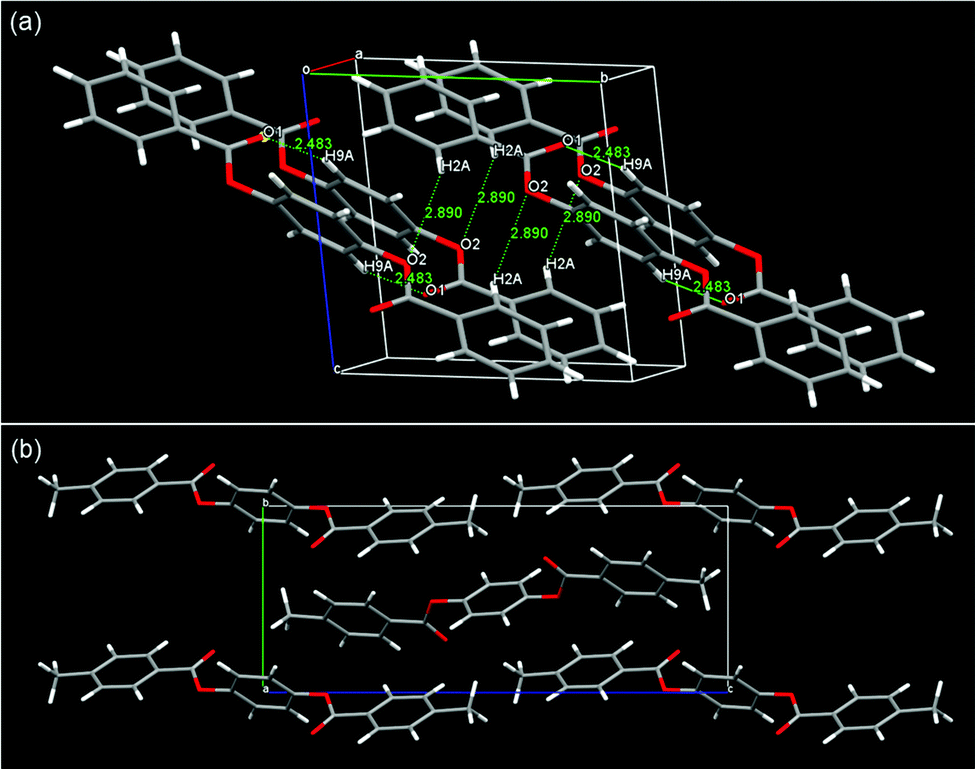
The LC phases of SO1 indicated strong tendency of PBB mesogenic core to form intercalated packing. If it really arises from core–core interactions, the pristine PBB molecules may exhibit similar packing manner even at low temperature limit: i.e., in its crystalline state. Indeed, it was evidenced by the single-crystal XRD analysis (Fig. 8). The intercalated LC structures were then considered in terms of molecular packing in the single-crystal structures of non-substituted PBB63 and 4,4′-dimethyl-substituted PBB-1.64 PBB exhibits a half-shifted packing arrangement between fully overlapped packing domains with an apparent edge alignment [Fig. 8(a)].63 The short interatomic contacts of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H in the packing structures are indicated by O(1)⋯H(9) and O(2)⋯H(2) (2.48 and 2.89 Å) between fully overlapped domains and half-shifted packing moieties, respectively. These contacts are ascribable to the CO⋯H hydrogen bonds, which are typically observed in phenyl benzoates.36,65,66 A similar half-slide packing structure was also observed in PBB-1 [Fig. 8(b)].64 The intermolecular CO⋯H contacts of O(2)⋯H(6) and O(2)⋯H(4) between the bis(benzoate) cores are 2.43 and 2.63 Å in length, respectively, suggesting the presence of CO⋯H hydrogen bonds. These intrinsic half-shifted packing structures of the PBB cores in the crystal structure may be associated with the intercalated structures of the NCybAint and SmAint phases of the homologs bearing short alkyl chains [Fig. 7(d)]. Nonetheless, in the case of SS1 which also show a similar SmA phase,34 PBB-core and methylthio moieties are clearly segregated into each domain in its crystalline state rather than such half-shifted packing.36 This microphase separation was interpreted by the large dispersion forces and enhanced affinities between more polarizable core π-systems, afforded by the sulfur atoms. This is also likely to be linked to that the influences of those attractive interactions from the sulfur atoms become more pronounced in denser states such as Cr phases.36,40 It is interesting to note that the formation of such an unexpected Sm phase by short tail homologs was also observed in other 1,4-bis(ethynyl)benzenes (the so-called bistolanes) bearing methoxy or methylthio groups.49 Their LC structures have not ever been reported due to their monotropic nature. This indicates that formation of the Sm phase by short-chain molecules could be characteristic of highly anisotropic molecular systems due to their strong core–core interactions.
O⋯H in the packing structures are indicated by O(1)⋯H(9) and O(2)⋯H(2) (2.48 and 2.89 Å) between fully overlapped domains and half-shifted packing moieties, respectively. These contacts are ascribable to the CO⋯H hydrogen bonds, which are typically observed in phenyl benzoates.36,65,66 A similar half-slide packing structure was also observed in PBB-1 [Fig. 8(b)].64 The intermolecular CO⋯H contacts of O(2)⋯H(6) and O(2)⋯H(4) between the bis(benzoate) cores are 2.43 and 2.63 Å in length, respectively, suggesting the presence of CO⋯H hydrogen bonds. These intrinsic half-shifted packing structures of the PBB cores in the crystal structure may be associated with the intercalated structures of the NCybAint and SmAint phases of the homologs bearing short alkyl chains [Fig. 7(d)]. Nonetheless, in the case of SS1 which also show a similar SmA phase,34 PBB-core and methylthio moieties are clearly segregated into each domain in its crystalline state rather than such half-shifted packing.36 This microphase separation was interpreted by the large dispersion forces and enhanced affinities between more polarizable core π-systems, afforded by the sulfur atoms. This is also likely to be linked to that the influences of those attractive interactions from the sulfur atoms become more pronounced in denser states such as Cr phases.36,40 It is interesting to note that the formation of such an unexpected Sm phase by short tail homologs was also observed in other 1,4-bis(ethynyl)benzenes (the so-called bistolanes) bearing methoxy or methylthio groups.49 Their LC structures have not ever been reported due to their monotropic nature. This indicates that formation of the Sm phase by short-chain molecules could be characteristic of highly anisotropic molecular systems due to their strong core–core interactions.
3.3. Birefringence
The temperature dependence of birefringence in the N phase of a uniaxially aligned sample of SO6 was measured to characterize its optical properties. The results were compared with those of SS6 and OO6. Thus, the values of Δn obtained at 550 nm upon cooling were plotted as a function of ΔT (Fig. 9), and the Δn values of SS6 and OO6 were taken from our previous work.34 The Δn values of all three molecules increase with decreasing T (i.e., increasing ΔT), which is related to their elevated orientational order parameters upon cooling. Replacing alkoxy groups with alkylthio groups increases the birefringence in the order SS6 > SO6 > OO6, which is consistent with the results of our previous work.4,32,34 More specifically, the Δn values of SS6, which contains two alkylthio groups, were found to be significantly higher than those of SO6 and OO6. Moreover, SO6 exhibited higher Δn values than OO6 from the middle range of ΔT upon cooling. These are thought to be due to the high molecular polarizability and anisotropy provided by the alkylthio groups. Birefringence is known to be related to the order parameter (S) by the equations Δn = Δn0S and S = (1 − T/Ti)β,67 where Δn0, T, Ti, and β are the Δn value at S = 1, the temperature, the approximate isotropic temperature, and the fitting constant, respectively. The Δn values of the three compounds fit this equation well, and the obtained Δn0, Ti, and β values are summarized in Table 2. The obtained Ti values are similar to the experimental TIN values upon cooling, while the β values are within the usual range for nematic phases. In addition, the Δn0 value decreases in the order SS6 (0.32) > SO6 (0.27) > OO6 (0.25), i.e., it increases upon replacing an alkoxy group with an alkylthio group. The S values of the sulfur-containing SO6 and SS6 homologs obtained from the Δn0 values tend to be smaller compared to those of the alkoxy-containing OO6 homolog. The narrower LC phase temperature ranges of alkylthio-based materials and their strongly temperature-dependent birefringence properties lead to a higher Δn0 than those of the alkyl- and alkoxy-containing counterparts.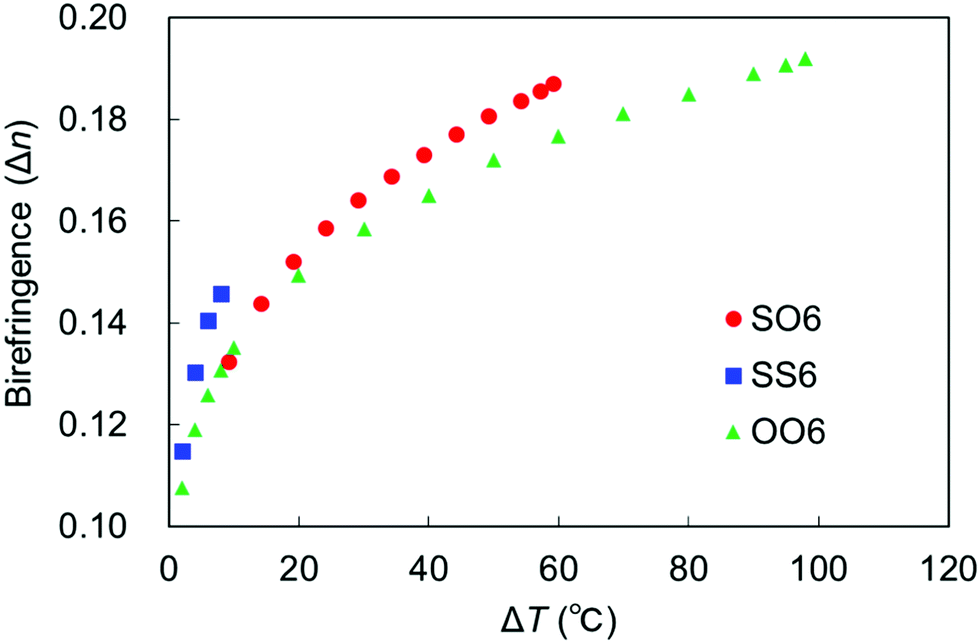 | ||
| Fig. 9 Birefringence of SO6 (red circles), SS6 (blue squares), and OO6 (green triangles) as a function of ΔT. | ||
| Code | Δn0 | T i | β |
|---|---|---|---|
| SO6 | 0.27 | 183.7 | 0.18 |
| OO6 | 0.25 | 210.7 | 0.16 |
| SS6 | 0.32 | 158.5 | 0.19 |
4. Conclusions
We developed a homologous series of alkylthio- and alkoxy-based asymmetric 1,4-phenylene bis(benzoate) LCs possessing different alkyl chain lengths (SOn, n = 1–8). Their phase transitions were evaluated and compared with those of the already known symmetric alkoxy and alkylthio analogues (OOn and SSn). It was found that all SOn homologs exhibited well-defined enantiotropic LC phases. The SO2–SO7 homologs only exhibited the N phase, but the longest homolog (SO8) showed a tilted SmC phase as well as an N phase. Interestingly, the shortest homolog (SO1) formed an enantiotropic orthogonal SmA phase and an N phase. Upon substituting an alkoxy group with an alkylthio group, the N–Iso phase transition temperature decreased for all n values, whereas the melting points of homologs with n ≥ 3 increased. The XRD results revealed that the N phases of longer SOn homologs (n = 6 and 8) were NCybC phases with SmC-like clusters and without alkyl chain intercalation, wherein the alkyl chains of SO8 intercalated into the neighboring layers in the SmC phase. However, the N and SmA phases of SO1 were identified as intercalated NCybAint and SmAint phases, respectively, which contained half-intercalated structures. The unexpected appearance of the SmA phase, in addition to the intercalated structures of SO1, was considered in terms of the molecular contacts and packings in the crystal structures. Furthermore, the birefringence of the 1,4-phenylene bis(benzoate) LCs could be increased by substituting their alkoxy groups with alkylthio groups. Overall, the present study provides not only a good understanding of the effects of alkylthio and alkoxy groups on the phase transitions and LC properties of such systems, but also insight into the remarkably different NCyb and Sm phase structures observed for the short- and long-chain homologs of highly anisotropic calamitic LCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (Grant Numbers 15H06285 and 17K14493) and research grants from Nitto Foundation, Toukai Foundation for Technology, and Toyohashi University of Technology. We would like to thank Ms Tsugumi Shiokawa and Dr Hiroko Tada in the Division of Instrumental Analysis of Okayama University for carrying out the mass spectrometry measurements.References
- A. J. Seed, K. J. Toyne, J. W. Goodby and D. G. McDonnell, J. Mater. Chem., 1995, 5, 1 RSC.
- A. J. Seed, K. J. Toyne and J. W. Goodby, J. Mater. Chem., 1995, 5, 2201 RSC.
- G. J. Cross, A. J. Seed, K. J. Toyne, J. W. Goodby, M. Hird and M. C. Artal, J. Mater. Chem., 2000, 10, 1555 RSC.
- Y. Arakawa, S. Kang, J. Watanabe and G. Konishi, RSC Adv., 2015, 5, 8056 RSC.
- Y. Arakawa, S. Kang, H. Tsuji, J. Watanabe and G. Konishi, RSC Adv., 2016, 6, 16568 RSC.
- D. Węgłowska, P. Kula and J. Herman, RSC Adv., 2016, 6, 403 RSC.
- Y. Arakawa, S. Kang, H. Tsuji, J. Watanabe and G. Konishi, RSC Adv., 2016, 6, 92845 RSC.
- J. Herman and P. Kula, Design of new super-high birefringent isothiocyanato bistolanes–synthesis and properties, Liq. Cryst., 2017, 44, 1462 CrossRef CAS.
- D. Adam, P. Schumacher, J. Simmerer, L. Haussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141 CrossRef CAS.
- M. Funahashi and J. Hanna, Jpn. J. Appl. Phys., 1996, 35, L703 CrossRef CAS.
- M. Funahashi and J. Hanna, Phys. Rev. Lett., 1997, 78, 2184 CrossRef CAS.
- K. Ban, K. Nishizawa, K. Ohta, A. M. van de Craats, J. M. Warman, I. Yamamoto and H. Shirai, J. Mater. Chem., 2001, 11, 321 RSC.
- K. Ohta, K. Hatsusaka, M. Sugibayashi, M. Ariyoshi, K. Ban, F. Maeda, R. Naito, K. Nishizawa, A. M. Van de Craats and J. M. Warman, Mol. Cryst. Liq. Cryst., 2003, 397, 25 CrossRef.
- F. Nekelson, H. Monobe, M. Shiro and Y. Shimizu, J. Mater. Chem., 2007, 17, 2607 RSC.
- E. F. Gramsbergen, H. J. Hoving, W. H. De Jeu, K. Praefcke and B. Kohne, Liq. Cryst., 1986, 1, 397 CrossRef CAS.
- P. Doppelt and S. Huille, New J. Chem., 1990, 14, 607 CAS.
- G. Morelli, G. Ricciardi and A. Roviello, Chem. Phys. Lett., 1991, 185, 468 CrossRef CAS.
- E. Keinan, S. Kumar, S. P. Singh, R. Ghirlando and E. J. Wachtel, Liq. Cryst., 1992, 11, 157 CrossRef CAS.
- S. Kumar, E. J. Wachtel and E. Keinan, J. Org. Chem., 1993, 58, 3821 CrossRef CAS.
- S. H. Kang, Y.-S. Kang, W.-C. Zin, G. Olbrechts, K. Wostyn, K. Clays, A. Persoons and K. Kim, Chem. Commun., 1999, 1661 RSC.
- G. Kestemont, V. de Halleux, M. Lehmann, D. A. Ivanov, M. Watson and Y. H. Geerts, Chem. Commun., 2001, 2074 RSC.
- P. H. J. Kouwer, W. F. Jager, W. J. Mijs and S. J. Picken, J. Mater. Chem., 2003, 13, 458 RSC.
- P. Kouwer, W. Jager, W. Mijs and S. Picken, Mol. Cryst. Liq. Cryst., 2004, 411, 305 CrossRef.
- S. Kumar, Liq. Cryst., 2004, 31, 1037 CrossRef CAS.
- S. K. Varshney, H. Nagayama, V. Prasad and H. Takezoe, Liq. Cryst., 2011, 38, 1321 CrossRef CAS.
- H. Eichhorn, D. W. Bruce and D. Wöhrle, Adv. Mater., 1998, 10, 419 CrossRef CAS.
- M. Alaasar, A. F. Darweesh, X. Cai, F. Liu and C. Tschierske, Chem. – Eur. J., 2021, 27, 14921 CrossRef CAS PubMed.
- J. W. Goodby, R. J. Mandle, E. J. Davis, T. Zhong and S. J. Cowling, Liq. Cryst., 2015, 42, 593 CrossRef CAS.
- E. Kleinpeter, H. Köhler, A. Lunow, C. Tschierske and H. Zaschke, Tetrahedron, 1988, 44, 1609 CrossRef CAS.
- Y. Arakawa, Y. Sasaki, K. Igawa and H. Tsuji, New J. Chem., 2017, 41, 6514 RSC.
- Y. Arakawa, Y. Sasaki and H. Tsuji, Chem. Lett., 2017, 46, 1657 CrossRef CAS.
- Y. Arakawa and H. Tsuji, Mol. Cryst. Liq. Cryst., 2017, 647, 422 CrossRef CAS.
- Y. Arakawa, S. Inui and H. Tsuji, Liq. Cryst., 2018, 45, 811 CrossRef CAS.
- Y. Arakawa, Y. Sasaki, N. Haraguchi, S. Itsuno and H. Tsuji, Liq. Cryst., 2018, 45, 821 CrossRef CAS.
- Y. Arakawa, Y. Sasaki and H. Tsuji, J. Mol. Liq., 2019, 280, 153 CrossRef CAS.
- Y. Arakawa, Y. Ishida, Y. Sasaki, K. Igawa, S. Sasaki and H. Tsuji, CrystEngComm, 2022 10.1039/D1CE01470F.
- X. Zong, Z. Fang and C. Wu, Liq. Cryst., 2018, 45, 1844 CrossRef CAS.
- Y. Arakawa, K. Komatsu and H. Tsuji, New J. Chem., 2019, 43, 6786 RSC.
- I. C. Pintre, J. L. Serrano, M. B. Ros, J. Ortega, I. Alonso, J. Martínez-Perdiguero, C. L. Folcia, J. Etxebarria, F. Goc, D. B. Amabilino, J. Puigmartí-Luis and E. Gomar-Nadal, Chem. Commun., 2008, 2523 RSC.
- Y. Arakawa, S. Inui and H. Tsuji, Polymer, 2021, 233, 124194 CrossRef CAS.
- M. J. S. Dewar and J. P. Schroeder, J. Org. Chem., 1965, 30, 2296 CrossRef CAS.
- M. J. S. Dewar and R. S. Goldberg, J. Org. Chem., 1970, 35, 2711 CrossRef CAS.
- M. J. S. Dewar and R. S. Goldberg, J. Am. Chem. Soc., 1970, 92, 1582 CrossRef CAS.
- M. J. S. Dewar and A. C. Griffin, J. Am. Chem. Soc., 1975, 97, 6662 CrossRef CAS.
- J. P. Schroeder, Mol. Cryst. Liq. Cryst., 1980, 61, 229 CrossRef CAS.
- W. Weissflog and D. Demus, Cryst. Res. Technol., 1984, 19, 55 CrossRef CAS.
- M. M. Naoum, R. I. Nessim, G. R. Saad and T. Y. Labeeb, Liq. Cryst., 2003, 30, 165 CrossRef CAS.
- T. Tasaka, H. Okamoto, Y. Morita, K. Kasatani and S. Takenaka, Liq. Cryst., 2003, 30, 961 CrossRef CAS.
- P. Nguyen, G. Lesley, C. Dai, N. J. Taylor, T. B. Marder, V. Chu, C. Viney, I. Ledoux and J. Zyss, Well-Defined Conjugated Rigid-Rods as Multifunctional Materials: Linear and Nonlinear Optical Properties and Liquid Crystalline Behavior, in Applications of Organometallic Chemistry in the Preparation and Processing of Advanced Materials, ed. J. F. Harrod and R. M. Laine, NATO ASI Series (Series E: Applied Sciences), Springer, Dordrecht, 1995, vol. 297, p. 333, DOI:10.1007/978-94-011-0337-4_19.
- Y. Ishida, Y. Sasaki, Y. Arakawa and H. Tsuji, Polym. Prepr., 2018, 67, 1 Search PubMed.
- Y. Arakawa, S. Nakajima, R. Ishige, M. Uchimura, S. Kang, G. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 8394 RSC.
- A. D. Vries, Mol. Cryst. Liq. Cryst., 1970, 10, 219 CrossRef.
- C. Tschierske and D. J. Photinos, J. Mater. Chem., 2010, 20, 4263 RSC.
- S. J. Picken, T. J. Dingemans, L. A. Madsen, O. Francescangeli and E. T. Samulski, Liq. Cryst., 2012, 39, 19 CrossRef CAS.
- S. Marčelja, J. Chem. Phys., 1974, 60, 3599 CrossRef.
- A. P. J. Emerson and G. R. Luckhurst, Liq. Cryst., 1991, 10, 861 CrossRef CAS.
- C. Tschierske, D. Joachimi, H. M. Vorbrodt, H. Zaschke, A. Wiegeleben, A. Hauser and D. Demus, Liq. Cryst., 1989, 5, 177 CrossRef CAS.
- Y. Arakawa, S. Inui, K. Igawa and H. Tsuji, Liq. Cryst., 2019, 46, 1621 CrossRef CAS.
- E. Cruickshank, G. J. Strachan, J. M. Storey and C. T. Imrie, J. Mol. Liq., 2021, 117094 Search PubMed.
- S. Kang, S. Nakajima, Y. Arakawa, M. Tokita, J. Watanabe and G. Konishi, Polym. Chem., 2014, 5, 2253 RSC.
- N. Vaupotič, J. Szydlowska, M. Salamonczyk, A. Kovarova, J. Svoboda, M. Osipov, D. Pociecha and E. Gorecka, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 80, 030701 CrossRef PubMed.
- K. Fodor-Csorba, A. Jákli, A. Vajda, S. K. Prasad, D. S. Shankar Rao, R. Y. Dong, J. Xu and G. Galli, ChemPhysChem, 2006, 7, 2184 CrossRef CAS PubMed.
- J. A. Ganaie, J. Kumar, R. J. Butcher, J. P. Jasinski and S. K. Gupta, J. Chem. Crystallogr., 2016, 46, 93 CrossRef CAS.
- M. R. Ciajolo, A. Sirigu and A. Tuzi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, C47, 106 CrossRef CAS.
- J. M. Adams and S. E. Morsi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, B32, 1345 CrossRef CAS.
- L. G. Kuz'mina, I. I. Konstantinov and A. V. Churakov, Mol. Cryst. Liq. Cryst., 2018, 664, 95 CrossRef.
- I. Haller, Prog. Solid State Chem., 1975, 10, 103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ma00050d |
| This journal is © The Royal Society of Chemistry 2022 |