
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly-dispersed and high-metal-density electrocatalysts on carbon supports for the oxygen reduction reaction: from nanoparticles to atomic-level architectures
Zesheng
Li
 *,
Bolin
Li
,
Yifan
Hu
,
Shaoyu
Wang
and
Changlin
Yu
*
*,
Bolin
Li
,
Yifan
Hu
,
Shaoyu
Wang
and
Changlin
Yu
*
College of Chemistry, Guangdong University of Petrochemical Technology, Maoming, 525000, China. E-mail: lzs212@163.com; yuchanglinjx@163.com
First published on 1st December 2021
Abstract
Electrocatalysts for the oxygen reduction reaction (ORR) are crucial for a variety of renewable energy applications (e.g., proton exchange membrane fuel cells, PEMFCs). The synthesis of highly-dispersed and high-metal-density ORR electrocatalysts (e.g., nanoscale and atomic-level structures) on carbon supports with strong durability is extremely desirable but remains challenging. Carbon-supported high-loading noble metal catalysts with nanoscale structures (e.g., Pt-based nanoparticles) are the most widely used catalysts with the best catalytic performances. Single atom catalysts (SACs) that integrate the merits of homogeneous and heterogeneous catalysts have attracted considerable attention in recent years. Aside from the manipulation of the geometric and electronic structures of active metal sites, another key challenge in this field is the development of strategies for preparing high-metal-density SACs, thus rendering atomic-level ORR electrocatalysts dramatically reactive, selective, and stable compared to their nanoscale counterparts. This review summarizes the recent advancements in carbon-supported nanoscale and atomic-level ORR electrocatalysts with high metal density (namely high loading) for fuel cells. Special emphasis is placed on the basic principles, preparation strategies and catalytic applications of these highly-dispersed and high-metal-density ORR electrocatalysts on carbon supports from nanoparticles to atomic-level architectures.
1 Introduction
In recent years, in order to deal with the problems of energy shortage and environmental pollution caused by traditional fossil fuels, numerous research institutions in many countries have committed to developing sustainable clean energy technologies.1 In this context, proton exchange membrane fuel cells (PEMFCs) have attracted extensive attention because of their high specific power density, high energy conversion efficiency, environmental friendliness, and low starting and working temperatures, and are thus considered to be one of the most ideal classes of energy conversion devices that can replace traditional internal combustion engines as automobile power sources.2 In PEMFCs, the design of cathode “oxygen reduction” electrocatalysts is more important and complex than that of anode “hydrogen oxidation” electrocatalysts. The main reasons are the following: (1) the kinetics of oxygen reduction is very slow, and its exchange current density is only 1/100th of that of anodic hydrogen oxidation, and thus oxygen reduction has become the restrictive step of the electrocatalytic reaction of PEMFCs; and (2) the process of oxygen reduction is relatively complex, involving multiple elementary steps and a variety of intermediates, which often leads to a decrease of energy conversion efficiency and an increase of oxygen reduction overpotential.3 Therefore, the design and development of high-performance cathode oxygen reduction electrocatalysts is of great significance in promoting the development of PEMFCs.The oxygen reduction reaction (ORR), that is, the electrochemical reduction of oxygen molecules, is a relatively complex process in fuel cells or metal–oxygen cells.4 The oxygen reduction reaction of cathodes has many possible reaction mechanisms; the main reaction pathways are as follows:
(i) Oxygen molecules can be reduced to form water through a direct “four-electron mechanism” (taking metal Pt as an example):
| 2Pt + O2 → 2Pt–O | (1) |
| 2Pt–O + 2H+ + 2e− → 2Pt–OH | (2) |
| 2Pt–OH + 2H+ + 2e− → Pt + 2H2O | (3) |
| O2 + 4H+ + 4e− → 2H2O E = 1.23 V vs. SHE (25 °C) | (4) |
(ii) Oxygen molecules can also be reduced by a “two-electron mechanism”; that is, two electrons can be obtained to be reduced to hydrogen peroxide:
| O2 + 2H+ + 2e− → H2O2 E = 0.69 V vs. SHE (25 °C) | (5) |
The intermediate product H2O2 can be further reduced to water:
| H2O2 + 2H+ + 2e− → 2H2O E = 1.77 V vs. SHE (25 °C) | (6) |
The development of ORR electrocatalysts with high performance and low cost is of great strategic significance in the development of advanced energy conversion devices. In essence, platinum (Pt) is still the most effective ORR electrocatalyst.6 However, its high price has restricted the development of PEMFCs. At present, effective countermeasures include: (i) reducing the use of Pt by optimizing the utilization of Pt catalyst, (ii) reducing the use of Pt by improving the performance of Pt catalyst, and (iii) developing other catalysts that can replace Pt metal.7 At present, the design direction of ORR electrocatalysts mainly includes “low-Pt electrocatalysts”, “Pt-free electrocatalysts” and “non-noble-metal electrocatalysts”. According to their action mechanism, low-Pt electrocatalysts can be divided into three types: (i) core–shell structure low-Pt electrocatalysts, (ii) alloy structure low-Pt electrocatalysts, and (iii) synergistic-effect low-Pt electrocatalysts (facilitated using co-catalysts).8 Pt-free electrocatalysts refer to other lower-price noble-metal electrocatalysts such as palladium (Pd), iridium (Ir) and silver (Ag).9 Non-noble-metal electrocatalysts refer to transition metal catalysts especially those of iron (Fe) and cobalt (Co), which are alternative electrocatalysts with rich resources and low price.10 These noble-metal or non-noble-metal ORR electrocatalysts have their own characteristics and advantages, and they are related to each other, and even complement each other in specific dimensions and environments.
Recently, several reviews have summarized the preparation methods and structural modification of noble-metal11,12 and non-noble-metal13,14 ORR electrocatalysts. They all are very much concerned with the relationship between the nanostructures, crystal structures, and electronic structures of metals and their electrochemical performances. Some of them only focus on the design of individual nano-morphologies and ignore the interaction with carbon supports. However, electrocatalysts for the actual application in PEMFCs are typically composed of highly dispersed active metal nanoparticles (NPs) on a variety of carbon supports, often with a high metal content (40–50 wt%), to guarantee the formation of thin catalyst layers along with metallically conducting interfaces.15–17 Therefore, it is necessary to pay attention to summarizing the synthetic strategies and structural properties of high-metal-loading ORR electrocatalysts, to meet the demands of practical applications of PEMFCs.
On the other hand, the dimensions and distribution of these high-loading active metal components on carbon supports have a great impact on the catalytic activity, product selectivity (i.e., two electron or four electron mechanism), and electrochemical stability of ORR electrocatalysts.18–20 In recent years, in order to improve the catalytic activity and selectivity and increase the utilization of active metals, ORR electrocatalysts have been developed from nanosize to sub-nanosize21 and even atomic-level22 architectures. Therefore, the development of highly-dispersed and high-metal-content (or high-metal-density) electrocatalysts with reduced dimensions (or atomic level structures) on carbon supports is very critical for the ORR in the practical application of fuel cells.
Single atom catalysts (SACs) usually consist of dispersed metal atoms and appropriate support materials, where the supports are employed to anchor onto, confine to, and/or coordinate with isolated metal atoms.23 Therefore, the features of SACs allow achieving maximum atom utilization (∼100%), which is of particular significance for the development of carbon-supported SACs for ORR electrocatalysis. In recent years, several reviews have discussed the synthesis strategies, coordination regulation, electrocatalytic applications, and structure–property relationships of carbon-supported SACs.24–26 However, these reviews to some extent have not addressed the regulation of metal density and site distance effects of SACs, or the synergistic effects of high-density single atoms, dual atoms, or atom clusters in ORR electrocatalysis. So, in order to deeply understand the ORR mechanisms and structure–property relationship of high-density SACs, a relevant review study is very necessary.
In this review, the preparation methods and ORR performances of carbon-supported high-metal-density ORR electrocatalysts (including nanosize NPs and atomic-level structures) are systematically summarized (see Scheme 1 for an overview of the topics covered here). The promising carbon supports for ORR electrocatalysts are introduced. The basic principles and applications of highly-dispersed and high-metal-density ORR electrocatalysts on different carbon supports are reviewed and analyzed. Particularly, the development process of carbon-supported SACs, the synthetic strategies of high-density SACs, and the synergistic effect of high-density SACs on carbon supports for the ORR are discussed. The fundamental understanding of the synthesis–construction–performance correlations for carbon-supported high-metal-density ORR electrocatalysts is preliminarily demonstrated. Finally, the challenges and prospects for the development of high-metal-density ORR electrocatalysts on carbon supports are highlighted.
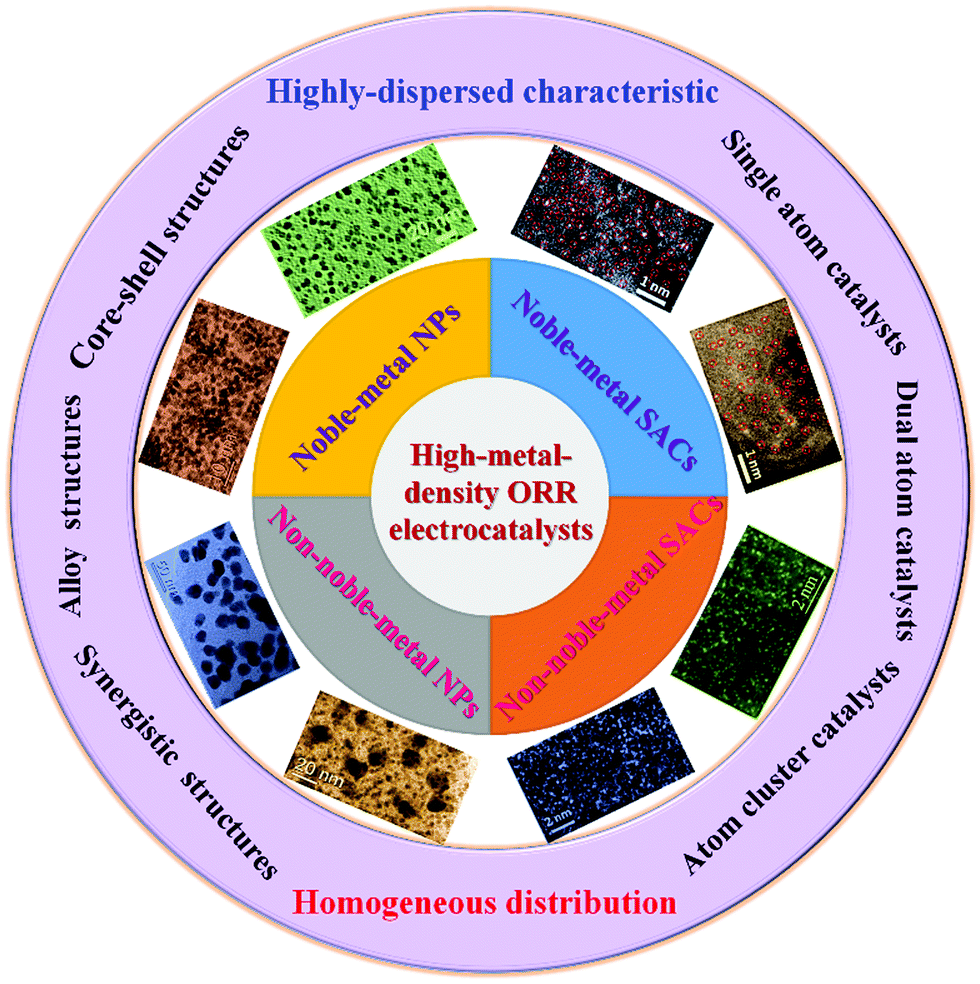 | ||
| Scheme 1 Overview of the topics covered in this review. | ||
2 Carbon supports for ORR electrocatalysts
In order to design high-performance ORR electrocatalysts, carbon supports should have the following three pivotal characteristics: (i) high specific surface area, accommodating high-content metal components with maximized catalytically active area; (ii) high electronic conductivity, minimizing the ohmic resistance-induced voltage loss of cell systems; and (iii) high chemical and electrochemical corrosion resistance, offering high stability and durability for electrocatalysis.27 Currently, the widely used carbon supports for ORR electrocatalysts include carbon black, carbon nanotubes, graphene, template-based porous carbon, and other pyrolytic carbon materials.2.1 Carbon black
Carbon black (CB) is a low-cost support for the cathode of PEMFCs. CB materials produced by pyrolysis or incomplete combustion of hydrocarbons have a turbine layered structure and high conductivity and specific surface area. Oxygen groups (such as hydroxyl, carbonyl and carboxyl) usually exist on the surface of carbon black, and play an important role in the dispersion and stability of the metal component. Modifying the hydrophilic or hydrophobic properties of carbon black by oxidizing or doping it with nitrogen, fluorine and other heteroatoms can also improve the service life and gas transport characteristics of carbon black. The surface functionalization of carbon black will affect the ionomer coverage in the catalyst layer and its performance and life in fuel cell devices.282.2 Carbon nanotubes
Carbon nanotubes (CNTs) have become one of the most widely used supports because of their large specific surface area, high conductivity, good thermal stability, and surface modification feasibility. CNTs often need to be oxidized with nitric acid or mixed acid to form oxygen-containing functional groups as nucleation centers to anchor metal nanoparticles or metal atoms, which will also introduce defects in the carbon matrix, increasing the surface-active sites. At present, the research work on CNTs mainly focuses on the methods of loading active metal components onto the surfaces of CNTs and the encapsulation of metal components into CNTs, and the influence of the surface properties of CNTs on metal dispersion and electrocatalytic performances.292.3 Graphene
Graphene is a recently-discovered carbon nanomaterial with a two-dimensional planar structure, which has many unique properties, such as large specific surface area, an excellent electron conduction ability and high chemical stability, making it as an ideal catalyst support for fuel cells. Graphene plays a very important role in reducing the use of noble metals and improving the catalytic performance of non-noble metals. The synthesis of graphene with a porous structure can effectively limit the size of metal and inhibit agglomeration, greatly improving the catalytic activity and stability of catalysts. By adjusting the three-dimensional structure of graphene, the transmission of fuels and products can be effectively controlled and the catalytic performance of catalysts can be improved.302.4 Template-based porous carbon
Porous carbon materials show bright prospects for energy conversion and storage devices due to their outstanding properties such as high surface area, accessible active sites, mass transport, and diffusion. Template-based (e.g., SiO2 and micelle based) porous carbon materials have more regular and controllable pore structures, which are more suitable for electrocatalyst supports. Template-based porous carbon materials include ordered mesoporous carbon (narrow mesoporous structure) and ordinary porous carbon (mesoporous or macroporous structure) materials. The controllable pore structures (2–500 nm) of template-based porous carbon materials provide adjustable void spaces for limiting metal catalysts and preventing agglomeration, as well as promoting the diffusion and migration of substances.312.5 Other pyrolytic porous carbon materials
Porous carbon materials can also be synthesized by direct pyrolysis of a variety of carbon precursors (e.g., biomass, polymer, and metal–organic frameworks (MOFs)) without extraneous templates.32 Biomass-derived porous carbon has some advantages, such as abundance in nature, sustainability, economic viability, and environmental friendliness. Polymer-derived porous carbon also has some advantages, such as high carbon content, controllable constituent elements, and low price. As a typical class of MOFs, zeolitic imidazolate frameworks (ZIFs) have the characteristics of both MOFs and zeolites. ZIF-derived porous carbon and that coupled with metal components have exhibited remarkable activity and stability for a broad range of electrocatalysis applications.33When preparing high-metal-density ORR electrocatalysts, nano-scale carbon materials with high specific surface area are preferentially selected as supports, so that the active sites of metal NPs (or metal atoms) are fully exposed and participate in the catalytic reaction to maximize the mass activity.34 Because of the high metal loading, the metal components can very easily agglomerate, so the high dispersion of the catalyst is particularly critical. The pore structure (defects and nanopores) and surface state (doped N, P, and S atoms) of carbon materials play important roles in anchoring metal nanoparticles and dispersing metal atoms by coordination.35 Therefore, it is demonstrated that carbon supports with favorable porous structures, optimized surface defects and suitable functional groups should be considered to fabricate high-metal-density ORR electrocatalysts for PEMFCs.
3 Fundamental basis of highly-dispersed and high-metal-density ORR electrocatalysts
3.1 Highly-dispersed ORR electrocatalysts
The dispersion degree refers to the ratio of the number of metal atoms exposed to the surface to the total number of metal atoms in a catalyst. The dispersion degree of a metal catalyst often affects its physical and chemical properties, which is directly related to its electroactive surface area.36 The finer the dispersion of a metal component, the higher the degree of dispersion, and the larger the surface area. The electrocatalytic reactions occur entirely on the surfaces of metal nanoparticles, so high dispersion is required to enhance the activity and reduce the amount of expensive noble metal used. Hence the better the dispersion of electrocatalysts, the higher their electrochemical performance.37 For example, highly dispersed Pt NPs on carbon black supports have been used as commercial ORR electrocatalysts in the practical application of PEMFCs. Notably, the high dispersion of metal components (including other Pt-free noble metals and transition metals) is also very important in ORR electrocatalysts for fuel cells.38 The wide variety of pore structures and chemical functional groups on the surfaces of carbon supports affects the dispersion of metal components. For this reason, various high-dispersion studies have been conducted by modifying the pore structure and chemical nature of carbon supports.39 In general, the development of highly-dispersed noble metal catalysts or non-noble metal catalysts on different supports is a key route to designing high-performance ORR electrocatalysts.3.2 High-metal density ORR electrocatalysts
The reaction kinetics of the ORR at the cathode of PEMFCs is poor, and thus requires the participation of high-efficiency catalysts. In practical fuel cell applications, carbon-supported Pt NP catalysts with high Pt loading (more than 40 wt%) have high practical value.17,40,41 On the one hand, the use of carbon-supported Pt based catalysts with high Pt loading can reduce the thickness of the catalytic layer in membrane electrode assemblies (MEAs), to reduce mass transfer loss at high current density.40 On the other hand, highly-dispersed Pt based catalysts with small particle size are recognized as one of the most promising classes of cathode catalysts, which have good activity in half cells and full cells.41 However, in recent years, there have been few reports on carbon-supported low-Pt electrocatalysts with high metal loading (such as core–shell, alloy, and synergistic effect low-Pt systems), which is mainly due to the severe challenges brought by particle agglomeration in the preparation of high load multi-metal catalysts and their poor stability in the electrochemical process.42 Therefore, it is of great significance to summarize the preparation methods and catalytic properties of existing high-loading (i.e., high metal NP density) ORR electrocatalysts, which helps in promoting the development of high-performance catalytic systems in the future.In recent years, single-atom catalysts (SACs) have been widely studied because of their nearly 100% utilization of metal active sites and accurate and controllable coordination structures.43 However, due to the lack of multiple-atom connected group sites, monatomic sites are not competent in some multi-step complex catalytic reactions. For example, in the ORR catalytic process, monatomic Pt catalyst often cannot effectively catalyze the fracture of the O–O bond, so it is difficult to effectively catalyze the ORR in the four-electron mechanism.44 Due to the inability of isolated Pt single atoms to break the O–O bond through lateral adsorption, the ORR tends to form the two-electron product (H2O2) rather than the four-electron product (H2O) required by fuel cells.45 High-metal density (high loading) Pt SACs show much better ORR performance (with much higher mass activity) than low-metal density (low loading) Pt SACs and Pt NPs, in which the selectivity of high-metal density Pt SACs can be greatly inclined to the four-electron path.46 Therefore, for atomically-dispersed ORR electrocatalysts, the high-metal density strategy appears to be particularly important in the design of high-efficiency catalytic systems, in view of their distance-tunable metal sites and variable coordination environments.
4 Preparation and application of highly-dispersed and high-metal-density ORR electrocatalysts
Based on the above analysis, the design and synthesis of highly-dispersed and high-metal-density ORR electrocatalysts on various carbon supports are of great theoretical and practical significance. In this section, we will introduce the preparation methods and catalytic performances of high-metal-density ORR electrocatalysts (including nanoscale and atomic-level architectures) by the analysis of several typical cases. For nanoscale ORR electrocatalysts, single metal NPs, metal alloy NPs, metal core–shell NPs, metal compound NPs, and metal compound-promoted metal (or its alloy) NPs are presented. On the other hand, for atomic-level ORR electrocatalysts, single-atom catalysts, dual-atom catalysts, atom cluster catalysts, single-atom/cluster catalysts and single-atom/NP mixed catalysts are introduced systematically.4.1 Nanoscale ORR electrocatalysts
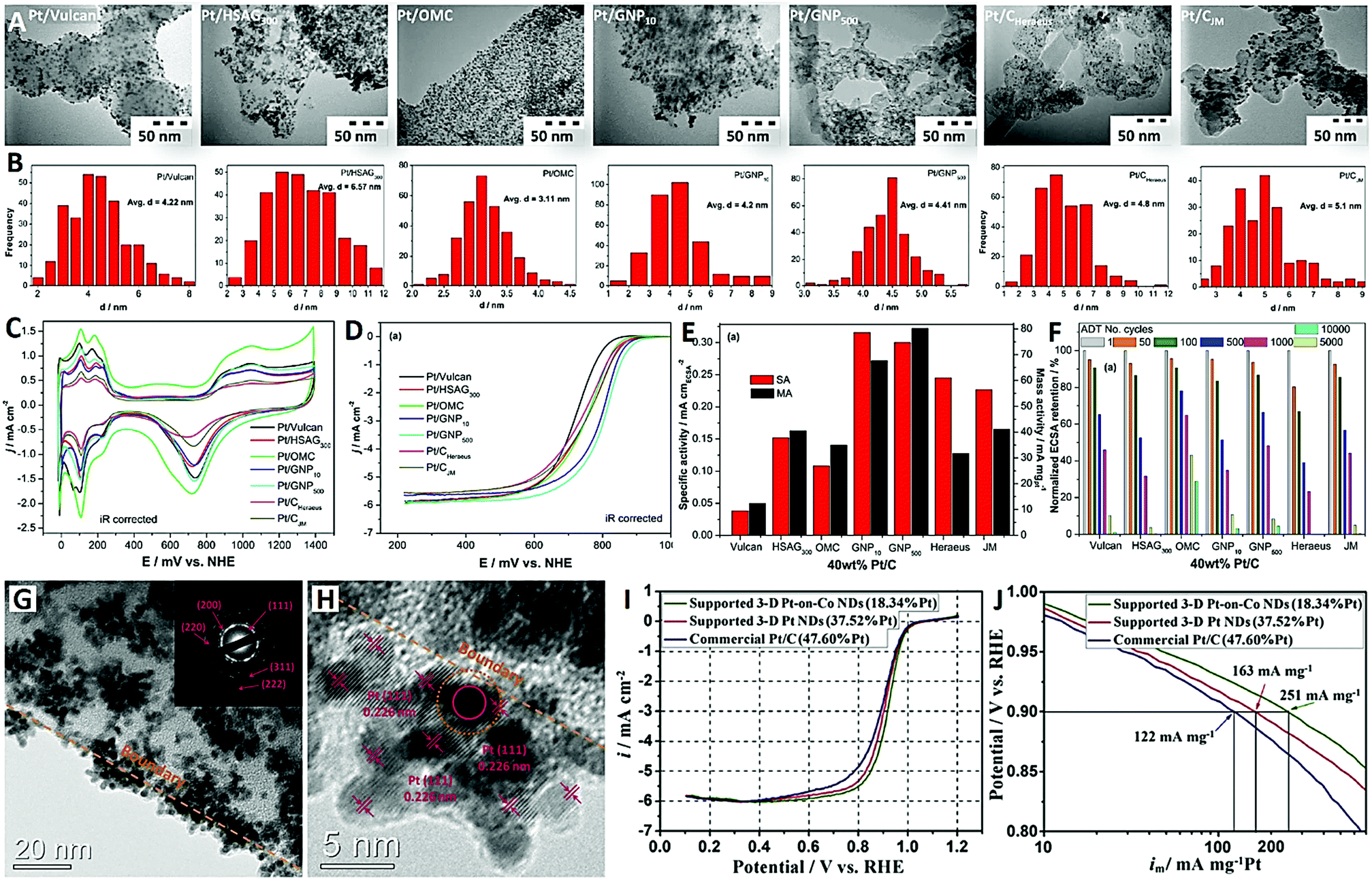 | ||
| Fig. 1 A series of carbon-supported Pt nanoparticles (reprinted with permission from ref. 50): (A) TEM images, (B) particle size distribution histograms of Pt, and (C–F) electrochemical performances; and typical carbon-supported Pt nanodendrites (reprinted with permission from ref. 54): (G and H) TEM images and (I and J) electrochemical performances. | ||
Fig. 1A and B show the TEM images of a variety of carbon-supported Pt catalysts (40 wt% Pt) and the corresponding particle size distribution histograms of Pt. The Pt NPs are uniformly dispersed on Vulcan, OMC and GNP500 supports, while some particle agglomerates are observed on HSAG300 and GNP10 supports. The highly graphitized structure and lack of oxygen functional groups in HSAG300 and GNP10 are not conducive to the uniform nucleation and dispersion of Pt NPs on their surfaces. The average particle sizes of Pt NPs were as follows: 3.11 nm (Pt/OMC) < 4.20 nm (Pt/GNP10) < 4.22 nm (Pt/Vulcan) < 4.41 nm (Pt/GNP500) < 6.57 nm (Pt/HSAG300). The ordered mesoporous carbon (OMC) with an internal porosity can offer a high surface area (1000 m2 g−1) for accommodating a large number of small Pt NPs compared with the solid carbon black (Vulcan) and graphitized carbons (HSAG300). The mesoporous graphitized nanoparticle (GNP500) support has favorable mesoporous structures (5–10 nm), moderately graphitized structures (namely moderate oxygen content), and small nanoparticle structures (∼50 nm), and should be a better support for Pt NPs than the OMC with amorphous structures and larger nanoscale structures (>500 nm). In addition, two commercial Pt/C catalysts (Pt/CJM and Pt/CHeraeus with 40 wt% Pt) also showed larger Pt NPs and poorer dispersion.
Fig. 1C shows the cyclic voltammograms (CVs) of the seven carbon-supported Pt catalysts (with 40 wt% Pt). In all these CVs, typical electrochemical potential sweep responses for Pt metal, such as Hads/des (0–300 mV), double-layer capacitance (300–750 mV) and oxide formation (750–1200 mV) regions, are clearly presented. The electrochemically active surface areas (ECSAs) of all these catalysts were evaluated by integrating Hdes peaks. This showed that Pt/OMC had the highest ECSA (34.3 m2 gPt−1) and the widest double-layer capacitance region. These are attributed to the smallest Pt NPs and the largest specific BET surface area of the carbon support for Pt/OMC. Fig. 1D shows the linear sweep voltammograms (LSVs) of the catalysts for the ORR in oxygen-saturated 0.5 M H2SO4 electrolyte. In the kinetic regime, the ORR activity was evaluated at a half-wave potential (2.5 mA cm−2), in which the two GNP-supported catalysts (Pt/GNP10 and Pt/GNP500) obviously showed better performance than the other catalysts. About 85 mV less overpotential for the ORR was obtained at Pt/GNP500 compared with that at Pt/Vulcan. Obviously, Pt/GNP500 showed the highest mass activity (Fig. 1E) and desirable stability (Fig. 1F) for the ORR. The results of this study demonstrated that the moderate graphitization level (namely moderate oxygen content and specific surface area), suitable mesoporous structure (5–10 nm), and interconnected small spherical morphology (∼50 nm) of carbon were found to be ideal properties for designing well-performing and highly-stable Pt NPs on carbon ORR electrocatalysts.50
On the other hand, three-dimensional (3-D) Pt-based nanostructures with a dendritic morphology have received much attention as ORR electrocatalysts.51–53 These unique Pt-based nanodendrites (NDs) generally have the advantages of high surface area and enhanced performances due to their dominant crystal planes and highly branched shapes.53 Previously, Li et al. reported carbon-supported Pt-on-Co NDs with high-Pt-content (37.52 wt% Pt) as an efficient ORR electrocatalyst for the first time (see Fig. 1G–J).54 These Pt-based NDs were spontaneously generated and deposited on polymer-derived carbon by a simple cobalt-induced and carbon-mediated galvanic cell reaction approach. The TEM images clearly reveal that the carbon-supported Pt NDs have distinct 3-D flower-like structures and an average diameter of 20 nm (with individual Pt nanoparticles of 3 nm) (Fig. 1G and H). The ORR LSV curves of the catalysts were recorded in oxygen-saturated 0.1 M HClO4 (Fig. 1I), and the mass activities at 0.9 V were calculated to be 122, 163 and 251 mA mg−1 Pt for commercial Pt/C (47.60 wt% Pt), supported 3-D Pt NDs (37.52 wt% Pt) and supported 3-D Pt-on-Co NDs (18.34 wt% Pt), respectively. More recently, Feng and co-workers synthesized branched Pt–Pd NDs supported on reduced graphene oxide (rGO), which exhibited enhanced ORR activity (onset potential of 0.05 V, half-wave potential of −0.15 V, electron transfer number >3.99) and good stability in alkaline medium (0.1 M KOH).55 Besides, high-density Au ND-loaded GO (Au ND-GO) also exhibited high electrocatalytic activity (in a near four-electron pathway) and superior stability toward the ORR in alkaline medium (0.1 M KOH).56 Overall, the results of these studies indicated that the construction of 3-D dendritic nanostructures is an attractive strategy for designing high-metal-density ORR catalysts with remarkable catalytic activity and durability.
The development of carbon-supported PtM (where M = Fe, Co, Ni, etc.) alloys with high dispersion and high metal density is an important research direction for high-performance ORR electrocatalysts.62–64 Achieving the homogeneous formation of ultrasmall PtM alloy NPs directly on carbon supports, with high catalyst loading, promises a facile and scalable production of alloy ORR electrocatalysts. However, the precise control of nucleation and alloying of two different metals on carbon supports is still a great challenge. Recently, Sung and co-workers63 reported a novel synthetic strategy to directly grow highly-dispersed MPt (where M = Fe, Co, and Ni) alloy NPs on various carbon supports with high catalyst loading, from a unique bimetallic compound composed of [M(bpy)3]2+ cations (bpy = 2,2′-bipyridine) and [PtCl6]2− anions. Thereinto, the representative rGO-supported FePt catalyst (37 wt%-FePt/rGO) with high homogeneity and narrow size distribution (5–6 nm) exhibited 18.8 times higher specific activity and 11.5 times higher mass specific activity than commercial Pt/C catalyst at 0.9 V (see Fig. 2A–C). It is noteworthy that the high metal loading catalyst (37 wt%-FePt/rGO) showed better electrocatalytic activity than the low metal loading catalyst (24 wt%-FePt/rGO), due to its high uniformity and high utilization efficiency. The high metal loading catalyst (37 wt%-FePt/rGO) also demonstrated excellent electrochemical stability without degradation over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.63
000 cycles.63
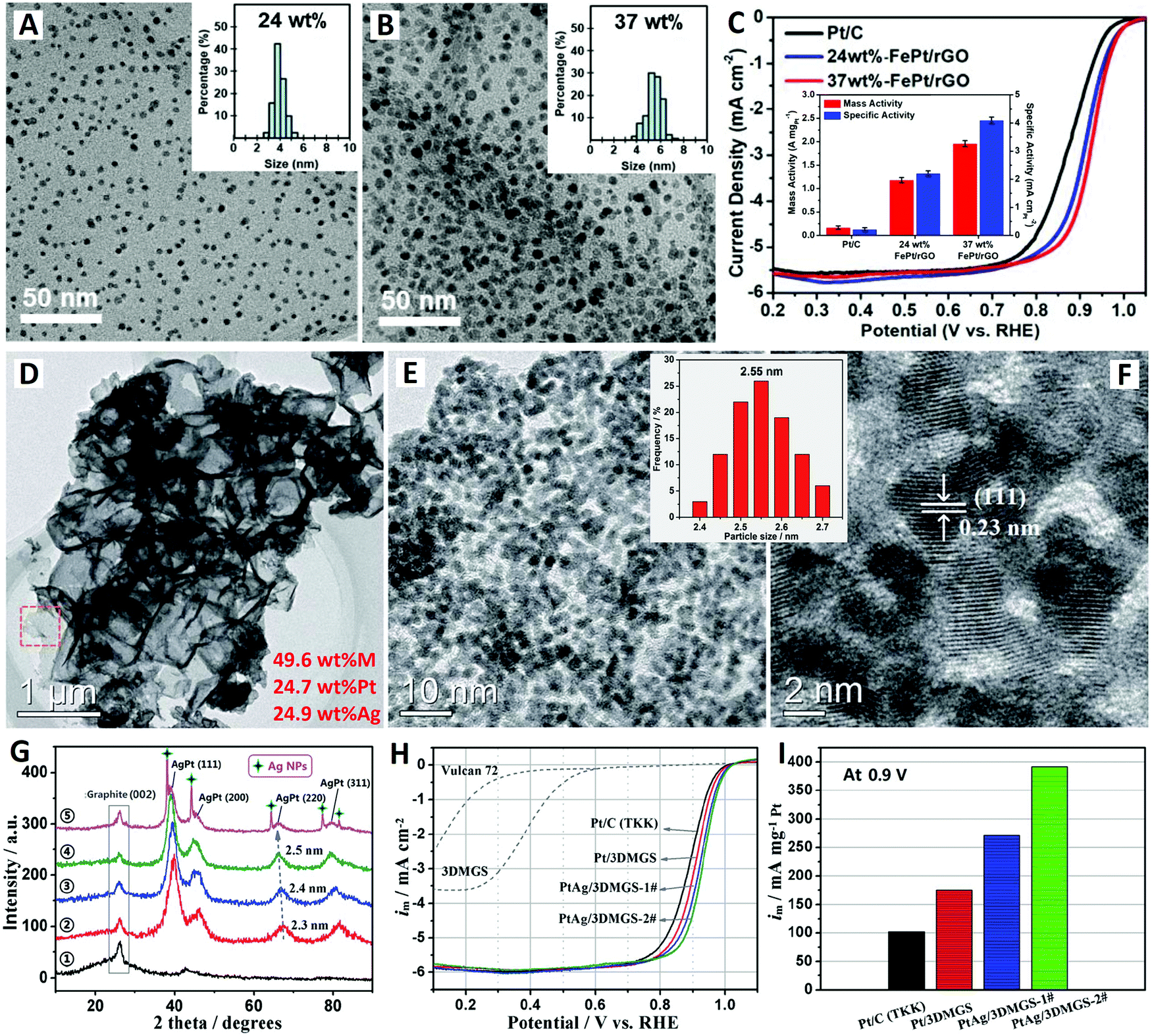 | ||
| Fig. 2 The rGO-supported FePt alloy catalyst (reprinted with permission from ref. 63): (A and B) TEM images and (C) electrochemical performances. The 3-D MGS-supported PtAg alloy catalyst (reprinted with permission from ref. 70): (D–F) TEM images, (G) XRD patterns, and (H and I) electrochemical performances. | ||
Fu and co-workers64 also demonstrated a general hydrogel-freeze drying and annealing joint strategy for the synthesis of rGO-supported Pt3M (where M = Mn, Cr, Fe and Co) alloy NPs (Pt3M/rGO) with ultrafine particle size (about 3 nm) and dramatic monodispersity. With Pt3Mn/rGO as an example, after the annealing process at high temperature (700 °C) for 12 h, ultrasmall Pt3Mn ordered intermetallic NPs can be formed due to the confinement effect of the porous structure of rGO. The catalytic performance of Pt3Mn/rGO (∼3 nm, 22.5 wt%) for the ORR was evaluated and compared with that of commercial Pt/C (∼3.3 nm, 20 wt%). The Pt3Mn/rGO catalyst exhibited greatly improved kinetic activity (1.37 times) compared to the commercial Pt/C at 0.9 V potential. The improved catalytic activity can be due to three factors: (i) the ultrafine particle size of Pt3Mn NPs affords rich active sites, (ii) the ordered intermetallic phase promotes the shift of the d-band of Pt3Mn NPs, and (iii) the high graphitization degree of rGO improves the conductivity of the catalyst.64
For traditional PtM (where M is transition metal) alloy ORR electrocatalysts, their electrochemical stability is far from meeting the requirements of practical applications, and the dissolution of transition metal is always a key problem affecting fuel cell systems.65 In order to design stable alloy electrocatalysts, the use of acid-soluble transition metals should be avoided or reduced as much as possible. Some noble metals with relatively low price or rich reserves, such as Ag, Pd and even Au, can be selected to prepare newly Pt-based alloy electrocatalysts.66–68 As a cheap noble metal with the highest conductivity, Ag can be used to design cost-effective AgPt alloy ORR electrocatalysts. In addition to the “structural adjustment” of alloys to improve their stability, the “support enhancement” of alloys can also be considered. For example, one can develop highly-stable support materials (e.g., 3-D porous graphene) to improve the stability of catalysts in terms of support enhancement.
Previously, Shen and co-workers prepared a new type of 3-D mesoporous graphene nanosheet conductive network (3-D MGS) material by an efficient synchronous graphitization-activation technology.69 The 3-D MGS material has highly mesoporous structures with high permeability and well graphitized structures with high conductivity. Recently, Li et al. further synthesized a high-metal-content AgPt alloy ORR electrocatalyst based on this novel 3-D MGS as a support material (see Fig. 2D–I).70 Typically, when the total metal content is 49.6 wt% (24.7 wt% Pt + 24.9 wt% Ag), highly-dispersed and highly-dense AgPt alloy NPs with ultrafine size (2.4–2.7 nm) are loaded onto the surface of the 3-D MGS support without any agglomeration (see Fig. 2D–F). The uniformity of the particle size distribution of PtAg/3DMGS (49.6 wt%) is much better than that of PtAg/Vulcan XC-72 (25.7 wt%),71 indicating that the 3DMGS with small mesoporous structures (2–10 nm) is more beneficial for the preparation of catalysts with high metal content. The average crystal sizes of Pt/3DMGS (2) (46.1 wt%), PtAg/3DMGS-1# (3) (45.3 wt%), and PtAg/3DMGS-2# (4) (49.6 wt%) are estimated to be 2.3, 2.4, and 2.5 nm based on (220) planes in XRD patterns (see Fig. 2G).
The ORR electrocatalytic activities of the series of catalysts were evaluated using the RRDE in oxygen-saturated 0.1 M HClO4 aqueous solution. The PtAg/3DMGS-2# catalyst exhibits better ORR performance; it shows 43 mV higher half-wave potential (3.0 mA cm−2) than commercial Pt/C catalyst (47.6 wt%, TKK) based on the same Pt loading (10 mgPt) on the electrode (see Fig. 2H). This promising PtAg/3DMGS-2# catalyst exhibits an ultrahigh mass activity (at 0.9 V) of 392 mA mgPt−1, which is nearly 4 times that of commercial Pt/C catalyst (102 mA mgPt−1) (see Fig. 2I). The PtAg/Vulcan XC-72 (25.7 wt%) catalyst shows a lower mass activity (at 0.9 V) of 316 mA mgPt−1 under the same test conditions.71 Moreover, comparable electrocatalytic stability (81.6% of the initial activity) is also obtained for PtAg/3DMGS-2# through an accelerated durability test with 1000 CV cycles. The excellent electrocatalytic performances of the PtAg/3DMGS catalysts are mainly attributed to the structural advantages and electronic effects of the ultrafine PtAg alloy NPs. Meanwhile, the convenient binary channels for both electron transport and ion diffusion of 3DMGS conductive networks are also supposed to be beneficial for their performance improvement.70
The M@Pt core–shell structure can be generally prepared by a seed-mediated galvanic replacement strategy by in situ displaced growth of a Pt shell on a M core (seed).74–76 Galvanic replacement usually takes place between a noble metal salt (high standard reduction potential) and transition metal (low standard reduction potential), which can be inhibited during the growth of Co@Pt NPs by using carbon monoxide as a stabilizing ligand and a reducing agent.75 However, these Co@Pt NPs need to be further loaded onto a carbon support before the measurement of electrochemical performances. Recently, Jia and co-workers developed a facile galvanic replacement method to achieve gradient Pt–Ni alloys (with Pt-rich surfaces), followed by a partial dealloying approach for the construction of core–shell (Pt–Ni alloy core with defective (D) Pt shell (≈3 monolayers)) NPs directly on a graphene carbon support (Pt–Ni@PtD/G) as a high-performance ORR electrocatalyst (see Fig. 3A–D).76
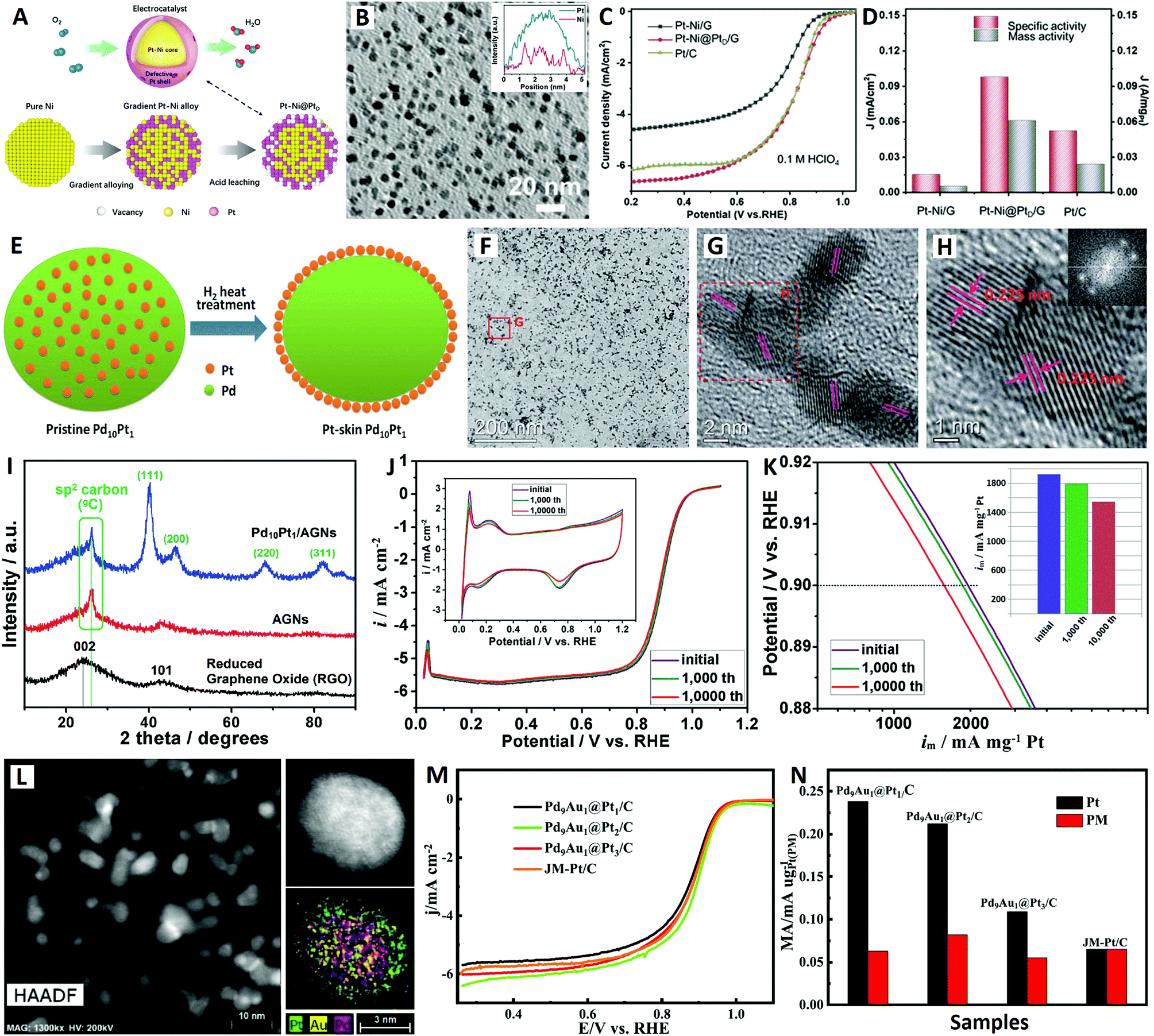 | ||
| Fig. 3 The Pt–Ni@PtD/G core–shell catalyst (reprinted with permission from ref. 76): (A) structural schematic diagram, (B) TEM image, and (C and D) electrochemical performances. The Pt-skin Pd10Pt1/AGN catalyst (reprinted with permission from ref. 79): (E) structural schematic diagram, (F–H) TEM images, (I) XRD patterns, and (J and K) electrochemical performances. The Pd9Au1@Pt/C core–shell catalyst (reprinted with permission from ref. 81): (L) TEM images and (M and N) electrochemical performances. | ||
The graphene (G)-supported Ni NPs were directly reacted in chloroplatinic acid to form gradient Pt–Ni alloys on G by galvanic replacement reaction. Then, the Pt–Ni@PtD/G core–shell catalyst was obtained by an acid leaching process to gradually etch surface Ni atoms in the Pt–Ni alloys (see Fig. 3A). The high-density Pt–Ni@PtD/G NPs were homogeneously dispersed on the graphene surface with particle sizes of around 5 nm (see Fig. 3B). The Pt–Ni@PtD/G core–shell NPs performed as a promising catalyst for the ORR in 0.1 M HClO4 electrolyte with a mass activity threefold higher than that of the Pt/C counterpart (see Fig. 3C and D), which is mainly due to the optimized electronic structure of Pt because of the synergetic effect between the Pt–Ni core and defective Pt shell. More importantly, the Pt–Ni@PtD/G core–shell NPs showed extremely enhanced stability arising from the Pt shell preventing the dissolution of the Ni core, which solve a long-standing issue of unstable Pt–Ni alloy in an acidic ORR.76
On the other hand, PtM (where M = Fe, Co, Ni, etc.) alloys with Pt-skin surfaces can be produced by thermally-induced surface segregation in specific atmospheres (CO, NO, O2, H2, etc.).77 Different PtM alloys require different atmospheres for the surface segregation of Pt atoms. For example, PtCo alloy NPs can be transformed into PtCo@Pt-skin NPs by heat treatment in a CO atmosphere.78 Recently, Li et al. successfully constructed an ultra-low-Pt Pd10Pt1 bimetallic catalyst (with a Pt-skin surface) by heat treatment in a H2 atmosphere for the ORR (see Fig. 3E–K).79 Particularly, a novel active graphene-like nanosheet (AGN) material with high specific surface area and high conductivity was developed as an efficient support to form a high-metal-density Pt-skin Pd10Pt1/AGN catalyst (the total metal content was 32.04 wt% (29.19 wt% Pd + 2.85 wt% Pt)) (see Fig. 3F–I). The Pd10Pt1/AGN catalyst exhibited excellent catalytic activity and superb durability for the ORR, giving a high mass activity of 1930 mA mgPt−1 (equal to 1185 mA mgPt−1 when the activity of Pd is taken into account) at 0.9 V in 0.1 M HClO4 electrolyte (see Fig. 3J and K). Relying on the remarkable supporting effect of AGNs and the favorable Pt-skin core–shell structure, a highly active and stable ORR electrocatalyst was demonstrated in this study.79
Previously, Adzic et al. reported a novel core-catalyzed coating strategy for epitaxial growth of Pt atomic layers on the surfaces of Pd NPs, inspired by aerobic alcohol oxidation catalyzed by Pd cores (where alcohol is employed as both a reducing agent and solvent).80 In addition, noble metal Au-incorporation is a promising strategy to retard composition-loss and boost the catalytic durability for Au-core/Pt-based-shell structured catalysts (via outward diffusion of Au atoms in the subsurface).61 More recently, inspired by the above-mentioned two design strategies, Qin and co-workers developed a Pd9Au1@Pt/C core–shell catalyst via a Pd9Au1-catalyzed coating route in ethanol solution with carbon black (Vulcan XC-72) as support for the ORR (see Fig. 3L–N).81 The as-synthesized Pd9Au1@Pt NP catalyst presents well-defined core–shell structures superposed on a carbon support (see Fig. 3L), and the thickness of the Pt layers can be controlled by tuning the amount of Pt precursor during synthesis. The Pd9Au1@Pt/C catalyst with 2 atomic Pt layers exhibits the best mass activity for the ORR (see Fig. 3M and N), and excellent stability as evidenced by its even increased half-wave potential after 10000 CV cycles in oxygen-saturated 0.1 M HClO4 electrolyte. This greatly enhanced ORR stability can be attributed to the compressive strain and stabilizing effect of the Pd9Au1 core on the Pt shell.81
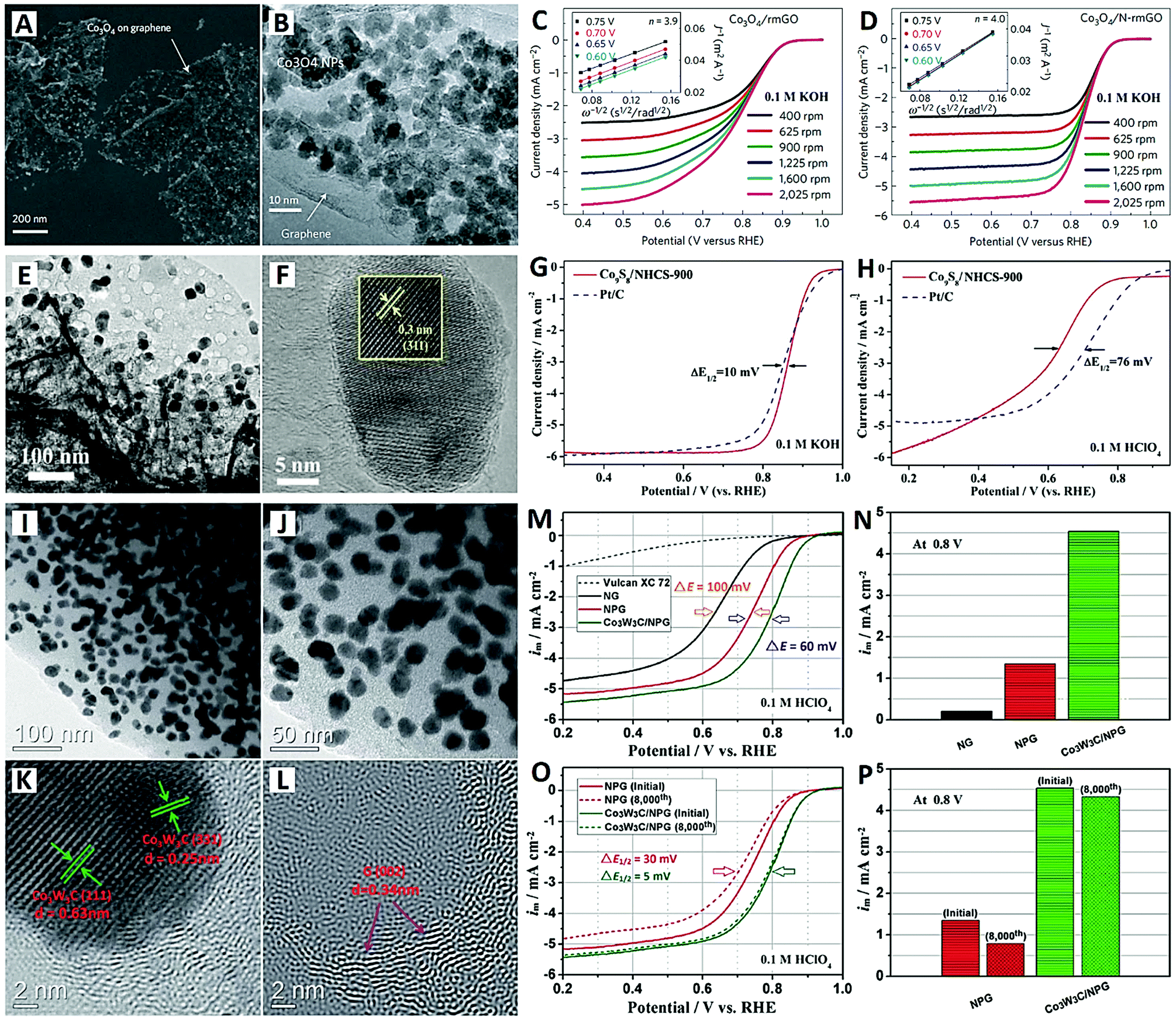 | ||
| Fig. 4 The Co3O4/N-rmGO catalyst (reprinted with permission from ref. 84): (A and B) SEM and TEM images, and (C and D) electrochemical performances. The Co9S8/NHCS-900 catalyst (reprinted with permission from ref. 85): (E and F) TEM images and (G and H) electrochemical performances. The Co3W3C/NPG catalyst (reprinted with permission from ref. 88): (I–L) TEM images and (M–P) electrochemical performances. | ||
To achieve both high catalytic efficiency and outstanding stability, Wang et al. designedly synthesized monodispersed Co9S8 NPs embedded in N-doped hierarchical carbon nanoflakes (denoted Co9S8/NHCS) as an ORR catalyst, by direct carbonization of metanilic anion-confined 2-D cobalt–aluminum layered double hydroxides (CoAl-LDHs) (see Fig. 4E–H).85 The electrochemical results showed that the Co9S8/NHCS material prepared at 900 °C exhibited superior ORR catalytic activity in both alkaline (0.1 M KOH) and acidic (0.1 M HClO4) electrolytes (see Fig. 4G and H). High onset and half-wave potentials of 0.97 and 0.86 V were obtained in 0.1 M KOH electrolyte, which are comparable to those of commercial Pt/C (1.00 V and 0.85 V). And in 0.1 M HClO4 electrolyte, the Co9S8/NHCS-900 catalyst showed only a 76 mV difference in half-wave potential when compared with commercial Pt/C. The average value of the electron number (n) is 3.95 and 3.65 for alkaline and acidic electrolytes respectively. The durability of Co9S8/NHCS-900 is much better than that of Pt/C in both electrolytes. The high-density Co9S8 NPs with small particle size provide abundant active sites and high catalytic activity. The synchronous carbonization of CoAl-LDHs makes the Co9S8 NPs embedded within the carbon nanoflakes, leading to strong anchoring force and outstanding stability.85
Carbide-based electrocatalysts (e.g., tungsten carbide, WC) have been intensively studied because of their Pt-like behavior and intrinsic catalytic activity for chemical catalysis.86 Especially, bimetallic carbides showed more enhanced electrocatalytic activity for the ORR due to their special electronic structure and the synergistic effect of the dual metal species.87 Recently, Li and co-workers proposed a composite of cobalt–tungsten bimetallic carbide NPs and N/P co-doped graphitized carbon (Co3W3C/NPG) as a highly active and stable non-noble-metal catalyst for the acidic ORR (see Fig. 4I–P).88 Obviously, extremely high-density Co3W3C NPs of about 20 nm size were loaded onto the graphitized carbon support (see Fig. 4I–L). The carbon content of Co3W3C/NPG was determined to be 39.8% by TGA; in other words, the Co3W3C loading onto the NPG support was up to 60.2%. The newly Co3W3C/NPG catalyst presented a high onset potential of 0.92 V, a large limiting current (5.3 mA cm−2) and an especially high half-wave potential (0.79 V) in 0.1 M HClO4 electrolyte (see Fig. 4M–P). The slightly decreased half-wave potential of 5 mV and the high electron transfer number of 3.95 after 8000 cycles indicated that the catalyst undergoes a steady four-electron process in acidic medium. The excellent performances can be attributed to two factors: (i) the Co element in the carbide endows it with Co/N/C and Co/P/C double active sites; and (ii) the W species in the carbide is a synergistic component that boosts the activity of the hybrid catalyst system.88
Recently, He et al. utilized large-surface-area graphene as a support to load high-density WC and Pt NPs, which showed high activity and high stability for the ORR in 0.1 M HClO4 (see Fig. 5A–C).93 A microwave-assisted method was used to synthesize truncated hexagonal pyramid (THP) WC with 5 nm in size on graphene (WCTHP/G) first, then Pt NPs on the WCTHP/G composite with close-contact Pt–WC interfaces were prepared by a chemical adsorption/reduction method (see Fig. 5A). Pt–WCTHP/G showed much higher ORR onset potential (1.052 vs. 0.993 V), half-wave potential (0.942 vs. 0.900 V) and mass activity (528 vs. 137 mA mgPt−1) values compared with commercial Pt/C (see Fig. 5B and C). The enhancement of ORR activity on Pt–WCTHP/G is mainly due to the synergistic effect of Pt and WC, which correlates with the change in the Pt surface d-band center caused by the EMSI on the Pt–WC interfaces. Furthermore, the EMSI can also increase the binding energy of oxygen on Pt–WCTHP/G, thereby enhancing its catalytic activity toward the ORR.93
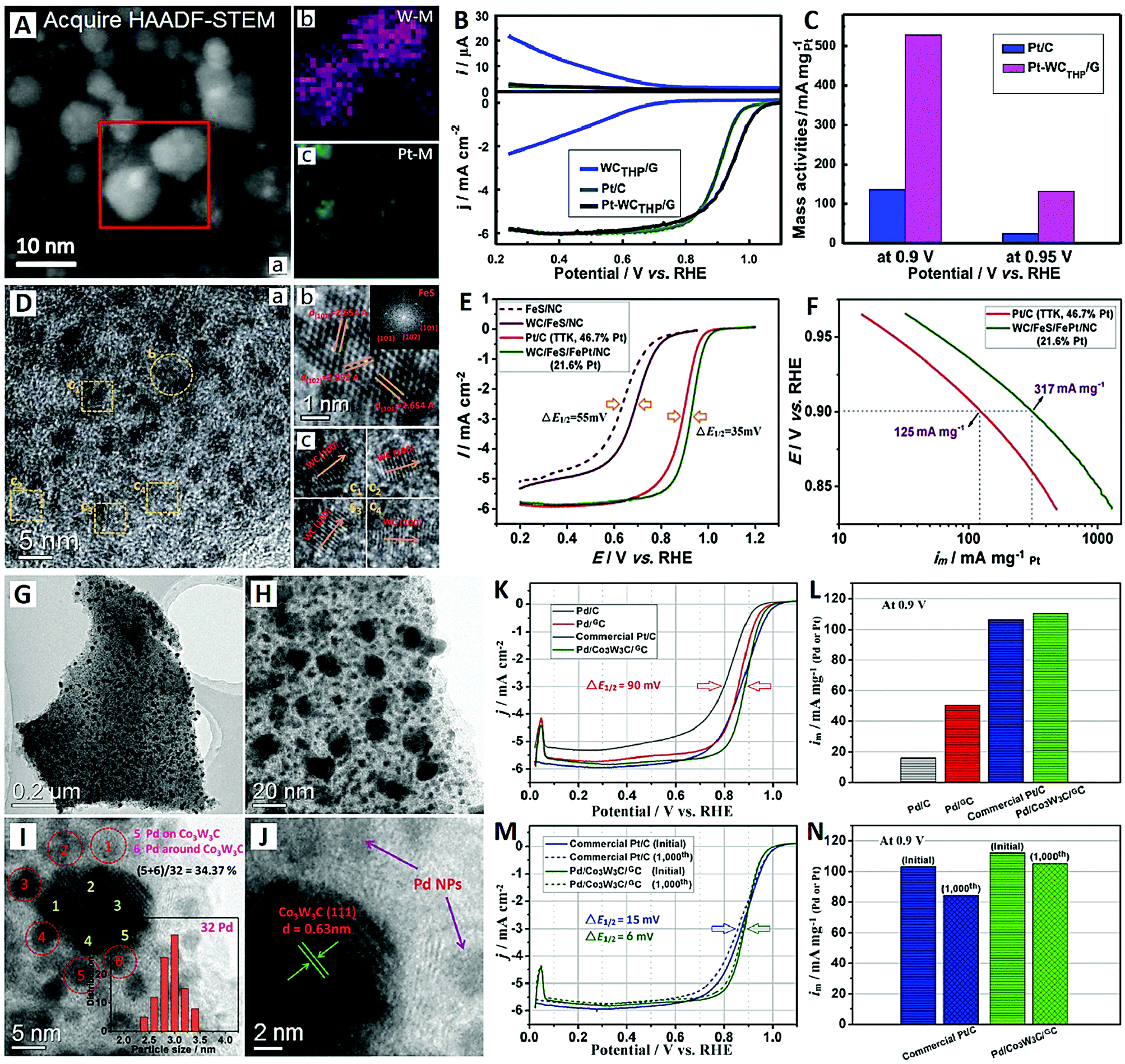 | ||
| Fig. 5 The Pt–WCTHP/G catalyst (reprinted with permission from ref. 93): (A) HAADF-STEM images and (B and C) electrochemical performances. The WC/FeS/FePt/NC catalyst (reprinted with permission from ref. 94): (D) TEM images and (E and F) electrochemical performances. The Pd/Co3W3C/GC catalyst (reprinted with permission from ref. 95): (G–J) TEM images and (K–N) electrochemical performances. | ||
The development of multi-component hybrid catalysts offers great promise to enhance catalytic performance for the ORR. Li and co-workers reported a quaternary hybrid material composed of WC, FeS, FePt alloy and N-doped carbon (NC), i.e. WC/FeS/FePt/NC hybrid architecture, as a high-performance electrocatalyst for the ORR (see Fig. 5D–F).94 Due to the efficient ternary promoting effects from WC, FeS and NC, the FePt alloy electrocatalyst exhibits an excellent mass activity of 317 mA mgPt−1, which is much higher than that of Pt/C catalyst (125 mA mgPt−1) (see Fig. 5E and F). Moreover, superior durability of the WC/FeS/FePt/NC is also demonstrated for the ORR in acidic electrolyte (0.1 M HClO4). This hybrid catalyst has three structural advantages over previous reports:89–91 (i) ultrafine 1-D WC nanorods are prepared by an FeS-regulated strategy; (ii) FePt alloy is formed by in situ transformation on FeS without an external Fe source; and (iii) multiple synergistic effects on the FePt alloy might be in operation relying on the WC, FeS and NC components.
Recently, Pt-free electrocatalysts based on Pd have been proposed as promising candidates for the ORR in acidic electrolyte, due to the inherent catalytic activity and lower cost of Pd compared to Pt. For example, Li and co-workers synthesized a high-performance Pd electrocatalyst (Pd/Co3W3C/GC) for the ORR, which was synergistically enhanced using Co3W3C and graphitic carbon (GC) (see Fig. 5G–N).95 The GC nanosheet was uniformly decorated with the ultrahigh-density and well-proportioned Co3W3C NPs (∼20 nm) and Pd NPs (∼3 nm) (see Fig. 5A and B). The Pd NPs were deposited not only onto the carbon substrate but also onto Co3W3C, indicating a strong interaction between the Co3W3C and Pd NPs (see Fig. 5C and D). It was estimated that more than 30% of the Pd NPs were deposited onto or closely around the Co3W3C, which provided necessary conditions for interface synergistic effects.91 Due to the synergistic effects of Co3W3C and acceleration by GC, the Pd/Co3W3C/GC electrocatalyst showed much higher activity than Pd/GC and Pd/C in a 0.1 M HClO4 electrolyte, and the mass activity of Pd/Co3W3C/GC (110 mA mgPd−1) was comparable to that of commercial Pt/C (107 mA mgPt−1) (see Fig. 5K and L). After the 1000th cycle, a high activity retention of 93% was achieved for Pd/Co3W3C/GC, which was higher than that of commercial Pt/C (82%) (see Fig. 5M and N). These excellent properties make it a highly active and stable Pt-free acidic ORR electrocatalyst. For the sake of convenience, the comparison parameters of different nanoscale ORR electrocatalysts are summarized in Table 1.
| Ref. | Name of the catalyst | Type of catalyst | Content of metals | Electrolyte | Onset potential | Half-wave potential | Limited diffusion current density | Specific activity/electron transfer number (n)/H2O2 yield/Tafel slope |
|---|---|---|---|---|---|---|---|---|
| [50] | Pt/GNP500 | Single metal NPs | 39.2 wt% Pt | 0.5 M H2SO4 | 0.91 V | 0.80 V | 5.9 mA cm−1 | 80 mA mg−1 Pt@0.8 V |
| [54] | Pt dendrites/C | Single metal NPs | 37.5 wt% Pt | 0.1 M HClO4 | 1.02 V | 0.92 V | 6.0 mA cm−1 | 251 mA mg−1 Pt@0.9 V |
| [63] | FePt/rGO | Metal alloy NPs | 37.0 wt% Pt | 0.5 M H2SO4 | 1.00 V | 0.93 V | 5.6 mA cm−1 | 1960 mA mg−1 Pt@0.9 V |
| [70] | PtAg/3DMGS | Metal alloy NPs | 49.6 wt% PtAg | 0.1 M HClO4 | 1.02 V | 0.93 V | 6.0 mA cm−1 | 392 mA mg−1 Pt@0.9 V |
| [76] | Pt–Ni@PtD/G | Metal core–shell NPs | — | 0.1 M HClO4 | 0.94 V | 0.83 V | 6.3 mA cm−1 | 610 mA mg−1 Pt@0.9 V |
| [79] | Pd10Pt1/AGNs | Metal core–shell NPs | 32.0 wt% PdPt | 0.1 M HClO4 | 1.00 V | 0.89 V | 5.7 mA cm−1 | 1185 mA mg−1 Pt@0.9 V |
| [81] | Pd9Au1@Pt/C | Metal core–shell NPs | 19.6 wt% PdAuPt | 0.1 M HClO4 | 0.94 V | 0.89 V | 6.1 mA cm−1 | 239 mA mg−1 Pt@0.9 V |
| [84] | Co3O4/N-rmGO | Metal compound NPs | 70.0 wt% Co3O4 | 0.1 M KOH | 0.93 V | 0.83 V | 5.0 mA cm−1 | n = 3.90 |
| [85] | Co9S8/NHCS | Metal compound NPs | 19.6 wt% Co9S8 | 0.1 M KOH/0.1 M HClO4 | 0.97 V/0.78 V | 0.86 V/0.63 V | 6.0 mA cm−1/5.0 mA cm−1 | n = 3.95/n = 3.65 |
| [88] | Co3W3C/NPG | Metal compound NPs | 60.2 wt% Co3W3C | 0.1 M HClO4 | 0.92 V | 0.79 V | 5.3 mA cm−1 | n = 3.95 |
| [93] | Pt-WCTHP/G | Synergistic hybrid NPs | — | 0.1 M HClO4 | 1.05 V | 0.94 V | 6.0 mA cm−1 | 528 mA mg−1 Pt@0.9 V |
| [94] | WC/FeS/FePt/NC | Synergistic hybrid NPs | 28.4 wt% Pt | 0.1 M HClO4 | 1.03 V | 0.92 V | 6.0 mA cm−1 | 317 mA mg−1 Pt@0.9 V |
| [95] | Pd/Co3W3C/GC | Synergistic hybrid NPs | 30.0 wt% Pd | 0.1 M HClO4 | 0.98 V | 0.87 V | 5.6 mA cm−1 | 110 mA mg−1 Pd@0.9 V |
| [118] | Fe–N–C/N-OMC | Single-atom catalysts | 2.9 wt% Fe | 0.1 M KOH | 1.08 V | 0.93 V | 8.1 mA cm−1 | n = 3.93 |
| [120] | SA-Fe-NHPC | Single-atom catalysts | 1.25 wt% Fe | 0.1 M KOH | 1.04 V | 0.93 V | 5.9 mA cm−1 | n = 3.92 |
| [121] | Commercial Fe–N–C | Single-atom catalysts | — | 0.1 M KOH | 0.97 V | 0.85 V | 7.1 mA cm−1 | n = 3.97 |
| [123] | Fe–N4 SACs | Single-atom catalysts | 8.02 wt% Fe | 0.1 M KOH | 1.00 V | 0.84 V | 5.8 mA cm−1 | 1570 mA mg−1http://mailto:Fe@0.85 V |
| [124] | Fe–N–C SACs | Single-atom catalysts | 1.5 at% Fe | 0.5 M H2SO4 | 1.03 V | 0.88 V | 4.0 mA cm−1 | H2O2 < 1% |
| [125] | Fe–N–C SACs | Single-atom catalysts | 2.5 wt% Fe | 0.5 M H2SO4 | 0.94 V | 0.82 V | 5.4 mA cm−1 | 0.047 A cm−2 at 0.88 ViR-free |
| [128] | Co–N4 SACs | Single-atom catalysts | 1.0 at% Co | 0.5 M H2SO4 | 0.93 V | 0.82 V | 4.0 mA cm−1 | H2O2 < 1.5% |
| [129] | Fe2N6 DACs | Dual-atom catalysts | 4.9 wt% Fe | 0.5 M H2SO4 | 0.92 V | 0.82 V | 5.0 mA cm−1 | n = 3.96 |
| [130] | Co2N5 DACs | Dual-atom catalysts | — | 0.1 M HClO4 | 0.90 V | 0.79 V | 6.0 mA cm−1 | n = 3.97; 7468 mA mgCo2−1 |
| [131] | Fe,Co N–C DACs | Dual-atom catalysts | — | 0.1 M HClO4 | 1.06 V | 0.86 V | 6.0 mA cm−1 | n = 3.98 |
| [132] | FeCoN6 DACs | Dual-atom catalysts | 10 wt% FeCo | 0.1 M KOH/0.1 M HClO4 | 0.95 V/0.91 V | 0.89 V/0.81 V | 5.7 mA cm−1/5.0 mA cm−1 | n = 3.97 |
| [133] | Fe–N4|Co–N4 DACs | Dual-atom catalysts | 3.27 wt% FeCo | 0.1 M KOH/0.5 M H2SO4 | 0.98 V/0.91 V | 0.86 V/0.75 V | 5.7 mA cm−1/5.2 mA cm−1 | n = 3.97; 52 mV dec−1 |
| [136] | Co/PC SCCs | Atom cluster catalysts | — | 0.1 M KOH | 1.00 V | 0.92 V | 6.0 mA cm−1 | 66 mV dec−1 |
| [137] | CuZn/NC SCCs | Atom cluster catalysts | 0.77 wt% CuZn | 0.1 M KOH | 1.00 V | 0.89 V | 5.2 mA cm−1 | 45 mV dec−1 |
| [138] | FeAC@FeSA–N–C | SACs + SCCs | 4.0 wt% Fe | 0.1 M KOH | 1.00 V | 0.91 V | 6.1 mA cm−1 | 61 mV dec−1 |
| [139] | Fe–N–C | SACs + SCCs | 2.9 wt% Fe | 0.1 M KOH | 0.98 V | 0.90 V | 3.8 mA cm−1 | n = 3.85 |
| [140] | FeSA/FeONC/NSC | SACs + SCCs | 0.25 wt% FeSA | 0.1 M KOH | 0.95 V | 0.85 V | 4.5 mA cm−1 | n = 3.87 |
| [142] | Co-SAC/SNPs@NC | SACs + NPs | 5.8 wt% Co | 0.1 M KOH | 0.97 V | 0.90 V | 6.0 mA cm−1 | n = 3.96 |
| [143] | CoNPs@Fe–N4–C | SACs + NPs | 0.1 wt% Co | 0.1 M KOH | 1.01 V | 0.92 V | 5.0 mA cm−1 | n = 3.95 |
| [144] | FeCo–N–C | SACs + NPs | 6.6 wt% FeCo | 0.1 M KOH | 0.98 V | 0.85 V | 4.8 mA cm−1 | n = 3.97 |
4.2 Atomic-level ORR electrocatalysts
4.2.1.1 Origin of carbon-supported single-atom catalysts (Me/N/C) for the ORR. Noble metal Pt catalysts are still the most efficient ORR electrocatalysts, but their scarcity, high price and unsatisfactory stability badly hinder their large-scale application.6–8 Therefore, on the premise of guaranteeing high performance, it is of great importance to develop cost-effective low-Pt, Pt-free, or even non-noble metal electrocatalysts. The introduction of heterogeneous element nitrogen (N) and active transition metal (Me) components on carbon materials is an extensive and efficient strategy for the engineering of non-noble metal (including Me/N/C and M–N4/C single-atom catalysts) ORR electrocatalysts.22,96
The initially developed non-noble metal ORR electrocatalysts are Me/N/C catalysts (where Me includes transition metals, such as Fe, Co, Ni and Cu, N is chelating atomic nitrogen, and C is sp2 carbon).97 The earliest non-noble metal electrocatalysts were macrocyclic compounds containing nitrogen transition metals, such as phthalocyanines and porphyrins. The common structure of these macrocyclic compounds is their planar configuration MeN4 structure, which is also the most likely active site in the Me/N/C catalyst. Some of the studies have shown that the structures of MeN2 and MeN2+2 may also be the active sites for oxygen reduction. Therefore, the structure formula of the active sites of Me/N/C non-noble metal electrocatalysts can be expressed as: Me/Nx/C (x = 2 and 4, or 2 + 2).98 The early Me/N/C non-noble metal catalysts were usually in the form of composites with transition metal NPs and N-doped carbon, laying more emphasis on the pyrolytic synthesis of catalysts, as well as the effect of the size of metal NPs, the content of N, and the thickness of the graphited layer on ORR performances.
In recent years, since the original concept of “single-atom catalysts (SACs)” (noble metal Pt single atoms on oxide supports) was proposed by the Zhang group,23 numerous noble metal (Pt, Pd, Ag, Ir, etc.) and transition metal (Fe, Co, Ni, Cu, etc.) SACs on carbon supports have been discovered in different electrocatalytic fields.22,99 The formation of M–N4 active moieties (where M includes noble or transition metals) from atomically dispersed metal (M) atoms and the coordination N atoms doped onto carbon supports is the main reason for the stabilization and high ORR activity of these carbon-supported SACs.25,100 In general, the research priorities of carbon-supported SACs (M–N4) ORR electrocatalysts are as follows: (i) adjusting the coordination environment and electronic structures of M centers, (ii) optimizing the nanoscale morphology and pore structures of carbon supports, (iii) increasing the number of metal active centers by increasing the metal density, and (iv) investigating the atomic interaction and synergistic effect of single atoms with different metal densities.
4.2.1.2 Synergistic effect of high-density single-atom catalysts on carbon supports. In the field of catalysis, when the distance between the two catalytically active centers is reduced to the atomic scale, the interaction between the active centers will greatly affect the catalytic performance of the catalyst (including catalytic selectivity and conversion).101 Due to the high surface energy of single atoms, the metal loading in carbon-supported SACs is generally low (<1.5 wt%) in order to avoid the agglomeration of single metal atoms. Therefore, the distance between two adjacent active sites is relatively far (≫1 nm), and the interaction between two single atoms is basically ignored.102 In fact, in order to improve the practicability of SACs, it is necessary to fix more metal atoms per unit area to realize the design of high-density single-atom active sites, which will make the metal atoms come close together, and the interaction between adjacent sites cannot be ignored.99 If the metal loading in carbon-supported SACs can be increased so that it not only maintains the feature of high dispersion of single atoms, but also increases the interaction between adjacent atoms, it is highly expected to realize the synergistic effect between these adjacent single atoms.103 The synergistic effect between metal atoms may exist in single or dual atom systems with the same metal or different metals, which plays roles in enhancing the adsorption of reaction substrates, reducing the reaction potential barrier, changing the reaction path, etc.104 Therefore, it is a very important topic to study the synergistic effect of single atoms. It enriches the regulation means of SACs, emphasizes the importance of site interaction, and has positive guiding significance for the design of carbon-supported SACs with high dispersion and high metal density.
However, some of the current carbon-supported SACs cannot provide enough reaction sites in ORR catalytic reactions due to their low metal loading.44–46 Generally, at least two adjacent metal atoms (i.e., two adjacent active centers) need to cooperate effectively to promote the efficient four-electron mechanism. Therefore, the existing noble metal SACs with low metal loading usually exhibit oxygen reduction by the two-electron mechanism because they cannot cooperate with each other.105 Many carbon nanocomposites with a M/N/C structure, especially those in atomically-dispersed M–N4 (where M = Fe, Co, Ni, etc.) moieties, exhibit ORR catalytic properties that are even comparable to commercial Pt based catalysts.25,99,100 The latest research shows that in M/N/C catalyst, when the dispersed M–N4 single sites are close enough (i.e., when the metal density is large enough), there will be strong interaction between two adjacent single atoms, so as to form the synergetic paired active centers of M–N4 and significantly improve the ORR catalytic performances.104,106 The specific reasons can be attributed to the following aspects: (i) adjacent metal atoms may change the adsorption energy and adsorption state of catalytic species; (ii) the synergistic effect may be conducive to the activation of chemical bonds and reduce the reaction energy barrier; and (iii) the adjacent paired active centers may also change the reaction route of the intermediate and lead to fast kinetics.
4.2.1.3 Preparation strategies of high-density single-atom catalysts on carbon supports. In recent years, carbon-supported SACs have become the hotspot due to their high atomic utilization, definite active centers, and high catalytic performances. So far, various synthesis strategies have been developed to prepare carbon-supported SACs:107 (i) SACs can be synthesized by impregnation in aqueous dispersion, where the metal precursors are anchored onto carbon by physical adsorption or chemical coordination;22 (ii) because bulk metal can be changed into gaseous species by high-temperature treatment, the thermal emission and atom trapping method has been developed to prepare sintering-resistant SACs on pyrolytic carbon supports;99 (iii) atomic layer deposition (ALD) and chemical vapor deposition (CVD) technologies are powerful and effective strategies for controllable synthesis of SACs and cluster catalysts;26 and (iv) some other physical and chemical synthesis methods are used to prepare SACs, such as ball milling and electrochemical deposition.107
However, due to the easy migration and aggregation of active atoms in the preparation process, the high loading of SACs onto carbon supports is still a great challenge.25 Common synthesis methods of M–N–C (in M–N4 moieties) SACs include the process of high-temperature pyrolysis. Increasing the density of active sites only by increasing the concentration of metals usually leads to inevitable metal clusters and metal NPs.99Researchers have achieved some impressive results by using the following methods to achieve highly-dispersed and high-density SACs on carbon supports: (i) using a large number of complexing agents to form coordination bonds with metal ions to physically isolate the metal sources at the molecular scale, ensuring the formation of highly-dispersed single atom sites by subsequent pyrolysis;108 (ii) nanoscale carbon supports with high specific surface area and rich functional groups are used to realize the high-density loading of metal complexes relying on their high pore volume and rich coordinated groups;26 and (iii) high-density M–N4 moieties can be generated by the synchronous pyrolysis of N-containing precursors and metal complexing compounds, where a porous carbon network is generated as an effective cascaded anchoring carrier.99 N-Doped carbon (N–C) is an ideal support for the preparation of high-metal-density SACs, where the N with lone pair electrons can form a M–N4 coordination bond with metal species, which greatly improves the thermal and chemical stability of SACs. The synthetic strategies of N–C materials mainly include: use of (1) nitrogen-containing organic compounds (such as phenanthroline, porphyrin and phthalocyanine); (2) MOFs and their derivatives; (3) N-doped graphene and CNTs; (4) g-C3N4 with high nitrogen content; and (5) rich, cheap and renewable nitrogen-containing biomass.34
In the following, we will introduce the design strategies and preparation methods of high-density SACs on carbon supports through several typical cases according to the “physical synthesis” and “chemical synthesis” categories. For the physical synthesis, ALD and CVD synthetic strategies are analyzed contrastively. Recently, Yan et al. devised a reliable ozone-assisted multicycle ALD technique for the preparation of high-metal-loading Co1/G SACs, which allows the precise tuning of the density of isolated Co single atoms (Co1) on the graphene (G) support (see Fig. 6A–C).109 During ALD cycles, the self-limiting surface reaction ensures that each Co precursor molecule is anchored onto a single active site of the graphene, and the active site (epoxy groups) can be re-generated by the secondary reaction between ozone and the graphene surface (see Fig. 6A). The expected result is that the ALD in different cycles can be used to accurately control the synthesis of SACs with different metal loadings. A series of Co1/G catalysts with Co loadings of 0.4, 0.8, 1.3, 2.0, and 2.5 wt% (see Fig. 6B1–B5) were synthesized by performing 1, 2, 3, 4, and 5 cycles of ozone-assisted Co ALD respectively (see Fig. 5C). The density of Co single atoms loaded onto graphene is closely correlated with the amount of epoxy groups on the support, which further supports the idea that the epoxy groups act as anchor sites for the Co precursors. The ozone-assisted multicycle ALD not only completes a single cycle of ALD, but also regenerates the active sites of chelating single atoms, conducive to the precise tuning of the density of Co single atoms.109
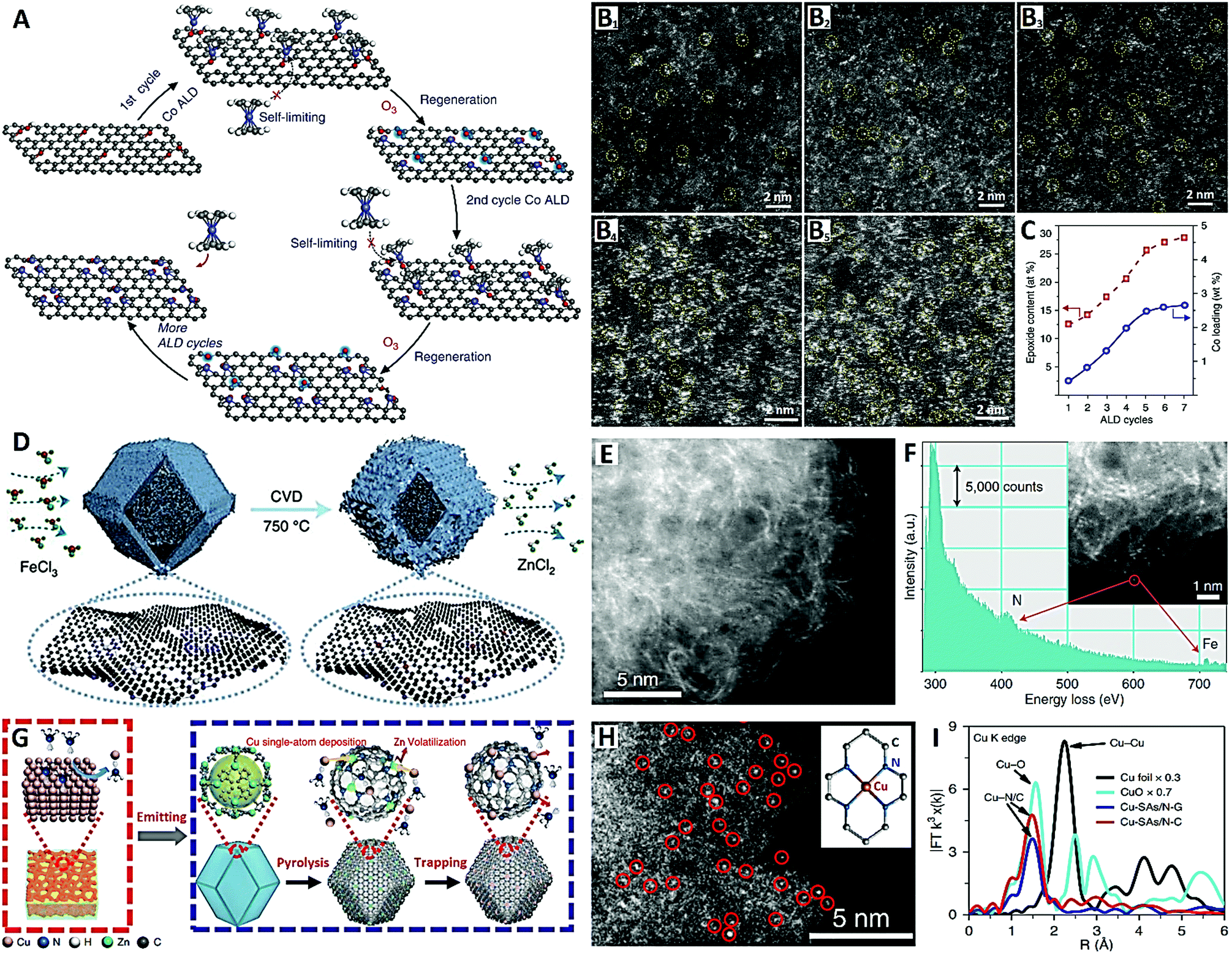 | ||
| Fig. 6 The Co1/G SACs prepared by ALD (reprinted with permission from ref. 109): (A) schematic diagram of the synthesis, (B) HAADF-STEM images and (C) ALD cycle relational graph. The Fe–N–C SACs prepared by CVD (reprinted with permission from ref. 110): (D) schematic diagram of the synthesis and (E and F) HAADF-STEM images. The Cu–N–C SACs prepared by CVD (reprinted with permission from ref. 112): (G) schematic diagram of the synthesis, (H) HAADF-STEM image and (I) EXAFS spectra. | ||
In order to increase active site density and site utilization, Jiao et al. proposed a metal-transferred CVD technique to synthesize Fe–N–C by flowing iron chloride (FeCl3) vapor over a Zn–N–C substrate at 750 °C, leading to in situ trans-metalation of Zn–N4 sites into Fe–N4 sites (see Fig. 6D–F).110 Zeolite imidazole framework (ZIF-8) nanocrystals with a uniform size of about 80 nm were first prepared to obtain a Zn–N–C substrate with high-density Zn–N4 sites (see Fig. 6D). After the in situ CVD trans-metalation, carbon-supported Fe-based SACs with high-density Fe–N4 sites were readily achieved (see Fig. 6E and F). These Fe–N4 sites formed by this transformation approach were in the gas-phase and electrochemically accessible, and the catalyst had a high active site density of Fe–N4 (1.92 × 1020 sites g−1) with 100% site utilization.110 Wang et al. lately reported Fe–N–C SACs (Fe–N4 sites) obtained by pyrolysis of ferric (Fe3+) ion-adsorbed porous N-doped carbon derived from ZIF-8, which also showed a high density of accessible surface Fe–N4 sites (2.63 × 1020 sites g−1).111 The high porosity of the ZIF-8-derived N-doped carbon support is a key factor in building Fe–N–C SACs with a high density of accessible-sites.
Because bulk copper can be changed into gaseous species of Cu(NH3)x at high temperature and in an ammonia environment, a thermal emitting and atom trapping strategy was developed to prepare Cu–N–C SACs (Cu–N4 sites) on a pyrolytic carbon support from ZIF-8 (see Fig. 6G–I) by the Li group.112 In the NH3 atmosphere, Cu(NH3)x species were trapped by Zn-vaporized defects in the N-doped carbon, forming isolated Cu sites, and then forming Cu–N4 catalysts (see Fig. 6G). Relatively high metal density and close atomic distance (∼1 nm) were demonstrated for these Cu–N4 catalysts (see Fig. 6H and I). Overall, this work developed an NH3-assisted gas migration strategy (i.e., facile CVD strategy) that enabled the direct conversion of bulk metal to M–N4 single atoms, bringing new hope for large-scale preparation and industrial applications of SACs on carbon supports.
For the chemical synthesis of carbon-supported SACs, the wet-chemistry and pyrolysis synthetic strategies are widely investigated.23 The wet-chemistry method usually includes impregnation, co-precipitation, and electrochemical deposition, and belongs to the “top-down approach” based on ready-made carbon supports with vacancy modification (see Fig. 7A-(1)). This synthetic approach can easily lead to inevitable metal clusters or metal NPs when increasing the concentration of metals, and the subsequent acid treatment is therefore a necessary step.113 On the other hand, the pyrolysis method belongs to the “bottom-up approach”, where SACs are directly prepared from metal node-containing nitrogen and carbon precursors (such as MOFs and COFs) by pyrolysis (see Fig. 7A-(2)). For example, Jiang and co-workers114 recently reported atomically-dispersed Ni metal on N-doped carbon nanotubes (CNTs) with a high Ni loading (20 wt%) via the direct pyrolysis “bottom-up approach” with Ni(acac)2 and C2H8N2 as precursors (see Fig. 7B). Wu and co-workers115 also reported high-density Fe SACs (<1 nm in distance) supported on N-doped carbon nanosheets by a Phen-molecule-confined pyrolysis “top-down approach” with FeCl2, polyetherimide (PI) and g-C3N4 as precursors (see Fig. 7C and D). Importantly, during pyrolysis, the closed Fe ions are directly reduced by carbonization of PEI to form isolated Fe atoms without additional acid treatment.
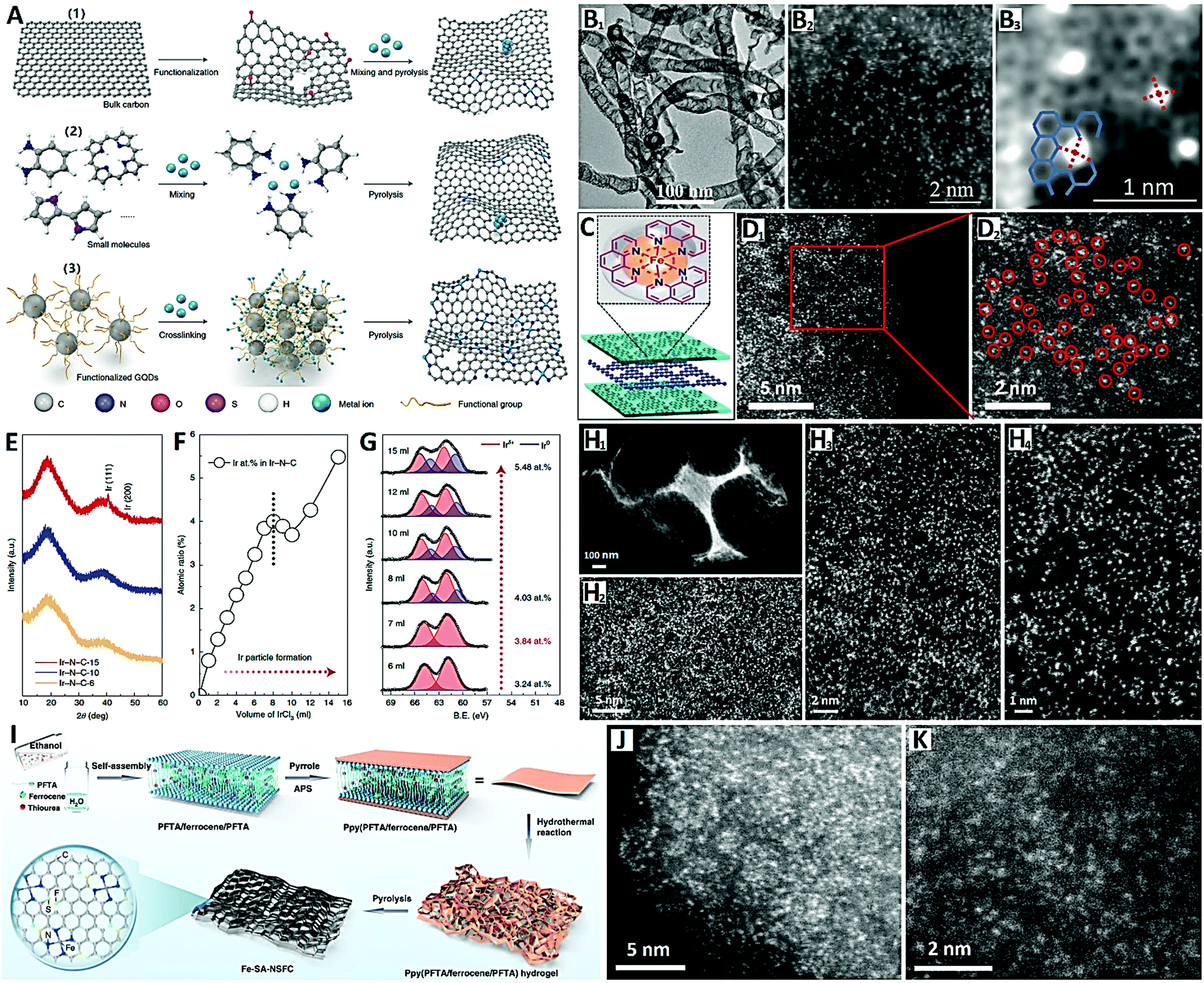 | ||
| Fig. 7 (A) Schematic diagram of carbon-supported SACs by chemical synthesis; (B) HAADF-STEM images of Ni–N–C SACs prepared by pyrolysis (reprinted with permission from ref. 114); (C) schematic diagram and (D) HAADF-STEM images of Fe–N–C SACs obtained by pyrolysis (reprinted with permission from ref. 115); (E) XRD patterns, (F) atomic ratios, (G) XPS spectra, and (H) HAADF-STEM images of Fe–N–C SACs obtained by pyrolysis (see (A) for the schematic diagram) (reprinted with permission from ref. 116); (I) schematic diagram of the synthesis and (J and K) HAADF-STEM images of M-SA-NSFC (reprinted with permission from ref. 117). | ||
Noble metal SACs have high catalytic activity per metal site, but it is usually difficult to achieve a high density of SACs (usually less than 3 wt%), so the overall catalytic activity is limited. Therefore, developing a general synthesis strategy to greatly improve the atom density of noble metal SACs and make the metal loading close to or even exceed the commercial benchmark (e.g., 20 wt% Ir/C) will play a key role in the field of electrocatalysis. In view of this, Xia and co-workers116 developed a universal method lately for synthesizing record high-metal-loading (41.6 wt%) Ir SACs on graphene quantum dots (GQDs) (see Fig. 7A-(3) and E–H). Specifically, when functionalized with amine groups and mixed with IrCl3, GQDs can stably and uniformly diffuse and limit Ir3+ on their surfaces (Ir3+/GQDs-NH2) due to the strong chelation effect between Ir3+and amine groups (see Fig. 7A-(3)). This strong interaction helps GQDs connect with each other and crosslink into a 3-D network structure during the freeze-drying process (see Fig. 7H1). Then pyrolysis was carried out in an ammonia rich atmosphere to obtain a GQD-supported Ir SAC (3.84 at% or 41.6 wt%) with 8 ml of IrCl3 (see Fig. 7E–G). The GQD-supported Ir SAC (Ir–N4 sites) reveals an extremely-uniform and highly-dense distribution of Ir atoms (∼0.5 nm in distance) on GQDs (see Fig. 7H2–H4).
Starting from amine-group functional graphene quantum dots (GQDs-NH2), the authors had the following motives: (i) compared with the carbon support in the “top-down approach”, GQDs are small enough to provide many N-doped active sites for anchoring a large number of isolated metal atoms; and (ii) compared with the organic precursors in the “bottom-up approach”, GQDs, as an intermediate carbon support, do not undergo significant structural evolution during pyrolysis, providing stable and large spacings between metal atoms to avoid aggregation.116 The authors also obtained other noble metal (Pt) SACs or transition metal (Ni) SACs with similarly high metal loadings (32.3 wt% and 15.0 wt% respectively) on GQDs, demonstrating the favorable universality and generality of this GQD-NH2-guided synthetic strategy for high-loading metal single-atom architectures.
Furthermore, the design of tunable coordination environments (e.g., second coordination regulation) for carbon-supported SACs with high metal loading has special significance for catalytic performance regulation. Recently, Zhou et al. reported a multilayer stabilization strategy for constructing M–N4 (where M = Fe, Co, Ru, Ir and Pt) SACs with high metal loading (∼16 wt%) on N, S and F co-doped porous graphitized carbons (i.e., M-SA-NSFC) (see Fig. 7I–K).117 The metal precursors (ferrocene) are embedded into perfluorotetradecanoic acid (PFTA) multilayers and further coated with polypyrrole (Ppy) prior to hydrothermal reaction and pyrolysis (see Fig. 7I). The confinement by the PFTA and Ppy multilayers can efficiently prevent metal precursors from migrating during the pyrolysis process, resulting in the efficient coordination of high-density metal atoms (∼0.5 nm in distance) with N atoms in graphitized carbon (see Fig. 7J and K). This multilayer stabilization bottom-up strategy coupled with the N, S and F co-doping conception is very promising, in terms of abundant metal species, the control of metal loading and second coordination regulation by S (or long-range regulation by F), to explore the potential of high-loading M-SACs and their diversified catalytic applications.117
4.2.1.4 Application of high-density single-atom catalysts on carbon supports for the ORR. Among many transition metal SACs, Fe–N–C catalyst has the most outstanding ORR performance. In alkaline electrolyte, the performance of Fe–N–C catalyst is even better than that of Pt/C catalyst.97 Fe–N–C catalysts are mainly prepared by pyrolysis of precursors containing Fe, N and C, where C and N precursors are transformed into an N-doped carbon skeleton, and Fe atoms coordinate with N atoms to obtain FeN4 active sites.100 The N-doped carbon skeleton obtained by direct pyrolysis contains many disordered micropores, which results in low utilization of FeN4 active sites due to the low mass transfer kinetics in the micropores. In addition, the activity of the catalyst depends on the intrinsic activity of single metal sites and the number of accessible active sites.118 Therefore, increasing the metal loading of Fe–N–C SACs and improving the accessibility of FeN4 active sites is an effective way to further improve the ORR performance of Fe–N–C catalyst.
Because ORR activity is controlled by kinetics and mass transfer processes, in order to better mass diffusion, optimizing the pore structure of catalysts can effectively affect their ORR activity. The rapid transfer of O2 from bulk solution to active centers is a key step in providing high current density.70 The three types of pore structures that can promote the mass transfer of O2 in porous catalysts are macropores (>50 nm), mesopores (2–50 nm) and micropores (<2 nm).69 Each type of pore has a unique function to improve ORR activity: (i) macropores are designed to ensure that reactants quickly pass through the entire dense catalyst layer; (ii) mesopores ensure that electrolytes and reactants enter the active centers (e.g., FeN4 active sites) deeply buried under the surface of the catalyst; and (iii) micropores help in increasing the number of active sites and the total surface area of the catalyst.119
Recently, Feng and co-workers120 designed a novel N-doped hierarchical micro/meso-porous carbon (NHPC)-supported Fe–N–C SAC with densely available FeN4 species by a Zn-mediated silica-template strategy, and applied it to a high-performance alkaline ORR (see Fig. 8A–E). Firstly, the DAP/ZnFe/SiO2 composite was prepared by drying colloidal silica containing 2,6-diaminopyridine (DAP), zinc nitrate and iron nitrate. The obtained DAP/ZnFe/SiO2 was heat treated at 900 °C under nitrogen for 2 hours, then etched in HF followed by secondary pyrolysis in nitrogen to obtain the final SA-Fe-NHPC SAC (see Fig. 8A). There are many mesopores on the Fe-NHPC SAC, with an average diameter of about 12 nm, which is consistent with the size of the silica template (see Fig. 8B). In addition, the HAADF-STEM image of the Fe-NHPC SAC shows that the dense single Fe atoms are well dispersed on the carbon matrix with rich micropores by Zn vaporization (see Fig. 8C). The Fe-NHPC SAC showed unprecedentedly high ORR activity in 0.1 M KOH electrolyte, and the half wave potential was 0.93 V (see Fig. 8D and E), which is much better than those of Pt/C catalyst (0.82 V) and commercial Fe–N–C catalyst (0.85 V) (see Fig. 8F).121 This work opens a new way to improve the density and accessibility of FeN4 species and develop high-performance Fe–N–C ORR catalysts. In addition, Fe–N–C/N-OMC catalysts with N-doped ordered mesoporous carbon (N-OMC) can also provide highly accessible FeN4 active sites and reduce mass transfer resistance.118 Therefore, the Fe–N–C/N-OMC catalysts show extremely high ORR activity in 0.1 M KOH electrolyte, with a half wave potential of 0.93 V and a limited current density of 8.14 mA cm−2. To sum up, the design of mesoporous carbon and high metal loading provide double guarantee for high-performance SACs.
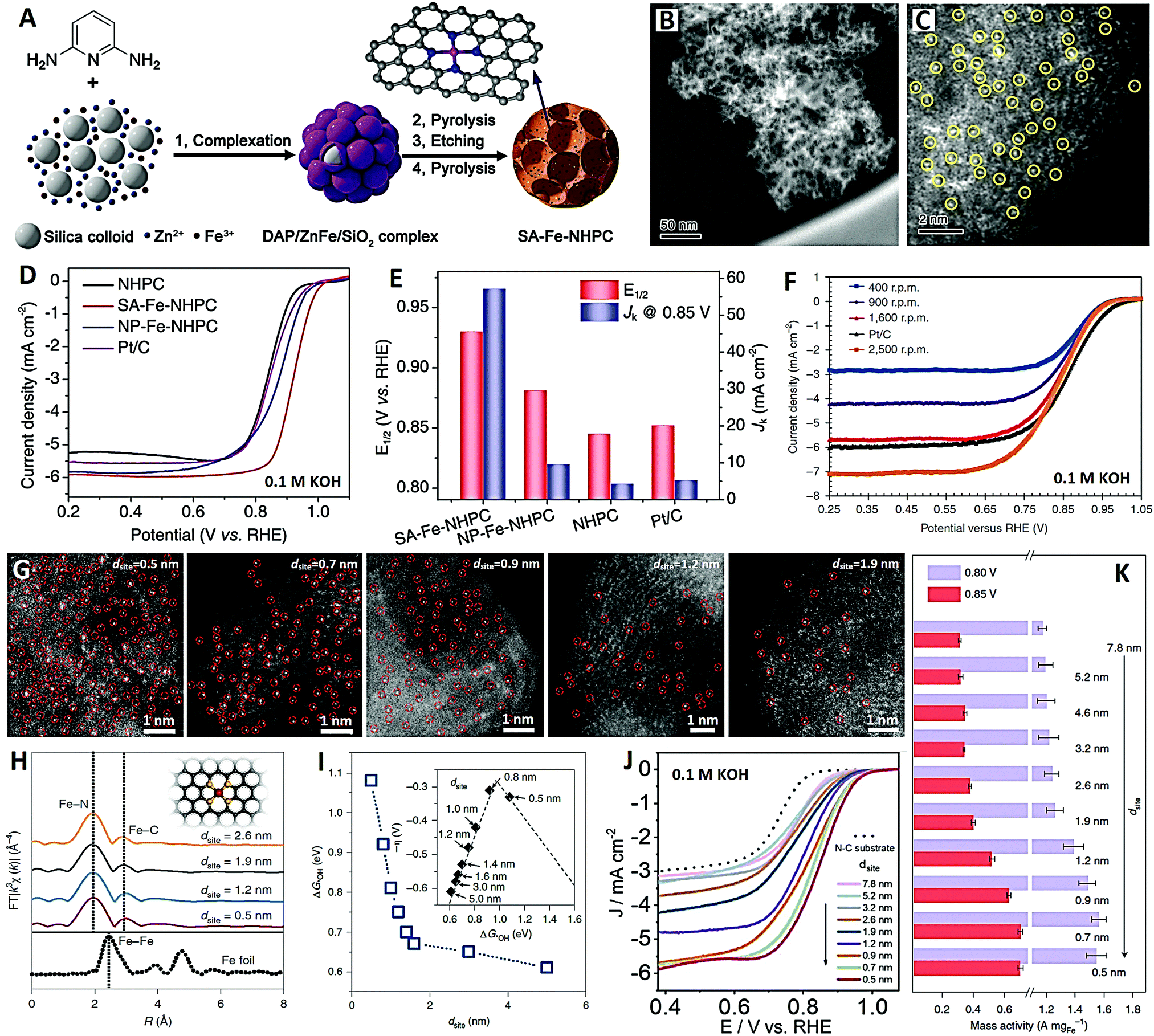 | ||
| Fig. 8 The SA-Fe-NHPC catalyst (reprinted with permission from ref. 120): (A) schematic diagram of synthesis, (B and C) HAADF-STEM images and (D and E) electrochemical performances. (F) ORR curves of commercial Fe–N–C catalyst (reprinted with permission from ref. 121). The Fe–N4 SACs with various site distances (dsite) (reprinted with permission from ref. 123): (G) HAADF-STEM images, (H) EXAFS spectra, (I) Gibbs free energy, (J) ORR curves and (K) mass activity for various dsite values. | ||
When the density of single-atom active sites increases to a certain extent, the distance between single atoms will become very small, and the interaction between adjacent active sites becomes a key factor affecting intrinsic activity. And the adjacent single-atom active sites can cooperate to produce an optimized electronic structure, which affects the catalytic activity and catalytic path of SACs in heterogeneous catalysis.122 Previous studies have shown that adjusting the active site density of SACs can significantly improve the electrocatalytic performance of the ORR due to the synergistic effect.104–106 At the same time, the recent breakthrough in the synthesis method of SACs has helped the metal loading to exceed 10 wt%, which exceeds the performance of Pt/C (20 wt%).23 Although high-density SACs have better overall activity for the ORR, the catalytic behavior controlled by the distance between single active sites has not been deeply studied.
With that in mind, Yu and Xiao recently studied and revealed the origin of the enhancement of ORR activity (in 0.1 M KOH electrolyte) of isolated Fe–N4 SACs with the different intersite distances at the sub-nanometer level, by integrating the experimental and theoretical methods (see Fig. 8G–K).123 Atomically-dispersed Fe–N4 sites with controllable density and various site distances (dsite) were prepared on an N-doped carbon substrate via the hydrogel anchoring strategy (see Fig. 8G). When the dsite was controlled at 0.5–2.6 nm, favorable Fe–N4 moieties were obtained without Fe clusters or nanoparticles (see Fig. 8H). The theoretical and experimental results indicate that, when the dsite value is less than 1.2 nm, the strong interaction between adjacent Fe–N4 sites changes the electronic structure and improves the inherent activity of the ORR. This distance-dependent enhanced activity could be maintained until the dsite value was close to 0.7 nm; when the dsite was further reduced, the ORR activity decreased slightly (see Fig. 8I–K). This study determined in detail the kinetic behavior of a single active site and the site distance effect of adjacent single metal atoms, which provides an important opportunity to further understand the inherent catalytic behavior of carbon-supported SACs. And understanding the site distance effect of Fe–N4 catalyst is of great significance to the mechanism of the ORR, which is helpful to realize the full potential of densely distributed SACs in electrocatalysis.123
Platinum group metal-free (PGM-free) catalysts for the ORR with atomically-dispersed Fe–N4 sites have emerged as potential catalysts for acidic polymer electrolyte fuel cells (PEFCs).26 Therefore, it is very important to study the acidic ORR performance (in H2SO4 or HClO4) of carbon-supported Fe–N–C SACs with high Fe–N4 site density. Previously, polyaniline/cyanamide-based Fe–N–C SACs with rich Fe–N4 sites at graphene edges exhibited a half-wave potential of 0.80 V and a limited current density of 3 mA cm−2 in 0.5 M H2SO4 electrolyte.100 The Fe–N–C catalyst with the highest density of Fe–N4 active sites achieved from ZIF-8 precursors also showed respectable ORR activity with a half-wave potential of 0.88 and a limited current density of 4 mA cm−2 in 0.5 M H2SO4 electrolyte.124 Mesoporous silica coated ZIF-8 derived Fe–N–C with dense active sites and efficient mass transport exhibited a desirable half-wave potential of 0.82 V and a larger limited current density of 5.4 mA cm−2 in 0.5 M H2SO4 electrolyte.125 Compared with ZIF-8-derived Fe–N–C prepared via wet chemical synthesis, the CVD method can reduce the pore size of the Kat-Zn (MeIm)2 (Kat) phase to reduce the formation of inert metal aggregates, resulting in a much higher density of Fe–N4 active sites and excellent catalytic performance in 0.5 M H2SO4 electrolyte.126 Besides, previously mentioned high-density Fe SACs on porous carbon nanosheets presented a half-wave potential of 0.80 V and a larger limited current density of 5.5 mA cm−2 in 0.1 M HClO4 electrolyte.115 In general, it is difficult for the ORR half-wave potential of Fe–N–C in acidic electrolyte to reach 0.9 V as in alkaline electrolyte,118,121 so the development of acidic PEFCs with PGM-free catalysts is a challenging task.
On the other hand, carbon-supported Co–N–C SACs with a high-density of Co–N4 active sites are also promising candidates for PGM-free catalysts, by virtue of their high intrinsic activity and immunity to Fenton reactions (for Fe–N–C SACs) that occurred in the proton exchange membrane of fuel cells.26,96 The research on the relationship between the site density of Co–N4 moieties and the activity of Co–N–C SACs is also very necessary for achieving optimized performances in fuel cells. In view of this, the Shui team synthesized a series of Co–N–C SACs with different densities of Co–N4 active sites for a quantitative study of the structure–property relationship.127 It was found that in the low-density region, the battery power density increased slowly and linearly with the Co–N4 site density, while in the high-density region, an accelerated growth trend in terms of power density was observed. These results confirm the fact that M–N–C SACs with limited intrinsic activity can effectively improve the performance of batteries by increasing the density of M–N4 active sites.97,100
More recently, the Shao team synthesized a high-performance atomically-dispersed Co(mIm)-NC catalyst by means of a microporous encapsulation-ligand exchange strategy, where ZIF-8, Co(acac)3, and 2-methylimidazole (mIm) were used as precursors.128 This strategy greatly increases the density of Co–N4 sites and therefore greatly increases ORR catalytic activity in 0.5 M H2SO4 electrolyte. The mass activity of the catalyst increased with the increase of Co content (0.28–1.0 at%), in which the Eonset and E1/2 values of Co(mIm)–NC (1.0 at% Co) were 0.93 V and 0.82 V, which are only 25 mV and 35 mV lower than those of Pt/C catalyst, respectively. In addition, the catalytic activity of this Co–N–C SAC is comparable to that of the Fe–N–C SACs, but the durability is increased by four times. This study provides a promising method to develop high activity and durable Fe-free and PGM-free ORR electrocatalysts (i.e., high-density single atom Co–N4/C) for PEM fuel cells.128
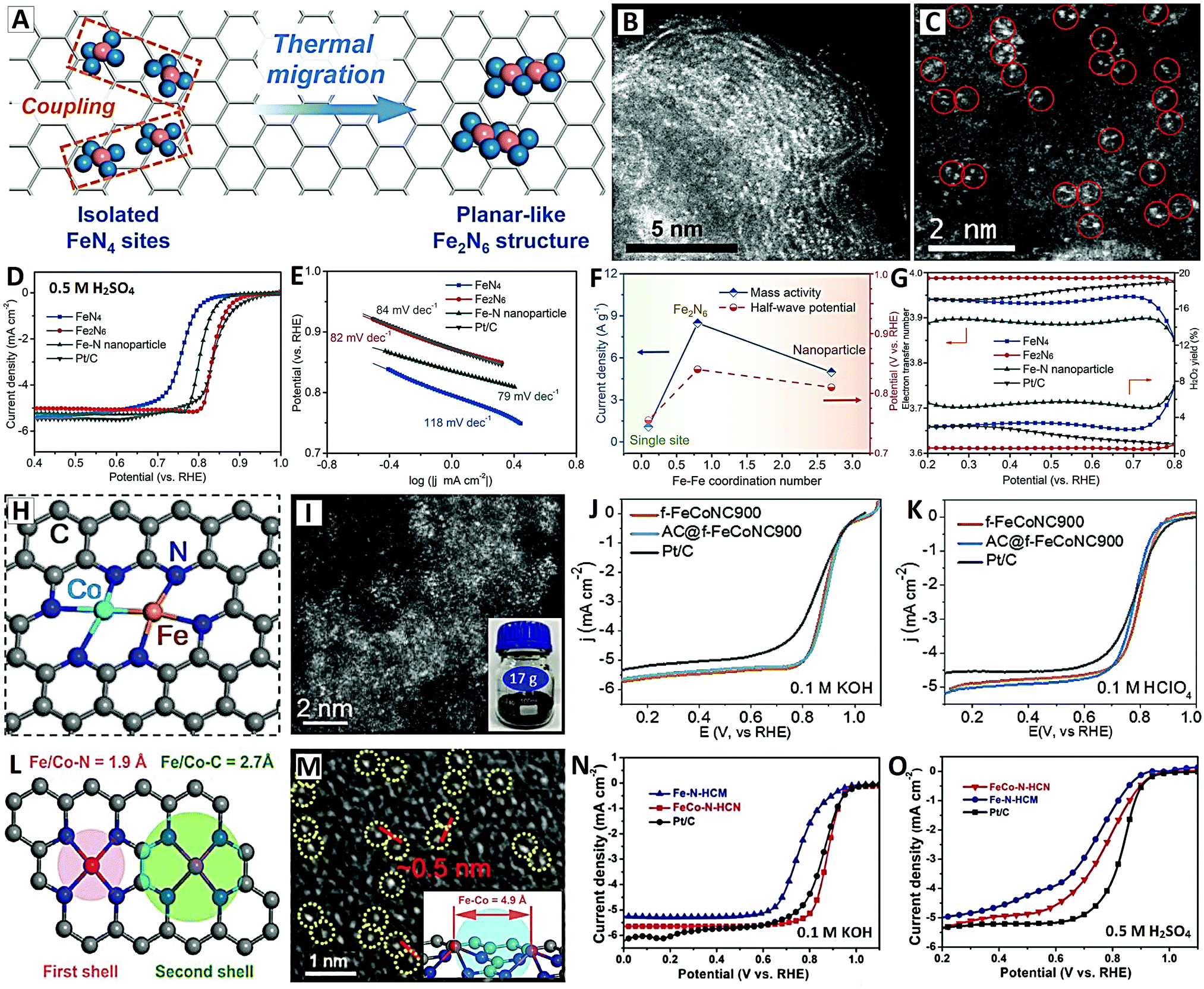 | ||
| Fig. 9 The Fe2N6 DACs (reprinted with permission from ref. 129): (A) structural schematic diagram, (B and C) HAADF-STEM images and (D–G) electrochemical performances; the FeCoN6 DAC (reprinted with permission from ref. 132): (H) structural schematic diagram, (I) HAADF-STEM image and (J and K) ORR curves in different electrolytes; and the FeN4|CoN4 DAC (reprinted with permission from ref. 133): (L) structural schematic diagram, (M) HAADF-STEM image and (N and O) ORR curves in different electrolytes. | ||
For example, Wu and co-workers unraveled planar-like Fe2N6 active sites (homonuclear DACs) as a highly efficient ORR catalyst, which showed greatly improved catalytic activity and excellent stability compared to isolated Fe–N4 sites (see Fig. 9A–G).129 The formation of planar-like Fe2N6 structures results from the thermal migration of isolated Fe–N4 sites in hemin molecules (see Fig. 9A). The planar-like Fe2N6 structure presents a high-density and homogeneous distribution of Fe atoms on the carbon framework (see Fig. 9B), and most of the Fe atoms get together to form many Fe2 dimer structures (see Fig. 9C). The planar-like Fe2N6 structure exhibits the highest catalytic activity with a high half-wave potential of 0.84 V (see Fig. 9D) and a small Tafel slope of 82 mV dec−1 (see Fig. 9E) in 0.5 M H2SO4 electrolyte. The Fe2N6 structure shows much enhanced mass activity and half-wave potential compared to single Fe–N4 and Fe NPs (see Fig. 9F). The electron transfer number of the Fe2N6 structure reaches 3.98 below 0.75 V, indicating an efficient four-electron reduction pathway (see Fig. 9G). The planar-like Fe2N6 follows a unique ORR redox transition from initial state Ox–Fe3+–Fe2+ to final state Fe2+–Fe2+, which triggers optimized oxygen intermediate adsorption and strong driving force for O–O bond breaking, thus promoting ORR kinetics and inhibiting side reactions.129
By tuning the Zn/Co ratio in a bimetallic MOF (ZnCo-ZIF8) and subsequent pyrolysis, non-planar binuclear Co2N5 sites with high density were also synthesized and identified (with a Co–Co distance of 0.212 nm and a protruding N between the two Co atoms).130 This homonuclear DAC shows optimized catalytic activity with a halfwave potential of 0.79 V, a limited current density of 6.0 mA cm−2, and an electron transfer number of 3.97 in 0.1 M HClO4 electrolyte. Excitingly, the intrinsic mass activity of Co2N5 sites is as high as 7468 mA mgCo2−1, which is about 12 times higher than that of isolated Co–N4 sites (560 mA mgCo1−1). Theoretical calculations reveal that this novel Co2N5 site exhibits a greatly reduced thermodynamic barrier towards the ORR, thus contributing to the much higher intrinsic activity. This finding opens up a new opportunity to design high-performance M–N–C catalysts, thus pushing the fuel cell industry one step ahead possibly.130
Recently, Wu and co-workers131 reported a host–guest synthesis of a high-density heteronuclear (Fe,Co)/N–C DAC (with FeCoN6 dual sites) on N-doped porous carbon from a Zn/Co bimetallic MOF (ZnCo-ZIF8) with adsorbed FeCl3 molecules. The bonding (Fe–Co) between the Co nodes (host) and adsorbed Fe ions (guest) can be precisely controlled within ZnCo-ZIF8 by a double-solvent method. Compared with Fe SAs/N–C, Co SAs/N–C, commercial Pt/C and those previously reported,129,130 this well-defined (Fe,Co)/N–C DAC exhibits a considerably high ORR activity with a record halfwave potential of 0.86 V, an onset potential of 1.06 V and a limited current density of 6.0 mA cm−2 in 0.1 M HClO4 electrolyte.131 Just like the catalytic behavior of Pt/C, the H2O2 yield of the (Fe,Co)/N–C DAC is below 1.17%, demonstrating the efficient four-electron mechanism (n = 3.98). Besides, excellent power density and long-term stability are also obtained in H2/air fuel cells, suggesting its great potential in shortening the gap between non-noble metal catalysts and commercial Pt/C catalyst.
Considering the high price of MOF-based precursors, it is of great significance to prepare bimetallic (Fe,Co)/N–C catalysts with cheap and extensive raw materials. Back in 2011, Wu et al.10 used commercial carbon black and polyaniline (PANI) as low-cost precursors for the high-temperature synthesis of (Fe,Co NPs)/N–C catalysts, which showed desirable activity with an onset potential of 0.93 V and a halfwave potential of 0.78 V in 0.5 M H2SO4 electrolyte. Recently, Zhang et al.132 reported an efficient synthesis of (Fe,Co)/N–C DACs (with FeCoN6 dual sites) based on a cost-effective route by condensation and carbonization of inexpensive formamide (FA) (see Fig. 9H–K). This general method can be used for mass production (16 g) of DACs with high-loading (∼10 wt% for Fe and Co) and atomically-dispersed FeCoN6 sites (see Fig. 9H and I). Under the same catalyst load, the ORR activity in alkaline (0.1 M KOH) and acidic (0.1 M HClO4) electrolytes is outstanding, and is better than that of commercial Pt/C (see Fig. 9J and K). The halfwave potential in alkaline and acidic electrolytes is 0.89 V and 0.81 V, respectively. More importantly, the extraordinary ORR activity was preserved when these FeCoN6 sites were deposited on inexpensive activated carbon (AC), presenting broad application prospects for implantable electrocatalysts or heterogeneous catalysts.132
The controllable preparation of adjacent dual active centers (M1–N4|M2–N4) is also a very promising approach to high-activity DACs for the ORR due to synergetic effects.134 More recently, Zhang et al.133 reported FeCo–N-doped hollow carbon nanocages (FeCo–N–HCN) with adjacent Fe–N4 and Co–N4 dual active centers as an efficient ORR electrocatalyst (see Fig. 9L–O). The theoretical calculations show that the Fe–Co distance in adjacent M1–N4|M2–N4 active centers is 0.49 nm, which is very close to the value (∼0.5 nm) obtained by experimental observation (see Fig. 9L and M). In 0.1 M KOH electrolyte, FeCo–N–HCN shows much higher ORR activity than Fe–N–HCN, even better than Pt/C, with halfwave potentials of 0.86 V, 0.76 V and 0.85 V, respectively. And the corresponding halfwave potentials are 0.75 V, 0.70 V and 0.84 V in 0.5 M H2SO4 electrolyte on FeCo–N–HCN, Fe–N–HCN and Pt/C. The synergistic effect of adjacent Fe–N4 and Co–N4 dual active centers reduces the energy barrier for the ORR by largely increasing the O–O bond on Fe–N4|Co–N4 DACs. This work provides new insights into and understandings about adjacent dual-active-centers at the atomic level.133
Recently, Zhou et al. devised porous carbon limited Co SCCs (Co/PC SCCs) as an efficient electrocatalyst for the ORR in alkaline electrolyte via a “top-down approach” from carbon black.136 Typically, Co(NO3)2 and phenanthroline were dissolved in ethanol and then a porous carbon black material (Ketjenblak) was added. Because this carbon black has a rich porous structure and Π–Π interaction with metal macrocyclic molecules, the Co-phenanthroline intermediate can be adsorbed in the nanopores. After sintering in an inert atmosphere at 750 °C for 2 h, Co clusters (0.5–1.0 nm) can be confined to the nanopores to form Co/PC SCCs (see Fig. 10A). The as-prepared Co/PC SCCs showed an extremely high initial potential (1.0 V), a half-wave potential (0.92 V), and a remarkable limited current density of 6.0 mA cm−2 in 0.1 M KOH electrolyte (see Fig. 10B). The Tafel slope of Co/PC was as low as 66 mV dec−1, indicating that the ORR kinetics was significantly enhanced, which was closely related to the rich active sites and higher O2 mass transfer effect (see Fig. 10C). In addition, the stability of Co/PC can be significantly improved by the nonporous confinement, where the attenuation of the half-wave potential and the limiting current density of the Co/PC catalyst can be ignored after 10000 CV cycles. Thanks to the water-resistance effect of the nanopores, the highly active Co clusters are confined in specific nanopores and have a stable gas–solid–liquid three-phase reaction region, which enhances the mass transfer capacity, makes full use of the active sites, and also has strong stability due to confinement.136
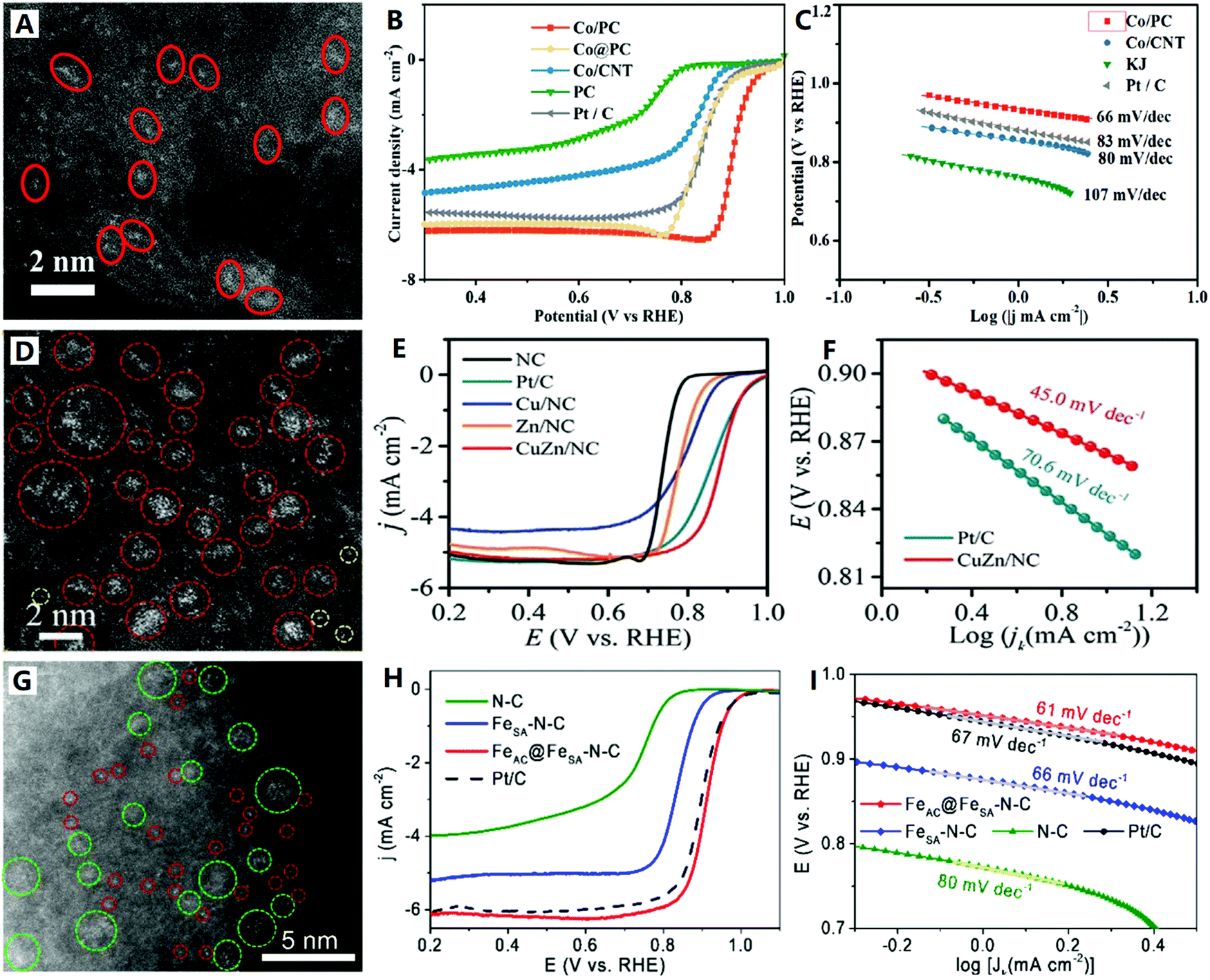 | ||
| Fig. 10 The Co/PC SCCs (reprinted with permission from ref. 136): (A) HAADF-STEM image and (B and C) electrochemical performances; the CuZn/NC SCCs (reprinted with permission from ref. 137): (D) HAADF-STEM image and (E and F) electrochemical performances; and the FeAC@FeSA–N–C (reprinted with permission from ref. 138): (G) HAADF-STEM image and (H and I) electrochemical performances. | ||
Zhou et al. also reported a simple one-step pyrolysis method (so-called “bottom-up approach”) for the preparation of bimetallic CuZn sub-nanoclusters (0.8 nm in mean size) anchored onto N-doped carbon (denoted CuZn/NC SCCs) with rhamnose (C6H12O5) as a carbon source.137 The formation of abundant homogeneous Cu and Zn sub-nanoclusters and a spot of single Cu/Zn sites on carbon carriers can be clearly observed in HAADF-STEM images (see Fig. 10D). An X-ray absorption fine structure test showed that the M–N bond (M = Cu or Zn) and M–M bond existed simultaneously in the catalyst. These N-coordinated Cu and Zn bimetallic structures make CuZn/NC SCCs exhibit high ORR activity in alkaline solution (0.1 M KOH), showing a half wave potential of 0.89 V (see Fig. 10E) and a Tafel slope of 45 mV dec−1 (see Fig. 10F). The ORR activity of CuZn/NC SCCs is even higher than that of commercial Pt/C catalyst. After a 30000 s continuous electrolysis, CuZn/NC SCCs showed a smaller current drop of 12.3% than Pt/C (49.4%), indicating a very high durability of these SCCs. Comprehensive experiments and theoretical calculations approved that the excellent performances of this catalyst can be attributed to the collaboration of the Cu/Zn–N4 sites with CuZn moieties on N-doped carbons.137
For example, Ao et al. recently designed and fabricated a multi-scale Fe–N–C ORR electrocatalyst, where abundant Fe atomic clusters and Fe–N4 single-atom sites are both embedded in an N-doped carbon support (FeAC@FeSA–N–C), by using a covalent organic framework (COF) as the precursor.138 The coexistence of Fe single atoms and Fe nanoclusters was realized by a spatial isolation strategy based on COFs (see Fig. 10G). The FeAC@FeSA–N–C catalyst exhibits excellent electrocatalytic performance for the ORR with a half-wave potential of 0.912 V in 0.1 M KOH electrolyte, which is 68 and 15 mV higher than those of the single-atom Fe–N4 catalyst (FeSA–N–C) (0.844 V) and commercial Pt/C (0.897 V), respectively (see Fig. 10H). The Tafel slope of FeAC@FeSA–N–C was as low as 61 mV dec−1 (see Fig. 10I), which is slightly lower than that of Co/PC SCCs (see Fig. 10C). The DFT calculation showed that Fe–N4 sites are the main active sites, and Fe nanoclusters could further enhance its activity. In addition to the formation of Fe–N bonds, Fe nanoclusters also promote the conversion of pyrrole nitrogen and nitrogen oxide to graphitic nitrogen and pyridine nitrogen due to metal catalysis, which is beneficial for improving the performance of the ORR.138
Lee and Yang also reported an Fe–N–C ORR electrocatalyst with Fe single atoms and Fe nanoclusters co-embedded in N-doped porous carbon by pyrolysis of metal–organic precursors and NaCl.139 The geometric structures of catalytic sites in Fe–N–C are revealed, where the isolated distribution of Fe–N4 sites and Fe nanoclusters with sizes less than 0.5 nm are well deposited on NC. The synergistic enhancement effect of Fe single atoms and Fe nanoclusters results in excellent ORR performances with a half-wave potential of 0.895 V in 0.1 M KOH electrolyte. The DFT calculation showed that, compared with the Fe–N4 site with single structures, the coexistence of Fe nanoclusters on the carbon layer will supply electrons to the Fe–N4 system. Therefore, both the d-band center of the Fe active site and the Fermi level of the system are adjusted, and the neighboring Fe nanoclusters are shown to weaken the binding energies of the ORR intermediates on Fe–N4 sites, hence enhancing both catalytic kinetics and thermodynamics. This study provides new insights into the understanding of the synergies of single atoms and clusters on N-doped carbon ORR electrocatalysts.139
More recently, Lei and co-workers reported a hybrid catalyst (FeSA/FeONC/NSC) with Fe single atoms (Fe–N4 sites) and Fe2O3 clusters (only a few Fe atomic centers) on N,S-co-doped porous carbon, which was obtained by the in situ transformation of Fe-distributed biomass (Spirulina platensis) based on a small molecular nitrogen source-mediated co-pyrolysis strategy.140 Due to the coupling effect with ultra-small Fe2O3 clusters and the favorable reaction site provided by the ultra-thin N,S-co-doped porous carbon skeleton, the FeSA/FeONC/NSC catalyst showed excellent ORR activity and catalytic stability comparable to Pt/C and. FeSA/FeONC/NSC has high Eonset (0.99 V) and E1/2 (0.86 V) values, which are better than those of Pt/C (Eonset = 0.98 V; E1/2 = 0.85 V). The ultra-thin porous structure of FeSA/FeONC/NSC is rich in edge defects, which not only provides a favorable site for the ORR, but also contributes to enhancing ORR activity. Crucially, the high ORR activity is mainly caused by the interaction between the atomic-level dispersed Fe–N4 sites and Fe2O3 clusters embedded in the N,S-mediated porous carbon layers. In this study, a highly efficient catalyst coupled with metal single atoms and homologous oxide clusters was obtained directly from biomass by in situ conversion, which provided inspiration for the synthesis of newly non-noble metal electrocatalysts.140
Recently, Wang et al.142 reported a mixed ORR catalyst with atomically-dispersed Co–N4 sites (Co SAC) and small Co NPs (Co SNPs) co-anchored in N-doped porous carbon nanocages (denoted Co-SAC/SNPs@NC). Co SNPs promote the formation of a graphited layer, which also acts as the support of Co–N4 sites without affecting the highly porous structure of the carbon nanocages. The synergy between Co SAC and Co SNPs and the enhanced graphitization matrix of carbon endowed this mixed catalyst with a high half-wave potential of 0.898 V and a high dynamic current density (JK) of 60.7 mV cm−2 at 0.85 V in alkaline medium. After 20000 cycles, the negative shift in half-wave potential was only 7 mV, which was better than that of Co-SAC@NC with only Co–N4 sites and commercial Pt/C. It may be a feasible method to solve the stability problem, by appropriately introducing metal small NPs into carbon-supported SACs. The small NPs and SCAs can work together to not only improve the activity of the ORR, but also improve the graphitization (high stability) of carbon supports.142
Recently, Jiang et al.143 also reported an Fe–N–C ORR catalyst (with Fe–N4 sites) enhanced by heterogeneous Co NPs (denoted Co@Fe–N4–C) prepared by an auxiliary thermal loading method. The characteristics showed that the Co@Fe–N4–C catalyst had larger specific surface area and higher graphitic N content than single Fe–N4–C. The Co@Fe–N4–C catalyst showed strong ORR activity in 0.1 M KOH electrolyte, where the half-wave potential (E1/2) was 0.92 V, which was much higher than those of single Fe–N4–C (E1/2 = 0.85 V) and Pt/C (E1/2 = 0.90 V). In 0.1 M HClO4 electrolyte, the E1/2 of the catalyst was 0.79 V, which was only 30 mV lower than that of Pt/C (E1/2 = 0.82 V). The DFT calculations confirmed that the strong synergistic effect between Co NPs and Fe–N4 sites provided Co@Fe–N4–C with favorable electronic structures and a local coordination environment. The introduction of Co NPs onto the surfaces of Fe–N4–C materials plays a key role in improving the catalytic activity and stability of the ORR, which provides a new way to prepare efficient Fe–N–C ORR catalysts.143
More recently, Sun and co-workers144 developed a bimetallic FeCo–N–C catalyst with highly active M–NPs and MN4 composite sites (M = FeCo) on N-doped porous carbon (denoted M/FeCo–SA–N–C). This FeCo–N–C catalyst showed the coexistence of Fe/Co NPs and Fe/Co single atoms, indicating that M/MN4 composite sites were successfully constructed. The M/FeCo–SA–N–C catalyst exhibited superior catalytic activity in terms of superhigh onset potential (0.981 V) and half-wave potential (0.851 V) in 0.1 M HClO4 electrolyte, which surpassed those of most M–N–C electrocatalysts in an acidic ORR.88,115,130,132,143 It was also successfully proved that the M/MN4 composite sites provided excellent stability to the M/FeCo–SA–N–C catalyst in acidic electrolyte. The synergistic effect between M–NPs and MN4 may play a crucial role in the boosted ORR performance through the construction of M/MN4 composite sites. The DFT calculations indicated that the M–NPs can increase the local charge density of MN4, and subsequently enhance O2 adsorption and elongate O–O bonds, resulting in the facile cleavage of the O–O bond. This work may present new highly active M/MN4 composite sites for catalyzing the ORR in acidic medium.144
On the other hand, transition metal SACs with MN4 sites can synergistically promote single noble metal (or its alloy) NPs for the ORR in acidic medium.145,146 For example, He and Mu145 reported a Pt@Co SAs-ZIF-NC catalyst by isolation of Pt NPs with CoN4 sites on ZIF-based N-doped carbon. In 0.1 M HClO4 electrolyte, the Pt@Co SAs-ZIF-NC showed a half wave potential (0.917 V) that is better than that of commercial Pt/C (0.868 V), and its mass activity (480 mA mgPt−1) at 0.9 V is three times that of Pt/C (160 mA mgPt−1). More recently, Wu and co-workers146 also reported that Fe SACs with FeN4 sites on N-doped carbon can significantly promote the ORR activity of Pt NPs and Pt3Co alloy NPs. Pt/FeN4–C and Pt3Co/FeN4–C showed mass activities of 451 mA mgPt−1 and 720 mA mgPt−1 at 0.9 V, respectively. The DFT calculation predicted that the synergy between Pt NPs and the surrounding FeN4 sites can weaken the adsorption of O2 on the Pt plane, thus enhancing the intrinsic activity of the Pt catalyst. In general, transition metal single atoms can improve the activity of noble metal catalysts, and finally achieve the goal of reducing the use of noble metal. For the sake of convenience, the comparison parameters of different atomic-level ORR electrocatalysts are also summarized in Table 1.
5 Proposed evaluation methods and site interaction rules for high-metal-density electrocatalysts
At present, “high dispersion” and “high density” are just general adjectives, and it is difficult to establish a unified evaluation standard for this type of high-metal-density electrocatalyst. Usually, the smaller particle size and the higher degree of dispersion can endow catalytic materials with larger surface catalytic activity. Meanwhile, it is of great significance to develop high-loading (i.e., high metal density of small NPs or atomic sites) electrocatalysts for the sake of high-mass-activity catalytic systems in practical industrial applications. In order to better elucidate the relationship between the metal density and electrocatalytic performance of catalysts, we propose two methods to calculate or identify the metal density or site density on the surface of the support: (i) the method based on the relative distance of metal particles or atomic sites (i.e., the ratio of actual distance to particle diameter or atom diameter) and (ii) that based on the average number of particles or atomic sites per unit area (i.e., particle number per 100 nm2 and atom number per 1 nm2) (see Fig. 11A for the schematic diagram). | ||
| Fig. 11 (A) The relative distance of metal particles or atomic sites, (B) the diffusion models of nanoparticle arrays at different potential sweep rates (reprinted with permission from ref. 147), and (C–E) schematic diagrams of atomic site electrostatic interaction (reprinted with permission from ref. 148). | ||
For nanoparticle catalysts, method (i) (relative distance method) is more objective and convenient to calculate or evaluate the metal density, due to their different particle diameters and easily distinguishable structures. Based on the previous case analysis, it can be found that when the relative distance is close to or less than 1, the distribution of nanoparticles on the carbon support is very close, and these catalysts also show high mass catalytic activity (see Fig. 2–5 for details). For atomic-level catalysts, both method (i) (relative distance method) and method (ii) (average number method) can be used under high resolution HAADF-STEM. Particularly, considering that the site sizes of metal atoms are very close, we can use the actual site distance to evaluate the metal density of atomic-level catalysts. In general, when the average distance of atomic sites is equal to or less than 1 nm, the distribution of metal atoms in the carbon carrier is very dense (see Fig. 7 for details) and the catalytic performance is obviously dependent on the site distance (see Fig. 8 for details).
For nanoparticle systems, high-metal-density electrocatalysts can lead to distinctive ion diffusion characteristics in extensive electrochemical processes. Fig. 11B shows the surface-density-dependent diffusion models of nanoparticle arrays at different potential sweep rates: (left) high sweep rate, (middle) medium sweep rate and (right) very low sweep rate.147 For the high sweep rate, the diffusion layer thickness is much smaller than the average distance between adjacent particles; for the medium sweep rate the diffusion layers of adjacent particles overlap for a high particle density; and for the very low sweep rate steady-state diffusion is reached for all density samples. These electrochemical characteristics have positive guiding significance in the design of high-performance electrocatalysis, where the overlap effect of the diffusion layer thickness of adjacent particles may induce unexpected electrocatalytic synergistic effects for high-metal-density electrocatalysts.
For atomic-level systems, high-metal-density electrocatalysts can also lead to distinctive electronic properties and catalytic activity. Fig. 11C–E show the structural model and surface-density-dependent atomic site electrostatic interaction of Pd1/CeO2 single atom catalyst.148 Due to the cumulative enhancement of CeO2 reducibility, the specific activity of a single Pd atom supported by CeO2 increases linearly with the increase of Pd atom density. With the increase of surface Pd density, the long-range electrostatic footprints (∼1.5 nm) around each Pd site overlap each other, resulting in a deviation from the constant specific activity observed. The results show that the specific catalytic activity of reducible oxide supported single atom catalysts can be adjusted by changing the surface density of a single metal atom. These theoretical models provide help with the development of high specific activity and high metal density atomic-level catalysts.
6 Conclusions and prospects
6.1 Conclusions
In summary, carbon-supported noble metal Pt NP (including their alloy and core–shell structure) catalysts with high loading and high dispersity are very beneficial to constructing ultra-thin catalyst layers for fast-diffusion-dynamics MEA application in PEMFCs. The easily available commercial carbon black supports and state-of-the-art graphene supports with moderately graphited structures and mesoporous structures are the best choices for structuring high-metal-loading ORR electrocatalysts. The densely-distributed NPs of transition metal compounds on carbon supports are also critical for designing non-noble metal ORR electrocatalysts, where the high metal content can be obtained by a synchronous pyrolysis process. Metal carbide-promoted noble metal NPs with a high-density dispersion of two components on carbon supports are conducive to the formation of close-contact synergistic interfaces. Therefore, the two-step synthesis by loading single noble metal (or its alloy) NPs onto a carbon-supported carbide is often adopted, where the highly-proportioned noble metal NPs can be grown tightly on or around the carbide component. High-density carbides synergistically enhanced low-Pt or Pd ORR electrocatalysts are of special importance due to the fact that strong EMSI can be easily formed on the dense interface of Pt/Pd and carbides.When the distance between two active metal atom centers is reduced to the subnanometer level, the catalytic behavior will be significantly affected because of the site interaction effect. High-density atomically-dispersed metal catalysts (including SACs, DACs, and SCCs) often exhibit more significant catalytic properties than the sparse counterparts due to the synergistic effect of dense atomic sites. M–N–C (where M includes transition metals and noble metals) ORR catalysts with high active site density can generate higher activity by exposing the inaccessible M–N4 active sites and strengthening the mass transport. In order to ensure the mass transport and high activity of high-metal-density SACs, it is very necessary to develop carbon supports with high surface area and mesoporous structure, which facilitates ion transfer and improves their catalytic performances. Besides, the high-density bimetal sites dispersed on N-doped carbon (M1M2Nx DACs) can form different adjacent active sites and improve the activity and stability of the catalyst through synergistic catalysis. The construction of dual active sites provides a new opportunity for high-performance ORR catalysts, which can not only develop more combinations of coordination environments, but also lead to the enhancement of two different metal sites of high-density atomically-dispersed catalysts. Preparation of cluster catalysts (SCCs) and the combination of SACs with SCCs/NPs are the significant complement strategies for the design of high-density ORR catalysts with improved catalytic performances.
6.2 Prospects
Usually, the low metal loading of catalysts will increase the thickness and impedance of the catalyst layer in the MEA, which causes more serious mass transfer loss, and reduces the performances of fuel cells at high current density. ORR electrocatalysts based on noble metal NPs with high metal loading (∼40 wt%) have attracted great attention, which can well solve the above technical problems of fuel cells. However, many studies ignore the loss of mass activity caused by high loading (such as the decrease of activity caused by dense stacking), and the electrochemical oxidation of carbon supports under high potentials (resulting in a decrease of catalyst stability). Therefore, in future work, one should strive to find effective methods to prepare high-loading catalysts and develop high stability support materials, to ensure the mass activity of the ORR and the stability of catalysts while improving the metal density on supports. At the same time, the as-prepared catalysts with high-loadings of noble metal NPs should be applied to the MEA, and the feasibility of high-metal-density catalysts should be confirmed by the measurement of impedance, maximum power density and other parameters in fuel cells, which will in turn guide the optimized preparation of high-loading ORR electrocatalysts and further improve the device performances of fuel cells.The ORR performance of high-metal-density M–N–C (M = Fe, Co, Ni, etc.) catalysts (including SACs, DACs, and SCCs) in the kinetic region is very close to that of Pt-based catalysts, but there are still great challenges for these non-noble metal catalysts, especially under acidic conditions. Because of their sensitive physical and chemical properties, especially in acidic environments, these transition metal elements have poor corrosion resistance. Therefore, high-density transition metal M–N–C catalysts (especially Fe–N4 SACs) still face great challenges in the practical application of PEMFCs, due to their under-stable characteristic and potential Fenton oxidation. More catalytic systems with high activity, high stability and four-electron selectivity still need to be found. Rational design of metal cluster catalysts, including dimers, trimers, and larger metal clusters, is also an important research direction to overcome the defects of SACs. At present, the central metal atom of most high-density atomically-dispersed metal catalysts only coordinates with the N atom in the carbon substrates. In the subsequent research, the coordination effect of other elements such as S, P, B and O can be considered to adjust the electronic structure of the central metal atom, so as to obtain a more efficient and stable ORR electrocatalyst. Particularly, some reliable synthesis strategies (e.g., mechanochemistry and heat migration strategies) need to be explored to realize the batch production of SACs with high metal density and high stability on different supports.
Carbon-supported ORR electrocatalysts (including nanoparticles and atomic-level architectures) are always faced with problems of carbon corrosion of the support and the resultant catalyst instability in practical application. Especially under the catalysis of ultrafine metal species and high-potential or acidic oxidation conditions, the amorphous component in conventional carbon supports (e.g., porous carbon and acetylene black) shows a fast electrochemical corrosion rate in the operating state of fuel cells. Based on this, it is necessary to develop new-type high-crystallinity carbon with or without ceramic support materials (e.g., conducting metal oxides, carbides, and borides) with high electrochemical and thermodynamic stability, in order to accelerate the progress of both nano-structured and atomic-level ORR electrocatalysts in practical application of fuel cells.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (22078071, 21777034), the Natural Science Foundation of Guangdong Province (2021A1515010125, 2020A1515010344), Maoming Science and Technology Project (mmkj2020032), the Science and Technology Innovation Project of Guangdong Province College Students (733330, 733336), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2019), Guangdong Basic and Applied Basic Research Foundation (2019A1515011249), the Key Research Project of Natural Science of Guangdong Provincial Department of Education (2019KZDXM010), and the Program for Innovative Research Team of Guangdong University of Petrochemical Technology.References
- D. F. Dominković, I. Bačeković, A. S. Pedersen and G. Krajačić, The future of transportation in sustainable energy systems: Opportunities and barriers in a clean energy transition, Renewable Sustainable Energy Rev., 2018, 82, 1823–1838 CrossRef.
- X. X. Wang, M. T. Swihart and G. Wu, Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation, Nat. Catal., 2019, 2(7), 578–589 CrossRef CAS.
- E. H. Majlan, D. Rohendi, W. R. W. Daud, T. Husaini and M. A. Haque, Electrode for proton exchange membrane fuel cells: A review, Renewable Sustainable Energy Rev., 2018, 89, 117–134 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Understanding catalytic activity trends in the oxygen reduction reaction, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- L. Zhang, L. Fan, P. Yang, M. Li, H. Zhang, Y. Tang, Z. Kang, H. Guo, R. Wang and D. Sun, Green synthesis of hierarchical carbon coupled with Fe3O4/Fe2C as an efficient catalyst for the oxygen reduction reaction, Materials Advances, 2020, 1, 2010–2018 RSC.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. HAbner and P. Strasser, Ionomer distribution control in porous carbon-supported catalyst layers for high-power and low Pt-loaded proton exchange membrane fuel cells, Nat. Mater., 2020, 19(1), 77–85 CrossRef CAS PubMed.
- J. Hou, M. Yang, C. Ke, G. Wei, C. Priest, Z. Qiao and J. Zhang, Platinum-group-metal catalysts for proton exchange membrane fuel cells: From catalyst design to electrode structure optimization, Energy Chem., 2020, 2(1), 100023 CrossRef.
- Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu and Y. Huang, Pt-Based nanocrystal for electrocatalytic oxygen reduction, Adv. Mater., 2019, 31(31), 1808115 CrossRef PubMed.
- G. Bampos, L. Sygellou and S. Bebelis, Oxygen reduction reaction activity of Pd-based bimetallic electrocatalysts in alkaline medium, Catal. Today, 2020, 355, 685–697 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt, Science, 2011, 332(6028), 443–447 CrossRef CAS PubMed.
- L. Pan, S. Ott, F. Dionigi and P. Strasser, Current challenges related to the deployment of shape-controlled Pt alloy oxygen reduction reaction nanocatalysts into low Pt-loaded cathode layers of proton exchange membrane fuel cells, Curr. Opin. Electrochem., 2019, 18, 61–71 CrossRef CAS.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Advanced platinum-based oxygen reduction electrocatalysts for fuel cells, Acc. Chem. Res., 2021, 54(2), 311–322 CrossRef CAS PubMed.
- L. Du, V. Prabhakaran, X. Xie, S. Park, Y. Wang and Y. Shao, Low-PGM and PGM-free catalysts for proton exchange membrane fuel cells: Stability challenges and material solutions, Adv. Mater., 2021, 33(6), 1908232 CrossRef CAS PubMed.
- U. Martinez, S. K. Babu, E. F. Holby and P. Zelenay, Durability challenges and perspective in the development of PGM-free electrocatalysts for the oxygen reduction reaction, Curr. Opin. Electrochem., 2018, 9, 224–232 CrossRef CAS.
- S. Song, Y. Wang and P. K. Shen, Pulse-microwave assisted polyol synthesis of highly dispersed high loading Pt/C electrocatalyst for oxygen reduction reaction, J. Power Sources, 2007, 170(1), 46–49 CrossRef CAS.
- Y. Xiong, L. Xiao, Y. Yang, F. J. DiSalvo and H. D. Abruña, High-loading intermetallic Pt3Co/C core–shell nanoparticles as enhanced activity electrocatalysts toward the oxygen reduction reaction (ORR), Chem. Mater., 2018, 30(5), 1532–1539 CrossRef CAS.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Recent advances in electrocatalysts for oxygen reduction reaction, Chem. Rev., 2016, 116(6), 3594–3657 CrossRef CAS PubMed.
- S. H. Joo, K. Kwon, D. J. You, C. Pak, H. Chang and J. M. Kim, Preparation of high loading Pt nanoparticles on ordered mesoporous carbon with a controlled Pt size and its effects on oxygen reduction and methanol oxidation reactions, Electrochim. Acta, 2009, 54(24), 5746–5753 CrossRef CAS.
- X. Xu, C. Tan, H. Liu, F. Wang, Z. Li, J. Liu and J. Ji, Carbon black supported ultra-high loading silver nanoparticle catalyst and its enhanced electrocatalytic activity towards oxygen reduction reaction in alkaline medium, J. Electroanal. Chem., 2013, 696, 9–14 CrossRef CAS.
- G. Han, Y. Zheng, X. Zhang, Z. Wang, Y. Gong, C. Du and X. Sun, High loading single-atom Cu dispersed on graphene for efficient oxygen reduction reaction, Nano Energy, 2019, 66, 104088 CrossRef CAS.
- J. F. Huang, J. R. Sie and R. H. Zeng, Engineering sub-nano structures with highly jagged edges on the Pt surface of Pt/C electrocatalysts to promote oxygen reduction reactions, Electrochim. Acta, 2021, 372, 137868 CrossRef CAS.
- L. Liu and A. Corma, Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles, Chem. Rev., 2018, 118(10), 4981–5079 CrossRef CAS PubMed.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chemical synthesis of single atomic site catalysts, Chem. Rev., 2020, 120(21), 11900–11955 CrossRef CAS PubMed.
- Y. Peng, B. Lu and S. Chen, Carbon-supported single atom catalysts for electrochemical energy conversion and storage, Adv. Mater., 2018, 30(48), 1801995 CrossRef PubMed.
- J. Wang, Z. Li, Y. Wu and Y. Li, Fabrication of single-atom catalysts with precise structure and high metal loading, Adv. Mater., 2018, 30(48), 1801649 CrossRef PubMed.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Progress in the development of Fe-based PGM-free electrocatalysts for the oxygen reduction reaction, Adv. Mater., 2019, 31(31), 1806545 CrossRef PubMed.
- A. Bharti and G. Cheruvally, Influence of various carbon nano-forms as supports for Pt catalyst on proton exchange membrane fuel cell performance, J. Power Sources, 2017, 360, 196–205 CrossRef CAS.
- A. Pal, M. Palashuddin Sk and A. Chattopadhyay, Recent advances in crystalline carbon dots for superior application potential, Materials Advances, 2020, 1, 525–553 RSC.
- C. Wei, H. Wang, K. Eid, J. Kim, J. H. Kim, Z. A. Alothman and L. Wang, A three-dimensionally structured electrocatalyst: Cobalt-embedded nitrogen-doped carbon nanotubes/nitrogen-doped reduced graphene oxide hybrid for efficient oxygen reduction, Chem. – Eur. J., 2017, 23(3), 637–643 CrossRef CAS PubMed.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir and X. Duan, Single atom electrocatalysts supported on graphene or graphene-like carbons, Chem. Soc. Rev., 2019, 48(20), 5207–5241 RSC.
- H. Tan, J. Tang, J. Kim, Y. V. Kaneti, Y. M. Kang, Y. Sugahara and Y. Yamauchi, Rational design and construction of nanoporous iron-and nitrogen-doped carbon electrocatalysts for oxygen reduction reaction, J. Mater. Chem. A, 2019, 7(4), 1380–1393 RSC.
- Z. Li, L. Zhang, X. Chen, B. Li, H. Wang and Q. Li, Three-dimensional graphene-like porous carbon nanosheets derived from molecular precursor for high-performance supercapacitor application, Electrochim. Acta, 2019, 296, 8–17 CrossRef CAS.
- X. F. Lu, B. Y. Xia, S. Q. Zang and X. W. Lou, Metal–organic frameworks based electrocatalysts for the oxygen reduction reaction, Angew. Chem., 2020, 132(12), 4662–4678 CrossRef.
- N. Cheng, L. Ren, X. Xu, Y. Du and S. X. Dou, Recent development of zeolitic imidazolate frameworks (ZIFs) derived porous carbon based materials as electrocatalysts, Adv. Energy Mater., 2018, 8(25), 1801257 CrossRef.
- J. F. Sun, Q. Q. Xu, J. L. Qi, D. Zhou, H. Y. Zhu and J. Z. Yin, Isolated single atoms anchored on N-doped carbon materials as a highly efficient catalyst for electrochemical and organic reactions, ACS Sustainable Chem. Eng., 2020, 8(39), 14630–14656 CrossRef CAS.
- Y. N. Chen, X. Zhang and Z. Zhou, Carbon-based substrates for highly dispersed nanoparticle and even single-atom electrocatalysts, Small Methods, 2019, 3(9), 1900050 CrossRef.
- Y. X. Xiao, J. Ying, G. Tian, Y. Tao, H. Wei, S. Y. Fan and C. Janiak, Highly dispersed PtPd on graphitic nanofibers and its heavy d-π effect, Appl. Catal., B, 2019, 259, 118080 CrossRef CAS.
- E. Zagoraiou, M. K. Daletou, L. Sygellou, S. Ballomenou and S. G. Neophytides, Highly dispersed platinum supported catalysts – Effect of properties on the electrocatalytic activity, Appl. Catal., B, 2019, 259, 118050 CrossRef CAS.
- Y. T. Kim and T. Mitani, Surface thiolation of carbon nanotubes as supports: a promising route for the high dispersion of Pt nanoparticles for electrocatalysts, J. Cat., 2006, 238(2), 394–401 CrossRef.
- J. E. Park, J. Lim, M. S. Lim, S. Kim, O. H. Kim, D. W. Lee and Y. E. Sung, Gas diffusion layer/flow-field unified membrane-electrode assembly in fuel cell using graphene foam, Electrochim. Acta, 2019, 323, 134808 CrossRef CAS.
- W. J. Lee, S. Bera, C. M. Kim, E. K. Koh, W. P. Hong, S. J. Oh and S. H. Kwon, Synthesis of highly dispersed Pt nanoparticles into carbon supports by fluidized bed reactor atomic layer deposition to boost PEMFC performance, NPG Asia Mater., 2020, 12(1), 1–13 CrossRef.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Stabilizing Pt-based electrocatalysts for oxygen reduction reaction: Fundamental understanding and design strategies, Adv. Mater., 2021, 33(20), 2006494 CrossRef CAS PubMed.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng and Y. Xiong, Precisely tuning the number of Fe atoms in clusters on N-doped carbon toward acidic oxygen reduction reaction, Chem, 2019, 5(11), 2865–2878 CAS.
- L. Zhang, T. Yang, W. Zang, Z. Kou, Y. Ma, M. Waqar and J. Wang, Quasi-paired Pt atomic sites on Mo2C promoting selective four-electron oxygen reduction, Adv. Sci., 2021, 2101344 CrossRef PubMed.
- X. Lu, X. Zhu, X. Tan, K. H. Wu, S. C. Haw, C. W. Pao and R. Amal, Intrinsic ORR activity enhancement of Pt atomic sites by engineering d-band center via local coordination tuning, Angew. Chem., Int. Ed., 2021, 60, 21911–21917 CrossRef PubMed.
- T. Zhang, X. Han, H. Yang, A. Han, E. Hu, Y. Li and B. Liu, Atomically dispersed nickel(i) on an alloy-encapsulated nitrogen-doped carbon nanotube array for high-performance electrochemical CO2 reduction reaction, Angew. Chem., 2020, 132(29), 12153–12159 CrossRef.
- Y. C. Park, H. Tokiwa, K. Kakinuma, M. Watanabe and M. Uchida, Effects of carbon supports on Pt distribution, ionomer coverage and cathode performance for polymer electrolyte fuel cells, J. Power Sources, 2016, 315, 179–191 CrossRef CAS.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu and A. Kongkanand, Boosting fuel cell performance with accessible carbon mesopores, ACS Energy Lett., 2018, 3(3), 618–621 CrossRef CAS.
- Z. Qiao, S. Hwang, X. Li, C. Wang, W. Samarakoon, S. Karakalos and G. Wu, 3D porous graphitic nanocarbon for enhancing the performance and durability of Pt catalysts: A balance between graphitization and hierarchical porosity, Energy Environ. Sci., 2019, 12(9), 2830–2841 RSC.
- M. Sakthivel and J. F. Drillet, An extensive study about influence of the carbon support morphology on Pt activity and stability for oxygen reduction reaction, Appl. Catal., B, 2018, 231, 62–72 CrossRef CAS.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu and Y. Xia, Pd–Pt bimetallic nanodendrites with high activity for oxygen reduction, Science, 2009, 324(5932), 1302–1305 CrossRef CAS PubMed.
- H. Tan, J. Kim, J. Lin, C. Li, S. M. Alsheri, T. Ahamad and Y. Yamauchi, A facile surfactant-assisted synthesis of carbon-supported dendritic Pt nanoparticles with high electrocatalytic performance for the oxygen reduction reaction, Microporous Mesoporous Mater., 2019, 280, 1–6 CrossRef CAS.
- C. Li, H. Tan, J. Lin, X. Luo, S. Wang, J. You and J. Kim, Emerging Pt-based electrocatalysts with highly open nanoarchitectures for boosting oxygen reduction reaction, Nano Today, 2018, 21, 91–105 CrossRef CAS.
- Z. Li, C. He, M. Cai, S. Kang and P. K. Shen, A strategy for easy synthesis of carbon supported Co@Pt core–shell configuration as highly active catalyst for oxygen reduction reaction, Int. J. Hydrogen Energy, 2012, 37(19), 14152–14160 CrossRef CAS.
- S. S. Li, J. N. Zheng, A. J. Wang, F. L. Tao, J. J. Feng, J. R. Chen and H. Yu, Branched platinum-on-palladium bimetallic heteronanostructures supported on reduced graphene oxide for highly efficient oxygen reduction reaction, J. Power Sources, 2014, 272, 1078–1085 CrossRef CAS.
- X. R. Li, X. L. Li, M. C. Xu, J. J. Xu and H. Y. Chen, Gold nanodendrities on graphene oxide nanosheets for oxygen reduction reaction, J. Mater. Chem. A, 2014, 2(6), 1697–1703 RSC.
- X. X. Wang, J. Sokolowski, H. Liu and G. Wu, Pt alloy oxygen-reduction electrocatalysts: Synthesis, structure, and property, Chin. J. Cat., 2020, 41(5), 739–755 CrossRef CAS.
- V. Colic and A. S. Bandarenka, Pt alloy electrocatalysts for the oxygen reduction reaction: From model surfaces to nanostructured systems, ACS Catal., 2016, 6(8), 5378–5385 CrossRef CAS.
- Y. J. Wang, W. Long, L. Wang, R. Yuan, A. Ignaszak, B. Fang and D. P. Wilkinson, Unlocking the door to highly active ORR catalysts for PEMFC applications: polyhedron-engineered Pt-based nanocrystals, Energy Environ. Sci., 2018, 11(2), 258–275 RSC.
- S. Sankarasubramanian, N. Singh, F. Mizuno and J. Prakash, Ab initio investigation of the oxygen reduction reaction activity on noble metal (Pt, Au, Pd), Pt3M (M = Fe, Co, Ni, Cu) and Pd3M (M = Fe, Co, Ni, Cu) alloy surfaces, for LiO2 cells, J. Power Sources, 2016, 319, 202–209 CrossRef CAS.
- L. M. Guo, D. F. Zhang and L. Guo, ). Structure design reveals the role of Au for ORR catalytic performance optimization in PtCo-based catalysts, Adv. Funct. Mater., 2020, 30(22), 2001575 CrossRef CAS.
- K. Jayasayee, J. R. Van Veen, T. G. Manivasagam, S. Celebi, E. J. Hensen and F. A. De Bruijn, Oxygen reduction reaction (ORR) activity and durability of carbon supported PtM (Co, Ni, Cu) alloys: Influence of particle size and non-noble metals, Appl. Catal., B, 2012, 111, 515–526 CrossRef.
- T. Y. Yoo, J. M. Yoo, A. K. Sinha, M. S. Bootharaju, E. Jung, H. S. Lee and T. Hyeon, Direct synthesis of intermetallic platinum–alloy nanoparticles highly loaded on carbon supports for efficient electrocatalysis, J. Am. Chem. Soc., 2020, 142(33), 14190–14200 CrossRef CAS PubMed.
- B. Zhang, G. Fu, Y. Li, L. Liang, N. S. Grundish, Y. Tang and Z. Cui, General strategy for synthesis of ordered Pt3M intermetallics with ultrasmall particle size, Angew. Chem., 2020, 132(20), 7931–7937 CrossRef.
- W. Wang, B. Lei and S. Guo, Engineering multimetallic nanocrystals for highly efficient oxygen reduction catalysts, Adv. Energy Mater., 2016, 6(17), 1600236 CrossRef.
- Y. T. Liang, S. P. Lin, C. W. Liu, S. R. Chung, T. Y. Chen, J. H. Wang and K. W. Wang, The performance and stability of the oxygen reduction reaction on Pt–M (M = Pd, Ag and Au) nanorods: An experimental and computational study, Chem. Commun., 2015, 51(30), 6605–6608 RSC.
- X. Deng, S. Yin, X. Wu, M. Sun, Z. Li, Z. Xie and Q. Huang, Scalable preparation of PtPd/carbon nanowires in the form of membrane as highly stable electrocatalysts for oxygen reduction reaction, Int. J. Hydrogen Energy, 2019, 44(5), 2752–2759 CrossRef CAS.
- Y. Deng, H. Xue, S. Lu, Y. Song, X. Cao, L. Wang and H. Gu, Trimetallic Au@PtPd mesoporous nanorods as efficient electrocatalysts for the oxygen reduction reaction, ACS Appl. Energy Mater., 2018, 1(9), 4891–4898 CrossRef CAS.
- Y. Li, Z. Li and P. K. Shen, Simultaneous formation of ultrahigh surface area and three-dimensional hierarchical porous graphene-like networks for fast and highly stable supercapacitors, Adv. Mater., 2013, 25(17), 2474–2480 CrossRef CAS PubMed.
- Z. Li, Y. Li, C. He and P. K. Shen, Bimetallic PtAg alloyed nanoparticles and 3-D mesoporous graphene nanosheet hybrid architectures for advanced oxygen reduction reaction electrocatalysts, J. Mater. Chem. A, 2017, 5(44), 23158–23169 RSC.
- Q. Zhao, C. Wang, H. Wang, J. Wang, Y. Tang, Z. Mao and K. Sasaki, Synthesis of a high-performance low-platinum PtAg/C alloyed oxygen reduction catalyst through the gradual reduction method, N. J. Chem., 2020, 44(9), 3728–3736 RSC.
- R. Wang, H. Wang, F. Luo and S. Liao, Core–shell-structured low-platinum electrocatalysts for fuel cell applications, Electrochem. Energy Rev., 2018, 1(3), 324–387 CrossRef CAS.
- R. Jiang, S. Tung, Z. Tang, L. Li, L. Ding, X. Xi and J. Zhang, A review of core–shell nanostructured electrocatalysts for oxygen reduction reaction, Energy Storage Mater., 2018, 12, 260–276 CrossRef.
- K. Sasaki, K. A. Kuttiyiel and R. R. Adzic, Designing high performance Pt monolayer core–shell electrocatalysts for fuel cells, Curr. Opin. Electrochem., 2020, 21, 368–375 CrossRef CAS.
- L. Wang, Z. Zeng, C. Ma, Y. Liu, M. Giroux, M. Chi and C. Wang, Plating precious metals on nonprecious metal nanoparticles for sustainable electrocatalysts, Nano Lett., 2017, 17(6), 3391–3395 CrossRef CAS PubMed.
- X. Lyu, Y. Jia, X. Mao, D. Li, G. Li, L. Zhuang and X. Yao, Gradient-concentration design of stable core–shell nanostructure for acidic oxygen reduction electrocatalysis, Adv. Mater., 2020, 32(32), 2003493 CrossRef CAS PubMed.
- Y. T. Pan and H. Yang, Design of bimetallic catalysts and electrocatalysts through the control of reactive environments, Nano Today, 2020, 31, 100832 CrossRef CAS.
- K. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Adsorbate-induced surface segregation for core–shell nanocatalysts, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef CAS PubMed.
- Z. Li, Y. Li, G. He and P. K. Shen, Novel graphene-like nanosheet supported highly active electrocatalysts with ultralow Pt loadings for oxygen reduction reaction, J. Mater. Chem. A, 2014, 2(40), 16898–16904 RSC.
- Y. Zhang, Y. C. Hsieh, V. Volkov, D. Su, W. An, R. Si and R. R. Adzic, High performance Pt monolayer catalysts produced via core-catalyzed coating in ethanol, ACS Catal., 2014, 4(3), 738–742 CrossRef CAS.
- J. Kong, Y. H. Qin, T. L. Wang and C. W. Wang, Pd9Au1@Pt/C core–shell catalyst prepared via Pd9Au1-catalyzed coating for enhanced oxygen reduction, Int. J. Hydrogen Energy, 2020, 45(51), 27254–27262 CrossRef CAS.
- M. Kiani, J. Zhang, Y. Luo, C. Jiang, J. Fan, G. Wang and R. Wang, Recent developments in electrocatalysts and future prospects for oxygen reduction reaction in polymer electrolyte membrane fuel cells, J. Energy Chem., 2018, 27(4), 1124–1139 CrossRef.
- H. Tan, Y. Li, J. Kim, T. Takei, Z. Wang, X. Xu and Y. Yamauchi, Sub-50 nm iron–nitrogen-doped hollow carbon sphere-encapsulated iron carbide nanoparticles as efficient oxygen reduction catalysts, Adv. Sci., 2018, 5(7), 1800120 CrossRef PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction, Nat. Mater., 2011, 10(10), 780–786 CrossRef CAS PubMed.
- J. Wang, L. Li, X. Chen, Y. Lu and W. Yang, Monodisperse cobalt sulfides embedded within nitrogen-doped carbon nanoflakes: An efficient and stable electrocatalyst for the oxygen reduction reaction, J. Mater. Chem. A, 2016, 4(29), 11342–11350 RSC.
- R. B. Levy and M. Boudart, Platinum-like behavior of tungsten carbide in surface catalysis, Science, 1973, 181(4099), 547–549 CrossRef CAS PubMed.
- Z. Yan, M. Zhang, J. Xie, J. Zhu and P. K. Shen, A bimetallic carbide Fe2MoC promoted Pd electrocatalyst with performance superior to Pt/C towards the oxygen reduction reaction in acidic media, Appl. Catal., B, 2015, 165, 636–641 CrossRef CAS.
- B. Li, J. Zhou, L. Zhang and Z. Li, Bimetallic carbide of Co3W3C enhanced non-noble-metal catalysts with high activity and stability for acidic oxygen reduction reaction, RSC Adv., 2018, 8(22), 12292–12299 RSC.
- S. Yin, M. Cai, C. Wang and P. K. Shen, Tungsten carbide promoted Pd–Fe as alcohol-tolerant electrocatalysts for oxygen reduction reactions, Energy Environ. Sci., 2011, 4(2), 558–563 RSC.
- X. Ma, H. Meng, M. Cai and P. K. Shen, Bimetallic carbide nanocomposite enhanced Pt catalyst with high activity and stability for the oxygen reduction reaction, J. Am. Chem. Soc., 2012, 134(4), 1954–1957 CrossRef CAS PubMed.
- D. Göhl, A. Garg, P. Paciok, K. J. Mayrhofer, M. Heggen, Y. Shao-Horn and M. Ledendecker, Engineering stable electrocatalysts by synergistic stabilization between carbide cores and Pt shells, Nat. Mater., 2020, 19(3), 287–291 CrossRef PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Electronic metal–support interaction of single-atom catalysts and applications in electrocatalysis, Adv. Mater., 2020, 32(49), 2003300 CrossRef CAS PubMed.
- C. He and P. K. Shen, Pt loaded on truncated hexagonal pyramid WC/graphene for oxygen reduction reaction, Nano Energy, 2014, 8, 52–61 CrossRef CAS.
- Z. Li, B. Li, Z. Liu, Z. Liu and D. Li, A tungsten carbide/iron sulfide/FePt nanocomposite supported on nitrogen-doped carbon as an efficient electrocatalyst for oxygen reduction reaction, RSC Adv., 2015, 5(128), 106245–106251 RSC.
- Z. Li, S. Ji, B. G. Pollet and P. K. Shen, A Co3W3C promoted Pd catalyst exhibiting competitive performance over Pt/C catalysts towards the oxygen reduction reaction, Chem. Commun., 2014, 50(5), 566–568 RSC.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang and L. Dai, Carbon nanocomposite catalysts for oxygen reduction and evolution reactions: From nitrogen doping to transition-metal addition, Nano Energy, 2016, 29, 83–110 CrossRef CAS.
- L. Osmieri, L. Pezzolato and S. Specchia, Recent trends on the application of PGM-free catalysts at the cathode of anion exchange membrane fuel cells, Curr. Opin. Electrochem., 2018, 9, 240–256 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J. P. Dodelet, Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells, Science, 2009, 324(5923), 71–74 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2(6), 65–81 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357(6350), 479–484 CrossRef CAS PubMed.
- D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao and J. R. Long, A spin transition mechanism for cooperative adsorption in metal–organic frameworks, Nature, 2017, 550(7674), 96–100 CrossRef CAS PubMed.
- Q. K. Li, X. F. Li, G. Zhang and J. Jiang, Cooperative spin transition of monodispersed FeN3 sites within graphene induced by CO adsorption, J. Am. Chem. Soc., 2018, 140(45), 15149–15152 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen and J. Zeng, Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation, Nat. Nanotechnol., 2018, 13(5), 411–417 CrossRef CAS PubMed.
- J. Wu, L. Xiong, B. Zhao, M. Liu and L. Huang, Densely populated single atom catalysts, Small Methods, 2020, 4(2), 1900540 CrossRef CAS.
- W. Chen, W. Gao, P. Tu, T. Robert, Y. Ma, H. Shan and J. Wu, Neighboring Pt atom sites in an ultrathin FePt Nanosheet for the efficient and highly CO-tolerant oxygen reduction reaction, Nano Lett., 2018, 18(9), 5905–5912 CrossRef CAS PubMed.
- J. Wu, H. Zhou, Q. Li, M. Chen, J. Wan, N. Zhang and L. Huang, Densely populated isolated single CoN site for efficient oxygen electrocatalysis, Adv. Energy Mater., 2019, 9(22), 1900149 CrossRef.
- J. Xi, H. S. Jung, Y. Xu, F. Xiao, J. W. Bae and S. Wang, Synthesis strategies, catalytic applications, and performance regulation of single-atom catalysts, Adv. Funct. Mater., 2021, 31(12), 2008318 CrossRef CAS.
- L. Zhao, Y. Zhang, L. B. Huang, X. Z. Liu, Q. H. Zhang, C. He and L. J. Wan, Cascade anchoring strategy for general mass production of high-loading single-atomic metal–nitrogen catalysts, Nat. Commun., 2019, 10(1), 1–11 CrossRef PubMed.
- H. Yan, X. Zhao, N. Guo, Z. Lyu, Y. Du, S. Xi and J. Lu, Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity, Nat. Commun., 2018, 9(1), 1–9 CrossRef CAS PubMed.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu and Q. Jia, Chemical vapour deposition of Fe–N–C oxygen reduction catalysts with full utilization of dense Fe–N4 sites, Nat. Mater., 2021 DOI:10.1038/s41563-021-01030-2.
- Q. Wang, Y. Yang, F. Sun, G. Chen, J. Wang, L. Peng and G. I. Waterhouse, Molten NaCl-assisted synthesis of porous Fe–N–C electrocatalysts with a high density of catalytically accessible FeN4 active sites and outstanding oxygen reduction reaction performance, Adv. Energy Mater., 2021, 2100219 CrossRef CAS.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang and Y. Li, Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms, Nat. Catal., 2018, 1(10), 781–786 CrossRef CAS.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Large-scale and highly selective CO2 electrocatalytic reduction on nickel single-atom catalyst, Joule, 2019, 3(1), 265–278 CrossRef CAS.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles and S. P. Jiang, Atomically dispersed transition metals on carbon nanotubes with ultrahigh loading for selective electrochemical carbon dioxide reduction, Adv. Mater., 2018, 30(13), 1706287 CrossRef PubMed.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei and Y. Li, Boosting oxygen reduction catalysis with Fe–N4 sites decorated porous carbons toward fuel cells, ACS Catal., 2019, 9(3), 2158–2163 CrossRef CAS.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang and H. Wang, General synthesis of single-atom catalysts with high metal loading using graphene quantum dots, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M. X. Chen and K. MAllen, Multilayer stabilization for fabricating high-loading single-atom catalysts, Nat. Commun., 2020, 11(1), 1–11 CrossRef PubMed.
- J. Han, H. Bao, J. Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, 3D N-doped ordered mesoporous carbon supported single-atom Fe–NC catalysts with superior performance for oxygen reduction reaction and zinc–air battery, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- C. Wan, X. Duan and Y. Huang, Molecular design of single-atom catalysts for oxygen reduction reaction, Adv. Energy Mater., 2020, 10(14), 1903815 CrossRef CAS.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong and X. Feng, Zinc-mediated template synthesis of Fe–N–C electrocatalysts with densely accessible Fe–Nx active sites for efficient oxygen reduction, Adv. Mater., 2020, 32(8), 1907399 CrossRef CAS PubMed.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov and W. E. Mustain, High-performing commercial Fe–N–C cathode electrocatalyst for anion-exchange membrane fuel cells, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- X. Wu, H. Zhang, S. Zuo, J. Dong, Y. Li, J. Zhang and Y. Han, Engineering the coordination sphere of isolated active sites to explore the intrinsic activity in single-atom catalysts, Nano-Micro Lett., 2021, 13(1), 1–28 CrossRef PubMed.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More and G. Wu, High-performance fuel cell cathodes exclusively containing atomically dispersed iron active sites, Energy Environ. Sci., 2019, 12(8), 2548–2558 RSC.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan and J. Shui, Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells, Nat. Catal., 2019, 2(3), 259–268 CrossRef CAS.
- S. Liu, M. Wang, X. Yang, Q. Shi, Z. Qiao, M. Lucero and G. Wu, Chemical vapor deposition for atomically dispersed and nitrogen coordinated single metal site catalysts, Angew. Chem., Int. Ed., 2020, 59(48), 21698–21705 CrossRef CAS PubMed.
- L. Chen, X. Liu, L. Zheng, Y. Li, X. Guo, X. Wan and J. Shui, Insights into the role of active site density in the fuel cell performance of Co–NC catalysts, Appl. Catal., B, 2019, 256, 117849 CrossRef CAS.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener and Y. Shao, Performance enhancement and degradation mechanism identification of a single-atom Co–N–C catalyst for proton exchange membrane fuel cells, Nat. Catal., 2020, 3(12), 1044–1054 CrossRef CAS.
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, W. Wang and Y. Xie, High-density planar-like Fe2N6 structure catalyzes efficient oxygen reduction, Matter, 2020, 3(2), 509–521 CrossRef.
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin and W. Xing, Identification of binuclear Co2N5 active sites for oxygen reduction reaction with more than one magnitude higher activity than single atom CoN4 site, Nano Energy, 2018, 46, 396–403 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li and Y. Li, Design of N-coordinated dual-metal sites: A stable and active Pt-free catalyst for acidic oxygen reduction reaction, J. Am. Chem. Soc., 2017, 139(48), 17281–17284 CrossRef CAS PubMed.
- G. Zhang, Y. Jia, C. Zhang, X. Xiong, K. Sun, R. Chen and H. Dai, A general route via formamide condensation to prepare atomically dispersed metal–nitrogen–carbon electrocatalysts for energy technologies, Energy Environ. Sci., 2019, 12(4), 1317–1325 RSC.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang and S. Luo, Understanding of neighboring Fe–N4–C and Co–N4–C dual active centers for oxygen reduction reaction, Adv. Funct. Mater., 2021, 2011289 CrossRef CAS.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang and S. Guo, Emerging dual-atomic-site catalysts for efficient energy catalysis, Adv. Mater., 2021, 2102576 CrossRef CAS PubMed.
- B. W. Zhang, L. Ren, Z. F. Xu, N. Y. Cheng, W. H. Lai, L. Zhang and S. X. Dou, Atomic structural evolution of single-layer Pt clusters as efficient electrocatalysts, Small, 2021, 2100732 CrossRef CAS PubMed.
- T. Zhou, H. Shan, H. Yu, C. A. Zhong, J. Ge, N. Zhang and Y. Xie, Nanopore confinement of electrocatalysts optimizing triple transport for an ultrahigh-power-density zinc–air fuel cell with robust stability, Adv. Mater., 2020, 32(47), 2003251 CrossRef CAS PubMed.
- D. Qi, Y. Liu, M. Hu, X. Peng, Y. Qiu, S. Zhang and X. Liu, Engineering atomic sites via adjacent dual-metal sub-nanoclusters for efficient oxygen reduction reaction and Zn–air battery, Small, 2020, 16(48), 2004855 CrossRef CAS PubMed.
- X. Ao, W. Zhang, Z. Li, J. G. Li, L. Soule, X. Huang and X. C. Zeng, Markedly enhanced oxygen reduction activity of single-atom Fe catalysts via integration with Fe nanoclusters, ACS Nano, 2019, 13(10), 11853–11862 CrossRef CAS PubMed.
- M. Liu, J. Lee, T. C. Yang, F. Zheng, J. Zhao, C. M. Yang and L. Y. S. Lee, Synergies of Fe single atoms and clusters on N-doped carbon electrocatalyst for pH-universal oxygen reduction, Small Methods, 2021, 5(5), 2001165 CrossRef CAS.
- Y. Lei, F. Yang, H. Xie, Y. Lei, X. Liu, Y. Si and H. Wang, Biomass in situ conversion to Fe single atomic sites coupled with Fe2O3 clusters embedded in porous carbons for the oxygen reduction reaction, J. Mater. Chem. A, 2020, 8(39), 20629–20636 RSC.
- X. Wei, X. Luo, N. Wu, W. Gu, Y. Lin and C. Zhu, Recent advances in synergistically enhanced single-atomic site catalysts for boosted oxygen reduction reaction, Nano Energy, 2021, 105817 CrossRef CAS.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. Xu, Y. Zhang and X. Lu, Understanding the synergistic effects of cobalt single atoms and small nanoparticles: Enhancing oxygen reduction reaction catalytic activity and stability for zinc–air batteries, Adv. Funct. Mater., 2021, 2104735 CrossRef CAS.
- T. Jiang, W. Luan, L. Turyanska and Q. Feng, Enhanced electrocatalytic oxygen reduction reaction for Fe–N4–C by the incorporation of Co nanoparticles, Nanoscale, 2021, 13(13), 6521–6530 RSC.
- S. H. Yin, J. Yang, Y. Han, G. Li, L. Y. Wan, Y. H. Chen and S. G. Sun, Construction of highly active metal-containing nanoparticles and FeCo–N4 composite sites for the acidic oxygen reduction reaction, Angew. Chem., 2020, 132(49), 22160–22163 CrossRef.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen and S. Mu, Cobalt single atom site isolated Pt nanoparticles for efficient ORR and HER in acid media, Nano Energy, 2021, 106221 CrossRef CAS.
- Z. Qiao, C. Wang, C. Li, Y. Zeng, S. Hwang, B. Li and G. Wu, Atomically dispersed single iron sites for promoting Pt and Pt3Co fuel cell catalysts: performance and durability improvements, Energy Environ. Sci., 2021, 14, 4948–4960 RSC.
- P. Diao, M. Guo and Q. Zhang, How does the particle density affect the electrochemical behavior of gold nanoparticle assembly?, J. Phys. Chem. C, 2008, 112(17), 7036–7046 CrossRef CAS.
- Y. Kim, G. Collinge, M. S. Lee, K. Khivantsev, S. J. Cho, V. A. Glezakou and J. H. Kwak, Rücktitelbild: Surface density dependent catalytic activity of single palladium atoms supported on ceria (Angew. Chem. 42/2021), Angew. Chem., 2021, 133(42), 23212 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |