
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported nanocatalysts: recent developments in microwave synthesis for application in heterogeneous catalysis†
Hasan
Ahmad
 *a and
Mohammad Kawsar
Hossain
ab
*a and
Mohammad Kawsar
Hossain
ab
aResearch Laboratory of Polymer Colloids and Nanomaterials, Department of Chemistry, Rajshahi University, Rajshahi 6205, Bangladesh. E-mail: samarhass@yahoo.com; hahmad@ru.ac.bd
bBiomedical and Toxicological Research Institute, Bangladesh Council for Science and Industrial Research, Kudrat-E-Khoda Road, Dhanmondi, Dhaka 1205, Bangladesh
First published on 2nd December 2021
Abstract
The synthesis of nanocatalysts on high surface area solid supports is gaining attention from synthetic chemists for application in heterogeneous catalysis due to their high catalytic performance and easy recoverability. However, the synthesis of nanocatalysts on solid supports mostly composed of metals/metal oxides/hybrids is challenging for researchers considering their durability and discrete nano-dispersibility. Microwave (MW)-assisted synthetic methods, attractive as fast and green approaches, are being widely studied for the synthesis of solid-supported nanocatalysts. This review article describes recent advances devoted towards MW-assisted synthetic strategies for creating nanostructured catalysts on solid supports, as well as their characterization and applications in catalyzing various organic transformations, including electrocatalysis. The sustainable use of solid-supported nanocatalysts is then discussed in comparison with similar supported nanocatalysts prepared by conventional methods. The pros and cons of MW-assisted strategies for the synthesis of solid-supported nanocatalysts are then summarized, and their limitations as well as future directions for further investigation are emphasized.
 Hasan Ahmad | Hasan Ahmad graduated from the Department of Applied Chemistry, Rajshahi University, Bangladesh. He obtained his Doctor of Engineering degree in 1997 from Kobe University, Japan with a specialization in Material Sciences. He did his postdoctoral research at different institutes including the Max Planck Institute of Colloid and Interfaces, Germany; Sheffield University, UK; and GIST, South Korea. Dr Hasan has published more than 100 research articles in journals from publishers such as Elsevier, Springer, the American Chemical Society, The Royal Society of Chemistry and Wiley InterScience. He is presently serving as a Professor and Chairman at the Department of Chemistry at Rajshahi University, Bangladesh. |
 Mohammad Kawsar Hossain | Md. Kawsar Hossain has completed BSc and MSc (Organic) degrees and achieved the 1st Merit Position from the Department of Chemistry, Rajshahi University, Bangladesh. He did his research for his MSc degree under the supervision of Professor Hasan Ahmad. His research is focused mainly on polymer colloid chemistry, nanochemistry and hybrid materials. He has published 6 articles in international journals and is presently working as a Scientific Officer in the Biomedical and Toxicological Research Institute, Bangladesh Council for Science and Industrial Research, Dhaka. |
1. Introduction
To meet the rising demand of fine chemicals and to reduce the dependency on the use of expensive and non-recyclable homogeneous catalysts, synthetic chemists are searching for more durable nanostructured heterogeneous catalysts. Compared to bulk materials, nanostructured materials have proved beneficial in catalysis because they offer improved physicochemical properties such as high surface-to-volume ratio, surface energy, active sites and better composition-based selectivity towards particular reaction pathways.1,2 Additionally, the vast improvement in characterization tools has also enabled researchers to follow catalytic reactions from nanoparticles (NPs) down to even single atoms.3Transition metals (Ti, Cr, Mn, Fe, Ni, Cu, Ru, Pd, Pt, Au and Ag), which can easily donate and accept electrons from molecules, have been considered ideal for heterogeneous catalysis in many organic and bioorthogonal syntheses.4,5 However, nanocatalysts derived from transition metals/metal oxides/blends are prone to aggregation due to stronger inter-particle dipolar forces compared to their bulk counterparts, thereby reducing their potential for long-time application and preservation. Some transition metals (Cu, Ni) are also unstable and readily oxidized in air, preventing their sustainable use.6 The modification of transition metal nanocatalysts with functionalizing agents such as surfactants7,8 or polymers9 has helped to improve their stability against aggregation, but such modification often deteriorates their catalytic properties via changes in the electronic structure of the metal nanocatalysts.10 An alternative way proposed to overcome aggregation and the deterioration of catalytic properties is the implanting or anchoring of nanocatalysts on inert low-density solid supports. Solid supports help to maintain the size, shape, and morphology of the nanocatalysts and also limit the leaching of metal ions, which is favorable for achieving durable and enhanced catalytic performance. The solid supports used prominently include graphene,11,12 zeolites,13 cellulose,14–16 activated coke/carbon,17,18 sepiolite,19 bentonite,20 SiO2,21,22 and Al2O3.23,24 In general, the preparation of supported nanocatalysts is usually conducted in two steps. In the first step, the deposition or impregnation of the metal precursor on a solid support is achieved via impregnation/precipitation/coprecipitation/electrostatic/co-ordination, depending on the support functional groups.25,26 The second step involves calcination/reduction/both by different techniques such as chemical reduction,24,27 sonochemical reduction,28,29 laser ablation,30,31 and thermal decomposition.32 These methods generally require high temperatures, long reaction times and inert atmosphere. All these factors adversely affect the large-scale synthesis of supported nanocatalysts, and therefore the search for simple and scalable synthetic methods has become imperative. Recently, microwave (MW) irradiation/heating has gained popularity for the synthesis of metal/metal oxide nanocatalysts due to the fast and uniform heating of the reaction medium, which results in fast reaction kinetics,33,34 uniform growth of the NPs,35–37 easy operation and green synthesis.38
In this review, we discuss the recent articles on supported nanocatalysts prepared using MW strategies. The nanocatalysts discussed herein include metals, metal oxides, multi-metals, mixed metal–metal oxides and metal carbides. However, the few studies available on metal sulfides, nitrides and phosphides are not included. Some review articles on the optimization of catalytic performance by controlling the size, shape and morphology of different metal nanocomposites, both supported and non-supported, are available.39–43 However, these reviews hardly considered all types of metals and metal oxides, rather focusing on a group of metals or metal oxides. Jamkhande et al. reviewed the different methods used for the preparation of metal NPs and focused on their advantages, disadvantages and applications.44 In a recent review, MW chemistry was discussed briefly to understand the formation mechanism and growth of particles, focusing on improving the performance of MW methods for future research.45 In this review, we discuss the latest articles on the MW-assisted synthesis of transition metals, metal oxides, hybrids and perovskite-type nanocatalysts supported on solid supports. Initially, the chemistry of MW synthesis is shortly outlined, and then the preparation of various types of nanocatalysts is discussed. Subsequently, an explicit review is presented on the applications of supported nanocatalysts in various organic transformations. Particular emphasis is given to the recyclability/durability of supported nanocatalysts and the comparison of their performance with that of similar nanocatalysts prepared via other methods. Finally, the future challenges and ways of further improving the catalytic performance of nanocatalysts prepared via the MW method are described in the Scope and outlook section.
2. MW chemistry and its applications
Since the 1950s, MW energy was found to be useful for heating foodstuffs, food processing, chemical drying, etc. Upon irradiation with MW, the dielectric heating creates a heating effect on matter and the heating efficiency depends on the ability of the substance to interact with MW radiation.46 In 1986, two research groups first reported the utilization of MW warming to speed up organic chemical changes.47,48 In the early days, a household oven was used to perform experiments in sealed Teflon or glass vessels with no mechanism for controlling the temperature or pressure. This fast and uncontrolled heating of organic solvents later encouraged researchers to use solvent-free MW chemistry.49 The solvent free methodology is based on the pre-adsorption of reagents and catalysts on MW transparent inorganic supports, enabling controlled reactions in MW-assisted organic synthesis. With continuous improvement in MW technology and the availability of online temperature and pressure control, in the mid-90s Strauss developed a method of organic synthesis in a sealed vessel using a conventional solvent.50 Consequently, MW heating/irradiation has now become a recognized tool in all areas of organic synthesis including both solvent-free and water-mediated reactions.51–54 Additionally, the further development of MW reactor technology now facilitates the synthesis of nanomaterials.55–58 Recently, the continuous flow microfluidic approach has been attempted in MW-assisted synthesis to improve the control of the temperature gradient, rate of heating and mixing, and also to enable fast quenching to stop prolonged nucleation events.59The frequency and wavelength of MW radiation (i.e., electromagnetic radiation) are in the range of 0.3–300 GHz and 1 mm–1 m, which are between the infrared and radio region in the electromagnetic range. Commercial and domestic MW ovens operate at a frequency of up to 2.45 GHz to eliminate interference from the communication frequency bands of cellular phones. MW radiation travels with the speed of light and provides energy that is insufficient to break a chemical bond, which in most cases does not affect the framework of chemical molecules.60 MW heating depends on the capacity of materials to absorb and transform MW radiation into heat. Three categories of materials including MW reflectors, MW transmitters and MW absorbers describe the interaction with MW. Metals classified as MW reflectors are not effectively heated by MW. Materials that are transparent to MW (e.g., Teflon and polystyrene) are classified as MW transmitters and ideal for use as MW reactor and vessel materials. Materials known as MW absorbers can absorb energy from the MW field (e.g., solvents/reactants) and are heated very quickly in the process.61 The addition of a small amount of ionic liquid, a strong MW absorbing agent, to a non-polar solvent generally enhances the heating rate, and hence increases the reaction rate and product yield.62
MW-assisted reactions proceed at lightning speed, reducing the reaction time from several hours to minutes due to the reduction in the activation energy barrier.63 For example, the MW-irradiated green synthesis of Ag nanoclusters (2 nm) was possible within just 70 s,63 whereas the sonochemical method required 1 h to produce AgNPs,28 and chemical reduction of the precursor AgNO3 required an even longer reaction time (6 h).64 The conventional heating process using hot plates or mantles is slow and inefficient for transferring energy to the reactants, given that it depends on convective currents and the conductive nature of various compounds and the materials to be penetrated. This often produces a temperature gradient between the reaction medium and vessel, which is responsible for non-uniform growth, hindering scaled-up production. In contrast, MW irradiation causes volumetric heating, i.e., uniform temperature rise throughout the liquid volume via direct coupling of MW energy to the molecules present in the reaction vessel.65–67 This volumetric heating in the synthesis of nanomaterials causes rapid and homogeneous heating, which reduces the temperature gradient and provides instantaneous nucleation, controlled and uniform growth with good reproducibility in terms of size, shape and morphology.45 The shorter reaction time reduces the probability of the formation of byproducts through side reactions and increases the product yield, homogeneity and crystallinity.38
Thus far, MW-assisted irradiation/heating has been attempted for the synthesis of a large variety of unsupported mono- and bi-metallic NPs as well as different types of simple to quaternary metal oxdes.68–76 The morphology and average size can be controlled by the precursor type, concentration, solvent type, pH and MW heating conditions such as power, pressure, temperature and time.
3. Synthesis and characterization
3.1. Simple metal catalysts
Chuang et al. observed changes in the morphology of a Cu/AC catalyst due to a variation in MW irradiation power (400, 700 and 900), irradiation time (45, 60 or 110 s), and pH (4, 7, 10 and 12) of the polyol precursor during its preparation from a homogeneous solution of Cu precursor salt in ethylene glycol (EG).81 The obtained solids from the MW reactor were washed and dried, and then calcined under an H2/He atmosphere at 475 °C for 4 h before characterization. The maximum surface area and pore size were observed when the MW power was set at 700 W for 60 s and the pH of the polyol precursor was 7 (Cu/AC-700 W-60 sec-7). Under these conditions, Cu0 was the dominant phase. In contrast, the minimum 16 nm-sized Cu cubes (some aggregated) were formed when the pH of the polyol precursor was 12. Ren et al. employed pre-impregnation of AC with an aqueous Cu(NO3)2·3H2O solution and carbothermic MW irradiation at three different temperatures of 150 °C, 410 °C and 540 °C.82 The characteristic diffraction signal due to Cu0 was the strongest in Cat-48-540 among the catalysts, and optimistically a decrease in the average size of the cubic Cu2O and Cu0 particles from 43 ± 12 to 37 ± 12 nm was observed with an increase in irradiation temperature from 410 °C to 540 °C. The higher irradiation temperature was favorable to produce a higher Cu0 content. The same group also prepared hollow Cu/AC via template-free carbothermic MW reduction using a homogeneous solution of Cu(NO3)2·3H2O.83 The formation mechanism of hollow CuNPs was explained based on the Ostwald ripening effect and self-assembly under MW-induced reduction (Fig. 1). Under the optimum conditions, hollow CuNPs with large cavities of ∼35 nm diameter and 50 nm outer diameter were obtained within 8 min when the precursor was heated from room temperature to 360 °C.
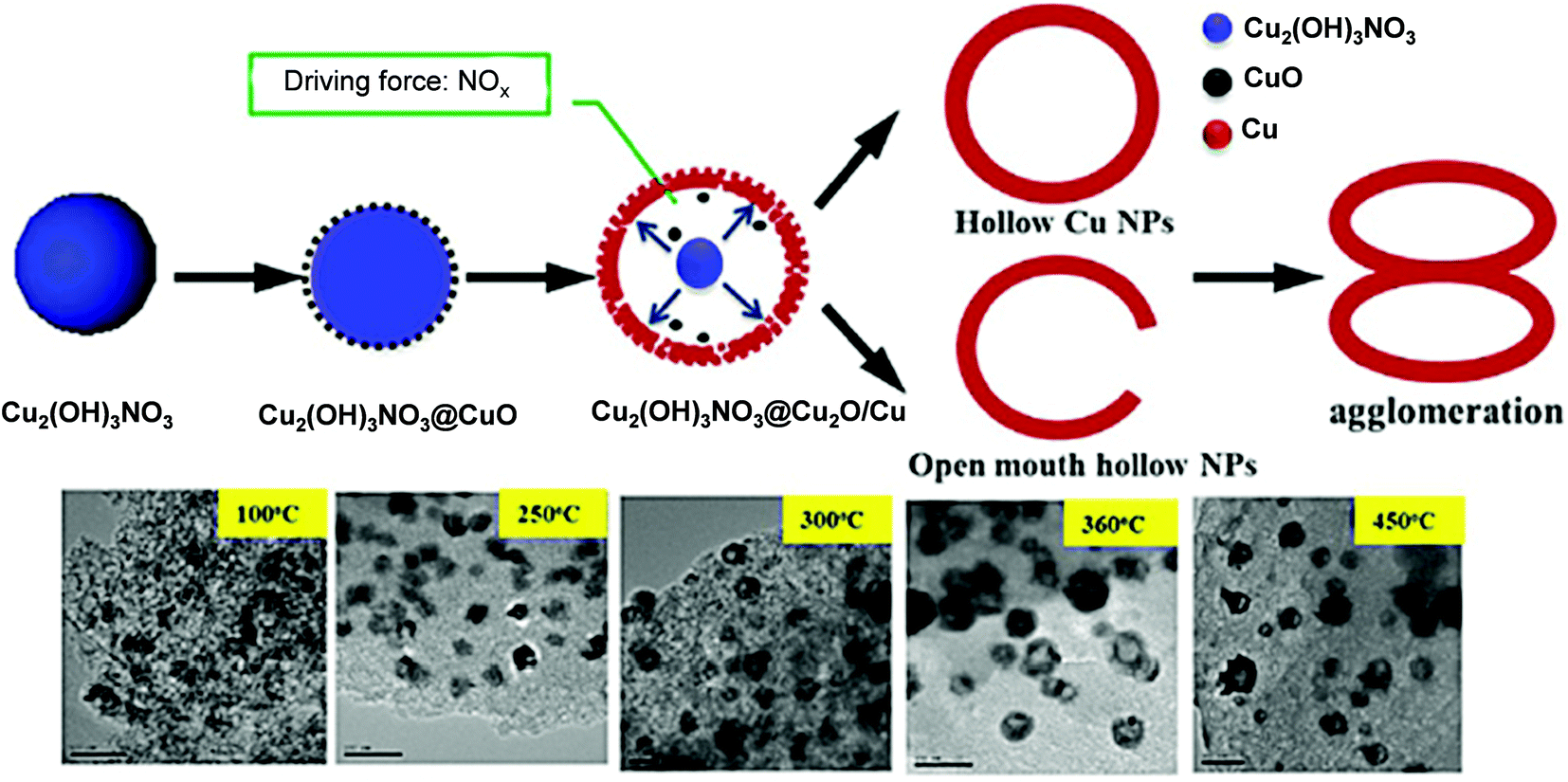 | ||
| Fig. 1 Schematic representation of the Ostwald ripening-coupled self-assembly process of the hollow Cu structures.83 (Copyright 2016, Wiley-VCH.) | ||
An Fe/AC catalyst was prepared via the impregnation of Fe(NO)3·9H2O in AC, drying and MW treatment for 2 h at 400 °C under an He flow.84 The theoretical ratio of Fe to AC was 10 wt%. The Brunauer–Emmett–Teller (BET) surface area of the catalyst obviously decreased in the Fe loading process; however, microscopic evidence was not provided.
Veerakumar et al. generated 3D ordered mesoporous carbon materials (CPMs) for supporting RuNPs by subjecting a cured mixture of polymer and Ru(acac)3 to MW thermal reduction and carbonization.85 The catalysts were labeled as Ru/CPM-1 and Ru/CPM-2 based on 0.5 and 1.0 wt% Ru loading, respectively. Hexagonal close-packed (hcp) Ru0NPs on a 3D CPM matrix with a crystallite size of 3–5 nm were detected and the average size of the RuNPs from transmission electron microscopy (TEM) was 2.6 ± 0.5 nm. The same group employed porous plastic-derived carbon (PDC) to prepare a PDC@RuNP catalyst by applying MW irradiation to a mixture of Ru(acac)3 and PDC in tetrahydrofuran at 300 W for 1 h.86 Very fine Ru0 crystallites with an average size of 5–6 nm were formed under these conditions.
The chemistry of supported Pt catalysts on carbon-based materials via MW synthesis started at the beginning of the 21st century.87,88 Here, they discussed the preparation of homogeneously distributed <4 nm sized PtNPs on either carbon or carbon nanotubes (CNT). In most cases Pt had a face centered cubic structure (fcc) and contained Pt0 as the major component. Later, Bo et al. used coal-derived granular AC (GAC) for the preparation of a Pt/GAC catalyst via MW irradiation.89 GAC was impregnated with an H2PtCL6·6H2O aqueous solution, evaporated to near-dryness in an MW oven at 500 W, and then calcined by MW at 500 W for 5 min. The Pt particles on the porous GAC support had an asymmetric dispersion and irregular shape with a size of 100–800 nm. Comparatively, Gan et al. obtained homogeneously distributed PtNPs (3–5 nm) on CNT via a one-pot MW polyol method using a mixture of H2PtCl6, EG and sodium dodecyl sulfate (SDS).90 The anchored Pt had an fcc crystalline structure and average crystallite size of 4.6 nm. A similar polyol method also produced well-dispersed spherical 2.7 nm-sized PtNPs in a Pt/carbon aerogel (CA) catalyst.91 Pt had an identical fcc structure and the average crystallite size (2.88 nm) matched the microscopic image diameter. Sharma et al. observed an improvement in the Pt loading up to 23.7 wt% in Pt/C electrocatalysts when the (NH4)2PtCl6 precursor was used in the MW-assisted polyol method.92 In contrast, the H2PtCl6-sourced and commercial 20% Pt/C catalysts contained 19.5 and 18.8 wt% Pt, respectively. Independent of the nature of the precursor, the average size of the PtNPs was around 2 nm. Thus, the precursor type is important during the MW synthesis of supported catalysts.
Pentsak et al. studied the deactivation process of Pd/C catalytic systems during catalysis under MW and conventional heat treatment (CHT).93 The catalyst preparation method was not detailed, but most likely wet impregnation and thermal decomposition were employed. Substantial transformation of the carbon support via the formation of pits, trenches, nanofibers and nanowalls during catalysis under both MW and CHT was observed. The narrow size distribution of PdNPs (6 nm) increased to 62 nm after 30 min of MW catalysis. The movement of hot metal particles on the carbon surface turned the carbon surface into a porous spongy framework. The initial Pd0 also turned into PdO (88%) after 30 min of MW treatment in air. These transformations of the metal support were more dominant upon MW treatment due to the easy formation of metal hot spots. The same research group further showed that the modification of the carbon support could be minimized if MW irradiation is carried out under vacuum and that the selection of an appropriate metal salt is also crucial.94
Antonetti et al. used a low-boiling point alcohol or a mixture of alcohol (isopropanol/ethanol)/water as the solvent and a reducing agent to prepare RuNPs/CNT under MW irradiation.95 The MW-assisted synthesis of unsupported RuNPs in isopropanol was first optimized using poly(N-vinyl-2-pyrrolidone), PVP, as a stabilizing agent, and finally the idea of MW synthesis was extended to the in situ preparation of RuNPs/CNT. Two types of CNTs were used, one was purified CNT (via treatment with HCl) and the other was HNO3-treated purified CNT (FCNT). Independent of the CNT type, the average diameter of the RuNPs was between 2.7 to 3.6 nm. However, the carboxylic and oxygenated groups on FCNT produced a more homogeneous distribution of RuNPs and sufficient to stabilize the NPs without the need for PVP. The same in situ approach using FCNT was also evaluated in aqueous ethanol (EtOH/H2O 20/80) at 150 °C under excess pressure. Again, small-sized particles with a homogeneous distribution were confirmed. In contrast, the solvent-free one-pot MW thermolytic route was found to be unsuitable for the production of well-dispersed Ru on FCNT.96,97 Hemraj-Benny et al. used non-functional single-walled CNT (SWCNT) to prepare an SWCNT-Ru catalyst via the reduction of RuCl3 in and ethanolic solution of NaBH4.98 The average size of the RuNPs was dependent on the MW temperature, where 100 °C yielded smaller (2.0 ± 0.5 nm) Ru0NPs. In contrast, a higher temperature caused partial agglomeration. In another investigation, the ethanol reduction of RuCl3 at 150 °C was examined to determine the effect of the weight percentage (0.05–1.00 wt%) of RuCl3 on the size distribution of the RuNPs in an SWCNT-Ru catalyst.99 An increase in RuCl3 content from 0.05 to 1.00 wt% resulted in an increase in particle size from 2.0 ± 0.5 nm to around 45 nm Ru clusters.
Graphene (G), an allotrope of carbon with a hexagonal atomic layer structure, is another important support material with unique properties such as high surface area and good thermal, chemical and mechanical stability.100–104 Fei et al. prepared a series of embedded monodispersed atomic transition metals in nitrogen-doped G (NG) via MW heating.105 The prepared nanocomposites were named M-NG-MW, where M represents Co/Ni/Cu. Initially, an aqueous solution of GO was reacted with ammonia solution at 70 °C to produce amine-functionalized GO (AGO). Typically, a certain amount of CoCl2 solution was well mixed with the AGO solution and the freeze-dried product was MW irradiated in an argon atmosphere. The amino N in Co-AGO was the dominant species, but in Co-NG-MW the amino N signal vanished due to its interaction with the uniformly distributed metal atoms.
In 2011, Siamaki et al. prepared Pd/G via batch-wise MW irradiation for 60 s.106 For this preparation, a mixture of dried graphite oxide (GrpO), requisite amount of Pd(NO3)2 and HNO3 was microwaved with N2H4 reducing agent following sonication. The complete reduction of GrpO to G by the reducing agent visibly improved the thermal stability of Pd/G compared to Pd/GrpO. Subsequently, the success of the continuous scalable flow approach was tested with respect to the reproducibility and yield of the Pd/G catalyst.107 A homogeneous suspension of GrpO in water containing Pd(NO3)2 and N2H4 solution was fed from two separate vessels at an equal rate into the MW reactor operating at 80 °C with an overall flow rate of 4 mL min−1. The produced Pd/G had an average particle diameter of 9.37 nm and contained 80% Pd0 with the rest being Pd(II). Alternatively, Yang et al. employed a batch MW-assisted reduction approach under 1.2 MPa H2 pressure to prepare Pd/G, Pd/AC, Pd/Grp and Pd/SiO2 catalysts using a mixture of support and [Pd(OAc)2]n in 0.1 M SDS.108 They found that the nature of the support influenced the Pd content in the catalyst.
Recently, Elazab et al. used a one-step MW irradiation approach to prepare magnetic Pd-Fe3O4/G from reduced GO sheets (rGO).109 In a typical procedure, Pd(NO3)2 was added to an aqueous solution of FeCl3 and reduced under MW condition using N2H4. Well-dispersed Fe3O4 particles were observed. A similar method was also tested to prepare Pd/G, Fe3O4/G nanocomposites using Pd(NO3)2 and FeCl3 precursors.
Sulfonated G and GrpO (100 g) were evaluated to prepare Pt/G and Pt/GrpO by heating a mixture of crushed K2PtCl6 (variable: 14.5, 355, and 15 mg) and ionic liquid, 2-hydroxyethanaminium formate, (5.0 g) in a MW oven.110 PtNPs with a size of 5 to 30 nm were formed on sulfonated G or GrpO. The shape of the PtNPs on G was spherical, whereas that on GrpO was cubic. A higher amount of functional oxygen groups on GrpO increased the anchoring efficiency of Pt and also influenced the shape and size of the produced NPs. The one-pot MW-assisted polyol synthesis of Pt/G nanocomposite and subsequent heating at 500 °C produced flaky crystalline G sheets.111 The Pt/G3 (0.50%) nanocomposite was comprised a comparatively uniform distribution of 7–11 nm-sized PtNPs.
3.1.2.1. Silica. Zuliani et al. used AlSBA-15, a mesoporous Al-coordinated SiO2 material, as a support for the preparation of MW-2%Ag@AlSBA-15 and MW-2%Au@AlSBA-15 via the simple MW in situ reduction of an ethanolic mixture of AgNO3 (0.04 mmol)/AuBr3 (0.02 mmol) at 150 °C for 3 min.112 The abundant hydroxyl groups on the support surface stabilized the metal NPs.113,114 Alternatively, the mechanochemical ball milling (BM) technique was used to prepare the BM-2%Ag@AlSBA-15 and BM-2%Au@AlSBA-15 nanocatalysts developed by other researchers.115 However, a detailed characterization in terms of size distribution, valence state and crystallinity was not presented. Manno et al. had two different MW synthetic approaches, i.e., batch synthesis (three different times: 70, 35 and 17 s) and continuous flow microfluidic synthesis (Fig. 2), to anchor Ag nanoclusters (AgNCs) on mesoporous amine-functionalized SBA-15 using an acrylic acid Na salt as a reducing and stabilizing agent.116,117 In the batch synthesis, an increase in irradiation time resulted in an increased in the mean particle size and the most uniform AgNCs (1.6 ± 0.7 nm) were obtained at 17 s. Slow cooling after MW heating in the batch system caused aggregation and size evolution of the clusters. In contrast, in a continuous flow reactor, rapid quenching to 4 °C within 21 s favored a reduction in the average diameter to 1.2 ± 0.3 nm. The continuous microfluidic approach produced better reproducibility and comparable inter-batch size distribution.
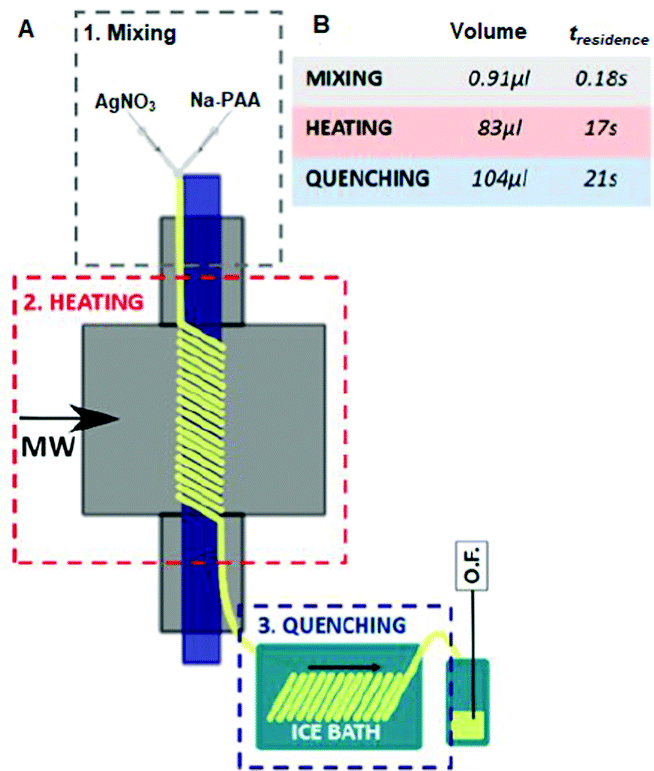 | ||
| Fig. 2 (A) Schematic representation of the flow diagram for the continuous synthesis of AgNCs. Three different zones: mixing stage (grey color), MW heating zone (red color) and quenching stage (blue color). (B) Volume and calculated residence time for each stage (total flow rate 0.292 mL min−1).116 (Copyright 2020, the American Chemical Society.) | ||
Jiang et al. used 40% H2SO4-treated bentonite (AB) to prepare an Ni/AB catalyst via a MW-assisted drying process (250 W, 10 min) and subsequent calcination (550 °C, 3 h).118 The impregnation slurry was made from an ethanol–water solution of Ni(NO3)2·6H2O, cetyltrimethylammonium bromide (CTAB) and AB suspension and the obtained catalyst was named Ni/AB-MW. Another batch of catalysts, named Ni/AB-CH, was prepared via conventional drying and calcination. MW-assisted drying improved the loading amount of Ni (16.1%) in Ni/AB-MW and also favored the formation of smaller Ni and NiO crystallites due to the better interaction of Ni with the AB support.
Akay et al. used the combination of MW radiation, heat treatment and plasma reduction under a stream of H2 for 24 h at 400 °C and 550 °C to prepare porous Co/SiO2 catalysts.119 The reduction under an H2 stream at 550 °C produced scattering signals due to the Co crystal lattice and CoO but reduction at 400 °C did not result in any detectable metallic Co. The crystallite size of metallic Co at 550 °C was 15.6 nm, larger than that of CoO (5.01 nm). Simultaneously, another group attempted to prepare Co/SiO2via a similar four-step process, including the impregnation of porous SiO2 in Co(NO3)2·6H2O, MW drying, furnace drying at 550 °C, and finally reduction under an H2 flow at 550 °C for 24 h.120 The scanning electron microscopy (SEM) and TEM images revealed that the MW catalyst had a better distribution and uniformity (crystallite size, 3–8 nm) and loading of CoNPs compared to that prepared via the conventional drying method. Similar to the previous literature,119 this investigation also suggested that the final reduction under an H2 flow is necessary to produce the Co metallic phase.
The MW in situ reduction of a dried SiO2-impregnated H2PtCl6·6H2O precursor in EG containing N2H4 produced fine well-dispersed ≤2 nm-sized PtNPs on an SiO2 support at the loading of 0.3 wt%.121 An increase in the Pt loading to 0.9 wt% slightly increased the particle size to <10 nm. Comparatively, the rotary chemical evaporation (RCE) method produced larger (≥20 nm)-sized PtNPs. The comparative study revealed that the MW-assisted reduction method is superior with respect to the organized distribution of smaller PtNPs on the surface of SiO2.
Wang et al. prepared a series of Ti-MCM-41 with different Si/Ti ratios (100 to 5) under MW irradiation using CTAB template, TEOS and tributyl titanate (TBOT).122 The obtained materials were calcined at 550 °C for 6 h in air to decompose the template. The Ti contents in the catalyst samples closely resembled the theoretical values. The arrangement of channels was slightly disturbed with an increase in the Ti content. Earlier, another research group prepared ZrMCM-41 mesoporous molecular sieves using Zr(SO4)2 and CTAB under MW irradiation.123 The BET specific surface area and pore volume decreased with an increase in the incorporated Zr. The mesoporous ordering deteriorated with an increase in the thermal treatment temperature.
3.1.2.2. Fe3O4 and analogous oxides. Fe3O4 NPs possess interesting application properties as a catalyst support because the presence of Fe2+ ions provides some reducible oxide characters.124 Additionally Co3O4 and Ni(OH)2 also show good activity in the oxidation of CO.125,126 Elazab et al. proposed a method for the preparation of PdNPs supported on Fe3O4, Co3O4 and Ni(OH)2 NPs and nanoplates via MW irradiation.127 In a typical process for the preparation of Pd/Fe3O4 containing 5, 20, 40 and 50 wt% Pd, mixtures of Fe(NO3)3·9H2O and Pd(NO3)2 solutions (10 wt% in 10 wt% HNO3) were sonicated for 1 h, and then irradiated in the presence of N2H4 in 60 s cycles for a total 7 min. Similarly, with slight modification, Pd/Co3O4 and Pd/Ni(OH)2 catalysts containing variable wt% of Pd were prepared. The majority of the PdNPs were deposited on the surface of nanoplates. In another study, Elazab et al. reported the synthesis of a Pd/Fe3O4 catalyst via continuous flow synthesis under MW irradiation.128 In this approach, two separate solutions, one containing the required amount of Fe(NO3)3·6H2O and another containing N2H4, were injected and mixed before being introduced in the reactor. Different operating temperatures (80 °C, 120 °C and 150 °C) and flow rates were studied. The XPS binding energy showed that a fraction of Pd was in PdO. An increase in MW temperature resulted in an increase in size and the saturation magnetization was reduced when the particle size was smaller. Continuous flow reaction was found to be advantageous for producing smaller (7–9 nm) PdNPs than that (12–14 nm) prepared under batch conditions.
Morad et al. prepared AuNPs on an Mn3O4 support by reducing an HAuCl4 hydrate solution using N2H4 under MW irradiation.129 Mn(OH)2 gel was prepared via direct precipitation from Mn(NO3)2·4H2O at pH 12. The obtained gel was divided into two parts. One part was subjected to hydrothermal treatment (HT) at 150 °C, filtered, washed, and then dried to obtain Mn3O4 HT. The other part of the gel, represented as Mn3O4, was directly filtered and dried at 100 °C. The TEM images indicated the formation of cubic-, spheroid- and plate-shaped nanocatalysts (∼33 nm) without any Au aggregates. Almost simultaneously, another research group reported identical cubic-, spheroid- and plate-shaped PdNPs/Mn3O4 nanostructures (38 nm) using an identical preparation method.130 The presence of PdNPs could not be confirmed from the TEM images, but the XPS results indicated the presence of Pd0 and Pd2+ species on the Mn3O4 support.
3.1.2.3. Other metal oxides. In an early attempt, Glaspell et al. observed that the use of a capping agent, poly(ethylene glycol), PEG, or PVP in the preparation of Au and Pd NPs on CeO2/CuO/ZnO support materials accelerated the nucleation process of metal NPs and produced a narrow size distribution.131 The respective support was derived from precursor salts (Ce(NO3)4, Cu(NO3)2 and Zn(NO3)2, respectively) and dopant Au or PdNPs were prepared via the addition of an appropriate amount of metal salt (HAuCl4 or Pd(NO3)2) to obtain the desired dopant concentration (2%, 5% and 10%). Comparatively, the 10% Pd/CeO2 catalyst had the narrowest size distribution with average size 3–4 nm. Chuang et al. used CeO2-TD, which was obtained via the thermal decomposition of Ce salt, to prepare Cu/CeO2-TD (Cu 5 wt%) by MW heating of an EG solution of Cu(NO3)·3H2O and PVP.132 Finally, the dried product was calcined at 475 °C for 4 h. For comparison, commercial ceria (CeO2-C) and AC were also used as support materials. The size of the Cu particles was dependent on the nature and size of the support materials, where the minimum size (33 nm) was found for the Cu/CeO2-TD catalyst. Elazab et al. used different amounts (347, 329, 292, and 256 mg) of Cu2H10N4O17 precursor salt to prepare a Pd/CuO catalyst with variable Pd loadings (5, 10, 20, and 30 wt%).133,134 A simple one-step MW process was applied in the presence of N2H4. The TEM images revealed the formation of a 20–40 nm-sized catalyst and X-ray photoelectron spectroscopy (XPS) confirmed the valence states as Cu2+ and Pd0. The estimated Pd content in the catalyst was much lower than the theoretical content calculated from its formula.
The different forms of Al2O3 (α, β, and γ) are perhaps the most important as catalyst supports due to their favorable combination of textural properties such as specific surface area, pore volume, pore size distribution, local microstructure, phase composition and acid/base characteristics.135 Song et al. prepared an Ni/Al2O3 catalyst using PVP as a stabilizer and MW absorbent.136 The catalysts were denoted as Ni20–Al2O3–M and Ni20–Al2O3–I, where 20 represents the loading amount of Ni, and M and I stand for the MW and impregnation techniques, respectively. The MW method produced smaller (∼10 nm) NiNPs in Ni20–Al2O3–M compared to that (15 nm) in Ni20–Al2O3–I. The BET surface area and pore volume of Ni20–Al2O3–M were also higher. This indirectly confirmed that the impregnation method (IM) produced slight agglomeration and blocking of the pore structure. In another investigation, Pd/Al2O3 catalysts were prepared via fixed-bed MW-induced plasma irradiation under three different conditions.137Fig. 3 illustrates the fixed-bed reactor with MW-induced plasma jet. The particles prepared under different conditions were named PF (fixed bed reactor). The distance between the waveguide and particle bed influenced the distribution and size of Pd (Fig. 3). Spherical-shaped tiny Pd particles and the highest dispersion (25.3%) were observed in sample PF-1 when a shorter distance was applied (3 cm). It is obvious that a short distance produced a higher temperature. The plasma treatment reduced PdO to Pd in a short time, which had an fcc structure. The crystalline phase of γ-Al2O3 was detected in all the catalysts. The same authors reported the use of an MW-induced plasma-spouted bed reactor for the preparation of Pd/Al2O3 under various operating conditions.138 The same catalyst was also prepared via CHT in an electric arc furnace. The MW-combined plasma treatment produced smaller PdNPs than that prepared by conventional heating at 500 °C. The rapid heat change induced by an increase in MW power reportedly produced surface cracks in Pd/Al2O3, whereas CHT at 500 °C did not produce surface cracks in the Pd/Al2O3 samples.
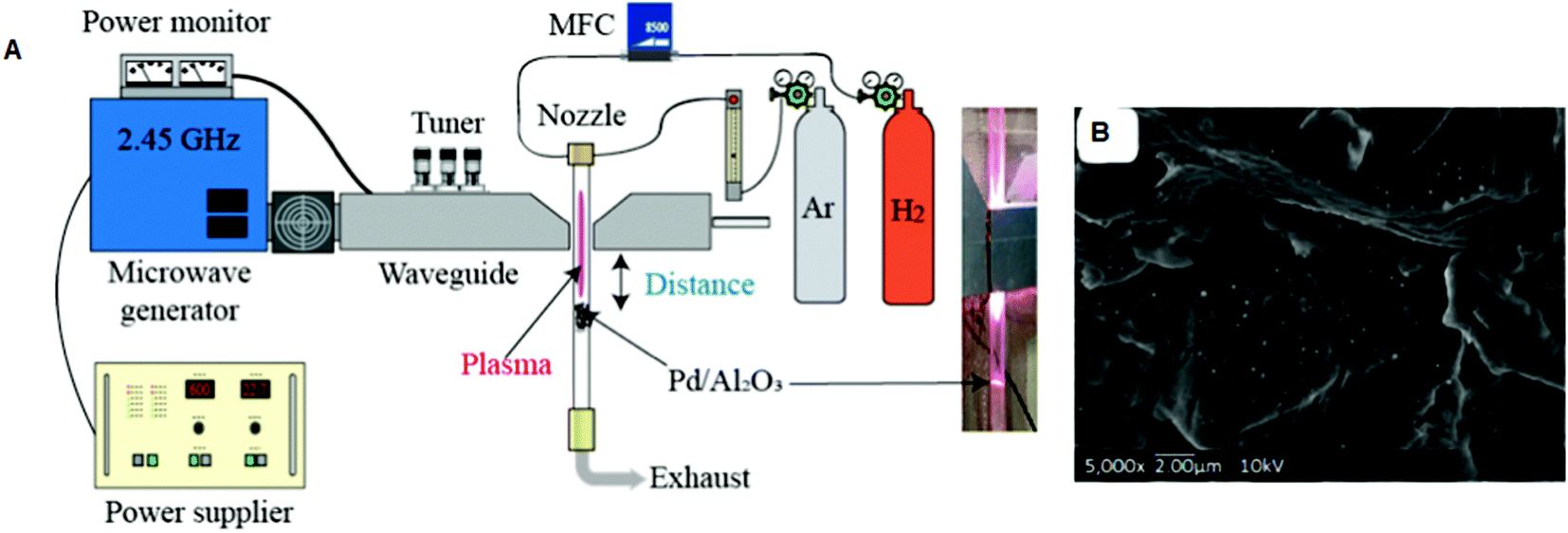 | ||
| Fig. 3 (A) Schematic representation of the fixed bed reactor with plasma irradiation. (B) A typical SEM image of the Pd/Al2O3 catalyst (PF-1).137 (Copyright 2019, MDPI.) | ||
Wang et al. observed that MW hydrothermal (MWH) treatment is superior to conventional IM in terms of the better dispersion of WO3 on an Al2O3 support and oxalic acid stabilizer had little effect on the dispersion.139 The catalyst prepared by IM also had a higher agglomeration of WO3. After MWH treatment, the dried WO3/Al2O3 was calcined at 550 °C for 4 h to produce W/Al2O3.
TiO2 as a catalyst support is known to possess high thermal and chemical stability and offer outstanding resistance to corrosion in electrolytic media.140 Howe et al. prepared a 1 wt% Ru/TiO2 catalyst in an MW reactor from stock solutions of Ru(acac)3 or RuCl3·xH2O in EG and TiO2 at 150 °C, 170 °C and 200 °C for 5, 10 and 15 min.141 The obtained catalysts were air dried at 110 °C for 16 h. The catalysts were represented by the reductant-preparation temperature (°C)-preparation time, e.g. Acac-200-10. Independent of the precursor salt, MW treatment for a longer time at 200 °C caused sintering of the RuNPs, given that their mean diameter increased slightly. However, despite the sintering effect, the average size of the RuNPs was still less than 3 nm. In either case, the catalysts exhibited a high surface carbon concentration.
To understand the detailed formation mechanism of various hybrids, Pt and AuNPs supported on CeO2, TiO2, ZnO, MgO, Al2O3, and SiO2 were prepared via the one-pot MW-assisted reduction of metal salts in EG.142 The size of the metal NPs was influenced by the efficiency of MW absorption by the support materials. Smaller and fine 2–3 nm-sized PtNPs were formed on CeO2, TiO2 and ZnO but not on the SiO2 particles. Comparatively, the low dielectric loss substrate SiO2 did not heat up rapidly. Likewise, on MgO and TiO2, the uniform decoration of AuNPs was observed, whereas on Al2O3 and Si substrates, very few particles were present. This is because the Al2O3 and Si substrates did not absorb MW radiation.
3.2. Metal carbide catalysts
In terms of application performance, metal carbides such as W and Mo carbides are considered alternatives to noble metal catalysts, particularly Pt.148 In 1973, Levy et al. showed that WC and Pt had similarities in electronic structure and catalysis behavior in water formation from H2 and O2 at room temperature.149 Following that groundbreaking work, these carbides were proven to be useful in a wide variety of catalytic reactions such as hydrogenation, hydrodeoxygenation, hydrodesulphurization, hydrodenitrogenation and isomerization.150–153 In this perspective, a few works carried out on the preparation and applications of supported metal carbides are discussed below.In 2009, Liang et al. described the preparation of WC/CNT via MW-assisted metal–organic chemical vapor decomposition (MOCVD) under atmospheric pressure in a fluidized reactor.157 A precursor mixture made from W(CO)6 and CNT was placed in a quartz-tube reactor and fluidized with an argon flow for 2 h before irradiation in an MW oven. CNTs without pre-oxidized and pre-oxidized were evaluated as a support. A uniform distribution of 2–5 nm-sized WC was found on the surface of the CNTs. Overall, the results indicated that preoxidation of CNTs is not essential in the MW-assisted MOCVD method. In the following year, Pang et al. described the preparation of Mo2C/CNTs utilizing the above-mentioned method.158 However, the CNTs were not pre-oxidized before use. In 16.7 wt% Mo2C/CNT, well-dispersed 5 nm-sized Mo2C was detected. The average crystallite size of Mo2C increased with an increase in the Mo loading. Pt-Mo2C/CNT was also prepared and 3–6 nm monodispersed PtNPs were formed.
3.3. Metal oxide catalysts
Zhong et al. demonstrated the suitability of MW-assisted 3D heating in air for the rapid in situ synthesis of metal oxides (CoOx, NiO and Fe3O4) using carbonized wood (C-wood) as a 3 D heating substrate.75 In a typical experiment, the Co(NO3)2 precursor was dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol-water mixture, and then a piece of C-wood was immersed in this mixture for 10 min and dried for 20 min to obtain the C-wood/Co(NO3)2 precursor. The C-wood/Co(NO3)2 precursor was then microwaved at 1000 W for 4 s to produce C-wood/CoOx. Around 11 nm-sized uniformly distributed CoOx NPs were formed on the C-wood substrate. The potential scalability of the 3D heating method was also verified with a larger-sized C-wood substrate.
:1 ethanol-water mixture, and then a piece of C-wood was immersed in this mixture for 10 min and dried for 20 min to obtain the C-wood/Co(NO3)2 precursor. The C-wood/Co(NO3)2 precursor was then microwaved at 1000 W for 4 s to produce C-wood/CoOx. Around 11 nm-sized uniformly distributed CoOx NPs were formed on the C-wood substrate. The potential scalability of the 3D heating method was also verified with a larger-sized C-wood substrate.
Motshekga prepared MWCNT/SnO2 NPs via the conventional and MW-assisted wet impregnation for application in optoelectronic devices.160 the SEM and TEM images confirmed the high loading of SnO2 NPs (2–4 nm) on MWCNT via MW method within 10 min. There is also a report on the preparation of TiO2/G from GrpO, titanium isopropoxide, TTIP (variable) in isopropyl alcohol and N2H4via MW irradiation in a closed Pyrex glass bottle.161 The obtained dried precipitate was finally calcined at 400 °C for 4 h.
Li et al. prepared an NiO/vermiculite or NiO/Vmt catalyst with 10 wt% loading from a dried precursor comprising Vmt in Ni(NO3)2·6H2O aqueous solution.163 The precursor was divided into two parts, where one part was subjected to CHT and the other part was subjected to MW treatment to obtain CHT-NiO/Vmt and MW-NiO/Vmt, respectively. The NiO NPs were well dispersed, but MW irradiation produced smaller particles (3 nm) compared to that from CHT (6 nm). The estimated crystallite size in the MW-NiO/Vmt catalyst was 2.4 nm, lower than that in the CHT-NiO/Vmt catalyst (2.7 nm). In a similar investigation, 2-D layered Vmt was used to prepare MW-assisted MW-NiO/2D-Vmt and compared with muffle furnace (MF) calcination.164 The overall size distribution and crystallinity again confirmed that the MW method is superior for producing smaller-grained highly crystalline NiO.
 | ||
| Fig. 4 (A) Proposed mechanism for the controlled synthesis of Co3O4@SUS. TEM images of (B) Co3O4@SUS and (C) Co3O4-LNS.165 (Copyright 2017, the American Chemical Society.) | ||
May-Masnou discussed the preparation of anatase TiO2 NPs attached to larger anisotropic Au via an MW-assisted method.166 Firstly, TiO2 NPs were synthesized from a homogeneous solution of titanium butoxide (TBOT) and PVP in benzyl alcohol (BA) in an MW reactor at 50 °C, and then again heated at 190 °C for 10 min. In the second step, two different sized AuNPs (Fig. 5) were prepared in the presence of TiO2 NPs dispersed in EG using (a) 100 and (b) 600 mg of PVP. Part of the sample was dried overnight and the other part was calcined in air at 400 °C, 450 °C, 550 °C, and 600 °C for 2 h to remove PVP and increase the contact between Au and TiO2. The PVP-capped TiO2 NPs possessed a pure crystalline anatase phase with an average size of 9 ± 2 nm. The TEM photographs (Fig. 5) indicated that 100 mg of PVP produced >50 nm-sized Au NPs, whereas 600 mg of PVP produced smaller (8 ± 2 nm) AuNPs. The larger AuNPs were randomly decorated with smaller TiO2 NPs but the smaller AuNPs were only surrounded by TiO2. Calcination had a larger effect on the shape of Au than that of the TiO2 NPs.
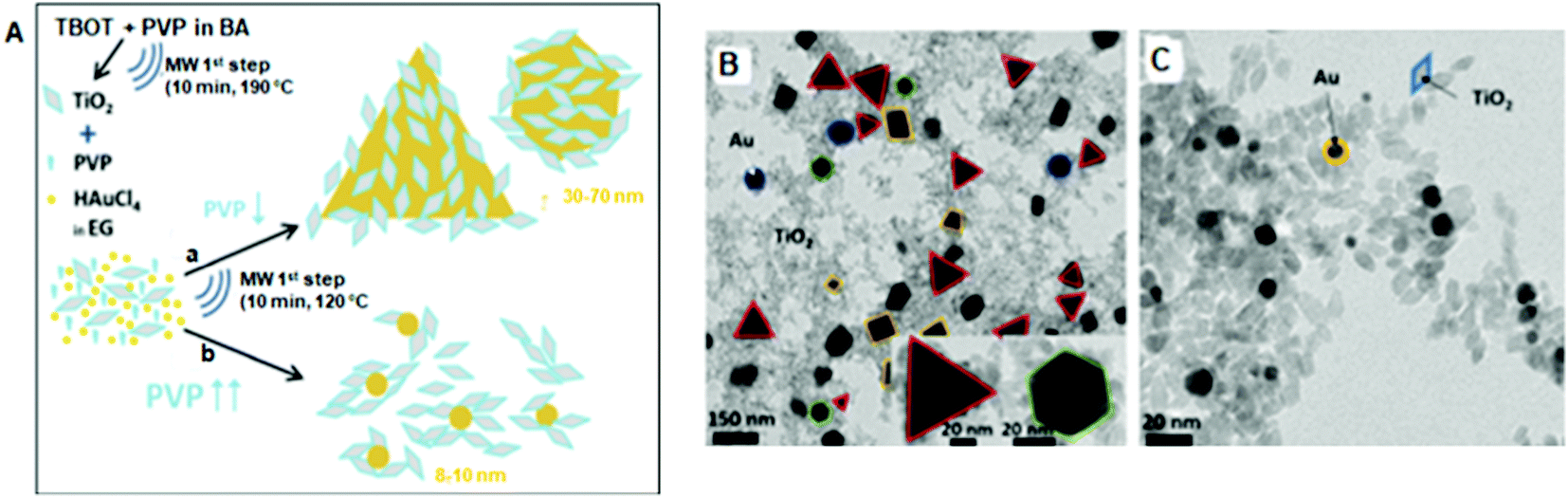 | ||
| Fig. 5 (A) Schematic diagram of the two-step MW-assisted synthesis of Au/TiO2 nanostructures. (a) TiO2:PVP:HAuCl4 = 1:0.097:0.155 and (b) TiO2:PVP:HAuCl4 = 1:0.583:0.155 (molar ratios). TEM images of the as-synthesized (B) sample ‘a’ and (C) sample ‘b’.166 | ||
3.4. Binary metal catalysts
Binary and ternary metal catalysts, which are considered more active compared to simple metal catalysts, are particularly useful as electrocatalysts in proton exchange membrane fuel cells (PEMFCs) and direct alcohol fuel cells.167,168 In early investigations, electrocatalysts based on precious metals such as Pt or Au were mostly investigated. However, the low durability and high cost Pt and Au metal catalysts hinder their commercial application. Additionally, supported Pt-based catalysts exhibit sluggish oxygen reduction reaction (ORR) activity at the cathode. Accordingly, doping with transition metals containing Pt or Au reportedly improved their durability.169,170 The search for alternative low-cost materials with high electrocatalytic activity and good durability has led to the development of ternary metal catalysts either by replacing or reducing the Pt or Au content.:5 and Pt–YOx/C(B):5:5, where A and B represent the cis-[Pt(NH3)2(NO2)2] and Pt(C5H7O2)2 precursor, respectively. To overcome the solubilization of the YOx component in acid fuel solution, the samples were treated with HNO3 to produce PtY/C alloy catalysts. After acid treatment, both samples contained a small amount of Y component and the catalyst size with a narrow distribution was retained. The addition of Y(CH3COO)3·4H2O was found to be beneficial for producing monodispersed Pt/C(A, B) particles. To enhance the binding between the PtNPs and carbon supports, a Pt3Cu/C catalyst was prepared with a theoretical Pt loading of 20 wt%.173 A mixture of metallic precursors (H2PtCl6·6H2O and CuCl2·2H2O) and carbon support in an EG–water mixture was microwaved at pH 10 for approximately 6 min at 125 °C. The Pt3Cu NPs (2.31 nm) were uniformly distributed on the support. Pt and Cu had an fcc crystalline alloy structure. The metallic state of Pt and Cu was also confirmed. By just changing the amount of Cu precursor in the recipe, Pt4Cu/C (2.14 ± 0.88 nm) and Pt2Cu/C (3.49 ± 1.097 nm) were also prepared and characterized. In the synthesis of an Pd–Au/C catalyst, relative to CHT, catalyst activation by MW irradiation produced a much higher degree of Pd–Au alloying and better homogeneity of the bimetallic phase.174 The use of acetone as a solvent also improved the homogeneity of the Pd–Au phase compared to the aqueous solution. The catalyst samples produced by MW irradiation also contained an increase Pd content at the surface of the NPs. The poor ORR activity of 20% CoSe/C encouraged the preparation of the corresponding CoSe2/C.175–177 Accordingly, a homogeneous precursor solution of Co(CH3COO)2 and SeO2 was placed in an MW oven at a power of 800 W for 180 s, and the product was washed and dried before mixing with carbon black. Diffraction signals of the CoSe2 phase were not observed up to an Se/Co ratio of 4.0 due to their uniform distribution. In another investigation, FeSe2/C NPs were prepared via an MW method under identical conditions with the FeSe2 loading adjusted to 36%.178 The average particle diameter was in the range of 3–12 nm.
Sulfonated MWCNT (SF-MWCNT) was used to prepare SF-MWCNT-PdSn and SF-MWCNT-PdNi via MW-assisted solvothermal co-reduction (1000 W, 170 °C, 60 s) of the precursor salts in EG.179 Pd had a well-defined fcc crystalline structure, but Ni and Sn possessed a partially amorphous structure. The alloy structure was confirmed and the XPS spectra suggested the presence of Pd in the metallic and oxide forms with Ni in the oxo-nickel species (i.e., NixOy and NiOOH). However, the uniform distribution of the bimetallic catalysts was not analyzed. Nassr et al. used oxygen-functionalized CNT (FCNT) to prepare a PtNi/FCNT catalyst via the polyol MW reduction of metal salts, H2PtCl6 and Ni(NO3)·6H2O, either continuously for a defined time or in pulse mode with a duration of 20 s.180 The transition of the NiO peaks to PtNi alloy occurred at 90 s. However, in pulse mode, the formation of the PtNi alloy was observed even at a lower pulse number of 6. An increase in the irradiation time enhanced the particle size of PtNi/CNT from 2.7 to 3.3 nm. Comparatively, the size of PtNi/CNT prepared in the pulse mode was almost independent of the pulse number given that the average size was around 2.5 nm. Rahsepar et al. prepared a PtRu/MWCNT catalyst using MW-assisted reduction at pH ∼12.5 from a water solution of metal precursors, pretreated MWCNT and HCHO reducing agent.181 After re-acidification and drying, additional heating at 300 °C under a flow of H2 helped to form well-dispersed PtRu alloy NPs (1.5–3.5 nm) on the surface of MWCNT. The XPS signals indicated the presence of a large amount of Pt0 (72.15%) and Ru0 (68.07%) with a small amount of Pt(II), Pt(IV), RuO2 and RuO2·xH2O species. Al-Tememy had MWCNT-doped G nanoplatelets (MWCNT-GNP) to support a PtPd bimetallic catalyst by microwaving a mixture of metal precursors and the support dispersed in an EG-isopropanol mixture at pH 12.182 For comparison, PtPd/GNP and PtPd/MWCNT catalysts were also investigated. The doping of G nanoplatelets by MWCNT improved the metal loading (35.1%), dispersity and crystallinity index of the PtPd NPs. Independent of the support material, all the catalysts contained graphitic carbon and fcc-structured metals. The average sizes were in the range of 2.47–2.70 nm.
3.4.2.1. Silica. Verma et al. discussed the preparation of Pd–Ag/SiO2 and Pd–Au/SiO2 bimetallic catalysts in two steps.183 Firstly, 1 wt% of either Au or Ag was incorporated on SBA-15 via the MW-assisted polyol method, and then decorated with PdNPs via a localized surface plasmon resonance-assisted deposition method under visible light irradiation. Spherical NPs of Pd-Ag and Pd–Au with diameters of 4.2 and 4.9 nm were formed. The formation of the alloy via the electron transfer mechanism was proved by XPS. Tripathi et al. demonstrated the preparation of Cu–Ag/MgO–SiO2via four different techniques including conventional impregnation, MW-assisted method, PVA/NaBH4-assisted method and benzoxazine-assisted method.184 In the MW method, drying was carried out at 80 °C in an MW oven before calcination at 400 °C for 4 h. MW drying and benzoxazine-assisted reduction efficiently produced a higher surface area and pore volume. Partial oxidation of Cu to CuO was observed in Cu–Ag/MgO–SiO2. However, other characterizations were not presented. In another investigation, Ti-inserted ordered mesoporous SiO2 (Ti-SBA-15) was used to support a CoMo catalyst.185 20Ti-SBA-15 (Si/Ti molar ratio 20) was prepared via hydrolysis and aging in an MW reactor from TEOS and TTIP in the presence of Pluronic P123 as a structure-directing agent. The crystallized product was further calcined at 450 °C for 6 h. As expected, the insertion of Ti reduced the surface area and pore size of SBA-15. The Ti loading in 20Ti-SBA-15 and CoMo/20Ti-SBA15 was 2.9 and 2.4 wt%, respectively, which was close to the theoretical value.
3.4.2.2. Metal oxides. In an early investigation, Ru–Sn/ZnO catalysts were synthesized by subjecting a mixture of RuCl3·3H2O dissolved in EG containing SnCl2·2H2O and ZnO (targeted metal loading 2%) to variable MW power.186 XPS analysis showed the formation of Sn0 in all the Ru–Sn/ZnO catalysts prepared at 450 W for 4 and 8 min and Ru0 was detected only when the MW power was set at 150 W for 20 min. The rest of the samples mainly contained Run+. The morphological data for the Ru–Sn/ZnO catalysts was not available.
 | ||
| Fig. 6 (A) Preparation scheme of the Ni3Fe/Co–N–C bifunctional electrocatalyst. TEM (B and C) and HRTEM (D) images of (B) Co–N–C and (C and D) Ni3Fe/Co–N–C.187 (Copyright 2020, Elsevier B.V.) | ||
3.5. Mixed metal oxide catalysts
:1) mixture containing anatase TiO2 and varying amounts of 0.01 mol L−1 AgNO3.190 The weight ratio of Ag to the TiO2–G composite was controlled at 0, 0.03, 0.06, 0.09, 0.12, and 0.15 and composites were named TG, ATG3, ATG6, ATG9, ATG12, and ATG15, respectively. A crystalline anatase TiO2 phase was found, but G and Ag could not be detected in Ag–TiO2/G owing to their low content. The valence states were confirmed to be Ti4+ and Ag0 in the composites. Pan et al. used N-doped coal-derived carbon to support Co/CoOx.191 Depending on the heat treatment temperature, the coal-derived carbons were labeled as MC800, MC950, MC1050 and MC1100. The high-temperature treatment of the carbon (MC1050) was found to be favorable for producing structural disorder and subsequent introduction of Co/CoOx. The Co nucleation was followed by strong electrostatic interaction between the oxygen functional groups on the surface and Co cations. The Co/CoOx particles were homogeneously distributed at a lower Co content. Galal et al. discussed the preparation of Ru-based perovskites supported on RGO.192 The preparation was carried out via the redox reaction of metal salts (Sr(NO3)2/Ba(NO3)2/Ca(NO3)2), RuCl3 and GO via either conventional or MW heating. Both conventional and MW heating allowed the formation of SrRuO3 in combination with RuO2 or RuNPs. Highly crystalline nanosized CaRuO3 was detected as the main phase using MW heating. In comparison, around 1 nm-sized BaRuO3 particles with RuO2 were formed via conventional heating, while MW heating produced amorphous BaRuO3.
Li et al. used carboxymethyl cellulose (CMC), an inexpensive biobased polymer, to support Ag–Fe3O4.193 Firstly, Ag@CMC was prepared via the MW heating of Ag(NH3)2+ salt in the presence of aqueous CMC sodium salt.194,195 Secondly, Fe3O4 was co-precipitated under MW irradiation and alkaline conditions to afford Ag–Fe3O4@CMC (Fig. 7). Monodispersed AgNPs (2.3 ± 0.3 nm) were formed in Ag@CMC, but the magnetic Fe3O4 NPs (15.3 ± 3.5 nm) were slightly polydispersed (Fig. 7).
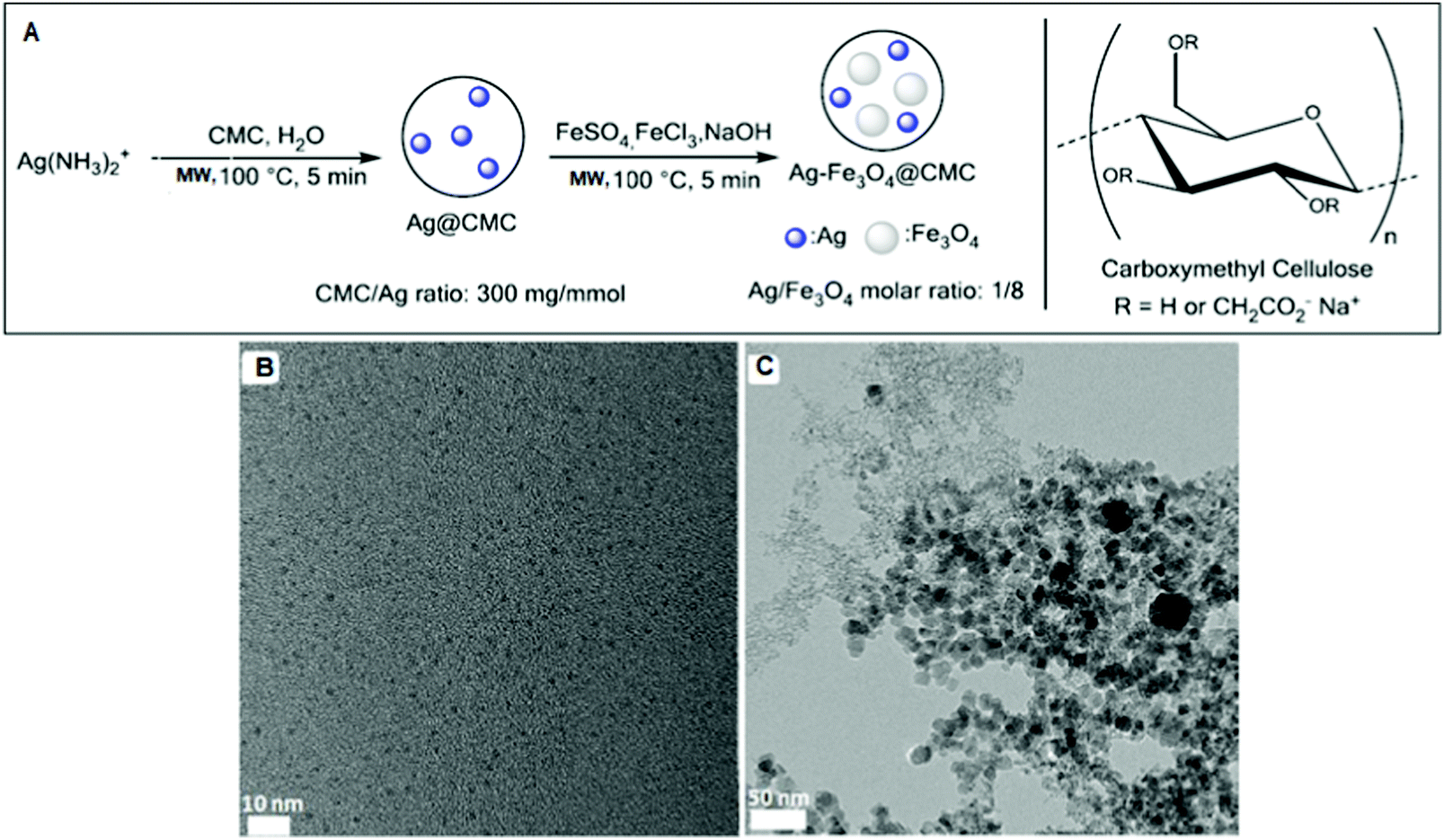 | ||
| Fig. 7 (A) Preparation scheme of Ag@CMC and Ag–Fe3O4@CMC catalysts. TEM images of (B) Ag@CMC and (C) Ag–Fe3O4@CMC catalysts.193 (Copyright 2016, the American Chemical Society.) | ||
3.5.2.1. Metal oxide. Jing et al. prepared a CuNiZnO/γ-Al2O3 catalyst via the MW-assisted thermal hydrolysis of urea.196 The required amounts of Ni(NO3)2·6H2O and γ-Al2O3 powder were taken in Cu(NO3)2·3H2O and Zn(NO3)2·6H2O aqueous solution, sonicated, and finally heated in an MW oven at 80 °C, 85 °C, 90 °C or 95 °C for 2 h. The recovered dried product was calcined at 350 °C for 5 h. The composition analysis of the four catalysts indicated that the Cu content gradually decreased with an increase in the MW temperature, whereas both the Ni and Zn contents increased. The surface area and pore volume of the γ-Al2O3 support decreased as usual following modification. A higher MW temperature produced uniform smaller particles with better dispersion.
A Ce0.8Zr0.2O2 solid solution was synthesized via a rather long process of citrate complexation (17 h) and supported on different loadings of Al2O3via the impregnation (CI) or MW combustion technique (CC), and finally calcined at 800 °C for 5 h.197 The CZ loading was found to be optimum at 40 wt%, and a greater CZ loading produced agglomeration. Accordingly, samples CZ40A-CC and CZ40A-CI contained relatively finer particles.
3.6. Ternary metal catalysts
 | ||
| Fig. 8 (A) Scheme of the preparation of ternary Pt–Sn–Rh USNP/C composites. TEM images of (B) Pt4.5Sn1.5Rh1 USNP/C, (C) Pt3Sn1Rh1 USNP/C and (D) Pt4.5Sn1Rh1.5 USNP/C composites.199 (Copyright 2020, Elsevier B.V.) | ||
4. Catalysis by supported simple metal catalysts
The previous sections revealed that a variety of supported nanocatalysts has been prepared via MW-assisted methods. The supported nanocatalysts include simple metal catalysts such as Cu,81–83,105,132 Ni,105,118,136 Co,105,119,120 Fe,84 Ru,85,86,95–99,141 Ag,112,116 Au,112,129,131,142 Pt,87–92,106,111,121,142,145,146 Ti,122 Zr,123 Pd,93,106–108,127,128,130,133,134,137,138,147 and W.139 Several attempts have also been made to prepare supported simple metal oxide (CoOx, Co3O4, NiO, Fe3O4, SnO2, CeO2, and TiO2) nanocatalysts for application in the petrochemical industry.75,160–166 To improve the catalytic activity and versatility of the application potential of simple metal or metal oxide nanocatalysts, investigations on supported bimetal,171–175,177–187 mixed metal oxide188–193,196,197 and ternary metal198,199 nanocatalysts have also been performed. MW-assisted synthesis has been claimed to be an economically viable, short, green method to produce supported nanocatalysts. However, to achieve high catalytic efficiency, strong bonding of the nanocatalyst to the support materials without damaging the support materials due to MW irradiation is essential. Moreover, the loading amount, nanocatalyst size and distribution, and reduction efficiency of the precursor salt during the preparation of catalysts are also known to contribute to their catalytic activity. Consequently, the following section covers the usefulness of various MW-synthesized supported nanomaterials in catalysis with emphasis on their catalytic performance, stability and recycling efficiency. The performance is compared with that prepared via conventional methods, if available. For better comparison, this section is divided into subsections under different reaction types such as oxidation, reduction/reductive degradation, hydrodesulfurization/hydrodearomatization, photocatalytic degradation, hydrogenation, cross couplings, synthetic reaction (cycloisomerization, dehydrogenation, transesterification, and photocatalytic hydrogen generation) and electrocatalysis. Some of these reactions are illustrated in Scheme S1 (ESI†). It is worth mentioning that only the articles which published after 2012 are considered in the following section.4.1. Oxidation reactions
Chuang et al. observed that smaller (16 nm) CuNPs and a cubic morphology in Cu/AC-700 W-60 s-12 prepared under the optimized MW conditions were favorable for obtaining the highest oxidation of CO.81 The same observation was made with the Cu/CeO2-TD catalyst during CO oxidation (95.8% conversion) due to the better dispersion and smaller CuNPs (33 nm).132 The catalytic efficiency of Cu/CeO2-TD was comparable to that of Cu/CeO2 nanorods prepared via the wet IM and hydrothermal method.200,201 A comparative study on Pd/Co3O4, Pd/Fe3O4 and Pd/Ni(OH)2 catalysts for the CO oxidation in a programmable flow tube furnace reactor indicated that both 30 wt% Pd/Co3O4 and 50 wt% Pd/Fe3O4 catalysts can produce 100% CO oxidation at 127 °C given that the Co3O4 support itself has significant conversion efficiency.127 In a related report, the Pd/Fe2O3-573 catalyst (Pd 1.87 wt%) prepared via coprecipitation followed by calcination at 573 K produced 100% CO oxidation efficiency at only 60 °C.202 This was due to the smaller (∼ 10 nm) crystalline size of the Fe2O3 support compared to Fe3O4 (25–30 nm). The deposition of AuNPs on the Mn3O4 support improved the catalytic oxidation of CO and the 4-Au/Mn3O4 catalyst (4 wt% Au) resulted in 100% conversion at 98 °C.129 The stability test of 4-Au/Mn3O4 at 50 °C and 125 °C exhibited its excellent stability up to 20 h, and then it started to deactivate slowly, losing 4% of its initial activity after 30 h. Likewise, the best catalytic activity was obtained over the 4-Pd/Mn3O4 (containing 4 wt% Pd) catalyst given that 100% CO oxidation occurred at only 20 °C, a much lower temperature than that of the previous 4-Au/Mn3O4 catalyst.130 This activity was almost on par with the meso-structured 2.7 wt% Pd-loaded Pd/Mn3O4 catalyst prepared via impregnation and reduction.203 The stability of 4-Pd/Mn3O4 was also better and found to be stable up to 30 h without any remarkable decrease in its activity.Both the loading method (e.g., complexation-MW assisted combustion, CC, and complexation-impregnation, CI) and weight percentage of Ce–Zr solid solution in the Ce0.8Zr0.2O2/Al2O3 catalyst influenced the oxidation of CO.197 Obviously, the conversion degree increased with the loading of Ce0.8Zr0.2O2 and the MW-assisted (CC) method resulted in better oxidation activity. CZ40A-CC converted 98% and 100% CO at 250 °C and 275 °C, respectively, but CZ40A-CI converted 100% CO at only the higher temperature (275 °C). The maximum conversion achieved by the CZ40A catalysts was due to the proper amount of Al2O3, reasonable surface area and higher dispersion of ceria and zirconia. However, the stability of the catalysts was not studied.
Ren et al. evaluated the performance of a hollow Cu/AC nanocatalyst in the oxidative carbonylation of methanol to dimethyl carbonate (DMC).83 Compared to solid Cu/AC-4, the hollow structure of CuNPs with exposed Cu(111) crystals improved the selectivity.204,205 An irradiation temperature of 360 °C for a longer duration during the preparation of catalyst was found to be undesirable given that the catalytic efficiency was reduced due to the change in initial morphology.206
An MW-NiO/2D-Vmt catalyst exhibited better activity than an MF-NiO/2D-Vmt catalyst for acetylene carbonylation in a constant volume sealed container for the synthesis of acrylic acid (AA).164 Again, smaller NiO grains in the MW-NiO/2D-Vmt catalyst produced a higher yield of AA (86%), whereas the MF-NiO/2D-Vmt catalyst produced 68% yield. However, the MW-NiO/2D-Vmt catalyst was unstable, demonstrating a drastic change in morphology and disappearance of the NiO signal after the first cycle.
An Fe/AC catalyst removed 93% of added phenol (concentration unknown) after 180 min at 30 °C during oxidative degradation but its stability was not that good given that the removal efficiency dropped to 20% after the fourth recycle due to severe leaching of its Fe ions.84 However, the Fe/AC catalyst (4 wt% Fe), prepared by heat treatment in air, was worse in terms of performance and stability.207
4.2. Reduction/reductive degradation reactions
Independent of the Ru content, both Ru/CPM-1 (0.5 wt%) and Ru/CPM-2 (1.0 wt%) catalysts completely reduced p-nitroaniline (p-NA) and crystal violet (CV) using NaBH4 reductant within 15 min and 30 s, respectively.85 However, a smaller size of embedded RuNPs in the PDC@RuNP catalyst and porous structure helped to attain 98% elimination of the yellow color of K3Fe(CN)6 within 30 s.86 Comparatively, commercial Ru/C took a longer time (>30 s) due to its insufficient porous structure. The PDC@RuNPs catalyst was fairly stable, retaining ∼85% catalytic activity after six consecutive cycles. The specific surface area decreased slightly after the 6th run but no leaching of Ru was detected. The complete degradation of new fuchsin (NF) dye was also possible over the PDC@RuNP catalyst.The smaller (2.0 ± 0.5 nm) size and dense distribution of RuNPs in the SWCNT-Ru catalyst produced through MW reduction at 100 °C for 15 min degraded Congo red (CR) dye at both low concentration (0.01 mM, 63.8%, 2.1 min) and high concentration (0.06 mM, 91.0%, 3.2 min).99 The reusability of the SWCNT-Ru catalyst did not show any significant drop in degradation after the third cycle. The same research group observed that the SWCNT-Ru catalyst prepared using ethanol as a reducing agent degraded up to 98.2% of 0.04 mM CR dye solution within 4 min.100 The formation of larger Ru clusters at a higher Ru loading reduced its activity.
Independent of the NP incorporation method (IM or DR), Pd–Au/C catalysts activated by MW irradiation exhibited better hydrodechlorination, HdCl, activity than that activated by thermal treatment in hydrogen.174 The improved catalytic activity was attributed to the better homogeneity in the Pd–Al alloy. The deactivation of the PdAu-DR-MW catalyst was also lower owing to the better homogeneity of the Pd–Au alloy phase.
4.3. Photocatalytic/thermal degradation
A smaller size and homogeneous distribution of PtNPs and Mo2C in Pt/G and Mo2C/C catalysts, respectively, again showed good to excellent methylene blue (MB) photodegradation and hydrous hydrazine decomposition activities.111,156 The MW-assisted catalytic decomposition with a power of 180 W produced complete decomposition over the Mo2C/C catalyst within 4 min. The WC/AC-H catalyst prepared by the rather long procedure of impregnation and carbothermal hydrogen reduction, produced complete hydrazine conversion at 50 °C.208 CeO2–SiO2 NPs abbreviated as M-30 (irradiation time 30 min), exhibited better MB decomposition activity under UV light, also due to their lower particle size and higher surface area and pore volume.162 In this case, the degradation efficiency was 99.9% at pH 7. On the contrary, the same CeO2–SiO2 catalyst (H-24 h) prepared via the hydrothermal process showed only 85% degradation efficiency.4.4. Hydrogenation reaction
The selective hydrogenation reaction of p-chloronitrobenzene (p-CNB) to p-chloroaniline (p-CAN) in the presence of either Ru/CNT or Ru/FCNT catalysts indicated that the Ru/FCNT catalysts prepared without/with a PVP stabilizer had better activity with 100% selectivity at 100% conversion after 30 min of reaction.95 These findings were consistent with the more homogeneous distribution of RuNPs and stronger anchoring of the RuNPs on the FCNT surface. Further, the selectivity (96%) of commercial Ru/CNT was comparatively lower. In a study on the effect of the precursor type (Ru(acac)3/RuCl3) on the catalytic activity of an Ru/TiO2 catalyst, it was found that an increase in the irradiation time resulted in an increase in the particle size.141 This reduced the yield of γ-valerolactone (GVL) from the hydrogenation of levulinic acid (LA).An MW-synthesized Ni/AB-MW catalyst was found to be superior to an Ni/AB-CH catalyst in the hydrogenation of nitrobenzene to aniline with respect to conversion (98.7%) and selectivity (93.3%).118 MW synthesis produced a smaller and uniform size as well as a higher loading of metallic Ni and NiO particles. The stability of the Ni/AB-MW catalyst was also better given that the Ni crystallite size indicated an increase in size by 27.4% after 60 h of reaction, while in the Ni/AB-CH catalyst, the size increase was more dominant (26.5% after 36 h).
Pd/Al2O3 catalysts prepared via plasma-induced MW irradiation, designated as PF (plasma fixed-bed reactor) and PS (plasma spouted bed reactor), were used for the hydrogenation of acetylene and compared with CM (conventional thermal treatments).137 In the plasma fixed-bed reactor, three catalysts named PF-1, PF-2 and PF-3 were prepared by controlling the distance between the waveguide and particle bed in the range of 3 to 7 cm. Overall, PF-1 possessed the maximum activity due to formation of the Pd (100) plane and a better dispersion. The highest selectivity for consecutive acetylene hydrogenation towards ethane was also possible in the presence of PF-1. In another investigation, the authors proved that although plasma spouted bed treatment produced a higher gas temperature than conventional heat treatment at 500 °C, the former method prevented Pd metal from sintering due to its shorter reaction time.138
4.5. Hydrodesulfurization/hydrodearomatization
Hydrodesulfurization (HDS) is an industrial process required for reducing the sulfide content in fuel. A W/Al2O3 catalyst (identified as MWH-B) prepared via the part-wise addition of oxalic acid showed enhanced HDS activity due to the good dispersion of WO3.139 The overall analysis suggested that the catalyst prepared via the MWH method had superior HDS activity relative to that prepared by IM. Nguyen et al. found that the HDS conversion of 4,6-dimethyldibenzothiophene (4,6-DMDBT) over the CoMo/Ti-SBA-15 catalyst was 1.35-times higher than that on the CoMo/SBA-15 catalyst.185 Alternatively, the insertion of Ti in the CoMo/SBA-15 catalyst reduced the hydrodearomatization (HDA) activity of 1-methylnaphthalene (1-MN) and phenanthrene by half. Hence, the inclusion of Ti promised control of the HDA activity over the CoMo/SBA-15 catalyst.4.6. Coupling reactions
To understand the deactivation process, Pd/C and Pd/Grp catalysts were subjected to MW irradiation for variable times (30 s, 5 min and 30 min) before the Suzuki–Miyaura (phenylboronic acid and p-nitrobromobenzene) and Mizoroki–Heck (iodobenzene, butyl acrylate, and triethylamine) coupling transformations.93 A longer MW irradiation time between 5 to 30 min produced a considerable loss in catalytic activity for the Pd/C catalyst, which was more severe for the Pd/Grp catalyst after irradiation for 30 s. The deactivation of the catalyst resulted from an increase in particle size, dissolution of carbon inside the metal NPs and formation of metal oxide. In another investigation, 1 mol% of Pd/G catalyst prepared by continuous flow synthesis produced 100% conversion in the MW-assisted batch Suzuki–Miyaura coupling reaction between bromobenzene and phenylboronic acid.107 However, the recyclability test was not promising given that conversion decreased from 100% to 77% in the fourth cycle of the coupling reaction. Comparatively, the MW-assisted Suzuki–Miyaura coupling reaction between bromobenzene and phenylboronic acid over a magnetic Pd–Fe3O4/G nanocomposite showed better stability with 100% conversion up to the 3rd cycle.109 Further recycling reduced the conversion to around 81% in the 9th cycle, which is still good. Controlling the MW temperature between 25–80 °C is important given that an increase in temperature to 100 °C reduced the conversion after 10 min of reaction, indicating destabilization of the catalyst.Elazab et al. observed that a Pd content of 20 wt% in Pd/CuO catalysts is optimum for obtaining 100% conversion in the MW-assisted Suzuki–Miyaura coupling reaction between aryl halides and phenylboronic acid.133 The application of the 20 wt% Pd/CuO catalyst for other substrates also produced good conversion (82–90%). The conversion drastically dropped to 60% in the sixth run due to the agglomeration of the PdNPs. Elazab et al. also investigated the applicability of Pd/CuO catalysts for the Mizoroki–Heck (aryl bromide and alkene) and Sonogashira–Hagihara (aryl bromide and alkyne) coupling reactions under MW irradiation.134 The Mizoroki–Heck coupling reaction between bromobenzene and alkene on 2 mol% of the 20 wt% Pd/CuO catalyst produced the maximum conversion of 93% at the optimized temperature of 150 °C. The catalyst was found to be fairly stable up to 5 cycles.
Interestingly, the Pd/SiC catalyst, labeled as MW3, exhibited high catalytic activity up to 20 reaction cycles with more than >90% yield in the Mizoroki–Heck (iodobenzene and methyl acrylate) coupling reaction.147 Comparatively, the OB3 catalyst, which was prepared via conventional heating, produced 100% yield in the first cycle, but this was drastically reduced to 75% yield. In contrast, the commercial Pd/C catalyst was usable for only one Mizoroki–Heck reaction cycle. The better catalytic activity, i.e., longer catalytic life of MW3 was due to larger amounts of PdNPs on the support. MW3 was also found applicable in the Suzuki–Miyaura coupling reaction.
In comparison to the Pd/Ag/SBA-15 catalyst, the Pd/Au/SBA-15 catalyst showed superior activity in the Suzuki–Miyaura coupling reaction between iodobenzene and aryl boronic acid and produced ∼70% conversion.183 The enhanced activity of aryl halides followed the order of I > Br > Cl. A comparative study among the Pd–Fe3O4/G, Pd–Co3O4/G and Pd–Ni(OH)2/G catalysts for the Suzuki–Miyaura coupling reaction (bromobenzene with phenyl boronic acid) showed the lowest (<60%) conversion over the Pd-Ni(OH)2/G catalyst but both the Pd–Fe3O4/G and Pd–Co3O4/G catalysts produced 100% conversion within 10 min at 120 °C.188 The good nano-size distribution of the catalyst on G nanosheets in both cases substantially improved the catalytic conversion. The Pd–Fe3O4/G catalyst was fairly stable, producing 90% conversion in the fifth cycle. In an early article, Elazab et al. demonstrated that 0.3 mol% of magnetically recyclable Pd–Fe3O4/G catalyst, labeled as catalyst 3 (Pd 7.6 wt%), produced 100% conversion within 45 min in the Suzuki–Miyaura coupling reaction.189 Despite having a low Pd content, catalyst 3 had the highest activity because it contained the smallest Pd (3–4 nm) and Fe3O4 NPs (12–16 nm).
4.7. Synthesis reactions
The catalytic cycloisomerization of substituted propargylic urea to produce substituted imidazolone under optimized reaction conditions was carried out over MW-2%Ag@AlSBA-15, MW-2%Au@AlSBA-15, BM-2%Ag@AlSBA-15 and BM-2%Au@AlSBA-15 catalysts (Scheme 1).112 Irrespective of their preparation methods, the supported Au nanocomposites were almost inactive with a yield of less than 5%. Alternatively, the ball-milled BM-2%Ag@AlSBA-15 catalyst was more active (yield, 56%) than the MW-synthesized MW-2%Ag@AlSBA-15 catalyst (yield 35%). However, despite the lower activity of the MW-synthesized catalyst, the authors claimed that the MW-assisted synthesis of the catalyst is favorable because it requires a few minutes for the one-step reduction compared to several hours required by ball milling-assisted synthesis. However, information on the size distribution, valence state and crystallinity of the nanocatalyst is not available, and thus it is difficult to predict the exact reason for the poor performance by the MW-2%Ag@AlSBA-15 catalyst. However, another author reported 99% yield of 2a (Scheme 2) with 100% conversion at 50 °C using a similar type of 0.96 wt% AgNCs/SBA-15 catalyst during the cyclization of propargylguanidine.116 This result is remarkable compared to that of Ag salts and AgNCs, although the metal loading is much lower.209,210 The conversion was retained at >90% conversion after 4 cycles, and no Ag leaching was detected and the morphology with the size distribution of the recovered catalyst remained unchanged. The cyclization of 2-(phenylethynyl)phenol in the presence of 0.96% AgNCs/SBA-15 catalyst also resulted in 100% conversion with 93% selectivity for 2-phenylbenzofuran (4a) at 70 °C (Scheme 2).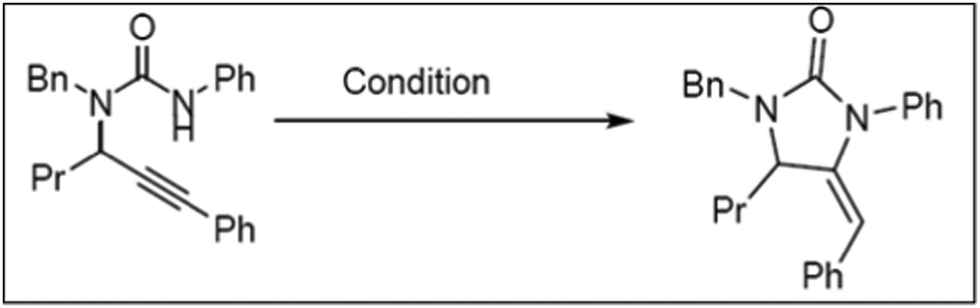 | ||
| Scheme 1 The catalytic cycloisomerization of substituted propargylic urea.112 (Copyright 2019, the American Chemical Society.) | ||
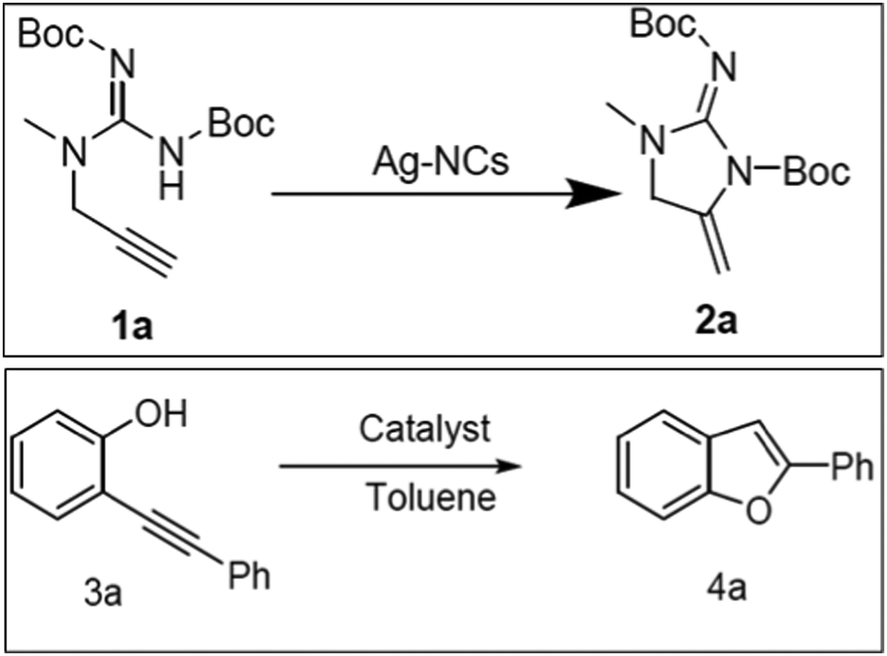 | ||
| Scheme 2 The catalytic cyclization of propargylguanidine (1a) and 2-(phenylethynyl)phenol (3a).116 (Copyright 2020, the American Chemical Society.) | ||
Song et al. observed superior CO2 methanation activity over an Ni–Al2O3–M catalyst compared to that over an Ni–Al2O3–I catalyst.136 The maximum conversion efficiency of 91.6% was possible at 325 °C over the Ni20–Al2O3–M catalyst (20 wt% Ni) and the selectivity for CH4 in the temperature range of 200–400 °C was around 99.3%. The catalyst was stable at 325 °C for up to 72 h given that the conversion remained high (91.6%). The anti-sintering stability of the Ni20–Al2O3–M catalyst was confirmed.
Akay et al. tested a Co/SiO2 catalyst in non-thermal plasma-promoted Fisher-Tropsch synthesis (FTS) involving CO and H2 gases.119 Catalyst-ABC-550, where metal oxide was reduced in a plasma reactor under a stream of H2 for 24 h at 550 °C, was found to be the best, producing 100% CO conversion. Catalyst-ABC-550 also possessed better stability given that the CO conversion of 100% remained constant over the complete test period of 175 h. It was possible to restore the complete catalytic activity of the deliberately deactivated Catalyst-ABC-550 by reduction with H2 (25 mL min−1) at a plasma power of 90 W for 24 h at 250 °C. In contrast, the Co/SiO2 catalyst prepared by depositing Co monolayers on a 10 nm-thick SiO2 film demonstrated poor stability in the FTS reaction.211 In another investigation, Co/SiO2 catalysts prepared via conventional drying offered better CO conversion (68.3%) compared to that prepared via the MW method (42.1%) but the selectivity towards liquid hydrocarbons of C5+ was higher (63.2%) for the catalyst prepared via the MW method.120 However, it is not clear whether the MW method caused any damage to the support or agglomeration.
A comparative study between MW-NiO/Vmt and CHT-NiO/Vmt catalysts for CO methanation in a stainless steel tubular microreactor in the temperature range of 250–500 °C showed the better CO conversion (99.6%) and CH4 selectivity (93.8%) by the MW-synthesized MW-NiO/Vmt catalyst at 400 °C.163 MW-NiO/Vmt was also found to be comparatively more stable.
The ethanol conversion in the range of 250–450 °C with a constant H2 flow over Pt/SiO2 catalysts, which were prepared with different Pt loadings via the MW-assisted solution and RCE methods, increased markedly with an increase in temperature, but decreased slightly with the Pt loading, which was affected by the dispersion behavior of the PtNPs.121 The selectivity toward ethylene (dehydration) at 450 °C for the 0.6 and 0.9 wt% Pt-loaded catalysts prepared via the MW method was higher (50–53%) compared to that (47%) for the samples prepared via the RCE method.
The photocatalytic activity of TiO2/Au NPs for H2 production from a mixture of ethanol and water in the gas phase through a tubular reactor suggested that the removal of the capping agent, PVP, via calcination is necessary to improve the efficiency.166 The optimal calcination temperature was 450 °C for the best H2 evolution of ∼5.3 H2 gcat−1 h−1 (sample A, 100 mg of PVP) and ∼3.5 H2 gcat−1 h−1 (sample B, 600 mg of PVP). A higher calcination temperature (>550 °C) reduced the catalytic performance via aggregation. The higher catalytic activity of sample A was due to the larger interface area between Au and TiO2, which favored the interaction between the Au and TiO2 NPs.212 A comparative study of the catalytic property between Pd/Ag/SBA-15 and Pd/Au/SBA-15 catalysts for hydrogen production from ammonia borane (AB) under both dark and visible light irradiation conditions indicated the superior catalytic activity of the Pd/Ag catalyst.183 Khalid et al. observed that the catalysis power of an Ag–TiO2/G composite for hydrogen production from methanol aqueous solution was superior to that of the pure TiO2, Ag–TiO2 and TiO2/G samples.190 Hence, the inclusion of a G support and Ag played a vital role in enhancing the photocatalytic activity. The hydrogen production efficiency from methanol over four different copper-based catalysts (CuNiZnO/γ-Al2O3), which were prepared at different MW temperatures (80 °C, 85 °C, 90 °C and 95 °C) indicated that the content of Cu, Ni and Zn had an effect on the catalyst power.196 The MW-Cu/Ni-80 catalyst contained a higher amount of Cu (47.92 wt%), resulting in higher activity at a lower reaction temperature (200 °C). In contrast, the other catalysts processed at higher MW temperatures contained larger amounts of Ni and Zn than the MW-Cu/Ni-80 catalyst, which played the main role for increasing methanol conversion at a higher temperature (≥225 °C). The catalytic selectivity with respect to H2 production increased with an increase in the reaction temperature for MW-Cu/Ni-80, but for MW-Cu/Ni-90 and MW-Cu/Ni-95 this trend could not be distinctly confirmed and the overall yield was relatively high for both MW-Cu/Ni-90 and MW-Cu/Ni-95. The formation of an increasing amount of Ni possibly inhibited the formation of methyl formate and increased the selectivity for H2 and CO in methanol decomposition.213,214 The MW-Cu/Ni-90 catalyst was fairly stable and methanol conversion was maintained at 100% for up to 26 h, only slightly decreasing to 98.8% after 30 h.
4.8. Electrocatalysis
In the last few decades, a considerable amount of research has been devoted towards the application of Pt NPs in PEMFCs,215,216 water splitting,217 and direct alcohol fuel cells.218 However, due to the high cost of Pt, its application is limited, and thus researchers are focusing more on the development of multicomponent (binary or ternary) catalysts to promote the catalytic performance. The combination of Pt with non-precious metals can also reduce the dosage of Pt and is beneficial for improving its resistance to poisoning.219,220Sharma et al. studied the electrocatalytic activity of Pt/C electrocatalysts prepared using different precursors.92 A comparative study of the cyclic voltammograms (CV) suggested that relative to the commercial Pt/C catalyst, the H2PtCl6- and (NH4)2PtCl6-sourced MW-synthesized Pt/C catalysts possessed a higher and almost identical electrochemical surface area (ECSA). The (NH4)2PtCl6-sourced Pt/C catalyst exhibited a comparable ORR performance with that of the commercial and H2PtCl6-sourced catalysts. The electrocatalytic activity of CoSe2/C catalysts prepared with different Se/Co ratios showed that an Se/Co ratio of 3.0 is the best for ORR activity and stability.177 An excess amount of Se oxide reduced the ORR activity due to the occurrence of severe aggregation of the CoSe2 NPs. Likewise, FeSe2/C NPs prepared with different Fe/Se ratios exhibited the best ORR activity at Se/Fe ratios of 2.0 and 4.0, respectively.178 These ratios were equivalent to 47.21% and 48.2% of the total amount of FeSe2 and FeSe, respectively. An increase in Co content in a series of ternary (CoxNi1−x)Se2/C catalysts improved their electrocatalytic activities towards the ORR.198 The beneficial effect of the incorporation of Co was further confirmed by the increase in the ORR potential (EORR). The incorporation of Co in binary NiSe2/C was also advantageous for the enhanced stability of the (Co0.75Ni0.25)Se2/C electrocatalyst.
The addition of Sn to a Pt/C catalyst greatly enhanced its electrocatalytic activity for ethanol oxidation (EO), whereas the addition of Rh only modestly improved the electrochemical performance.171 Chronoamperometric tests suggested that PtSn/C is considerably less poisoned compared to the Pt/C or PtRh/C catalysts. Similarly, the inclusion of Cu in Pt/C (Pt3Cu/C, Pt4Cu/C, and Pt2Cu/C) catalysts improved their electrocatalytic performances compared to the commercial Pt/C catalyst for EO and MO.173 The Pt3Cu/C catalyst had the highest mass activity, MA, (1.61 A mg−1) and surface specific activity, SA, (3.23 mA cm−2) towards EO. The Pt3Cu/C catalyst also displayed the highest MA (1.92 A mg−1) towards MO. The inclusion of Cu also improved the durability of PtCu/C towards the EO and MO reactions. SF-MWCNT-Pd and its mixed bimetallic SF-MWCNT-PdSnmix and SF-MWCNT-PdNimix nanocatalysts were found to be more active towards EO compared to alloyed SF-MWCNT-PdNi and SF-MWCNT-PdSn catalysts produced via MW assisted co-reduction.179 The addition of Sn to Pd produced the maximum influence on electrocatalytic property. However, chronopotentiometric experiments showed that SF-MWCNT-PdNimix was more tolerant to CO poisoning than SF-MWCNT-PdSnmix. The electrocatalytic activity of PtNi/FCNT analyzed in MO was dependent on the metal loading and mode of irradiation.180 The catalysts prepared at different irradiation times had almost the same stability, but higher than that of the reference Pt/C catalyst. Three different ternary catalysts labeled as Pt4.5Sn1.5Rh1 USNP/C, Pt3Sn1Rh1 USNP/C and Pt4.5Sn1Rh1.5 USNP/C also showed an improvement in electrocatalytic performance towards both MO and EO than 20 wt% Pt/C.199 Notably, the highest MA and SA were observed for the Pt4.5Sn1.5Rh1 USNP/C catalyst. The stability tests also confirmed the superiority of the Pt4.5Sn1.5Rh1 USNP/C catalyst. The structures and morphologies after 1000 sweeping cycles did not change for all the Pt–Sn–Rh/C catalysts, whereas for the 20 wt% Pt/C catalyst, severe aggregation was observed. A trimetallic AuPdPt@WC/C catalyst also showed an improvement in electrocatalytic MO reaction compared to the commercial Pt/C catalyst.200 The anti-poisoning ability of trimetallic AuPdPt@WC/C was 5-times better than that of the commercial Pt/C catalyst. The formation of highly dispersed AuPdPt metals on a WC support and the coupling effect between NPs enhanced the electrocatalytic performance.
The MW synthesis of PtRu/MWCNT NPs resulted in an improvement in poisoning resistance to CO following the formation of a homogeneously dispersed alloy phase.181 In a similar investigation, the PtPd/MWCNT-GNP catalyst was found to possess the highest electrochemical stability and durability compared to PtPd/MWCNT and PtPd/GNP catalysts.182 This behavior was ascribed to its better dispersion, smallest NP size and highest specific surface area (164 m2 g−1).
The electrochemical activity of the Co3O4@SUS electrode towards the OER in water splitting was compared with reference materials including commercial RuO2@SUS and Co3O4-LNS@SUS catalysts.165 Co3O4@SUS exhibited the most favorable OER kinetics due to its rod-like morphology and highest surface to volume ratio. The stability of the Co3O4@SUS catalyst was good towards the OER with 78% current retention after 24 h and the initial morphology of the catalyst was maintained. Meanwhile, a metal organic framework, Co–N–C, supported Ni3Fe catalyst offered the best OER activity and best reaction kinetics compared to Ni3Fe/N–C, IrO2, and Co–N–C.187 A negligible decay in current density was observed for the Ni3Fe/Co–N–C catalyst after 3000 potential sweeps. Subsequently, the Ni3Fe/Co–N–C showed a comparable ORR performance. Hence, the Ni3Fe/Co–N–C catalyst can be an efficient bifunctional electrocatalyst for the OER and ORR. Pan et al. also demonstrated an improvement in OER activity for a supported Co/CoOx catalyst following the inclusion of Co.191
5. Scope and outlook
This review article focused on evaluating the feasibility of MW-assisted methods over conventional methods for the preparation of durable and effective supported heterogeneous metal/metal oxide/their hybrid nanocatalysts. It was already mentioned that unsupported nanocatalysts are prone to aggregation, are unstable in air, and promote leaching, and thus deactivate easily during catalysis. In this regard, the anchoring or mobilization of nanocatalysts on an inert or catalytically active support could help to improve the catalyst lifetime by inhibiting leaching, enhancing its stability, reducing its poisoning and maintaining its nano-size distribution.Numerous variations in MW-assisted routes have been developed and evaluated for the preparation of supported nanocomposite catalysts. The predominantly used methods studied include MW-assisted polyol/alcohol reduction of metal salts,81,87,88,90–92,95,99,111,112,132,141,142,145,146,158,162,166,171–173,175–180,182,183,186,190,198,199 MW-assisted reduction in the presence of reducing agents or under H2 pressure,98,106–109,116,121,127–130,133,134,161,181,188,189 MW heating with the aid of an MW absorber (ionic liquid),110 carbothermic MW reduction of impregnated metal salts,82,83 MW thermal reduction of impregnated dried precursors,75,84,85,96,97,105,154,163,164 drying and calcination under MW conditions followed by impregnation,89 MW drying followed by impregnation,118,120,184 MW thermal decomposition of metal salts with or without subsequent drying and/or calcination,86,122,123,131,136,139,147,155,157,158,165,187,191,193,196 MW activation followed by wet impregnation of metal NPs,174,197 and MW-induced plasma irradiation.119,137,138 Most of these methods are carried out in a batch-wise fashion, except a few in continuous flow mode.107,116,128 The comparative study of MW-assisted methods and conventional methods, whenever investigated, demonstrated that MW methods promoted faster nucleation, produced more homogeneous smaller-size NPs within a shorter time, improved the catalyst loading and better alloying in the binary/ternary metallic phase because of the high loss tangent values, strong MW absorption, and thus faster heating and lower activation energy.112,118,120,121,136,138,139,160,162,163,174,184
The usefulness of MW-assisted methods is dependent on the ability to produce a perfectly homogeneous distribution of uniform nanosized catalyst with a controlled morphology on a solid support. The intimate contact with strong fixation is also a desirable requirement from the view point of stability and good dispersity of the nanocatalyst on the support. In almost all the above-mentioned articles, it was found that MW-assisted batch methods under optimum conditions favorably produced a well-dispersed, uniform, nano-sized distribution of metal/metal oxide/their hybrid catalyst, reportedly down to even 1.6 ± 0.7 nm, which was observed for AgNCs.116 The addition of stabilizer was considered not essential to improve the size distribution, and thus the surface property of the nanocatalyst was easily maintained. The MW power, irradiation time, temperature, loading amount, functionality/porosity of the support materials, pH of the reaction medium, nature of the precursor salt and reducing agent all influenced the size distribution of the supported nanocatalysts. The use of a too high MW irradiation power and too long irradiation time sometimes caused sintering of the nanocatalysts116,141,157,162,180 and even damaged the support materials.94 Thus, a balance is needed between MW power and irradiation time to achieve the optimum nanosize distribution. MW irradiation under vacuum and the selection of an appropriate precursor salt were also found to be crucial in minimizing damage to the support.94 In some instances, functionality or porosity in the support materials was found beneficial for improving the metal loading, dispersibility and crystallinity index of the anchored nanocatalyst.86,95,110,116,120,121,162,180,191,193 Interestingly, a variation in the shape and morphology (e.g. spheroid, cubic, rod, plate, hollow, and porous) of nanocatalysts was also achieved by controlling the MW reaction conditions such as pH, irradiation temperature and time, precursor and support materials.81–83,110,129,130,162,165 However, despite the successful preparation of various supported, well-dispersed nanocatalysts, the wide variations in MW-assisted methods needed to be optimized in terms of simplicity, scalability, economic feasibility, processability and reproducibility. In this regard, methods such as the one-pot MW polyol/alcohol reduction method, one-pot MW-assisted reduction in the presence of reducing agents and one-pot thermal decomposition of the precursor without additional calcination can be preferentially suitable for mass production. These particular methods have been proven beneficial for preparing a large variety of supported well-dispersed nanocatalysts (including Pd, Ag, Cu, Pt, Ru, Au, Pd–Fe3O4, Pd–Co3O4, Pt–Mo2C, PtRh, PtSn, CoSe2, and PdNi) without aggregation. In the above-mentioned in situ methods, the addition of a solvent is desirable because the solvent-free MW method was not found to be conducive for obtaining a homogeneous distribution due to the aggregation of the particles.96,97 Although scarcely studied, the continuous flow MW-assisted method, which is favorable for improving the reproducibility, batch consistency and yield, was found to be promising for preparing supported, well-dispersed finely tuned, uniform nanocatalysts on a mass scale.107,116,128 Importantly, the average size of the nanocatalysts produced in the continuous flow mode was smaller compared to that prepared via the batch method.116,128
The second point is the analysis of the catalytic power of various MW-synthesized supported nanocatalysts, tested for different organic transformation reactions including electrocatalysis. In general, the MW irradiation conditions, particle size, dispersibility, morphology, loading amount, valence state of the active catalyst and nature of the support materials affect the catalyst efficiency. For example, irradiation for longer time reduced the catalyst efficiency due to a change in morphology, dispersity, damage to the support and formation of metal oxide.83,94,141 Morphology also plays a key role, given that cubic-shaped smaller nanocatalyst particles showed better catalytic activity, which is ascribed to their higher surface area.81 Table S1 (ESI†) summarizes the activity of various supported nanocatalysts prepared under MW irradiation and conventional reaction conditions. Interestingly, in most cases, the MW-assisted-synthesized supported nanocatalysts possessed better catalytic activity compared to the commercial95,101,147,162,186 and/or corresponding nanocatalysts prepared via conventional methods.118,120,121,136,139,147,162,163,174,221–223 Although the comparative data on the corresponding catalysts prepared by conventional methods is still inadequate, it is reasonable to conclude based on the available results that the MW-assisted-synthesized supported nanocatalysts possess good prospects for industrial use. Some authors even carried out the catalysis reactions under MW reaction conditions over supported nanocatalysts and found fairly good conversion and/or yield.112,119,120,128,156 The better catalytic activity of MW-synthesized supported nanocatalysts can be assumed to be due to the improved homogeneous distribution of smaller nanocatalyst particles, larger volume to surface ratio, surface heterogeneity, synergistic interaction between metals and stronger interaction between the nanocatalyst and support materials. The data on the stability of MW-synthesized nanocatalysts during recycle is scarce, and thus more investigation is needed in the area. However, according to the limited available data, the stability and recyclability were found to be comparable to both the commercial and corresponding nanocatalysts prepared by conventional methods. In some instances, even better recycle stability was observed for MW-assisted synthesized supported nanocatalysts.118,164,174 However, although the data on the stability and recyclability of MW-synthesized nanocatalysts is encouraging, much more effort is necessary to further improve their performance, given that in some cases the activity decreased significantly after a few reaction cycles.85,86,95,98,99,107,116,133,174 The decrease in activity during recycle in either case occurred mostly due to the change in morphology, aggregation, graphitization and loss of the active catalyst via leaching of metal ions.84,86,118,133,134,155,164 In this regard, the one-pot synthesis of supported nanocatalysts under mild MW conditions without additional calcination and use of a functional support for strong fixation can be attempted to further improve the recycle stability, thus reducing both the aggregation and leaching of the metal ions. It is noteworthy to mention that in batch catalysis reactions in a constant volume reactor, the activity of the nanocatalyst decreases sharply compared to that under continuous flow reaction conditions in a flow tube reactor (Table S1, ESI†). The shorter residence time, more efficient heat transfer from the heating/cooling source due to faster mixing, faster reaction kinetics than their batch counterpart and reduction in frequent abrasion/collision due to the absence of additional stirring may have improved the catalyst life in the continuous flow reaction condition.
A considerable volume of research is also available on the testing of the electrocatalytic activity of MW-synthesized supported binary and ternary metal nanocatalysts, with the aim to reduce the cost/dosage of precious Pt-based supported nanocatalysts for applications in PEMFCs, water splitting and alcohol fuel cells. In many cases, improved electrochemical performances with improved durability were observed for MW-synthesized electrocatalysts compared to the reference/commercial Pt/C electrocatalyst.92,145,165,171,173,178,180–182,187,199 The synergistic coupling effect, smaller size, and electronic and ionic effect helped to improve the electrocatalytic performance of binary and ternary metal nanocatalysts. Continuous irradiation rather than pulsed mode of irradiation was found to result in better electrocatalytic activity and better CO anti-poisoning ability.180 The electrocatalytic activity suggested that a porous support enhanced the property due to the improved mass-transport property.105 Overall analyses suggested that bimetallic: FeSe2/C, Pt–WC/C–F, PtSn/C, PtRh/C, Pt3Cu/C, PtNi/FCNT, PtRu/MWCNT, PtPd/MWCNT-GNP, Ni3Fe/Co–N–C, and Co/CoOx40-MC1050 and trimetallic: Pt–Sn–Rh/C, AuPdPt@WC/C can be good replacement for the commercial Pt/C catalyst.
6. Conclusion
The MW-assisted methods available for the preparation of supported metal/metal oxide/their blend nanocatalysts were reviewed with the aim to evaluate their suitability and present suggestions for improving their performance, if any. Numerous variations in MW-assisted methods have been developed for the preparation of a broad spectrum of supported nanocatalysts. In almost all the methods, well-dispersed, smaller, uniform supported nanocatalysts were successfully prepared with good catalytic properties. The limited data on durability and recyclability also indicated the better performance of MW-synthesized nanocatalysts compared to commercial and the corresponding supported nanocatalysts prepared via conventional methods. Thus, it can be concluded that MW-assisted methods can be used on an industrial scale for the preparation of supported nanocatalysts, and from now, efforts should be made to optimize the various processes into a few that can be used for mass scale production. Accordingly, the wide variations in MW-assisted methods need to be simplified and optimized into scalable, one-step in situ methods, where all the reactants (precursor salt, solvent, MW absorber, and polyol/alcohol/reducing agent) in the presence of the support materials can be MW treated. There is also scope to further improve the stability/recyclability of the used nanocatalyst, at least for up to 10 recycles. Presently only 5–6 recycles are possible, which are insufficient from an economic point. This can be done by eliminating high temperature calcination, which is employed on many occasions after MW synthesis, using a functional support to improve anchoring and by developing MW reactors for efficient heat transfer so that the MW power can be reduced to the minimum. Continuous flow MW reactors have also been tried on a limited scale, and this idea is interesting for mass scale production but more investigation is needed to check the efficiency of the reactor for synthesizing a wide variety of supported nanocatalysts with durable properties.Author contributions
Hasan Ahmad: conceptualization, data curation, formal analysis, resources, writing – original draft, writing – review & editing, supervision. Mohammad Kawsar Hossain: visualization, software.Conflicts of interest
The authors declare that there are no conflicts to declare.References
- M. Nasrollahzadeh, M. Sajjadi, S. Iravani and R. S. Varma, Green-Synthesized Nanocatalysts and Nanomaterials for Water Treatment: Current Challenges and Future Perspectives, J. Hazard. Mater., 2021, 401, 123401, DOI:10.1016/j.jhazmat.2020.123401.
- G. A. Ozin, A. C. Arsenault and L. Cademartiri, Nanochemistry: A Chemical Approach to Nanomaterials, Royal Society of Chemistry, London, United Kingdom, 2009 Search PubMed.
- C.-Y. Wu, W. J. Wolf, Y. Levartovsky, H. A. Bechtel, M. C. Martin, F. D. Toste and E. Gross, High-Special-Resolution Mapping of Catalytic Reactions on Single Particles, Nature, 2017, 541, 511–515, DOI:10.1038/nature20795.
- M. O. N. van de ĽIsle and M. C. Orgega-Liebana, A. Unciti-Broceta, Transition Metal Catalysts for the Bioorthogonal Synthesis of Bioactive Agents, Curr. Opin. Chem. Biol., 2021, 61, 32–42, DOI:10.1016/j.cbpa.2020.10.001.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines, Chem. Rev., 2020, 120, 2613–2692, DOI:10.1021/acs.chemrev.9b00462.
- M. V. Cañamares, J. V. Garcia-Ramos, J. D. Domingo and S. Sanchez-Cortes, Comparative Study of the Morphology, Aggregation, Adherence to Glass, and Surface-Enhanced Raman Scattering Activity of Silver Nanoparticles Prepared by Chemical Reduction of Ag+ Using Citrate and Hydroxlamine, Langmuir, 2005, 21(18), 8546–8553, DOI:10.1021/la050030l.
- A. Chakraborty, T. Chakraborty, M. I. Menendez and T. Chattopadhyay, Surfactant-Mediated Solubilization of Magnetically Separable Nanocatalysts for the Oxidation of Alcohols, ACS Omega, 2019, 4(7), 11558–11565, DOI:10.1021/acsomega.9b01143.
- T. Song, F. Gao, S. Guo, Y. Zhang, S. Li, H. You and Y. Du, A Review of the Role and Mechanism of Surfactants in the Morphology Control of Metal Nanoparticles, Nanoscale, 2021, 13, 3895–3910, 10.1039/D0NR07339C.
- G. I. Dzhardimalieva, A. K. Zharmagambetova, S. E. Kudaibergenov and I. E. Uflyand, Polymer-Immobilized Clusters and Metal Nanoparticles in Catalysis, Kinet. Catal., 2020, 61, 198–223, DOI:10.1134/S0023158420020044.
- M. Shukla and I. Sinha, Catalytic Activation of Nitrobenzene on PVP Passivated Silver Cluster: a DFT Investigation, Int. J. Quantum Chem., 2018, 118(3), e25490, DOI:10.1002/qua.25490.
- N. Sharma, A. Choudhary, M. Kaur, C. Sharma, S. Paul and M. Gupta, Modified Graphene Supported Ag–Cu NPs with Enhanced Bimetallic Synergistic Effect in Oxidation and Chan–Lam Coupling Reactions, RSC Adv., 2020, 10, 30048–30061, 10.1039/D0RA01540G.
- S. Prabu and K.-Y. Chiang, Improved Catalytic Effect and Metal Nanoparticle Stability using Graphene Oxide Surface Coating and Reduced Graphee Oxide for Hydrogen Generation from Ammonia-Borane Dehydrogenation, Mater. Adv., 2020, 1, 1952–1962, 10.1039/D0MA00441C.
- M. Nasrollahzadeh, T. Baran, N. Y. Baran, M. Sajjadi, M. R. Tahsili and M. Shokouhimehr, Pd Nanocatalyst Stabilized on Amine-Modified Zeolite: Antibacterial and Catalytic Activities for Environmental Pollution Remediation in Aqueous Medium, Sep. Purif. Technol., 2020, 239, 116542, DOI:10.1016/j.seppur.2020.116542.
- L. Muthulakshmi, N. Rajini, H. Nellaiah, T. Kathiresan, M. Jawaid and A. V. Rajulu, Preparation and Properties of Cellulose Nanocomposite Film with In Situ Generated Copper Nanoparticles unsing Terminalia catappa Leaf Extract, Int. J. Biol. Macromol., 2017, 95, 1064–1071, DOI:10.1016/j.ijbiomac.2016.09.114.
- S. Keshipour, S. Shojaei and A. Shaabani, Palladium Nano-Particles Supported on Ethylenediamine-Functionalized Cellulose as a Novel and Efficient Catalyst for the Heck and Sonogashira Couplings in Water, Cellulose, 2013, 20, 973–980, DOI:10.1007/s10570-012-9852-8.
- M. Niakan, M. Masteri-Farahani, H. Shekaari and S. Karimi, Pd Supported on Clicked Cellulose-Modified Magnetite-Graphene Oxide Nanocomposite for C-C Coupling Reactions in Deep Eutectic Solvent, Carbohydr. Polym., 2021, 251, 117109, DOI:10.1016/j.carbpol.2020.117109.
- Y. Fu, D. Huang, G. Zheng, C. Lai, L. Qin, B. Li, J. He, H. Yi, M. Cheng and C. Zhang, Au Nanoparticles Decorated on Activated Coke via a Facile Preparation for Efficient Catalytic Reduction of Nitrophenols and Azo Dyes, Appl. Surf. Sci., 2019, 473, 578–588, DOI:10.1016/j.apsusc.2018.12.207.
- M. Tariq, M. Muhammad, J. Khan, A. Raziq, M. K. Uddin, A. Niaz, S. S. Ahmed and A. Rahim, Removal of Rhodamine B Dye from Aqueous Solutions using Photo-Fenton Processes and Novel Ni-Cu@MWCNTs Photocatalyst, J. Mol. Liq., 2020, 312, 113399, DOI:10.1016/j.molliq.2020.113399.
- P. Wang, C. Qi, L. Hao, P. Wen and X. Xu, Sepiolite/Cu2O/Cu Photocatalyst: Preparation and High Performance for Degradation of Organic Dye, J. Mater. Sci. Technol., 2019, 35(3), 285–291, DOI:10.1016/j.jmst.2018.03.023.
- M. Sajjadi, N. Y. Baran, T. Baran, M. Nasrollahzadeh, M. R. Tashili and M. Shokouhimehr, Palladium Nanoparticles Stabilized on a Novel Schiff Base Modified Unye Bentonite: Highly Stable, Reusable and Efficient Nanocatalyst for Treating Wastewater Contaminants and Inactivating Pathogenic Micobes, Sep. Purif. Technol., 2020, 237, 116383, DOI:10.1016/j.seppur.2019.116383.
- Y. Chi, Q. Yuan, Y. Li, J. Tu, L. Zhao, N. Li and X. Li, Synthesis of Fe3O4@SiO2–Ag Magnetic Nanocomposite Based on Small-Sized and Highly Dispersed Silver Nanoparticles for Catalytic Reduction of 4-Nitrophenol, J. Colloid Interface Sci., 2012, 383(1), 96–102, DOI:10.1016/j.jcis.2012.06.027.
- M. Z. Sarker, M. M. Rahman, H. Minami, T. Suzuki, M. K. Hossain and H. Ahmad, Mesoporous Amine Functinalized SiO2 Supported Cu Nanocatalyst and a Kinetic-Mechanistic Degradation Study of Azo Dyes, Colloids Surf., A, 2021, 617, 126403, DOI:10.1016/j.colsurfa.2021.126403.
- Z. X. Li, F. B. Shi, L. L. Li, T. Zhang and C. H. Yan, A Facile Route to Ordered Mesoporous-Alumina-Supported Catalysts, and Their Catalytic Activities for CO Oxidation, Phys. Chem. Chem. Phys., 2011, 13, 2488–2491, 10.1039/C0CP01309A.
- K. Sato, A. Ito, H. Tononaga, H. Kanematsu, Y. Wada, H. Asakura, S. Hosokawa, T. Tanaka, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, Pt–Co Alloy Nanoparticles on a γ-Al2O3 Support: Synergistic Effect between Isolated Electron-Rich Pt and Co for Automotive Exhaust Purification, ChemPlusChem, 2019, 84(5), 447–456, DOI:10.1002/cplu.201800542.
- M. A. Rabbi, M. M. Rahman, H. Minami, N. Yamashita, M. R. Habib and H. Ahmad, Magnetically Responsive Antibacterial Nanocrystalline Jute Cellulose Nanocomposites with Moderate Catalytic Activity, Carbohydr. Polym., 2021, 251, 117024, DOI:10.1016/j.carbpol.2020.117024.
- S. Sarkar, E. Guibal, F. Quignard and A. K. SenGupta, Polymer-Supported Metals and Metal Oxide Nanoparticles: Synthesis, Characterization, and Applications, J. Nanopart. Res., 2012, 14, 715, DOI:10.1007/s11051-011-0715-2.
- T. C. Damato, C. C. S. de Oliveira, R. A. Ando and P. H. C. Camargo, A Facile Approach to TiO2 Colloidal Spheres Decorated with Au Nanoparticles Displaying Well-Defined Sizes and Uniform Dispersion, Langmuir, 2013, 29(5), 1642–1649, DOI:10.1021/la3045219.
- H. Xu and K. S. Suslick, Sonochemical Synthesis of Highly Fluorescent Ag Nanoclusters, ACS Nano, 2010, 4(6), 3209–3214, DOI:10.1021/nn100987k.
- R. G. Bai, K. Muthoosamy, M. Zhou, M. A. Kumar, N. M. Huang and S. Manickam, Biosens. Sonochemical and Sustainable Synthesis of Graphene-Gold (G-Au) Nanocomposites for Enzymeless and Selective Electrochemical Detection of Nitric Oxide, Biosensors Bioelectronics, 2017, 87, 622–629, DOI:10.1016/j.bios.2016.09.003.
- V. Rico, C. López-Gascón, J. P. Espinós, R. Lahoz, M. Laguna, A. R. González-Elipe and G. F. de la Fuente, Metallization of Ceramic Substrates by Laser Induced Decomposition of Coordination Complexes, J. Eur. Ceramic Soc., 2016, 36(11), 2831–2836, DOI:10.1016/j.jeurceramsoc.2016.04.016.
- M. I. Zhilnikova, V. V. Voronov and G. A. Shafeev, Laser Abalation of Metals in Salts Melts, Chem. Phys. Lett., 2020, 755, 137778, DOI:10.1016/j.cplett.2020.137778.
- N. Nityashree, U. K. Gautam and M. Rajamathi, Synthesis and Thermal Decomposition of Metal Hydroxide Intercalated Saponite, Appl. Clay Sci., 2014, 87, 163–169, DOI:10.1016/j.clay.2013.10.026.
- J. A. Gerbec, D. Magana, A. Washington and G. F. Strouse, Microwave-Enhanced Reaction Rates for Nanoparticle Synthesis, J. Am. Chem. Soc., 2005, 127(45), 15791–15800, DOI:10.1021/ja052463g.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. Khder, K. M. AbouZeid, J. Terner, M. S. El- Shall, S. I. Al-Resayes and A. El-Azhary, Microwave Synthesis of Graphene Sheets Supporting Metal Nanocrystals in Aqueous and Organic Media, J. Mater. Chem., 2009, 19, 3832–3837, 10.1039/b906253j.
- M. B. Mohamed, K. M. AbouZeid, V. Abdelsayed, A. A. Alijarash and M. S. El-Shall, Growth Mechanism of Anisotropic Gold Nanocrystals via Microwave Synthesis: Formation of Diolemamide by Gold Nanocatalysis, ACS Nano, 2011, 4(5), 2766–2772, DOI:10.1021/nn9016179.
- V. Abdelsayed, A. Aljarash, M. S. El-Shall, Z. A. Al Othman and A. H. Alghamdi, Microwave Synthesis of Bimetallic Nanoalloys and CO Oxidation on Ceria-Supported Nanoalloys, Chem. Mater., 2009, 21(13), 2825–2834, DOI:10.1021/cm9004486.
- J. A. Hachtel, S. Yu, A. R. Lupini, S. T. Pantelides, M. Gich, A. Laromaine and A. Roig, Gold Nanotriangles Decorated with Superparamagnetic Iron Oxide Nanoparticles: a Compositional and Microstructural Study, Faraday Discuss., 2016, 191, 215–227, 10.1039/C6FD00028B.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Microwave-Assisted Chemistry: Synthetic Applications for Rapid Assembly of Nanomaterials and Organics, Acc. Chem. Res., 2014, 47(4), 1338–1348, DOI:10.1021/ar400309b.
- C. Ray and T. Pal, Retracted Article: Recent Advances of Metal-Metal Oxide Nanocomposites and Their Tailored Nanostructures in Numerous Catalytic Applications, J. Mater. Chem. A, 2017, 5, 9465–9487, 10.1039/C7TA02116J.
- T. S. Rodrigues, A. G. M. da Silva and P. H. C. Camargo, Nanocatalysis by Noble Metal Nanoparticles: Controlled Synthesis for the Optimization and Understanding of Activities, J. Mater. Chem. A, 2019, 7, 5857–5874, 10.1039/C9TA00074G.
- K. Hong, M. Sajjadi, J. M. Suh, K. Zhang, M. Nasrollahzadeh, H. W. Jang, R. S. Varma and M. Shokouhimehr, Palladium Nanoparticles on Assorted Nanostructured Supports: Application for Suzuk, Heck, and Sonogashira Cross-Coupling Reactions, ACS Appl. Nano Mater., 2020, 3(3), 2070–2103, DOI:10.1021/acsanm.9b02017.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. H. Hutchings, Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts, Chem. Rev., 2020, 120, 3890–3938, DOI:10.1021/acs.chemrev.9b00662.
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, Porous Organic Polymer Material Supported Palladium Nanoparticles, J. Mater. Chem. A, 2020, 8, 17360–17391, 10.1039/D0TA05175F.
- P. G. Jamkhande, N. W. Ghule, A. H. Bamer and M. G. Kalaskar, Metal Nanoparticles Synthesis: a Overview on Methods of Preparation Advantages and Disadvantages, and Applications, J. Drug Delivery Sci. Technol., 2019, 53, 101174, DOI:10.1016/j.jddst.2019.101174.
- A. Kumar, Y. Kuang, Z. Liang and X. Sun, Microwave Chemistry, Recent Advancements, and Eco-Friendly Microwave-Assisted Synthesis of Nanoarchitechtures and Their Applicadtions: a Review, Mater. Today Nano, 2020, 11, 100076, DOI:10.1016/j.mtnano.2020.100076.
- J. Hoffmann, M. Nüchter, B. Ondruschka and P. Wasserscheid, Ionic Liquids and Their Heating Behavior during Microwave Irradiation – a State of the Art Report and Challange to Assessment, Green Chem., 2003, 5, 296–299, 10.1039/B212533A.
- R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, The Use of Microwave Ovens for Rapid Synthesis, Tetrahedron Lett., 1986, 27(3), 279–282, DOI:10.1016/S0040-4039(00)83996-9.
- R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Application of Commercial Microwave Ovens to Organic Synthesis, Tetrahedron Lett., 1986, 27(41), 4945–4948, DOI:10.1016/S0040-4039(00)85103-5.
- T. E. Muñoz, R. T. Giberson, R. Demaree and J. R. Day, Microwave-Assisted Immunostaining: a New Approach Yields Fast and Consistent Results, J. Neurosci. Methods, 2004, 137(2), 133–139, DOI:10.1016/j.jneumeth.2004.02.020.
- A. K. Bose, M. S. Manhas, S. N. Ganguly, A. H. Sharma and B. K. Banik, MORE Chemistry for Less Pollution: Applications for Process Development, Synthesis, 2002,(11), 1578–1591, DOI:10.1055/s-2002-33344.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Benign by Design: Catalyst-Free In-Water, On-Water Green Chemical Methodologies in Organic Synthesis, Chem. Soc. Rev., 2012, 42, 5522–5551, 10.1039/c3cs60025d.
- M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Solvent-Free and Catalysts-Free Chemistry: a Benign Pathway to Sustainability, ChemSusChem, 2014, 7, 24–44, DOI:10.1002/cssc.201300485.
- V. Polshettiwar and R. S. Varma, Aqueous Microwave Chemistry: a Clean and Green Synthetic Tool for Rapid Drug Discovery, Chem. Soc. Rev., 2008, 37, 1546–1557, 10.1039/b716534j.
- M. B. Gawande and P. S. Branco, A Efficient and Expeditious Fmoc Protection of Amines and Amino Acids in Aqueous Media, Green Chem., 2011, 13, 3355–3359, 10.1039/c1gc15868f.
- M. N. Nadagouda, T. F. Speth and R. S. Varma, Microwave-Assisted Green Synthesis of Silver Nanostructures, Acc. Chem. Res., 2011, 44(7), 469–478, DOI:10.1021/ar1001457.
- B. Baruwati and R. S. Varma, High Value Products from Waste: Grape Pomace Extract—a Three-in-One Package for the Synthesis of Metal Nanoparticles, ChemSusChem, 2009, 2(11), 1041–1044, DOI:10.1002/cssc.200900220.
- J. Kou and R. S. Varma, Beet Juice-Induced Green Fabrication of Plasmonic AgCl/Ag Nanoparticles, ChemSusChem, 2012, 5(12), 2435–2441, DOI:10.1002/cssc.201200477.
- J. Virkutyte and R. S. Varma, Green Synthesis of Metal Nanoparticles: Biodegradable Polymers and Enzymes in Stabilization and Surface Functionalization, Chem. Sci., 2011, 2, 837–846, 10.1039/C0SC00338G.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L.-Z. Wu, C.-H. Tung, Y. Yin and T. Zhang, Graphene-Supported Ultrafine Metal Nanoparticles Encapsulated by Mesoporous Silica: Robust Catalysts for Oxidation and Reduction Reactions, Angew. Chem., Int. Ed., 2014, 53(1), 250–254, DOI:10.1002/anie.201306863.
- A. K. Tagantsev, V. O. Sherman, K. F. Astafiev, J. Venkatesh and N. Setter, Ferroelectric Materials for Microwave Tunable Applications, J. Electroceram., 2003, 11, 5–66, DOI:10.1023/B:JECR.0000015661.81386.e6.
- I. Bilecka, I. Djerdj and M. Niederberger, One-Minute Synthesis of Crystalline Binary and Ternary Metal Oxide Nanoparticles, Chem. Commun., 2008, 886–888, 10.1039/B717334B.
- R. Martínez-Palou, Microwave-Assisted Synthesis using Ionic Liquids, Mol. Diversity, 2010, 14, 3–25, DOI:10.1007/s11030-009-9159-3.
- S. Liu, F. Lu and J.-J. Zhu, Highly Fluorescent Ag Nanoclusters: Microwave-Assisted Green Synthesis and Cr3+ Sensing, Chem. Commun., 2011, 47, 2661–2663, 10.1039/c0cc04276e.
- M. A. Rabbi, M. M. Rahman, H. Minami, M. R. Habib and H. Ahmad, Ag Impregnated Sub-Micrometer Crystalline Jute Cellulose Particles: Catalytic and Antibacterial Properties, Carbohydr. Polym., 2020, 233, 115742, DOI:10.1016/j.carbpol.2020.115842.
- I. Bilecka and M. Niederberger, Microwave Chemistry for Inorganic Nanomaterials Synthesis, Nanoscale, 2010, 2, 1358–1374, 10.1039/b9nr00377k.
- G. Garnweitner and M. Niederberger, Organic Chemistry in Inorganic Nanomaterials Synthesis, J. Mater. Chem., 2008, 18, 1171–1182, 10.1039/B713775C.
- G. Cravotto and P. Cintas, The Combined Use of Microwaves and Ultrasound: Improved Tools in Process Chemistry and Organic Synthesis, Chemistry, 2007, 13(7), 1902–1909, DOI:10.1002/chem.200601845.
- S. S. Rao, K. S. Jayachandran and V. P. D. Rekha, Microwave-Assisted Rapid Synthesis of Silver Nanoparticles using Fucoidan: Characterization with Assessment of Biocompatibility and Antimicrobial Activity, Int. J. Biol. Macromol., 2020, 163, 745–755, DOI:10.1016/j.ijbiomac.2020.06.230.
- P. Kunal, H. Li, B. L. Dewing, L. Zhang, K. Jarvis, G. Henkelman and S. M. Humphrey, Microwave-Assisted Synthesis of PdxAu100–x Alloy Nanoparticles: a Combined Experimental and Theoretical Assessment of Synthetic and Compositional Effects upon Catalytic Reactivity, ACS Catal., 2016, 6(8), 4882–4893, DOI:10.1021/acscatal.6b01014.
- C. Gutiérrez-Wing, R. Esparza, C. Vargas-Hernández, M. E. F. García and M. José-Yacamán, Microwave-Assisted Synthesis of Gold Nanoparticles Self-Assembled into Self-Supported Superstructures, Nanoscale, 2012, 4, 2281–2286, 10.1039/c2nr12053d.
- A. J. Nozik, Multiple Exciton Generation in Semiconductor Quantum Dots, Chem. Phys. Lett., 2008, 457(1–3), 3–11, DOI:10.1016/j.cplett.2008.03.094.
- A. Mirzaei and G. Neri, Microwave-Assisted Synthesis of Metal Oxide Nanostructures for Gas Sensing Application: a Review, Sens. Actuators, B, 2016, 237, 749–775, DOI:10.1016/j.snb.2016.06.114.
- A. Verma, R. Dwivedi, R. Prasad and K. S. Bartwal, Microwave-Assisted Synthesis of Mixed Metal-Oxide Nanoparticles, J. Nanopart., 2013, 737831, DOI:10.1155/2013/737831.
- K. Kannan, D. Radhika, M. P. Nikolova, V. Andal, K. K. Sadasivuni and S. Krishna, Facile Microwave-Assisted Synthesis of Metal Oxide CdO-CuO Nanocomposite: Photocatalytic and Antimicrobial Enhancing Properties, Optik, 2020, 218, 165112, DOI:10.1016/j.ijleo.2020.165112.
- G. Zhong, S. Xu, C. Chen, D. J. Kline, M. Giroux, Y. Pei, M. Jiao, D. Liu, R. Mi, H. Xie, B. Yang, C. Wang, M. R. Zachariah and L. Hu, Synthesis of Metal Oxide Nanoparticles by Rapid, High-Temperature 3D Microwave Heating, Adv. Funct. Mater., 2019, 29(48), 1904282, DOI:10.1002/adfm.201904282.
- M. B. Schütz, L. Xiao, T. Lehnen, T. Fischer and S. Mathur, Microwave-Assisted Synthesis of Nanocrystalline Binary and Ternary Metal Oxides, Int. Mater. Rev., 2017, 63, 341–374, DOI:10.1080/09506608.2017.1402158.
- W. Tu and H. Liu, Continuous Synthesis of Colloidal Metal Nanoclusters by Microwave Irradiation, Chem. Mater., 2000, 12(2), 564–567, DOI:10.1021/cm990637l.
- T.-Y. Ma, L. Liu and Z.-Y. Yuan, Direct Synthesis of Ordered Mesoporous Carbons, Chem. Soc. Rev., 2013, 42, 3977–4003, 10.1039/C2CS35301F.
- E. Lam and Y. H. T. Luong, Carbon Materials as Catalyst Supports and Catalysts in the Transformation of Biomass to Fuels and Chemicals, ACS Catal., 2014, 4(10), 3393–3410, DOI:10.1021/cs5008393.
- Y. Chen and Y. Liu, Preparation of Porous Carbon with High Dispersion of Ru Nanoparticles by Sol–Gel Method and Its Application in Hydrogen Storage, J. Mater. Chem. A, 2014, 2, 9193–9199, 10.1039/C4TA01126K.
- K.-H. Chuang, K. Shih and M.-Y. Wey, The Influences of Microwave Irradiation and Polyol Precursor pH on Cu/AC Catalyst and Its CO Oxidation Performance, J. Nanopart. Res., 2012, 14, 1178, DOI:10.1007/s11051-012-1178-9.
- J. Ren, M. Ren, D. Wang, J. Lin and Z. Li, Mechanism of Microwave-Induced Carbothermic Reduction and Catalytic Performance of Cu/Activated Carbon Catalysts in the Oxidative Carbonylation of Methanol, J. Therm. Anal. Calorim., 2015, 120, 1929–1939, DOI:10.1007/s10973-015-4519-y.
- M. Ren, J. Ren, P. Hao, J. Yang, D. Wang, Y. Pei, J.-Y. Lin and Z. Li, Influence of Microwave Irradiation on the Structural Properties of Carbon-Supported Hollow Copper Nanoparticles and Their Effect on the Synthesis of Dimethyl Carbonate, ChemCatChem, 2016, 8(4), 861–871, DOI:10.1002/cctc.201501182.
- X. Liu, H. Yin, A. Lin and Z. Guo, Effective Removal of Phenol by Using Activated Carbon Supported Iron Prepared Under Microwave Irradiation as a Reusable Heterogeneous Fenton-Like Catalyst, J. Environ. Chem. Eng., 2017, 5(1), 870–876, DOI:10.1016/j.jece.2017.01.010.
- P. Veerakumar, N. Dhenadhayalan, K.-C. Lin and S.-B. Liu, Highly Stable Ruthenium Nanoparticles on 3D Mesoporous Carbon: an Excellent Opportunity for Reduction Reactions, J. Mater. Chem. A, 2015, 3, 23448–23457, 10.1039/C5TA06875D.
- P. Veerakumar, K. Salamalai, P. Thanasekaran and K.-C. Lin, Simple Preparation of Porous Carbon-Supported Ruthenium: Propitious Catalytic Activity in the Reduction of Ferrocyanate(III) and a Cationic Dye, ACS Omega, 2018, 3(10), 12609–12621, DOI:10.1021/acsomega.8b01680.
- W. X. Chen, J. Y. Lee and Z. Liu, Microwave-Assisted Synthesis of Carbon Supported Pt Nanoparticles for Fuel Cell Applications, Chem. Commun., 2002,(21), 2588–2589, 10.1039/b208600j.
- Z. Liu, L. M. Gan, L. Hong, W. Chen and J. Y. Lee, Carbon-Supported Pt Nanoparticles as Catalysts for Proton Exchange Membrane Fuel Cells, J. Power Sources, 2005, 139(1–2), 73–78, DOI:10.1016/j.jpowsour.2004.07.012.
- L. Bo, X. Quan, X. Wang and S. Chen, Preparation and Characteristics of Carbon-Supported Platinum Catalyst and Its Application iIn The Removal of Phenolic Pollutants in Aqueous Solution by Microwave-Assisted Catalytic Oxidation, J. Hazard. Mater., 2008, 157(1), 179–186, DOI:10.1016/j.jhazmat.2007.12.111.
- Z. Gan, X. Zheng, D. Wei, Q. Hu, A. Zhao, X. Zhang and G. Li, One-Pot Synthesis of One-Dimensional Array Pt Nanoparticles on Carbon Nanotubes via a Facile Microwave Polyol Method, Superlattices Microstruct., 2010, 47(6), 705–709, DOI:10.1016/j.spmi.2010.04.001.
- Z. Guo, H. Zhu, X. Zhang, F. Wang, Y. Guo and Y. Wei, Microwave-Assisted Synthesis of High-Loading, Highly Dispersed Pt/Carbon Aerogel Catalyst for Direct Methanol Fuel Cell, Bull. Mater. Sci., 2011, 34, 577–581, DOI:10.1007/s12034-011-0082-8.
- R. Sharma, Y. Wang, F. Li, J. Chamier and S. M. Anderson, Synthesis of a Pt/C Electrocatalyst from a User-Friendly Pt Precursor (Ammonium Hexachloroplatinate) through Microwave-Assisted Polyol Synthesis, ACS Appl. Energy Mater., 2019, 2(9), 6875–6882, DOI:10.1021/acsaem.9b01336.
- E. O. Pentsak, V. A. Cherepanova and V. P. Ananikov, Dynamic Behavior of Metal Nanoparticles in Pd/C and Pt/C Catalytic Systems under Microwave and Conventional Heating, ACS Appl. Mater. Interfaces, 2017, 9(42), 36723–36732, DOI:10.1021/acsami.7b09173.
- E. O. Penstak, E. G. Gordeev and V. P. Ananikov, Noninnocent Nature of Carbon Support in Metal/Carbon Catalysts: Etching/Pitting vs Nanotube Growth under Microwave Irradiation, ACS Catal., 2014, 4(11), 3806–3814, DOI:10.1021/cs500934g.
- C. Antonetti, M. Oubenali, A. M. R. Galletti, P. Serp and G. Vannucci, Novel Microwave Synthesis of Ruthenium Nanoparticles Supported on Carbon Nanotubes Active in the Selective Hydrogenation of p-Chloronitrobenzene to p-Chloroaniline, Appl. Catal., A, 2012, 421–422, 99–107, DOI:10.1016/j.apcata.2012.02.003.
- X. Ni, B. Zhang, C. Li, M. Pang, D. Su, C. T. Williams and C. Liang, Microwave-Assisted Green Synthesis of Uniform Ru Nanoparticles Supported on Non-Functional Carbon Nanotubes for Cinnamaldehyde Hydrogenation, Catal. Commun., 2012, 24, 65–69, DOI:10.1016/j.catcom.2012.03.035.
- B. Zhang, X. Ni, W. Zhang, L. Shao, Q. Zhang, F. Girgsdies, C. Liang, R. Schlögi and D. S. Su, Structural Rearrangements of Ru Nanoparticles Supported on Carbon Nanotubes Under Microwave Irradiation, Chem. Commun., 2011, 47, 10716–10718, 10.1039/c1cc13858h.
- T. Hemraj-Benny, N. Tobar, N. Carrero, R. Sumner, L. Pimentel and G. Emeran, Microwave-Assisted Synthesis of Single-Walled Carbon Nanotube-Supported Ruthenium Nanoparticles for the Catalytic Degradation of Congo Red Dye, Mater. Chem. Phys., 2018, 216, 72–81, DOI:10.1016/j.matchemphys.2018.05.081.
- T. Hemraj-Benny, L. Pimentel and G. Emeran, Formation of Single-Walled Carbon Nanotube-Ruthenium Nanoparticles in Ethanol upon Microwave Radiation, Inorg. Chem. Commun., 2020, 112, 107707, DOI:10.1016/j.inoche.2019.107707.
- R. Samiee-Zafarghandi, A. Hadi and J. Karimi-Sabet, Graphene-Supported Metal Nanoparticles as Novel Catalysts for Syngas Production Using Supercritical Water Gasification of Microalgae, Biomass Bioenergy, 2019, 121, 13–21, DOI:10.1016/j.biombioe.2018.11.035.
- S. Ren, Q. Yu, X. Yu, P. Rong, L. Jiang and J. Jiang, Graphene-Supported Metal Single-Atom Catalysts: a Concise Review, Sci. China Mater., 2020, 63, 903–920, DOI:10.1007/s40843-019-1286-1.
- K. M. Yam, N. Guo, Z. Jiang, S. Li and C. Zhang, Graphene-Based Heterogeneous Catalysis: Role of Graphene, Catalysts, 2020, 10(1), 53, DOI:10.3390/catal10010053.
- M. Nasrollahzadeh, Z. Issaabadi, M. M. Tohidi and S. M. Sajadi, Recent Progress in Application of Graphene Supported Metal Nanoparticles in C−C and C−X Coupling Reactions, Chem. Rec., 2018, 18(2), 165–229, DOI:10.1002/tcr.201700022.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L.-Z. Wu, C.-H. Tung, Y. Yin and T. Zhang, Graphene-Supported Ultrafine Metal Nanoparticles Encapsulated by Mesoporous Silica: Robust Catalysts for Oxidation and Reduction Reactions, Angew. Chem., 2014, 126(1), 254–258, DOI:10.1002/ange.201306863.
- H. Fei, J. Dong, C. Wan, Z. Zhao, X. Xu, Z. Lin, Y. Wang, H. Liu, K. Zang, J. Luo, S. Zhao, W. Hu, W. Yan, I. Shakir and Y. Huang, Microwave-Assisted Rapid Synthesis of Graphene-Supported Single Atomic Metals, Adv. Mater., 2018, 30(35), 1802146, DOI:10.1002/adma.201802146.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, Microwave-Assisted Synthesis of Palladium Nanoparticles Supported on Graphene: A Highly Active and Recyclable Catalyst for Carbon–Carbon Cross-Coupling Reactions, J. Catal., 2011, 279(1), 1–11, DOI:10.1016/j.jcat.2010.12.003.
- K. W. Brinkley, M. Burkholder, A. R. Siamaki, K. Belecki and B. F. Gupton, The Continuous Synthesis and Application of Graphene Supported Palladium Nanoparticles: a Highly Effective Catalyst for Suzuki-Miyaura Cross-Coupling Reactions, Green Process. Synth., 2015, 4, 241–246, DOI:10.1515/gps-2015-0021.
- J.-H. Yang and D. Ma, Graphene-Supported Pd Nanoparticles: Microwave-Assisted Synthesis and as Microwave-Active Selective Hydrogenation Catalysts, RSC Adv., 2013, 3, 10131–10134, 10.1039/c3ra41340c.
- H. A. Elazab, M. A. Radwan and M. A. Sadek, Microwave-Assisted Synthesis of Graphene Supported Hexagonal Magnetite for Applications in Catalysis, Int. J. Innov. Technol. Exploring Eng., 2019, 8(12), 5511–5513, DOI:10.35940/ijitee.L2535219.
- J.-Y. Lee, T.-Y. Yung and L.-K. Liu, The Microwave-Assisted Ionic Liquid Nanocomposite Synthesis: Platinum Nanoparticles on Graphene and the Application on Hydrogenation of Styrene, Nanoscale Res. Lett., 2013, 8, 414, DOI:10.1186/1556-276X-8-414.
- K. Ullah, S. Ye, L. Zhu, Z.-D. Meng, S. Sarkar and W.-C. Oh, Microwave Assisted Synthesis of a Noble Metal-Graphene Hybrid Photocatalyst for High Efficient Decomposition of Organic Dyes Under Visible Light, Mater. Sci. Eng., B, 2014, 180, 20–26, DOI:10.1016/j.mseb.2013.10.014.
- A. Zuliani, P. Ranjan, R. Luque and E. V. V. Eycken, Heterogeneously Catalyzed Synthesis of Imidazolones via Cycloisomerizations of Propargylic Ureas Using Ag and Au/Al SBA-15 Systems, ACS Sustainable Chem. Eng., 2019, 7(5), 5568–5575, DOI:10.1021/acssuschemeng.9b00198.
- M. B. Gawande, Y. Monga, R. Zboril and R. K. Sharma, Silica-Decorated Magnetic Nanocomposites For Catalytic Applications, Coord. Chem. Rev., 2015, 288, 118–143, DOI:10.1016/j.ccr.2015.01.001.
- X. Deng, K. Chen and H. Tüysüz, Protocol for the Nanocasting Method: Preparation of Ordered Mesoporous Metal Oxides, Chem. Mater., 2017, 29(1), 40–52, DOI:10.1021/acs.chemmater.6b02645.
- J. M. Campelo, T. D. Conesa, M. J. Gracia, M. J. Jurado, R. Luque, J. M. Marinas and A. A. Romero, Microwave Facile Preparation of Highly Active and Dispersed SBA-12 Supported Metal Nanoparticles, Green Chem., 2008, 10, 853–858, 10.1039/b801754a.
- R. Manno, P. Ranjan, V. Sebastian, R. Mallada, S. Irusta, U. K. Sharma, E. V. V. Eycken and J. Santamaria, Continuous Microwave-Assisted Synthesis of Silver Nanoclusters Confined in Mesoporous SBA-15: Application in Alkyne Cyclizations, Chem. Mater., 2020, 32(7), 2874–2883, DOI:10.1021/acs.chemmater.9b04935.
- L. Uson, J. L. Hueso, V. Sebastian, R. Arenal, I. Florea, S. Irusta, M. Arruebo and J. Santamaria, In Situ Preparation of Ultra-Small Pt Nanoparticles Within Rod-Shaped Mesoporous Silica Particles: 3-D Tomography and Catalytic Oxidation of n-Hexane, Catal. Commun., 2017, 100, 93–97, DOI:10.1016/j.catcom.2017.06.022.
- Y. Jiang, X. Li and H. Ji, Acid-Treated Bentonite-Supported Ni Catalysts via Rapid Microwave-Assisted Drying for Nitrobenzene Hydrogenation, Chem. Eng. Commun., 2018, 205, 624–636, DOI:10.1080/00986445.2017.1409737.
- G. Akay, K. Zhang, W. S. S. Al-Harrasi and R. R. Sankaran, Catalytic Plasma Fischer–Tropsch Synthesis Using Hierarchically Connected Porous Co/SiO2 Catalysts Prepared by Microwave-Induced Co-assembly, Ind. Eng. Chem. Res., 2020, 59(26), 12013–12027, DOI:10.1021/acs.iecr.0c01585.
- T. Mukhriza, K. Zhang and A. N. Phan, Microwave Assisted Co/SiO2 Preparation for Fischer-Tropsch Synthesis, J. Nat., 2020, 20(2), 42–48, DOI:10.24815/jn.v20i2.16889.
- S. A. Hassan, H. M. Gobara, M. M. Gomaa, R. S. Mohamed and F. H. Khalil, Can Microwave Assisted In Situ Reduction of Supported Pt Nanoparticles Challenge The Chemical Method in Controlling the Dispersion Profile-Catalytic Performance Relationship?, RSC Adv., 2015, 5(67), 54460–54470, 10.1039/C5RA08116E.
- S. Wang, Y. Shi and X. Ma, Microwave Synthesis, Characterization and Transesterification Activities of Ti-MCM-41, Microporous Mesoporous Mater., 2012, 156, 22–28, DOI:10.1016/j.micromeso.2012.02.011.
- T. S. Jiang, Y. H. Li, X. P. Zhou, Q. Zhao and H. B. Yin, Thermal and Hydrothermal Stability of Zrmcm-41 Mesoporous Molecular Sieves Obtained by Microwave Irradiation, J. Chem. Sci., 2010, 122, 371–379, DOI:10.1007/s12039-010-0042-7.
- S. Chen, R. Si, E. Taylor, J. Janzen and J. Chen, Synthesis of Pd/Fe3O4 Hybrid Nanocatalysts with Controllable Interface and Enhanced Catalytic Activities for CO Oxidation, J. Phys. Chem. C, 2012, 116(23), 12969–12976, DOI:10.1021/jp3036204.
- C.-J. Jia, M. Schwickardi, C. Weidenthaler, W. Schmidt, S. Korhonen, B. M. Weckhuysen and F. Schüth, Co3O4–SiO2 Nanocomposite: A Very Active Catalyst for CO Oxidation with Unusual Catalytic Behavior, J. Am. Chem. Soc., 2011, 133(29), 11279–11288, DOI:10.1021/ja2028926.
- W. Zhou, M. Yao, L. Guo, Y. Li, J. Li and S. Yang, Hydrazine-Linked Convergent Self-Assembly of Sophisticated Concave Polyhedrons of β-Ni(OH)2 and NiO from Nanoplate Building Blocks, J. Am. Chem. Soc., 2009, 131(8), 2959–2964, DOI:10.1021/ja808784s.
- H. A. Elazab, S. Moussa, B. F. Gupton and M. S. El-Shall, Microwave-Assisted Synthesis of Pd Nanoparticles Supported on Fe3O4, Co3O4, and Ni(OH)2 Nanoplates and Catalysis Application for CO Oxidation, J. Nanopart. Res., 2014, 16, 2477, DOI:10.1007/s11051-014-2477-0.
- H. A. Elazab, S. Moussa, K. W. Brinkley, B. F. Gupton and M. S. El-Shall, The Continuous Synthesis of Pd Supported on Fe3O4 Nanoparticles: a Highly Effective and Magnetic Catalyst for CO Oxidation, Green Process. Synth., 2017, 6, 413–424, DOI:10.1515/gps-2016-0168.
- M. Morad, M. A. Karim, H. M. Altass and A. E. R. S. Khder, Microwave-Assisted Synthesis of Gold Nanoparticles Supported on Mn3O4 Catalyst for Low Temperature CO Oxidation, Environ. Technol., 2021, 42, 2680–2689, DOI:10.1080/09593330.2019.1709988.
- A. E. R. S. Khder, H. M. Altass, M. I. Orif, S. S. Ashour and L. S. Almazroai, Preparation And Characterization of Highly Active Pd Nanoparticles Supported Mn3O4 Catalyst for Low-Temperature CO Oxidation, Mater. Res. Bull., 2019, 113, 215–222, DOI:10.1016/j.materresbull.2019.02.011.
- G. Glaspell, L. Fuoco and M. S. El-Shall, Microwave Synthesis of Supported Au and Pd Nanoparticle Catalysts for CO Oxidation, J. Phys. Chem. B, 2005, 109(37), 17350–17355, DOI:10.1021/jp0526849.
- K.-H. Chuang, K. Shih, C.-Y. Lu and M.-Y. Wey, Copper Catalysts Prepared via Microwave-Heated Polyol Process for Preferential Oxidation of CO in H2-Rich Streams, Int. J. Hydrogen Energy, 2013, 38(1), 100–108, DOI:10.1016/j.ijhydene.2012.10.039.
- H. A. Elazab, M. A. Sadek and T. T. El-Idreesy, Microwave-Assisted Synthesis of Palladium Nanoparticles Supported on Copper Oxide in Aqueous Medium as an Efficient Catalyst for Suzuki Cross-Coupling Reaction, Adsorpt. Sci. Technol., 2018, 36(5-6), 1352–1365, DOI:10.1177/0263617418771777.
- H. A. Elazab, M. A. Radwan and T. T. El-Idreesy, Facile Microwave-Assisted Synthetic Approach to Palladium Nanoparticles Supported on Copper Oxide as an Efficient Catalyst for Heck and Sonogashira Cross-Coupling Reactions, Int. J. Nanosci., 2019, 18(5), 1850032, DOI:10.1142/S0219581X18500321.
- M. Trueba and S. P. Trasatti, γ-Alumina as a Support for Catalysts: a Review of Fundamental Aspects, Eur. J. Inorg. Chem., 2005,(17), 3393–3403, DOI:10.1002/ejic.200500348.
- F. Song, Q. Zhong, Y. Yu, M. Shi, Y. Wu, J. Hu and Y. Song, Obtaining Well-Dispersed Ni/Al2O3 Catalyst for CO2 Methanation with a Microwave-Assisted Method, Int. J. Hydrogen Energy, 2017, 42(7), 4174–4183, DOI:10.1016/j.ijhydene.2016.10.141.
- J. Y. Chung, S. Khdama and H. Sekiguchi, Preparation of a Pd/Al2O3 Catalyst with Microwave-Induced Plasma Jet Irradiation under Atmospheric Pressure, Nanomaterials, 2019, 9(12), 1734, DOI:10.3390/nano9121734.
- J. Y. Chung, S. Kodama and H. Sekiguchi, Preparation of Catalyst with Microwave Induced Plasma Jet Combined with Spouted Bed, J. Nanosci. Nanotechnol., 2019, 19(10), 6849–6855, DOI:10.1166/jnn.2019.17122.
- H. Wang, Z. Liu, Y. Wu, Z. Yao, W. Zhao, W. Duan and K. Guo, Preparation of Highly Dispersed W/Al2O3 Hydrodesulfurization Catalysts via a Microwave Hydrothermal Method: Effect of Oxalic Acid, Arabian J. Chem., 2016, 9(1), 18–24, DOI:10.1016/j.arabjc.2014.11.023.
- S. Bagheri, N. M. Julkapli and S. B. A. Hamid, Titanium Dioxide as a Catalyst Support in Heterogeneous Catalysis, Sci. World J., 2014, 2014, 727496, DOI:10.1155/2014/727496.
- A. G. R. Howe, R. Maunder, D. J. Morgan and J. K. Edwards, Rapid Microwave-Assisted Polyol Synthesis of TiO2-Supported Ruthenium Catalysts for Levulinic Acid Hydrogenation, Catalysts, 2019, 9(9), 748, DOI:10.3390/catal9090748.
- E. A. Anumol, P. Kundu, P. A. Deshpande, G. Madras and N. Ravishankar, New Insights into Selective Heterogeneous Nucleation of Metal Nanoparticles on Oxides by Microwave-Assisted Reduction: Rapid Synthesis of High-Activity Supported Catalysts, ACS Nano, 2011, 5(10), 8049–8061, DOI:10.1021/nn202639f.
- Z. Li, L. Yu, C. Milligan, T. Ma, L. Zhou, Y. Cui, Z. Qi, N. Libretto, B. Xu, J. Luo, E. Shi, Z. Wu, H. Xin, W. N. Delgass, J. T. Miller and Y. Wu, Two-Dimensional Transition Metal Carbides as Supports for Tuning the Chemistry of Catalytic Nanoparticles, Nat. Commun., 2018, 9, 5258, DOI:10.1038/s41467-018-07502-5.
- J. A. Rodriguez and F. Illas, Activation of Noble Metals on Metal-Carbide Surfaces: Novel Catalysts for CO Oxidation, Desulfurization and Hydrogenation Reactions, Phys. Chem. Chem. Phys., 2012, 14, 427–438, 10.1039/C1CP22738F.
- M. Shi, L. Kang, Y. Jiang and C. Ma, Microwave-Assisted Synthesis of Mesoporous Tungsten Carbide/Carbon for Fuel Cell Applications, Catal. Lett., 2014, 144, 278–284, DOI:10.1007/s10562-013-1132-1.
- K. Wang, Z. Pan, F. Tzorbatzoglou, Y. Zhang, Y. Wng, P. Tsiakaras and S. Song, An Investigation of WC Stability During The Preparation of Pt@WC/OMC via a Pulse Microwave Assisted Polyol Method, Appl. Catal., B, 2015, 166–167, 224–230, DOI:10.1016/j.apcatb.2014.11.025.
- H. Asahara, Y. Kuribayashi, P. Wang, K. Kobiro and N. Nishiwaki, An Effect of Microwave Irradiation on Pd/SiC Catalyst for Prolonging the Catalytic Life, Curr. Microwave Chem., 2014, 1(1), 142–147, DOI:10.2174/2213335601666140703184655.
- M. Führer, T. van Haasterecht and J. H. Bitter, Molybdenum and Tungsten Carbides Can Shine Too, Catal. Sci. Technol., 2020, 10, 6089–6097, 10.1039/D0CY01420F.
- R. B. Levy and M. Boudart, Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis, Science, 1973, 181, 547–549, DOI:10.1126/science.181.4099.547.
- E. Furimsky, Metal Carbides and Nitrides as Potential Catalysts for Hydroprocessing, Appl. Catal., A, 2003, 240(1–2), 1–28, DOI:10.1016/S0926-860X(02)00428-3.
- C. E. Chan-Thaw and A. Villa, Metal Carbides for Biomass Valorization, Appl. Sci., 2018, 8(2), 259, DOI:10.3390/app8020259.
- M. M. Sullivan, C. J. Chen and A. Bhan, Catalytic Deoxygenation on Transition Metal Carbide Catalysts, Catal. Sci. Technol., 2016, 6, 602–616, 10.1039/C5cy01665g.
- S. Posada-Pérez, F. Viñes, J. A. Rodriguez and F. Illas, Fundamentals of Methanol Synthesis on Metal Carbide Based Catalysts: Activation of CO2 and H2, Top. Catal., 2015, 58, 159–173, DOI:10.1007/s11244-014-0355-8.
- S. J. Ardakani, X. Liu and K. J. Smith, Hydrogenation and Ring Opening of Naphthalene on Bulk and Supported Mo2C Catalysts, Appl. Catal., A, 2007, 324, 9–19, DOI:10.1016/j.apcata.2007.02.048.
- M. Pang, C. Liu, W. Xia, M. Muhler and C. Liang, Activated Carbon Supported Molybdenum Carbides as Cheap and Highly Efficient Catalyst in The Selective Hydrogenation of Naphthalene to Tetralin, Green Chem., 2012, 14, 1272–1276, 10.1039/c2gc35177c.
- R. Mnatsakanyan, A. R. Zhurnachyan, V. A. Matyshak, K. V. Manukyan and A. S. Mukasyan, Microwave-Assisted Synthesis of Carbon-Supported Carbides Catalysts for Hydrous Hydrazine Decomposition, J. Phys. Chem. Solids, 2016, 96–97, 115–120, DOI:10.1016/j.jpcs.2016.05.008.
- C. Liang, L. Ding, A. Wang, Z. Ma, J. Qiu and T. Zhang, Microwave-Assisted Preparation and Hydrazine Decomposition Properties of Nanostructured Tungsten Carbides on Carbon Nanotubes, Ind. Eng. Chem. Res., 2009, 48(6), 3244–3248, DOI:10.1021/ie801591x.
- M. Pang, C. Li, L. Ding, J. Zhang, D. Su, W. Li and C. Liang, Microwave-Assisted Preparation of Mo2C/CNTs Nanocomposites as Efficient Electrocatalyst Supports for Oxygen Reduction Reaction, Ind. Eng. Chem. Res., 2010, 49(9), 4169–4174, DOI:10.1021/ie901741c.
- J. C. Védrine, Importance, Features and Uses of Metal Oxide Catalysts in Heterogeneous Catalysis, Chin. J. Catal., 2019, 40(11), 1627–1636, DOI:10.1016/S1872-2067(18)63162-6.
- S. Motshekga, S. K. Pillai and S. S. Ray, Conventional Wet Impregnation versus Microwave-Assisted Synthesis of SnO2/CNT Composites, J. Nanopart. Res., 2011, 13, 1093–1099, DOI:10.1007/s11051-010-0098-9.
- S. Gayathri, M. Kottaisamy and V. Ramakrishnan, Facile Microwave-Assisted Synthesis of Titanium Dioxide Decorated Graphene Nanocomposite for Photodegradation of Organic Dyes, AIP Adv., 2015, 5, 127219, DOI:10.1063/1.4938544.
- R. M. Mohamed and E. S. Aazam, Synthesis and Characterization of CeO2-SiO2 Nanoparticles by Microwave-Assisted Irradiation Method for Photocatalytic Oxidation of Methylene Blue Dye, Int. J. Photoenergy, 2012, 928760, DOI:10.1155/2012/928760.
- P. Li, B. Wen, F. Yu, M. Zhu, X. Guo, Y. Han, L. Kang, X. Huang, J. Dan, F. Ouyang and B. Dai, High Efficient Nickel/Vermiculite Catalyst Prepared via Microwave Irradiation-Assisted Synthesis for Carbon Monoxide Methanation, Fuel, 2016, 171, 263–269, DOI:10.1016/j.fuel.2015.12.076.
- G. Hu, D. Guo, H. Shang, Y. Sun, J. Zeng and J. Li, Microwave-Assisted Rapid Preparation of Vermiculite-Loaded Nano-Nickel Oxide as a Highly Efficient Catalyst for Acetylene Carbonylation to Synthesize Acrylic Acid, ChemistrySelect, 2020, 5(10), 2940–2948, DOI:10.1002/slct.201904667.
- A. R. Jadhav, J. M. C. Puguan and H. Kim, Microwave-Assisted Synthesis of a Stainless Steel Mesh-Supported Co3O4 Microrod Array As a Highly Efficient Catalyst for Electrochemical Water Oxidation, ACS Sustainable Chem. Eng., 2017, 5(11), 11069–11079, DOI:10.1021/acssuschemeng.7b03027.
- A. May-Masnou, L. Soler, M. Torras, P. Salles, J. Llorca and A. Roig, Fast and Simple Microwave Synthesis of TiO2/Au Nanoparticles for Gas-Phase Photocatalytic Hydrogen Generation, Front. Chem., 2018, 6, 110, DOI:10.3389/fchem.2018.00110.
- S.-S. Chen, Z.-Z. Yang, A.-J. Wang, K.-M. Fang and J.-J. Feng, Facile Synthesis of Bimetallic Gold-Palladium Nanocrystals As Effective and Durable Advanced Catalysts for Improved Electrocatalytic Performances of Ethylene Glycol and Glycerol Oxidation, J. Colloid Interface Sci., 2018, 509, 10–17, DOI:10.1016/j.jcis.2017.08.063.
- G. Sheng, J. Chen, H. Ye, Z. Hu, X.-Z. Fu, R. Sun, W. Huang and C.-P. Wong, Hollow PdCo Alloy Nanospheres with Mesoporous Shells as High-Performance Catalysts for Methanol Oxidation, J. Colloid Interface Sci., 2018, 522, 264–271, DOI:10.1016/j.jcis.2018.03.039.
- S. Dutta, C. Ray, A. K. Sasmal, Y. Negishi and T. Pal, Fabrication of Dog-Bone Shaped Au NRcore–Pt/Pdshell Trimetallic Nanoparticle-Decorated Reduced Graphene Oxide Nanosheets for Excellent Electrocatalysis, J. Mater. Chem. A, 2016, 4, 3765–3776, 10.1039/C6TA00379F.
- J. Mao, W. Chen, D. He, J. Wan, J. Pei, J. Dong, Y. Wang, P. An, Z. Jin, W. Xin, H. Tang, Z. Zhuang, X. Liang, Y. Huang, G. Zhou, L. Wang, D. Wang and Y. Li, Design of Ultrathin Pt-Mo-Ni Nanowire Catalysts for Ethanol Electrooxidation, Sci. Adv., 2017, 3(8), e1603068, DOI:10.1126/sciadv.1603068.
- S. Stevanović, D. Tripković, J. Rogan, K. Popović, J. Lović, A. Tripković and V. M. Jovanović, Microwave-Assisted Polyol Synthesis of Carbon-Supported Platinum-Based Bimetallic Catalysts for Ethanol Oxidation, J. Solid State Electrochem., 2012, 16, 3147–3157, DOI:10.1007/s10008-012-1755-y.
- M. Tsuji, K. Uto, T. Nagami, A. Muto, H. Fukushima and J. Hayashi, Synthesis of Carbon-Supported Pt–YOx and PtY Nanoparticles with High Catalytic Activity for the Oxygen Reduction Reaction Using a Microwave-based Polyol Method, ChemCatChem, 2017, 9(6), 962–970, DOI:10.1002/cctc.201601479.
- P. Song, Y. Lei, X. Hu, C. Wang, J. Wang and Y. Tang, Rapid One-Step Synthesis of Carbon-Supported Platinum–Copper Nanoparticles with Enhanced Electrocatalytic Activity via Microwave-Assisted Heating, J. Colloid Interface Sci., 2020, 574, 421–429, DOI:10.1016/j.jcis.2020.04.041.
- M. Bonarowska, Z. Kaszkur, K. Matus, A. Drelinkiewicz, T. Szumelda and A. Kubas, Towards High Efficacy of Pd–Au/C Catalyst for Tetrachloromethane Hydrodechlorination, Chemistry, 2021, 3(1), 338–359, DOI:10.3390/chemistry3010025.
- P. Nekooi, M. Akbari and M. K. Amini, CoSe Nanoparticles Prepared by the Microwave-Assisted Polyol Method as an Alcohol and Formic Acid Tolerant Oxygen Reduction Catalyst, Int. J. Hydrogen Energy, 2010, 35(12), 6392–6398, DOI:10.1016/j.ijhydene.2010.03.134.
- L. Zhu, P. C. Wong, K. C. Wong, F. Ernst, K. A. R. Mitchell and S. A. Campbell, Synthesis, Characterization of a CoSe2 Catalyst for the Oxygen Reduction Reaction, Appl. Catal., A, 2010, 386(1–2), 157–165, DOI:10.1016/j.apcata.2010.07.048.
- H. Li, D. Gao and X. Cheng, Simple Microwave Preparation of High Activity Se-Rich CoSe2/C for Oxygen Reduction Reaction, Electrochim. Acta, 2014, 138, 232–239, DOI:10.1016/j.electacta.2014.06.065.
- Q. Zheng, X. Cheng and H. Li, Microwave Synthesis of High Activity FeSe2/C Catalyst toward Oxygen Reduction Reaction, Catalysts, 2015, 5(3), 1079–1091, DOI:10.3390/catal5031079.
- T. Ramulifho, K. I. Ozoemena, R. M. Modibedi, C. J. Jafta and M. K. Mathe, Fast Microwave-Assisted Solvothermal Synthesis of Metal Nanoparticles (Pd, Ni, Sn) Supported on Sulfonated MWCNTs: Pd-Based Bimetallic Catalysts for Ethanol Oxidation in Alkaline Medium, Electrochim. Acta, 2012, 59, 310–320, DOI:10.1016/j.electacta.2011.10.071.
- A. B. A. A. Nassr, I. Sinev, M.-M. Pohl, W. Grünert and M. Bron, Rapid Microwave-Assisted Polyol Reduction for the Preparation of Highly Active PtNi/CNT Electrocatalysts for Methanol Oxidation, ACS Catal., 2014, 4(8), 2449–2462, DOI:10.1021/cs401140g.
- M. Rahsepar and H. Kim, Microwave-Assisted Synthesis and Characterization of Bimetallic PtRu Alloy Nanoparticles Supported on Carbon Nanotubes, J. Alloys Compds., 2015, 649, 1323–1328, DOI:10.1016/j.jallcom.2015.07.224.
- M. G. H. Al-Tememy and Y. Devrim, Development of Effective Bimetallic Catalyst for High-Temperature PEM Fuel Cell to Improve CO Tolerance, Int. J. Energy Res., 2021, 45(2), 3343–3357, DOI:10.1002/er.6032.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Pd/Ag and Pd/Au Bimetallic Nanocatalysts on Mesoporous Silica for Plasmon-Mediated Enhanced Catalytic Activity Under Visible Light Irradiation, J. Mater. Chem. A, 2016, 4, 10142–10150, 10.1039/C6TA01664B.
- A. Tripathi, K. Faungnawakij, A. Laobuthee, S. Assabumrungrat and N. Laosiripojna, Catalytic Activity of Bimetallic Cu-Ag/MgO-SiO2 Toward the Conversion of Ethanol to 1,3-Butadiene, Int. J. Chem. React. Eng., 2016, 14, 945–954, DOI:10.1515/ijcre-2015-0164.
- T. T. Nguyen and E. W. Qian, Synthesis of Mesoporous Ti-Inserted SBA-15 and CoMo/Ti-SBA-15 Catalyst for Hydrodesulfurization and Hydrodearomatization, Microporous Mesoporous Mater., 2018, 265, 1–7, DOI:10.1016/j.micromeso.2018.01.026.
- A. S. Rini, S. Radiman and M. A. Yarmo, Microwave-assisted Synthesis of Ru–Sn/ZnO for Catalysis Application, AIP Conf. Proc., 2010, 1284, 129, DOI:10.1063/1.3515535.
- J. Tan, T. Thomas, J. Liu, L. Yang, L. Pan, R. Cao, H. Shen, J. Wang, J. Liu and M. Yang, Rapid Microwave-Assisted Preparation of High-Performance Bifunctional Ni3Fe/Co–N–C for Rechargeable Zn–Air Battery, Chem. Eng. J., 2020, 395, 125151, DOI:10.1016/j.cej.2020.125151.
- H. A. Elazab, S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, The Effect of Graphene on Catalytic Performance of Palladium Nanoparticles Decorated with Fe3O4, Co3O4, and Ni (OH)2: Potential Efficient Catalysts Used for Suzuki Cross—Coupling, Catal. Lett., 2017, 147, 1510–1522, DOI:10.1007/s10562-017-1990-z.
- H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Highly Efficient and Magnetically Recyclable Graphene-Supported Pd/Fe3O4 Nanoparticle Catalysts for Suzuki and Heck Cross-Coupling Reactions, Appl. Catal., A, 2015, 491, 58–69, DOI:10.1016/j.apcata.2014.11.033.
- N. R. Khalid, E. Ahmed, M. Ahmed, N. A. Niaz, M. Ramzan, M. Shakil, T. Iqbal and A. Majid, Microwave-Assisted Synthesis of Ag–TiO2/Graphene Composite for Hydrogen Production Under Visible Light Irradiation, Ceram. Int., 2016, 42(16), 18257–18263, DOI:10.1016/j.ceramint.2016.08.149.
- H. Pan, D. Wu, X. Huang, K. Xie, B. He, Z. Lu, P. Liu, J. Cheng, X. Zhao and J. Masa, Microwave-Assisted Synthesis of Co/CoOx Supported on Earth-Abundant Coal-Derived Carbon for Electrocatalysis of Oxygen Evolution, J. Electrochem. Soc., 2019, 166(8), F479–F486, DOI:10.1149/2.0281908jes.
- A. Galal, H. K. Hassan, N. F. Atta, A. M. Abdel-Mageed and T. Jacob, Synthesis, Structural and Morphological Characterizations of Nano-Ru-Based Perovskites/RGO Composites, Sci. Rep., 2019, 9, 7948, DOI:10.1038/s41598-019-43726-1.
- A. Y. Li, M. Kaushik, C.-J. Li and A. Moores, Microwave-Assisted Synthesis of Magnetic Carboxymethyl Cellulose-Embedded Ag–Fe3O4 Nanocatalysts for Selective Carbonyl Hydrogenation, ACS Sustainable Chem. Eng., 2016, 4(3), 965–973, DOI:10.1021/acssuschemeng.5b01048.
- B. Liu, X. Li, C. Zheng, X. Wang and R. Sun, Facile and Green Synthesis of Silver Nanoparticles in Quaternized Carboxymethyl Chitosan Solution, Nanotechnology, 2013, 24(23), 235601, DOI:10.1088/0957-4484/24/23/235601.
- S. Horikoshi, H. Abe, K. Torigoe, M. Abe and N. Serpone, Access to Small Size Distributions of Nanoparticles by Microwave-Assisted Synthesis. Formation of Agnanoparticles in Aqueous Carboxymethylcellulose Solutions in Batch and Continuous-Flow Reactors, Nanoscale, 2010, 2, 1441–1447, 10.1039/c0nr00141d.
- J. Jing, L. Li, W. Chu, Y. Wei and C. Jiang, Microwave-Assisted Synthesis of High Performance Copper-Based Catalysts for Hydrogen Production from Methanol Decomposition, Int. J. Hydrogen Energy, 2018, 43(27), 12059–12068, DOI:10.1016/j.ijhydene.2018.04.104.
- L. Rezaee and M. Haghighi, Citrate Complexation Microwave-Assisted Synthesis of Ce0.8Zr0.2O2 Nanocatalyst over Al2O3 Used in CO Oxidation for Hydrogen Purification: Influence of Composite Loading and Synthesis Method, RSC Adv., 2016, 6, 34055–34065, 10.1039/C6RA02973F.
- J. Zheng, Q. Gong, X. Cheng, S. Gong, W. Yang and L. Huang, Microwave Synthesis of Carbon-Supported Cobalt Nickel Selenide Ternary Catalyst Toward the Oxygen Reduction Reaction, ChemElectroChem, 2018, 5(14), 2019–2028, DOI:10.1002/celc.201800190.
- X. Hu, P. Song, X. Yang, C. Wang, J. Wang, Y. Tang, J. Zhang and Z. Mao, One-Step Microwave-Assisted Synthesis of Carbon-Supported Ternary Pt–Sn–Rh Alloy Nanoparticles for Fuel Cells, J. Taiwan Inst. Chem. Eng., 2020, 115, 272–278, DOI:10.1016/j.jtice.2020.10.008.
- C. Wu, Z. Guo, X. Chen and H. Liu, Cu/CeO2 as Efficient Low-Temperature CO Oxidation Catalysts: Effects of Morphological Structure and Cu Content, React. Kinet., Mech. Catal., 2020, 131, 691–706, DOI:10.1007/s11144-020-01870-0.
- Y. Li, Y. Cai, X. Xing, N. Chen, D. Deng and Y. Wang, Catalytic Activity for CO Oxidation of Cu–Ceo2 Composite Nanoparticles Synthesized by a Hydrothermal Method, Anal. Methods, 2015, 7, 3238–3245, 10.1039/C5AY00261C.
- F. Wang, Y. Xu, K. Zhao and D. He, Preparation of Palladium Supported on Ferric Oxide Nano-catalysts for Carbon Monoxide Oxidation in Low Temperature, Nano-Micro Lett., 2014, 6, 233–241, DOI:10.1007/BF03353787.
- G. Li, L. Li, Y. Yuan, Y. Yuan, Y. Li, W. Zhao and J. Shi, Mesostructured Pd/Mn3O4 Catalyst for Efficient Low-Temperature CO Oxidation Especially Under Moisture Condition, RSC Adv., 2014, 4, 35762–35768, 10.1039/C4RA01764A.
- G. Zhang, Z. Li, H. Zheng, T. Fu, Y. Ju and Y. Wang, Influence of the Surface Oxygenated Groups of Activated Carbon on Preparation of a Nano Cu/AC Catalyst and Heterogeneous Catalysis in the Oxidative Carbonylation of Methanol, Appl. Catal., B, 2015, 179, 95–105, DOI:10.1016/j.apcatb.2015.05.001.
- B. Yan, S. Huang, S. Wang and X. Ma, Catalytic Oxidative Carbonylation over Cu2O Nanoclusters Supported on Carbon Materials: The Role of the Carbon Support, ChemCatChem, 2014, 6(9), 2671–2679, DOI:10.1002/cctc.201402201.
- Q. Hua, T. Cao, X. K. Gu, J. Lu, Z. Jiang, X. Pan, L. Luo, W. X. Li and W. Huang, Crystal-Plane-Controlled Selectivity of Cu2O Catalysts in Propylene Oxidation with Molecular Oxygen, Angew. Chem., Int. Ed., 2014, 53(19), 4856–4861, DOI:10.1002/anie.201402374.
- A. Rey, M. Faraldos, J. A. Casas, J. A. Zazo, A. Bahamonde and J. J. Rodríguez, Catalytic Wet Peroxide Oxidation of Phenol Over Fe/AC Catalysts: Influence of Iron Precursor and Activated Carbon Surface, Appl. Catal., B, 2009, 86(1-2), 69–70, DOI:10.1016/j.apcatb.2008.07.023.
- J. Sun, M. Zheng, X. Wang, A. Wang, R. Cheng, T. Li and T. Zhang, Catalytic Performance of Activated Carbon Supported Tungsten Carbide for Hydrazine Decomposition, Catal. Lett., 2008, 123, 150–155, DOI:10.1007/s10562-008-9409-5.
- V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van Der Eycken, Reactions of Secondary Propargylamines with Heteroallenes for the Synthesis of Diverse Heterocycles, Chem. Soc. Rev., 2018, 47, 3861–3898, 10.1039/C7CS00065K.
- Z. Chang, X. Jing, C. He, X. Liu and C. Duan, Silver Clusters as Robust Nodes and π–Activation Sites for the Construction of Heterogeneous Catalysts for the Cycloaddition of Propargylamines, ACS Catal., 2018, 8(2), 1384–1391, DOI:10.1021/acscatal.7b02844.
- Z. Yan, Z. Wang, D. B. Bukur and D. W. Goodman, Fischer–Tropsch Synthesis on a Model Co/SiO2 Catalyst, J. Catal., 2009, 268(2), 196–200, DOI:10.1016/j.jcat.2009.09.015.
- M. Haruta, Catalysis of Gold Nanoparticles Deposited on Metal Oxides, CATTECH, 2002, 6, 102–115, DOI:10.1023/A:1020181423055.
- J. Y. Xi, W. P. Wang and G. X. Lu, Mechanism of Cu/ZnO Based Catalysts for Methanol Decomposition Promoted by Nickel, Acta Chim Sinica, 2002, 60(3), 419–426 CAS.
- W. H. Cheng, Reaction and XRD Studies on Cu Based Methanol Decomposition Catalysts: Role of Constituents and Development of High-Activity Multicomponent Catalysts, Acta Chim. Sin., 1995, 130(1), 13–30, DOI:10.1016/0926-860X(95)00102-6.
- S. Chen, M. Li, M. Gao, J. Jin, M. A. V. Spronsen, M. B. Salmeron and P. Yang, High-Performance Pt–Co Nanoframes for Fuel-Cell Electrocatalysis, Nano Lett., 2020, 20(3), 1974–1979, DOI:10.1021/acs.nanolett.9b05251.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Culllen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Hard-Magnet-L10-CoPt Nanoparticles Advance Fuell Cell Catalysis, Joule, 2019, 3(1), 124–135, DOI:10.1016/j.joule.2018.09.016.
- J. Ding, Q. Shao, Y. Feng and X. Huang, Ruthenium-Nickel Sandwiched Nanoplates for Efficient Water Splitting Electrocatalysis, Nano Energy, 2018, 47, 1–7, DOI:10.1016/j.nanoen.2018.02.017.
- Z. Yang, Y. Shi, X. Wang, G. Zhang and P. Cui, Boron As A Superior Activator for Pt Anode Catalyst in Direct Alcohol Fuel Cell, J. Power Sources, 2019, 431, 125–134, DOI:10.1016/j.jpowsour.2019.05.052.
- T. V. Reshetenko and J. St-Pierre, Effects of Propylene, Methyl Methacrylate and Isopropanol Poisoning on Spatial Performance of A Proton Exchange Membrane Fuel Cell, J. Power Sources, 2018, 378, 216–224, DOI:10.1016/j.jpowsour.2017.12.038.
- W. Wang, W. Wang and S. Chen, The Effects of Hydrogen Dilution, Carbon Monoxide Poisoning for a Pt–Ru Anode in a Proton Exchange Membrane Fuel Cell, Int. J. Hydrogen Energy, 2016, 41(45), 20680–20692, DOI:10.1016/j.ijhydene.2016.09.151.
- J. W. Sun, Y. S. Fu, G. Y. He, X. Q. Sun and X. Wang, Green Suzuki–Miyaura Coupling Reaction Catalyzed by Palladium Nanoparticles Supported on Graphitic Carbon Nitride, Appl. Catal., B, 2015, 165, 661–667, DOI:10.1016/j.apcatb.2014.10.072.
- H. A. Elazab, M. A. Gadalla, M. A. Sadek and T. T. El-Idreesy, Hydrothermal Synthesis of Graphene Supported Pd/Fe3O4 Nanoparticles as Efficient Magnetic Catalysts for Suzuki Cross – Coupling, Biointerface Res. Appl. Chem., 2019, 9, 3906–3911, DOI:10.33263/BRIAC92.906911.
- W. Fu, Y. Cao, Q. Feng, W. R. Smith, P. Dong, M. Ye and J. Shen, Pd–Co Nanoalloys Nested on Cuo Nanosheets for Efficient Electrocatalytic N2 Reduction and Room-Temperature Suzuki–Miyaura Coupling Reaction, Nanoscale, 2019, 11, 1379–1385, 10.1039/C8NR08724E.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00840d |
| This journal is © The Royal Society of Chemistry 2022 |