
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in the development of electrocatalysts for the electrochemical N2 reduction reaction
Kousik
Bhunia
 *a,
Sanjeev Kumar
Sharma
a,
Biraj Kanta
Satpathy
b and
Debabrata
Pradhan
*a
*a,
Sanjeev Kumar
Sharma
a,
Biraj Kanta
Satpathy
b and
Debabrata
Pradhan
*a
aMaterials Science Centre, Indian Institute of Technology Kharagpur, Kharagpur 721302, West Bengal, India. E-mail: kkousikmodabhunia@gmail.com; deb@matsc.iitkgp.ac.in
bSchool of Nanoscience and Technology, Indian Institute of Technology Kharagpur, Kharagpur 721302, West Bengal, India
First published on 25th November 2021
Abstract
Ammonia is the second-most produced chemical throughout the world to maintain the global food supply and other chemical stocks. The annual worldwide ammonia production is currently more than 200 million tons through the Haber–Bosch process, which consumes an enormous amount of energy due to the requirement of high pressure (>10 MPa) and relatively high temperature (400–500 °C). In recent years, electrochemical N2 reduction reaction (ENRR) under ambient conditions has received paramount attention in the scientific community. However, large-scale production of ammonia from the ENRR is limited by the lack of efficient cost-effective catalysts. The success of ENRR firmly depends on the efficiency of the electrocatalyst in a suitable electrolyte. However, identification and generation of the active sites in the electrocatalysts for ENRR remain elusive, impeding the development of the catalysts. In this review article, recent progress made in the development of efficient electrocatalysts for ENRR under ambient conditions is focused on with special attention on the physicochemical properties and active sites of the catalyst towards the NH3 production rate by considering experimental as well as theoretical aspects. This review elaborates on key aspects for the development of an efficient and stable electrocatalyst for NH3 production. In addition, the role of electrolytes and different sources of errors in the ENRR measurement for NH3 production are outlined briefly.
1. Introduction
Over the last few decades, the industrialization of populous developing countries has been demanding huge energy that primarily comes from fossil fuels leading to increased greenhouse gas emissions, not only resulting in depletion of non-renewable fossil fuel resources but also rapidly disturbing the climate. It has thus become an imperative objective of the scientific community to develop cleaner energy resources and technologies to mitigate fossil fuel use and maintain environmental sustainability. Ammonia (NH3) is the second-most manufactured synthetic chemical, which is extensively used as an agricultural fertilizer, chemical feedstock, and hydrogen carrier as well. For more than a century, the Haber–Bosch method has been the primary process of NH3 production worldwide using nitrogen (N2) and hydrogen (H2) gas as precursors. The N2 to NH3 conversion is a multi-step complex process, which requires a large amount of energy due to the high bond dissociation energy of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (940.95 kJ mol−1).1 In particular, the Haber–Bosch process requires temperature and pressure in the range of 350–550 °C and 150–350 atm, respectively, to convert N2 to NH3 (N2 + 3H2 = 2NH3). Most importantly, the H2 consumed for NH3 production is exclusively produced from steam reforming of natural gas (CH4 + 2H2O = 4H2 + CO2), which emits around 450 million metric tons of CO2 to the earth's atmosphere. Hence, it is one of the primary sources of environmental pollution and cause of rising global temperature. Therefore, an alternative energy-efficient and sustainable route for NH3 production without hampering its supply chain is urgently required to protect our environment. The electrochemical nitrogen reduction reaction (ENRR) at ambient temperature and pressure is one such highly promising alternative method that requires only N2 and H2O for NH3 production, making the process completely green if solar/renewable energy is used. Hence, a great amount of effort has been directed toward ENRR research in the past couple of years indicating the prospects of this method.
N bond (940.95 kJ mol−1).1 In particular, the Haber–Bosch process requires temperature and pressure in the range of 350–550 °C and 150–350 atm, respectively, to convert N2 to NH3 (N2 + 3H2 = 2NH3). Most importantly, the H2 consumed for NH3 production is exclusively produced from steam reforming of natural gas (CH4 + 2H2O = 4H2 + CO2), which emits around 450 million metric tons of CO2 to the earth's atmosphere. Hence, it is one of the primary sources of environmental pollution and cause of rising global temperature. Therefore, an alternative energy-efficient and sustainable route for NH3 production without hampering its supply chain is urgently required to protect our environment. The electrochemical nitrogen reduction reaction (ENRR) at ambient temperature and pressure is one such highly promising alternative method that requires only N2 and H2O for NH3 production, making the process completely green if solar/renewable energy is used. Hence, a great amount of effort has been directed toward ENRR research in the past couple of years indicating the prospects of this method.
In any chemical reaction, the catalyst plays a central role not only in increasing the yield and selectivity of the products but also in seamlessly driving the reaction with minimum inputs. Catalysts are thus the key and core ingredient for any electrocatalytic reaction. In heterogeneous catalysis, solid materials are majorly used as catalysts and thus their physicochemical properties have paramount importance to catalytic performance. The microstructure, especially steps, kinks, edge atoms, and surface electronic structures of the solid catalyst play significant roles in the efficiency and selectivity of the reaction. The literature reveals that a greater percentage of solid catalysts are noble and rare earth elements that have less abundance and are expensive. For those catalysts, support materials play an equally important role in reducing their amount for use in catalytic reactions while enhancing the overall performance. In particular, the support material controls the catalyst distribution and nucleation, and protects the nanostructure-based catalysts from agglomeration.
The morphology and composition of the catalysts play primary roles in the catalytic reactions including ENRR. The suitably designed surface controls the active sites for the adsorption/desorption of the reactive species while composition determines the surface oxidation state and electronic structure. Therefore, the design of efficient electrocatalysts is highly desirable for ENRR. Electrocatalysts for ENRR can be categorically placed in three groups: (i) metallic catalysts, (ii) metal-based catalysts including oxides, carbides, phosphides, and nitrides, and (iii) metal-free electrocatalysts. Understanding the reaction mechanism at these catalyst surfaces is important for developing and increasing their efficiency. For example, competitive hydrogen evolution reaction (HER) is an obstacle associated with ENRR, which leads to a decrease in its Faradaic efficiency (FE). The suppression of HER and the enhancement of the ENRR process are thus obvious fundamental objectives in the development of ENRR catalysts. So far, noble metal-based electrocatalysts such as Au,2 Pd,3 and Ru4 have shown promising results towards ENRR. Similarly, non-noble metal-based electrocatalysts have also been explored. As compared to a sole electrocatalyst, a nanostructured electrocatalyst integrated with a suitable support material shows better activity.5 The high electrocatalytic activity arises from the synergistic effect between the support and the catalyst, which mitigates the competitive HER. Therefore, the rational design of stable, active, and cost-effective catalysts to obtain a high yield rate and selectivity of NH3 is the prime requirement for ENRR.
1.1 ENRR versus HER
To synthesize highly efficient ENRR electrocatalysts, it is important to understand the challenge associated with the ENRR kinetics at the catalyst surface. The electrochemical heterogeneous conversion process of N2 to NH3 at room temperature occurs via multiple proton-coupled electron transfer (PCET) reactions. In particular, electrochemical conversion requires six protons and six electrons for one N2 molecule to produce NH3 as per the following equations:6,7| N2 (g) + 2H2O (l) + 6H+ (aq.) + 6e− = 2NH3 E0 = 0.148 V vs. RHE (pH = 0) |
| N2 (g) + 6H2O (l) + 6e− = 2NH3 + 6OH− E0 = −0.763 V vs. SHE (pH = 14). |
On the other hand, the competitive HER reaction requires only two electrons to produce one H2 molecule. In addition, the potential required for HER is much closer (or lower than) that of the ENRR. Thus HER becomes a complementary reaction at the cathode for ENRR, which leads to poor selectivity towards NH3 production. The HER at the cathode occurs as per the following equations:8
| 2H+ (aq.) + 2e− = H2 (g) E0 = 0.00 V vs. NHE in an acidic electrolyte (pH = 0) |
| 2H2O + 2e− = H2 (g) + 2OH− E0 = −0.828 V vs. SHE in an alkaline electrolyte (pH = 14) |
As ENRR proceeds via multiple electron and proton transfer processes, various intermediates are formed as presented in Fig. 1. Even though ENRR appears to be very favourable for NH3 production as per the standard reduction potential chart, the achieved FE for ENRR remains mostly below 10%, which is far lower than the expected results. This is believed to be due to the kinetically preferred HER, which requires only two electrons whereas ENRR proceeds through a multi-step six electron process. This competing HER thus leads to a comprehensive FE loss for ENRR. The performance of ENRR further depends significantly on the electrolyte medium and its pH. Therefore, elimination of the unwanted HER from the electrolyte reduction becomes the most concerning issue for ENRR as it is difficult to avoid the protic medium. Furthermore, ENRR is highly affected by the mass transport of N2. The low solubility of N2 in water, i.e., only about 2 vol%, is another hurdle for ENRR. Therefore, the choice of electrolyte is another important factor to be considered in which ENRR should be preferred over HER.
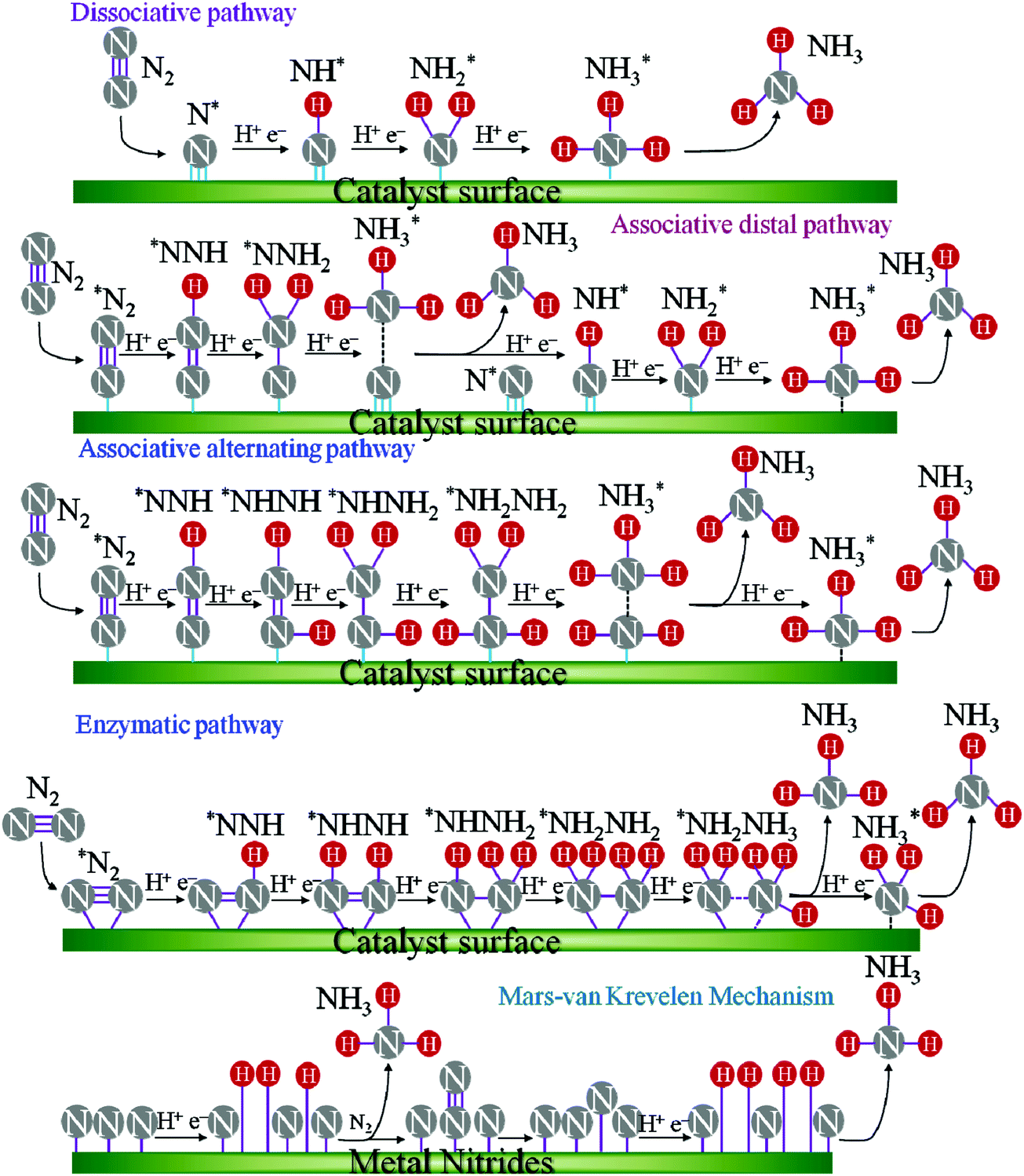 | ||
| Fig. 1 The possible reaction mechanisms of ENRR. * represents an adsorption site. | ||
1.2 ENRR mechanisms
ENRR over a heterogeneous catalyst follows two distinct pathways, named dissociative and associative pathways, with different intermediates and rate-limiting steps in terms of free energy.9–11 According to the dissociative pathway (Fig. 1),11 the adsorbed N2 molecule undergoes dissociation at the catalyst surface followed by hydrogenation of the adsorbed N atom to form NH3. However, because of the high binding energy of the NN bond, the dissociative path requires more energy. On the other hand, the associative pathway is subdivided into two pathways: (i) associative distal pathway and (ii) associative alternating pathway. The ENRR mechanism usually follows the associative distal pathway or alternating pathway and/or both as shown in Fig. 1. The density functional theory (DFT) calculation reveals that the associative mechanism during the ENRR is governed by the specific active site of the catalyst.12 The active site controls the adsorption of different active intermediate species during the progression of the reaction. In the distal pathway, the end atoms are attached to the active site, where the terminal atoms get hydrogenated by the PCET process through the formation of *NNH and *NNH2 intermediates, and then desorb from the catalyst surface as NH3. In the alternating pathway, both the terminal and the end atoms are hydrogenated simultaneously by a PCET through the formation of *NNH, *NHNH, and *NH2NH2 intermediates and desorb from the catalyst surface as an NH3 molecule consequently. Apart from that, in nature, nitrogenase enzymes perform biological N2 fixation under ambient conditions, where active sites have Fe and S along with Mo atoms, and the detailed mechanism of it remains elusive.13 Nevertheless, both enzymatic and ENRR mechanisms require six protons and six electrons for the production of NH3, which is the common point in all the reaction mechanisms.13 The ENRR mechanism was theoretically studied in an acid medium over transition metal surfaces by Nørskov and co-workers.14 The free energy change for the N2ads molecule and Nads atoms towards catalytic NH3 conversion was calculated. Fig. 2 shows the volcano diagram of N2 reduction reaction considering a Heyrovsky-type reaction, which includes both dissociative (solid lines) and associative (dashed lines) mechanisms on both flat (black) and stepped (red) surfaces. The H-bonding effect is presented without (solid lines) and with (dotted lines). The DFT calculated negative free energy (−ΔG) values are presented with data points for a given reaction step. The right-hand side of the volcano plot represents the first proton transfer step, i.e., N2(g) + H+ + e− → *N2H, indicating the activity of the metals for the associative mechanism whereas the N2 splitting is the rate-determining step for the dissociative mechanism. The metals on the left side have the same rate-determining steps for both the mechanisms (associative and dissociative). The x-axis represents the binding energy of N-adatoms. The large grey area inside the plot suggests the surface that is more favourable to be covered with H-adatoms. Mo, Fe, Rh, and Ru can be found at the top of the volcano plot suggesting their higher activity for ENRR. However, the surfaces of these catalysts are also predicted to be active sites for HER thereby leading to low FE for ENRR.
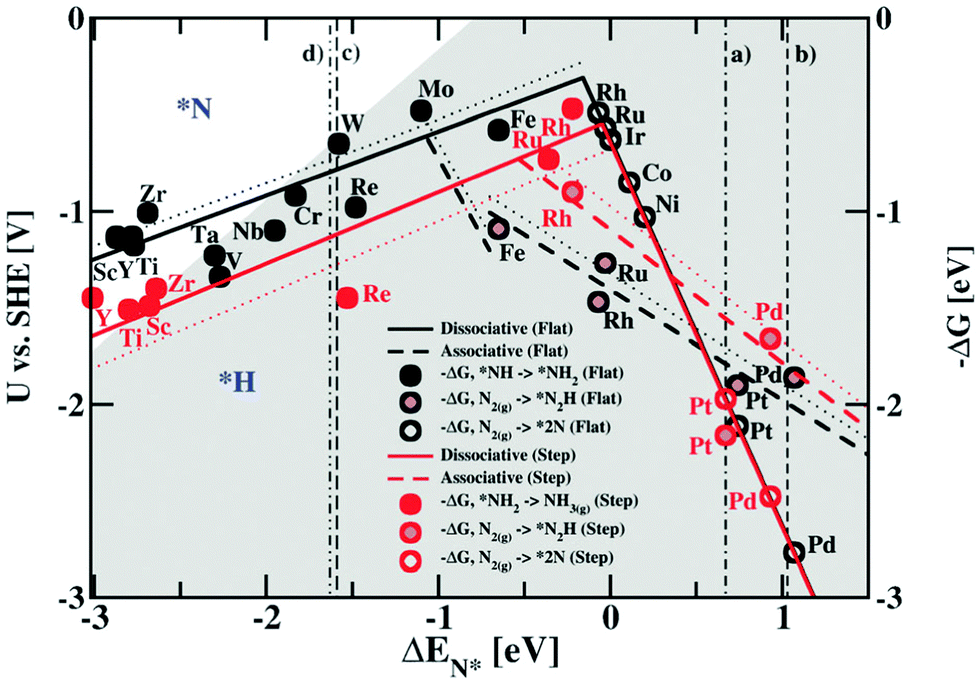 | ||
| Fig. 2 Combined volcano diagrams (lines) for the flat (black) and stepped (red) transition metal surfaces for reduction of nitrogen with a Heyrovsky type reaction, without (solid lines) and with (dotted lines) the H-bond effect. Reproduced with permission from ref. 14. Copyright 2012, RSC. | ||
A completely different pathway named the Mars–van Krevelen (MvK) mechanism is proposed for transition metal nitride (TMN) catalysts which require relatively small overpotentials.15 Different from the associative and dissociative pathways, here lattice N atoms are hydrogenated to produce NH3 from the surface of the TMNs and thus creating N vacancies. Subsequently, these N vacancies react with the dissolved N2 and generate a second NH3 molecule. Recently, Nash et al. experimentally proved the MvK mechanism over the Cr2N catalyst.16 Detailed bulk and surface analyses using XRD and XPS, respectively, reveal that the bulk of the catalyst is a pure Cr2N phase whereas the surface possesses CrN, CrNxOy, and CrOx along with Cr2N. The bulk CrN phase exhibits negligible ENRR activity compared with Cr2N indicating that the latter is the active phase for ENRR. The isotopic batch cell study using 15N2 indicates the formation of 14NH3 and 15NH3 as identified from NMR analysis, which suggests active participation of surface N of Cr2N to activate dinitrogen through the MvK mechanism as presented in Fig. 1. The authors also identified two possible reasons for the deactivation of the catalyst. Those are leaching of surface N from Cr2N at lower potentials (<−0.4 V) and silent conversion of the active Cr2N to the inactive CrN at −0.2 V. Using advanced instrumental tools such as surface-enhanced infrared absorption spectroscopy (SEIRAS) and differential electrochemical mass spectrometry (DEMS) Yao et al. studied ENRR on a Rh catalyst.17 The authors identified the formation of N2Hx (0 ≤ x ≤ 2) species during the progression of ENRR with an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode at ∼2020 cm−1 from SEIRAS analysis and the DEMS signal at m/z = 29. The study proposed a new two-step ENRR reaction pathway on the Rh surface, in which the ENRR involved two-electron transfer to N2 to form N2H2 first and subsequent decomposition to NH3.
N stretching mode at ∼2020 cm−1 from SEIRAS analysis and the DEMS signal at m/z = 29. The study proposed a new two-step ENRR reaction pathway on the Rh surface, in which the ENRR involved two-electron transfer to N2 to form N2H2 first and subsequent decomposition to NH3.
1.3 Performance evaluation of electrocatalysts for NRR
The yield rate and Faradaic efficiency (FE) are the important indicators to define the catalytic conversion efficiency of an electrocatalyst. Therefore, it is important to describe the procedure for yield rate and FE calculation along with the factors that affect them. In an ideal condition, FE is supposed to be one or 100%. However, practically obtained FE for ENRR is much lower than the theoretical value. The Faradaic loss is a prominent issue, which is associated with the catalyst. Two major factors that strongly influence the FE of ENRR are applied potential and competitive HER. The ENRR at a lower potential leads to a higher FE. However, a higher reduction potential facilitates HER at the catalyst surface and thus loss of FE for ENRR. Another important point is the selectivity of ENRR. So far only two nitrogen-containing products are identified from ENRR, i.e., NH3 and N2H4. The product selectivity of ENRR is another criterion to obtain high FE for NH3 conversion.The yield rate is usually calculated from the salicylate method. The concentration of ammonia and hydrazine produced from ENRR is estimated from the calibration curve obtained from the UV-Vis absorption data. The ammonia yield rate is estimated by using the following equation:18
| r[NH3] = ([NH3] × V)/(t × A) |
The hydrazine yield is estimated from the following equation:
| r[N2H4] = ([N2H4] × V)/(t × A) |
Taking into account that three electrons are required for one ammonia molecule synthesis and four electrons for one N2H4 from N2, FE is calculated from the following equation:
| FENH3 = (3 × F × [NH3] × 10−6 × V)/(17 × Q) + (4 × F × [N2H4] × 10−6 × V)/(32 × Q) |
2. Noble metal catalysts
2.1 Platinum (Pt)
Among the different noble metals, Pt and Pt-based materials are studied in almost every branch of catalyst science due to their high intrinsic conversion efficiency. Especially in fuel cell technology, Pt is considered as the benchmark catalyst due to its inherent HER activity, with HER being a competitive reaction for ENRR. As a result, Pt shows very poor FE towards ENRR as evident from the experimental results.19,20 A few reports on Pt-based materials as electrocatalysts reveal their poor selectivity towards ENRR and very high activity towards HER.19 For example, Lan et al. studied ENRR using commercial Pt/C in a 0.5 M H2SO4 electrolyte in a membrane electrode based assembly, where a mixed NH4+/H+ pre-exchanged Nafion membrane was used. The report shows a maximum NH3 yield rate of 1.14 × 10−5 mol m−2 s−1 with a FE of ∼0.52% only at a cell voltage of 1.6 V.19 Later on, Lan and Tao modified the experimental setup using a 0.1 M Li2SO4 electrolyte with a mixed Li+/NH4+/H+ Nafion membrane and achieved an NH3 yield rate of 9.37 × 10−6 mol m−2 s−1 with an improved FE of 0.83% at 80 °C temperature.202.2 Other noble metals (Rh and Ir)
Rhodium (Rh) and iridium (Ir) have been explored for ENRR. Like Pt, Ir is an important catalyst for the electrochemical water-splitting reaction. Ir has very limited performance towards ENRR due to its dominating HER activity over ENRR activity.21,22 Recently, Sheet and Botte reported an Ir/C loaded gas-diffusion electrode as the cathode for ENRR and implemented it in a PEM-type electrochemical cell using a strong alkaline polymer-based gel electrolyte developed with a combination of polyacrylic acid and 6 M KOH. The best performance of Ir/C was achieved with an NH3 yield of 2.763 × 10−11 mol cm−2 s−1 and a FE of 0.108% at 60 °C temperature and 0.25 V cell voltage.22 Rh, another noble metal, has also been tested in the search for an efficient catalyst towards ENRR for NH3 production.14,23 Liu et al. reported a surfactant-free approach to synthesize atomic-level ultrathin two-dimensional Rh nanosheet (1 nm thick) nanoassemblies (Rh NNs) as an electrocatalyst for ENRR from the inorganic cyanogel polymer precursor RhCl3–K3Co(CN)6.24 The Rh NNs exhibited an impressively high NH3 yield rate of 23.88 μg h−1 mgcat−1 in a 0.1 M KOH electrolyte at −0.2 V. The as-synthesized electrocatalyst showed good selectivity (no hydrazine detected) and higher NH3 yield rate compared to Rh nanoparticles (11.45 μg h−1 mgcat−1). However, the FE was found to be only 0.217% at −0.2 V due to vigorous hydrogen generation. Chen et al. fabricated unusual 3D Rh particles with nanowires as the subunits using a reactive ionic liquid.25n-Octylammonium formate was used as a reaction medium, reducing agent, and template simultaneously for the successful construction of 3D Rh particles. Under ambient conditions, the as-prepared 3D Rh particles exhibited an excellent activity for ENRR with a high NH3 yield of 35.58 μg h−1 mgcat.−1 at −0.2 V and a FE of only 0.52% in 0.1 M KOH.2.3 Gold (Au)
Au-based nanostructures are extensively explored in the field of nanoscience and nanotechnology.26 Due to their specific electronic, optical, plasmonic, and catalytic properties, Au and Au-based materials are extensively studied in the field of biosensors, optoelectronics, and heterogeneous catalysis.26,27 The intrinsic catalytic as well as the electronic properties of the catalyst can be manipulated by tuning the microstructure shape, size, and surrounding environment of the Au nanostructures.28,29 The theoretical study revealed that the ENRR on the Au surface follows associative mechanisms that precede adsorption of the N2 molecule at the electrode surface followed by hydrogenation of the N atoms. The success of the Au-based electrocatalysts towards ENRR is due to the strong N2 adsorption ability of the Au surface composed of multifaceted facets with various steps and/or nanoclustering that facilitate the adsorption and reduction of N2.30,31 Wang et al. synthesized flower-like Au nanostructures for ENRR with an NH3 yield rate of 25.57 μg h−1 mgcat−1 and a FE 6.05% at −0.2 V (Table 1).32 Zhang et al. correlated the coordination of the surface gold atoms with ENRR.33 They found that the nanoporous Au film exhibits 5.8 times higher ENRR activity than the {111} facet exposed octahedral Au nanocrystal with an NH3 production rate of 30.5 μg h−1 mg−1. The higher ENRR activity of the nanoporous Au film was further theoretically confirmed by the binding energy of the intermediate species which depends on the coordination number of the surface atoms. Unlike Au (111) surface, the Au film favours the adsorption of the *NNH intermediate species due to low corrdination number Au atoms and thus facilitates the NH3 production. Later on, Chen et al. reported a size-dependent ENRR rate over Au NPs.34 Au NPs of 8 nm size among the as-synthesized Au NPs in the range of 4 nm to 10 nm showed the highest ENRR activity with FE and NH3 production rate at 5.79% and 17.49 μg h−1 mgAu−1, respectively (Fig. 3). The theoretical analysis revealed that the 8 nm size Au NPs have an optimum number of surface edge sites to suppress the HER and hence facilitate the ENRR. The rate-limiting step for NH3 production was reported to be the formation of *NNH from *NN.34 The free energy calculation revealed that Au(211) has the potential active site for ENRR.34 The density of state of different Au models suggested that the 5d band of the Au(211) facet has a higher tendency to overlap with the 2p orbital of Nads in *NNH than that of the Au(111) facet. Such stronger binding stabilizes the intermediate species and hence increases the NH3 production rate.34| Catalyst | Electrolyte | Temperature | Yield rate (μg h−1 mgcat.−1) | FE (%) | Potentials vs. RHE (V) | Ref. |
|---|---|---|---|---|---|---|
| Au | LiClO4 | 20 °C | 3.9 (μg cm−2 h−1) | 30.2 | −0.4 | 35 |
| Au/Ni | Ambient | 7.4 | 60 | −0.14 | 42 | |
| Au | HCl | Ambient | 25.57 | 6.05 | −0.20 | 32 |
| npPdH | 0.1 M PBS | Ambient | 20.4 | 43.6 | −0.15 | 56 |
| B | Na2SO4 | Ambient | 13.22 | 4.04 | −0.80 | 81 |
| Ru SA/N–C | H2SO4 | 120.9 | 29.6 | −0.2 | 46 | |
| Rh | KOH | Ambient | 23.88 | 0.217 | −0.2 | 24 |
| Mo | KOH | Room temp. | 34.0 | 14.6 | −0.3 | 82 |
| Cu | HCl | Ambient | 25.63 | 15.12 | −0.40 | 83 |
| Au1Cu1 | H2SO4 | Ambient | 154.91 | 54.96 | −0.2 | 84 |
| Ag3Cu | Na2SO4 | Ambient | 24.59 | 13.28 | −0.5 | 79 |
| RhCu | KOH | Ambient | 95.06 | 1.5 | −0.2 | 85 |
| Pt6Cu | Na2SO4 | Ambient | 16.5 | 6.15 | −0.2 | 86 |
| Pt3Fe | KOH | Ambient | 18.3 | 7.3 | −0.05 | 87 |
| a-FeB2 | LiClO4 | Ambient | 39.8 (−0.3 V) | 16.7 | −0.2 | 88 |
| Rh2Sb | Na2SO4 | Room temp. | 228.85 | 1.54 | −0.45 | 89 |
| Pd | PBS | Ambient | 4.5 | 8.2 | 0.1 | 53 |
| Pd1Ag1 | KOH | Ambient | 24.1 | ∼1.15 | −0.2 | 90 |
| Pd3Cu1 | KOH | Ambient | 39.9 | 1.22 | −0.25 | 78 |
| PdCuIr | Na2SO4 | Ambient | 13.43 | 5.29 | −0.30 | 91 |
| Pd3Pb | Na2SO4 | Ambient | 18.2 | 21.46 | −0.2 | 92 |
| PdRu | HCl | Ambient | 25.92 | 1.53 | −0.1 | 93 |
| PdRu | KOH | Ambient | 37.23 | 1.85 | −0.2 | 77 |
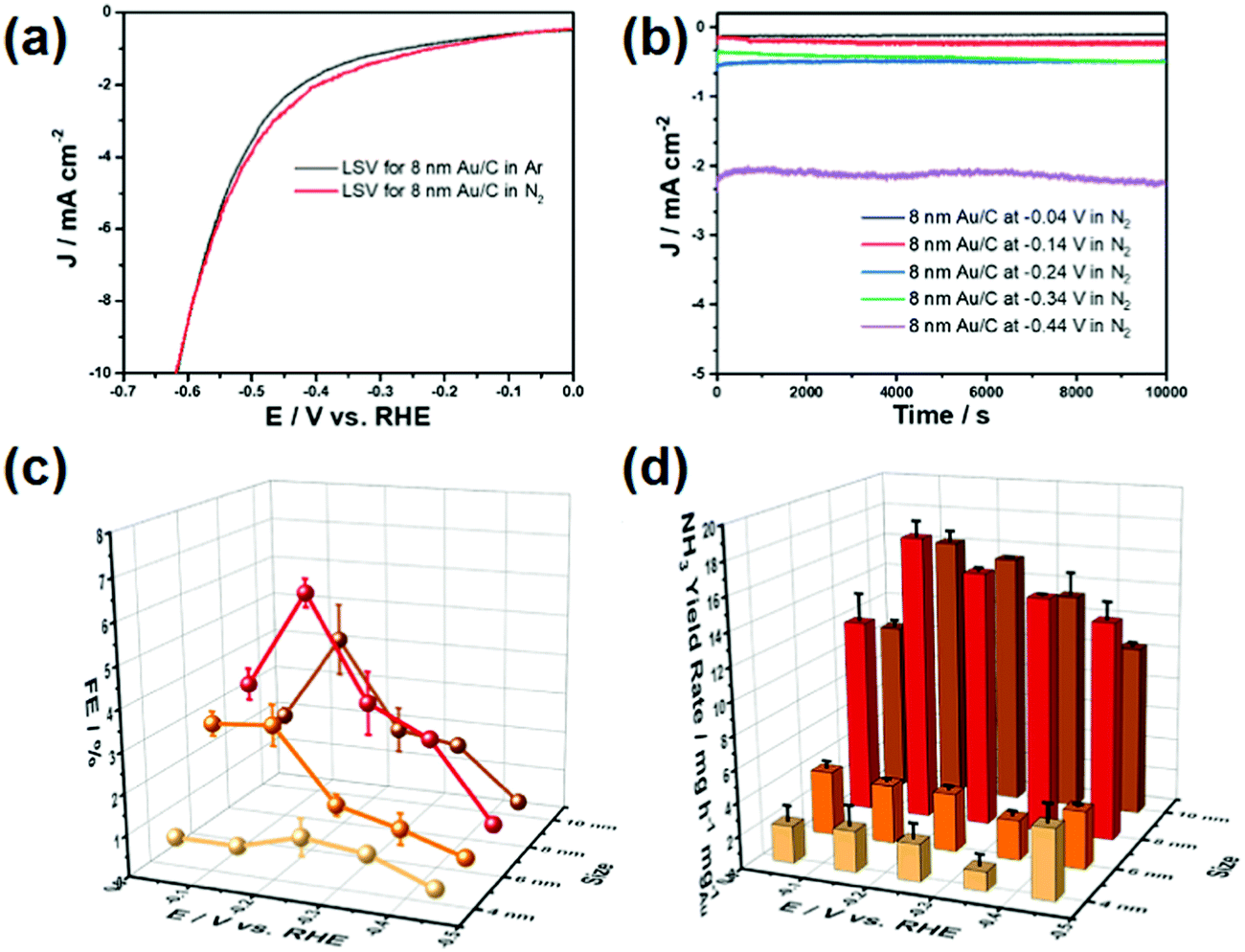 | ||
| Fig. 3 (a) LSV curves of 8 nm Au/C. (b) Chronoamperometric curves of the 8 nm Au/C catalyst at different potentials. (c) Faradaic efficiency of the Au/C catalyst with different Au particle sizes. (d) Ammonia yield of the Au/C catalyst with different Au particle sizes. The error bar represents standard deviations of the results from three independent experiments. (Error bars in the figures represent standard deviations determined from three individual experiments.) Reproduced with permission from ref. 34. Copyright 2020, Elsevier. | ||
Nazemi et al. synthesized hollow Au nanocages to enhance ENRR under ambient conditions.35 The hollow Au nanocages showed a FE of 30.2% at −0.4 V (vs. RHE) with an NH3 yield of 3.9 μg cm−2 h−1 at −0.5 V (Table 1). The FE increased from 30.2% to 40.5% with the temperature increasing from 20 °C to 50 °C at −0.4 V indicating the role of temperature. The increase in FE with temperature is due to the high mass transportation rate at higher temperatures even though the solubility of N2 decreases. The hollow cages offer a large surface area for the reactant molecules than a solid one leading to three times more ENRR activity compared to the latter one. This suggests the urgency to develop an advanced microstructure for ENRR. In addition, a suitable support material for catalysts plays an important role. Support materials not only increase the stability of the catalyst but also facilitate adsorption and diffusion of the reactant molecules.36 Qin et al. synthesized single Au atom supported hierarchical N-doped porous carbon to enhance ENRR. The N-doped porous carbon (NDPC) supported Au catalyst showed a FE of 12.3% with an NH3 yield of 2.32 μg h−1 cm−2 at −0.2 V. Both the NDPCs and Au single-atom sites participated in ENRR under ambient conditions. The metal-free NDPCs reached a FE of only 2.1% at a potential of −0.6 V further highlighting the significance of Au-NDPC heterojunction catalyst synthesis. The highly porous architecture of NDPCs provided more active sites and enhanced mass transportation during the ENRR process. The N and C sites stabilized the single atomic Au catalyst and retained the durability of the catalyst. The catalyst also selectively yielded NH3 without the signature of N2H4. Li et al. reported amorphous Au NPs supported by the CeOx–RGO hybrid (a-Au/CeOx–RGO) for ENRR under ambient conditions.2 Amorphous Au has higher chemical reactivity than the crystalline nanostructure towards small molecules. The a-Au/CeOx–RGO showed a FE of 10.10% with an NH3 yield of 8.3 μg h−1 mgcat−1 at −0.2 V, which is much higher than its crystalline counterpart (c-Au/RGO, FE: 3.67%, NH3 yield: 3.5 μg h−1 mgcat−1). The role of CeOx was to increase the amorphous nature of Au NPs leading to increased catalyst performance. The surface oxidation state (Mδ+) of metal catalysts is another factor that plays a significant role in the electrochemical reaction by the redistribution of the surface electronic structure to enhance the adsorption ability of the catalyst towards reactant and radical species.37–39 Zheng et al. introduced the CoOx layer to manipulate the local electronic structures of Au NPs with a positive valence site (Au+) to enhance the NH3 production rate.37 By using the vapour deposition method, small-sized Au and Co islands were homogeneously deposited onto the Si surface, followed by fast annealing at 800 °C forming a Au NPs/CoOx layer (Au/CoOx) (Fig. 4a). The CoOx layer created Au+ active sites due to the strong charge exchange between them.40 High-resolution transmission electron microscopy (HRTEM) images (Fig. 4b) indicated that the Au NPs had penetrated into the CoOx layer on the Si substrate. The cross-section analysis of the Au/CoOx sample (Fig. 4c) using scanning transmission electron microscopy (STEM) coupled with EDS mapping revealed the intimate contact between Au and CoOx. Elemental mapping showed the thin CoOx layer surrounding the bottom of the Au NPs while the top portion of the Au NPs was directly exposed to the external environment. Quasi in situ XPS after 10 h electrolysis showed a positive chemical shift of about 0.4 and 0.2 eV compared to the original Au/CoOx sample. The down-field shift of the f-band centre of Au suggests the hybridized band formation through a strong electronic effect by Au NPs within an atomic dimension during ENRR.41 The as-synthesized catalyst with the highest average oxidation state (ca. 40%) achieved an NH3 yield of 15.1 μg cm−2 h−1 and a FE of 19% at −0.5 V. On the other hand, the Au catalyst showed much lower NH3 yield and FE in the range of 2.0–6.1 μg cm−2 h−1 and 0.4–5.4%, respectively, in the potential range of −0.3 to −0.7 V.
 | ||
| Fig. 4 Synthesis and structural characterization of Au-based samples. (a) Schematic of the vapour deposition and annealing to fabricate the Au-based samples. (b) Cross-sectional TEM and HRTEM images of the Au/CoOx sample. Inset: Magnified image of the specified area by HRTEM. High-magnification images on the right match the marked area in the inset. (c) STEM image with the corresponding elemental distribution maps for the Au/CoOx sample. Reproduced with permission from ref. 37. Copyright 2019, Wiley-VCH. | ||
Unlike the Au NP and oxide combination, Xue et al. reported the Ni and Au NP combination by a galvanic replacement method where Au was deposited over Ni for ENRR.42 The atomic ratio of the Au–Ni catalyst was tuned by controlling the Au3+ precursor concentration. The pristine Ni catalyst produced a negligible amount of NH3. The NH3 yield rate significantly increased after the incorporation of Au. The highest NH3 yield of 7.4 μg h−1 mgcat−1 and FE of 67.8% at −0.14 V were obtained with Au6/Ni. In this metallic couple, Ni acts as an electron donor and enriches the Au surface electron density by a combination of support and size effects. XPS analysis provided clear evidence for the gradual shift of the Au 4f spectra towards lower energy binding, whereas Ni 2p shifted towards higher binding energy confirming the acceptor behaviour of the former. Moreover, the optimal Ni–Au donor–acceptor pair enhanced the pre-adsorption and activation of N2 and the desorption of NH3 from the catalyst surface.
2.4 Ruthenium (Ru)
Ruthenium (Ru), known for its use in the Haber–Bosch process, is another example of an efficient catalyst for ENRR.43 The theoretical analysis revealed favourable nitrogen adsorption energy leading to a lower ENRR overpotential in both associative and dissociative mechanisms compared to that of Pt and Pd electrocatalysts.14 Moreover, Ru is placed almost at the top of the volcano plot (Fig. 2) suggesting it to be one of the most active catalysts for ENRR. Thus, Ru-based nanostructured catalysts have been explored in recent years for ENRR. The fundamental role of the Ru catalyst towards the ENRR mechanism is highlighted here based on experimental and theoretical findings. Back et al. studied the mechanism of ENRR through both associative and dissociative pathways and compared with the energy state of Ru theoretically.44 The calculated free energy diagram revealed that the kinetically facile intermediate of the dissociative pathway required a thermodynamic limiting potential of −0.71 V, which is comparable to the associative pathway (−0.68 V). The study highlighted that both associative and dissociative reaction pathways are probable during the ENRR. Previously, it was believed that the associative pathway is energetically more feasible than the dissociative pathway because of the high energy requirement to dissociate the NN bond (9.79 eV).45 In addition, NH3 formation is energetically more favourable than N2H4 formation making Ru nanostructures more selective for NH3 synthesis. However, the competitive HER prohibits the ENRR by blocking the active sites due to the energetically preferential adsorption of *H.14 Geng et al. developed Ru single atoms on nitrogen-doped carbon (Ru SAs/N–C), which showed very high activity toward ENRR.46 At −0.2 V, Ru SAs/N–C attained a FE of 29.6% with a partial current density of −0.13 mA cm−2 and a yield rate of 120.9 μgNH3 mgcat−1 h−1. The high electrocatalytic activity of Ru SAs/N–C arises from the strong chemical bond between Ru and N2 as confirmed by the N2-temperature program desorption analysis. Zhang et al. demonstrated the synthesis of Ru dispersed N-doped carbon by the carbonization of Ru dispersed ZIF-8. The catalyst showed a maximum NH3 yield rate at about 16.68 μgNH3 h−1 mgcat−1 at −0.4 V and a FE of 14.23% at −0.3 V for the 16 wt% Ru loaded catalyst.47 The N-doped carbon frameworks not only ensured uniform dispersion of the Ru NPs but also protected the catalyst from dissolution during electrocatalysis. The residual hydrophobic nature of ZIF-8 remained in the carbon matrix which prohibited competitive HER. Furthermore, among the different N-doped carbons, the pyridinic N-type carbon moiety facilitated the adsorption of N2 in which Ru atoms accelerated dissociation.
Yao et al. studied the ENRR kinetics on a Ru thin film using a combination of surface-enhanced infrared absorption spectroscopy and electrochemical measurements.48Fig. 5a shows the voltammetry plots of the Ru thin film performed in a N2 and Ar-saturated HClO4 electrolyte. The cathodic scan reveals the onset of NRR and HER at 0 V and an increase in reduction current with decreasing potential. It is worth noting that the NRR current (solid red line) is slightly lower than the HER current (solid black line), suggesting that some active species adsorbed on the Ru surface get oxidized in the backward scan and thus showing a small peak between −0.1 and 0 V. To identify the probable intermediate, surface-enhanced infrared absorption spectroscopy was used during the ENRR measurements. The *N2Hx (0 ≤ x ≤ 2) species was identified and the stability of the species was further studied. The *N2Hx species was detected at a potential below 0.2 V in an N2 saturated HClO4 solution (Fig. 5b). The NN stretching band at 1940 cm−1 was observed at −0.1 V and remained constant above 0.1 V. The NN stretching band intensity was accompanied by the oxidation current in between the potential of −0.1 and 0 V during the positive scan (Fig. 5a). The oxidation current is believed to arise from the electrochemical oxidation of the *N2Hx species. The results (Fig. 5a and b) reveal that the oxidation current (dashed line) below 0 V potential arises from the oxidation of the *N2Hx species whereas in the potential range of 0 and 0.4 V, the oxidation of *N2Hx and Hads surface species occurred. They also studied the ENRR kinetics in alkaline electrolytes and revealed that the N-related signal at the electrode surface is very weak in an alkaline medium. This observation highlights the feasibility of the Ru-based catalyst in an alkaline medium. Recently, Ru NP loaded Ti3C2 MXene (Ru@MXene) was tested for ENRR which showed an NH3 yield of 2.3 μmol h−1 cm−2 at −0.4 V with a FE of 13.13%.49
 | ||
| Fig. 5 (a) Cyclic voltammograms of a Ru film electrode deposited on the Si prism in Ar- (black line) and N2-saturated (red line) 0.1 M HClO4 aqueous solutions. An Ag/AgCl electrode was used as the reference. Potential scan rate: 2.5 mV s−1. (b) FTIR spectra recorded during the potential scan from −0.2 to 0.4 V on the Ru film electrode in an N2-saturated 0.1 M HClO4 solution. The reference spectrum was taken at −0.4 V. Reproduced with the permission of ref. 48. Copyright 2019 ACS. | ||
2.5 Palladium (Pd)
The Pd-based materials are widely explored as electrocatalysts in the field of the fuel cell, especially for ORR,50 OER,51 and HER,52 as well as anode catalysts for alcohol electrooxidation reaction. Pd has thus been studied as an electrocatalyst for ENRR. Wang et al. reported carbon-supported Pd NPs (Pd/C) for ENRR under ambient conditions.53 Under a potential, Pd can form Pd hydride, which promotes the surface hydrogenation reaction. The Pd/C catalyst showed an NH3 yield rate of around 4.5 μg mgPd−1 h−1 and a high FE of 8.2% at 0.1 V. The Pd/C catalyst showed higher catalytic activity than Pt/C and Au/C with similar wt% mass loading. Interestingly, the HER activity of Pd was effectively suppressed in the neutral PBS electrolyte and N2 hydrogenation followed the Grotthuss-like hydride transfer mechanism. Free energy calculation suggested that the *N2 to *N2H hydrogenation is the rate-limiting step for ENRR. Lv et al. reported a PdO/Pd heterojunction for ENRR.54 The desired compositions of PdO/Pd decorated carbon nanotubes (PdO/Pd/CNTs) were synthesized through laser irradiation of PdO/CNTs in distilled water. The optimized PdO (82%)–Pd (18%) interface provided maximum active sites for N2 activation and proton transportation. In PdO/Pd/CNTs, both Pd and PdO sites were exposed for electrocatalysis where the Pd site strongly bound the N2 molecule while the PdO site facilitates the subsurface activated H protons to imitate α-PdH and hydrogenate the adsorbed N2 molecule. The synergy of Pd and PdO contributed to the efficiency of the catalyst. They further studied defect-engineered titanium dioxide and the interfacial effect of supported palladium or platinum for ENRR, which demonstrated 1847.3 μg mgcat.−1 h−1 (Pd/TiO2) and 2520.5 μg mgcat.−1 h−1 (Pt/TiO2) yield at potential −0.1 V, respectively.55 Interestingly, the FE for NRR was higher on Pd/TiO2 (2.63%) as compared to Pt/TiO2 (1.57%). Xu et al. reported nanoporous palladium hydride (np-PdH) as an electrocatalyst for ENRR under ambient conditions, which showed a high NH3 yield rate of 20.4 μg h−1 mg−1 with a FE of 43.6% at a low overpotential of −150 mV.56 Isotopic hydrogen labelling studies suggested that the lattice hydrogen atoms present in the np-PdH acted as the active hydrogen source. In situ Raman analysis and DFT calculations further revealed that the *N2 to *N2H reduction energy barrier is the rate-limiting step of ENRR. The unique protonation nature of the np-PdH could provide a new dimension for designing efficient and robust electrocatalysts. To study the ENRR mechanism, the authors studied the isotopic labelling experiments on np-PdH0.43, as presented in Fig. 6. In the presence of 15N2, two peaks with a coupling constant of 73.2 Hz were detected in the baseline-subtracted 1H NMR spectra (Fig. 6b), which is consistent with a heteronuclear coupling constant between 1H and 15N of 15NH4+. The FTIR band at 1439 cm−1 corresponds to the N–H bending, which further provides evidence for NH3 generation. Three weak FTIR bands that appeared at 1177, 1241, and 1325 cm−1 were ascribed to the bending modes of NH2D2+ and NH3D+ (Fig. 6c). XRD data suggest that there is no shift in the characteristic (111) peak at different potentials which underlines the structural durability of the catalyst. In situ Raman spectra of np-PdH0.43 indicate a broad peak at 1644.4 cm−1 that relates to the overlapping of H–N–H/H–O–H bending and the peak intensity increase with potential indicates the adsorption of ammonia on the catalyst surface (Fig. 6d and e). Recently, Wang et al. achieved a record-high FE of up to 97% for ENRR in aqueous solution using a Pd/activated carbon cloth.57 The high electrocatalytic activity is demonstrated by adjusting the three-phase interface, i.e., gas/solid catalyst/liquid electrolyte, which favours ENRR over HER. The complete suppression of HER is facilitated by the three-interface contact due to high N2 coverage on the catalyst surface that weakens H adsorption as evidenced by the DFT calculations and experimental results. The authors further confirm the merits of a three-phase interface strategy for selective electrochemical reduction of N2 to NH3 using Ir- and RuPd-based catalysts, which are known to have strong H adsorption ability.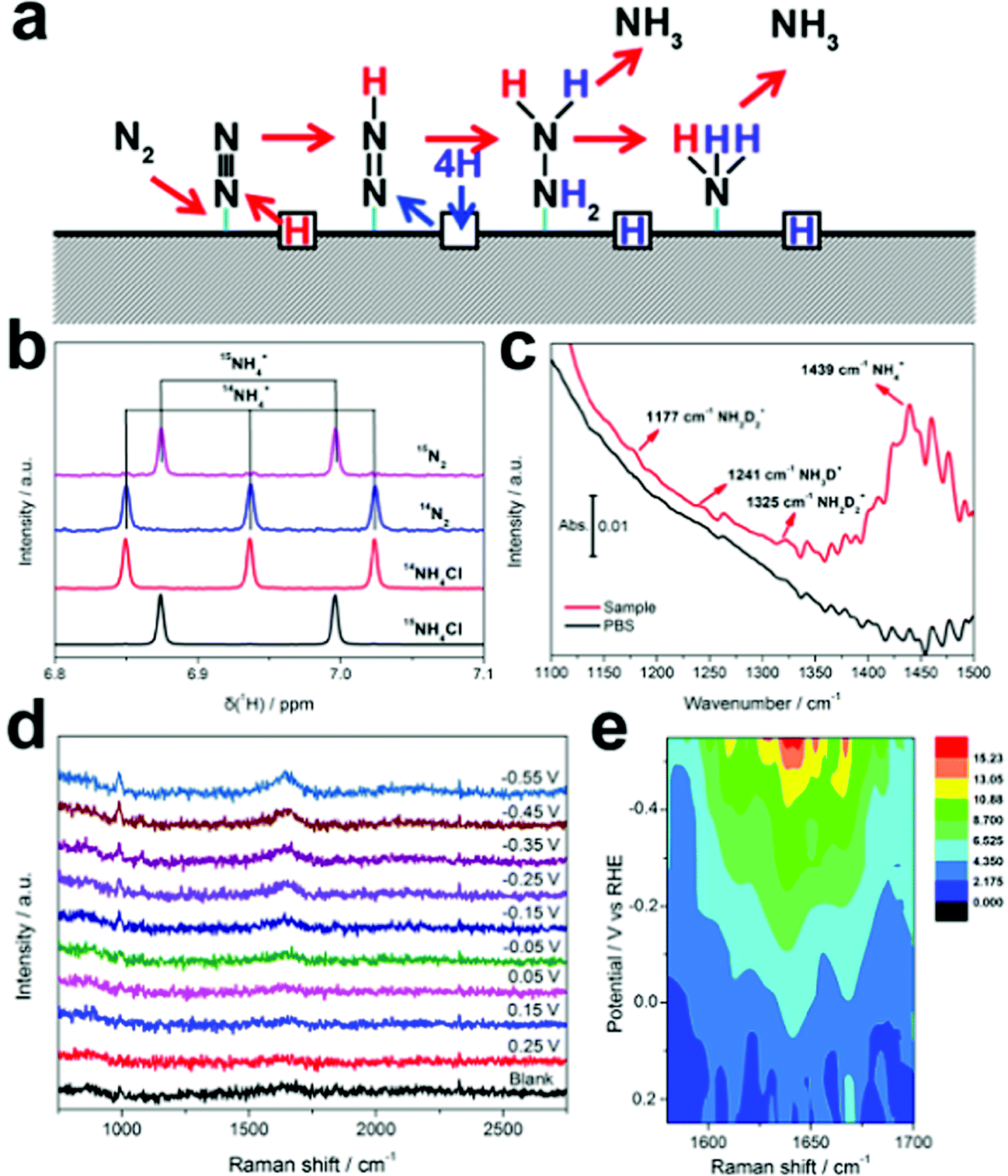 | ||
| Fig. 6 (a) Proposed lattice hydrogen-involved reaction pathway for nitrogen reduction on palladium hydride. (b) Baseline-subtracted 1H NMR spectra of the post-electrolyte with 15N2 and 14N2 and the reference 14NH4Cl and 15NH4Cl. (c) FTIR spectra of the post-electrolyte with a deuterium substituted catalyst. (d) In situ Raman spectra and (e) the corresponding contour plots of np-PdH0.43 at various potentials in a N2-saturated electrolyte. Reproduced with permission from ref. 56. Copyright 2020, Wiley-VCH. | ||
2.6 3d- and 4d-transition metal catalysts
Even though noble metals such as Au, Pd, and Ru show promising electrocatalytic activity towards ENRR, the scarcity and cost-ineffectiveness associated with these prompt the researchers to explore alternate materials without compromising the performance. In recent years, a variety of noble metal-free transition metal electrocatalysts have been studied for ORR,58 OER,59 and HER.60 These were also extensively employed for ENRR. Transition metals are largely abundant in the mother earth and inexpensive, making them suitable alternatives to noble metal-based catalysts. Mukherjee et al. reported an atomically dispersed Ni site on N-doped carbon (Ni–Nx–C), which exhibited an optimal NH3 yield of 115 μg cm−2 h−1 at −0.8 V in a neutral medium.61 The as-prepared Ni–Nx−C catalyst showed a FE of 21 ± 1.9% at −0.2 V under alkaline conditions although the NH3 yield was lower. The atomically dispersed Ni sites were stabilized with nitrogen in the carbon matrix. The active site and probable reaction pathways through the DFT calculations reveal that the Ni–N3 sites are responsible for the experimentally observed catalytic activity and selectivity. The N2 molecule adsorbs on the top of the central Ni atom in an end-on configuration (Fig. 7) in which one N atom is chelated with Ni by a bond length of 1.76 Å, whereas another one is tilted along with a Ni–N bond of the Ni–N3 site. The proximal N of the adsorbed N2 undergoes two hydrogenation steps to form *NHNH2 and *NH2NH2. The NN bond length in *NH2NH2 is increased about 28% than that of the N2 gas molecule. Therefore, subsequent hydrogenation on the distal N is believed to break the NN bond and release one NH3.
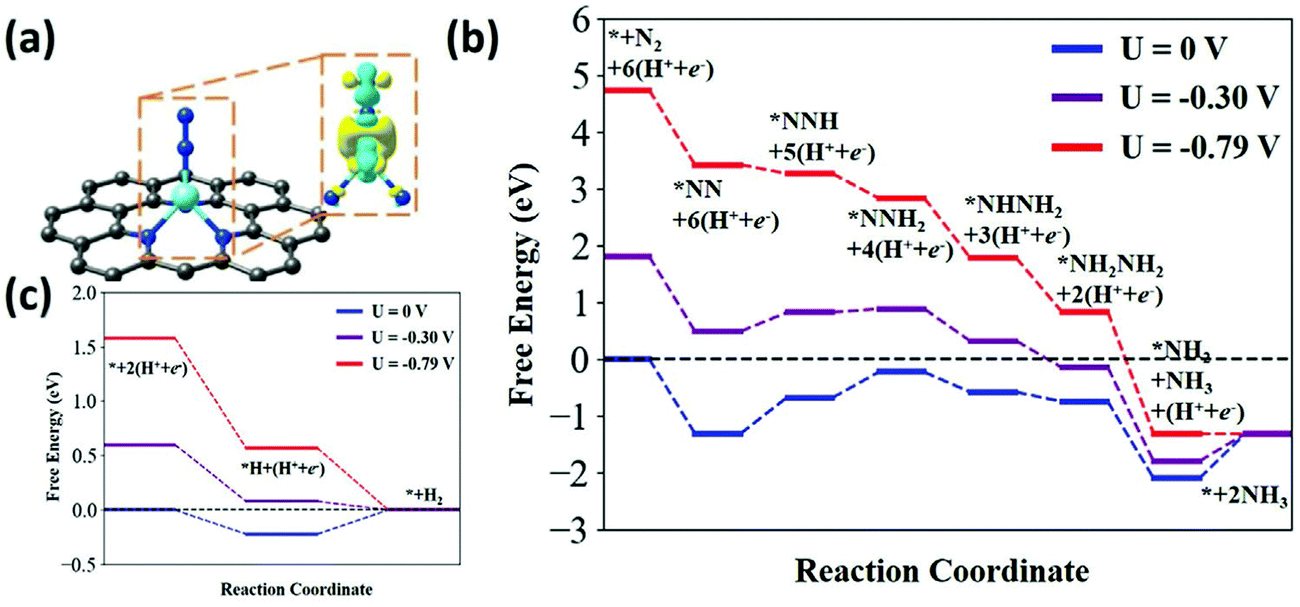 | ||
| Fig. 7 (a) Atomistic structure and charge distribution of N2 adsorption on the Ni–N3 active site. In this figure the cyan and yellow clouds show the charge depletion and accumulation; the gray, blue, cyan and white balls represent the C, N, Ni, and H atoms, respectively. The isosurface was set to be 0.006 e Å−3. (b) Predicted energy evolution of ENRR on a Ni–N3 site under an applied potential of 0, −0.3, and −0.79 V versus RHE and (c) calculated free energy evolution for HER on the Ni–N3 active site at 0, −0.3, and −0.79 V versus RHE. Reproduced with permission from ref. 61. Copyright 2020, Wiley-VCH. | ||
Zang et al. reported on a single copper atom attached porous N-doped carbon network (NC-Cu SA) for ENRR by the combination of experimental and theoretical calculation.62 The single-atom copper site exhibits high-density active sites and the porosity of the catalyst leads to an NH3 yield rate of ∼53.3 μg(NH3) h−1 mgcat−1 with a FE of 13.8% in 0.1 M KOH, and these were decreased to ∼49.3 μg(NH3) h−1 mgcat−1 and 11.7% in the acidic medium (0.1 M HCl). The N2 temperature-programmed desorption (N2-TPD) on NC-Cu SAC indicates a stronger desorption peak at higher temperature due to its higher binding energy for N2 and ability towards N2 fixation. The theoretical study revealed that the Cu–N2 sites are more active than any Cu–N4/N3 site energetically. The Gibbs free energy change for carbon vacant and two N atoms connected Cu is favourable. To improve the electrocatalytic activity of Cu NPs towards ENRR, Lin et al. reported electron-deficient Cu nanocrystals for ENRR.63 The Cu nanocrystals were interfaced with polyimide (Cu/PI). This strategy was found to be very efficient in boosting the ENRR activity of Cu nanocrystals by nurturing the electron density over the Cu nanocrystals and simultaneously suppressing the HER activity. Cu/PI showed a high yield rate of NH3 (17.2 μg h−1 cm−2) in addition to a turnover frequency (TOF) value of 0.26 h−1. Moreover, tuning of the Cu work function leads to improved N2 reduction and could achieve a high TOF value of 0.26 h−1.
Molybdenum (Mo) is another transition metal used for N2 reduction because the d-electrons of Mo can weaken the NN triple bond. In the early experiments, Yandulov et al. reported the use of a single Mo centre for N2 reduction with efficiencies second to Fe/Mo nitrogenase.64 Chen et al. successfully synthesized super small-sized Mo species through reduction of large-sized molybdenum oxide in a H2 atmosphere on a carbon support (carbon cloth).65 The particle size decreased with increasing synthesis temperature. Remarkably, up to an average particle size of 0.95 nm was demonstrated using the developed method. The synthesized Mo catalyst showed an excellent activity towards ENRR with FE as high as 22.3% and a yield rate of 7.02 μg h−1 mgcat−1 at 0 V vs. RHE under ambient conditions using a 0.1 M KOH electrolyte. The high electrocatalytic activity is attributed to large active sites of super small-sized Mo NPs.
2.7 Bismuth (Bi)
The potential of the Bi metal towards ENRR arises from its poor HER activity due to the high Gibbs free energy barrier for hydrogen adsorption on its surface.66 Thus Bi nanostructures have been widely investigated for ENRR. N2 molecules selectively adsorb on the Bi surface due to the overlap of the Bi 6p band and the N 2p orbital, which enhances the ENRR.67,68 Moreover, due to the weak binding of Had atoms on the Bi surface, it can selectively promote N2 to N2H* reduction, and restrict surface electron accessibility for the HER process thus enhancing the NH3 production rate.67,69 Li et al. reported two-dimensional mosaic Bi nanosheets (NSs) fabricated by an in situ electrochemical method as shown in Fig. 8.70 The Bi NSs exhibited an average NH3 yield of 2.54 ± 0.16 μgNH3 cm−2 h−1 (∼13.23 μg mgcat.−1 h−1) with a FE of 10.46 ± 1.45% at −0.8 V in a 0.1 M Na2SO4 electrolyte. The enhanced ENRR electrocatalytic activity was due to the semiconducting nature of the mosaic bismuth NSs, which limits surface electron accessibility and exposes effective p-orbital electron delocalized edge sites. Wang et al. successfully synthesized defect-rich Bi nanoplates by a low-temperature plasma bombardment approach as an ENRR electrocatalyst for NH3 production. The defect-rich Bi(110) nanoplates showed an 15NH3 yield rate of 5.453 μg mgBi−1 h−1 at −0.9 V and a FE of 11.68% at −0.6 V in aqueous solution under ambient conditions.71 Xia et al. synthesized carbon fiber paper supported single-crystalline Bi NSs by in situ electrocatalytic topotactic transformation of BiOI NSs.72 These Bi NSs exhibited an NH3 yield of 12.49 μg h−1 mgcat.−1 and a FE of 7.09% at −0.3 V in a 0.1 M NaHCO3 electrolyte. Although the basal (001) plane of Bi NSs is relatively inert, the engineered exposed edge sites/planes (010) are conducive toward the adsorption and activation of N2 molecules as confirmed by the DFT calculations. Recently, Qiu and co-workers prepared multi-yolk–shell bismuth@porous carbon (MB@PC) composites via a facile simple hydrothermal reaction followed by pyrolyzation. This MB@PC composite showed an excellent performance with an NH3 yield of 28.63 μg h−1 mgcat.−1 and a FE of 10.58% at −0.5 V in N2-saturated 0.1 M HCl solution. The synergetic effect of good conductivity, the highly porous feature derived from the carbon framework, and the intrinsic electrocatalytic ENRR activity of Bi NPs was the primary reason for excellent ENRR performance.73 | ||
| Fig. 8 (a) XRD patterns of the BiOI precursor and Bi NSs; (b) SEM and (c) dark-field TEM images showing the nanosheet feature of Bi NSs; (d) TEM image of Bi NSs showing the mosaic structure; (e) high-resolution TEM image of Bi NSs; (f) SAED pattern of mosaic Bi NSs. Reproduced with permission from ref. 70. Copyright 2020, ACS. | ||
Xu et al. demonstrated the ENRR activity using ultrathin Bi NSs prepared through the in situ electrochemical reduction-assisted exfoliation of BiOCl nanoplates (BiOCl NPs). This method facilitated an increase in electrochemically active surface area and under-coordinated Bi sites which accelerate electron transport capability. With these unique features, the as-converted Bi NSs exhibited a FE of 14.14% and an NH3 yield of 11.11 mg h−1 mgcat.−1 at −0.5 V (vs. RHE) in 0.1 M Na2SO4, and high NH3 selectivity (nearly 100%).74 One of the most significant breakthroughs has been achieved by Yan's group, attaining a very high FE of 66% and an NH3 yield of 200 mmol g−1 h−1 in aqueous solution and under ambient conditions by utilizing a unique strategy of combining Bi nanocrystals (NCs) and K+-supporting cations in water.67 These studies suggest the potential of Bi-based electrocatalysts towards ENRR for producing a sizable amount of NH3.
3. Bimetallic catalysts
Nanostructured metal catalysts such as Ru, Au, and Pd show significant electrocatalytic responses towards ENRR as discussed earlier.32 However, the electrocatalytic activity of bimetallic alloy nanostructured catalysts is superior to their monometallic counterparts. The electrocatalytic performances of bimetallic nanostructured electrocatalysts depend on their surface electronic structure and thus on the shape, size, surface area, exposed facets, and compositions.75 The high electrocatalytic activity of the bimetallic or alloy nanostructures arises from the synergistic effect of the component metals.76 Thus bimetallic nanostructures have also been explored towards ENRR in recent years. Wang et al. reported ENRR using bimetallic PdRu nano tripods (TPs).77 The highest average NH3 yield of 37.23 mg h−1 mgcat−1 and a corresponding FE of 1.85% was achieved at −0.2 V for PdRu TPs, which is higher than that of PdRu dendrite nanostructures and Pd NPs. This is attributed to the microstructures and alloying of Pd and Ru. Porous bimetallic alloys were synthesized and employed for ENRR. Bimodal porous Pd3Cu1 exhibited an NH3 yield rate of 39.9 μg h−1 mgcat−1.78 Yu et al. reported bimetallic Ag3Cu porous networks which exhibited excellent ENRR performance with an NH3 yield of 24.59 μg h−1 mgcat−1 and a FE of 13.28% at −0.5 V for ENRR.79 The bimetallic catalyst showed higher activity than monometallic Cu and Ag in which its composition played a significant role. Wang et al. reported bullet-like M–Te (M = Ru, Rh, Ir) porous nanorods (PNRs) for ENRR.80 Among the MTe, optimized IrTe4 showed the highest FE and NH3 yield rate of 15.3% and 51.1 μg h−1 mgcat−1 at −0.2 V, respectively. Strong chemical adsorption between N2 and IrTe4 PNRs was indicated by N2 temperature-programmed desorption (N2-TPD) and valence band X-ray spectroscopy measurements, which is the key feature towards the high ENRR of IrTe4 and suppression of HER. The high adsorption arises from the synergistic effect between the electron-rich Ir and highly electroactive surroundings of the Te atom.Table 1 compares the ENRR performance of single metal and bimetallic/trimetallic alloys under ambient conditions.
4. Transition metal oxides and defect-based catalysts
Over the years, transition metal oxides (TMOs) are explored almost in every type of energy conversion and storage device with reasonably satisfying results.94,95 The catalytic activity of TMO-based materials arises from a combination of inherent electronic structure, microstructure, edges, defects, and interfacial structure in heterojunction and/or with support materials. Several TMOs such as TiO2,96 Fe2O3,97 Mn3O4,98 NiO,99 Co3O4,100 and ZnO65 have been studied as electrocatalysts for ENRR, which have demonstrated quite comparable performances with metal NP based catalysts (Au, Pd, and Ru). In oxides, oxygen vacancy defects play a crucial role in ENRR. An oxygen vacancy is created in an oxide-based semiconductor to form a defect centre.101 A defect-rich oxygen vacancy exposes co-ordinately unsaturated metal sites, which enhances the chemisorption and activates inert N2 molecules. Fang et al. reported oxygen vacancy-contained TiO2 nanosheets as an electrocatalyst towards ENRR.102 Oxygen vacancies were created by annealing the as-prepared TiO2 nanostructures under a H2/Ar atmosphere. The high activity of the as-prepared oxygen vacant TiO2 is attributed to the synergistic effect between the structural features and microstructure of the catalyst. The DFT calculations suggest that the oxygen vacancies (OVs) can significantly lower the activation energy barrier of NN bond dissociation during N2 fixation (Fig. 9). Without OVs, TiO2 required a free energy of 4 eV for N2 activation, which makes TiO2 (without OV) inert to ENRR. The surface of the OV TiO2 site has much higher catalytic activity than the perfect TiO2 surface. The uphill energy of the first hydrogenation step is only 0.25 eV on the OV site, whereas as large as 2.29 eV is required for the perfect surface. After the first hydrogenation step, free energy changes for all steps toward the formation of NH3 are thermodynamically favourable on the OV site except for the desorption of NH3. The two-dimensional TiO2 nanosheets provide a large number of active sites on their surface. The oxygen vacant TiO2 shows a 2.83 times higher NH3 yield rate than that of the as-prepared TiO2 nanostructures. Li et al. reported Ti3+ self-doped TiO2−x nanowires on Ti mesh (Ti3+–TiO2−x/TM) as the catalyst for ENRR.103 The theoretical calculation further suggests that the presence of Ti3+ decreases the ENRR energy barrier and facilitates more number of active sites on the TiO2 surface. In 0.1 M Na2SO4, Ti3+–TiO2−x/TM achieves a FE of 14.62% with an NH3 yield of 3.51 × 10−11 mol s−1 cm−2 at −0.55 V. Qin et al. reported a carbon-doped TiO2/C (C–TixOy/C) material synthesized from the MIL-125(Ti) metal–organic framework (MOF) with an impressive FE of 17.8%. The high catalytic activity arises from the combination of C–Ti bond formation and oxygen vacancy.104 The C–Ti binding is more favourable for N2 activation than the non-substituted OV in TiO2 catalysts.
 | ||
| Fig. 9 (a and b) Adsorption configurations of the N2 molecule on the perfect TiO2 surface and at the OV site of OV-TiO2. (c) Free energy profile of the NRR process on the anatase(010) surface with and without OV at a potential of 0.0 V versus RHE. (d) Free energy profile of the NRR process on the anatase(010) surface with and without OV at a potential of −0.8 V versus RHE. The asterisks denote the adsorption. Reproduced with permission from ref. 102. Copyright 2020, Wiley-VCH. | ||
Huang et al. reported the Mn3O4 NPs@rGO composite (Mn3O4@rGO) for ENRR.105 The theoretical study revealed that the N2 molecule specifically adsorbed to a particular facet over the other. Mn3O4@rGO showed ENRR activity with excellent selectivity for NH3 formation in 0.1 M Na2SO4 solution with an NH3 yield of 17.4 μg h−1 mgcat−1 and a FE of 3.52% at −0.85 V. The rGO significantly improved the catalytic performance of Mn3O4 by increasing the conductivity, surface area, and charge transfer kinetics across the electrode. The calculated binding energies (BE) of the N2 molecule on the (221), (112), and (103) surface were −0.035, −0.32, and 0.29 eV, respectively, which suggest very weak adsorption of the N2 molecule on the (221) surface whereas no adsorption on the (103) surface. On the other hand, the (112) surface exhibited decent adsorption of the N2 molecule (Ebinding = −0.32 eV). The calculated binding energies also well agreed with the bond length between the N2 molecule and the Mn atoms on different surfaces. Liu et al. reported ZnO quantum dot supported rGO (ZnO/rGO) for ENRR.106 The DFT calculation revealed that the synergistic electronic coupling between ZnO and rGO significantly reduces the activation energy barrier for *N2H species stabilization, which is the rate-limiting step for ENRR. The as-synthesized ZnO/RGO showed an NH3 yield of 17.7 μg h−1 mg−1 and a FE of 6.4% in 0.1 M Na2SO4 at −0.65 V, which is higher than that of pure ZnO quantum dots and RGO.
5. Perovskite-based catalysts
Materials having the general formula ABO3 are regarded as perovskite-type oxide materials where A is a rare-earth or alkaline earth element and B is a transition metal element. Though most of them have been used as electrolytes in ENRR, earlier studies have also used them as catalysts for NH3 synthesis.107,108 Co-based perovskites such as Sm0.5Sr0.5CoO3−δ, Ba0.5Sr0.5Co0.8Fe0.2O3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ have been tested as cathodes for electrochemical ENRR. However, the high thermal expansion coefficient, easy reduction of the active site, and evaporation of cobalt at higher temperatures raise a concern about their practical applicability.109,110 Therefore it is highly desirable to develop cobalt-free perovskite oxide catalysts for NH3 synthesis. In this regard, Amar et al. developed a new perovskite oxide La0.6Sr0.4Fe0.8Cu0.2O3−δ as a cathode catalyst for the electrochemical synthesis of NH3 with a maximum production rate of 5 × 10−9 mol s−1 cm−2 at 450 °C.111 Similarly, cobalt-free perovskite oxide SmFe0.7Cu0.3−xNixO3 as the cathode material, a Nafion membrane as the electrolyte, and Ni-doped Ce0.8Sm0.2O2−δ as the anode have been tested for ENRR. The obtained ammonia yield rate was 1.13 × 10−8 mol cm−2 s−1 at 80 °C.112 Recently, a different effective approach was demonstrated by Liu et al. The authors introduced oxygen vacancies into perovskite oxide LaCoO3 (denoted as VO-LaCoO3) that remarkably increased the yield rate for NH3.113 The Co atom and its adjacent oxygen vacancy created an active centre that facilitated N2 activation. The obtained NH3 yield rate reached a maximum value of 182.2 μgNH3 mgcat.−1 h−1, which was 2.8 times higher than that of pristine LaCoO3 (65.3 μgNH3 mg−1 h−1) leading to a FE of 7.6%. Further computational studies were carried out by the authors to understand the mechanism. The DFT calculation revealed that the oxygen vacancy increased the charge density around the valence band edge of VO-LaCoO3 resulting in increased activation of N2 at the VO-LaCoO3 surface. The thermodynamic limiting potential for ENRR as compared to HER is also favourable for VO-LaCoO3 in comparison to pristine LaCoO3.6. Spinel-based catalysts
Spinels, having the general formula AB2O4, have been explored to some extent as electrolytes and catalysts for ENRR. The first report on a spinel oxide, i.e., CoFe2O4 prepared by a co-precipitation method was investigated by Amar et al., which showed a maximum ammonia production rate of 2.32 × 10−10 mol s−1 cm−2 at 400 °C at 0.8 V.114 A cost-effective sol–gel technique was also utilized to synthesize the same spinel oxide CoFe2O4, which produced 6.5 × 10−11 mol s−1 cm−2 ammonia through ENRR at 400 °C and 1.6 V, twice that of the value reported previously, using water vapour and nitrogen under atmospheric pressure.115 This suggests that the synthesis techniques of catalyst materials have a strong impact on their ENRR performance. Ahmed et al. employed a one-pot solvothermal approach to synthesize CoFe2O4 nanoclusters anchored on reduced graphene oxide as a nanocomposite catalyst for ENRR. They obtained a FE of 6.2% with a high NH3 yield rate of 4.2 × 10−11 mol s−1 cm−2 in 0.1 M Na2SO4 under ambient conditions along with excellent stability and durability for the long term ENRR performance.116 Besides that, a low-cost material, i.e., Mn3O4, known to be naturally abundant has been reported as an efficient electrocatalyst for ENRR. Sun and co-workers prepared Mn3O4 nanocubes through a hydrothermal route and tested for ENRR in 0.1 M Na2SO4 solution. The catalyst delivered a FE of 3.0% with an NH3 yield of 11.6 μg mgcat.−1 h−1 at −0.8 V.98 So far Mn3O4 has been successfully tested as an ENRR electrocatalyst.105 To further improve the performance of Mn3O4 and suppress the HER, Li+ ions were incorporated into the Mn3O4 spinel. The spinel LiMn2O4 was prepared by electrospinning and tested in 0.1 M HCl at −0.50 V. The as-synthesized catalyst showed a FE of 7.44% and an NH3 yield rate of 15.83 μg mgcat.−1 h−1, which are much higher than those of pristine Mn3O4 reported earlier.117Oxygen vacancies (OVs) were created in spinels to enhance the ENRR activity. Various OV-rich nanostructured spinel wrapped hollow nitrogen-doped carbon polyhedra such as OV-rich NiCo2O4@HNCP, OV-rich ZnCo2O4@HNCP, and OV-rich Co3O4@HNCP were reported to facilitate the ENRR. Among these, OV-rich NiCo2O4@HNCP showed a high NH3 production yield of 4.1 μg h−1 cm−2/17.8 μg mg−1 h−1 and a FE of 5.3%.118 However, there is still a vast opportunity to explore these spinel-based materials with rational design for enhancing the efficiency of ENRR.
The ENRR performance of more oxides, perovskites, and spinels under ambient conditions is presented in Table 2 for comparison.
| Catalyst | Electrolyte | Temperature | Yield rate (μg h−1 mgcat.−1) | FE (%) | Potentials vs. RHE (V) | Ref. |
|---|---|---|---|---|---|---|
| B–TiO2 | Na2SO4 | Ambient | 14.4 | 3.4 | −0.8 | 119 |
| V–TiO2 | LiClO4 | Ambient | 17.73 | — | −0.50 | 120 |
| — | 15.3 | −0.40 | ||||
| C–TiO2 | Na2SO4 | Ambient | 16.22 | 1.84 | −0.7 | 121 |
| Fe–TiO2 | LiClO4 | Ambient | 25.47 | 25.6 | −0.40 | 122 |
| VO2 | Na2SO4 | Ambient | 14.85 | 3.97 | −0.7 | 123 |
| Cr2O3 | HCl | Ambient | 28.13 | 8.56 | −0.75 | 124 |
| Mn3O4 | Na2SO4 | Ambient | 11.6 | 3.0 | −0.8 | 98 |
| Mo–MnO2 | Na2SO4 | Ambient | 36.6 | 7.9 | −0.5 | 125 |
| Fe2O3 | Na2SO4 | Ambient | 15.9 | 0.94 | −0.8 | 126 |
| NiO | Na2SO4 | Ambient | 29.1 | 10.8 | −0.5 | 127 |
| NiWO4 | HCl | Ambient | 40.05 | 19.32 | −0.3 | 128 |
| Na2SO4 | 23.14 | 10.18 | ||||
| FeMoO4 | Na2SO4 | Ambient | 17.51 | 10.53 | −0.6 | 129 |
| Fe–SnO2 | HCl | Ambient | 82.7 | 20.4 | −0.3 | 130 |
| β-Bi2O3 | Na2SO4 | Ambient | 19.92 | 4.3 | −0.8 | 131 |
| r-CeO2 | Na2SO4 | Ambient | 16.4 | ∼3.5 | −0.5 | 132 |
| Cr–CeO2 | Na2SO4 | Ambient | 16.82 | 3.84 | −0.7 | 133 |
| Ta2O5 | HCl | Ambient | 15.9 | 8.9 | −0.7 | 134 |
| La2O3 | Na2SO4 | Ambient | 17.04 | 4.76 | −0.8 | 135 |
| La2Ti2O7 | HCl | Ambient | 25.15 | 4.55 | −0.55 | 136 |
| LaCrO3 | Na2SO4 | Ambient | 24.8 | 15 | −0.8 | 137 |
| LaFeO3 | HCl | Ambient | 18.59 | 8.77 | −0.55 | 138 |
7. Phosphide-based catalysts
Transition metal phosphides (TMPs) have emerged as highly active and inexpensive electrocatalysts as well as cocatalysts for HER with performance comparable to that of the state-of-the-art Pt/C. A wide range of TMPs such as CoP,139 Ni2P,140 MoP,141 Cu3P,142 and FeP143 have thus been studied for HER. Over the years, different compositions, microstructures, hetero-microstructures, and doped TMPs have been synthesized to enhance the performance. A review article by Wang et al. provides the details on the development of TMPs and their potential application for (photo) electrochemical water splitting.144 However, TMPs for ENRR have not been largely explored. The same challenge, i.e., suppression of HER activity, remains the limiting factor to enhance ENRR with TMP-based catalysts. Zhu et al. reported phosphorus as a modulating agent of the FeP2 catalyst towards ENRR performance.145 The synthesized P rich FeP2-reduced graphene oxide (FeP2–rGO) showed a FE and NH3 yield rate of 21.99% and 35.26 μg h−1 mgcat−1 at −0.40 V, respectively. On the other hand, the FeP–rGO catalyst showed a lower FE and NH3 yield of 8.57% and 17.13 μg h−1 mgcat−1 at −0.40 V, respectively. The ENRR suffers from the competitive HER at the electrode surface due to the low H adsorption energy to the catalyst surface in the latter case. ΔG(*H) is used as a parameter to quantify the metal hydrogen bond strength. The theoretical calculation of ΔG(*H) for FeP(211) and FeP2(101) indicates that FeP(101) is more active towards HER than FeP2(211). The ENRR mechanism follows the distal pathways, which are energetically more favourable than the alternating pathways. The rate-determining step for ENRR is *N2 to *NNH. The N2 adsorption free energy for FeP2(101) is higher than that for FeP(211), which suggests the former to be more selective towards ENRR. The P-rich FeP2–rGO decreases HER activity, increases N2 adsorption, and facilitates a large number of active sites towards ENRR compared with FeP. Jin et al. reported partially electrochemically reduced oxygen-doped Cu3P nanosheets (RO-Cu3P) with rich surface phosphorus vacancies for the ENRR process.146 Both NH3 and N2H4 were formed during the ENRR process indicating that the reaction follows the alternating pathways. An NH3 yield of 28.12 μg h−1 cm−2 with a FE of 17.5% was achieved under ambient conditions for the RO-Cu3P catalyst with abundant phosphorus vacancies, which is 3.4 times higher than that of the pure Cu3P catalyst. The surface phosphorus vacancy acts as an active site for ENRR, which is proven by controlled experimental as well as DFT calculations. The free energy change (ΔG*NNH) on a phosphorus vacancy on the surface of Cu3P(110) is found to be 0.97 eV (at 0 V, pH = 0) for *N2 to *NNH. The ΔG*NNH on the surface phosphorus vacancy of O-Cu3P structures was calculated to be 1.25 eV for Cu3P0.98O0.02 and 2.47 eV for Cu3P0.96O0.04, which is higher than that required on the surface phosphorus vacancy of Cu3P. This demonstrates that the superfluous oxygen doped in the phosphide crystal phase is not conducive to ENRR activity. The experimental and theoretical investigations suggest that the phosphorus vacancies on the surface of Cu3P and O-Cu3P are the highly active species that lead to enhanced ENRR activity. Yang et al. synthesized IrP2 anchored P, N co-doped porous carbon nanofilms (IrP2@PNPC-NFs) by the plasma-assisted method.147 The IrP2@PNPC-NFs showed a FE and NH3 yield rate of 17.8% and 94.0 μg h−1 mgcat−1, respectively. The edge site of IrP2 was found to be very irregular and the low coordinated step atoms at the edge site of the IrP2 nanocrystals lowered the reaction energy barrier towards the ENRR while inhibiting the HER activity. The plasma-assisted strategy also makes it possible to synthesize other high-melting-point noble-metal phosphides (such as OsP2@PNPC-NFs, Re3P4@PNPC-NFs) at lower temperatures.8. Nitride-based catalysts
Transition metal nitrides (TMNs) are another class of materials that have high electronic conductivity with rich Nad atoms. Theoretical as well as experimental investigations suggest that TMNs have huge potential for ENRR.148,149 However, the chemical stability of the nitride-based materials is an obstacle in addition to competent HER.150 Moreover the generation of NH3 from the nitrogen atom of the catalyst can give false-positive results (discussed later). Abghoui and Skúlason theoretically investigated TMNs for ENRR under ambient conditions and provided possible mechanistic insights considering several nitrides such as ZrN, MoN, CrN, MnN, NbN, and VN with a rocksalt structure.151 The free energy diagrams were constructed on the rocksalt (100) structure using the associated mechanism to evaluate their performance in terms of onset potential for ENRR. The free energy of each intermediate was calculated at room temperature, pH = 0, and an applied potential of 0.0 V, which indicates that the potential determining step (PDS) is the first protonation of *N2 to *NNH for all the candidates except for VN and NdN where the formation of *NNH3 is the PDS. Among these nitrides, MoN showed promising results with ΔGPDS = 0.83 eV.151 Zhang et al. demonstrated a MoN nanosheet array on carbon cloth (MoN NA/CC) as an ENRR electrocatalyst in 0.1 M HCl under ambient conditions.152 This catalyst showed an NH3 yield of 3.01 × 10−10 mol s−1 cm−2 and a FE of 1.15% at −0.3 V with high electrochemical durability and selectivity. DFT calculations suggested that MoN NA/CC catalyzes ENRR via the Mars–van Krevelen mechanism rather than the associative/dissociative Heyrovsky mechanism. Jin et al. reported nitrogen vacant 2D layered W2N3 (Fig. 10) as an efficient and stable electrocatalyst for ENRR with a steady NH3 yield rate of 11.66 ± 0.98 μg h−1 mgcat.−1 (3.80 ± 0.32 × 10−11 mol cm−2 s−1) and FE of 11.67 ± 0.93% at −0.2 V for 12 cycles (24 h).153 The ex situ synchrotron-based characterization of the catalyst before and after the catalytic cycle revealed the stability of 2D W2N3, which was attributed to the vacancies on 2D W2N3 with the combinational effect of the high valence state of tungsten atoms and 2D morphology induced surface distortion. The DFT calculations suggested that the electron-deficient environment in the 2D layered W2N3 effectively facilitates the electron acceptance from N2 and enhances the subsequent reduction rate. | ||
| Fig. 10 (a) TEM image of NV-W2N3. (b) XRD patterns of W2N3 and NV-W2N3. (c) HAADF-STEM images of NV-W2N3 that show the single-layered thickness and interlayer spacing of NV-W2N3. (d) HAADF-STEM image and the corresponding SAED pattern (inset) of NV-W2N3. Reproduced with permission from ref. 153. Copyright 2020, Wiley-VCH. | ||
Ren et al. synthesized Mo2N nanorods and studied the same as a catalyst for ENRR.149 Mo2N/GCE delivered an NH3 yield of 78.4 μg h−1 mgcat.−1 with a FE of 4.5% at −0.3 V. The as-synthesized catalyst was found to be durable up to 10 cycles as confirmed by XPS and XRD analysis after the stability test. However, Hu et al. found that the decomposition of the tetragonal Mo2N catalyst led to NH3 evolution, which was confirmed by isotope experiments and thus contradicts the previous report entirely.154 The authors of the latter report suggested that isotope experiments are needed to evaluate the catalytic activity of the TMNs. Yang et al. reported VN nanoparticles as a stable electrocatalyst for ENRR with a yield rate of 3.3 × 10−10 mol s−1 cm−2 and a FE of 6.0% at −0.1 V within 1 h.155 The ex situ XPS measurement indicated the presence of the oxide and oxynitride species in the catalyst. However, the conversion of VN0.7O0.45 to the VN phase leads to catalyst deactivation. The study highlights that the N site adjacent to the surface O atoms is the active site towards ENRR. Zhang et al. reported VN nanosheets on Ti mesh for ENRR as an active and selective electrocatalyst for ENRR in 0.1 M HCl at room temperature and atmospheric pressure and obtained an NH3 yield of 8.40 × 10−11 mol s−1 cm−2 at −0.50 V with a FE of 2.25%.156
9. Carbide-based catalysts
Transition metals carbides (TMCs) have also been explored in the search for a noble metal-free catalyst for ENRR. TMCs exhibit specific properties which are desirable for electrocatalysis, such as corrosion resistance, and high stability, melting point, and mechanical strength.157–159 These specific properties have attracted increasing research interest for their potential application as electrocatalysts,160 catalyst supports,161 lithium-ion battery materials,162 and solar cell materials,163 particularly for energy generation and storage purpose.164 The carbides are referred as an interstitial alloy. The electronic structure of the host metal of the carbide is altered due to the inclusion of the carbon atoms through the charge transfer process. The distinct electrocatalytic activity of the TMC arises from the electronic structure, due to the presence of carbon atoms in the metal lattice. For example, the excellent HER performance of the tungsten carbide arises from the filling of the d-states at the Fermi level of tungsten by alloying it with carbon.165 Thereby, TMCs are investigated as electrocatalysts without certain disadvantages such as chemical susceptibility and durability. So far, TMCs are employed as HER, OER, and ORR cathode catalysts with excellent activity.165,166 However, there are relatively few reports on the use of TMC materials for ENRR through nanostructuring,167 combination with another material,168 and exploiting hybrid structures.The DFT calculation suggests that the metal site of the carbide can activate the N2 molecule and stabilize the intermediate NxHy species while destabilizing the –NH2 species during the desorption as NH3.82 Although the carbide-based materials show promising ENRR activity theoretically, the experimental performance is not up to the mark due to the competitive HER under similarly applied potentials.169 Therefore different fabrication methods have been adopted to enhance the ENRR performance of the carbide-based catalysts by creating heterojunction interfaces with the generation of active sites for various elementary steps. DFT analysis indicates that all the crystallographic surfaces of the cubic MoC catalyst have the ability for adsorption and dissociation.170 Ramaiyan et al. reported an origami-like Mo2C cathode catalyst for ENRR and achieved a maximum NH3 synthesis rate of 2.16 × 10−11 mol cm−2 s−1 with a FE of 1.8% at 30 °C using Nafion-212 as an electrolyte.171 The authors revealed that numerous kinks at the surface of the origami-like structures are responsible for the catalytic ENRR. However, the synthesized origami-like Mo2C catalyst was found to be unstable during the ENRR process. XPS analysis after the electrolysis process indicates the formation of molybdenum oxycarbide which raises concern about catalyst susceptibility for long term uses. Qu et al. reported oxygen doped molybdenum carbide (O-MoC) embedded in nitrogen-doped carbon layers (N-doped carbon) for ENRR.172 The O-MoC embedded N-doped carbon was synthesized from dopamine and molybdate. The catalyst showed a FE of 25.1% and an NH3 yield rate of 22.5 μg h−1 mgcat−1 in 0.1 mM HCl + 0.5 M Li2SO4. The competitive HER was suppressed due to the interaction between O-MoC and N-doped carbon. The catalyst retained its performance after continuous electrolysis for 30 h. The carbon shelling around O-MoC provides physicochemical protection to O-MoC in the acidic electrolyte from structural dissolution. Liu et al. reported a Mo/Mo2C/MoC (Mo/MoxC) heterostructure supported rGO for ENRR.173 The catalyst showed an excellent yield rate of 20.4 μg h−1 mg−1 at −0.2 V and a FE up to 18.9% at −0.3 V. By controlling the precursor, well-dispersed Mo, Mo2C, binary MoC/Mo2C (MoxC), and ternary Mo/MoC/Mo2C (Mo/MoxC) NPs were synthesized on rGO and the authors experimentally and theoretically studied the influence of composition and hetero-interface on the ENRR performance (Fig. 11). The heterostructured MoxC and Mo/MoxC showed significantly enhanced ENRR activity. The heterojunction facilitates a higher number of available active sites compared to that of Mo and Mo2C only catalysts. DFT calculations further confirmed that the introduction of metallic Mo into binary and ternary carbides appreciably lowered the energy demand for NH3 desorption from the catalyst surface.
 | ||
| Fig. 11 (a and b) SEM, (c) TEM, (d) STEM, and (e) EDS mapping images of the Mo/MoxC heterostructure. (f) TEM and (g) STEM images of the MoxC heterostructure. Reproduced with permission from ref. 173. Copyright 2020, RSC. | ||
Peng et al. reported the bio-inspired synthesis of a Fe3C@C core–shell structured catalyst for ENRR.174 The core–shell structure acts as an active site and selectively contributes toward the NH3 synthesis at low potentials. A FE of 9.15% and an NH3 yield rate of 8.53 μg h−1 mgcat.−1 at −0.2 V were achieved with the same catalyst. The degree of graphitization during the synthesis of the catalyst plays a prominent role in the ENRR. The catalyst prepared at 900 °C showed higher ENRR activity than the catalyst prepared at 700 °C. With increasing temperature, the degree of graphitization increases, whereas on further increasing the temperature to 1100 °C, the performance significantly decreases. The graphitization at 900 °C improved the electron transportation with optimum Fe content. With a further increase in graphitization, the Fe contribution towards ENRR decreases. DFT analysis suggested that the free energy change of the rate-limiting step over the Fe3C@C core–shell towards formation and desorption of NH3 is more favourable than that of Fe3C. The observation indicates that the interaction between the carbon shell and the Fe3C core facilitates the charge transport in the catalyst.
MXene represents another group of novel 2D materials with the general formula Mn+1Cn, where M represents transition metals such as Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, and W with n = 1, 2, 3 along with the terminal functional groups (F, OH, and/or O).175,176 Azofra et al. computationally investigated the possibility of d2–d4 transition metal-based MXene (M3C2) as a model material for N2 capture and reduction. The DFT calculation revealed that the transition metal atoms present at the terminal surface can activate the adsorbed N2 and facilitate the ENRR under mild conditions.175 Zheng et al. theoretically investigated the possible prospect of boron-doped MXene towards ENRR.176 The B-doped Mo2CO2 and W2CO2 MXenes exhibited decent catalytic activity and selectivity with limiting potentials of −0.20 and −0.24 V, respectively. The boron centre mimics the empty and filled d-orbital electronic structure of transition metals and activates the N2 molecule effectively. In addition, a high B-to-adsorbate electron donation tendency facilitates the hydrogenation of *N2 to *N2H during the ENRR. A recent study further suggested that the terminal oxygen groups act as active sites for the HER and CO2 reduction.160 Even though there is a huge prospect for MXene materials toward ENRR, competitive HER mitigation is the main issue that needs to be addressed to unveil the higher efficiency of MXene towards ENRR. Luo et al. reported Ti3C2O2 2D MXene for ENRR.177 The surface Ti atoms act as active sites for N2 adsorption in the single-layer T–Ti–C–Ti–C–T–T (T = terminal atoms) sandwich structure with maximum binding energy. The N2 adsorption energy indicates that the middle Ti atoms are energetically preferable to the C and O sites. The terminal O atoms cannot overcome the high energy barrier for ENRR whereas the Ti site at the edge plane is preferred. Ti3C2Tx MXene with a smaller sheet size shows higher performance than that with a large sheet size under −0.1 V. The study also revealed that vertically aligned Ti3C2Tx MXene has more active sites than the randomly oriented one. Vertically aligned Ti3C2Tx MXene with a FeOOH nanosheet host was synthesized to overcome the thermodynamic obstacles. The FE of MXene/FeOOH reached 5.78% under −0.2 V, which is 1.25 times higher than the maximum value of the MXene. The improved performance of the MXene was ascribed to a higher number of active sites of the vertically aligned MXene and the sluggish nature of FeOOH towards HER.
10. Sulfide-based catalysts
Over the past few years, metal sulfides have been widely studied as a potential candidate for various electrocatalytic activities.178–180 Inspired by the natural nitrogenase enzymes (Mo–Fe protein), MoS2 has been investigated as a catalyst for ENRR.181 Sun and co-workers were the first to introduce and experiment on MoS2 as a catalyst under room temperature and atmospheric pressure. The catalyst showed a FE of 1.17% and an NH3 yield of 8.08 × 10−11 mol s−1 cm−1 at −0.5 V in 0.1 M Na2SO4.182 Later on, they further improved the FE to 8.34% with an NH3 yield of 29.28 μg mgcat.−1 h−1 at −0.40 V, by designing defect-rich MoS2 nanoflowers.183 Zeng et al. developed nanoflower-like N-doped MoS2 electrocatalysts and introduced rich sulfur vacancies to further enhance the FE to 9.14% with an NH3 yield of 69.82 mg mgcat.−1 h−1 at −0.3 V in 0.1 M Na2SO4 solution.184 Suryanto et al. developed MoS2 nanosheets decorated with Ru clusters and obtained a FE of 17.6% and an NH3 yield rate of 1.14 × 10−10 mol s−1 cm−2 attributed to the polymorphic engineering in Ru/2H-MoS2.185 Thereafter several other studies based on MoS2 have been reported to date achieving FE from as low as 4.58% to 20.6% using different strategies.186–190 Apart from MoS2, other transition metal dichalcogenides such as WS2 have been tested as an ENRR catalyst with the lowest potential of −0.24 V through an enzymatic mechanism using DFT.191 Flower-like SnS2 and forest-like ZnS nanoarrays directly grown on Ni foam showed certain abilities towards ENRR. The NH3 yield and FE of 15N2 with flower-like SnS2 were 9.08 × 10−10 mol s−1 cm−2 and 10.3%, whereas for ZnS, they were 5.27 × 10−10 mol s−1 cm−2 and 5.62%, at −0.5 V, respectively.192 Similarly, cobalt disulfide (CoS2) NP-embedded N-doped carbon nanoboxes (CoS2@NC) derived from ZIF-67 have shown effective electrochemical NH3 production under ambient conditions. The catalyst showed an NH3 yield of 17.45 μg mgcat.−1 h−1 and a high FE of 4.6% at −0.15 V vs. RHE.19311. Metal-free electrocatalysts: heteroatom doped carbon-based catalysts
Metal and metal-based oxide, phosphide, carbide, nitride, and sulfide catalysts have been explored as potential ENRR electrocatalysts as discussed earlier. In addition to these, metal-free electrocatalysts have also been studied as ENRR electrocatalysts due to their N2 adsorption ability and weak interaction with H+ ions that specifically suppressed the HER.194–196 However, pure carbon-based materials are inactive towards ENRR.197 This led to the synthesis of a variety of heteroatom doped carbons for ENRR that exhibit considerable ENRR performance. In particular, N-doped porous carbon (NC) exhibited ENRR activity, where pyridinic and pyrrolic N-sites are identified as active sites for N2 adsorption and subsequent dissociation.198 Polymeric carbon nitride (PCN),199 N-doped carbon synthesized from biomass,200 and zeolitic imidazolate frameworks (ZIF-8) have shown considerable ENRR activity.201 The ENRR performance of these catalysts is attributed to the defects arising from the heteroatom doping. However, a recent study has shown that heteroatom-doped carbon materials are also efficient HER electrocatalysts.202 Thus, suppression of HER is the fundamental task to obtain efficient carbon-based catalysts for ENRR.Yu et al. studied boron-doped graphene for ENRR.203 Boron doping redistributed the electron density in the graphene framework, where the electron-deficient boron sites enhanced the binding capability of N2 molecules. DFT calculations based on different boron carbon models revealed that the BC3 site facilitates the lowest energy barrier for ENRR. At 6.2% of boron doping on graphene, an NH3 yield of 9.8 mg h−1 cm−2 and a FE of 10.8% were achieved at −0.5 V in aqueous solutions under ambient conditions. Qiao et al. revealed that the addition of boron (B) to nitrogen-doped graphene yields a higher Gibbs free energy of hydrogen adsorption (DGH*) indicating that *H species were not easily adsorbed onto the active sites and hence suppressed HER.204 BC3 sites in the boron-doped graphene exhibit as active centres for better ENRR activity compared to that of raw graphene.203 Song et al. reported ENRR using N and P co-doped porous carbon with a trace of iron (0.028 wt%) (Fe-NPC).205 The Fe-NPC catalyst was synthesized from solid-state pyrolysis of polyaniline aerogels with phytic acids and FeCl3, which showed significantly enhanced electrocatalytic activity in an alkaline medium as compared to NPC. The Fe sites acted as active sites towards ENRR. In the presence of SCN−, the ENRR was significantly decreased due to the strong affinity of the SCN− ions towards the Fe site. Fe-NPC showed a FE and NH3 yield rate of 5.3% and 4.36 μg h−1 mg−1 at −0.1 V. The in situ FTIR spectra at −0.1 V for the Fe-NPC catalyst showed four peaks at 1126, 1273, 1412, and 3256 cm−1. The peak at 1126 cm−1 indicates the cleavage of NN whereas the peaks at 1273, 1412, and 3256 cm−1 correspond to –NH2 wagging and H–N–H bending, suggesting the intermediate species formed during the ENRR process. It is to be noted that the ENRR process over the Fe-NPC catalyst follows associative pathways.
Wu et al. reported P-doped graphene (PG) for ENRR with a FE and NH3 yield of 20.82% and 32.33 μg h−1 mg−1 at −0.65 V, respectively.197 Two different types of bonded P atoms were present in PG, namely P–O and P–C, with an atomic percent of 0.47 wt%. The DFT calculation showed that the O functional group prefers to adsorb on P than the OH functional group energetically. Therefore, the O functional group is considered to be active for ENRR, and N2 adsorbs preferentially at the C site adjacent to the P–O bonding. This study also revealed that the PDS is *N2 to *NNH hydrogenation. Song et al. modified reduced graphene oxide (rGO) with tannic acid (TA) and studied the ENRR.206 TA-rGO showed a yield rate and FE of 17.02 μg h−1 mg−1 and 4.83% at −0.75 V, respectively. Qiu et al. reported boron carbide (B4C) nanosheets as an electrocatalyst for ENRR with an NH3 yield rate of 26.57 μg h−1 mgcat.−1 and a FE of 15.95% at −0.75 V.196 The N2 atom adsorption on the B4C nanosheets (110) follows either end-on terminal N atom adsorption or adsorption to the adjacent B4C site. Both configurations show a similar energy profile for N2 adsorption on the B4C nanosheet. Free energy calculations showed that hydrogenation of *NN to *NH2NH2 proceeds almost without a barrier. The *NH2–*NH2 → *NH2 + *NH3 step is the rate-limiting step of the ENRR on the B4C(110) surface, which requires a free energy of 0.34 eV at a potential of 0 V.
Liu et al. developed a metal-free F-doped carbon catalyst by introducing F atoms into a 3D porous carbon framework for ENRR.207 The F-doped carbon catalyst exhibited the highest FE of 54.8% for NH3 at −0.2 V, which is 3.0 times higher than that of pristine carbon frameworks (18.3%), and a yield rate of 197.7 μgNH3 mg−1cat. h−1 at −0.3 V. The improved performance of the F-doped carbon catalyst is due to the binding strength of N2 which facilitates dissociation of N2 into *N2H as revealed by mechanistic studies. Due to the different electronegativity between the F and C atoms, a Lewis acid site is created when the F atom is bonded to the C atom. As a result, the HER activity is suppressed significantly. On the other hand, the selectivity of N2 electroreduction into NH3 is enhanced because of the repulsive interaction between the Lewis acid site and proton H.207 Wang and co-workers identified oxygen-doped graphene (O–G) derived from sodium gluconate as a new promising catalyst for the effective production of NH3 by catalyzing ambient electro-hydrogenation of N2.208 They obtained a sizable NH3 yield of 21.3 μg h−1 mgcat.−1 at −0.55 V (vs. RHE) and a high FE of 12.6% at −0.45 V in 0.1 M HCl solution with electrochemical and structural durability. The DFT calculations revealed that the CO and O–CO groups primarily contributed to the catalytic mechanism of ENRR as compared to the C–O group. Xia et al. reported sulfur-doped graphene (S–G) as an electrocatalyst towards ENRR under ambient conditions.209 In 0.1 M HCl, an NH3 yield of 27.3 μg h−1 mgcat.−1 at −0.6 V (vs. RHE) and a high FE of 11.5% at −0.5 V (vs. RHE), which are significantly higher compared to the undoped graphene catalyst (6.25 μg h−1 mgcat.−1; 0.52%), were achieved. The ENRR mechanism was further explored by DFT calculations, which revealed that carbon atoms nearest to substituted sulfur atoms act as underlying active sites for the ENRR on S–G. Further, they prepared S doped carbon nanospheres (S-CNSs) as an electrocatalyst with excellent selectivity towards ENRR, demonstrating an NH3 yield of 19.07 μg h−1 mgcat.−1 and a FE of 7.47% at −0.7 V in a 0.1 M Na2SO4 electrolyte.210 The S-doped CNS showed much higher catalytic activity than that of the undoped CNS (3.70 μg h−1 mgcat.−1, 1.45%) which is similar to the previous discussion.209 Importantly, this S-CNS catalyst also showed high stability for ambient N2 to NH3 conversion.
Cheng et al. adopted a novel one-step synthesis protocol to synthesize defect-rich S and N co-doped carbon cloth (CC) by using ammonium persulfate (APS) as the source of nitrogen and sulfur. The S/N co-doped CC catalyst, prepared at 800 °C (CC-APS 800), exhibited a higher number of defects and heteroatom sites and acted as an active and stable electrocatalyst for ENRR with an NH3 yield of 9.87 × 10−10 mol s−1 cm−2 and a FE of 8.11% at −0.3 V in 0.05 M H2SO4 solution.211 Kong et al. synthesized B and N co-doped porous carbon nanofibers (B/N-CNFs) that exhibited much higher electrocatalytic performance than the only N- or B-doped carbon materials towards ENRR with a FE of 13.2% at −0.5 V and an NH3 yield of 32.5 mg h−1 mgcat.−1 at −0.7 V in an alkaline electrolyte. The synergistic effect of the B–N bond enhances the ENRR process, where N atoms increase the electronic conductivity and B atoms enhance the adsorption of N2 and charge transfer.212 Boron and nitrogen dual-doped carbon nanospheres (BNC-NSs), prepared by another group, exhibited ENRR activity with an NH3 yield rate of 15.7 μgNH3 h−1 mgcat.−1 and a FE of 8.1% at −0.4 V (vs. RHE) in a 0.05 M H2SO4 electrolyte.213 Recently, Zhang et al. reported a rationally synthesized boron, nitrogen, and fluorine ternary-doped carbon (BNFC) from the carbonization of cigarette butts saturated with the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate (C8H15N2BF4).214 In this case, the high electrocatalytic activity toward the ENRR was due to the capability of N atoms in BNFC for the adsorption of hydrogen ions while the adjacent B or F heteroatoms offered Lewis acid sites for chemisorption of N2. An NH3 yield rate of 41 mgNH3 h−1 mgcat.−1 and a FE of 14% at −0.4 V (vs. RHE) in a 0.05 M H2SO4 electrolyte were recorded for BNFC electrocatalysts. However, in a 0.1 M NaOH electrolyte, a relatively high NH3 yield rate of 39 mgNH3 h−1 mgcat.−1 with a FE of 16% at −0.4 V (vs. RHE) was achieved. This is due to the capability of water dissociation and adsorption of generated protons by the doped nitrogen atoms present in BNFC electrocatalysts.214
Table 3 presents the ENRR performance of recently reported metal phosphide, nitride, carbide, sulfide, and metal-free electrocatalysts, indicating their potential.
| Catalyst | Electrolyte | Temperature | Yield rate (μg h−1 mgcat.−1) | FE (%) | Potentials vs. RHE (V) | Ref. |
|---|---|---|---|---|---|---|
| BP | HCl | Ambient | 26.42 | 12.7 | −0.60 | 215 |
| CoP | KOH | Ambient | 10.78 | 0.03 | −0.4 | 216 |
| AuPdP | Na2SO4 | Ambient | 18.78 | 15.44 | −0.3 | 217 |
| Mo–FeP | HCl | Ambient | 13.1 | — | −0.3 | 218 |
| — | 7.49 | −0.2 | ||||
| Fe–Ni2P | HCl | Ambient | 88.51 | 7.92 | −0.3 | 219 |
| BN | HCl | Ambient | 22.4 | 4.7 | −0.75 | 220 |
| FeS2 | Li2SO4 | Ambient | 11.5 | 14.6 | −0.2 | 221 |
| O-doped carbon | HCl | Ambient | 18.03 | — | −0.55 | 222 |
| — | 10.3 | −0.45 | ||||
| B–N enriched carbon | KCl | Ambient | 7.75 | 13.79 | −0.3 | 223 |
| N,P-co-doped carbon | HCl | Ambient | 1.08 | 0.0754 | −0.1 | 224 |
12. Effect of electrolytes
To achieve ENRR activity with high FEs, several other factors are needed to be considered to tackle the challenges in addition to suppressing the competing HER. In particular, besides having an efficient electrocatalyst, the choice of a suitable electrolyte (in terms of ions and pH) is one of the fundamentally important factors to enhance ENRR activity. For instance, acidic, neutral, and alkaline electrolytes have been tested taking into consideration pH adjustment. The pH value for ENRR systems is needed to be well adjusted using dilute H2SO4, HCl, Na2SO4, KHCO3, and KOH aqueous solutions. Chen et al. tested ENRR using a 30% Fe2O3–CNT electrocatalyst in different electrolytes with different pH values at a constant potential.225 The highest NH3 yield rate of 1.06 × 10−11 mol cm−2 s−1 was obtained in the 0.5 M KOH electrolyte, which was higher than that obtained in 0.25 M KHSO4 (7.87 × 10−12 mol cm−2 s−1). In acidic electrolytes, due to the higher proton concentration, HER is significantly increased and thereby suppressing ENRR. In contrast, HER activity is significantly reduced in alkaline electrolytes. Recently, the effective suppression of HER activities was found for the ENRR using the neutral phosphate buffer solution (PBS) electrolyte.53,226 PBS has a higher barrier for mass and charge transfer, which prohibits HER kinetics. Upon using Pd/C as an electrocatalyst, the authors were able to obtain a FE of 2.4% in 0.1 M PBS (pH = 7.2) as compared to <0.1% with 0.05 M H2SO4 (pH = 1.2) and 0.1 M NaOH (pH = 12.9) electrolytes.53 All these electrolytes were Ar-saturated. Zhang et al. showed a higher ENRR selectivity and a FE of 1.17% with MoS2/CC in 0.1 M Na2SO4 as compared to 0.09% in 0.1 M HCl due to the strong HER activity of the latter.182 Moreover, to suppress HER and increase N2 solubility in the electrolyte, a mixture of water and low-proton (alcohols) solvents was also taken.227 Kim et al. studied the water and 2-propanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v) mixture as an electrolyte along with H2SO4 as the supporting electrolyte.228 The mixed solvent gave rise to a FF of 0.89% as compared to 0.07% for pure water. The observed result is far from being optimal as the FE could not exceed more than 1%. Alkali metal ions are also known to suppress the HER and have a strong ability to interact with molecular N2 in an electrolyte. Selection of counterions in the aqueous electrolyte was found to have a significant role with an order of Li+ > Na+ > K+ in terms of NH3 yield rate, which suggests a most favourable role of the smallest counterions. The steric effect and the relatively strong interaction between counterions and N2 play important roles in ENRR.35,229 To enhance the FE, 0.1 M LiCl/ethylenediamine (EDA) and 0.05 M H2SO4 solution were used as an electrolyte in the cathode and anode compartment, respectively, to study the effect of EDA towards ENRR. A much higher FE of 17.2% was obtained under ambient conditions under this condition, possibly due to the wide electrochemical window of EDA in the negative potential region.230 Similarly, a mixture of water/alcohols and aprotogenic (tetrahydrofuran, dimethyl sulfoxide) solvents were used as an electrolyte for ENRR.231,232 In this regard, Lee et al. used a hydrophobic MOF (ZIF) coated Ag–Au catalyst to accumulate N2 molecules at the catalyst surface to suppress competitive HER. A mixed solvent containing ∼1% ethanol in dry tetrahydrofuran (THF) was used as an electrolyte to enhance ENRR selectivity. Ag–Au@ZIF showed a FE of 18 ± 4% under ambient conditions, far superior to many other ENRR studies having <1% FE at elevated temperature/pressure.233 Electrolytes consisting of a mixture of water and an ionic liquid (IL) were also reported.234,235 MacFarlane and co-workers reported a combination of hydrophobic and high nitrogen-solubility IL electrolytes, namely [C4mpyr][eFAP], [P6,6,6,14][eFAP], and [HMIM][NTf2], as electrolytes and tested ENRR using a nanostructured Fe-based electrocatalyst.236 The Fe-based catalyst deposited on a FTO substrate in the presence of [P6,6,6,14][eFAP] showed the highest FE of 60%, which is much higher than that obtained with [C4mpyr][eFAP] (30%) and [HMIM][NTf2] (0.64%). The high efficiency with [P6,6,6,14][eFAP] is attributed to the high solubility of N2 in ILs due to the strong interaction between N2 and the IL as confirmed by the DFT calculations. The DFT calculation suggests that the N2 molecules interact with ions such as Cl− and fluorinated anions BF4− and PF6− weakly, whereas they strongly interact with [eFAP]−. Two modes of favourable interactions were identified, namely in complex 1, the N2 atoms interact with the F atoms of the alkaline chains, and in complex 2, the N2 atoms interact with F atoms bonded with phosphorus as presented in Fig. 12. The N2 binding energy depends strongly on the delocalization of the negative charge. The interaction further increases with the addition of the cation.236
:9, v/v) mixture as an electrolyte along with H2SO4 as the supporting electrolyte.228 The mixed solvent gave rise to a FF of 0.89% as compared to 0.07% for pure water. The observed result is far from being optimal as the FE could not exceed more than 1%. Alkali metal ions are also known to suppress the HER and have a strong ability to interact with molecular N2 in an electrolyte. Selection of counterions in the aqueous electrolyte was found to have a significant role with an order of Li+ > Na+ > K+ in terms of NH3 yield rate, which suggests a most favourable role of the smallest counterions. The steric effect and the relatively strong interaction between counterions and N2 play important roles in ENRR.35,229 To enhance the FE, 0.1 M LiCl/ethylenediamine (EDA) and 0.05 M H2SO4 solution were used as an electrolyte in the cathode and anode compartment, respectively, to study the effect of EDA towards ENRR. A much higher FE of 17.2% was obtained under ambient conditions under this condition, possibly due to the wide electrochemical window of EDA in the negative potential region.230 Similarly, a mixture of water/alcohols and aprotogenic (tetrahydrofuran, dimethyl sulfoxide) solvents were used as an electrolyte for ENRR.231,232 In this regard, Lee et al. used a hydrophobic MOF (ZIF) coated Ag–Au catalyst to accumulate N2 molecules at the catalyst surface to suppress competitive HER. A mixed solvent containing ∼1% ethanol in dry tetrahydrofuran (THF) was used as an electrolyte to enhance ENRR selectivity. Ag–Au@ZIF showed a FE of 18 ± 4% under ambient conditions, far superior to many other ENRR studies having <1% FE at elevated temperature/pressure.233 Electrolytes consisting of a mixture of water and an ionic liquid (IL) were also reported.234,235 MacFarlane and co-workers reported a combination of hydrophobic and high nitrogen-solubility IL electrolytes, namely [C4mpyr][eFAP], [P6,6,6,14][eFAP], and [HMIM][NTf2], as electrolytes and tested ENRR using a nanostructured Fe-based electrocatalyst.236 The Fe-based catalyst deposited on a FTO substrate in the presence of [P6,6,6,14][eFAP] showed the highest FE of 60%, which is much higher than that obtained with [C4mpyr][eFAP] (30%) and [HMIM][NTf2] (0.64%). The high efficiency with [P6,6,6,14][eFAP] is attributed to the high solubility of N2 in ILs due to the strong interaction between N2 and the IL as confirmed by the DFT calculations. The DFT calculation suggests that the N2 molecules interact with ions such as Cl− and fluorinated anions BF4− and PF6− weakly, whereas they strongly interact with [eFAP]−. Two modes of favourable interactions were identified, namely in complex 1, the N2 atoms interact with the F atoms of the alkaline chains, and in complex 2, the N2 atoms interact with F atoms bonded with phosphorus as presented in Fig. 12. The N2 binding energy depends strongly on the delocalization of the negative charge. The interaction further increases with the addition of the cation.236
 | ||
| Fig. 12 (a)–(c) N2 binding energies (in kcal mol−1) and bond distances in Å for various anions. The interaction with [eFAP]− in panel (c) shows two distinct modes: complex 1 exhibits C–F interactions and complex 2 exhibits P–F interactions. Panel (d) introduces the cation to show the interaction with an ion pair of [C4mpyr][eFAP]; the interaction is the strongest of all those studied. Reproduced with permission from ref. 236. Copyright 2017, RSC. | ||
Licht et al. used a molten hydroxide suspension of nano-Fe2O3 named the NaOH–KOH eutectic electrolyte, which showed high rates of ammonia generation upon addition of nano-Fe2O3 at modest temperatures of 200–250 °C.5 As per the recent reports on ENRR, electrolytes play a vital role in HER suppression and enhancement of ENRR. Nevertheless, more experimental and theoretical studies could further improve ENRR under ambient conditions.
12.1 Lithium-mediated N2 reduction reaction
As mentioned in the previous section, not only the catalyst but also the electrolyte plays an important role in ENRR. Among the different electrolytes tested for ENRR, a lithium-mediated ENRR is found to be the most promising one. Typically the lithium-mediated ENRR is performed in tetrahydrofuran solvent that can dissolve a high amount of N2 (as high as ∼6 mM) at 25 °C. The mechanism of Li-mediated N2 reduction is hypothesized as the reduction of Li+ ions in the presence of N2 forming lithium nitride (Li3N) at the cathode. The Li3N species react with a proton source and produce NH3. Ethanol has been used as a proton source for nitride to NH3 conversion. In 1994, Tsuneto et al. reported a Li-mediated ammonia synthesis with a yield rate of ∼2 to 4 nmol s−1 cm−2 at 50 bar of N2 pressure.231 McEnaney et al. experimentally proposed Li-recycling as a tool to produce NH3 using renewable energy.237 First, a highly active Li metal surface was synthesized from LiOH electrolysis in a proton-free environment and then exposed to N2 to form Li3N. Suryanto et al. recently demonstrated a phosphonium based cation such as trihexyltetradecylphosphonium ([P6,6,6,14]+) as a proton source for lithium-mediated NH3 synthesis.238 [P6,6,6,14]+ is known to have excellent thermal, chemical, and electrochemical durability. The schematic reaction of electrochemical synthesis of NH3 is presented in Fig. 13. Herein, electrochemically generated Li3N is reacted with the phosphonium cation to produce NH3 and form a ylide as a derivative. The original phosphonium cation is regenerated from the protonation of phosphonium ylide which is a strong base. The author and his group have achieved the best performance at −0.75 V versus Li0/+ with N2 to NH3 conversion FE as high as 69 ± 7% and an NH3 yield rate of 58 ± 14 nmol s−1 cm−2, at an average current density of about −22.5 mA cm−2. The high reduction potential for the generation of the Li plate simultaneously leads to degradation of the electrode interface over time. The continuous regeneration from the Li salt thus limits the scalability of the process. Moreover, Schwalbe et al. observed the development of a solid electrolyte interface (SEI) layer as well as the accumulation of lithium and lithium-containing species at the electrode surface.239 In this regard, Andersen et al. succeeded in developing a stable cycle of Li-mediated NRR through the implementation of the pulse deposition strategy as presented in Fig. 14.240 Using this method, the authors achieved a FE of ∼30%. They revealed that the SEI layer not only prevents the catalyst from poisoning but also maintains its long-term Li cycling ability. The SEI layer is further found to control the diffusion rate during the electrolysis process. | ||
| Fig. 13 Schematic illustration of sustainable electrosynthesis of ammonia from H2 and N2. The cathode process involves the reduction of Li+ to the lithium metal that rapidly reacts with N2 to produce Li3N, which is protonated to release the target product, NH3. The proton carrier is the phosphonium cation, which forms an ylide structure in its deprotonated state; this cycles to the anode where it is regenerated back to the phosphonium form through the reaction with H+ derived from the oxidation of H2. Reproduced with permission from ref. 238. Copyright 2021, AAAS. | ||
 | ||
| Fig. 14 (a) Cycling method between −2.0 and 0.0 mA cm−2 (red), for a total of 100C of charge passed (black). The WE potential (blue) and the CE potential (green) are stable across the entire experiment by varying the resting time. (b) A close-up of the cycling. Immediately after switching to a deposition current, the absolute value of the WE potential increases for the entire 1 min duration. When switching back to resting, the WE potential is initially stable just above 0 V vs. Li+/Li, until it eventually starts becoming anodic after some minutes, suggesting dissolution of all surface Li species that were deposited in the preceding deposition pulse. At this point, another Li depositing pulse is applied. As the experiment did not have constant supervision, there are occasional points where the resting WE potential becomes very positive (e.g. around 22 and 40 hours) before the deposition pulse is applied. This is simply changed by shortening the resting time. Reproduced with permission from ref. 240. Copyright 2020, RSC. | ||
13. Sources of false positives in ENRR and their elimination
Electrochemical N2 reduction has gained momentum in recent years due to its potential feasibility under ambient conditions, unlike the Haber–Bosch process. However, the large difference in yield rate and FE raised serious concerns about its actual applicability. Therefore, it is important to evaluate the pitfalls of ENRR, in particular, the inconvenient sources of error and their elimination from ENRR during the estimation of FE and yield rate. The recent developments of the catalyst for ENRR have been discussed in detail in the previous sections. However, a question may arise whether all the catalysts catalyze ENRR under ambient conditions? As we have seen, the conversion rates are in the range of nano or micromolar range with most of the catalysts. Such a low value could be contributed from other sources and/or impurities. For example, Ru is considered as one of the prominent candidates for ENRR in aqueous electrolytes. Theoretical as well as experimental evidence for the same is discussed earlier. However, Andersen et al. reported false-positive results with Ru-based catalysts.241 Interestingly, the same catalyst does not reduce N2 at all under ambient conditions in aqueous electrolytes. The author and his group carried out a thorough investigation and revealed that the false-positives arise from NOx impurities present in the N2 feed stream. Thus it is very important to eliminate such impurities to find the true nature of the catalyst in ENRR. In this regard, the authors have further proposed modified methods to carry out ENRR as presented in Fig. 15. The readers are urged to go through the modified methods for accurate measurement of NH3 production through ENRR using a suitable catalyst. In this regard, Suryanto et al. and Choi et al. reported excellent perspectives on the steps and missteps made in the ENRR study and suggested the directions to minimize missteps during the ENRR under ambient conditions.242,243 A few recent studies thus clearly suggest sources of error for the estimation FE and yield rate of ENRR under ambient conditions in an aqueous electrolyte. It is important to identify such errors and eliminate those for correct measurements of NH3 yield and FE. In the following sections, the sources of errors that could potentially provide false positives in ENRR are elaborated. | ||
| Fig. 15 Suggested protocol for the benchmarking of electrochemical nitrogen reduction. Reproduced with permission from ref. 241. Copyright 2019, Nature Research. | ||
13.1 Nitrogen-containing catalyst
Nitrogen-containing electrocatalysts, especially metal nitrides have been explored as efficient catalysts for ENRR. The mechanism of ENRR is generally explained by the Mars van Krevelen (MvK) mechanism which is discussed earlier. Recently Du et al. investigated the ENRR on vanadium nitride (VN) and niobium nitride (Nb4N5) as model electrocatalysts to validate the ENRR at different pH and raised serious questions about the feasibility of these catalysts toward ENRR.150 The authors revealed that VN and Nd4N5 do not catalyze N2 under ambient conditions. The false-positive for NH3 arises from the nitrogen atoms present in the catalyst instead of true ENRR. The N-species present in the catalyst surface may lead to an erroneous conclusion. Therefore, it is important to take necessary precautions to avoid such missteps during ENRR. One simple strategy that could be implemented to avoid and remove the contaminants from the catalyst is rigorous prewashing and following the pre-treatment method several times before studying the ENRR performance. The obtained pretreatment results must be subtracted from the final FE and yield rate.13.2 Electrolyte
The false-positive detection of NH3 can arise from the electrolyte. Li et al. reported the identification process and elimination of contamination in an electrolyte which provides the false-positive.244 The investigation found that a trace amount of nitrate and nitrite salts exist in the lithium-based electrolyte. Thus a significant amount of NH3 is produced with excellent reproducibility in the presence of bare electrodes (Ti foil, carbon paper, or copper foam) without loading any catalyst in both N2 and Ar-saturated Li2SO4 solution. The detected NH3 arises from the trace amount of nitrate and nitrite present in the electrolyte which is electrochemically reduced to NH3 rather than N2. In this regard, the author studied the ENRR using bare Ti foil as the working electrode and Li2SO4 as the electrolyte. The false ENRR to NH3 as measured using the indophenol method is presented in Fig. 16. In particular, an apparent increase in absorbance is found in the experiments performed in both N2 and Ar atmospheres, which confirms that the NH3 yield is not from ENRR but due to contamination in the electrolyte. To eliminate the NOx contamination from the electrolyte, the author annealed the Li2SO4 salt at 800 °C in an argon atmosphere. After annealing the electrolyte, the NH3 yield was found to be significantly reduced. This suggests that the presence of contamination in the electrolyte contributed to NH3 formation, thus giving the false-positive test. Therefore it is important to identify the source of contamination in aqueous electrolytes and eliminate it to estimate the correct NH3 yield rate. | ||
| Fig. 16 False positive NRR performance observed in both N2 and Ar atmospheres using a bare Ti foil without loading any catalyst in a 0.5 M Li2SO4 solution. (a) UV-Vis spectra for the indophenol blue indicator stained electrolytes after electrolysis at −0.1, −0.5, −1.0, and −2.0 mA cm−2 in N2 for 1 h and (b) the corresponding ammonia yield rates at each current density. (c) UV-Vis spectra for the indophenol blue indicator stained electrolytes after electrolysis at −1.0 mA cm−2 in N2 for 1, 2, and 3 h. (d) UV-Vis spectra for the indophenol blue indicator stained electrolytes after electrolysis at −0.5 and −1.0 mA cm−2 in Ar for 1 h. (e) UV-Vis spectra for the indophenol blue indicator stained electrolytes after electrolysis at −1.0 mA cm−2 in Ar for 1–16 h and (f) the corresponding accumulative ammonia yield. In (a) and (d), the solid lines represent the results for the second cycle and the dashed lines show the results for the third cycle. In (a), (c), (d), and (e), the lowest line shows the result for the fresh electrolyte before electrolysis. Reproduced with permission from ref. 244. Copyright 2019, ACS. | ||
13.3 Membrane separator
Currently, most of the reported ENRR studies are carried out in an H-cell with Nafion membrane as a separator. However, the Nafion membrane can absorb and release NH3 during the ENRR as reported by Liu et al.245 This suggests that the Nafion membrane is a possible source of error in the ENRR process. On the other hand, Celgard 3401 and 3501 has poor ability to absorb NH3 as compared to Nafion and other membranes and thus must be considered for use in ENRR studies.13.4 Gas purity
The feed gas must be ultrapure and devoid of any atmospheric contamination. The main source of contamination is NOx species present in the N2 feed gas (14N2 as well as 15N2). Dabundo et al. reported that the trace amount of NOx in the N2 feedstock is enough to give a false positive test.246 It must be noted that the electrochemical reduction of NOx species to N2 is energetically favourable.243 Thus enough care must be taken to eliminate any signal due to the contamination in the gas feed. Choi et al. suggested NOx scrubbing to eliminate NOx from the N2 feed gas.243 To evaluate the FE and yield rate of NH3, it is thus necessary to subtract the background signal from the ENRR signal obtained in the presence of N2.13.5 Background subtraction
To eliminate false positives from the experimental results, it is necessary to perform control experiments at open circuit potential and in the absence/presence of a catalyst in the absence and presence of Ar/N2. The control experiments will provide clarity on the actual performance of the catalyst. However, many studies either ignore or do not perform sufficient and satisfactory control experiments.242,243 This leads to potential errors in the assessment of the FE and yield rate of ENRR. Likewise, some reports have presented control experiments where the background NH3 is significant, and thus it cannot be ignored.242,243 Therefore the researchers are suggested to perform control experiments as prescribed above to eliminate the potential error from the results.14. Conclusions
For a sustainable global energy economy, persistent effort is needed through research and development to generate products from renewable resources through different strategies. The present context reveals that ENRR could serve as a potential alternative to synthesize NH3 against the conventional fossil fuel-based energy economy even though ENRR is an energy-intensive reaction. In the present review, we have detailed the roles of the electrocatalysts in facilitating the ENRR at room temperature, which is the ultimate goal of the scientific community. To date, noble metal electrocatalysts such as Pd, Au, and Ru along with a few non-noble metal electrocatalysts have exhibited promising ENRR efficiency while keeping a lot of space for further improvement. This review highlights several key aspects that are important to develop efficient catalysts while taking into account the key features that control the catalyst efficiency and selectivity including nicks, steps, facets, porosity, and coordination number of the surface atoms. Designing nanostructured electrocatalysts with an optimized surface structure facilitates N2 adsorption and thereby ENRR. In particular, nanostructuring increases the active site density with a specific structure. To obtain enormous active sites on the catalyst surface, various synthesis routes have been developed in recent years. Furthermore, to improve the mass transport kinetics across the electrode/electrolyte interface, the catalyst can be anchored on a suitable support material, which allows minimal use of the catalyst while improving the product yield. Herein, we summarize the key achievements in catalyst development for ENNR.(1) The catalytic activity significantly increases especially for the single atomic electrocatalyst supported and/or stabilized by the N-site over the pure bulk catalyst. The single atomic catalyst site at the surface is coordinated by different numbers of N-sites, which act as active sites to control the N2 reduction kinetics steps. The metal to N site coordination number strongly influences the rate-limiting step for ENRR.
(2) The oxygen vacancy in the oxide-based catalysts intrinsically enhances the catalytic conversion rate of N2 to NH3. The oxygen vacant site of the catalysts plays a predominant role in the activation of N2 during the progression of the reaction and hence decreases the activation energy barrier for ENRR.
(3) Doping is another approach to obtain efficient catalysts that generate active sites for ENRR. The doping sites facilitate the adsorption as well as activation of N2.
(4) Support materials integrated with catalysts play a crucial role in the suppression of HER. Especially, carbon shelling around the catalyst is found to play a prominent role in suppressing HER activity at the catalyst surface under a potential and facilitate the ENRR. Support materials also increase the catalyst distribution in the synthesis process and enhance mass transfer kinetics during the electrolysis process.
15. Challenges
Although impressive progress has been made in ENRR, there are still several major challenges that need to be addressed for the commercialization of the process. One of the major challenges associated with ENRR is the yield rate and FE. So far, the yield rate and FE are not up to the mark. In particular, the yield rate is mostly in micrograms per hour per milligram of catalyst or per centimetre square and FE is mostly less than 10% with a few exceptions. Therefore, more efforts are needed for the enhancement of the yield rate and FE of ENRR. The requirement of higher negative potential for ENRR is another major obstacle that is associated with HER with almost every catalyst. Thus, efficient electrocatalysts must be explored which could show ENRR at lower negative potentials to make it viable for commercial application. Hence, efforts should be directed to suppress the HER activity while increasing the FE of the catalyst for ENRR. The theoretical calculation provides a certain model to mitigate HER; however, the feasibility of this active model is still unclear. Identification of suitable electrolytes is another feature that can increase the solubility of N2 while reducing the mass transport for HER and thereby increase the ENRR efficiency. Another important challenge associated with ENRR is measuring the evolved NH3 through the ENRR correctly and precisely. Considering a very small yield of NH3 (in microgram quantity per hour) through ENRR, it could be from other sources such as the atmosphere, supplied N2, and nitrogen-containing chemicals or catalysts used in ENRR. Among these, the supplied N2 source is the main source of error as it often contains NOx species that are easily reducible to NH3. Similarly, nitrogen-containing catalysts and electrolytes are other potential sources of NH3 release through ENNR. Thus precautions must be taken to avoid these errors which show erroneous results. Furthermore, the most commonly used indophenol blue method is not suitable for low NH3 yield measurement. For detecting small quantities of NH3 yield, the need for sophisticated spectroscopy techniques including isotope labelling is another challenge. To meet these challenges more attention is required from the scientific community.16. Future directions
As highlighted in the previous sections, the development of catalysts, effective methods of measurement, and improvement of NH3 yield are the immediate needs to make progress in realizing the ENRR in future. Although several electrocatalysts have been studied and they were suitably modified, the research on the same remains not substantial. In particular, the small-sized or single atomic catalytic centre could be a possible candidate to increase the active sites drastically and thus increase the NH3 yield and FE. The synthesis of desired reactive facets and forming uniform defects such as oxygen vacancies appear to be a couple of promising strategies for the development of catalysts for ENRR. Not only an electrocatalyst but also an appropriate electrolyte is another important aspect to make the ENRR process practically viable. The theoretical investigation along with experimental study can rationalize the ENRR and provide further insights to improve the performance. As ENRR is a more complex process than the HER and not well understood at the molecular level, computational studies could be highly beneficial. Considering that the active intermediates form during the ENRR, the role of the active site in the stabilization of active intermediates remains ambiguous. Some investigations suggested that the N2 adsorption ability of the catalyst is the key step whereas others suggested that co-ordinately unsaturated atoms are primarily responsible. Therefore, further experimental as well as theoretical investigation is necessary to understand the active species of the catalysts under the given conditions. The adsorption of N2 and electron-coupled proton transfer to adsorbed N2 are energetically uphill processes. Therefore, in situ measurements could provide clues to precisely identify the active site for ENRR. Finally, it is very important to follow the correct steps to measure the effective reduction of N2 to NH3 and measure the evolved NH3 accurately. The possible sources of NH3 contamination must be avoided and appropriate spectroscopic techniques must be followed to measure the low yield NH3. In summary, the ENRR to form NH3 has strong potential for the production of fertilizer and for use as an energy carrier after addressing the shortcomings which might take a few years from now. If progress is made in the right direction, reduction of N2 to NH3 will significantly reduce the global energy demand and carbon emission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Science and Engineering Research Board (SERB), New Delhi, India, through the research grant EMR/2017/000697.References
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- S.-J. Li, D. Bao, M.-M. Shi, B.-R. Wulan, J.-M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- S. Dahl, E. Törnqvist and I. Chorkendorff, J. Catal., 2000, 192, 381–390 CrossRef CAS.
- K. Chu, Y.-P. Liu, J. Wang and H. Zhang, ACS Appl. Energy Mater., 2019, 2, 2288–2295 CrossRef CAS.
- J. Deng, J. A. Iñiguez and C. Liu, Joule, 2018, 2, 846–856 CrossRef CAS.
- A. Chen and B. Y. Xia, J. Mater. Chem. A, 2019, 7, 23416–23431 RSC.
- X. Wen and J. Guan, Nanoscale, 2020, 12, 8065–8094 RSC.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- X. Chen, Y. Guo, X. Du, Y. Zeng, J. Chu, C. Gong, J. Huang, C. Fan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903172 CrossRef CAS.
- J. Wang, Y. Liu, H. Zhang, D. Huang and K. Chu, Catal. Sci. Technol., 2019, 9, 4248–4254 RSC.
- X.-F. Li, Q.-K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X.-H. Zhang, Z.-Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- Y. Abghoui, A. L. Garden, J. G. Howalt, T. Vegge and E. Skúlason, ACS Catal., 2016, 6, 635–646 CrossRef CAS.
- J. Nash, X. Yang, J. Anibal, M. Dunwell, S. Yao, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Phys. Chem. C, 2019, 123, 23967–23975 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed.
- H. Xian, H. Guo, Z. Chen, G. Yu, A. A. Alshehri, K. A. Alzahrani, F. Hao, R. Song and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2445–2451 CrossRef CAS PubMed.
- R. Lan, J. T. S. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- R. Lan and S. Tao, RSC Adv., 2013, 3, 18016–18021 RSC.
- J. Nash, X. Yang, J. Anibal, J. Wang, Y. Yan and B. Xu, J. Electrochem. Soc., 2017, 164, F1712 CrossRef CAS.
- L. Sheets and G. G. Botte, Chem. Commun., 2018, 54, 4250–4253 RSC.
- K. Kugler, M. Luhn, J. A. Schramm, K. Rahimi and M. Wessling, Phys. Chem. Chem. Phys., 2015, 17, 3768–3782 RSC.
- H.-M. Liu, S.-H. Han, Y. Zhao, Y.-Y. Zhu, X.-L. Tian, J.-H. Zeng, J.-X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- T. Chen, S. Liu, H. Ying, Z. Li and J. Hao, Chem. – Asian J., 2020, 15, 1081–1087 CrossRef CAS PubMed.
- T. Xiao, J. Huang, D. Wang, T. Meng and X. Yang, Talanta, 2020, 206, 120210 CrossRef CAS PubMed.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS PubMed.
- Y. Yu, F. Cui, J. Sun and P. Yang, Nano Lett., 2016, 16, 3078–3084 CrossRef CAS PubMed.
- R. Omar, A. En Naciri, S. Jradi, Y. Battie, J. Toufaily, H. Mortada and S. Akil, J. Mater. Chem. C, 2017, 5, 10813–10821 RSC.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- Z. Wang, Y. Li, H. Yu, Y. Xu, H. Xue, X. Li, H. Wang and L. Wang, ChemSusChem, 2018, 11, 3480–3485 CrossRef CAS PubMed.
- K. Zhang, R. Guo, F. Pang, J. He and W. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 10214–10220 CrossRef CAS.
- C. Chen, C. Liang, J. Xu, J. Wei, X. Li, Y. Zheng, J. Li, H. Tang and J. Li, Electrochim. Acta, 2020, 335, 135708 CrossRef CAS.
- M. Nazemi, S. R. Panikkanvalappil and M. A. El-Sayed, Nano Energy, 2018, 49, 316–323 CrossRef CAS.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- J. Zheng, Y. Lyu, M. Qiao, J. P. Veder, R. D. Marco, J. Bradley, R. Wang, Y. Li, A. Huang, S. P. Jiang and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 18604–18609 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., 2015, 127, 5268–5271 CrossRef.
- J. Fester, A. Makoveev, D. Grumelli, R. Gutzler, Z. Sun, J. Rodríguez-Fernández, K. Kern and J. V. Lauritsen, Angew. Chem., Int. Ed., 2018, 57, 11893–12073 CrossRef CAS PubMed.
- C. Li, Q. Yuan, B. Ni, T. He, S. Zhang, Y. Long, L. Gu and X. Wang, Nat. Commun., 2018, 9, 3702 CrossRef PubMed.
- Z.-H. Xue, S.-N. Zhang, Y.-X. Lin, H. Su, G.-Y. Zhai, J.-T. Han, Q.-Y. Yu, X.-H. Li, M. Antonietti and J.-S. Chen, J. Am. Chem. Soc., 2019, 141, 14976–14980 CrossRef CAS PubMed.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202, 420–448 CrossRef CAS.
- S. Back and Y. Jung, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- T. H. Rod, A. Logadottir and J. K. Nørskov, J. Chem. Phys., 2000, 112, 5343–5347 CrossRef CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- Z. Zhang, K. Yao, L. Cong, Z. Yu, L. Qu and W. Huang, Catal. Sci. Technol., 2020, 10, 1336–1342 RSC.
- Y. Yao, H. Wang, X. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, Q. Yang, W. Guan, L. Gao, X. Liang and T. Ma, Nanoscale, 2020, 12, 10933–10938 RSC.
- M. Shao, T. Yu, J. H. Odell, M. Jin and Y. Xia, Chem. Commun., 2011, 47, 6566–6568 RSC.
- C. Alegre, A. Stassi, E. Modica, C. Lo Vecchio, A. S. Aricò and V. Baglio, RSC Adv., 2015, 5, 25424–25427 RSC.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Nano Energy, 2016, 29, 198–219 CrossRef CAS.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- J. Lv, S. Wu, Z. Tian, Y. Ye, J. Liu and C. Liang, J. Mater. Chem. A, 2019, 7, 12627–12634 RSC.
- J. Lv, Z. Tian, K. Dai, Y. Ye and C. Liang, J. Colloid Interface Sci., 2019, 553, 126–135 CrossRef CAS PubMed.
- W. Xu, G. Fan, J. Chen, J. Li, L. Zhang, S. Zhu, X. Su, F. Cheng and J. Chen, Angew. Chem., 2020, 132, 3539–3544 CrossRef.
- H. Wang, Y. Chen, R. Fan, J. Chen, Z. Wang, S. Mao and Y. Wang, Research, 2019, 2019, 1401209 CAS.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- S. Mukherjee, X. Yang, W. Shan, W. Samarakoon, S. Karakalos, D. A. Cullen, K. More, M. Wang, Z. Feng, G. Wang and G. Wu, Small Methods, 2020, 4, 1900821 CrossRef CAS.
- W. Zang, T. Yang, H. Zou, S. Xi, H. Zhang, X. Liu, Z. Kou, Y. Du, Y. P. Feng, L. Shen, L. Duan, J. Wang and S. J. Pennycook, ACS Catal., 2019, 9, 10166–10173 CrossRef CAS.
- Y.-X. Lin, S.-N. Zhang, Z.-H. Xue, J.-J. Zhang, H. Su, T.-J. Zhao, G.-Y. Zhai, X.-H. Li, M. Antonietti and J.-S. Chen, Nat. Commun., 2019, 10, 4380 CrossRef PubMed.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- J. Chen, H. Wang, Z. Wang, S. Mao, J. Yu, Y. Wang and Y. Wang, ACS Catal., 2019, 9, 5302–5307 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Y.-C. Hao, Y. Guo, L.-W. Chen, M. Shu, X.-Y. Wang, T.-A. Bu, W.-Y. Gao, N. Zhang, X. Su, X. Feng, J.-W. Zhou, B. Wang, C.-W. Hu, A.-X. Yin, R. Si, Y.-W. Zhang and C.-H. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- Q. Hao, C. Liu, G. Jia, Y. Wang, H. Arandiyan, W. Wei and B.-J. Ni, Mater. Horiz., 2020, 7, 1014–1029 RSC.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- Y. Wang, M.-M. Shi, D. Bao, F.-L. Meng, Q. Zhang, Y.-T. Zhou, K.-H. Liu, Y. Zhang, J.-Z. Wang, Z.-W. Chen, D.-P. Liu, Z. Jiang, M. Luo, L. Gu, Q.-H. Zhang, X.-Z. Cao, Y. Yao, M.-H. Shao, Y. Zhang, X.-B. Zhang, J. G. Chen, J.-M. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2019, 58, 9464–9469 CrossRef CAS PubMed.
- L. Xia, W. Fu, P. Zhuang, Y. Cao, M. O. L. Chee, P. Dong, M. Ye and J. Shen, ACS Sustainable Chem. Eng., 2020, 8, 2735–2741 CrossRef CAS.
- Y. Qiu, S. Zhao, M. Qin, J. Diao, S. Liu, L. Dai, W. Zhang and X. Guo, Inorg. Chem. Front., 2020, 7, 2006–2016 RSC.
- Y. Xu, T. Ren, S. Yu, H. Yu, S. Yin, Z. Wang, X. Li, L. Wang and H. Wang, Sustainable Energy Fuels, 2020, 4, 3334–3339 RSC.
- K. Bhunia, M. Chandra and D. Pradhan, J. Nanosci. Nanotechnol., 2018, 19, 332–355 CrossRef PubMed.
- K. Bhunia, S. Khilari and D. Pradhan, ACS Sustainable Chem. Eng., 2018, 6, 7769–7778 CrossRef CAS.
- H. Wang, Y. Li, C. Li, K. Deng, Z. Wang, Y. Xu, X. Li, H. Xue and L. Wang, J. Mater. Chem. A, 2019, 7, 801–805 RSC.
- F. Pang, Z. Wang, K. Zhang, J. He, W. Zhang, C. Guo and Y. Ding, Nano Energy, 2019, 58, 834–841 CrossRef CAS.
- H. Yu, Z. Wang, D. Yang, X. Qian, Y. Xu, X. Li, H. Wang and L. Wang, J. Mater. Chem. A, 2019, 7, 12526–12531 RSC.
- J. Wang, B. Huang, Y. Ji, M. Sun, T. Wu, R. Yin, X. Zhu, Y. Li, Q. Shao and X. Huang, Adv. Mater., 2020, 32, 1907112 CrossRef CAS PubMed.
- X. Zhang, T. Wu, H. Wang, R. Zhao, H. Chen, T. Wang, P. Wei, Y. Luo, Y. Zhang and X. Sun, ACS Catal., 2019, 9, 4609–4615 CrossRef CAS.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed.
- C. Li, S. Mou, X. Zhu, F. Wang, Y. Wang, Y. Qiao, X. Shi, Y. Luo, B. Zheng, Q. Li and X. Sun, Chem. Commun., 2019, 55, 14474–14477 RSC.
- Y. Liu, L. Huang, X. Zhu, Y. Fang and S. Dong, Nanoscale, 2020, 12, 1811–1816 RSC.
- J. Bai, H. Huang, F.-M. Li, Y. Zhao, P. Chen, P.-J. Jin, S.-N. Li, H.-C. Yao, J.-H. Zeng and Y. Chen, J. Mater. Chem. A, 2019, 7, 21149–21156 RSC.
- C. Fang, J. Hu, X. Jiang, Z. Cui, X. Xu and T. Bi, Inorg. Chem. Front., 2020, 7, 1411–1419 RSC.
- W. Tong, B. Huang, P. Wang, Q. Shao and X. Huang, Natl. Sci. Rev., 2021, 8, nwaa088 CrossRef CAS PubMed.
- K. Chu, W. Gu, Q. Li, Y. Liu, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 82–89 CrossRef.
- N. Zhang, L. Li, J. Wang, Z. Hu, Q. Shao, X. Xiao and X. Huang, Angew. Chem., 2020, 132, 8143–8148 CrossRef.
- F. Pang, F. Wang, L. Yang, Z. Wang and W. Zhang, Chem. Commun., 2019, 55, 10108–10111 RSC.
- R. D. Kumar, Z. Wang, C. Li, A. V. Narendra Kumar, H. Xue, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2019, 7, 3190–3196 RSC.
- J. Guo, H. Wang, F. Xue, D. Yu, L. Zhang, S. Jiao, Y. Liu, Y. Lu, M. Liu, S. Ruan, Y.-J. Zeng, C. Ma and H. Huang, J. Mater. Chem. A, 2019, 7, 20247–20253 RSC.
- Z. Wang, C. Li, K. Deng, Y. Xu, H. Xue, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 2400–2405 CrossRef CAS.
- J. Desilvestro and O. Haas, J. Electrochem. Soc., 1990, 137, 5C–22C CrossRef CAS.
- G. Maduraiveeran, M. Sasidharan and W. Jin, Prog. Mater. Sci., 2019, 106, 100574 CrossRef CAS.
- R. Zhang, X. Ren, X. Shi, F. Xie, B. Zheng, X. Guo and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- M.-T. Nguyen, N. Seriani and R. Gebauer, Phys. Chem. Chem. Phys., 2015, 17, 14317–14322 RSC.
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Small, 2018, 14, 1803111 CrossRef PubMed.
- X. Wang, J. Wang, Y. Li and K. Chu, ChemCatChem, 2019, 11, 4529–4536 CrossRef CAS.
- X.-W. Lv, Y. Liu, R. Hao, W. Tian and Z.-Y. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 17502–17508 CrossRef CAS PubMed.
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, Cambridge, 1994 Search PubMed.
- C. Fang, T. Bi, X. Xu, N. Yu, Z. Cui, R. Jiang and B. Geng, Adv. Mater. Interfaces, 2019, 6, 1901034 CrossRef CAS.
- B. Li, X. Zhu, J. Wang, R. Xing, Q. Liu, X. Shi, Y. Luo, S. Liu, X. Niu and X. Sun, Chem. Commun., 2020, 56, 1074–1077 RSC.
- Q. Qin, Y. Zhao, M. Schmallegger, T. Heil, J. Schmidt, R. Walczak, G. Gescheidt-Demner, H. Jiao and M. Oschatz, Angew. Chem., Int. Ed., 2019, 58, 13101–13106 CrossRef CAS PubMed.
- H. Huang, F. Gong, Y. Wang, H. Wang, X. Wu, W. Lu, R. Zhao, H. Chen, X. Shi, A. M. Asiri, T. Li, Q. Liu and X. Sun, Nano Res., 2019, 12, 1093–1098 CrossRef CAS.
- Y. Liu, Y. Li, D. Huang, H. Zhang and K. Chu, Chem. – Eur. J., 2019, 25, 11933–11939 CrossRef CAS PubMed.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- G. Marnellos, S. Zisekas and M. Stoukides, J. Catal., 2000, 193, 80–87 CrossRef CAS.
- X. Dong, Z. Xu, X. Chang, C. Zhang and W. Jin, J. Am. Ceram. Soc., 2007, 90, 3923–3929 CAS.
- C. Yu and S. B. Adler, Chem. Mater., 2005, 17, 4537–4546 CrossRef.
- I. A. Amar, R. Lan and S. Tao, J. Electrochem. Soc., 2014, 161, H350–H354 CrossRef CAS.
- G. Xu, R. Liu and J. Wang, Sci. China, Ser. B: Chem., 2009, 52, 1171 CrossRef CAS.
- Y. Liu, X. Kong, X. Guo, Q. Li, J. Ke, R. Wang, Q. Li, Z. Geng and J. Zeng, ACS Catal., 2020, 10, 1077–1085 CrossRef CAS.
- I. A. Amar, R. Lan, C. T. G. Petit, V. Arrighi and S. Tao, Solid State Ionics, 2011, 182, 133–138 CrossRef CAS.
- I. A. Amar, C. T. G. Petit, G. Mann, R. Lan, P. J. Skabara and S. Tao, Int. J. Hydrogen Energy, 2014, 39, 4322–4330 CrossRef CAS.
- M. I. Ahmed, S. Chen, W. Ren, X. Chen and C. Zhao, Chem. Commun., 2019, 55, 12184–12187 RSC.
- C. Li, J. Yu, L. Yang, J. Zhao, W. Kong, T. Wang, A. M. Asiri, Q. Li and X. Sun, Inorg. Chem., 2019, 58, 9597–9601 CrossRef CAS PubMed.
- F. Lai, J. Feng, X. Ye, W. Zong, G. He, C. Yang, W. Wang, Y.-E. Miao, B. Pan, W. Yan, T. Liu and I. P. Parkin, J. Mater. Chem. A, 2020, 8, 1652–1659 RSC.
- Y. Wang, K. Jia, Q. Pan, Y. Xu, Q. Liu, G. Cui, X. Guo and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 117–122 CrossRef CAS.
- T. Wu, W. Kong, Y. Zhang, Z. Xing, J. Zhao, T. Wang, X. Shi, Y. Luo and X. Sun, Small Methods, 2019, 3, 1900356 CrossRef CAS.
- K. Jia, Y. Wang, Q. Pan, B. Zhong, Y. Luo, G. Cui, X. Guo and X. Sun, Nanoscale Adv., 2019, 1, 961–964 RSC.
- T. Wu, X. Zhu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 18449–18453 CrossRef CAS PubMed.
- R. Zhang, H. Guo, L. Yang, Y. Wang, Z. Niu, H. Huang, H. Chen, L. Xia, T. Li, X. Shi, X. Sun, B. Li and Q. Liu, ChemElectroChem, 2019, 6, 1014–1018 CrossRef CAS.
- H. Du, X. Guo, R.-M. Kong and F. Qu, Chem. Commun., 2018, 54, 12848–12851 RSC.
- K. Chu, Y. Liu, Y. Li, Y. Guo, Y. Tian and H. Zhang, Appl. Catal., B, 2020, 264, 118525 CrossRef CAS.
- X. Xiang, Z. Wang, X. Shi, M. Fan and X. Sun, ChemCatChem, 2018, 10, 4530–4535 CrossRef CAS.
- Y. Li, Y. Liu, J. Wang, Y. Guo and K. Chu, Inorg. Chem. Front., 2020, 7, 455–463 RSC.
- J. Wang, H. Jang, G. Li, M. G. Kim, Z. Wu, X. Liu and J. Cho, Nanoscale, 2020, 12, 1478–1483 RSC.
- J. Wu, Z. Wang, S. Li, S. Niu, Y. Zhang, J. Hu, J. Zhao and P. Xu, Chem. Commun., 2020, 56, 6834–6837 RSC.
- L. Zhang, M. Cong, X. Ding, Y. Jin, F. Xu, Y. Wang, L. Chen and L. Zhang, Angew. Chem., 2020, 132, 10980–10985 CrossRef.
- B. Chang, Q. Liu, N. Chen and Y. Yang, ChemCatChem, 2019, 11, 1884–1888 CrossRef CAS.
- B. Xu, L. Xia, F. Zhou, R. Zhao, H. Chen, T. Wang, Q. Zhou, Q. Liu, G. Cui, X. Xiong, F. Gong and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2889–2893 CrossRef CAS.
- H. Xie, H. Wang, Q. Geng, Z. Xing, W. Wang, J. Chen, L. Ji, L. Chang, Z. Wang and J. Mao, Inorg. Chem., 2019, 58, 5423–5427 CrossRef CAS PubMed.
- W. Fu, P. Zhuang, M. OliverLam Chee, P. Dong, M. Ye and J. Shen, ACS Sustainable Chem. Eng., 2019, 7, 9622–9628 CrossRef CAS.
- B. Xu, Z. Liu, W. Qiu, Q. Liu, X. Sun, G. Cui, Y. Wu and X. Xiong, Electrochim. Acta, 2019, 298, 106–111 CrossRef CAS.
- J. Yu, C. Li, B. Li, X. Zhu, R. Zhang, L. Ji, D. Tang, A. M. Asiri, X. Sun, Q. Li, S. Liu and Y. Luo, Chem. Commun., 2019, 55, 6401–6404 RSC.
- M. Ohrelius, H. Guo, H. Xian, G. Yu, A. A. Alshehri, K. A. Alzahrani, T. Li and M. Andersson, ChemCatChem, 2020, 12, 731–735 CrossRef CAS.
- C. Li, D. Ma, S. Mou, Y. Luo, B. Ma, S. Lu, G. Cui, Q. Li, Q. Liu and X. Sun, J. Energy Chem., 2020, 50, 402–408 CrossRef.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef CAS PubMed.
- J. Tian, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., 2014, 126, 9731–9735 CrossRef.
- P. Jiang, Q. Liu, Y. Liang, J. Tian, A. M. Asiri and X. Sun, Angew. Chem., 2014, 126, 13069–13073 CrossRef.
- Y. Wang, B. Kong, D. Zhao, H. Wang and C. Selomulya, Nano Today, 2017, 15, 26–55 CrossRef CAS.
- X. Zhu, T. Wu, L. Ji, Q. Liu, Y. Luo, G. Cui, Y. Xiang, Y. Zhang, B. Zheng and X. Sun, Chem. Commun., 2020, 56, 731–734 RSC.
- M. Jin, X. Zhang, M. Han, H. Wang, G. Wang and H. Zhang, J. Mater. Chem. A, 2020, 8, 5936–5942 RSC.
- X. Yang, F. Ling, X. Zi, Y. Wang, H. Zhang, H. Zhang, M. Zhou, Z. Guo and Y. Wang, Small, 2020, 16, 2000421 CrossRef CAS PubMed.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Björgvinsdóttir, H. Ólafsdóttir and E. Skúlason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- X. Ren, G. Cui, L. Chen, F. Xie, Q. Wei, Z. Tian and X. Sun, Chem. Commun., 2018, 54, 8474–8477 RSC.
- H.-L. Du, T. R. Gengenbach, R. Hodgetts, D. R. MacFarlane and A. N. Simonov, ACS Sustainable Chem. Eng., 2019, 7, 6839–6850 CrossRef CAS.
- Y. Abghoui and E. Skúlason, Catal. Today, 2017, 286, 69–77 CrossRef CAS.
- L. Zhang, X. Ji, X. Ren, Y. Luo, X. Shi, A. M. Asiri, B. Zheng and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9550–9554 CrossRef CAS.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- B. Hu, M. Hu, L. Seefeldt and T. L. Liu, ACS Energy Lett., 2019, 4, 1053–1054 CrossRef CAS.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, X. Ren, G. Cui, A. M. Asiri, B. Zheng and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9545–9549 CrossRef CAS.
- D. J. Ham and J. S. Lee, Energies, 2009, 2, 873–899 CrossRef CAS.
- T. G. Kelly and J. G. Chen, Chem. Soc. Rev., 2012, 41, 8021–8034 RSC.
- Z. Yan, M. Cai and P. K. Shen, Sci. Rep., 2013, 3, 1646 CrossRef PubMed.
- G. Gao, A. P. O’Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- Y. Liu, T. G. Kelly, J. G. Chen and W. E. Mustain, ACS Catal., 2013, 3, 1184–1194 CrossRef CAS PubMed.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P.-L. Taberna, P. Simon, M. W. Barsoum and Y. Gogotsi, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- J. S. Jang, D. J. Ham, E. Ramasamy, J. Lee and J. S. Lee, Chem. Commun., 2010, 46, 8600–8602 RSC.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef PubMed.
- L. I. Johansson, Surf. Sci. Rep., 1995, 21, 177–250 CrossRef CAS.
- H. Jiang, Y. Yao, Y. Zhu, Y. Liu, Y. Su, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 21511–21520 CrossRef CAS PubMed.
- M. Kuang, W. Huang, C. Hegde, W. Fang, X. Tan, C. Liu, J. Ma and Q. Yan, Mater. Horiz., 2020, 7, 32–53 RSC.
- H. Cheng, L.-X. Ding, G.-F. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed.
- I. Matanovic and F. H. Garzon, Phys. Chem. Chem. Phys., 2018, 20, 14679–14687 RSC.
- K. P. Ramaiyan, S. Ozden, S. Maurya, D. Kelly, S. K. Babu, A. Benavidez, F. G. Garzon, Y. S. Kim, C. R. Kreller and R. Mukundan, J. Electrochem. Soc., 2020, 167, 44506 CrossRef CAS.
- X. Qu, L. Shen, Y. Mao, J. Lin, Y. Li, G. Li, Y. Zhang, Y. Jiang and S. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 31869–31877 CrossRef CAS PubMed.
- Y. Liu, X. Zhu, Q. Zhang, T. Tang, Y. Zhang, L. Gu, Y. Li, J. Bao, Z. Dai and J.-S. Hu, J. Mater. Chem. A, 2020, 8, 8920–8926 RSC.
- M. Peng, Y. Qiao, M. Luo, M. Wang, S. Chu, Y. Zhao, P. Liu, J. Liu and Y. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 40062–40068 CrossRef CAS PubMed.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- S. Zheng, S. Li, Z. Mei, Z. Hu, M. Chu, J. Liu, X. Chen and F. Pan, J. Phys. Chem. Lett., 2019, 10, 6984–6989 CrossRef CAS PubMed.
- Y. Luo, G.-F. Chen, L. Ding, X. Chen, L.-X. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- B. Dong, X. Zhao, G.-Q. Han, X. Li, X. Shang, Y.-R. Liu, W.-H. Hu, Y.-M. Chai, H. Zhao and C.-G. Liu, J. Mater. Chem. A, 2016, 4, 13499–13508 RSC.
- C. Tan, Z. Luo, A. Chaturvedi, Y. Cai, Y. Du, Y. Gong, Y. Huang, Z. Lai, X. Zhang, L. Zheng, X. Qi, M. H. Goh, J. Wang, S. Han, X.-J. Wu, L. Gu, C. Kloc and H. Zhang, Adv. Mater., 2018, 30, 1705509 CrossRef PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- L. Zeng, S. Chen, J. van der Zalm, X. Li and A. Chen, Chem. Commun., 2019, 55, 7386–7389 RSC.
- B. H. R. Suryanto, D. Wang, L. M. Azofra, M. Harb, L. Cavallo, R. Jalili, D. R. G. Mitchell, M. Chatti and D. R. MacFarlane, ACS Energy Lett., 2019, 4, 430–435 CrossRef CAS.
- X. Li, X. Ren, X. Liu, J. Zhao, X. Sun, Y. Zhang, X. Kuang, T. Yan, Q. Wei and D. Wu, J. Mater. Chem. A, 2019, 7, 2524–2528 RSC.
- X. Zhao, X. Zhang, Z. Xue, W. Chen, Z. Zhou and T. Mu, J. Mater. Chem. A, 2019, 7, 27417–27422 RSC.
- L. Zeng, X. Li, S. Chen, J. Wen, F. Rahmati, J. van der Zalm and A. Chen, Nanoscale, 2020, 12, 6029–6036 RSC.
- L. Zeng, X. Li, S. Chen, J. Wen, W. Huang and A. Chen, J. Mater. Chem. A, 2020, 8, 7339–7349 RSC.
- J. Liang, S. Ma, J. Li, Y. Wang, J. Wu, Q. Zhang, Z. Liu, Z. Yang, K. Qu and W. Cai, J. Mater. Chem. A, 2020, 8, 10426–10432 RSC.
- X. Yao, Z. Chen, Y. Wang, X. Lang, W. Gao, Y. Zhu and Q. Jiang, J. Mater. Chem. A, 2019, 7, 25961–25968 RSC.
- X. Chen, Y.-T. Liu, C. Ma, J. Yu and B. Ding, J. Mater. Chem. A, 2019, 7, 22235–22241 RSC.
- P. Wei, H. Xie, X. Zhu, R. Zhao, L. Ji, X. Tong, Y. Luo, G. Cui, Z. Wang and X. Sun, ACS Sustainable Chem. Eng., 2020, 8, 29–33 CrossRef CAS.
- L. Zhang, L.-X. Ding, G.-F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- P. Song, H. Wang, L. Kang, B. Ran, H. Song and R. Wang, Chem. Commun., 2019, 55, 687–690 RSC.
- W. Qiu, X.-Y. Xie, J. Qiu, W.-H. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- T. Wu, X. Li, X. Zhu, S. Mou, Y. Luo, X. Shi, A. M. Asiri, Y. Zhang, B. Zheng, H. Zhao and X. Sun, Chem. Commun., 2020, 56, 1831–1834 RSC.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., 2018, 130, 10403–10407 CrossRef.
- C. Zhao, S. Zhang, M. Han, X. Zhang, Y. Liu, W. Li, C. Chen, G. Wang, H. Zhang and H. Zhao, ACS Energy Lett., 2019, 4, 377–383 CrossRef CAS.
- S. Mukherjee, D. A. Cullen, S. Karakalos, K. Liu, H. Zhang, S. Zhao, H. Xu, K. L. More, G. Wang and G. Wu, Nano Energy, 2018, 48, 217–226 CrossRef CAS.
- Q. Zhang, F. Luo, Y. Ling, L. Guo, K. Qu, H. Hu, Z. Yang, W. Cai and H. Cheng, ChemCatChem, 2018, 10, 5194–5200 CrossRef CAS.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- P. Song, H. Wang, X. Cao, N. Liu, Q. Wang and R. Wang, ChemElectroChem, 2020, 7, 212–216 CrossRef CAS.
- Y. Song, T. Wang, J. Sun, Z. Wang, Y. Luo, L. Zhang, H. Ye and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 14368–14372 CrossRef CAS.
- Y. Liu, Q. Li, X. Guo, X. Kong, J. Ke, M. Chi, Q. Li, Z. Geng and J. Zeng, Adv. Mater., 2020, 32, 1907690 CrossRef CAS PubMed.
- T. Wang, L. Xia, J.-J. Yang, H. Wang, W.-H. Fang, H. Chen, D. Tang, A. M. Asiri, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 7502–7505 RSC.
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC.
- L. Xia, X. Wu, Y. Wang, Z. Niu, Q. Liu, T. Li, X. Shi, A. M. Asiri and X. Sun, Small Methods, 2019, 3, 1800251 CrossRef.
- S. Cheng, C. Li, Z. Yu, Y. Sun, L. Li and J. Yang, RSC Adv., 2020, 10, 9814–9823 RSC.
- Y. Kong, Y. Li, B. Yang, Z. Li, Y. Yao, J. Lu, L. Lei, Z. Wen, M. Shao and Y. Hou, J. Mater. Chem. A, 2019, 7, 26272–26278 RSC.
- S. Xiao, F. Luo, H. Hu and Z. Yang, Chem. Commun., 2020, 56, 446–449 RSC.
- Q. Zhang, F. Luo, Y. Ling, S. Xiao, M. Li, K. Qu, Y. Wang, J. Xu, W. Cai and Z. Yang, J. Mater. Chem. A, 2020, 8, 8430–8439 RSC.
- X. Zhu, T. Wu, L. Ji, C. Li, T. Wang, S. Wen, S. Gao, X. Shi, Y. Luo, Q. Peng and X. Sun, J. Mater. Chem. A, 2019, 7, 16117–16121 RSC.
- W. Guo, Z. Liang, J. Zhao, B. Zhu, K. Cai, R. Zou and Q. Xu, Small Methods, 2018, 2, 1800204 CrossRef.
- H. Wang, D. Yang, S. Liu, S. Yin, Y. Xu, X. Li, Z. Wang and L. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15772–15777 CrossRef CAS.
- Y.-X. Luo, W.-B. Qiu, R.-P. Liang, X.-H. Xia and J.-D. Qiu, ACS Appl. Mater. Interfaces, 2020, 12, 17452–17458 CrossRef CAS PubMed.
- C. Guo, X. Liu, L. Gao, X. Kuang, X. Ren, X. Ma, M. Zhao, H. Yang, X. Sun and Q. Wei, Appl. Catal., B, 2020, 263, 118296 CrossRef CAS.
- Y. Zhang, H. Du, Y. Ma, L. Ji, H. Guo, Z. Tian, H. Chen, H. Huang, G. Cui, A. M. Asiri, F. Qu, L. Chen and X. Sun, Nano Res., 2019, 12, 919–924 CrossRef CAS.
- D. Feng, X. Zhang, Y. Sun and T. Ma, Nano Mater. Sci., 2020, 2, 132–139 CrossRef.
- J. Chen, H. Huang, L. Xia, H. Xie, L. Ji, P. Wei, R. Zhao, H. Chen, A. M. Asiri and X. Sun, ChemistrySelect, 2019, 4, 3547–3550 CrossRef CAS.
- C. Chen, D. Yan, Y. Wang, Y. Zhou, Y. Zou, Y. Li and S. Wang, Small, 2019, 15, 1805029 CrossRef PubMed.
- P. Song, L. Kang, H. Wang, R. Guo and R. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 12408–12414 CrossRef CAS PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, ACS Sustainable Chem. Eng., 2017, 5, 7393–7400 CrossRef CAS.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- F. Köleli and T. Röpke, Appl. Catal., B, 2006, 62, 306–310 CrossRef.
- K. Kim, N. Lee, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F610–F612 CrossRef CAS.
- Y. Song, D. Johnson, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, J. Huang, F. Yang, F. Zhang, R. Qiao, A. P. Baddorf, T. J. Tschaplinski, N. L. Engle, M. C. Hatzell, Z. Wu, D. A. Cullen, H. M. Meyer, B. G. Sumpter and A. J. Rondinone, Sci. Adv., 2018, 4, e1700336 CrossRef PubMed.
- K. Kim, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F1523–F1526 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, Chem. Lett., 1993, 851–854 CrossRef CAS.
- H. K. Lee, C. S. L. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. Han, C.-K. Tsung and X. Y. Ling, Sci. Adv., 2018, 4, eaar3208 CrossRef PubMed.
- T. M. Pappenfus, K. Lee, L. M. Thoma and C. R. Dukart, ECS Trans., 2019, 16, 89–93 CrossRef.
- A. Katayama, T. Inomata, T. Ozawa and H. Masuda, Electrochem. Commun., 2016, 67, 6–10 CrossRef CAS.
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- J. A. Schwalbe, M. J. Statt, C. Chosy, A. R. Singh, B. A. Rohr, A. C. Nielander, S. Z. Andersen, J. M. McEnaney, J. G. Baker, T. F. Jaramillo, J. K. Norskov and M. Cargnello, ChemElectroChem, 2020, 7, 1542–1549 CrossRef CAS.
- S. Z. Andersen, Michael J. Statt, V. J. Bukas, S. G. Shapel, J. B. Pedersen, K. Krempl, M. Saccoccio, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. Nørskov and I. Chorkendorff, Energy Environ. Sci., 2020, 13, 4291–4300 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. Mcenaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- J. Choi, B. H. R. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. F. Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- L. Li, C. Tang, D. Yao, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 2111–2116 CrossRef CAS.
- H. Liu, Y. Zhang and J. Luo, J. Energy Chem., 2020, 49, 51–58 CrossRef.
- R. Dabundo, M. F. Lehmann, L. Treibergs, C. R. Tobias, M. A. Altabet, P. H. Moisander and J. Granger, PLoS One, 2014, 9, e110335 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |