
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microfluidics for antibiotic susceptibility testing
Witold
Postek
 *ab,
Natalia
Pacocha
a and
Piotr
Garstecki
*a
*ab,
Natalia
Pacocha
a and
Piotr
Garstecki
*a
aInstitute of Physical Chemistry of the Polish Academy of Sciences, ul. Kasprzaka 44/52, 01-224 Warszawa, Poland. E-mail: pgarstecki@ichf.edu.pl
bBroad Institute of MIT and Harvard, Merkin Building, 415 Main St, Cambridge, MA 02142, USA. E-mail: wpostek@broadinstitute.org
First published on 7th September 2022
Abstract
The rise of antibiotic resistance is a threat to global health. Rapid and comprehensive analysis of infectious strains is critical to reducing the global use of antibiotics, as informed antibiotic use could slow down the emergence of resistant strains worldwide. Multiple platforms for antibiotic susceptibility testing (AST) have been developed with the use of microfluidic solutions. Here we describe microfluidic systems that have been proposed to aid AST. We identify the key contributions in overcoming outstanding challenges associated with the required degree of multiplexing, reduction of detection time, scalability, ease of use, and capacity for commercialization. We introduce the reader to microfluidics in general, and we analyze the challenges and opportunities related to the field of microfluidic AST.
1. Antibiotic resistance crisis
Once called ‘miracle drugs’, antibiotics help humanity keep infectious diseases at bay. Since the dawn of antibiotics, strains of bacteria resistant to antibiotic effects have been reported – Alexander Fleming, famous for discovering the effect of penicillin on bacteria, mentioned the irresponsible use of antibiotics and the threat of drug-resistant bacteria as early as 1945 in his Nobel Lecture.1 The number of new antibiotics introduced to the clinic decreases over time, while the number of resistant strains increases.2 Antibiotic-resistant infections are responsible for ca. 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year in the European Union3 and around 35000 deaths per year in the USA,4 with the most deadly infection in the USA being caused by Clostridioides difficile, methicillin-resistant Staphylococcus aureus (MRSA), and extended-spectrum beta-lactamase (ESBL) producing Escherichia coli.4 Although most deaths caused by bacterial agents could be treated with antibiotics,5 the number of fatalities caused by antibiotic resistant bacteria is predicted to increase to a stagerring 10 million death per year in 2050 as predicted by WHO.5 This prediction is sometimes criticized,6 but even these critics agree that the antibiotic resistance of bacteria is a major health challenge now and in the predictable future. A wide-scale statistical analysis based on Global Burden of Disease Study,7 showed that the 2016 estimate of 700000 deaths8,9 per year by antibiotic resistant bacteria might be too shy: research showed that 1.27 million deaths per year can be directly attributed to antibiotic-resistant bacteria (if all antibiotic-resistant infections were replaced by no infection, 4.95 million deaths would have been avoided in 2019, while if all antibiotic-resistant infections were replaced by antibiotic-susceptible infections, 1.27 million deaths could have been prevented8). The data from the statistical analysis inform that it is not only the lack of sanitation or access to antibiotics that drive bacteria-related deaths, as antibiotic resistance plays a large role there. The geographic distribution of deaths per capita attributable to antibiotic resistant bacteria shows an expected tilt towards low-to-medium income countries, but the difference in per capita deaths by antibiotic resistant microbes between those and high income countries is not huge: antibiotic resistant bacteria are causing death and suffering worldwide.8
000 deaths per year in the European Union3 and around 35000 deaths per year in the USA,4 with the most deadly infection in the USA being caused by Clostridioides difficile, methicillin-resistant Staphylococcus aureus (MRSA), and extended-spectrum beta-lactamase (ESBL) producing Escherichia coli.4 Although most deaths caused by bacterial agents could be treated with antibiotics,5 the number of fatalities caused by antibiotic resistant bacteria is predicted to increase to a stagerring 10 million death per year in 2050 as predicted by WHO.5 This prediction is sometimes criticized,6 but even these critics agree that the antibiotic resistance of bacteria is a major health challenge now and in the predictable future. A wide-scale statistical analysis based on Global Burden of Disease Study,7 showed that the 2016 estimate of 700000 deaths8,9 per year by antibiotic resistant bacteria might be too shy: research showed that 1.27 million deaths per year can be directly attributed to antibiotic-resistant bacteria (if all antibiotic-resistant infections were replaced by no infection, 4.95 million deaths would have been avoided in 2019, while if all antibiotic-resistant infections were replaced by antibiotic-susceptible infections, 1.27 million deaths could have been prevented8). The data from the statistical analysis inform that it is not only the lack of sanitation or access to antibiotics that drive bacteria-related deaths, as antibiotic resistance plays a large role there. The geographic distribution of deaths per capita attributable to antibiotic resistant bacteria shows an expected tilt towards low-to-medium income countries, but the difference in per capita deaths by antibiotic resistant microbes between those and high income countries is not huge: antibiotic resistant bacteria are causing death and suffering worldwide.8
Bacteria resistant to multiple drugs at once, or even to all clinically available antibiotics, are of serious concern to researchers.10,11 It is estimated that more than 50% of antibiotics prescribed2,12 around the world is used either unnecessarily (e.g., for viral infections) or wrongly (either wrong narrow-spectrum antibiotic or blindly used broad-spectrum antibiotic that leads to intense growth of antibiotic-resistant strains that are usually kept in check by the rest of microbiota of a healthy human).13 Antibiotic resistance is also emerging due to antibiotic pollution in the environment,14 which suggests that combating antibiotic resistance should involve collaboration of multiple sectors, such as public health, animal health, plant health – as specified in the WHO's One Health approach.15 To curb the emergence of antibiotic resistance, it is imperative to reduce the overuse of antibiotics. We need more cheap and easy-to-use platforms for the identification of pathogens and for assessment of antibiotic resistance to stop the inappropriate use of antibiotics and perhaps limit the incorrect use of antibiotics – physicians with access to a point-of-care platform for identification of pathogens and their resistance are less likely to prescribe antibiotics when they are not needed.16–18 As a prolonged diagnosis of a pathogen and its antibiotic resistance profile decreases the chance of survival of an infected patient, the time from sampling to diagnosis should be reduced to a minimum. Therefore any new platform that could be used to diagnose bacterial infections should be as rapid as possible.
Quantitatively, bacterial drug resistance is established by measuring a parameter called minimum inhibitory concentration (MIC), the lowest concentration of a drug that prevents the growth of bacteria.19 MIC measurements are the basis for establishing resistance breakpoints by agencies such as CLSI or EUCAST.20 A breakpoint is a drug concentration against which a patient sample is tested – if there is growth, the bacterial strain is resistant; if there is no growth, the strain is susceptible. Setting breakpoint values by medical agencies is based on MIC and pharmacokinetics and pharmacodynamics (PK/PD) of an antibiotic.21 Clinicians use breakpoint values based on the MIC, but not the MIC itself. Breakpoint values, although highly useful, do not convey the information that an MIC screen personalized to a given patient would: e.g., a breakpoint does not necessarily take into account a wild-type resistance distribution, which can lead to both false positives and false negatives, or there is a possibility that a tested bacterium does have a resistance mechanism but is still below the breakpoint.22
The gold standard assays for establishing MIC are broth dilution and e-test. The most commonly used automated platform for quantifying MIC is VITEK® 2 by bioMérieux, based on the microbroth dilution method. VITEK® 2 uses cassettes with a set of compartments with a diluted drug and with the tested bacteria. The compartments are monitored constantly to generate a growth curve which is compared to a growth curve of bacteria with a known reference MIC. All the upcoming automated AST solutions have to be compared to this or similar devices (e.g., BD Phoenix by BD Diagnostics, Sensititre by Thermo Scientific, MicroScan by Siemens HC Diagnostics) as a standard if they are to be used in a clinical setting on a wide scale. A potent source of innovation in the area of automated broth dilution is microfluidics, and this is visible in various emerging start-up companies that try and develop disruptive technologies for AST.
Due to poor profit prospects on new antibiotics, big pharmaceutical companies are not inclined to spend money on antibiotic research and clinical trials.23 To tackle the growing levels of antimicrobial resistance, governments and international bodies are implementing push and pull strategies to incentivize the development of new antibiotics by private companies.24–26 Push strategies include direct grants and tax incentives, while pull strategies consist of milestone rewards or market exclusivity extensions.23 As an example of a pull strategy, the Longitude Prize of £10 million was set for a competitor that develops a point-of-care diagnostic test that will help conserve the usage of antibiotics. A few essential criteria are required to be considered for the prize: the test must be needed, accurate, affordable, rapid, easy to use, safe, and scalable. These requirements, relatively easy to achieve in separation, pose a significant technical challenge when combined. In recent years, there has been a surge of scientific publications about point-of-care, rapid, easy-to-use platforms for a number of biological applications. A considerable proportion of the presented solutions is based on microfluidics and tackle the antibiotic resistance problem.
In this review, we comprehensively describe many of the microfluidic systems that were applied to quantitative antibiotic resistance assessment. There is a need for quantitative antibiotic susceptibility testing systems27 as they provide more information than just the detection of resistance or susceptibility, and we identify assay multiplexing and signal detection time as the bottlenecks to the development of rapid AST with an MIC output. We therefore focus on the microfluidic technologies applied to AST to inform the reader which technology suits different multiplexing or detection methods. We put the most effort here to describe microfluidic solutions that not only identify antibiotic resistance, but also generate the MIC which we consider highly informative in medical diagnostics. We recommend that the reader reaches for other review articles that describe in detail different aspects of microfluidics applied to antibiotic susceptibility screens or are related to AST, such as strictly single-cell assays,28 technical challenges to manufacturing AST devices at scale,29 microfluidics put in a broader AST context,27,30,31 or barriers to the development of rapid AST platforms.32 For a recent list of innovative companies in the field of AST we send the reader elsewhere.27 We begin with a description of microfluidic systems in general that will allow the reader to understand how microfluidic systems for biological applications work and where to seek technical improvements. We divide the microfluidic platforms for AST into categories by the approach to liquid handling (Fig. 1), as this determines the assay's capabilities and challenges in the assay's potential development: static chamber arrays, flow chamber arrays, droplet arrays, and systems based on flowing droplets. A separate category is the systems for phenotypic-molecular (“pheno-molecular”) assessment of MIC in microfluidics. Among all the microfluidic platforms we describe, we highlight platforms that allow for single-cell level studies, as these platforms allow for either rapid AST or heterogeneity studies. We conclude this review with a discussion on the challenges that researchers face when working on microfluidic platforms for analysis of antibiotic resistance, and with identification of research opportunities in application of microfluidic techniques to AST.
 | ||
| Fig. 1 Schematic representation of microfluidic approaches used in antibiotic susceptibility testing. A given microfluidic solution is easier to use for an end-user at the cost of the process scale and the flexibility of the assay, i.e., multiplexing capabilities or on-chip operations. Static microfluidic chambers are, in principle, an array of wells in a well plate with automated filling and separating operations added. Static chambers provide for some multiplexing: antibiotics in different concentrations can be printed to separate chambers, and the monitoring of cells is easy as the chambers are not numerous and they do not move, so the cells are immobile for the sake of microscopy. Static chambers are feasible for transfer to commercial applications as they are easy to produce at scale. Flow chambers are similar to static chambers, albeit their content can be gradually changed over the course of an experiment, usually through diffusion from the main channel to the side chambers. Hybrid droplet arrays offer greater scale and multiplexing than chambers, and they still allow for easy monitoring of cells, as the droplets are immobile. Complex operations on liquids can be done on-chip before generation and immobilization of droplets. However, after immobilizing the droplets, it is difficult to introduce changes to reaction conditions controllably for each droplet. The use of droplets requires consideration of the transport of matter, e.g., antibiotics or dyes, between droplets, although in stationary arrays the droplets are not moving, so the surfactant-based transfer of molecules should be limited. Flowing droplets provide superb scale and multiplexing capabilities, as complex liquid handling operations can be done even at the level of single droplets. Droplets are usually incubated for the growth of encapsulated bacteria for long times, so the transfer of matter between droplets must be considered. Monitoring of cell growth dynamics is not trivial, as droplets move during incubation due to mixing or convection, so following the same droplet over the course of the experiment would pose a considerable challenge. Flowing droplet arrays are complex to automate from end to end; therefore, they are not easily transferable to commercial settings. However, flowing droplet arrays provide for vast research possibilities due to the scale of possible experiments. Pheno-molecular ASTs are different in that they can in principle be performed in droplets or static chambers. Although the currently used number of reactors for pheno-molecular assays is smaller than in droplet-based assays, this approach has great potential due to short sample-to-result time and high multiplexing capabilities. | ||
2. Microfluidic methods of studying bacteria
2.1. Introduction
Microfluidics is now a matured field of science that deals with the flows of fluids at microscopic scales. The flows are realized mainly within the confinement of microchannels that tend to have hydraulic diameters in the range of tens to hundreds of micrometers – such small diameters guarantee the laminar flow regime inside the channels, implications of which we discuss below. The flows are generated either by positive pressure – syringe pumps and compressed air systems – or negative pressure – vacuum pumps. Channels are usually made with soft lithography33 or CNC milling,34 and the resulting microfluidic devices, or chips, are made of elastomers (most notably polydimethylsiloxane, PDMS35,36), glass, thermoplastics, or hybrids of these materials. PDMS is often used for devices housing living organisms as this polymer is permeable to gases, thus granting good oxidation of the contained cells. Numerous review articles are available detailing the technical side of the microfluidic chip, covering fabrication of devices,37 generation of droplets,38 and physics of flow.39 Microfluidic systems are broadly divided into the ones that contain only one liquid phase (single-phase microfluidics) or two and more phases.A separate branch of multiphase microfluidic devices contains lab-on-a-disc systems, i.e., discus-shaped microfluidic devices with embedded channels specially engineered so that when the disc is spun on a centrifuge, the samples move sequentially between the chambers on the disc. The sequential release of liquids is based on capillary valves designed to break when a specific centrifugal force is reached in the device. We encourage the reader to find a detailed description of centrifugal microfluidic devices and recent developments in biomedical research in lab-on-a-disc devices elsewhere.64,65 Although not trivial to engineer, lab-on-a-disc diagnostic devices have great commercialization potential due to the ease of use for the end-user when the device is assembled.
2.2. Microfluidic static chambers for quantitative AST
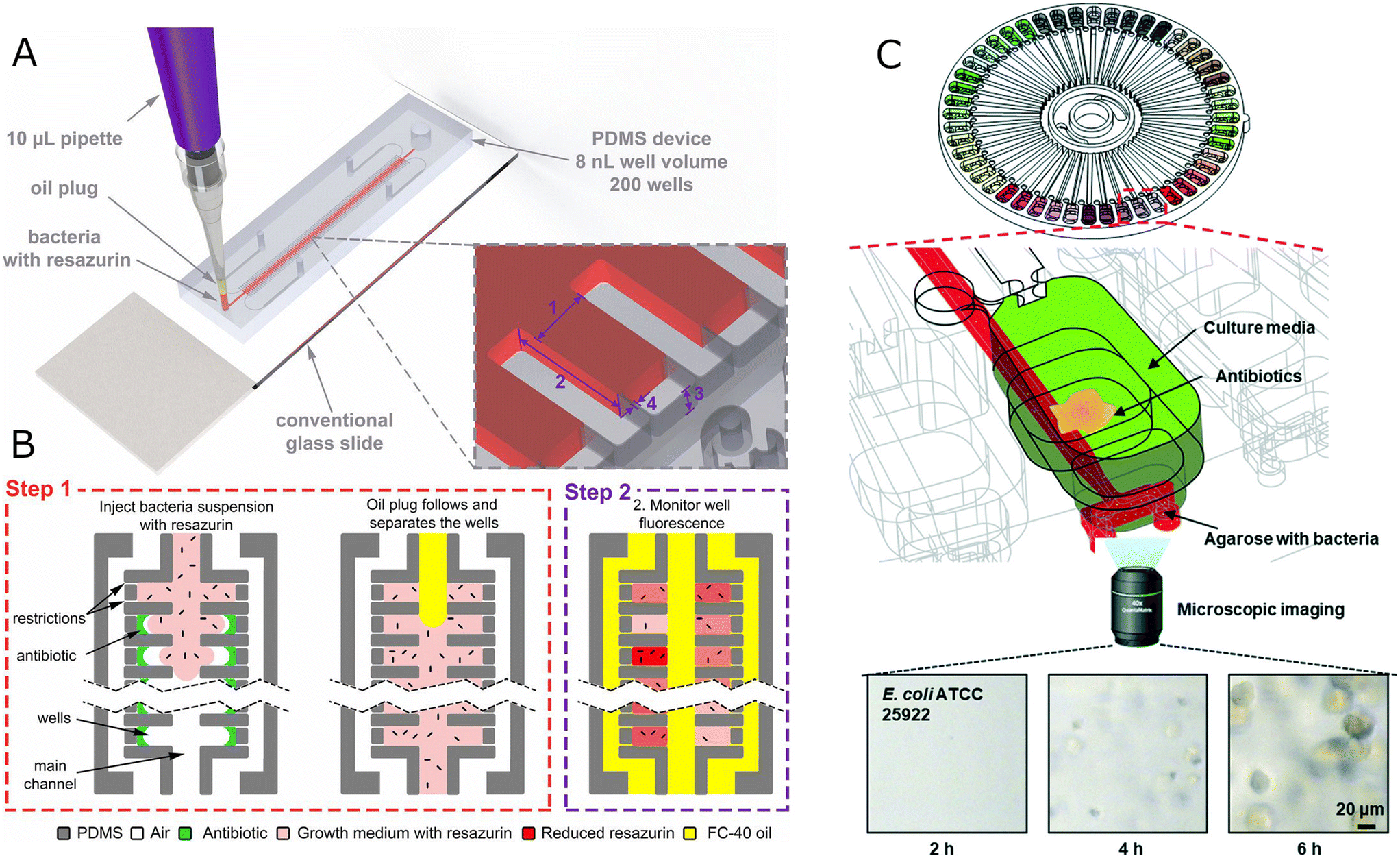 | ||
| Fig. 2 Microfluidic static chambers for population-level studies. A) Schematic of the stationary nanoliter droplet array (SNDA)-AST system. The SNDA-AST device is placed on a standard microscope slide and operated by a 10 μl pipette. It consists of two rows of 8 nl chambers connected with the main channel delivering bacteria suspension or oil phase. Each well is equipped with 3 μm restrictions to let air escape to the surrounding channels. B) The SNDA-AST system is operated in two steps. In step 1, the device is loaded with a single-step injection of two plugs, bacterial suspension with 10% resazurin, followed by a plug of FC-40 oil. The oil separates the wells creating discrete, isolated chambers, delivering oxygen, and preventing evaporation. In step 2, the fluorescence intensity is measured every 30 min, which indicates the level of bacteria metabolic activity, and it is proportional to the amount of bacteria/metabolism in the chamber. Bacteria are not illustrated to scale. Reproduced from ref. 52. C) Schematic of a capillary and centrifugal-based AST (C2-AST) system containing dried antibiotics, agarose with bacteria, and culture media. The bottom time-lapsed microscope images show growing E. coli cells after 2, 4, and 6 h of incubation for left, central, and right images, respectively. Reproduced from ref. 60. | ||
Upscaling static chambers for MIC has to tackle the problem of multiplexing the assay. One of approaches to the issue is by serial dilution of the sample on chip, which was recently done by Osaid et al., leading to an AST device integrated with a multiplexing module – this is a great feat of engineering; however, might be difficult to commercialize or use in clinic as the dilution module is rather complicated.74 Further upscaling of chamber-based devices for AST resulted in systems containing over 1000 chambers.75 Such an increase in throughput allowed for analysis of the efficacy of cocktails of antibiotics in search of synergistic, additive, or antagonistic relations. Pipetting over 1000 antibiotic samples into an array of microscopic wells is not feasible manually, so the authors used a droplet spotting machine. A spotting machine (a non-contact inkjet printer) allows for precise injection of very small volumes into wells that are microns apart from each other. However, such a machine is rather costly. If one were to commercialize such a system to put it to clinical use, they would need to consider the high maintenance cost of a spotting machine – this might be viable as the end chip is simple to use by non-experts. On the other hand, perhaps a different way of filling the chambers should be designed for commercialization, as PDMS used for degas filling of channels with oil to separate chambers is not ideal for mass production. It is possible to use syringes to generate suction within an array of chambers, and this approach does not necessarily require PDMS-made devices to work.76 Another instance of prefilling microfluidic chambers with antibiotics and later filling all the chambers with bacteria was shown in a hybrid PDMS-paper device,77 where small pieces of paper with antibiotic were placed in chambers before the experiment. Such an approach is undoubtedly more convenient than spotting the antibiotics to chambers with a spotting machine, and although only 21 chambers were placed in the device, the system was used to for breakpoint analysis of bacteria from clinical samples. In a separate approach, growth chambers were equipped with pressure valves that allowed for separate loading of chambers with bacteria and with antibiotic without drying the antibiotic, and antibiotic resistance of polymicrobial colonies was considered.78,79 It is also possible to prepare a combinatorial gradient of antibiotic concentrations with microfluidic channels that allow for diffusion of different amount of antibiotic molecules into different regions of a hydrogel on which bacteria would grow – this simplistic approach could be applied to single-cell studies if bacteria isolating chambers were layered on top of the hydrogel.80 Overall, the greatest scalability potential for microfluidic static chambers for AST lies in the antibiotic contact-free printing, even though this particular approach is technically demanding.
A distinct way of filling the chambers with a preloaded antibiotic was presented recently with centrifugal force within a lab-on-a-disc device.81,82 With devices presented in both works, it was possible to prepare a series of chambers with a gradually changing antibiotic concentration and bacteria added separately to the antibiotic chambers (Fig. 2C). The devices are rather complex, with 3 layers of PDMS mounted on a plastic base. However, when assembled, these devices should be easy to use for an operator, as usual with the ab-on-a-disc technology. The samples in the systems are metered based on the size of metering chambers, which is advantageous due to high dosage precision of metering in chambers, but might be limiting as the gradient (not the concentrations of antibiotic but rather the range of concentrations) is encoded in the device. This feature limits the flexibility of the device, but on the other hand, precise metering and reproducibility are desired when commercialization is considered.
A distinct technology for static chamber preparation relies on changes in the microchannel alignment after addition of subsequent reagents to the microfluidic device. This branch of microfluidics is called SlipChip and was first demonstrated in 2009.92 SlipChip was used for AST based on blood samples and entropy-based image analysis of single cells allowed for assay results within 3–8 hours.93 The applicability of this type of device is questionable for high throughput assays as leaks might be difficult to prevent while shifting two large plates with microstructures; however, smaller size SlipChips might be used in resource limited settings for their ease of use.
A distinct use of hydrogel trapping was demonstrated recently,94 where authors formed dried hydrogel plugs that were placed in a conventional 96-well plate. Bacteria and antibiotics were pipetted to the wells causing the dried plugs to absorb liquid and swell, causing mechanical separation of individual cells at the bottom of the well. However, the gel is prone to antibiotic diffusion, so the separated chambers do not show an inoculum effect, which means that scMIC cannot be measured in the presented device.
An impedance-based device was used to track the growth of individual bacteria based on their motion within a closed microfluidic system. Bacteria would swim into a narrow channel, changing its effective channel diameter and the electrical conductance, resulting in voltage fluctuations.95
2.3. Microfluidic flow chambers for quantitative AST
In this section, we discuss microfluidic systems that separate the tested reaction conditions (antibiotic concentrations) for AST but that do not forbid the efficient exchange of medium. Such property is the effect of either culturing bacteria within a flow channel or by not adding long and narrow connector channels in between the device's main channel and the bacteria culturing chambers. Flow systems in which bacteria grow in the main channel are not often used for AST as the swimming bacteria are not shielded from the medium flow, meaning that the cells might be washed out of the chamber before the measurement is completed.One of the most prevalent microfluidic single-phase designs for microbiology is the mother machine,102 which has been used on numerous occasions to study bacteria in an antibiotic context or close to it: e.g., mother machine was used for the analysis of mutation frequency in E. coli,103 for studying viable but non-culturable bacteria during antibiotic treatment,104 for describing the accumulation of polar multidrug efflux pumps that grant antibiotic resistance in older cells and less so in daughter cells,105 for studying bacterial persistance,106,107 uptake and efflux of antibiotics by bacteria108 with fluorescently labelled antibiotics,109 and measuring the efficacy of bacteria clearance with phages.110,111 The basic mother machine consists of a large main channel through which growth medium flows and of multiple shallow and narrow dead-end channels that are perpendicular to the main channel, forming a comb-like structure. The bacteria are placed in the main channel, and they fill the shallow channels one by one. When a sufficient number of channels contains a single cell, the flow is changed to a bacteria-free medium in the main channel. The bacteria at the dead ends of narrow channels divide, and their daughter cells move closer to the main channel and are eventually washed away, but the mother cell at the dead-end of a shallow channel does not move. The first use of the mother machine in AST was presented by Lu et al. where the authors positioned the bacteria in the shallow channels electrokinetically, so that the bacteria are always lined up next to an electrode, which facilitates optical imaging112 (Fig. 3A and B). The authors followed the length of colonies in time (growth rates) in channels starting from a single cell and analyzed the growth rate changes in the presence of antibiotics, and showed that MIC could be determined within one hour in their system. They also demonstrate distributions of growth rates within a population of bacteria when untreated or treated with an antibiotic. Baltekin et al. elaborate on this solution by following the growth rates of individual bacteria at a large scale in populations (Fig. 3C) where antibiotic was added to the flowing medium and compared the measured values to growth rates of cells in antibiotic-free conditions. After 30 minutes, the authors were able to determine with high confidence that a given strain is either susceptible or resistant to a given antibiotic in a given concentration.113 Li et al. used a similar approach and also measured growth rates (or lengths of bacterial colonies formed in an individual microchannel), but they also implemented channels perpendicular to shallow bacteria channels on top of the shallow bacteria channels114 (Fig. 3D). These top channels resemble a classical microfluidic module called Quake valves (in memory of the inventor):41 the top channel is pressurized, and the PDMS layer between the top channel and bottom channel deforms as the pressure is applied to the top channel (PDMS is elastic). Higher applied pressure means larger deformation of the channel, and, consequently, smaller height of the bottom bacteria channel. This behavior was used by Li et al. to stop bacteria of the desired size in the bottom channel through trapping the cells with the controlled channel height. The separation of differently sized cells allowed the authors to identify the trapped bacteria species from a sample (Fig. 3E). Size-based differentiation of the most common infectious agents might be helpful when planning an antibiotic therapy or tracking the infection onset (Fig. 3F).
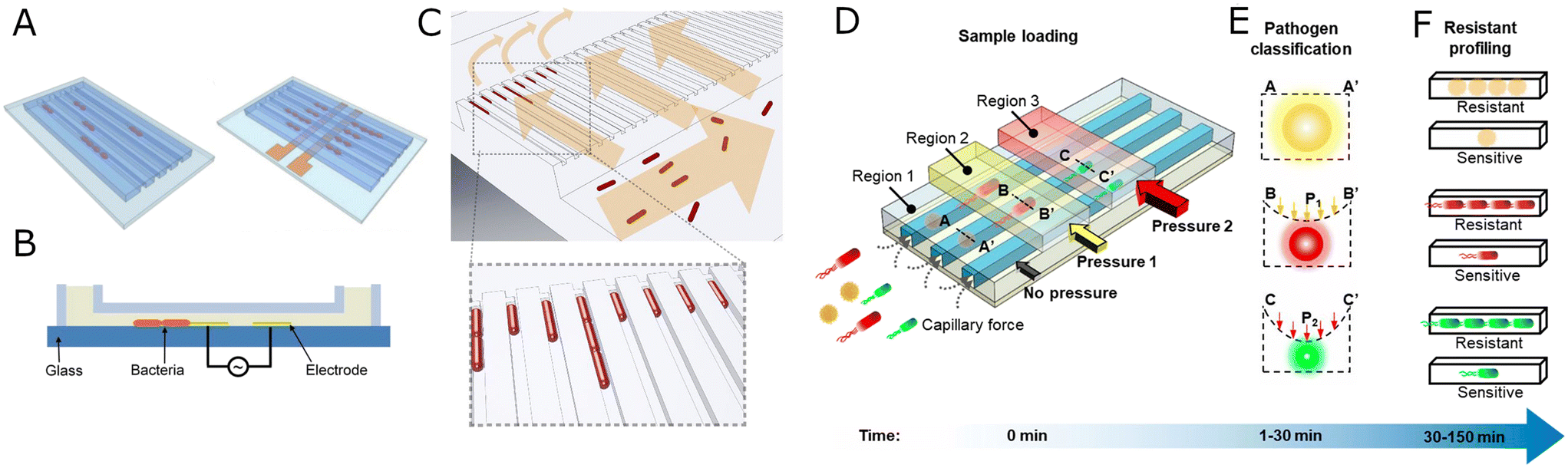 | ||
| Fig. 3 Evolution of mother machine systems. A) Electrode-based ordering of bacterial cells. Schematic representation of bacteria trapping in microchannels for single-cell AST without (left) and with electrokinetic positioning (right). B) A cross-section view of the microchannel with a pair of electrodes for bacterial cells (orange) trapping by dielectrophoretic force. A and B reproduced from ref. 81. C) Sieve-like design of the mother machine. Bacteria (red) are loaded from the front channel, and they are prevented from passing to the backchannel by a constriction at the end of each trap. The arrows indicate the direction of flow during chip loading. Reproduced from ref. 82. D) Adaptable mother machine system for bacteria identification and AST. Bacteria are loaded into region 1 (undeformed) and regions 2 and 3 (differently deformed with pressure) of the channels automatically by capillary force. Pressure might be applied to channels marked as regions 2 and 3 to deform the bacteria channels and to sieve out cells of different size. E) A cross-section view of microfluidic channels with trapped bacteria under different pneumatic pressures. The cells are trapped in different regions of microchannels and classified according to the applied pressure. A, A′, B, B′, C, C′ – positions in the bacteria channel profiles as seen in D. P1, P2 – different pressures used to deform the bacterial channels. F) Bacteria susceptibility to antibiotics is assessed by monitoring bacteria growth in the presence of antibiotics. D–F reproduced from ref. 83. | ||
In a separate effort to study antibiotic resistance by tracking divisions of single cells in a microfluidic device, Choi et al. and then Jung et al. presented a series of fast AST solutions based on tracking divisions of individual cells immobilized in agarose.83–85 The first iteration of their system is a flow chamber: Choi et al. had prepared a channel that they filled with bacteria in liquid agarose, and after the agarose solidified, a solution of antibiotic in a culture medium was injected into a side channel of the system, leading to diffusion of the antibiotic from the solution into the agarose pores. The authors then imaged the neighborhood of the side channel and assessed the growth of bacteria. The device was multiplexed into 6 sets of channels and side channels – this allowed for AST of 6 antibiotic concentrations at once. This system was rather complicated, and it required a syringe pump to be dosing antibiotics during the whole experiment. Another group of authors used a simple agarose-based diffusion design to generate an antibiotic concentration gradient and correlate the cell length with the antibiotic abundance,115 an approach similar to another microfluidic AST solution.90 They pointed to heterogeneity in cell length at intermediate antibiotic concentrations, suggesting different levels of resistance across the tested population. Their device is simpler in use than the early endeavors from Choi et al.83 However, it falls behind the newer design iterations from Choi et al. that use a 96-well plate as an assay base, as the well-plates offer great multiplexing opportunities.
A system that immobilized cells in narrow channels similarly to how it was achieved in the system by Baltekin et al.113 was used to test for antibiotic susceptibility of bacteria within 2 hours by means of electric impedance.116 The cells that produced growth gave different impedance readings that the cells that were susceptible to antibiotics. The research showed that the time-resolved changes of electrical impedance are different for different antibiotics, possibly opening doors for mechanistic studies of antibiotic resistance.
2.4. Hybrid droplet-chamber devices for quantitative AST
Most of the systems that rely on trapping the previously prepared droplets for AST are capable of testing antibiotic susceptibility of single cells as an end-point measurement: tracking of growth dynamics of bacteria swimming in droplets has not yet been presented; however, it is possible to trap a single cell in a droplet, let the cell divide and capture the cell growth signal (OD change, fluorescence change, etc.) after several hours. It is possible to track growth rates of cells in droplets (or rather beads) of hydrogel, as cells are immobilized in such a medium. If only population-level studies were performed, we note it and comment on the possibility of single-cell tracking. Recently, a system that uses electrowetting to control the movements of droplets on an AST device was reported; however at a limited throughput.117 The presented electrowetting device is fully integrated (dilution of antibiotics, dispension of bacteria, growth analysis), therefore it presents an opportunity for commercialization. The usual goal of using droplet arrays is to get the best of both worlds: the immovable or easy to track bacteria as in single-phase microfluidics and the large scale and ease of production of thousands of droplets and even thousands of reaction conditions per experiment.In an early attempt to run AST in arrayed droplets, Sinn and colleagues118 encapsulated single asynchronous magnetic beads in droplets with bacteria and pre-diluted antibiotics. The bacteria attached themselves to the bead, changing its rotational frequency. At a sufficiently high antibiotic concentration, there were no bacteria to reduce the rotational frequency, and MIC was thus established. Amselem et al.119 developed a system in which a gradient of concentrations is established by diffusion during the laminar flow of antibiotic containing liquid parallel to an antibiotic-lacking medium, similarly to solutions presented in single-phase microfluidic systems. In their system, Amselem et al. used a chamber with 1500 wells filled with a bacteria solution. Then the chamber was filled with oil that separated the wells, trapping individual droplets with bacteria in the wells (Fig. 4A–C). The device works with either liquid droplets for swimming bacteria or with gel beads for immobile colonies. The dynamic range of this system is limited. However, the authors later presented a modification of the system that allows for firstly trapping droplets of one sample and then adding precisely measured portions of another sample to the trapped droplets120 – one can imagine using such a modified platform to run AST with a broad dynamic range by firstly trapping droplets with bacteria and then adding droplets of pre-diluted antibiotic. Sabhachandani et al. demonstrated a droplet array for AST with such a pre-dilution where they trapped 1000 small droplets (1 pl to 10 nl). The droplets were generated with an embedded flow-focusing geometry, and the droplets were later imaged on a chip (Fig. 4D and E). The device integration is similar to the work of Amselem et al.119 A redesigned system of Sabhachandani et al.121 was demonstrated by researchers from the same laboratory as Kang et al.,122 where the authors use a device with 4 arrays of 8000 droplets each for AST and find subpopulations of highly resistant bacteria. The device is scalable, so it is possible to house more antibiotic concentrations on a single chip. However it would require more inlet and outlet ports, making the device relatively complex. An easy-to-use device for AST in droplet arrays was presented by Kao et al.55 The authors used a passive method of generation of droplets (microfluidic step emulsification123) to emulsify samples that had been pipetted on the chip. Each sample (bacteria with added different concentrations of antibiotics) was emulsified to a separate chamber with emulsification driven by gravity, thus making the system relatively cheap (no pumping setup for flow control) and easy to operate. The platform relied on fluorescence readout, thus limiting its clinical application. Derzsi et al. developed a system in which the antibiotic dilution series was prepared in droplets on a chip passively and merged with bacteria droplets also prepared passively on the same chip. The passive nature of the assay was possible due to special engineering of the microfluidic channels and allowed for preparation of a microdroplet-based AST in five pipetting steps.124 This system could in principle be used for single cell studies, however the small number of assayed droplets limits the information gained in the experiments. All the droplet arrays described to this point relied on separate droplet chambers for each antibiotic concentration. In a system developed by Kulesa and colleagues,53 the authors applied a method known as ‘color coding’:125 Kulesa et al. used combinations of three fluorescent dyes that emit fluorescence at distinct wavelengths to mark droplets with different antibiotic concentrations – each concentration received a unique set of concentrations of dyes (Fig. 4F). This approach allowed the authors to pool the droplets into one large droplet array, thus significantly reducing the engineering complexity of the assay, on the other hand making the optical analysis more complex. However, once the image analysis protocols were established, the authors were able to use this platform to assess dose–response interactions of 10 antibiotics with over 4000 adjuvants. The authors studied only populations of cells trapped in droplets, however single-cell analysis in their device is feasible.
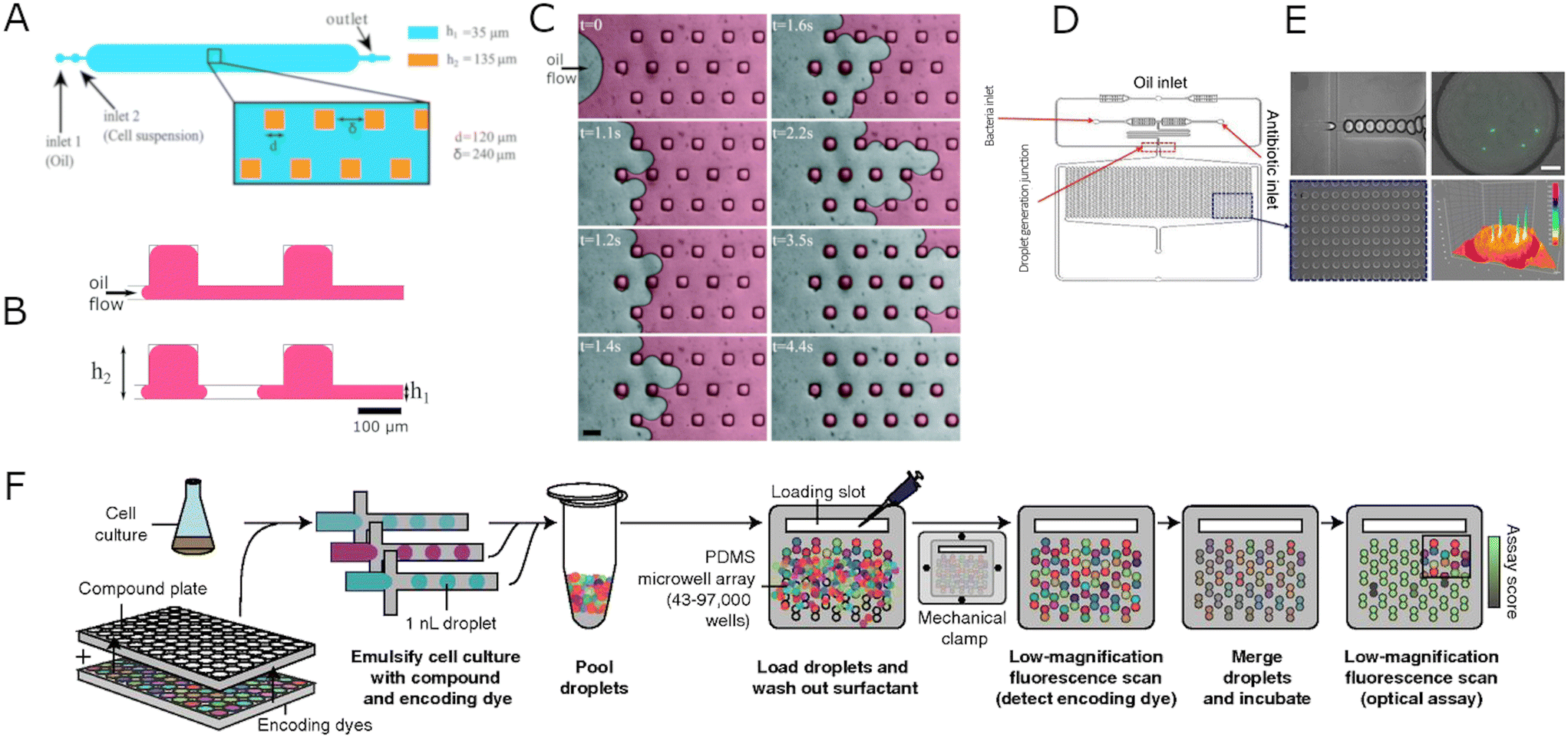 | ||
| Fig. 4 Schematic representations of hybrid droplet-chamber systems. A) Design of a microdroplet multiwell device. The main central chamber contains a 2D array of 113 × 15 surface-tension anchors. The side dimension of a single square anchor is d = 120 μm, and δ = 240 μm spaces it. The height of the chamber and the anchor are h1 = 35 and h2 = 135 μm, respectively. B) Cross-section scheme of anchored droplet generation. The aqueous sample initially covers the entire surface and then breaks on anchors creating isolated droplets. C) Time-lapse images presenting the droplet formation process. At t = 0, the sample colored in red for better visualization is introduced to the microfluidic chip and pushed by the oil phase (FC40 + 0.5% surfactant) using a hand-pushed syringe. The arrow indicates the direction of the oil flow. When the interface penetrates between two anchors, it deforms and breaks, creating a well-calibrated droplet in the anchor. Scale bar: 200 μm. A–C Reproduced from ref. 86. D) Schematic representation of the microfluidic chip for micro-droplet-based phenotypic AST. E) Micrographs showing droplet formation process with flow-focusing junction (top left), the docking array filled with droplets containing bacteria and antibiotic at various concentrations (bottom left), a green fluorescent protein (GFP) expressing E. coli in the droplet (top right), and fluorescent intensity profile of GFP E. coli (bottom right). Scale bar: 20 μm. D and E reproduced from ref. 88. F) Droplet-based platform for combinatorial drug screening. Cells, compounds, and encoding dyes are mixed, emulsified into nanoliter droplets, and pooled together. The droplets are paired by introducing them to a microwell array. A free surfactant is washed out to limit the compound exchange. The compounds are identified in each droplet using low-magnification epifluorescence microscopy. Then the pairs of droplets in each microwell are merged and incubated. The last step is an optical scan measuring cell growth inhibition in all microwells. Reproduced from ref. 37. | ||
2.5. Flowing droplets for quantitative AST
A separate category of microfluidic devices is based on droplets generated at high throughputs and collected for incubation in a test tube. After incubation, the droplets can be moved to a detection system, where they are scanned for optical signals at hundreds or thousands of droplets per second. Contrary to the droplet array systems described above, none of the systems based on the analysis of flowing droplets can be used for rapid AST based on tracking divisions of individual cells. This stems from the fact that the optical read-out of bacterial growth in flowing droplets is an end-point measurement – it is until now not possible to conveniently track thousands of individual droplets in an emulsion for hours to check if bacteria in a given droplet are dividing – such tracking was done only for at most hundreds of large (hundreds of nanoliters) droplets.126,127 Jakiela et al.128 presented the first droplet system supporting chemostat-like conditions for long-term incubation of bacteria and demonstrated the use of this system in tracking adaptation of bacteria to antibiotics. Apart from that, the bacteria in droplets need culturing so that the optical signal from droplets is strong enough to be detected. This instantly leads to a conclusion that systems based on flowing droplets are not suited for clinical practice as they cannot inform a diagnosis as rapidly as the systems for immobile bacterial cultures. Flowing droplet systems, however, offer large scaling possibilities, which we discuss in the ‘opportunities’ section.The flowing droplets can be used for single-cell level analysis: firstly, one needs to stochastically confine cells in droplets so that each droplet contains at most a single cell; then, after incubating the emulsion, the colonies formed in droplets from single cells are detectable. This was first demonstrated by Boedicker and colleagues129 when the authors prepared multiple series of 50 droplets, each droplet 1 nanoliter in volume, each series of droplets with different antibiotic concentrations. The droplet series were separated from each other in a piece of tubing with air bubbles. Due to stochastic confinement, only a few of the 50 droplets per antibiotic concentration contained cells, so the throughput of the method was limited. The throughput of this method was improved a decade later by Postek et al., where authors were able to semi-automatically generate dozens of emulsions of ca. 2000 droplets each where each emulsion contained a different antibiotic concentration130 (Fig. 5A–C). In single-cell level experiments by Postek et al., a single emulsion contained ca. 200 droplets with individually encapsulated bacteria inside, opening possibilities for studies of population heterogeneity in isolated cells. A system similar to that of Postek et al.130 was presented recently with a different approach to droplet generation and emulsion handling.131 Single-cell level studies have also been performed in droplets by Liu et al.,132 Lyu et al.,133 Scheler et al.,134 and Kaushik et al.135 Kaushik and colleagues manually (in a non-automated fashion) generated emulsions with different antibiotic concentrations and with single cells encapsulated in droplets, then measured end-point signals from droplets to establish whether the cells grew or not. Scheler et al. run similar experiments, however, on a larger scale: to identify emulsions with different antibiotic concentrations after pooling the droplets, the authors color-coded them before pooling the droplets for incubation and detection.134 The authors demonstrated that individual bacteria within an isogenic population have different levels of antibiotic resistance. Liu et al. also generated droplets with bacteria and antibiotics manually, incubated each emulsion with different antibiotic concentrations in separate test tubes, and later performed a high-throughput measurement of optical density inside droplets to verify if a given droplet contained growing bacteria.132 As optical density was screened, this technique is suitable for testing the resistance of clinical strains of bacteria. Lyu et al. used a similar strategy of manual emulsion generation133 (Fig. 5D–F). After incubation, the authors used a pico-injector136 to seed each droplet with a dye resazurin to detect fluorescence from droplets containing living cells. As the authors generated 107 droplets per antibiotic concentration, they were analyzing 106 individual bacteria per antibiotic concentration, thus being able to identify rare resistant subpopulations of cells.
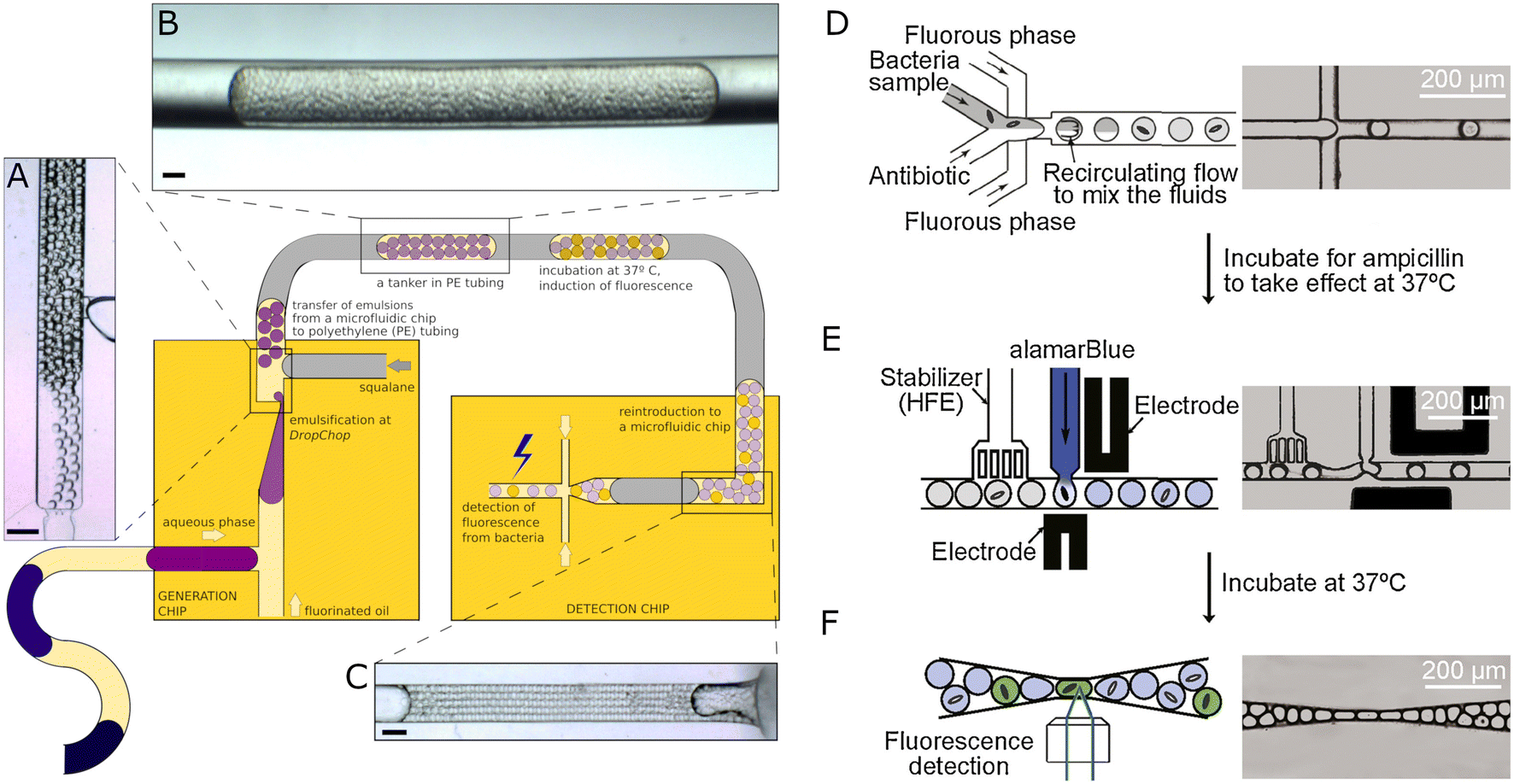 | ||
| Fig. 5 Antimicrobial susceptibility testing using flowing droplet systems. A–C) Semi-automatic droplet-based system for single-cell AST. Large droplets containing bacteria and various antibiotic concentrations generated before with T-junction or through aspiration by a positioning system for tubing are split into nanoliter droplets using vertically oriented DropChop emulsifying geometry (A). The subsequent libraries are separated by immiscible squalene oil (gray) and collected in PE tubing (B). The emulsions (tankers) are then incubated to allow bacteria to grow and increase the droplets' fluorescence intensity, which is measured in the detection chip after incubation (C). Scale bars: 400 μm. A–C reproduced from ref. 96. D–F) Droplet-based method for single-cell quantification of phenotypic heteroresistance. Bacteria sample with an antibiotic is split into droplets (D) and incubated at 37 °C. After incubation, the droplets are reintroduced to the microfluidic device. A viability probe alamarBlue is pico-injected into each drop I), and the droplets are collected for additional incubation. In the last step, the fluorescence intensity is registered in each droplet, and the heteroresistance of the bacterial population can be quantified. D–F Reproduced from ref. 99. | ||
In population-level studies, Funfak et al. demonstrated a system in which the authors screened for bacterial response to toxins by inserting pH-sensitive polymer sensor particles in droplets with bacteria and tested compounds.137 Churski and colleagues138 demonstrated an automated system for the generation of gradients of two antibiotics in droplets with the addition of bacteria to each droplet. The authors used the device to screen for interactions between antibiotics in multiple concentrations at a narrow concentration range. Due to the technical aspect of droplet generation, this particular solution is not fit to screen broad concentration ranges, limiting its use in medical diagnostics. In a different approach, Baraban et al.127 and Jiang et al.139 demonstrated platforms that use droplets of 100 nl or 1 nl, respectively, to screen for MIC. Baraban et al. used a system with 1000 media droplets of 100 nl each, each droplet being separated from its neighbor media droplets by a portion of mineral oil. This indexing of droplets in a piece of tubing is the same as in the earlier work of Boedicker et al.129 and later by Churski et al.,138 Postek et al.,130 and Li et al.131 Apart from establishing MIC with a fluorescence-based detector, Baraban et al. measured the saturation number of bacteria per droplet volume, being 27 × 104 cells per 100 nl droplet for the conditions used. The authors showed that their platform is more precise than a gold standard platform for clinical AST, the VITEK® 2 by bioMérieux,139 due to the large numbers of droplets screened per single experimental run. Jiang et al.139 used resazurin fluorescent dye, which allowed the authors to expand to clinical strains. However, resazurin does not work with anaerobic bacteria. Both systems by Baraban et al. and by Jiang et al. suffer from a narrow range of antibiotic concentrations that can be tested due to technical limitations of the droplet generation method used – in the same way that limited the system by Churski and colleagues.138
3. Signal detection in microfluidic AST
Here we describe the methods of bacterial growth detection in microfluidics in the context of AST. We focus on the two methods we find the most important: i) optical detection which is still the golden standard in the clinic; ii) pheno-molecular detection which combines uses quantitative measurements of bacterial nucleic acids as a proxy for cell growth, and is a major improvement over classical molecular AST detection assays. The only assay covered in this review that produced AST results within 30 minutes of the patient sample collection is pheno-molecular. We also highlight other important methods of bacterial growth detection that were used in microfluidic AST and show promise in cutting the signal detection time.3.1. Optical detection of bacterial growth for microfluidic AST
Eucast140 and CLSI141 are both recommending that an inoculum (bacteria density) of 5 × 105 CFU ml−1 should be used in an AST assay. This brings up a problem of the inoculum effect discussed later in this article. However, the inoculum recommendation also means that AST is generally measured for a population of cells. Studying whole populations masks the effects that might arise from a single mutated cell or a small set of interacting cells, such as perister cells142 or viable but non culturable bacteria.104 On the technical level, testing large numbers of bacteria implies relatively easy use of optical detection of signals: as the test subjects are numerous, the signal produced by the bacteria population is high compared to the background. It is possible to run AST coupled stimulated Raman scattering143 for detection. Analysis of metabolism143–145 allows for AST within 30 minutes or within 2–3 hours by measuring the dissolved oxygen levels in a bacteria sample during cultivation145 by tracking luminescence of oxygen-sensing probes.OD-based bacterial growth detection in flowing droplets is under development as well, and the first deployed systems promise good prospects for AST measurements of clinical samples in droplets.155 The first reports on label-free detectors of bacterial growth in droplets concern large compartments of ca. microliter volume126,156 – such volumes do not allow for high-throughput applications, which is the greatest advantage of droplet-based systems. High-throughput screening requires using small droplets of the order of nanoliters and smaller. Boitard and colleagues presented a method for bacteria growth detection in picodroplets based on the change of osmotic pressure and the droplets' size due to bacteria growth,157 although this method does not provide high-throughput droplet analysis. Similarly, Liao et al. showed an exciting approach for label-free and sensitive detection of bacterial cells in picodroplets using micro-Raman spectroscopy, but without the possibility of high-throughput droplet screening.42 More promising approaches are presented by Zang et al., who showed bacteria detection based on real-time image processing at a frequency of 100 Hz,158 and by Liu et al., where the measurement relies on detecting light scattered by bacterial cells at a frequency of 243 Hz.132 Similarly, Hengoju et al. showed an optofluidic detection system based on absorbance readout and scattered light in picoliter droplets with a frequency of approx. 40 Hz.155 The highest throughput of screening of droplets containing bacteria for bacteria growth to date was presented by Pacocha et al., where the growth of bacteria based on the intensity of scattered light was measured at a frequency of 1200 Hz, promising good prospects for AST measurements of clinical samples in droplets. We see flowing droplets not as tools for clinical use, but rather for academic research, and the throughput of signal detection from flowing droplets is not critical for non-clinical ASTs.
3.2. Pheno-molecular detection of bacterial growth for microfluidic AST
Molecular methods are a promising path in microfluidic AST development: molecular tests can identify a pathogen, verify if it is resistant to an antibiotic, and finally measure the MIC of a given antibiotic with the identified bacterium, also at the single-cell level. The molecular AST in microfluidics stems from digital PCR:159,160 similarly to confining individual bacteria in droplets, a sample containing multiple DNA target molecules can be divided into numerous separate droplets, so that each droplet contains either no DNA molecules or at most a single DNA target molecule.59 Then the amplification is performed as in classical PCR. The answer is a signal coming from only the droplets that contained DNA. Since each droplet stored at most one DNA molecule, counting the compartments that provide a positive signal allows counting the number of DNA molecules before the reaction with absolute precision and without the need for calibration curves as in real-time PCR. Molecular detection of antibiotic resistance is applied to microdroplets;161 however, the issue in incorporating molecular tests to AST is that the lack of resistance genes is not a reliable predictor of susceptibility to a given antibiotic162 apart from a few resistance genes (e.g., mecA, vanA, vanB163), which is why the hybrid phenotypic–genotypic approach (“pheno-molecular”, as called by Athamanolap et al.164) was developed. This hybrid approach uses the number of DNA or RNA molecules in the sample as a proxy for the growth of bacteria. The founding research for microfluidic pheno-molecular AST was published in 2015 by Hou et al.,165 who used inertial microfluidics to separate bacteria from whole blood samples and screened with non-enzymatic nucleic acid detection assay (NanoString) for antibiotic-response-related mRNA. A first DNA-based pheno-molecular assay was presented in 2016 by Schoepp and colleagues,166 where authors compared the changes in concentration of pathogen-specific DNA sequences after exposure to antibiotics. The resistant bacteria would show similar DNA concentrations over time in both antibiotic-treated sample and in antibiotic-free control, while susceptible strains would show a slower increase or no increase in DNA concentration in a treated sample when compared to an antibiotic-free control. The authors demonstrated that droplet digital PCR allows for faster detection of changes in concentration of pathogen-specific DNA after exposure to antibiotics compared to qPCR. In their subsequent development, Schoepp and colleagues167 optimized the droplet digital LAMP (loop-mediated isothermal amplification168) reaction, the sample handling protocol, and the signal detection software so that they were able to distinguish between antibiotic-susceptible and antibiotic-resistant bacteria from unprocessed clinical samples within 30 minutes (Fig. 6A–D). The detection this fast is impressive as even the fast optics-based AST methods that follow individual cells are slower than the presented method. The same group later showed that it is possible to rapidly identify antibiotic-resistant or susceptible slow-growing bacteria by measuring the levels of the RNA markers of bacteria exposed to antibiotics169 – possibly, this or another research team will try and run such RNA measurements with clinical samples as well. Athamanolap and colleagues developed a system for pheno-molecular AST based on 5000 1 nl wells for partitioning the sample instead of using droplets164 (Fig. 6E). The authors analyzed DNA melt curves from each well and used machine learning to teach their program to distinguish between species-specific melt curves. The number of species-specific melt curves was counted and compared between samples treated and untreated with an antibiotic to identify antibiotic susceptibility, albeit slower than in the report by Shoepp and colleagues. Kaushik and colleagues used a pheno-molecular approach to identify if bacteria from urine samples are E. coli, Enterobacteriales but not E. coli or non-Enterobacteriales Gram-negative cells. Additionally, the authors analyzed the antibiotic resistance of the tested cells by merging PNA fluorescent probes for 16S RNA with fluorescence detector. The system was shown to identify bacteria and their antibiotic resistance from urine samples in 30 minutes170 (Fig. 6F–H).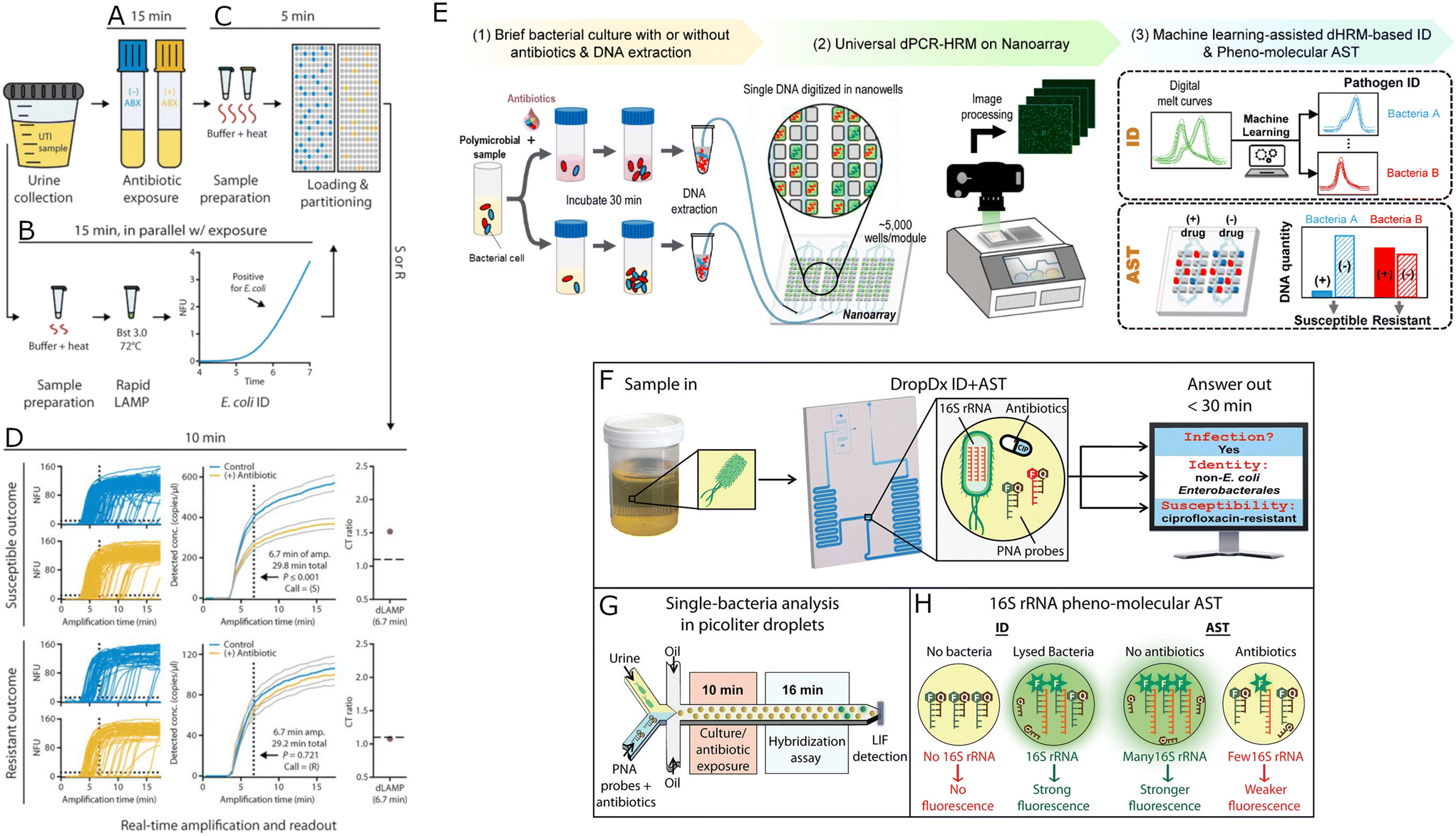 | ||
| Fig. 6 Microfluidic pheno-molecular AST assays. A–D) Phenotypic susceptibility testing using digital LAMP quantification. The UTI (urinary tract infection) sample is incubated in the presence and absence of antibiotic for 15 min (A). In parallel, the nucleic acids (NAs) prepared from an aliquot of the urine sample are amplified by bulk LAMP assay indicating E. coli at clinically relevant concentrations (B). Then, NAs are extracted from control and antibiotic-treated samples using extraction buffer and partitioned using SlipChips for dLAMP quantification (C). The process of NAs amplification is monitored in real-time, and after 6.7 min, the susceptibility can be determined (D). The presented data concerns one resistant and one susceptible bacteria. Gray lines represent 95% confidence intervals. A–D Reproduced from ref. 113. E) Digital PCR and melt platform for rapid bacteria identification and AST. The workflow starts with the incubation of bacteria with and without antibiotics for 30 min. Bacterial DNA is extracted from both samples, mixed with a universal PCR mixture, and loaded into a nanoarray device for dPCR-HRM (digital PCR and high-resolution melt). Then, the ‘temperature lapse’ fluorescence images are analyzed, and digital melt curves for all nanowells are created. Bacterial species are identified using a machine-learning-based melt curves identification algorithm. The DNA copy number is determined by enumeration of species-specific digital melt curves. Finally, the antibiotic susceptibility is assessed by comparing the DNA copy numbers of both samples (treated and not treated with antibiotics). Reproduced from ref. 110. F–H) Droplet-based pheno-molecular ID and AST of UTIs. The system allows for a single-cell detection of 16S rRNA, providing confirmation of Gram-negative bacterial infection, uropathogen identification, and determination of its antimicrobial susceptibility (F). Urine sample, 16S rRNA-specific fluorogenic PNA probes, and/or antibiotics are split into picoliter droplets. Then, bacteria are exposed to culture or antibiotic for 10 min followed by a probe hybridization process for 16 min, and target a two-color laser-induced fluorescence (LIF) detector (G). The identification of uropathogenic bacteria is based on detecting specific 16S rRNA sequences using the fluorescence color and intensity of droplets. The difference in probe fluorescence intensities between antibiotic-dosed and antibiotic-free droplets is used to measure the relative production of 16S rRNA in single cells, which is used to determine antimicrobial resistance. F–H Reproduced from ref. 116. | ||
3.3. Mechanical detection of bacterial growth for microfluidic AST
Mechanical detection of bacterial growth in microfluidics is done with cantilever sensors that resinate at different frequencies depending on the cantilever mass. The cantilever mass is changed when bacterial cells are added to the sensor.171–176 Microfluidic cantilever-based sensors173 allow for determining antibiotic resistance within 45 minutes,174 which is short enough time to validate deployment of the method to the clinic. There are start-up companies such as Resistell that are exploiting cantilever-based sensors in their products for AST.27 There is a recent review article available focusing on mechanical detection of bacterial growth for AST and we direct the reader there for more information.1773.4. Electrical detection of bacterial growth for microfluidic AST
Electrical impedance measurements for fast detection of bacterial growth has been a viable concept for many years.178 The advantage of electrical signal detection over the optical or molecular detection methods is the fact that no expensive miscroscope or no expensive molecular biology reagents are necessary for AST. In the past decade, the electrical detection of bacterial growth has been adapted to microfluidics, leading to rapid detection times.95,116,179 The electrical resistance of the microchannels change in proportion to the number of bacteria in the microchannels. Optimal electrode placement around a microchannel in a impedance micro-cytometer have allowed for identifying susceptibility or resistance in ca. 30 minutes180 after analyzing ca. 1000 cells per second. Microfluidics impedance cytometry was recently covered in an informative review article.1813.5. CRISPR detection of bacterial growth for microfluidic AST
Here we mention molecular (not pheno-molecular detection) of bacterial growth that is based on a CRISPR screen. Recently, CARMEN, an assay based on SHERLOCK182 was demonstrated in droplets to differentiate viruses from a vast collection of 169 human-associated viruses.183 A modified CARMEN assay was deployed to identify 52 clinically relevant bacterial pathogens in a single assay with additional identification of major antibiotic resistance genes.184 Although this assay is molecular, not pheno-molecular, it could in principle be combined with a pheno-molecular approach to achieve a platform that would both identify a pathogen and also rapidly establish the pathogen's antibiotic resistance levels – which is the holy grail of clinical AST.4. Challenges and opportunities
4.1. Challenges
The first main approach is to follow the growth of individual cells in the presence of antibiotics. It has been shown that such tests can provide MIC rapidly, even within 30 minutes113 with clinical isolates (not with clinical samples). However, approaches based on single-cell imaging require high-resolution optics and time-lapse imaging of multiple locations, which makes these methods expensive and, until today, unfit for clinical practice.
Another major approach to reducing detection type is “pheno-molecular” assays, where methods of nucleic acid quantification are used to quantify the number of DNA or RNA of interest in the bacterial lysate. The DNA/RNA molecule count is a proxy for bacterial growth and has been shown to be faster than optical growth-based assays, even for analysis of clinical samples such as urine (not the bacteria isolates from such samples).167 There are other paths to run rapid ASTs in microfluidic devices: analysis of metabolism143–145 allows for AST within 30 minutes, albeit with expensive stimulated Raman scattering,143 or within 2–3 hours by measuring the dissolved oxygen levels in a bacteria sample during cultivation;145 microfluidic cantilever-based sensors173 allow for determining antibiotic resistance within 45 minutes.174 A microfluidic impedance-based assay was demonstrated recently, identifying susceptibility or resistance in ca. 30 minutes.180
Obtaining AST results within 30–60 minutes from a clinical sample (not bacterial isolate: isolates require additional time to prepare) is enough for a physician to inform the patient of the nature of an infection during a single visit, and we do not see any need to improve on the detection time below what has already been shown. At the time of writing of this article, only microfluidic pheno-molecular assays were shown to achieve AST results from clinical samples in such a short time; however, optical or sensor-based based microfluidic methods are lagging not far behind.
4.1.2.1. Printing antibiotics. From systems that follow single cell division presented to date, only one is capable of multiplexing for many antibiotics or many concentrations of antibiotics per experimental run,84 and this is because the test is based on a modified 96 well-plate. Similar multiplexing could be easily achievable with chamber-based devices where antibiotic is placed into chambers and dried before the bacteria sample is flown through the device and segmented into chambers. Both of these approaches are possible to automate with either pipetting robots for 96 well-plates or with spotters (droplet printers) in case of drying antibiotics on chips. The well-plate method, however, requires the application of the sample separately to each well, therefore automation could only be done with the help of a specialized diagnostics facility, which is counter to the commercial goal of point-of-care microfluidic ASTs. Printing antibiotics into chambers is more difficult technically than pipetting samples to well-plates, as the chambers are usually much smaller than wells. However, the to-date presented chamber-based solutions require the end-user to perform only a single pipetting step or two steps at most to run AST.75
4.1.2.2. Serial dilution in droplets. Apart from the systems based on antibiotic printing, droplet-based systems are the most suitable systems for multiplexing AST and for being automated in general. The downside of such droplet-based AST is that flowing droplets do not support rapid AST. However, single-cell level (suggesting possible rapid AST) imaging in immobilized droplets has already been presented also with antibiotic multiplexing121,122 – the drawback of these systems is that they are rather complicated, and each new antibiotic tested would require changes in the device design. A possible solution to this issue is repurposing the already existing platform for multiplexed AST in trapped droplets53 for single-cell analysis – this would, however, require the preparation of all antibiotic solutions in a BioRad's QX200 cartridge (similar to a well plate) before the experiment which is impractical and on a large scale would require a specialized diagnostics laboratory. We suggest that in terms of multiplexing microfluidic ASTs, it would be best to pair a droplet-based dilution systems with a hybrid droplet-chamber array.
4.1.2.2.1. Microfluidic dilution systems. Automated microfluidic devices for the generation of concentration gradients are plentiful,185 and some of the AST systems described in this manuscript contain such a generator. We are not going to describe in detail all such systems. Rather, we will briefly outline the challenges for the main classes of concentration gradient generators (dilutors) that are used or could be used in droplet microfluidic AST. These main classes are i) diffusion-based dilutors;186 ii) dilutors based on a coalescence of flowing droplets;138 iii) dilutors based on trapped droplets.130 The diffusion-based dilutors rely on the passive mixing of fluids flowing in microchannels before the droplet generation, the fluids in AST being an antibiotic stock solution, an antibiotic-free medium, and a bacterial sample. The main challenge in this kind of devices is the limited range of concentrations they can produce and limited multiplexing capabilities due to the large channel networks necessary for the systems to operate. A diffusion-based method that overcomes these issues was presented by Miller et al.187 where the authors inject a portion of the drug (for ASTs, it would be an antibiotic), and the gradient is being generated through Taylor–Arys dispersion before droplet generation. This device was shown to operate at 3 orders of magnitude of drug concentrations, which is still not ideal for medical diagnostics. The droplet dilutors based on the coalescence of flowing droplets also suffer from a limited dynamic range: a droplet-based system for AST in droplets presented by Churski et al. contained a dilutor in which droplets of antibiotic and droplets of pure medium were coalesced in series.138 Each coalescence event comprised droplets of different volume so that the ratio of antibiotic to pure medium droplet sizes was different each time, but the final merged droplet was always the same size. This heavily limits the dynamic range of the method as to cover a wide range of concentrations, an impractically small droplet would have to be merged with an impractically large droplet to achieve a highly diluted sample – Churski et al. report only two orders of magnitude dynamic range, similarly to a more recent integrated dilutor-AST system by Osaid et al.74 Trap-based droplet dilutors offer, in theory, an infinite dynamic range of concentrations, as a trapped droplet can be diluted by subsequent diluent droplets indefinitely.188 However, trap-based dilutors require stable flow of droplets to operate properly, as mixing in droplets during dilution depends on the flow rates of liquids in a dilution system.189 There are reports, however, of successful integration of trap-based dilutors with droplet arrays190,191 and also running AST in such devices.124 Considering dilution errors, trap-based dilutors cumulate these errors: if the dilutions are done with 5% precision each, then the 10th dilution would have a concentration error of 50%, while dilutors as presented by Churski et al. generate sample concentrations with the same error for every dilution.
4.2. Opportunities
The most clinically relevant application of microfluidics in antibiotics studies is AST. However, there are other antibiotic-related research paths that can be followed using microfluidic methods. Some of these possible research areas could be explored even with the existing technologies designed for ASTs or for other applications e.g., in analytical chemistry. Here, we highlight some of the most exciting and achievable antibiotic-related research topics available for microfluidics.New approaches to this problem take advantage of droplet microfluidics: Sun et al. presented a digital droplet PCR to detect resistance genes and point mutations in H. pylori cells in stool samples.206 Scheler et al.134 and Lyu et al.133 demonstrated a digital quantification of the heteroresistance based on single-cell MIC determination. Techniques combining microfluidic devices with microscopy, where the growth of many individual cells is observed over time in the presence and absence of antibiotics113 are promising as well. There is still room for improvement in microfluidic detection of heteroresistance, especially in multiplexing and in label-free readout.
The standard method for quantifying persister cells or small colony variants is a colony counting assay, which is a tedious and expensive approach.212 Innovative microfluidic technology combines a mother machine with live-cell imaging, which allows for bacteria growth observation in various conditions.104 It provides a sensitive analysis of the antibiotic susceptibility distribution of cells in the bacterial population. However, it produces a massive amount of data for analysis, making the application of this method difficult in clinical microbiology. High-throughput methods are required to detect persisting tolerant to antibiotics cells in the bacterial population as persisters are rare, and such an assay could benefit from the large scale of droplet microfluidics.
Measurements of metabolism level of bacteria in droplets156 were recently used for microfluidic AST based on analysis of metabolism levels of cells as a phenotype.144,145 Such methods could be further developed to, e.g., sort out cells with high metabolism rates and identify their rates of emergence of antibiotic resistance with a single-cell microchemostat we propose in the previous paragraph. Is a higher metabolism level correlated with a higher rate of resistance emergence?
5. Concluding remarks
Antibiotic susceptibility testing research has gained considerable momentum thanks to microfluidic technologies. A wide variety of approaches to the AST field, such as single-cell optical tracking, sampling bacteria to chambers pre-filled with antibiotics, complex droplet-based systems, or pheno-molecular microfluidic assays, show the robustness of microfluidics in microbiology. The AST sample-to-detection time is already superb in some microfluidic assays and now it is mostly multiplexing of different antibiotics and different antibiotic concentrations that needs to be added on top of short assay times that is a technological bottleneck for microfluidic AST. If this bottleneck is overcome, then most requirements for an ideal AST assay would be achieved. The problem that will remain unchanged is the fact that the largest market for AST platforms is the US and EU,218 while the regions with the largest antibiotic-resistance-related problems in the world are Africa and Asia. Although the road to a highly informative multiplexed and fast microfluidic AST assay with the clinical application is not long, the challenge that looms on the horizon lies in introducing such a platform to countries that suffer the most from infectious diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. P. was supported by the Polish National Agency for Academic Exchange (NAWA) through the Bekker Programme, grant no. PPN/BEK/2020/1/00333/U/00001. W. P. was supported by the Foundation for Polish Science (FNP) with the START 069.2021 scholarship. N. P. and P. G. acknowledge the support from the National Science Centre, Poland, funding based on decision 2018/30/A/ST4/00036, Maestro 10.References
- A. Fleming, Nobel Prize.
- C. L. Ventola, Pharmacol. Ther., 2015, 40, 277–283 Search PubMed.
- ECDC/EMEA, The bacterial challenge: time to react, 2009.
- CDC, Antibiotic Resistance Threats in the United States, 2019, 2019.
- N. Daulaire, A. Bang, G. Tomson, J. N. Kalyango and O. Cars, J. Law Med. Ethics., 2015, 43, 17–21 CrossRef PubMed.
- M. E. A. de Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184 CrossRef PubMed.
- C. Abbafati, K. M. Abbas, M. Abbasi-Kangevari, F. Abd-Allah, A. Abdelalim, M. Abdollahi, I. Abdollahpour, K. H. Abegaz, H. Abolhassani, V. Aboyans, L. G. Abreu, M. R. M. Abrigo, A. Abualhasan, L. J. Abu-Raddad, A. I. Abushouk, M. Adabi, V. Adekanmbi, A. M. Adeoye, O. O. Adetokunboh, D. Adham, S. M. Advani, A. Afshin, G. Agarwal, S. M. K. Aghamir, A. Agrawal, T. Ahmad, K. Ahmadi, M. Ahmadi, H. Ahmadieh, M. B. Ahmed, T. Y. Akalu, R. O. Akinyemi, T. Akinyemiju, B. Akombi, C. J. Akunna, F. Alahdab, Z. Al-Aly, K. Alam, S. Alam, T. Alam, F. M. Alanezi, T. M. Alanzi, B. W. Alemu, K. F. Alhabib, M. Ali, S. Ali, G. Alicandro, C. Alinia, V. Alipour, H. Alizade, S. M. Aljunid, F. Alla, P. Allebeck, A. Almasi-Hashiani, H. M. Al-Mekhlafi, J. Alonso, K. A. Altirkawi, M. Amini-Rarani, F. Amiri, D. A. Amugsi, R. Ancuceanu, D. Anderlini, J. A. Anderson, C. L. Andrei, T. Andrei, C. Angus, M. Anjomshoa, F. Ansari, A. Ansari-Moghaddam, I. C. Antonazzo, C. A. T. Antonio, C. M. Antony, E. Antriyandarti, D. Anvari, R. Anwer, S. C. Y. Appiah, J. Arabloo, M. Arab-Zozani, A. Y. Aravkin, F. Ariani, B. Armoon, J. Ärnlöv, A. Arzani, M. Asadi-Aliabadi, A. A. Asadi-Pooya, C. Ashbaugh, M. Assmus, Z. Atafar, D. D. Atnafu, M. M. D. W. Atout, F. Ausloos, M. Ausloos, B. P. Ayala Quintanilla, G. Ayano, M. A. Ayanore, S. Azari, G. Azarian, Z. N. Azene, A. Badawi, A. D. Badiye, M. A. Bahrami, M. H. Bakhshaei, A. Bakhtiari, S. M. Bakkannavar, A. Baldasseroni, K. Ball, S. H. Ballew, D. Balzi, M. Banach, S. K. Banerjee, A. B. Bante, A. G. Baraki, S. L. Barker-Collo, T. W. Bärnighausen, L. H. Barrero, C. M. Barthelemy, L. Barua, S. Basu, B. T. Baune, M. Bayati, J. S. Becker, N. Bedi, E. Beghi, Y. Béjot, M. L. Bell, F. B. Bennitt, I. M. Bensenor, K. Berhe, A. E. Berman, A. S. Bhagavathula, R. Bhageerathy, N. Bhala, D. Bhandari, K. Bhattacharyya, Z. A. Bhutta, A. Bijani, B. Bikbov, M. S. Bin Sayeed, A. Biondi, B. M. Birihane, C. Bisignano, R. K. Biswas, H. Bitew, S. Bohlouli, M. Bohluli, A. S. Boon-Dooley, G. Borges, A. M. Borzì, S. Borzouei, C. Bosetti, S. Boufous, D. Braithwaite, M. Brauer, N. J. K. Breitborde, S. Breitner, H. Brenner, P. S. Briant, A. N. Briko, N. I. Briko, G. B. Britton, D. Bryazka, B. R. Bumgarner, K. Burkart, R. T. Burnett, S. Burugina Nagaraja, Z. A. Butt, F. L. Caetano Dos Santos, L. E. Cahill, L. A. Cámera, I. R. Campos-Nonato, R. Cárdenas, G. Carreras, J. J. Carrero, F. Carvalho, J. M. Castaldelli-Maia, C. A. Castañeda-Orjuela, G. Castelpietra, F. Castro, K. Causey, C. R. Cederroth, K. M. Cercy, E. Cerin, J. S. Chandan, K. L. Chang, F. J. Charlson, V. K. Chattu, S. Chaturvedi, N. Cherbuin, O. Chimed-Ochir, D. Y. Cho, J. Y. J. Choi, H. Christensen, D. T. Chu, M. T. Chung, S. C. Chung, F. M. Cicuttini, L. G. Ciobanu, M. Cirillo, T. K. D. Classen, A. J. Cohen, K. Compton, O. R. Cooper, V. M. Costa, E. Cousin, R. G. Cowden, D. H. Cross, J. A. Cruz, S. M. A. Dahlawi, A. A. M. Damasceno, G. Damiani, L. Dandona, R. Dandona, W. J. Dangel, A. K. Danielsson, P. I. Dargan, A. M. Darwesh, A. Daryani, J. K. Das, R. Das Gupta, J. das Neves, C. A. Dávila-Cervantes, D. V. Davitoiu, D. De Leo, L. Degenhardt, M. DeLang, R. P. Dellavalle, F. M. Demeke, G. T. Demoz, D. G. Demsie, E. Denova-Gutiérrez, N. Dervenis, G. P. Dhungana, M. Dianatinasab, D. Dias da Silva, D. Diaz, Z. S. Dibaji Forooshani, S. Djalalinia, H. T. Do, K. Dokova, F. Dorostkar, L. Doshmangir, T. R. Driscoll, B. B. Duncan, A. R. Duraes, A. W. Eagan, D. Edvardsson, N. El Nahas, I. El Sayed, M. El Tantawi, I. Elbarazi, I. Y. Elgendy, S. I. El-Jaafary, I. R. F. Elyazar, S. Emmons-Bell, H. E. Erskine, S. Eskandarieh, S. Esmaeilnejad, A. Esteghamati, K. Estep, A. Etemadi, A. E. Etisso, J. Fanzo, M. Farahmand, M. Fareed, R. Faridnia, A. Farioli, A. Faro, M. Faruque, F. Farzadfar, N. Fattahi, M. Fazlzadeh, V. L. Feigin, R. Feldman, S. M. Fereshtehnejad, E. Fernandes, G. Ferrara, A. J. Ferrari, M. L. Ferreira, I. Filip, F. Fischer, J. L. Fisher, L. S. Flor, N. A. Foigt, M. O. Folayan, A. A. Fomenkov, L. M. Force, M. Foroutan, R. C. Franklin, M. Freitas, W. Fu, T. Fukumoto, J. M. Furtado, M. M. Gad, E. Gakidou, S. Gallus, A. L. Garcia-Basteiro, W. M. Gardner, B. S. Geberemariyam, A. A. A. Ayalew Gebreslassie, A. Geremew, A. Gershberg Hayoon, P. W. Gething, M. Ghadimi, K. Ghadiri, F. Ghaffarifar, M. Ghafourifard, F. Ghamari, A. Ghashghaee, H. Ghiasvand, N. Ghith, A. Gholamian, R. Ghosh, P. S. Gill, T. G. Ginindza, G. Giussani, E. V. Gnedovskaya, S. Goharinezhad, S. V. Gopalani, G. Gorini, H. Goudarzi, A. C. Goulart, F. Greaves, M. Grivna, G. Grosso, M. I. M. Gubari, H. C. Gugnani, R. A. Guimarães, R. A. Guled, G. Guo, Y. Guo, R. Gupta, T. Gupta, B. Haddock, N. Hafezi-Nejad, A. Hafiz, A. Haj-Mirzaian, A. Haj-Mirzaian, B. J. Hall, I. Halvaei, R. R. Hamadeh, S. Hamidi, M. S. Hammer, G. J. Hankey, H. Haririan, J. M. Haro, A. I. Hasaballah, M. M. Hasan, E. Hasanpoor, A. Hashi, S. Hassanipour, H. Hassankhani, R. J. Havmoeller, S. I. Hay, K. Hayat, G. Heidari, R. Heidari-Soureshjani, H. J. Henrikson, M. E. Herbert, C. Herteliu, F. Heydarpour, T. R. Hird, H. W. Hoek, R. Holla, P. Hoogar, H. D. Hosgood, N. Hossain, M. Hosseini, M. Hosseinzadeh, M. Hostiuc, S. Hostiuc, M. Househ, M. Hsairi, V. C. R. Hsieh, G. Hu, K. Hu, T. M. Huda, A. Humayun, C. K. Huynh, B. F. Hwang, V. C. Iannucci, S. E. Ibitoye, N. Ikeda, K. S. Ikuta, O. S. Ilesanmi, I. M. Ilic, M. D. Ilic, L. R. Inbaraj, H. Ippolito, U. Iqbal, S. S. N. Irvani, C. M. S. Irvine, M. M. Islam, S. M. S. Islam, H. Iso, R. Q. Ivers, C. C. D. Iwu, C. J. Iwu, I. O. Iyamu, J. Jaafari, K. H. Jacobsen, H. Jafari, M. Jafarinia, M. A. Jahani, M. Jakovljevic, F. Jalilian, S. L. James, H. Janjani, T. Javaheri, J. Javidnia, P. Jeemon, E. Jenabi, R. P. Jha, V. Jha, J. S. Ji, L. Johansson, O. John, Y. O. John-Akinola, C. O. Johnson, J. B. Jonas, F. Joukar, J. J. Jozwiak, M. Jürisson, A. Kabir, Z. Kabir, H. Kalani, R. Kalani, L. R. Kalankesh, R. Kalhor, T. Kanchan, N. Kapoor, B. K. Matin, A. Karch, M. A. Karim, G. M. Kassa, S. V. Katikireddi, G. A. Kayode, A. Kazemi Karyani, P. N. Keiyoro, C. Keller, L. Kemmer, P. J. Kendrick, N. Khalid, M. Khammarnia, E. A. Khan, M. Khan, K. Khatab, M. M. Khater, M. N. Khatib, M. Khayamzadeh, S. Khazaei, C. Kieling, Y. J. Kim, R. W. Kimokoti, A. Kisa, S. Kisa, M. Kivimäki, L. D. Knibbs, A. K. S. Knudsen, J. M. Kocarnik, S. Kochhar, J. A. Kopec, V. A. Korshunov, P. A. Koul, A. Koyanagi, M. U. G. Kraemer, K. Krishan, K. J. Krohn, H. Kromhout, B. Kuate Defo, G. A. Kumar, V. Kumar, O. P. Kurmi, D. Kusuma, C. La Vecchia, B. Lacey, D. K. Lal, R. Lalloo, T. Lallukka, F. H. Lami, I. Landires, J. J. Lang, S. M. Langan, A. O. Larsson, S. Lasrado, P. Lauriola, J. V. Lazarus, P. H. Lee, S. W. H. Lee, K. E. Legrand, J. Leigh, M. Leonardi, H. Lescinsky, J. Leung, M. Levi, S. Li, L. L. Lim, S. Linn, S. Liu, S. Liu, Y. Liu, J. Lo, A. D. Lopez, J. C. F. Lopez, P. D. Lopukhov, S. Lorkowski, P. A. Lotufo, A. Lu, A. Lugo, E. R. Maddison, P. W. Mahasha, M. M. Mahdavi, M. Mahmoudi, A. Majeed, A. Maleki, S. Maleki, R. Malekzadeh, D. C. Malta, A. A. Mamun, A. L. Manda, H. Manguerra, F. Mansour-Ghanaei, B. Mansouri, M. A. Mansournia, A. M. Mantilla Herrera, J. C. Maravilla, A. Marks, R. V. Martin, S. Martini, F. R. Martins-Melo, A. Masaka, S. Z. Masoumi, M. R. Mathur, K. Matsushita, P. K. Maulik, C. McAlinden, J. J. McGrath, M. McKee, M. M. Mehndiratta, F. Mehri, K. M. Mehta, Z. A. Memish, W. Mendoza, R. G. Menezes, E. W. Mengesha, A. Mereke, S. T. Mereta, A. Meretoja, T. J. Meretoja, T. Mestrovic, B. Miazgowski, T. Miazgowski, I. M. Michalek, T. R. Miller, E. J. Mills, G. K. Mini, M. Miri, A. Mirica, E. M. Mirrakhimov, H. Mirzaei, M. Mirzaei, R. Mirzaei, M. Mirzaei-Alavijeh, A. T. Misganaw, P. Mithra, B. Moazen, D. K. Mohammad, Y. Mohammad, N. Mohammad Gholi Mezerji, A. Mohammadian-Hafshejani, N. Mohammadifard, R. Mohammadpourhodki, A. S. Mohammed, H. Mohammed, J. A. Mohammed, S. Mohammed, A. H. Mokdad, M. Molokhia, L. Monasta, M. D. Mooney, G. Moradi, M. Moradi, M. Moradi-Lakeh, R. Moradzadeh, P. Moraga, L. Morawska, J. Morgado-Da-Costa, S. D. Morrison, A. Mosapour, J. F. Mosser, S. Mouodi, S. M. Mousavi, A. M. Khaneghah, U. O. Mueller, S. Mukhopadhyay, E. C. Mullany, K. I. Musa, S. Muthupandian, A. F. Nabhan, M. Naderi, A. J. Nagarajan, G. Nagel, M. Naghavi, B. Naghshtabrizi, M. D. Naimzada, F. Najafi, V. Nangia, J. R. Nansseu, M. Naserbakht, V. C. Nayak, I. Negoi, J. W. Ngunjiri, C. T. Nguyen, H. L. T. Nguyen, M. Nguyen, Y. T. Nigatu, R. Nikbakhsh, M. R. Nixon, C. A. Nnaji, S. Nomura, B. Norrving, J. J. Noubiap, C. Nowak, V. Nunez-Samudio, B. Oancea, C. M. Odell, F. A. Ogbo, I. H. Oh, E. W. Okunga, M. Oladnabi, A. T. Olagunju, B. O. Olusanya, J. O. Olusanya, M. O. Omer, K. L. Ong, O. E. Onwujekwe, H. M. Orpana, A. Ortiz, O. Osarenotor, F. B. Osei, S. M. Ostroff, A. Otoiu, N. Otstavnov, S. S. Otstavnov, S. Øverland, M. O. Owolabi, P. A. Mahesh, J. R. Padubidri, R. Palladino, S. Panda-Jonas, A. Pandey, C. D. H. Parry, M. Pasovic, D. K. Pasupula, S. K. Patel, M. Pathak, S. B. Patten, G. C. Patton, H. P. Toroudi, A. E. Peden, A. Pennini, V. C. F. Pepito, E. K. Peprah, D. M. Pereira, K. Pesudovs, H. Q. Pham, M. R. Phillips, C. Piccinelli, T. M. Pilz, M. A. Piradov, M. Pirsaheb, D. Plass, S. Polinder, K. R. Polkinghorne, C. D. Pond, M. J. Postma, H. Pourjafar, F. Pourmalek, A. Poznañska, S. I. Prada, V. Prakash, D. R. A. Pribadi, E. Pupillo, Z. Q. Syed, M. Rabiee, N. Rabiee, A. Radfar, A. Rafiee, A. Raggi, M. A. Rahman, A. Rajabpour-Sanati, F. Rajati, I. Rakovac, P. Ram, K. Ramezanzadeh, C. L. Ranabhat, P. C. Rao, S. J. Rao, V. Rashedi, P. Rathi, D. L. Rawaf, S. Rawaf, L. Rawal, R. Rawassizadeh, R. Rawat, C. Razo, S. B. Redford, R. C. Reiner, M. B. Reitsma, G. Remuzzi, V. Renjith, A. M. N. Renzaho, S. Resnikoff, N. Rezaei, N. Rezaei, A. Rezapour, P. A. Rhinehart, S. M. Riahi, D. C. Ribeiro, D. Ribeiro, J. Rickard, J. A. Rivera, N. L. S. Roberts, S. Rodríguez-Ramírez, L. Roever, L. Ronfani, R. Room, G. Roshandel, G. A. Roth, D. Rothenbacher, E. Rubagotti, G. M. Rwegerera, S. Sabour, P. S. Sachdev, B. Saddik, E. Sadeghi, M. Sadeghi, R. Saeedi, S. Saeedi Moghaddam, Y. Safari, S. Safi, S. Safiri, R. Sagar, A. Sahebkar, S. M. Sajadi, N. Salam, P. Salamati, H. Salem, M. R. Salem, H. Salimzadeh, O. M. Salman, J. A. Salomon, Z. Samad, H. Samadi Kafil, E. Z. Sambala, A. M. Samy, J. Sanabria, T. G. Sánchez-Pimienta, D. F. Santomauro, I. S. Santos, J. V. Santos, M. M. Santric-Milicevic, S. Y. I. Saraswathy, R. Sarmiento-Suárez, N. Sarrafzadegan, B. Sartorius, A. Sarveazad, B. Sathian, T. Sathish, D. Sattin, S. Saxena, L. E. Schaeffer, S. Schiavolin, M. P. Schlaich, M. I. Schmidt, A. E. Schutte, D. C. Schwebel, F. Schwendicke, A. M. Senbeta, S. Senthilkumaran, S. G. Sepanlou, B. Serdar, M. L. Serre, J. Shadid, O. Shafaat, S. Shahabi, A. A. Shaheen, M. A. Shaikh, A. S. Shalash, M. Shams-Beyranvand, M. Shamsizadeh, K. Sharafi, A. Sheikh, A. Sheikhtaheri, K. Shibuya, K. D. Shield, M. Shigematsu, J. Il Shin, M. J. Shin, R. Shiri, R. Shirkoohi, K. Shuval, S. Siabani, R. Sierpinski, I. D. Sigfusdottir, R. Sigurvinsdottir, J. P. Silva, K. E. Simpson, J. A. Singh, P. Singh, E. Skiadaresi, S. T. Skou, V. Y. Skryabin, E. U. R. Smith, A. Soheili, S. Soltani, M. Soofi, R. J. D. Sorensen, J. B. Soriano, M. B. Sorrie, S. Soshnikov, I. N. Soyiri, C. N. Spencer, A. Spotin, C. T. Sreeramareddy, V. Srinivasan, J. D. Stanaway, C. Stein, D. J. Stein, C. Steiner, L. Stockfelt, M. A. Stokes, K. Straif, J. L. Stubbs, M. B. Sufiyan, H. A. R. Suleria, R. Suliankatchi Abdulkader, G. Sulo, I. Sultan, R. Tabarés-Seisdedos, K. M. Tabb, T. Tabuchi, A. Taherkhani, M. Tajdini, K. Takahashi, J. S. Takala, A. T. Tamiru, N. Taveira, A. Tehrani-Banihashemi, M. H. Temsah, G. A. Tesema, Z. T. Tessema, G. D. Thurston, M. V. Titova, H. R. Tohidinik, M. Tonelli, R. Topor-Madry, F. Topouzis, A. E. Torre, M. Touvier, M. R. Tovani-Palone, B. X. Tran, R. Travillian, A. Tsatsakis, L. T. Tudor Car, S. Tyrovolas, R. Uddin, C. D. Umeokonkwo, B. Unnikrishnan, E. Upadhyay, M. Vacante, P. R. Valdez, A. van Donkelaar, T. J. Vasankari, Y. Vasseghian, Y. Veisani, N. Venketasubramanian, F. S. Violante, V. Vlassov, S. E. Vollset, T. Vos, R. Vukovic, Y. Waheed, M. T. Wallin, Y. Wang, Y. P. Wang, A. Watson, J. Wei, M. Y. W. Wei, R. G. Weintraub, J. Weiss, A. Werdecker, J. J. West, R. Westerman, J. L. Whisnant, H. A. Whiteford, K. E. Wiens, C. D. A. Wolfe, S. S. Wozniak, A. M. Wu, J. Wu, S. Wulf Hanson, G. Xu, R. Xu, S. Yadgir, S. H. Yahyazadeh Jabbari, K. Yamagishi, M. Yaminfirooz, Y. Yano, S. Yaya, V. Yazdi-Feyzabadi, T. Y. Yeheyis, C. S. Yilgwan, M. T. Yilma, P. Yip, N. Yonemoto, M. Z. Younis, T. P. Younker, B. Yousefi, Z. Yousefi, T. Yousefinezhadi, A. Y. Yousuf, C. Yu, H. Yusefzadeh, T. Z. Moghadam, M. Zamani, M. Zamanian, H. Zandian, M. S. Zastrozhin, Y. Zhang, Z. J. Zhang, J. T. Zhao, X. J. G. Zhao, Y. Zhao, P. Zheng, M. Zhou, K. Davletov, A. Ziapour, S. Mondello, S. S. Lim, C. J. L. Murray, T. Wiangkham and S. Amini, Lancet, 2020, 396, 1204–1222 CrossRef.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS.
- J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report And Recommendations, 2016 Search PubMed.
- H. Nikaido, Annu. Rev. Biochem., 2009, 78, 119–146 CrossRef CAS PubMed.
- K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed., 2013, 52, 10706–10733 CrossRef PubMed.
- C.-E. Luyt, N. Bréchot, J.-L. Trouillet and J. Chastre, Crit. Care, 2014, 18, 480 CrossRef PubMed.
- C. Casals-Pascual, A. Vergara and J. Vila, Hum. Microbiome J., 2018, 9, 11–15 CrossRef.
- D. G. J. Larsson and C. F. Flach, Nat. Rev. Microbiol., 2021, 20(5), 257–269 CrossRef.
- T. Buschhardt, T. Günther, T. Skjerdal, M. Torpdahl, J. Gethmann, M. E. Filippitzi, C. Maassen, S. Jore, J. Ellis-Iversen and M. Filter, One Healt, 2021, 13, 100263 CrossRef PubMed.
- G. Worrall, J. Hutchinson, G. Sherman and J. Griffiths, Can. Fam. Physician, 2007, 53, 666–671 Search PubMed.
- J.-P. Humair, S. A. Revaz, P. Bovier and H. Stalder, Arch. Intern. Med., 2006, 166, 640 CrossRef PubMed.
- W. J. McIsaac, J. D. Kellner, P. Aufricht, A. Vanjaka and D. E. Low, JAMA, 2004, 291, 1587 CrossRef CAS PubMed.
- J. M. Andrews, J. Antimicrob. Chemother., 2001, 48, 5–16 CrossRef CAS PubMed.
- G. Kahlmeter, D. F. J. Brown, F. W. Goldstein, A. P. MacGowan, J. W. Mouton, A. Österlund, A. Rodloff, M. Steinbakk, P. Urbaskova and A. Vatopoulos, J. Antimicrob. Chemother., 2003, 52, 145–148 CrossRef CAS PubMed.
- J. Turnidge and D. L. Paterson, Clin. Microbiol. Rev., 2007, 20, 391–408 CrossRef CAS PubMed.
- D. L. Paterson, W.-C. Ko, A. Von Gottberg, J. M. Casellas, L. Mulazimoglu, K. P. Klugman, R. A. Bonomo, L. B. Rice, J. G. McCormack and V. L. Yu, J. Clin. Microbiol., 2001, 39, 2206–2212 CrossRef CAS PubMed.
- J. Cama, R. Leszczynski, P. K. Tang, A. Khalid, V. Lok, C. G. Dowson and A. Ebata, ACS Infect. Dis., 2021, 7, 2029–2042 CrossRef CAS PubMed.
- I. A. Dutescu and S. A. Hillie, Infect. Drug Resist., 2021, 14, 415–434 CrossRef.
- A. Mullard, Nat. Rev. Drug Discovery, 2022, 21, 406 Search PubMed.
- M. J. Renwick, D. M. Brogan and E. Mossialos, J. Antibiot., 2016, 69, 73–88 CrossRef CAS.
- A. van Belkum, C. A. D. Burnham, J. W. A. Rossen, F. Mallard, O. Rochas and W. M. Dunne, Nat. Rev. Microbiol., 2020, 18, 299–311 CrossRef CAS PubMed.
- K. Zhang, S. Qin, S. Wu, Y. Liang and J. Li, Chem. Sci., 2020, 11, 6352–6361 RSC.
- N. Qin, P. Zhao, E. A. Ho, G. Xin and C. L. Ren, ACS Sens., 2021, 6, 3–21 CrossRef CAS PubMed.
- A. Vasala, V. P. Hytönen and O. H. Laitinen, Front. Cell. Infect. Microbiol., 2020, 10, 308 CrossRef CAS PubMed.
- Z. A. Khan, M. F. Siddiqui and S. Park, Diagnostics, 2019, 9, 49 CrossRef CAS PubMed.
- A. van Belkum, T. T. Bachmann, G. Lüdke, J. G. Lisby, G. Kahlmeter, A. Mohess, K. Becker, J. P. Hays, N. Woodford, K. Mitsakakis, J. Moran-Gilad, J. Vila, H. Peter, J. H. Rex and W. M. Dunne, Nat. Rev. Microbiol., 2018, 17(1), 51–62 CrossRef PubMed.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- D. J. Guckenberger, T. E. de Groot, A. M. D. Wan, D. J. Beebe and E. W. K. Young, Lab Chip, 2015, 15, 2364–2378 RSC.
- J. M. K. Ng, I. Gitlin, A. D. Stroock and G. M. Whitesides, Electrophoresis, 2002, 23, 3461–3473 CrossRef CAS PubMed.
- K. M. Raj and S. Chakraborty, J. Appl. Polym. Sci., 2020, 137, 48958 CrossRef.
- B. Gale, A. Jafek, C. Lambert, B. Goenner, H. Moghimifam, U. Nze and S. Kamarapu, Inventions, 2018, 3, 60 CrossRef.
- P. Zhu and L. Wang, Lab Chip, 2017, 17, 34–75 RSC.
- S. L. Anna, Annu. Rev. Fluid Mech., 2016, 48, 285–309 CrossRef.
- S. K. W. Dertinger, D. T. Chiu, N. L. Jeon and G. M. Whitesides, Anal. Chem., 2001, 73, 1240–1246 CrossRef CAS.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- D. S. Liao, J. Raveendran, S. Golchi and A. Docoslis, Sens. Bio-Sens. Res., 2015, 6, 59–66 CrossRef.
- Q. Zhu, L. Qiu, B. Yu, Y. Xu, Y. Gao, T. Pan, Q. Tian, Q. Song, W. Jin, Q. Jin and Y. Mu, Lab Chip, 2014, 14, 1176–1185 RSC.
- R. H. A. Q. Zhang, G. Lambert, D. Liao, H. Kim, K. Robin, C.-k. Tung, N. Pourmand and R. H. Austin, Science, 2011, 1764, 1764–1767 CrossRef PubMed.
- J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef CAS.
- P. Garstecki, A. M. Gañán-Calvo and G. M. Whitesides, Bull. Pol. Acad. Sci.: Tech. Sci., 2005, 53, 361–372 CrossRef CAS.
- L. Shui, A. Van Den Berg and J. C. T. Eijkel, Microfluid. Nanofluid., 2011, 11, 87–92 CrossRef.
- S. L. Anna and H. C. Mayer, Phys. Fluids, 2006, 18, 1–13 Search PubMed.
- H.-H. Jeong, V. R. Yelleswarapu, S. Yadavali, D. Issadore and D. Lee, Lab Chip, 2015, 15, 4387–4392 RSC.
- V. R. Yelleswarapu, H.-H. Jeong, S. Yadavali and D. Issadore, Lab Chip, 2017, 17, 1083–1094 RSC.
- E. Amstad, M. Chemama, M. Eggersdorfer, L. R. Arriaga, M. P. Brenner and D. A. Weitz, Lab Chip, 2016, 16, 4163–4172 RSC.
- J. Kehe, A. Kulesa, A. Ortiz, C. M. Ackerman, S. G. Thakku, D. Sellers, S. Kuehn, J. Gore, J. Friedman and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12804–12809 CrossRef CAS PubMed.
- A. Kulesa, J. Kehe, J. E. Hurtado, P. Tawde and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6685–6690 CrossRef PubMed.
- M. N. Hatori, S. C. Kim and A. R. Abate, Anal. Chem., 2018, 90, 9813–9820 CrossRef CAS PubMed.
- Y.-T. Kao, T. S. Kaminski, W. Postek, J. Guzowski, K. Makuch, A. Ruszczak, F. von Stetten, R. Zengerle and P. Garstecki, Lab Chip, 2020, 20, 54–63 RSC.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angile, C. H. J. Schmitz, S. Koster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano, D. A. Weitz, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- M. S. Chowdhury, W. Zheng, S. Kumari, J. Heyman, X. Zhang, P. Dey, D. A. Weitz and R. Haag, Nat. Commun., 2019, 10, 4546 CrossRef PubMed.
- D. Liu, G. Liang, X. Lei, B. Chen, W. Wang and X. Zhou, Anal. Chim. Acta, 2012, 718, 58–63 CrossRef CAS PubMed.
- A. S. Basu, SLAS Technol., 2017, 22, 369–386 CrossRef PubMed.
- L. Cao, X. Cui, J. Hu, Z. Li, J. R. Choi, Q. Yang, M. Lin, L. Ying Hui and F. Xu, Biosens. Bioelectron., 2017, 90, 459–474 CrossRef CAS PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed.
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed.
- F. Lan, B. Demaree, N. Ahmed and A. R. Abate, Nat. Biotechnol., 2017, 35, 640–646 CrossRef CAS PubMed.
- O. Strohmeier, M. Keller, F. Schwemmer, S. Zehnle, D. Mark, F. von Stetten, R. Zengerle and N. Paust, Chem. Soc. Rev., 2015, 44, 6187–6229 RSC.
- M. Tang, G. Wang, S.-K. Kong and H.-P. Ho, Micromachines, 2016, 7, 26 CrossRef PubMed.
- C. H. Chen, Y. Lu, M. L. Y. Sin, K. E. Mach, D. D. Zhang, V. Gau, J. C. Liao and P. K. Wong, Anal. Chem., 2010, 82, 1012–1019 CrossRef CAS PubMed.
- N. J. Cira, J. Y. Ho, M. E. Dueck and D. B. Weibel, Lab Chip, 2012, 12, 1052–1059 RSC.
- J. Shemesh, T. Ben Arye, J. Avesar, J. H. Kang, A. Fine, M. Super, A. Meller, D. E. Ingber and S. Levenberg, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11293–11298 CrossRef CAS PubMed.
- J. Avesar, D. Rosenfeld, M. Truman-Rosentsvit, T. Ben-Arye, Y. Geffen, M. Bercovici and S. Levenberg, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5787–E5795 CrossRef CAS.
- C.-W. Tsao, W. Li, S. H. Tan and N.-T. Nguyen, Micromachines, 2016, 7, 225 CrossRef PubMed.
- M. Azizi, M. Zaferani, B. Dogan, S. Zhang, K. W. Simpson and A. Abbaspourrad, Anal. Chem., 2018, 90, 14137–14144 CrossRef CAS PubMed.
- O. Scheler, T. S. Kaminski, A. Ruszczak and P. Garstecki, ACS Appl. Mater. Interfaces, 2016, 8, 11318–11325 CrossRef CAS PubMed.
- S. J. Lin, P. H. Chao, H. W. Cheng, J. K. Wang, Y. L. Wang, Y. Y. Han and N. T. Huang, Lab Chip, 2022, 22, 1805–1814 RSC.
- M. Osaid, Y. S. Chen, C. H. Wang, A. Sinha, W. Bin Lee, P. Gopinathan, H. Bin Wu and G. Bin Lee, Lab Chip, 2021, 21, 2223–2231 RSC.
- A. S. Opalski, A. Ruszczak, Y. Promovych, M. Horka, L. Derzsi and P. Garstecki, Micromachines, 2020, 11, 142 CrossRef.
- W. Zeng, P. Chen, S. Li, Q. Sha, P. Li, X. Zeng, X. Feng, W. Du and B. F. Liu, Biosens. Bioelectron., 2022, 205, 114100 CrossRef CAS PubMed.
- B. Xu, Y. Du, J. Lin, M. Qi, B. Shu, X. Wen, G. Liang, B. Chen and D. Liu, Anal. Chem., 2016, 88, 11593–11600 CrossRef CAS PubMed.
- R. Mohan, C. Sanpitakseree, A. V. Desai, S. E. Sevgen, C. M. Schroeder and P. J. A. Kenis, RSC Adv., 2015, 5, 35211–35223 RSC.
- R. Mohan, A. Mukherjee, S. E. Sevgen, C. Sanpitakseree, J. Lee, C. M. Schroeder and P. J. A. Kenis, Biosens. Bioelectron., 2013, 49, 118–125 CrossRef CAS PubMed.
- Z. Liu, H. Sun and K. Ren, ChemPlusChem, 2017, 82, 792–801 CrossRef CAS PubMed.
- T. Lim, E.-G. Kim, J. Choi and S. Kwon, Lab Chip, 2020, 20, 4552–4560 RSC.
- M. Tang, X. Huang, Q. Chu, X. Ning, Y. Wang, S.-K. Kong, X. Zhang, G. Wang and H.-P. Ho, Lab Chip, 2018, 18, 1452–1460 RSC.
- J. Choi, Y. G. Jung, J. Kim, S. Kim, Y. Jung, H. Na and S. Kwon, Lab Chip, 2013, 13, 280–287 RSC.
- J. Choi, J. Yoo, M. Lee, E.-G. Kim, J. S. Lee, S. Lee, S. Joo, S. H. Song, E.-C. Kim, J. C. Lee, H. C. Kim, Y.-G. Jung and S. Kwon, Sci. Transl. Med., 2014, 6, 1–13 Search PubMed.
- J. Choi, H. Y. Jeong, G. Y. Lee, S. Han, S. Han, B. Jin, T. Lim, S. Kim, D. Y. Kim, H. C. Kim, E. C. Kim, S. H. Song, T. S. Kim and S. Kwon, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- K. P. Smith and J. E. Kirby, Antimicrob. Agents Chemother., 2018, 62, 1–9 Search PubMed.
- Y.-G. Jung, H. Kim, S. Lee, S. Kim, E. Jo, E.-G. Kim, J. Choi, H. J. Kim, J. Yoo, H.-J. Lee, H. Kim, H. Jung, S. Ryoo and S. Kwon, Sci. Rep., 2018, 8, 8651 CrossRef PubMed.
- T. Artemova, Y. Gerardin, C. Dudley, N. M. Vega and J. Gore, Mol. Syst. Biol., 2015, 11, 822 CrossRef PubMed.
- C.-C. Chung, I.-F. Cheng, W.-H. Yang and H.-C. Chang, Biomicrofluidics, 2011, 5, 021102 CrossRef PubMed.
- C.-C. Chung, I.-F. Cheng, H.-M. Chen, H.-C. Kan, W.-H. Yang and H.-C. Chang, Anal. Chem., 2012, 84, 3347–3354 CrossRef CAS PubMed.
- I.-H. Su, W.-C. Ko, C.-H. Shih, F.-H. Yeh, Y.-N. Sun, J.-C. Chen, P.-L. Chen and H.-C. Chang, Anal. Chem., 2017, 89, 4635–4641 CrossRef CAS PubMed.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC.
- Q. Yi, D. Cai, M. Xiao, M. Nie, Q. Cui, J. Cheng, C. Li, J. Feng, G. Urban, Y. C. Xu, Y. Lan and W. Du, Biosens. Bioelectron., 2019, 135, 200–207 CrossRef CAS PubMed.
- J. Yoon, Y. Kim, J.-W. Suh, Y.-Y. Jin, Y.-G. Jung and W. Park, ACS Appl. Bio Mater., 2020, 3, 4798–4808 CrossRef CAS PubMed.
- V. Kara, C. Duan, K. Gupta, S. Kurosawa, D. J. Stearns-Kurosawa and K. L. Ekinci, Lab Chip, 2018, 18, 743–753 RSC.
- M. Kalashnikov, J. C. Lee, J. Campbell, A. Sharon and A. F. Sauer-Budge, Lab Chip, 2012, 12, 4523–4532 RSC.
- H. Shi, Z. Hou, Y. Zhao, K. Nie, B. Dong, L. Chao, S. Shang, M. Long and Z. Liu, Chem. Eng. J., 2019, 359, 1327–1338 CrossRef CAS.
- S. Kim, S. Lee, J.-K. Kim, H. J. Chung and J. S. Jeon, Biomicrofluidics, 2019, 13, 014108 CrossRef PubMed.
- N. C. Parsley, A. L. Smythers and L. M. Hicks, Front. Cell. Infect. Microbiol., 2020, 10, 503 Search PubMed.
- M. Kalashnikov, M. Mueller, C. McBeth, J. C. Lee, J. Campbell, A. Sharon and A. F. Sauer-Budge, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- I. Peitz and R. Van Leeuwen, Lab Chip, 2010, 10, 2944–2951 RSC.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- L. Robert, J. Ollion, J. Robert, X. Song, I. Matic and M. Elez, Science, 2018, 359, 1283–1286 CrossRef CAS PubMed.
- R. A. Bamford, A. Smith, J. Metz, G. Glover, R. W. Titball and S. Pagliara, BMC Biol., 2017, 15, 121 CrossRef PubMed.
- T. Bergmiller, A. M. C. Andersson, K. Tomasek, E. Balleza, D. J. Kiviet, R. Hauschild, G. Tkačik and C. C. Guet, Science, 2017, 356, 311–315 CrossRef CAS PubMed.
- N. Q. Balaban, Science, 2004, 305, 1622–1625 CrossRef CAS PubMed.
- O. Goode, A. Smith, A. Zarkan, J. Cama, B. M. Invergo, D. Belgami, S. Caño-Muñiz, J. Metz, P. O’neill, A. Jeffries, I. H. Norville, J. David, D. Summers and S. Pagliara, MBio, 2021, 12, 1–18 CrossRef PubMed.
- U. Łapińska, M. Voliotis, K. K. Lee, A. Campey, M. R. L. Stone, B. Tuck, W. Phetsang, B. Zhang, K. Tsaneva-Atanasova, M. A. Blaskovich and S. Pagliara, eLife, 2022, 11, 1–33 CrossRef PubMed.
- M. Rhia, L. Stone, U. Łapin'ska, Ł. Łapin'ska, S. Pagliara, M. Masi, J. T. Blanchfield, M. A. Cooper and M. A. T. Blaskovich, RSC Chem. Biol., 2020, 1, 395–404 RSC.
- I. E. Kepiro, I. Marzuoli, K. Hammond, X. Ba, H. Lewis, M. Shaw, S. B. Gunnoo, E. De Santis, U. Łapińska, S. Pagliara, M. A. Holmes, C. D. Lorenz, B. W. Hoogenboom, F. Fraternali and M. G. Ryadnov, ACS Nano, 2020, 14, 1609–1622 CrossRef CAS PubMed.
- E. L. Attrill, R. Claydon, U. Łapińska, M. Recker, S. Meaden, A. T. Brown, E. R. Westra, S. V. Harding and S. Pagliara, PLoS Biol., 2021, 19, e3001406 CrossRef CAS PubMed.
- Y. Lu, J. Gao, D. D. Zhang, V. Gau, J. C. Liao and P. K. Wong, Anal. Chem., 2013, 85, 3971–3976 CrossRef CAS PubMed.
- Ö. Baltekin, A. Boucharin, E. Tano, D. I. Andersson and J. Elf, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9170–9175 CrossRef.
- H. Li, P. Torab, K. E. Mach, C. Surrette, M. R. England, D. W. Craft, N. J. Thomas, J. C. Liao, C. Puleo and P. K. Wong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10270–10279 CrossRef CAS PubMed.
- B. Li, Y. Qiu, A. Glidle, D. McIlvenna, Q. Luo, J. Cooper, H. C. Shi and H. Yin, Anal. Chem., 2014, 86, 3131–3137 CrossRef CAS PubMed.
- Y. Yang, K. Gupta and K. L. Ekinci, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10639–10644 CrossRef PubMed.
- A. A. Sklavounos, C. R. Nemr, S. O. Kelley and A. R. Wheeler, Lab Chip, 2021, 21, 4208–4222 RSC.
- I. Sinn, P. Kinnunen, T. Albertson, B. H. McNaughton, D. W. Newton, M. A. Burns and R. Kopelman, Lab Chip, 2011, 11, 2604–2611 RSC.
- G. Amselem, C. Guermonprez, B. Drogue, S. Michelin and C. N. Baroud, Lab Chip, 2016, 16, 4200–4211 RSC.
- A. Saint-Sardos, S. Sart, K. Lippera, E. Brient-Litzler, S. Michelin, G. Amselem and C. N. Baroud, Small, 2020, 16, 2002303 CrossRef CAS.
- P. Sabhachandani, S. Sarkar, P. C. Zucchi, B. A. Whitfield, J. E. Kirby, E. B. Hirsch and T. Konry, Microchim. Acta, 2017, 184, 4619–4628 CrossRef CAS.
- W. Kang, S. Sarkar, Z. S. Lin, S. McKenney and T. Konry, Anal. Chem., 2019, 91, 6242–6249 CrossRef CAS PubMed.
- Z. Liu, C. Duan, S. Jiang, C. Zhu, Y. Ma and T. Fu, J. Ind. Eng. Chem., 2020, 92, 18–40 CrossRef CAS.
- L. Derzsi, T. S. Kaminski and P. Garstecki, Lab Chip, 2016, 16, 893–901 RSC.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- S. Jakiela, T. S. Kaminski, O. Cybulski, D. B. Weibel and P. Garstecki, Angew. Chem., Int. Ed., 2013, 52, 8908–8911 CrossRef CAS PubMed.
- L. Baraban, F. Bertholle, M. L. M. Salverda, N. Bremond, P. Panizza, J. Baudry, J. A. G. M. De Visser and J. Bibette, Lab Chip, 2011, 11, 4057–4062 RSC.
- S. Jakiela, T. S. Kaminski, O. Cybulski, D. B. Weibel and P. Garstecki, Angew. Chem., 2013, 125, 9076–9079 CrossRef.
- J. Q. Boedicker, L. Li, T. R. Kline and R. F. Ismagilov, Lab Chip, 2008, 8, 1265–1272 RSC.
- W. Postek, P. Gargulinski, O. Scheler, T. S. Kaminski and P. Garstecki, Lab Chip, 2018, 18, 3668–3677 RSC.
- H. Li, P. Zhang, K. Hsieh and T.-H. Wang, Lab Chip, 2022, 22, 621–631 RSC.
- X. Liu, R. E. Painter, K. Enesa, D. Holmes, G. Whyte, C. G. Garlisi, F. J. Monsma, M. Rehak, F. F. Craig and C. A. Smith, Lab Chip, 2016, 16, 1636–1643 RSC.
- F. Lyu, M. Pan, S. Patil, J.-H. Wang, A. C. Matin, J. R. Andrews and S. K. Y. Tang, Sens. Actuators, B, 2018, 270, 396–404 CrossRef CAS.
- O. Scheler, K. Makuch, P. R. Debski, M. Horka, A. Ruszczak, N. Pacocha, K. Sozański, O.-P. Smolander, W. Postek and P. Garstecki, Sci. Rep., 2020, 10, 3282 CrossRef CAS.
- A. M. Kaushik, K. Hsieh, L. Chen, D. J. Shin, J. C. Liao and T. H. Wang, Biosens. Bioelectron., 2017, 97, 260–266 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS.
- A. Funfak, R. Hartung, J. Cao, K. Martin, K.-H. Wiesmüller, O. S. Wolfbeis and J. M. Köhler, Sens. Actuators, B, 2009, 142, 66–72 CrossRef CAS.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- L. Jiang, L. Boitard, P. Broyer, A. C. Chareire, P. Bourne-Branchu, P. Mahé, M. Tournoud, C. Franceschi, G. Zambardi, J. Baudry and J. Bibette, Eur. J. Clin. Microbiol. Infect. Dis., 2016, 35, 415–422 CrossRef CAS PubMed.
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Determination of minimum inhibitory concentrations (MICs) ofantibacterial agents by broth dilution, Clin. Microbiol. Infect., 2003, 9, ix–xv CrossRef.
- Clinical and Laboratory Standards Institute, CLSI Doc. M100-S16CLSI, Wayne, PA.
- K. Lewis, Annu. Rev. Microbiol., 2010, 64, 357–372 CrossRef CAS PubMed.
- W. Hong, C. W. Karanja, N. S. Abutaleb, W. Younis, X. Zhang, M. N. Seleem and J.-X. Cheng, Anal. Chem., 2018, 90, 3737–3743 CrossRef CAS PubMed.
- Y. Liu, T. Lehnert and M. A. M. Gijs, Lab Chip, 2020, 20, 3144–3157 RSC.
- P. Jusková, S. Schmitt, A. Kling, D. G. Rackus, M. Held, A. Egli and P. S. Dittrich, ACS Sens., 2021, 6, 2202–2210 CrossRef PubMed.
- F. Courtois, L. F. Olguin, G. Whyte, A. B. Theberge, W. T. S. Huck, F. Hollfelder and C. Abell, Anal. Chem., 2009, 81, 3008–3016 CrossRef CAS PubMed.
- Y. Chen, A. Wijaya Gani and S. K. Y. Tang, Lab Chip, 2012, 12, 5093–5103 RSC.
- T. S. Kaminski, O. Scheler and P. Garstecki, Lab Chip, 2016, 16, 2168–2187 RSC.
- P. Gruner, B. Riechers, B. Semin, J. Lim, A. Johnston, K. Short and J.-C. Baret, Nat. Commun., 2016, 7, 10392 CrossRef CAS.
- N. Pacocha, J. Bogusławski, M. Horka, K. Makuch, K. Liżewski, M. Wojtkowski and P. Garstecki, Anal. Chem., 2021, 93, 843–850 CrossRef CAS PubMed.
- I. P. Alves and N. M. Reis, Biosens. Bioelectron., 2019, 145, 111624 CrossRef CAS PubMed.
- X. Cui, L. Ren, Y. Shan, X. Wang, Z. Yang, C. Li, J. Xu and B. Ma, Analyst, 2018, 143, 3309–3316 RSC.
- E. Um, E. Rha, S.-L. Choi, S.-G. Lee and J.-K. Park, Lab Chip, 2012, 12, 1594 RSC.
- P. Sharma, P. Pratim Pandey, S. Jain, X. Liu, H. Li, M. Zhan, P. Greulich, J. Doležal, D. Taylor, N. Verdon, P. Lomax, R. J. Allen and S. Titmuss, Phys. Biol., 2022, 19, 026003 CrossRef PubMed.
- S. Hengoju, S. Wohlfeil, A. S. Munser, S. Boehme, E. Beckert, O. Shvydkiv, M. Tovar, M. Roth and M. A. Rosenbaum, Biomicrofluidics, 2020, 14, 024109 CrossRef CAS PubMed.
- M. Horka, S. Sun, A. Ruszczak, P. Garstecki and T. Mayr, Anal. Chem., 2016, 88, 12006–12012 CrossRef CAS PubMed.
- L. Boitard, D. Cottinet, C. Kleinschmitt, N. Bremond, J. Baudry, G. Yvert and J. Bibette, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7181–7186 CrossRef CAS PubMed.
- E. Zang, S. Brandes, M. Tovar, K. Martin, F. Mech, P. Horbert, T. Henkel, M. T. Figge and M. Roth, Lab Chip, 2013, 13, 3707–3713 RSC.
- A. C. Hatch, J. S. Fisher, A. R. Tovar, A. T. Hsieh, R. Lin, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 3838 RSC.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- T. J. Abram, H. Cherukury, C. Y. Ou, T. Vu, M. Toledano, Y. Li, J. T. Grunwald, M. N. Toosky, D. F. Tifrea, A. Slepenkin, J. Chong, L. Kong, D. V. Del Pozo, K. T. La, L. Labanieh, J. Zimak, B. Shen, S. S. Huang, E. Gratton, E. M. Peterson and W. Zhao, Lab Chip, 2020, 20, 477–489 RSC.
- J. D. Bard and F. Lee, Clin. Microbiol. Newsl., 2018, 40, 87–95 CrossRef PubMed.
- F. C. Tenover, Clin. Lab. Med., 1989, 9, 341–347 CrossRef CAS PubMed.
- P. Athamanolap, K. Hsieh, C. M. O'Keefe, Y. Zhang, S. Yang and T.-H. Wang, Anal. Chem., 2019, 91, 12784–12792 CrossRef CAS PubMed.
- H. W. Hou, R. P. Bhattacharyya, D. T. Hung and J. Han, Lab Chip, 2015, 15, 2297–2307 RSC.
- N. G. Schoepp, E. M. Khorosheva, T. S. Schlappi, M. S. Curtis, R. M. Humphries, J. A. Hindler and R. F. Ismagilov, Angew. Chem., Int. Ed., 2016, 55, 9557–9561 CrossRef CAS PubMed.
- N. G. Schoepp, T. S. Schlappi, M. S. Curtis, S. S. Butkovich, S. Miller, R. M. Humphries and R. F. Ismagilov, Sci. Transl. Med., 2017, 9, eaal3693 CrossRef PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, 63e CrossRef PubMed.
- T. Khazaei, J. T. Barlow, N. G. Schoepp and R. F. Ismagilov, Sci. Rep., 2018, 8, 11606 CrossRef PubMed.
- A. M. Kaushik, K. Hsieh, K. E. Mach, S. Lewis, C. M. Puleo, K. C. Carroll, J. C. Liao and T. Wang, Adv. Sci., 2021, 2003419 CrossRef CAS PubMed.
- G. Longo, L. Alonso-Sarduy, L. Marques Rio, A. Bizzini, A. Trampuz, J. Notz, G. Dietler and S. Kasas, Nat. Nanotechnol., 2013, 8(7), 522–526 CrossRef CAS PubMed.
- N. Cermak, S. Olcum, F. F. Delgado, S. C. Wasserman, K. R. Payer, M. A. Murakami, S. M. Knudsen, R. J. Kimmerling, M. M. Stevens, Y. Kikuchi, A. Sandikci, M. Ogawa, V. Agache, F. Baléras, D. M. Weinstock and S. R. Manalis, Nat. Biotechnol., 2016, 34, 1052–1059 CrossRef CAS PubMed.
- H. Etayash, M. F. Khan, K. Kaur and T. Thundat, Nat. Commun., 2016, 7, 12947 CrossRef CAS PubMed.
- I. Bennett, A. L. B. Pyne and R. A. McKendry, ACS Sens., 2020, 5, 3133–3139 CrossRef CAS PubMed.
- M. Godin, F. F. Delgado, S. Son, W. H. Grover, A. K. Bryan, A. Tzur, P. Jorgensen, K. Payer, A. D. Grossman, M. W. Kirschner and S. R. Manalis, Nat. Methods, 2010, 7(5), 387–390 CrossRef CAS PubMed.
- P. Stupar, O. Opota, G. Longo, G. Prod'hom, G. Dietler, G. Greub and S. Kasas, Clin. Microbiol. Infect., 2017, 23, 400–405 CrossRef CAS PubMed.
- F. Pujol-Vila, R. Villa and M. Alvarez, Front. Mech. Eng., 2020, 6, 44 CrossRef.
- P. Cady, S. W. Dufour, J. Shaw and S. J. Kraeger, J. Clin. Microbiol., 1978, 7, 265–272 CrossRef CAS PubMed.
- M. S. Mannoor, S. Zhang, A. J. Link and M. C. McAlpine, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19207–19212 CrossRef CAS PubMed.
- D. C. Spencer, T. F. Paton, K. T. Mulroney, T. J. J. Inglis, J. M. Sutton and H. Morgan, Nat. Commun., 2020, 11, 5328 CrossRef CAS PubMed.
- C. Honrado, P. Bisegna, N. S. Swami and F. Caselli, Lab Chip, 2021, 21, 22–54 RSC.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- C. M. Ackerman, C. Myhrvold, S. G. Thakku, C. A. Freije, H. C. Metsky, D. K. Yang, S. H. Ye, C. K. Boehm, T.-S. F. Kosoko-Thoroddsen, J. Kehe, T. G. Nguyen, A. Carter, A. Kulesa, J. R. Barnes, V. G. Dugan, D. T. Hung, P. C. Blainey and P. C. Sabeti, Nature, 2020, 582, 277–282 CrossRef CAS PubMed.
- S. G. Thakku, C. M. Ackerman, C. Myhrvold, R. P. Bhattacharyya, J. Livny, P. Ma, G. I. Gomez, P. C. Sabeti, P. C. Blainey and D. T. Hung, PNAS Nexus, 2022, 1, 1–10 CrossRef PubMed.
- X. Wang, Z. Liu and Y. Pang, RSC Adv., 2017, 7, 29966–29984 RSC.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- O. Miller, A. Harrak, T. Mangeat, J.-C. Baret, L. Frenz, B. Debs, E. Mayot, M. Samuels, E. Rooney, P. Dieu, M. Galvan and A. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS PubMed.
- X. Niu, F. Gielen, J. B. Edel and A. J. deMello, Nat. Chem., 2011, 3, 437–442 CrossRef CAS PubMed.
- W. Postek, T. S. Kaminski and P. Garstecki, Analyst, 2017, 142, 2901–2911 RSC.
- M. Sun, S. S. Bithi and S. A. Vanapalli, Lab Chip, 2012, 11, 3949–3952 RSC.
- M. Sun and S. A. Vanapalli, Anal. Chem., 2013, 85, 2044–2048 CrossRef CAS PubMed.
- K. I. Udekwu, N. Parrish, P. Ankomah, F. Baquero and B. R. Levin, J. Antimicrob. Chemother., 2009, 63, 745–757 CrossRef CAS PubMed.
- C. Tan, R. P. Smith, J. K. Srimani, K. A. Riccione, S. Prasada, M. Kuehn and L. You, Mol. Syst. Biol., 2012, 8, 617 CrossRef PubMed.
- W. A. Craig, S. M. Bhavnani and P. G. Ambrose, Diagn. Microbiol. Infect. Dis., 2004, 50, 229–230 CrossRef PubMed.
- Clinical and Laboratory Standards Institute, CLSI Doc. M07-A10, Clin. Lab. Stand. Institute, Wayne, PA.
- J. Q. Boedicker, M. E. Vincent and R. F. Ismagilov, Angew. Chem., Int. Ed., 2009, 48, 5908–5911 CrossRef CAS.
- D. I. Andersson, H. Nicoloff and K. Hjort, Nat. Rev. Microbiol., 2019, 17(8), 479–496 CrossRef CAS PubMed.
- S. S. Y. Wong, P. L. Ho, P. C. Y. Woo and K. Y. Yuen, Clin. Infect. Dis., 1999, 29, 760–767 CrossRef CAS PubMed.
- W. Yau, R. J. Owen, A. Poudyal, J. M. Bell, J. D. Turnidge, H. H. Yu, R. L. Nation and J. Li, J. Infect., 2009, 58, 138–144 CrossRef PubMed.
- H. Nicoloff, K. Hjort, B. R. Levin and D. I. Andersson, Nat. Microbiol., 2019, 4, 504–514 CrossRef CAS PubMed.
- D. M. Hermes, C. Pormann Pitt, L. Lutz, A. B. Teixeira, V. B. Ribeiro, B. Netto, A. F. Martins, A. P. Zavascki and A. L. Barth, J. Med. Microbiol., 2013, 62, 1184–1189 CrossRef.
- J. D. Sun, S. F. Huang, S. S. Yang, S. L. Pu, C. M. Zhang and L. P. Zhang, Clin. Microbiol. Infect., 2015, 21(5), 469 Search PubMed.
- A. E. B. da Silva, A. F. Martins, C. S. Nodari, C. M. Magagnin and A. L. Barth, Eur. J. Clin. Microbiol. Infect. Dis., 2017, 37(1), 185–186 CrossRef PubMed.
- T. Pelaez, E. Cercenado, L. Alcala, M. Marin, A. Martin-Lopez, J. Martinez-Alarcon, P. Catalan, M. Sanchez-Somolinos and E. Bouza, J. Clin. Microbiol., 2008, 46, 3028–3032 CrossRef CAS PubMed.
- O. M. El-Halfawy and M. A. Valvano, Clin. Microbiol. Rev., 2015, 28, 191–207 CrossRef CAS PubMed.
- L. Sun, S. Talarico, L. Yao, L. He, S. Self, Y. You, H. Zhang, Y. Zhang, Y. Guo, G. Liu, N. R. Salama and J. Zhang, J. Clin. Microbiol., 2018, 6(9) DOI:10.1128/JCM.00019-18.
- A. Raj and A. van Oudenaarden, Cell, 2008, 135, 216–226 CrossRef CAS PubMed.
- A. Brauner, O. Fridman, O. Gefen and N. Q. Balaban, Nat. Rev. Microbiol., 2016, 14, 320–330 CrossRef CAS PubMed.
- N. Balaban, Curr. Opin. Genet. Dev., 2011, 21, 768–775 CrossRef CAS PubMed.
- B. P. Conlon, S. E. Rowe, A. B. Gandt, A. S. Nuxoll, N. P. Donegan, E. A. Zalis, G. Clair, J. N. Adkins, A. L. Cheung and K. Lewis, Nat. Microbiol., 2016, 1, 16051 CrossRef CAS PubMed.
- E. A. Zalis, A. S. Nuxoll, S. Manuse, G. Clair, L. C. Radlinski and B. P. Conlon, J. Adkins and K. Lewis, MBio, 2019, 10(5), e-01930-19 CrossRef PubMed.
- L. Tuchscherr, M. Bischoff, S. M. Lattar, M. Noto Llana, H. Pförtner, S. Niemann, J. Geraci, H. Van de Vyver, M. J. Fraunholz, A. L. Cheung, M. Herrmann, U. Völker, D. O. Sordelli, G. Peters and B. Löffler, PLoS Pathog., 2015, 11(4), e1004870 CrossRef PubMed.
- P. Debski, K. Sklodowska, J. Michalski, P. Korczyk, M. Dolata and S. Jakiela, Micromachines, 2018, 9, 469 CrossRef PubMed.
- M. Abolhasani and K. F. Jensen, Lab Chip, 2016, 16, 2775–2784 RSC.
- D. Cottinet, F. Condamine, N. Bremond, A. D. Griffiths, P. B. Rainey, J. A. G. M. de Visser, J. Baudry and J. Bibette, PLoS One, 2016, 11, e0152395 CrossRef PubMed.
- M. Pousti, M. P. Zarabadi, M. Abbaszadeh Amirdehi, F. Paquet-Mercier and J. Greener, Analyst, 2019, 144, 68–86 RSC.
- C. B. Chang, J. N. Wilking, S.-H. Kim, H. C. Shum and D. A. Weitz, Small, 2015, 11, 3954–3961 CrossRef CAS PubMed.
- MarketsandMarkets, Antimicrobial Susceptibility Testing Market by Product (Manual, Automated Susceptibility Testing System), Type (Antibacterial, Antifungal), Application (Clinical Diagnostics), Method (ETEST, Disk Diffusion), End-User - Global Forecasts to 2025, 2020.
| This journal is © The Royal Society of Chemistry 2022 |