
Printed microfluidic sweat sensing platform for cortisol and glucose detection†
Aditi R.
Naik‡
 a,
Yiliang
Zhou‡
a,
Anita A.
Dey‡
b,
D. Leonardo González
Arellano
a,
Uzodinma
Okoroanyanwu
a,
Ethan B.
Secor
c,
Mark C.
Hersam
cd,
Jeffrey
Morse
a,
Jonathan P.
Rothstein
b,
Kenneth R.
Carter
a and
James J.
Watkins
*a
a,
Yiliang
Zhou‡
a,
Anita A.
Dey‡
b,
D. Leonardo González
Arellano
a,
Uzodinma
Okoroanyanwu
a,
Ethan B.
Secor
c,
Mark C.
Hersam
cd,
Jeffrey
Morse
a,
Jonathan P.
Rothstein
b,
Kenneth R.
Carter
a and
James J.
Watkins
*a
aDepartment of Polymer Science and Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: watkins@polysci.umass.edu
bDepartment of Mechanical and Industrial Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA
cDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA
dDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA
First published on 9th December 2021
Abstract
Wearable sweat biosensors offer compelling opportunities for improved personal health monitoring and non-invasive measurements of key biomarkers. Inexpensive device fabrication methods are necessary for scalable manufacturing of portable, disposable, and flexible sweat sensors. Furthermore, real-time sweat assessment must be analyzed to validate measurement reliability at various sweating rates. Here, we demonstrate a “smart bandage” microfluidic platform for cortisol detection and continuous glucose monitoring integrated with a synthetic skin. The low-cost, laser-cut microfluidic device is composed of an adhesive-based microchannel and solution-processed electrochemical sensors fabricated from inkjet-printed graphene and silver solutions. An antibody-derived cortisol sensor achieved a limit of detection of 10 pM and included a low-voltage electrowetting valve, validating the microfluidic sensor design under typical physiological conditions. To understand effects of perspiration rate on sensor performance, a synthetic skin was developed using soft lithography to mimic human sweat pores and sweating rates. The enzymatic glucose sensor exhibited a range of 0.2 to 1.0 mM, a limit of detection of 10 μM, and reproducible response curves at flow rates of 2.0 μL min−1 and higher when integrated with the synthetic skin, validating its relevance for human health monitoring. These results demonstrate the potential of using printed microfluidic sweat sensors as a low-cost, real-time, multi-diagnostic device for human health monitoring.
Introduction
Wearable sweat-based devices for personal health monitoring have received tremendous attention due to potential healthcare, athletic, and military applications.1–4 Sweat offers a compelling target for accurate, continuous, and real-time assessment of key biomarkers through noninvasive measurements. Wearable sweat sensors can be placed comfortably on the skin, in contrast to devices designed for other noninvasive biofluids such as tears,5 saliva,6 and urine.7 Wireless wearable sweat devices can also be paired with smartphone apps for ease of monitoring, allowing the user to modify lifestyle or behavior habits to achieve an improved health state.37 This real-time assessment is particularly important for diabetes management, pathogen detection, athletic training, drug measurement, and stress monitoring.While sweat is principally composed of water (99%),9 it also contains electrolytes, metabolites, amino acids, small molecules, and proteins, secreted from sweat glands. Two key biomarkers are cortisol, a steroid hormone indicative of physiological stress and fatigue within the body,10–12 and glucose, a monosaccharide requiring careful monitoring for effective diabetes treatment.13–15 These biomarkers are present in low concentrations within sweat and diluted at increased sweating rates, requiring sweat sensors engineered for low sample volumes and dynamic flow rates. Electrochemical sensors offer compelling opportunities for wearable sweat applications due to measurement precision, ease of miniaturization, and inexpensive fabrication.16–18 Recent studies have demonstrated impressive advances for electrochemical sweat sensors, including multi-sensing wearable arrays,1 temperature monitoring,19 stretchable and textile pH detection,20,21,22 electrolyte monitoring,23,24 and low-volume sweat collection.25
The exponential growth of research around wearable sweat sensors offers exciting opportunities towards flexible noninvasive biosensors with real-time and dynamic results for personalized health monitoring applications. Sweat sensors have been demonstrated using microfluidic devices,26,27 washable textile sensors,28 colorimetric sensors,29 touch-based devices,30 single-use disposable sensors,31 and many more recent advances. Furthermore, researchers have worked to develop compact sensors integrated with wireless capabilities,32 miniaturized potentiostats,45 real-time displays,34 and self-powered devices.35 The development of wearable sensors for sweat applications still face key challenges to achieve accurate sensing measurements for healthcare use, including mixing of old and new sweat, measurements at low sweating rates and low analyte concentrations, efficient sample collection, and scalable and inexpensive manufacturing.36,37 Javey et al. highlighted that a limitation of current sweat sensors was a lack of data around how sweat volume and flow rate affect sensor accuracy, especially for in situ monitoring.38 In particular, the contamination of old and new sweat and the presence of cellular and microbial byproducts at the interface between the sensor and the skin can limit sensor reliability.39 Sensor systems integrated with microfluidic channels allow for sweat evaporation through the constant renewal of sweat at the sensor electrodes. Microfluidic sweat sensors are explored in a limited number of studies, indicating the need for further research, particularly using an inexpensive assembly for scalability.2,39–41 Several wearable sweat sensors require complex and expensive fabrication, such as cleanroom photolithography to produce microfluidics using polydimethylsiloxane (PDMS) and vacuum evaporation of precious metals for electrode fabrication. Low-cost microfluidic sensors utilize alternative materials, such as adhesive-based or paper-based microfluidics using direct printing of electrodes to enable inexpensive and scalable devices.42–44 Mohan et al. successfully developed an adhesive-based microfluidic sweat sensor with screen-printed electrodes for continuous monitoring. However, this method is constrained by not exploring flow rate dependence of the printed sensors and microfluidic device.45 Sweat sensors must function at dynamic flow rates to accommodate the wide range of sweating rates encountered during daily activities.46–49
Wearable sweat sensors are commonly characterized using on-body measurements without an understanding of the sweat flow rates of the user or sweat pore density on the skin.38,36,37 Sweat sample acquisition from clinical trials on human participants causes the execution of these studies to be time-consuming and expensive. Furthermore, systematic testing of a sweat-based biomarker sensor requires accurate time-stamped sweat sample acquisition and delivery; however, human sweat rates are dependent on a variety of factors such as location on the body, age, sex, fitness levels, and environmental conditions, limiting comparison between wearable sensor studies.50 In order to circumvent these challenges, there have been recent advances in the development of artificial skin models.51–56 Hou et al. first presented an artificial sweat simulator with a micro replicated skin texture with flow rates comparable to human sweat rates. This study used a gravity-fed flow control, limiting pore size and preventing dynamic variations in sweat concentration and flow rate.57 Some of the advancements in the area of microfluidic skin models include an automated computer-controlled fluid mixing and dispensing system,55 ultra-low sample volumes through implementation of open nanofluidic films,56 a moisture-sensitive artificial skin model to simulate dry and moist in vivo human skin and the development of electronic skins, which have been designed to incorporate different types of sensors in ultrathin films.53 One key limitation is that these skin models have not been integrated with a sweat sensor for repeatable and dynamic measurements.
Here, we demonstrate the fabrication of an inexpensive microfluidic architecture with printed electrodes for single-use cortisol detection and continuous glucose monitoring. The flexible microfluidic system consists of a pressure-sensitive adhesive microchannel between polyimide (PI) and hydrophilic polyethylene terephthalate (PET) substrates for fast capillary-driven flow, integrated with an absorbent pad for sweat evaporation. Electrochemical three-electrode and two-electrode systems are inkjet-printed with graphene and silver.18,41,58 For the chronoamperometric cortisol device, the working electrode is further modified with electrodeposited gold and immobilized with anti-cortisol antibodies. A low voltage, portable, and printed electrowetting valve is also included in this design to illustrate the simplicity of customization for additional reagents.59–63 The printed cortisol sensor achieved a dynamic detection range and a limit of detection of 10 pM, relevant for typical physiological conditions. This microfluidic device is also evaluated for time-stamped sweat delivery of glucose through a polymer-based synthetic skin. This skin was designed to resemble eccrine sweat pores that cover the human body and was assessed for glucose diffusion and residence time of fluid within the skin. The amperometric glucose sensor is produced by electrodeposition of Prussian blue (PB) on printed graphene and immobilized with glucose oxidase in a chitosan layer. The glucose sensor demonstrated a reproducible response within a range of 0.2 to 1.0 mM and a 10 μM limit of detection. The effect of perspiration rate on sensor performance is evaluated by adhering the microfluidic device onto the synthetic skin and testing at flow rates of 1–5 μL min−1. A flexible synthetic skin composed of PDMS was developed using standard photolithography and soft lithography techniques.64 The key advantages of the synthetic skin were to mimic the dimensions and density of sweat pores as observed on human skin, achieve relevant sweat flow rates, and obtain a known biomarker concentration within sweat at the synthetic skin pores. Overall, this microfluidic sweat sensor offers an inexpensive and customizable platform to accommodate single-use and continuous biomarker measurements at various sweating rates for personalized health monitoring.
Results and discussion
Printed electrodes & microfluidic assembly
The fabrication of the microfluidic device is shown in Fig. 1 with printed electrodes and a laser cut design. Graphene and silver inks were inkjet-printed on a polyimide (PI) substrate for high-temperature stability. A polyethylene terephthalate (PET) substrate with a SiO2-based hydrophilic coating was used for the top substrate to promote capillary-driven flow through the device. A pressure-sensitive adhesive (PSA) layer was laser cut to produce a 1 mm wide channel, and an absorbent pad was placed at the end of the channel for constant evaporation of sweat through the device. The completed microfluidic sweat sensor measured 78 mm across, comparable to the size of a standard band-aid, and was able to withstand bending and twisting as shown in Fig. 1d–f, enabling conformal contact under mechanical deformation. Fig. 1b and c demonstrate the printed electrodes used for the cortisol and glucose sweat sensors. The electrochemical glucose sensor in Fig. 1b used a two-electrode system: graphene for the working electrode, and Ag/AgCl for a combined reference and counter electrode. In Fig. 1c, the chronoamperometric cortisol sensor used a three-electrode system: a graphene/Au nanoparticle (NP) working electrode, a graphene counter electrode, and a Ag/AgCl reference electrode.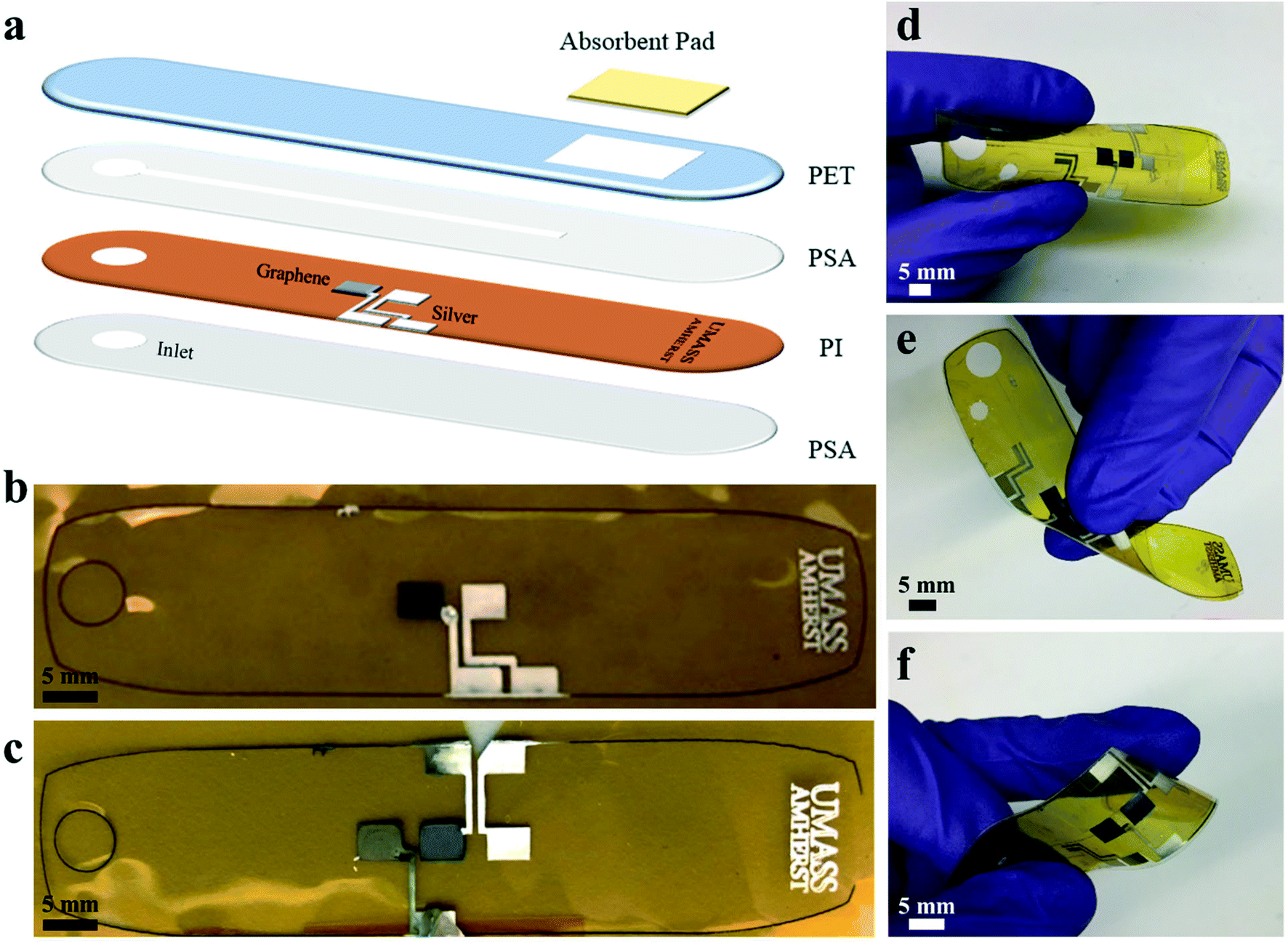 | ||
| Fig. 1 Printed electrodes and microfluidics fabrication. (a) Schematic of microfluidic device fabrication and assembly (polyimide (PI), polyethylene terephthalate (PET), pressure-sensitive adhesive (PSA)) of electrode and connector layout for (b) printed glucose sensor with graphene (left) and silver (right) electrodes and (c) printed cortisol sensor with graphene/Au NP (left), graphene (middle) and silver (right) electrodes, (d–f) assembled cortisol sensor under bending and twisting mechanical deformation. | ||
To decrease fabrication time and improve scalability, the graphene and silver printing parameters were optimized for a common jetting waveform and thermal treatment profile. Inkjet-printing both inks at the same voltages, drop size spacings, and nozzle cleaning conditions decreased individual electrode printing time and complexity. A jetting voltage of 28 V was used to print graphene and silver inks on a PI substrate, and silver ink on a hydrophilic PET substrate. Four nozzles were used to print the silver ink to prevent clogging and sixteen nozzles to print the graphene ink at 20 μm drop spacing (1270 dpi resolution) to enable clean, precise, and uniform print quality of each ink on the particular substrate, as shown in Fig. 1b and c. The inkjet printer and vials of silver and graphene ink are shown in Fig. S1.† For the microfluidic sensing device, electrodes were printed with dimensions of 4 mm by 3 mm with a 2 mm spacing between electrodes on a PI substrate. The electrode sizes and laser cut layer dimensions are shown in Fig. S2.† Two graphene electrodes and one silver electrode were printed for the cortisol sensor design, and one graphene electrode and one silver electrode were printed for the glucose sensor design, as shown in Fig. 1d and e. Silver interconnects were printed from the electrode pads to the edges of the PI substrate to allow for electrical connections. The printed graphene and silver patterns on PI were thermally annealed for 1 hour at 350 °C to optimize conductivity.
The silver electrodes for both sensors were further modified to be used as the reference electrode for the electrochemical sensors and the electrowetting valve. Printed silver was modified to Ag/AgCl to serve as a reference electrode for the cortisol sensor and a combined reference/counter electrode for the glucose sensor and fabricated by dropcasting ferric chloride solution over the silver electrode. The electrowetting valve was prepared for the cortisol sensor from a printed silver electrode, referred to as a hydrophilic electrode, and a perfluorodecanethiol (PFDT)-modified hydrophobic silver electrode to prevent fluid flow through the channel prior to actuation. The surfaces of the silver and graphene electrodes were characterized by scanning electron microscopy (SEM) and energy-dispersive x-ray spectroscopy (EDS) before and after each modification step. SEM images of printed graphene flakes following thermal treatment, an annealed printed silver electrode, a silver electrode modified with ferric chloride, and a silver electrode modified with PFDT are shown in Fig. S3.† Elemental analysis by EDS mapping, as shown in Fig. S4,† was performed on the electrodes to validate the electrode composition qualitatively for each modification step. For example, following the modification of silver to Ag/AgCl, chlorine is measured on the surface of the modified electrode as shown in Fig. S4f,† but not measured on the surface of unmodified silver (Fig. S4d†).
Printed cortisol sensor
Cortisol is a steroid hormone used as an indicator of stress, and produced mainly by the adrenal gland for the regulation of blood pressure, cardiovascular function, and many metabolic activities. The body is able to control cortisol secretion and production based on the cortisol level in the bloodstream.6 Cortisol levels follow a circadian rhythm in individuals and are very low around midnight, peak in the morning, and then slowly decrease during the day, recently demonstrated to be measureable using a wearable cortisol sensing device.32,65 An excess of cortisol within the body can lead to a variety of medical complications, including Cushing's syndrome with symptoms of obesity, fatigue, and skin and bone fragility.10 Therefore, the detection of cortisol levels in the body has become an important diagnostic indicator.Various methods have been developed for the detection of cortisol, including liquid chromatography, mass spectrometry, and enzyme-linked immunosorbent assays.8 Among these detection methods, electrochemical immunosensors have emerged as the most promising approach to cortisol detection. Due to the adsorption of cortisol onto surfaces functionalized with anti-cortisol antibodies, the electrical properties of a conductive material will change proportionally with concentration. Various conductive materials have been investigated for cortisol immunosensors, including gold,10,31 zinc oxide,11,12,76 graphene oxide,6,13 and carbon nanotubes.77
In this study, inkjet-printed graphene electrodes are used for the working and counter electrodes of the cortisol sensor and an inket-printed Ag/AgCl electrode is utilized as the reference electrode. Fabrication steps to produce the cortisol sensor are illustrated in Fig. 2a. Gold nanoparticles (Au NPs) were electrodeposited onto the graphene electrode, resulting in a well-decorated surface as shown by SEM in Fig. 2b and characterized by EDS in Fig. S4c.† The incorporation of Au NPs is beneficial in two ways: Au NPs enhance the charge transfer between electrodes, yielding better sensing performance, and the nanoparticles can be further functionalized as immobilization sites for cortisol antibodies. The procedure used for antibody immobilization was similar to that discussed in previous literature.31,33 Dithiobis(succinimidyl propionate) (DTSP) was applied as a linker between the cortisol antibody and the Au NPs on the graphene surface. The cortisol antibodies were covalently attached to the DTSP self-assembled monolayer through an incubation process. Finally, the electrodes were incubated with ethanolamine solution to block unreacted succinimidyl groups and remove unbound antibodies from the electrode surface.
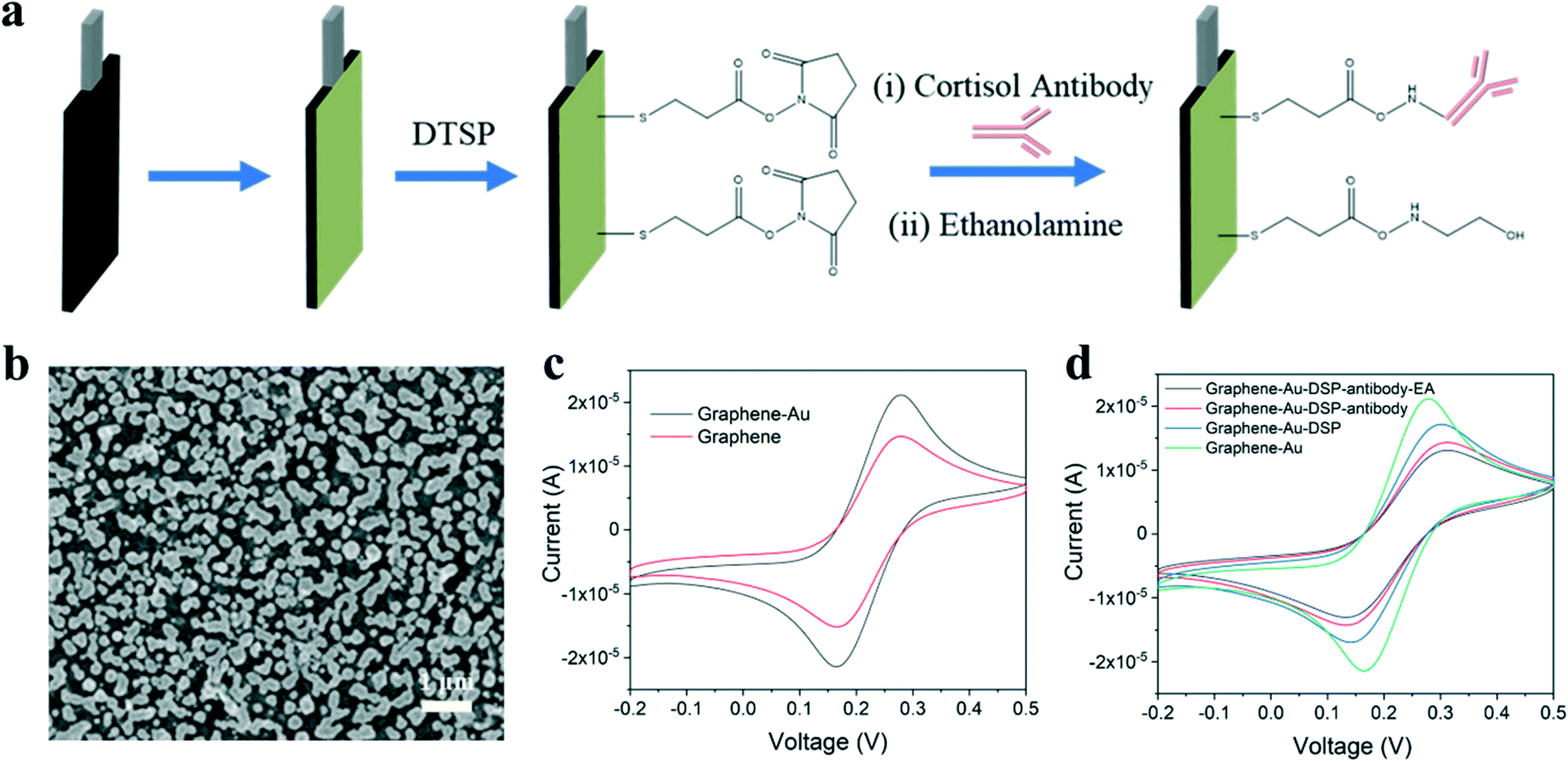 | ||
| Fig. 2 Cortisol sensor fabrication. (a) Schematic of bioelectrode fabrication using inkjet-printed graphene electrode modified with ethanolamine (EA), anti-cortisol, dithiobis(succinimidyl propionate) (DTSP), and gold nanoparticles (Au NPs). (b) SEM image of Au NPs on the graphene electrode. (c) CV curves of Au NPs/graphene and graphene electrodes in 5 mM Fe(CN)63−/4−. (d) CV curves of Au NPs/graphene in 5 mM Fe(CN)63−/4− during different stages of the immobilization process. | ||
Cyclic voltammetry (CV) was used to monitor the graphene electrodes at increasing voltages with redox peaks. Compared to the unmodified graphene electrode, the peak current of the graphene electrode with Au NPs increased due to charge transfer enhancement as shown in Fig. 2c. During the immobilization process, the magnitude of the peak current gradually decreased after each step as seen in Fig. 2d. This decrease was due to the antibody and ethanolamine layers hindering electron transfer from the medium to the electrode.
An electrochemical chronoamperometric technique is applied for cortisol detection using modified inkjet-printed graphene electrodes. This work builds on the cortisol sensor developed by Tuteja et al. using screen-printed electrodes via this immobilization method.17,78 The detection of cortisol by chronoamperometry involved two key steps. First, the antibody-modified graphene electrodes were incubated with analytes. Second, the redox agent solution, Fe(CN)63−/4−, was introduced, and the charge transfer performance was examined by measuring the current change for the electrodes. During sensing, cortisol is captured by the antibody and adhered to the electrode surface, producing a diffusion barrier to reduce the flow of ions. The extent of the current decrease with redox agent reflects the concentration of cortisol present in the sample solution. Fig. 3a exhibits the chronoamperometric curves of the inkjet-printed cortisol sensor using various concentrations of cortisol within artificial sweat. As shown in the calibration plots in Fig. 3b, the observed current values at the 30 s time point in the plateau region can be further correlated with the concentration of cortisol. The detection range of the inkjet-printed cortisol sensor is from 10 pM to 100 nM. The selectivity of the developed immunosensor was further evaluated by conducting chronoamperometric studies using non-specific interferents. As shown in Fig. 3c, the interferents used are ascorbic acid, uric acid, and glucose. The introduction of these interferents at 1.0 mM did not decrease the sensor current following incubation, demonstrating high selectivity of the sensor for cortisol.
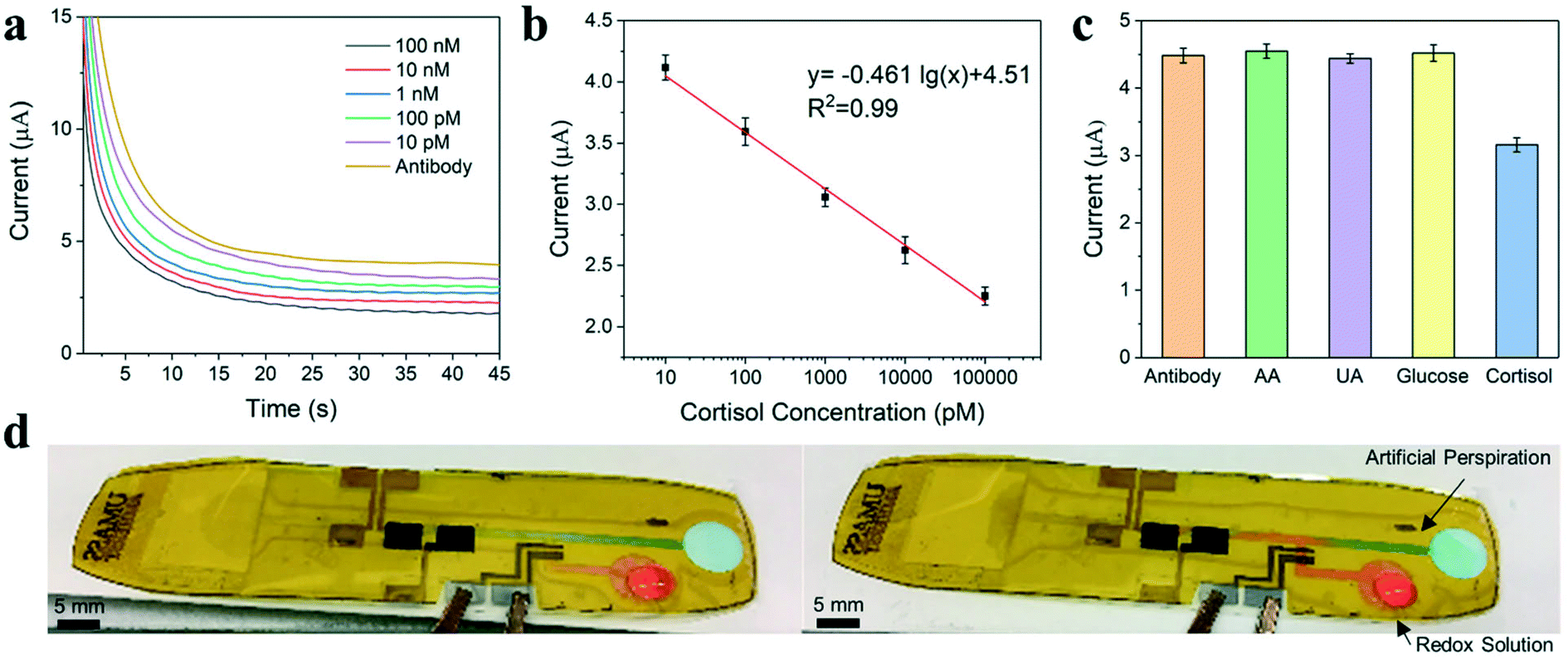 | ||
| Fig. 3 Cortisol sensor results. (a) Chronoamperometric curves of the cortisol sensor with decreasing concentrations for cortisol. (b) Calibration curve for cortisol detection. (c) Assessment of sensor specificity using 1.0 mM of non-specific interferents: antibody, ascorbic acid (AA), uric acid (UA), and glucose. (d) Closed electrowetting valve (left) and opened valve (right) where blue solution is artificial perspiration and red solution is redox agent solution, Fe(CN)63−/4−. Sensor electrodes from left to right are Ag/AgCl (reference), graphene (counter), graphene/Au NPs (working). | ||
The cortisol sweat sensor was assembled as shown in Fig. 1 using a laser cut PSA adhered over the inkjet-printed electrodes and sealed with a PET layer. An electrowetting valve was incorporated on the PET substrate for the cortisol sensor design to enhance ease-of-use as shown in Fig. 3d.63 Electrowetting valves are portable, inexpensive, and printable microfluidic valves based on the phenomenon of electrowetting-on-a-dielectric, which can actuate to open within seconds and provide dynamic fluid control. The valve was used to precisely control the injection of the redox agent solution, Fe(CN)63−/4−, into the microchannel leading to the sensor. The redox solution (red solution) is prevented from entering the main channel until the valve is actuated using a low voltage of 4 V. Once open, the redox solution flows forward to meet the artificial sweat (blue solution) within the channel. Furthermore, a microfluidic device with multiple valves and sensors could be used to achieve multi-point detection of cortisol concentrations within sweat at distinct time points.
Synthetic skin development
The human skin has a variable density of sweat pores, with the highest density of 700 pores per cm2 on the palms and soles, and the lowest density of 64 pores per cm2 on the back.80,81 The average sweat pore density has been found to be 200 sweat pores per cm2.79 The synthetic skin was designed to have 72 sweat pores distributed over a circular region of radius 3 mm, shown schematically in Fig. 4a, resulting in a sweat pore density of 254 pores per cm2. Additionally, human sweat rates have been found to vary across body locations and during times of rest or exercise, with a full-body average sweat rate of 0.72–3.65 mg cm−2 min−1 during exercise.49 To ensure applicability of these results, the synthetic skin was tested over a broader range of flow rates, between 0.35 mg cm−2 min−1 (0.1 μL min−1) and 17.8 mg cm−2 min−1 (5.0 μL min−1).The PDMS synthetic skin was fabricated as an assembly of a base and top layer of sweat pores, shown in Fig. 4a and b. A schematic of this assembly process is illustrated in Fig. S5.† The base layer was designed to distribute fluid from the inlet to an array of sweat pores aligned under an identical array of sweat pores in the top layer. The synthetic skin flow path was designed to allow for efficient mixing of the fluid at the maximum and minimum flow rates. Beyond the mixing region, the flow is divided into a network of channels that lead to the end points. The synthetic sweat pores were designed with a diameter of 50 μm, similar to human sweat pores.82 By varying the ratio of the flow rates at the two inlets in the base layer, the concentration of glucose released by the sweat pores varied with time. All measurements for the synthetic skin started when fluid entered the synthetic skin inlet and time lag related to syringe pump operation was not included. The microfluidic sensor device was then placed over the synthetic skin, allowing for continuous measurements to examine time-stamped delivery of sweat biomarkers at different sweating rates. | ||
| Fig. 4 Synthetic skin development. (a) Base and (b) top layers of the microfluidic synthetic skin. (c) Schematic showing assembly of the synthetic skin under the sensor. (d) Sweat sampling area of the synthetic skin visualized by pink dye. (e) Time lag of a plug of fluid to exit the synthetic skin pores as a function of flow rate. (f) Degree of spreading of a plug of fluid as a function of flow rate. | ||
The synthetic skin was shown to be an effective model for both steady-state and time-varying conditions. Fig. 4e demonstrates the time lag, tlag, from when sweat is introduced into the base layer of the microfluidic synthetic skin until it arrives at the sensor. The tlag was measured experimentally and shown alongside the calculated
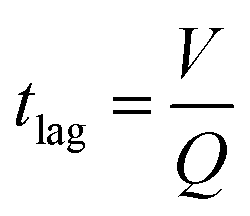
As shown in Fig. 4f, the effect of biomarker diffusion was similarly experimentally and theoretically verified using the Taylor–Aris analysis, accounting for axial spreading and radial diffusion within the microfluidic channels. The Taylor–Aris dispersion coefficient is

![[u with combining low line]](https://www.rsc.org/images/entities/i_char_0075_0332.gif) is the average channel velocity.83DTA is then used to approximate the spreading of a concentration plug of fluid over a time interval of tspread,
is the average channel velocity.83DTA is then used to approximate the spreading of a concentration plug of fluid over a time interval of tspread,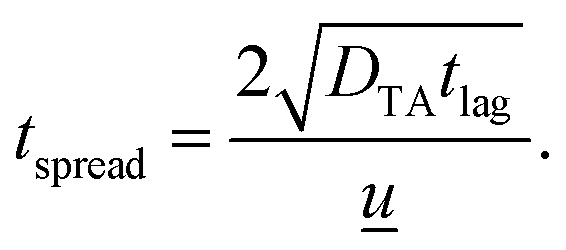
Therefore, if a plug of fluid of known concentration, Cinlet, is introduced into the inlet of the synthetic skin over a time interval of tinlet, then a plug of fluid with a time interval of approximately of tsensor = tinlet + tspread will pass through the skin pores. The average concentration at the pores will be

There will be a minimum initial plug width that can be effectively transported from the inlet of the synthetic skin to the sensor. For these experiments, we chose Csensor = 0.9 Cinlet to avoid complications. The results show that at a plug of fluid introduced over a time interval tinlet of 5 min at flow rates higher than 1 μL min−1 showed minimal spreading of the concentration profile. Using the synthetic skin in conjunction with the microfluidic sensor device, continuous sweat monitoring can be experimentally evaluated for significant health biomarkers.
Continuous glucose monitoring
Continuous glucose monitoring through sweat flow would be an invaluable resource for effective diabetes treatment and management. An electrochemical glucose sensor is fabricated on the microfluidic device and attached to the synthetic skin for continuous measurements. The working electrode of the sensor is produced from graphene ink and further modified with mediator layers immobilized with biorecognition elements for sensing. The graphene electrode was inkjet-printed onto the PI substrate of the microfluidic device, as discussed previously.Prussian blue (PB) was electrodeposited over the conductive graphene electrode to catalyze the reduction of hydrogen peroxide produced from glucose oxidase (GOx), a common configuration as a highly-stable mediator layer for enzyme-based sensors.66,67 A PB redox mediator layer decreases the necessary applied voltage to the working electrode to be close to 0 V, results in increased sensor sensitivity and reduces undesired side reactions.57Fig. 5a illustrates the mechanism of the PB-coated graphene electrode during glucose detection. Glucose is oxidized by GOx, yielding δ-gluconolactone and hydrogen peroxide, which is reduced by the PB mediator and electrochemically detected at a low potential. The electrodeposition solution for PB contained 2.5 mM FeCl3, 100 mM KCl, 2.5 mM K3Fe(CN)6, and 100 mM HCl. The surface of the graphene electrode was observed by SEM before and after the electrodeposition of PB as shown in Fig. 5b, demonstrating that PB nanoparticles were well-coated over the graphene surface. The deposition of PB was further verified by EDS, detecting the presence of iron and nitrogen (see Fig. S3e†). The CV curves of the PB-coated graphene electrode were measured in 0.1 M KCl solution, shown in Fig. 5d and e. The cathodic and anodic peaks of the CV curves demonstrate the redox reaction between PB and Prussian white. The CV curves were measured at various scan rates (10–50 mV s−1) and are typical of a quasi-reversible redox process. The peak currents linearly increase with the square root of scan rate, indicating this reaction is a diffusion-controlled process.66,68 In Fig. 5e, the stability of the PB-coated graphene electrode is confirmed by a multi-cycle CV measurement, where the peak currents maintained a similar value after 50 cycles.
 | ||
| Fig. 5 PB-coated graphene working electrode. (a) Mechanism for GOx/PB/graphene in glucose sensing. SEM images of (b) inkjet-printed graphene electrode and (c) PB-coated graphene electrode. (d) CV curves of PB/graphene electrode in 0.1 M KCl over 10–50 mV s−1 scan rates. (e) CV curves of PB-graphene electrode in 0.1 M KCl over 50 cycles. | ||
To complete the glucose sensor, a GOx enzyme layer was deposited over the PB layer. This enzyme solution was prepared from GOx mixed with chitosan and glutaraldehyde in a buffer solution, and 5 μL of solution was drop-cast over the PB-coated graphene electrode. The completed glucose sensor consisted of a working electrode of GOx/PB/graphene and an inkjet-printed Ag/AgCl electrode as both the counter and reference electrodes. The performance of the sensor was tested in a standard electrochemical setup using an applied potential of −0.05 V and various concentrations of a glucose/sweat solution. The glucose sensor was calibrated within the concentration range of 0.2 to 1.0 mM glucose, which is within the range of typical glucose concentration found in human sweat, and shown in Fig. S6a.†69 The inkjet-printed sensor also exhibited acceptable reproducibility for glucose sensing, demonstrated in Fig. S6b.† The limit of detection of the glucose sensor was measured to be 10 μM at a signal to noise ratio of 3, and the sensitivity of the sensor was measured to be ∼18 μA cm−2 mM−1 through benchtop analysis within a beaker.
Real-time monitoring of glucose in sweat was achieved by integrating the microfluidic glucose sensor with the synthetic skin developed with two inlet ports. For the measurements, simulated sweat solution was injected into one inlet and sweat with a specific concentration of glucose was injected into the second inlet. Initially, sweat was injected into the synthetic skin and through the microfluidic device to measure a baseline response, and then a glucose/sweat solution was injected to detect the glucose concentration. Fig. 6a shows the multiple injections of glucose solution with the same concentration in which the current signal achieved a similar value, demonstrating good sensor stability. The response curve of the sweat-based microfluidic device with various concentrations of glucose is shown in Fig. 6b. At the same flow rate, the response times are similar, and the current value of the plateau region increases appropriately at higher glucose concentrations.
 | ||
| Fig. 6 Continuous glucose sensor results. (a) Multiple injections of 0.2 mM glucose sweat solution into microfluidic device. (b) Response curves of microfluidic device at various concentrations of glucose. (c) Response and (d) decay curves of sweat-based glucose sensor for 1.0 mM glucose at various injection flow rates. (e) Linear relationship between response time and sweat flow rate. (f) Relationship between total injection volume and sweat flow rate. | ||
The microfluidic sensor device was tested at sweating flow rates from 1.0 to 5.0 μL min−1 to examine the effect of flow rate on the results of the glucose sensor. The response and decay curves for these flow rates are shown in Fig. 6c and d, respectively. Fig. 6c demonstrates that for flow rates equal to or greater than 2 μL min−1, the current signal achieved a similar value. However, lower flow rates, such as 1 μL min−1, showed a decreased current signal. This is likely a result of poor contact between the glucose/sweat solution and the working electrode of the glucose sensor due to the limited amount of fluid within the device.
The response and decay times for the glucose sensor were seen to increase as the sweat flow rate decreased. To understand this result, the relationship between flow rate and the inverse response rate was plotted and demonstrated to be linear, as shown in Fig. 6e. The total injection volume within the device was calculated and shown to decrease as flow rate increased in Fig. 6f, meaning the device has a required volume of sweat that must pass over the sensor to achieve an accurate measurement. When the fluid within the device is switched from sweat to a glucose/sweat solution, the earlier sweat solution must be replaced with glucose/sweat solution, which takes additional time at lower flow rates resulting in slower response and decay results. Therefore, the dynamic data of the response and decay curves demonstrates that the volume between the sweat entering the device and the sensor location within the device is critical for improving sensor response time. Notably, the distribution of fluid in the synthetic skin adds a constraint in this experimental demonstration that would not be present for actual implementation, and minimization of this volume in practice will shorten the response and decay times for real-time glucose sensing in continuous sweat analysis. Furthermore, optimization of the microfluidic device to reduce the internal volume from the device inlet to the sensor electrodes by printing electrodes closer to the inlet and decreasing channel size, would improve signal performance at low flow rates.
Conclusion
In summary, a flexible microfluidic device with inkjet-printed electrodes was developed for sweat analysis and tested using a synthetic skin. Silver and graphene electrodes were printed on polymeric substrates and utilized as biosensing materials via electrochemical measurements. Graphene electrodes were successfully developed into cortisol and glucose sensor working electrodes with further functionalization. For cortisol sensing, Au nanoparticles were deposited on the surface of graphene electrodes, acting as immobilization sites for cortisol antibodies. The cortisol detection in synthetic sweat can be achieved with chronoamperometric techniques, and electrowetting valves can be incorporated to improve the versatility of the devices. Moreover, this valve strategy could enable devices to achieve multiple detection using a multi-valve system. For glucose sensing, a thin layer of PB was deposited on the printed graphene electrode as a mediator layer. Using a PDMS-based synthetic skin, the effect of perspiration rate on sensor performance for real-time glucose monitoring applications was studied in detail. The relationship between response time and flow rate showed that microfluidic design optimization in minimizing volume from the inlet to the sensor is vital for biomarker monitoring through sweat analysis at low sweat flow rates. The development and fabrication of the cortisol and glucose sensors was specifically designed to be similar to demonstrate the capability of printed methods to create inexpensive wearable and flexible devices. These types of printed devices can serve as a platform for multiple biosensing mechanisms, regardless of whether the sensor is designed for a single-use cortisol measurement or continuous glucose measurements. As demonstrated for the key analytes of glucose and cortisol, these flexible and printed smart bandage microfluidic sensors provide a platform for low-cost biomarker measurements in sweat with potential extensions to additional analytes and electrochemical sensing mechanisms.Materials and methods
Ink preparation and deposition
Graphene was prepared by liquid-phase exfoliation from graphite powder by high shear mixing in the presence of the stabilizing polymer ethyl cellulose (Sigma Aldrich, 4 cP, 200646) following previously established protocols.70 Centrifugation and flocculation of the graphene dispersion yielded a graphene/ethyl cellulose powder containing 45 wt% graphene. This powder was dispersed at a concentration of 20 mg ml−1 in ethyl lactate (Sigma Aldrich, W244015) by bath sonication. The ethyl cellulose acted as a polymer stabilizer for the graphene flakes to prevent flake-to-flake aggregation, resulting in a well-dispersed ink containing primarily 1–5 nm thick graphene flakes with 100–500 nm lateral size. Nitrocellulose (Scientific Polymer Products, Cat. 714) was added at a concentration of 5 mg ml−1 as an adhesion promoter. Following thermal treatment, the nitrocellulose decomposes to form an amorphous carbon residue for improved mechanical stability and high electrical conductivity.71 Alpha-terpineol (Alfa-Aesar, 16285) was added to the ink in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volume ratio prior to inkjet printing to improve latency time and modulate drying characteristics, resulting in a nominal ink composition of 7.2 mg ml−1 graphene, 8.8 mg ml−1 ethyl cellulose, and 4 mg ml−1 nitrocellulose in 4:1 ethyl lactate:terpineol. Graphene ink viscosity was measured to be 8.2 cP using an m-VROC Viscometer by RheoSense. Metalon JSB-25 HV (Novacentrix) nanosilver ink with ∼25 wt% silver in an aqueous dispersion was used for printing. The silver ink was composed of ∼25 wt% silver particles (60–80 nm) in an aqueous dispersion stabilized with ethylene glycol and polyethylene glycol 4-(tert octylphenyl) ether.
:4 volume ratio prior to inkjet printing to improve latency time and modulate drying characteristics, resulting in a nominal ink composition of 7.2 mg ml−1 graphene, 8.8 mg ml−1 ethyl cellulose, and 4 mg ml−1 nitrocellulose in 4:1 ethyl lactate:terpineol. Graphene ink viscosity was measured to be 8.2 cP using an m-VROC Viscometer by RheoSense. Metalon JSB-25 HV (Novacentrix) nanosilver ink with ∼25 wt% silver in an aqueous dispersion was used for printing. The silver ink was composed of ∼25 wt% silver particles (60–80 nm) in an aqueous dispersion stabilized with ethylene glycol and polyethylene glycol 4-(tert octylphenyl) ether.
A Dimatix DMP-2850 (Fujifilm) inkjet printer with a 10 pL cartridge (Fujifilm, 2100201146) was used to print the graphene and silver patterns on polyimide (PI, Kapton, American Durafilm) and polyethylene terephthalate (PET, Novele, Novacentrix, 910007002) films, respectively. The disposable ink cartridge had sixteen piezoelectric-actuated nozzles.72 For graphene patterns, twenty-five layers of ink were printed using sixteen nozzles and annealed at 350 °C for 1 h to enhance electrical conductivity. For silver patterns, two layers of ink were printed using four nozzles for the sweat sensors and one layer for the electrowetting valve. Four nozzles were used when printing silver ink to prevent clogging and allow for improved monitoring of the printing process due to system constraints. The printed silver was sintered for 5 min at 125 °C on PET and 1 h at 350 °C on PI. Both graphene and silver inks were printed using 20 μm drop spacing (1270 dpi resolution) and 28 V jetting voltage using a common jetting waveform.
Cortisol sensor development
For the working electrode, Au NPs were electrodeposited on the inkjet-printed graphene surface. The deposition was achieved in 5 mM hydrogen tetrachloroaurate(III) trihydrate (HAuCl4, Thermo Fisher Scientific, AA3640003) solution via applying a voltage at −0.2 V (vs. Ag/AgCl) for 60 s. For antibody immobilization, the graphene/Au NPs electrode was immersed in a solution of 10 mM dithiobis(succinimidyl propionate) (DTSP, Thermo Fisher Scientific, 22585) in dimethyl sulfoxide (DMSO, Sigma Aldrich, 472301) for 2 h for the self-assembled monolayer formation. Then 20 μL monoclonal cortisol antibody (Lifespan Biosciences, LS-C79806) at 10 μg mL−1 was immobilized on the graphene/Au/DTSP electrode for 2 h followed by careful washing with 10 mM phosphate buffered saline (PBS, pH 7.4, Sigma Aldrich, P4417100TAB) to remove unbound antibody. Finally, the antibody modified graphene/Au NP electrode was immobilized with 20 μL 5% ethanolamine (Sigma Aldrich, E9508) for 10 min, blocking unreacted succinimidyl groups. The cortisol working electrode was stored at 4 °C prior to testing, and shelf-life stability of the sensors was not tested. The counter electrode was fabricated from printed graphene and the reference electrode was Ag/AgCl, as discussed above. For testing, 1.0 mg mL−1 cortisol stock solution in methanol (Sigma-Aldrich, 322415) was diluted with stabilized eccrine artificial perspiration (Pickering Laboratories, pH 4.5, 1700–0020) to achieve the desired concentration. The artificial perspiration solution is formulated to mimic adult human eccrine sweat and includes nineteen amino acids, seven minerals, and four metabolites at relevant concentrations based on published data, as described by the manufacturer. Three cortisol sweat sensors were tested to obtain results.Glucose sensor development
For the working electrode, a Prussian blue (PB) thin film was electrodeposited on the inkjet-printed graphene electrode by applying a constant potential of 0.4 V (vs. Ag/AgCl) for 120 s. The deposition bath was composed of 2.5 mM iron(III) chloride (FeCl3, Sigma Aldrich, 157740), 100 mM potassium chloride (KCl, Sigma Aldrich, P3911), 2.5 mM potassium hexacyanoferrate (III) (K3Fe(CN)6, Strem Chemicals, 931920), and 100 mM hydrochloric acid (HCl, Fisher Chemical, A142212). The glucose sensor was obtained by drop-casting 5 μL of glucose oxidase (Sigma Aldrich, G612550KU) and chitosan (Sigma Aldrich, C364625G) solution onto the graphene/PB electrode. The preparation of glucose oxidase/chitosan solution was performed as follows: 1 wt% chitosan solution was first prepared by dissolving chitosan in 2 wt% acetic acid (Sigma Aldrich, A6283) with magnetic stirring. The chitosan solution was mixed with glucose oxidase (Sigma Aldrich, G6125) solution diluted to 100 mg ml−1 in stabilized eccrine artificial perspiration (Pickering Laboratories, pH 4.5, 1700–0020) and 0.25 wt% glutaraldehyde/water solution (Sigma Aldrich, 340855) in the volume ratio of 1:2:1. The glucose sensor working electrode was stored at 4 °C prior to testing. An inkjet-printed Ag/AgCl electrode was used as a combined counter and reference electrode system, as demonstrated in previous literature.73 Ferric chloride (FeCl3, Copper Etchant Type CE-100, Chemtrec) was diluted with DI water to produce a 50 mM solution and a 30 μL droplet was dropcast over the annealed silver electrode, converting the top layer of silver to silver chloride. The reaction was quenched with DI water after 1 min and the Ag/AgCl electrode was air-dried. For sensor testing, a 0.1 M glucose (D-(+)-glucose, Sigma Aldrich, G7021) stock solution was prepared in artificial perspiration and kept at room temperature for 24 h prior to use to ensure the formation of β-D-glucose. Five glucose sweat sensors were tested to obtain results.
Microfluidic device assembly
The microfluidic devices were fabricated from 25 μm polyimide (PI, Kapton, American Durafilm), 48 μm pressure-sensitive adhesive (PSA, ARCare 92712), and 140 μm polyethylene terephthalate (PET, Novele, Novacentrix, 910007002) layers, which were individually laser cut (VLS3.50, Universal Laser Systems) according to computer-aided design (CAD) designs. The three layers were laser cut to 78 mm by 18 mm dimensions, similar to the size of a standard band-aid or adhesive bandage. A 1 mm wide microchannel was cut into the PSA layer for sweat flow over the sensor region. An absorbent pad (Whatman, Grade 1, WHA1001090) was placed at the outlet of the device to absorb excess sweat flow through the device. The absorbent pad was monitored visually during microfluidic testing and replaced prior to saturation to prevent changes to flow parameters within the device. To avoid absorbent pad replacement during testing, a superabsorbent material may be used in place of filter paper. A wicking pad at the entrance of sweat into the microfluidic device was examined and found to hinder flow, and was therefore not used. The microfluidic device was pre-wet with sweat prior to testing to remove air pockets within the channel. The microfluidic glucose sensor device placed on the synthetic skin inlet during operation is shown in Fig. S7.†Electrowetting valve fabrication
The valve was fabricated as detailed in earlier work.63 Printed silver electrodes on polyethylene terephthalate (PET, Novele, Novacentrix, 910007002) were exposed to UV/ozone for 10 min and then immersed in 12 mM solution of 1H,1H,2H,2H-perfluorodecanethiol (PFDT, Sigma Aldrich, 660493) in ethanol (Pharmco-Aaper, TSI111PL05M) for 40 min until the surface of the silver was hydrophobic. The modified electrode was then rinsed with ethanol for 2 minutes and dried under nitrogen. Following the PFDT modification, a second silver electrode was printed alongside the hydrophobic electrode and annealed. To actuate-to-open, the valve was supplied with 4 V from a voltage supply box (80 W Switching DC Power Supply, Extech).Electrochemical sensor measurements
All electrochemical measurements were carried out using an electrochemical analyzer (CHI 660E, CH Instruments). The detection of cortisol was achieved using a chronoamperometric three-electrode setup: graphene/Au NPs as the working electrode, graphene as the counter electrode, and Ag/AgCl as the reference electrode. Cyclic voltammetry (CV) curves were recorded in PBS solution (10 mM, pH 7.4, Sigma Aldrich, P4417100TAB) containing a 5 mM potassium hexacyanoferrate(II) trihydrate (Fe(CN)63−/4−) redox probe (Sigma Aldrich, P3289). For cortisol detection, the modified graphene electrodes were incubated with cortisol solution for 30 min, ensuring proper binding.33 The electrodes were then immersed in a redox solution (5 mM Fe(CN)63−/4− in PBS). The electrochemical chronoamperometric measurements at 0.4 V were recorded to monitor the change in the current response of these printed electrodes. The oxidation peak of Fe(CN)63−/4− with the graphene/Au electrode was measured to be ∼0.3 V using cyclic voltammetry as shown in Fig. 2c, demonstrating that a voltage above this value would trigger a redox reaction. Based on this result, a voltage of 0.4 V was selected as a proper voltage to activate the redox reaction. The detection of glucose was achieved using an amperometric two-electrode setup: graphene/PB as the working electrode and Ag/AgCl as the reference and counter electrode. The current between the electrodes was monitored in artificial perspiration while applying a voltage of −0.05 V versus the Ag/AgCl electrode.Synthetic skin development
The base and top layers of the synthetic skin were fabricated using standard photolithography techniques.64 A negative photoresist (Micro Chem, SU-82050) was spin-coated onto a master silicon wafer etched with the base and top layer design to yield a feature height of 50 μm. The details of the mask design are shown in Fig. S8.† The features of the photoresist were transferred onto a polydimethylsiloxane (PDMS) elastomer (Dow Corning, Sylgard 184) for fabricating the synthetic skin. This was achieved in two different methods for the two layers. A 10:1 ratio of base and curing agent of PDMS were mixed thoroughly and any bubbles in the mixture were removed by degassing within a vacuum chamber for 30 min. In the case of the base layer, this mixture was poured over the patterned silicon wafer and then allowed to cure in an oven at 70 °C over 24 h. After curing, the PDMS layer was peeled off of the silicon wafer to obtain the base layer of the synthetic skin. For the top layer, which consisted of an array of through-holes of 50 μm diameter in a 50 μm thin membrane, peeling off such a thin membrane of PDMS would result in the inadvertent damage of the silicon wafer. A sacrificial micro-molding technique was employed for the efficient peel-off of the thin PDMS membrane. The patterned silicon wafer was first spin coated with a 10:1 ratio of deionized water and polyvinyl alcohol (PVA) (Sigma-Aldrich). Then the wafers were baked at 95 °C for 5 min to remove any residual water. The resulting PVA sacrificial layer thickness was ∼750 nm.74 A PDMS mixture was then spin-coated over this sacrificial layer and allowed to cure. After curing at 70 C, the wafer was immersed in water and the edges of the PDMS were gently peeled, exposing the sacrificial PVA layer to the water. As PVA is soluble in water, the sacrificial layer was dissolved and the thin PDMS membrane could be lifted off the silicon wafer without any further peeling near the through-hole pattern area. This PDMS membrane was allowed to dry completely before being attached to the base layer. At the start of this assembly, the surfaces of the two layers were treated with a high-frequency electronic corona discharge (BD-20 AC, Electro Technic Products) for 5 min. This surface corona treatment produced a chemically active surface with increased adhesive properties.75 The base and pore layers were aligned under an optical microscope and cured together at 70 °C for 15 min. A schematic of the steps followed for the synthetic skin assembly is shown in Fig. S5.† The synthetic skin had 72 sweat pores distributed over a circular area with a radius of 3 mm, resulting in a sweat pore density of 254 pores per cm2.79–81 The base layer consisted of a network of microfluidic channels leading up to an array of sweat pores. The base layer also included two inlets connected to independently controlled syringe pumps: one with artificial sweat, and one with glucose dissolved in artificial sweat. To promote mixing of the two inlet streams, a serpentine channel was inserted in the base just downstream of the inlets. The serpentine channel was designed to fully mix the two inlet streams over the entire range of flow rates tested. The pore layer was fabricated from a 50 μm thick PDMS membrane designed to seal the microchannels in the base layer, while providing 50 μm diameter through-holes to act as synthetic sweat pores in the same pattern as on the circular terminal points of each microchannel in the base layer. The synthetic skin was tested over a range of flow rates from 0 to 5 μL min−1. For all experiments, the sweat-based biomarker sensor inlet was placed directly over the circular sampling area of the synthetic skin. A sequence of images captured at the start of sweat production at 1 μL min−1 in Fig. S9† is used to demonstrate the uniform circular sweating area produced. A programmable multi-syringe pump (Masterflex) was used to pump fluid through syringe needle tips inserted into the inlets of the synthetic skin device. A USB CMOS machine vision camera (Edmund Optics) was placed above the setup to record time-lapse videos of the fluid in the device. To visualize the fluid flow within the synthetic skin and sensor device, artificial perspiration dyed with food coloring was used.
Analysis techniques
Scanning electron microscopy (SEM) and energy dispersive x-ray spectroscopy (EDS, FEI Magellan 400 XHR) were used to characterize the surface of the printed and modified electrodes.Author contributions
Aditi R. Naik: investigation, validation, data curation, writing – original draft, writing – review & editing. Yiliang Zhou: investigation, validation, data curation, writing – original draft, writing – review & editing. Anita A. Dey: investigation, validation, data curation, writing – original draft, writing – review & editing. D. Leonardo González Arellano: investigation. Uzodinma Okoroanyanwu: investigation, writing – review & editing. Ethan B. Secor: resources, writing – review & editing. Mark C. Hersam: resources, funding acquisition, writing – review & editing. Jeffrey Morse: conceptualization, supervision, funding acquisition. Jonathan P. Rothstein: conceptualization, supervision, funding acquisition, writing – review & editing. Kenneth R. Carter: conceptualization, supervision, funding acquisition. James J. Watkins: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors also thank Dr. Karen Kalmakis, Dr. Jerry Meyer, and Dr. Lisa Chiodo from the UMass College of Nursing for their contributions. A. R. N., Y. Z., A. A. D., D. L. G. A., U. O., J. M., J. P. R., K. R. C., and J. J. W. acknowledge funding from the National Science Foundation (NSF) Center for Hierarchical Manufacturing at the University of Massachusetts at Amherst (CMMI1025020), and the Nano-Bio Manufacturing Consortium (NBMC) and Air Force Research Laboratory (AFRL) (FA8650-13-2-7311). E. B. S. and M. C. H. acknowledge funding from the National Science Foundation (CMMI-1727846) and the U.S. Department of Commerce, National Institute of Standards and Technology (Award 70NANB19H005) as part of the Center for Hierarchical Materials Design (CHiMaD).References
- W. Gao, S. Emaminejad, H. Y. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki, D. Kiriya, D.-H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nature, 2016, 529, 509–514 CrossRef CAS PubMed
.
- A. Koh, D. Kang, Y. Xue, S. Lee, R. M. Pielak, J. Kim, T. Hwang, S. Min, A. Banks, P. Bastien, M. C. Manco, L. Wang, K. R. Ammann, K. Jang, P. Won, S. Han, R. Ghaffari, U. Paik, M. J. Slepian, G. Balooch, Y. Huang and J. A. Rogers, Sci. Transl. Med., 2016, 8, 1–14 Search PubMed
- J. Heikenfeld, Nature, 2016, 529, 475–476 CrossRef CAS PubMed
- M. Bariya, H. Y. Y. Nyein and A. Javey, Nat. Electron., 2018, 1, 160–171 CrossRef
- D. Pankratov, E. González-Arribas, Z. Blum and S. Shleev, Electroanalysis, 2016, 28, 1250–1266 CrossRef CAS
- V. Mani, T. Beduk, W. Khushaim, A. E. Ceylan, S. Timur, O. S. Wolfbeis and K. N. Salama, TrAC, Trends Anal. Chem., 2021, 135, 116164 CrossRef CAS
- W.-C. Tai, Y.-C. Chang, D. Chou and L.-M. Fu, Biosensors, 2021, 11, 260 CrossRef CAS PubMed
- J. Heikenfeld, Electroanalysis, 2016, 28(6), 1242–1249 CrossRef CAS
- L. B. Baker, Temperature, 2019, 6(3), 211–259 CrossRef PubMed
- A. Kaushik, A. Vasudev, S. K. Arya, S. K. Pasha and S. Bhansali, Biosens. Bioelectron., 2014, 53, 499–512 CrossRef CAS PubMed
- A. Singh, A. Kaushik, R. Kumar, M. Nair and S. Bhansali, Appl. Biochem. Biotechnol., 2014, 174(3), 1115–1126 CrossRef CAS PubMed
- K. S. Kim, S. R. Lim, S.-E. Kim, J. Y. Lee, C.-H. Chung, W.-S. Choe and P. J. Yoo, Sens. Actuators, B, 2017, 242, 1121–1128 CrossRef CAS
- H. Lee, C. Song, Y. S. Hong, M. S. Kim, H. R. Cho, T. Kang, K. Shin, S. H. Choi, T. Hyeon and D.-H. Kim, Sci. Adv., 2017, 3, e1601314 CrossRef PubMed
- H. Lee, Y. J. Hong, S. Baik, T. Hyeon and D.-H. Kim, Adv. Healthcare Mater., 2018, 7, 1701150 CrossRef PubMed
- J. Kim, A. S. Campbell and J. Wang, Talanta, 2018, 177, 163–170 CrossRef CAS PubMed
- M. Bariya, H. Yin, Y. Nyein and A. Javey, Nat. Electron., 2018, 1, 160–171 CrossRef
- M. Li, L. Wang, R. Liu, J. Li, Q. Zhang, G. Shi, Y. Li, C. Hou and H. Wang, Biosens. Bioelectron., 2021, 174, 112828 CrossRef CAS PubMed
- A. J. Bandodkar and J. Wang, Trends Biotechnol., 2014, 32(7), 363–371 CrossRef CAS PubMed
- J. K. Sim, S. Yoon and Y.-H. Cho, Sci. Rep., 2018, 8, 1181 CrossRef PubMed
- S. Y. Oh, S. Y. Hong, Y. R. Jeong, J. Yun, H. Park, S. W. Jin, G. Lee, J. H. Oh, H. Lee, S.-S. Lee and J. S. Ha, ACS Appl. Mater. Interfaces, 2018, 10, 13729–13740 CrossRef CAS PubMed
- S. Coyle, D. Morris, K.-T. Lau, D. Diamond, N. Taccini, D. Costanzo, P. Salvo, F. D. Francesco, M. G. Trivella, J.-A. Porchet and J. Luprano, 3rd International Conference on Pervasive Computing for Heathcare, 2009, pp. 1–6 Search PubMed.
- F. Mazzara, B. Patella, C. D'Agostino, M. G. Bruno, S. Carbone, F. Lopresti, G. Aiello, C. Torino, A. Vilasi, A. O'Riordan and R. Inguanta, Chemosensors, 2021, 9, 169 CrossRef CAS
- D. P. Rose, M. E. Ratterman, D. K. Griffin, L. Hou, N. Kelley-Loughnane, R. R. Naik, J. A. Hagen, I. Papautsky and J. C. Heikenfeld, IEEE Trans. Biomed. Eng., 2015, 62(6), 1457–1465 Search PubMed
- S. Emaminejad, W. Gao, E. Wu, Z. A. Davies, H. Y. Y. Nyein, S. Challa, S. P. Ryan, H. M. Fahad, K. Chen, Z. Shahpar, S. Talebi, C. Milla, A. Javey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(18), 4625–4630 CrossRef CAS PubMed
- N. B. Twine, R. M. Norton, M. C. Brothers, A. Hauke, E. F. Gomez and J. Heikenfeld, Lab Chip, 2018, 18, 2816–2825 RSC
- A. Koh, D. Kang, Y. Xue, S. Lee, R. M. Pielak, J. Kim, T. Hwang, S. Min, A. Banks, P. Bastien, M. C. Manco, L. Wang, K. R. Ammann, K.-I. Jang, P. Won, S. Han, R. Ghaffari, U. Paik, M. J. Slepian, G. Balooch, Y. Huang and J. A. Rogers, Sci. Transl. Med., 2016, 8, 366ra165 Search PubMed
- J. Choi, Y. Xue, W. Xia, T. R. Ray, J. T. Reeder, A. J. Bandodkar, D. Kang, S. Xu, Y. Huang and J. A. Rogers, Lab Chip, 2017, 17, 2572–2580 RSC
- M. Jose, G. Oudebrouckx, S. Bormans, P. Veske, R. Thoelen and W. Deferme, ACS Sens., 2021, 6, 896–907 CrossRef CAS PubMed
- X. He, T. Xu, Z. Gu, W. Gao, L.-P. Xu, T. Pan and X. Zhang, Anal. Chem., 2019, 91(7), 4296–4300 CrossRef CAS PubMed
- J. R. Sempionatto, J.-M. Moon and J. Wang, ACS Sens., 2021, 6(5), 1875–1883 CrossRef CAS PubMed
- S. K. Arya, G. Chornokur, M. Venugopal and S. Bhansali, Analyst, 2010, 135, 1941–1946 RSC
- R. M. Torrente-Rodríguez, J. Tu, Y. Yang, J. Min, M. Wang, Y. Song, Y. Yu, C. Xu, C. Ye, W. W. IsHak and W. Gao, Matter, 2020, 2(4), 921–937 CrossRef PubMed
- A. Kaushik, A. Yndart, R. D. Jayant, V. Sagar, V. Atluri, S. Bhansali and M. Nair, Int. J. Nanomed., 2015, 10, 677–685 CrossRef PubMed
- J. Zhao, Y. Lin, J. Wu, H. Y. Y. Nyein, M. Bariya, L.-C. Tai, M. Chao, W. Ji, G. Zhang, Z. Fan and A. Javey, ACS Sens., 2019, 4(7), 1925–1933 CrossRef CAS PubMed
- Y. Song, D. Mukasa, H. Zhang and W. Gao, Acc. Mater. Res., 2021, 2, 184–197 CrossRef CAS
- J. Heikenfeld, Electroanalysis, 2016, 28, 1242–1249 CrossRef CAS
- R. Ghaffari, D. S. Yang, J. Kim, A. Mansour, J. A. Wright, J. B. Model, D. E. Wright, J. A. Rogers and T. R. Ray, ACS Sens., 2021, 6(8), 2787–2801 CrossRef CAS PubMed
- Y. Yu, H. Y. Y. Nyein, W. Gao and A. Javey, Adv. Mater., 2019, 1902083 Search PubMed
- H. Y. Y. Nyein, L. C. Tai, Q. P. Ngo, M. Chao, G. B. Zhang, W. Gao, M. Bariya, J. Bullock, H. Kim, H. M. Fahad and A. Javey, ACS Sens., 2018, 3, 944–952 CrossRef CAS PubMed
- S. Anastasova, B. Crewther, P. Bembnowicz, V. Curto, H. M. Ip, B. Rosa and G.-Z. Yang, Biosens. Bioelectron., 2017, 93, 139–145 CrossRef CAS PubMed
- A. Martín, J. Kim, J. F. Kurniawan, J. R. Sempionatto, J. R. Moreto, G. Tang, A. S. Campbell, A. Shin, M. Y. Lee, X. Liu and J. Wang, ACS Sens., 2017, 2, 1860–1868 CrossRef PubMed
- D. I. Walsh, D. S. Kong, S. K. Murthy and P. A. Carr, Trends Biotechnol., 2017, 35(5), 383–392 CrossRef CAS PubMed
- K. Yamada, T. G. Henares, K. Suzuki and D. Citterio, Angew. Chem., Int. Ed., 2015, 54, 5294–5310 CrossRef CAS PubMed
- B. Gao, A. Elbaz, Z. He, Z. Xie, H. Xu, S. Liu, E. Su, H. Liu and Z. Gu, Adv. Mater. Technol., 2018, 3, 1700308 CrossRef
- R. Vinoth, T. Nakagawa, J. Mathiyarasu and A. M. V. Mohan, ACS Sens., 2021, 6, 1174–1186 CrossRef CAS PubMed
- O. Shido, N. Sugimoto, M. Tanabe and S. Sakurada, Am. J. Physiol., 1999, 276(4), R1095-101 Search PubMed
- T. Amano, S. Kai, M. Nakajima, T. Ichinose-Kuwahara, N. Gerrett, N. Kondo and Y. Inoue, Exp. Physiol., 2017, 102(2), 214–227 CrossRef PubMed
- L. B. Baker, Sports Med., 2017, 47, S111–S128 CrossRef PubMed
- N. A. S. Taylor and C. A. Machado-Moreira, Extrem. Physiol. Med., 2013, 2, 4 CrossRef PubMed
- C. J. Smith and G. Havenith, Med. Sci. Sports Exercise, 2012, 44(12), 2350–2361 CrossRef PubMed
- E. Sutterby, P. Thurgood, S. Baratchi, K. Khoshmanesh and E. Pirogova, Small, 2020, 2002515 CrossRef CAS PubMed
- G. Rabost-Garcia, J. Farré-Lladós and J. Casals-Terré, Membranes, 2021, 11, 150 CrossRef CAS PubMed
- A. S. Dolbashid, M. S. Mokhtar, F. Muhamad and F. Ibrahim, Bioinspired, Biomimetic Nanobiomater., 2018, 7, 53–64 CrossRef
- M. Nachman and S. E. Franklin, Tribol. Int., 2016, 97, 431–439 CrossRef
- A. Brueck, K. Bates, T. Wood, W. House, Z. Martinez, S. Peters, B. Root, K. Yelamarthi and T. Kaya, Electron, 2019, 8, 606 CrossRef CAS
- N. B. Twine, R. M. Norton, M. C. Brothers, A. Hauke, E. F. Gomez and J. Heikenfeld, Lab Chip, 2018, 18, 2816–2825 RSC
- L. Hou, J. Hagen, X. Wang, I. Papautsky, R. Naik, N. Kelley-Loughnane and J. Heikenfeld, Lab Chip, 2013, 13, 1868–1875 RSC
- Y. Wan, Y. Su, X. Zhu, G. Liu and C. Fan, Biosens. Bioelectron., 2013, 47, 1–11 CrossRef CAS PubMed
- T. Mérian, F. He, H. Yan, D. Chu, J. N. Talbert, J. M. Goddard and S. R. Nugen, Colloids Surf., A, 2012, 414, 251–258 CrossRef
- F. He and S. R. Nugen, Microfluid. Nanofluid., 2014, 16, 879–886 CrossRef CAS
- C. K. W. Koo, F. He and S. R. Nugen, Analyst, 2013, 138, 4998–5004 RSC
- J. Chen, Y. Zhou, D. Wang, F. He, V. M. Rotello, K. R. Carter, J. J. Watkins and S. R. Nugen, Lab Chip, 2015, 15, 3086–3094 RSC
- A. R. Naik, B. Warren, A. Burns, R. Lenigk, J. Morse, A. Alizadeh and J. J. Watkins, Microfluid. Nanofluid., 2021, 25(2), 1–9 Search PubMed
- M. Lake, C. Narciso, K. Cowdrick, T. Storey, S. Zhang, J. Zartman and D. Hoelzle, Protoc. Exch., 2015, 10, 1–27 Search PubMed
- S. Chan and M. Debono, Ther. Adv. Endocrinol. Metab., 2010, 1(3), 129–138 CrossRef PubMed
- M. Aller-Pellitero, J. Fremeau, R. Villa, G. Guirado, B. Lakard, J.-Y. Hihn and F. J. del Campo, Sens. Actuators, B, 2019, 290, 591–597 CrossRef CAS
- F. Ricci and G. Palleschi, Biosens. Bioelectron., 2005, 21, 389–407 CrossRef CAS PubMed
- G. A. Mabbott, J. Chem. Educ., 1983, 60(9), 697–702 CrossRef CAS
- D. Bruen, C. Delaney, L. Florea and D. Diamond, Sensors, 2017, 17, 1866 CrossRef PubMed
- E. B. Secor, B. Y. Ahn, T. Z. Gao, J. A. Lewis and M. C. Hersam, Adv. Mater., 2015, 27, 6683–6688 CrossRef CAS PubMed
- E. B. Secor, T. Z. Gao, A. E. Islam, R. Rao, S. G. Wallace, J. Zhu, K. W. Putz, B. Maruyama and M. C. Hersam, Chem. Mater., 2017, 29, 2332–2340 CrossRef CAS
- V. Forsberg, R. Zhang, H. Andersson, J. Bäckström, C. Dahlström, M. Norgren, B. Andres and H. Olin, J. Imaging Sci. Technol., 2016, 60(4), 040405-7 CrossRef
- P. Khullar, J. V. Badilla and R. G. Kelly, ECS Electrochem. Lett., 2015, 4(10), C31–C33 CrossRef CAS
- N. Ferrell, J. Woodard and D. Hansford, Biomed. Microdevices, 2007, 9, 815–821 CrossRef CAS PubMed
- K. Haubert, T. Drier and D. Beebe, Lab Chip, 2006, 6, 1548–1549 RSC
- P. K. Vabbina, A. Kaushik, N. Pokhrel, S. Bhansali and N. Pala, Biosens. Bioelectron., 2015, 63, 124–130 CrossRef CAS PubMed
- C. Tlili, N. V. Myung, S. Shetty and A. Mulchandani, Biosens. Bioelectron., 2011, 26(11), 4382–4386 CrossRef CAS PubMed
- S. K. Tuteja, C. Ormsby and S. Neethirajan, Nano-Micro Lett., 2018, 10, 41 CrossRef PubMed
- K. Wilke, M. L. Terstegen and S. S. Biel, Int. J. Cosmet. Sci., 2007, 29, 169–179 CrossRef CAS PubMed
- K. Sato, W. H. Kang, K. Saga and K. T. Sato, J. Am. Acad. Dermatol., 1989, 20(4), 537–563 CrossRef CAS PubMed
-
T. M. Mauro, Fitzpatrick’s Dermatology in General Medicine, McGraw Hill, 8th edn, 2012 Search PubMed
- Z. Sonner, E. Wilder, J. Heikenfeld, G. Kasting, F. Beyette, D. Swaile, F. Sherman, J. Joyce, J. Hagen, N. Kelley-Loughnane and R. T. Naik, Biomicrofluidics, 2015, 9(3), 031301 CrossRef CAS PubMed
-
L. G. Leal, Advanced Transport Phenomena: Fluid Mechanics and Convective Transport Processes, Cambridge University Press, Cambridge, 2007, vol. 7 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Inkjet printer and ink; electrode SEM images; EDS spectra; sensor mask dimensions; synthetic skin assembly; glucose sensor characterization; sensor and skin assembly; skin mask dimensions; skin sweat production. See DOI: 10.1039/d1lc00633a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |