
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decarboxylative oxygenation of carboxylic acids with O2via a non-heme manganese catalyst†
Renpeng
Guan
,
Elliot L.
Bennett
 ,
Zhiliang
Huang
* and
Jianliang
Xiao
*
,
Zhiliang
Huang
* and
Jianliang
Xiao
*
Department of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: Z.Huang49@liverpool.ac.uk; J.Xiao@liverpool.ac.uk
First published on 10th March 2022
Abstract
Reported here is a new protocol for the decarboxylative oxygenation of carboxylic acids using a non-heme manganese catalyst under blue light irradiation with O2 as the sole oxidant. Featuring mild reaction conditions, the protocol allows readily available carboxylic acids to be converted into a wide variety of valuable aldehydes, ketones and amides. Mechanistic studies indicate that the decarboxylation and oxygenation involves the formation of active Mn-oxygen species.
Introduction
Carboxylic acids are one of the most important feedstocks in organic, polymer and material synthesis.1,2 They are widely available in nature and industry, inexpensive, stable, and non-toxic in general.3–5 Moreover, many carboxylic acids, such as amino acids, fatty acids and sugar acids, can be derived directly from natural resources. Therefore, a great number of transformations of carboxylic acids have been reported in the past few decades.6–9 In particular, the construction of useful carbon–carbon and carbon–heteroatom bonds through decarboxylation has received significant attention.10–24 However, direct decarboxylative oxygenation of carboxylic acids to the corresponding carbonyl compounds, i.e. aldehydes and ketones, with high efficiency and wide substrate scope under mild conditions remains limited. Aldehydes and ketones are the fundamental building blocks of organic chemistry,25–30 and they can be synthesized via a variety of methods, such as carbonylation of alkenes and oxidation of hydrocarbons.31,32 However, these methods often rely on harsh conditions such as high temperature, high pressure, and toxic reagents. For instance, whilst hydroformylation is widely used in industry to prepare aldehydes, the cobalt catalyst necessitates >200 atm syngas and >100 °C temperature and although much milder conditions are employed for Rh-based catalysts, the metal is expensive and toxic.33 Therefore, exploring rapid synthesis of aldehydes and ketones under mild conditions with cheap substrates and catalysts is a worthy endeavor. Given the easy availability of various carboxylic acids, they are well suited for this endeavor.Decarboxylative oxygenation of carboxylic acids is a challenging reaction, requiring a catalyst not only capable of promoting decarboxylation but also oxidation. So far, there have been only three reports, demonstrating the reaction via photoredox catalysis using molecular oxygen (O2) under mild conditions. Two of these require precious Ru or Ir photocatalysts,34,35 while the third produces a mixture of alcohols and ketones/aldehydes as the products (Scheme 1a).36 Other methods have been reported; however, they make use of stoichiometric strong oxidants, e.g. NaIO4,37n-Bu4NIO4,38 HgF2,39 Pb(OAc)4,40 PhI(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 and K2S2O842 (Scheme 1b), or rely on a high reaction temperature (Scheme 1c).25,43 Herein, we report a highly selective decarboxylative oxygenation of carboxylic acids to aldehydes or ketones by O2 (1 atm), promoted by a non-heme Mn(II) catalyst and visible light under mild conditions (Scheme 1d).
41 and K2S2O842 (Scheme 1b), or rely on a high reaction temperature (Scheme 1c).25,43 Herein, we report a highly selective decarboxylative oxygenation of carboxylic acids to aldehydes or ketones by O2 (1 atm), promoted by a non-heme Mn(II) catalyst and visible light under mild conditions (Scheme 1d).
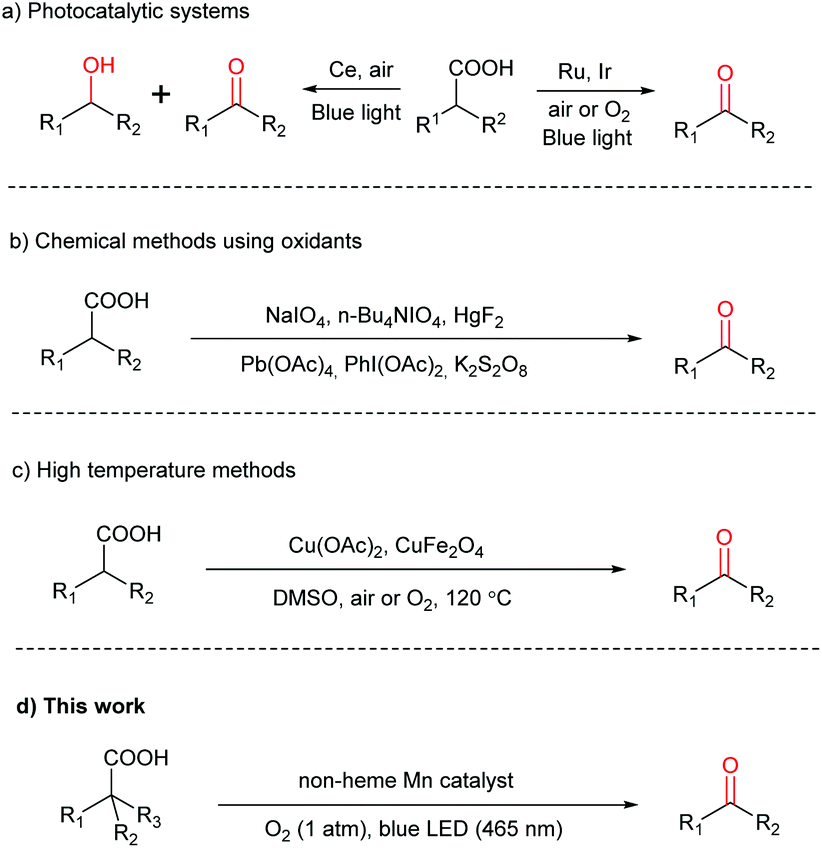 | ||
| Scheme 1 Decarboxylative oxygenation of carboxylic acids. | ||
The use of benign and inexpensive O2 for selective oxidation with first-row, biologically relevant metal complexes as catalysts remains a significant challenge for the synthetic chemist.44–46 Over the past a few decades, some biomimetic Fe and Mn complexes bearing heme and non-heme polydentate ligands have been developed to catalyze the oxidation of hydrocarbons, alcohols as well as carboxylic acids with O2.37,44,47–51 However, significant drawbacks remain in most cases, e.g. the need for stoichiometric co-reductants and unsatisfactory selectivity due to poorly controlled radical-type pathways. To date, only a few biomimetic metal complexes are known that can catalyze the aerobic oxidation in a controlled manner without using a co-reductant.52–57 Following on from our recent study of aerobic cleavage of alkenes with a non-heme Mn(II) complex,58 we investigated the decarboxylative oxygenation of carboxylic acids to aldehydes/ketones by O2 with this and related complexes. Our findings are detailed below.
Results and discussion
We commenced our study by examining the decarboxylative oxygenation of 4-methoxybenzeneacetic acid (1a) with O2 (1 atm) (Table 1). Based on our previous work,58 the photocatalytic system comprised of Mn(OTf)2 and L4 was initially tested, as it was proven to be effective for O2 activation in the presence of blue light. To our delight, the desired oxygenation product 2a could be obtained in 90% yield (Table 1, entry 6). [Mn(dtbpy)2(OTf)2] formed in situ is likely to be the catalytic species, as the isolated [Mn(dtbpy)2(OTf)2] complex displayed a similar activity (Table 1, entry 1). Unsurprisingly, the ligand-free Mn(OTf)2, which is less likely to produce activated oxygen species from O2 (E0Mn(III)/Mn(II) = 1.56 V/SHE, E0O2/H2O = 0.82 V/SHE, neutral conditions), showed no catalytic activity for the target reaction (Table 1, entry 2). The effect of ligand was further explored by testing a range of substituted bipyridines (L1–L8), revealing L4 to give the best yield (Table 1, entries 3–10). Meanwhile, evaluation of the solvents showed that the aerobic oxygenation reaction presented the highest reactivity in acetonitrile (ESI, Table S1†). The combinations of L4 with other metal salts, such as Cu(OTf)2, Fe(OTf)2, and CoCl2, were ineffective, affording much lower yields of the target product (Table 1, entries 11–13). In addition, MnCl2 is less effective than Mn(OTf)2, as can be seen in Table 1, entry 14. As is clear, both O2 and blue light play important roles in this transformation; in their absence, no target product was observed (Table 1, entry 15, and Fig. S3 in ESI†). The screening established the following optimized conditions: Mn(OTf)2 (5 mol%), L4 (10 mol%) as ligand, O2 as oxidant in CH3CN at 45 °C with blue light irradiation.|
|
|||
|---|---|---|---|
| Entry | [M] | Ligand | Yield (%) |
a Reaction conditions: 1a (0.5 mmol), [M] (5 mol%), ligand (10 mol%), CH3CN (2 mL), blue light (465 nm), 45 °C, O2 (1 atm), 12 h.
b NMR yields, determined using mesitylene (20 μL) as internal standard.
c N2 (1 atm).
d Air instead of O2. 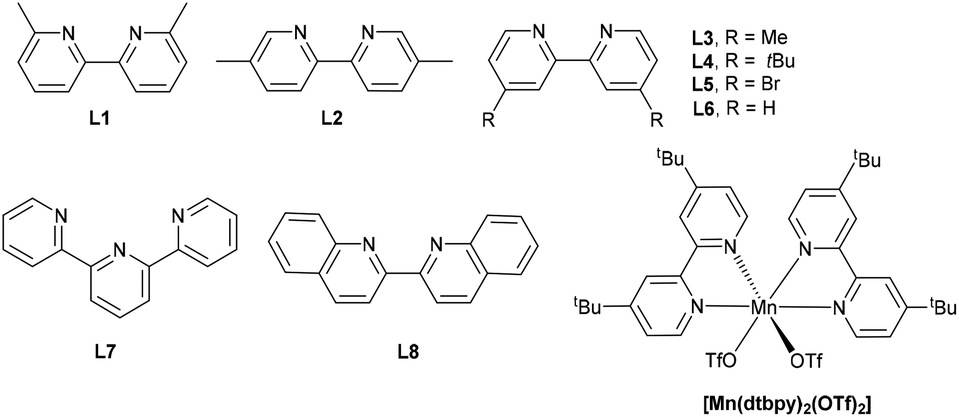
|
|||
| 1 | [Mn(dtbpy)2(OTf)2] | — | 87 |
| 2 | Mn(OTf)2 | — | 0 |
| 3 | Mn(OTf)2 | L1 | 33 |
| 4 | Mn(OTf)2 | L2 | 46 |
| 5 | Mn(OTf)2 | L3 | 61 |
| 6 | Mn(OTf)2 | L4 | 90 |
| 7 | Mn(OTf)2 | L5 | 41 |
| 8 | Mn(OTf)2 | L6 | 64 |
| 9 | Mn(OTf)2 | L7 | 14 |
| 10 | Mn(OTf)2 | L8 | 41 |
| 11 | Cu(OTf)2 | L4 | 4 |
| 12 | Fe(OTf)2 | L4 | 16 |
| 13 | CoCl2 | L4 | 42 |
| 14 | MnCl2 | L4 | 53 |
| 15c | [Mn(dtbpy)2(OTf)2] | — | 0 |
| 16d | Mn(OTf)2 | L4 | 62 |
Under the optimized reaction conditions, a series of benzylic carboxylic acids were first examined to test the generality of this decarboxylative oxygenation protocol. As shown in Scheme 2, phenyl acetic acids bearing electron-withdrawing or electron-donating groups on the phenyl ring are all suitable substrates (2a–2o), affording the desirable aldehyde products in good to excellent yields. During our investigation, we noticed that phenyl acetic acids bearing electronic-withdraw groups were usually less reactive. For example, para-F-substituted acid 1d and para-CF3-substituted acid 1e showed low reactivity for the oxidation under above standard conditions, giving the corresponding aldehydes in 35% and 17% yields, respectively. However, the oxidation was improved by introducing a catalytic amount of sodium acetate as an additive (Scheme 2. See ESI for the detailed reaction optimization, Table S2†). 2-Naphthyl acetic acid and S-heterocycle acetic acid can be oxidized selectively to afford 2p and 2q in moderate yields, respectively. Secondary benzylic carboxylic acids are also efficiently converted to the corresponding ketone products in good to excellent yields under the standard conditions (2r–2u). It is worth noting that a range of amino acids including a dipeptide can be oxidized to the corresponding amide products in a highly selective manner, showing potential applications in bio-conjugate chemistry (2v–2y).
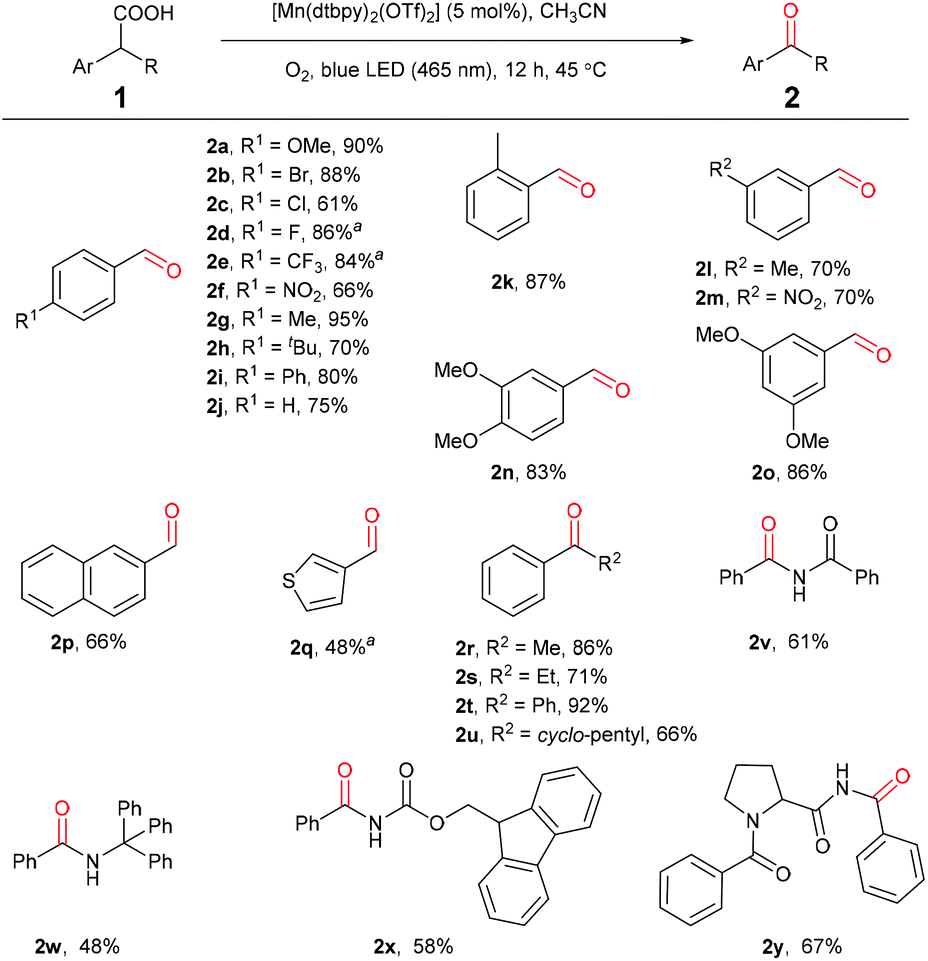 | ||
| Scheme 2 Oxidation of mono- and di-substituted benzylic carboxylic acids. Reaction conditions: 1 (0.5 mmol), [Mn(dtbpy)2(OTf)2] (5 mol%) (in situ prepared), CH3CN (2 mL), blue light (465 nm), 45 °C, O2 (1 atm), 12 h. Isolated yields are given. aCH3COONa (30 mol%) was added. | ||
Notably, a variety of drug molecules with functionalized aryl acetic acid scaffolds were also selectively oxidized by O2 under the conditions developed. As can be seen in Scheme 3, the oxidation of naproxen, ibuprofen, flubiprofen, ketoprofen, ioxoprofen, isoxepac and indomethacin all proceeded successfully, affording the corresponding aldehydes or ketones in good yields (3a–3g). Moreover, this photo-Mn protocol showed its ability in late-stage decarboxylative oxygenation of vitamin E, affording the aldehyde product 3h in 74% yield. The relatively active benzylic C–H bonds, tertiary C–H bonds, and indole ring, which are prone to oxidation, remained intact, showing the high chemoselectivity of this oxidation protocol. The success in late-stage decarboxylative oxygenation of drugs or natural products demonstrates the practical applicability of this oxidation protocol in accessing new bioactive molecules.
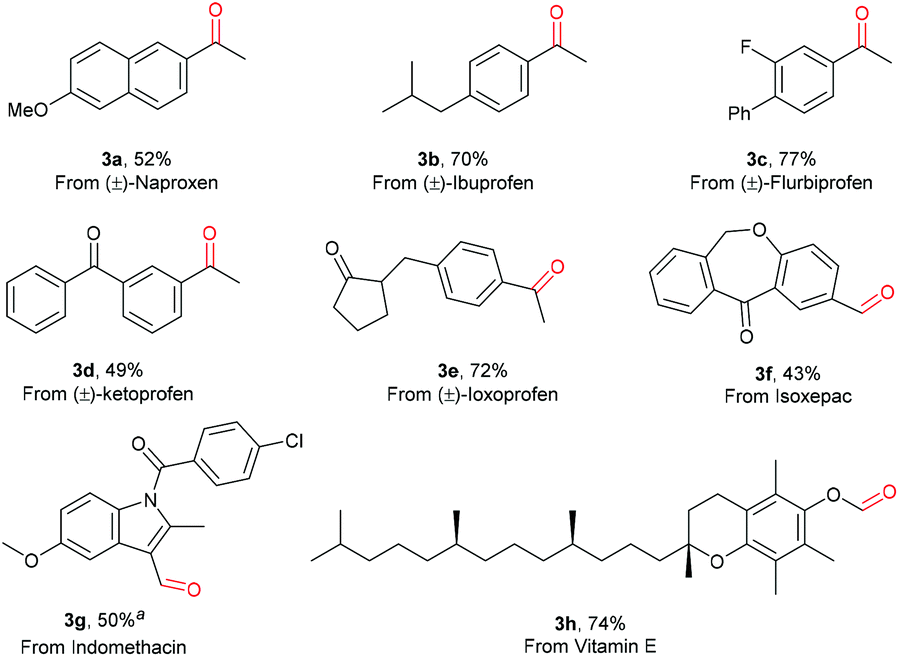 | ||
| Scheme 3 Oxidation of pharmaceutical molecules. Reaction conditions: Carboxylic acid (0.5 mmol), [Mn(dtbpy)2(OTf)2] (5 mol%), CH3CN (2 mL), blue light (465 nm), 45 °C, O2 (1 atm), 12 h. Isolated yields are given. aCH3COONa (30 mol%) was added. | ||
The more challenging enoic acids and aliphatic carboxylic acids were also tested.59,60 As can be seen in Scheme 4, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in enoic acids, usually sensitive to oxidation conditions, remained intact during the oxidation (5a). Notably, long-chain and cyclic aliphatic acids with different functionalities, including ether, carbamate, and amide, are suitable to give the corresponding carbonyl products with blue or UV light irradiation (5b–5e), highlighting the high chemoselectivity and hence great application potential of this method. Secondary aliphatic acids without other functional groups were also oxidatively decarboxylated to furnish the target carbonyls in 50%–63% yields (5f–5h) under the irradiation of UV-light. Notably, the primary stearic acid and oleic acid could be oxidized to the corresponding aldehydes; however, low yields, as well as low conversions were observed (5i and 5j). In comparison with Macmillan's work in 2019,35 this protocol shows a wider substrate scope. Not only can the cyclic aliphatic acids be oxidized selectively, but also long-chain fatty acids, saturated and unsaturated, could be converted, which are usually challenging substrates.61
C double bonds in enoic acids, usually sensitive to oxidation conditions, remained intact during the oxidation (5a). Notably, long-chain and cyclic aliphatic acids with different functionalities, including ether, carbamate, and amide, are suitable to give the corresponding carbonyl products with blue or UV light irradiation (5b–5e), highlighting the high chemoselectivity and hence great application potential of this method. Secondary aliphatic acids without other functional groups were also oxidatively decarboxylated to furnish the target carbonyls in 50%–63% yields (5f–5h) under the irradiation of UV-light. Notably, the primary stearic acid and oleic acid could be oxidized to the corresponding aldehydes; however, low yields, as well as low conversions were observed (5i and 5j). In comparison with Macmillan's work in 2019,35 this protocol shows a wider substrate scope. Not only can the cyclic aliphatic acids be oxidized selectively, but also long-chain fatty acids, saturated and unsaturated, could be converted, which are usually challenging substrates.61
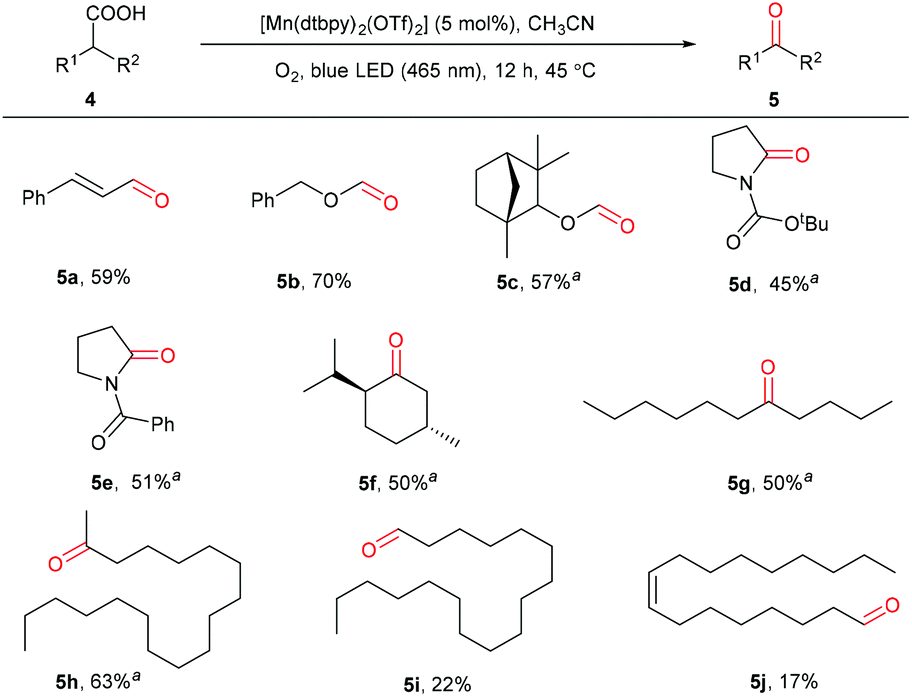 | ||
| Scheme 4 Oxidation of aliphatic acids. Reaction conditions: 4 (0.5 mmol), [Mn(dtbpy)2(OTf)2] (5 mol%), CH3CN (2 mL), blue LED light (465 nm), 45 °C, O2 atmosphere (1 atm), 12 h. Isolated yields are given. aDCE/CH3CN (1:1, 2 mL), ultraviolet light (365 nm). | ||
Although the detailed mechanism of the decarboxylative oxygenation reaction is unclear, a series of experiments were performed to shed light on possible reaction pathways. As reported by Boger's group,62 singlet oxygen (1O2) can efficiently transfer pyrrole-2-carboxylic acids to the corresponding ketones in the presence of rose bengal. Therefore, it is important to determine whether 1O2 plays a role in the photo-Mn enabled oxidative decarboxylation. As shown in Table 2, a range of well-known photosensitizers (Table 2, entries 1–4), such as Eosin Y disodium salt, Ru(bpy)3, Ir(dFppy)3 and rose bengal,58,63 which can generate 1O2 under blue light, were used individually as replacement catalysts for the decarboxylative oxygenation of 1a under standard reaction conditions. However, these photosensitizers showed a significantly lower catalytic effect. Furthermore, when an 1O2 trap, 9,10-diphenylanthracene (DPA) which is known to react rapidly with 1O2 to give an endoperoxide product (k ≈ 1.3 × 106 M−1 s−1),64 was added to the oxidative decarboxylation of 1a under the standard conditions (Table 2, entry 5), 2a was obtained in 70% yield, and no endoperoxide was detected. These observations appear to rule out the involvement of 1O2 in the reaction pathway.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
| a Reaction conditions: Acid (0.5 mmol), catalyst (5 mol%), CH3CN (2 mL), blue light (465 nm), 45 °C, O2 (1 atm), 12 h. b NMR yields, determined using mesitylene as internal standard. c Conversion was observed under the conditions reported in ref. 35 (see Scheme S2, ESI†). d DPA (0.1 mmol) was added. | ||
| 1 | Eosin Y disodium salt | 37 |
| 2 | [Ru(bpy)3·6H2O] | 19 |
| 3 | [Ir(dFppy)3] | 0c |
| 4 | Rose bengal | 4 |
| 5d | [Mn(dtbpy)2(OTf)2] | 70 |

Previous studies on decarboxylative oxygenation of phenylacetic acids by Mn-based enzymes indicate that the hydroxylation of C–H bond in the benzylic position is the initiation step of this oxidation reaction, which affords 2-hydroxy-2-phenylacetic acid as the key intermediate for the further oxidative decarboxylation process.65–67 Knowing this, several control experiments were performed to determine whether a similar hydroxylation process occurs as the key step. Firstly, the potential intermediate 2-hydroxy-2-phenylacetic acid (7) was subjected to the standard reaction conditions, affording 2j in 93% yield (Scheme 5, eqn (1)). This result indicates that 7 could be involved in the oxidative decarboxylation reaction under the current conditions. However, when the corresponding benzylic C–H bonds were substituted by two alkyl or phenyl groups, the desired decarboxylative oxygenation proceeded smoothly as well (Scheme 5, eqn (2)–(4)). In all cases, the ketone products were obtained, apparently via a C–C bond cleavage process due to the formation of the strong CO bond. This indicates the involvement of a radical fragmentation pathway. Additionally, no reactions happened when ethyl 2-phenylacetate (8) was employed (Scheme 5, eqn (5)). Meanwhile, 2r was afforded when (1-hydroperoxyethyl)benzene (9) was employed (Scheme 5, eqn (6)). These observations indicate that the acid moiety is involved and peroxide intermediates may be formed in the decarboxylative oxygenation reaction.
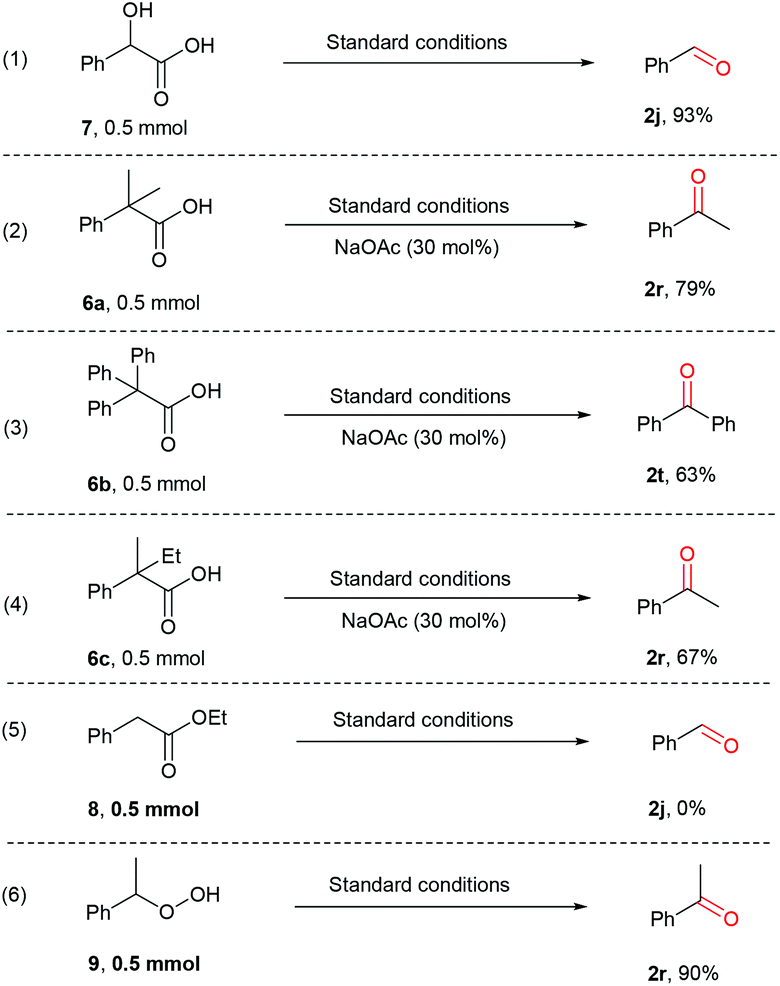 | ||
| Scheme 5 Control experiments to shed light on the mechanism. Conditions: Substrate (0.5 mmol), [Mn(dtbpy)2(OTf)2] (5 mol%), CH3CN (2 mL), blue light (465 nm), 45 °C, O2 (1 atm), 12 h, CH3COONa (30 mol%) where applicable; NMR yields, determined using mesitylene as internal standard. | ||
Further insight was obtained by following the kinetic isotope effect (KIE) (Scheme 6). Parallel reactions using acids 1a and 1a-d2 afforded the initial rates kHobs = 9.3 and kDobs = 8.8 mol L−1 h−1, respectively. Clearly, the cleavage of the C–H bond on the benzylic position is less likely to be involved in the turnover limiting step, considering the KIE value of ca. 1.1. This result lends further support to the notion that the oxidative decarboxylation is not initiated by the benzylic C–H bond hydroxylation.
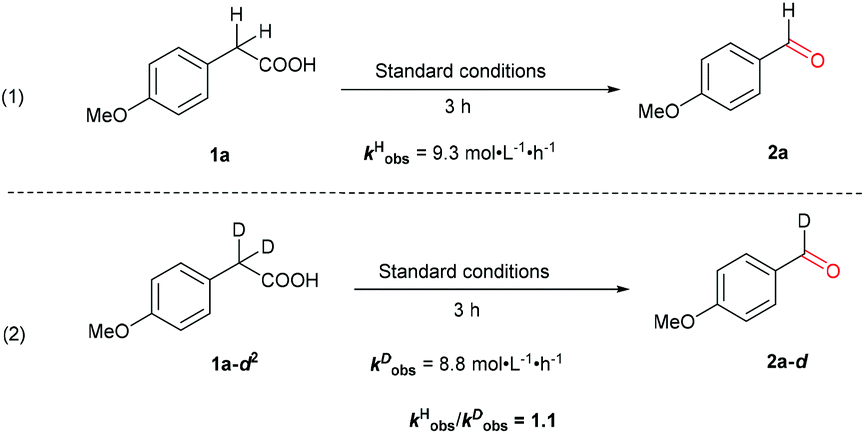 | ||
| Scheme 6 Kinetic isotope effect studied under the optimized conditions. | ||
Subsequently, the reaction of carboxylic acids with the [Mn(dtbpy)2(OTf)2] complex was studied with UV-Vis spectroscopy. As shown in Fig. 1, there is an obvious decrease of the absorbance of 1a at 230 nm when 1 equivalent of [Mn(dtbpy)2(OTf)2] was added. In the meantime, compared with that of [Mn(dtbpy)2(OTf)2], the spectrum of the mixture of [Mn(dtbpy)2(OTf)2] and 1a showed a significant absorption decrease at around 295 nm and 306 nm. These observations indicate that [Mn(dtbpy)2(OTf)2] reacts with 1a to form a new Mn(II) species. Further structural information of the new Mn(II) species was obtained by HRMS, which suggests that the carboxylate-coordinated [Mn(dtbpy)2(1a-H)(OTf)] is likely to be formed by losing a HOTf from the parent complex. Fig. 2 shows the experimental and calculated mass spectra of the molecular ion resulting from the loss of a −OTf fragment from the [Mn(dtbpy)2(1a-H)(OTf)] complex.
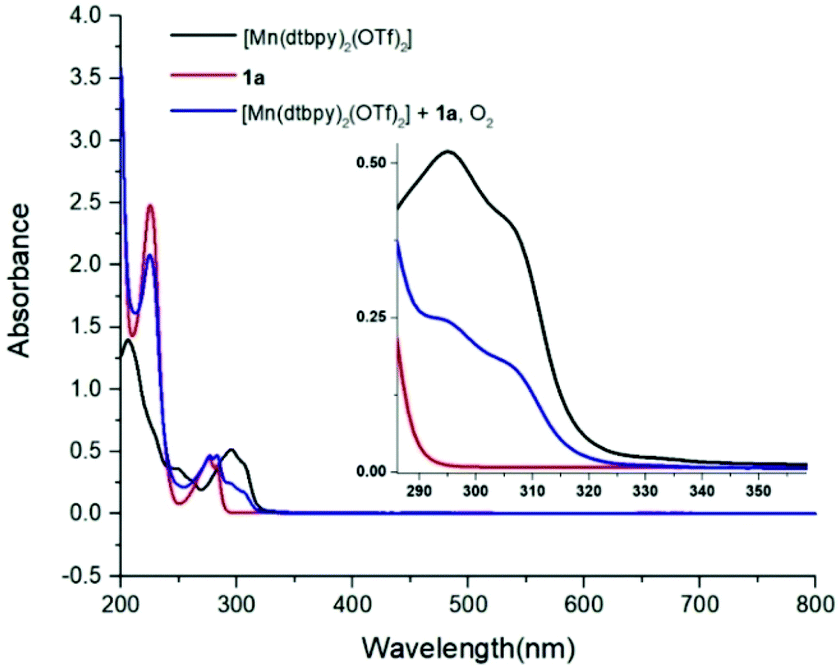 | ||
| Fig. 1 UV-Vis spectra of [Mn(dtbpy)2(OTf)2], 1a and the mixture of [Mn(dtbpy)2(OTf)2] and 1a without blue light irradiation (concentration of [Mn(dtbpy)2(OTf)2] and 1a: 25 μM in CH3CN). | ||
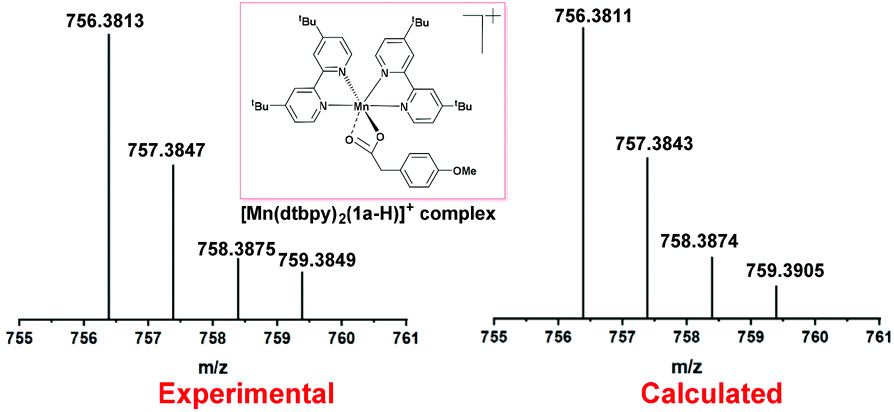 | ||
| Fig. 2 HRMS spectrum of [Mn(dtbpy)2(1a-H)(OTf)] formed in the in situ reaction of [Mn(dtbpy)2(OTf)2] with 1a. | ||
We envisioned that a Mn-peroxide intermediate may be formed by the reaction of [Mn(dtbpy)2(1a-H)(OTf)] with O2 in the decarboxylative oxidation under blue light.34,67 Thus, a mixture of [Mn(dtbpy)2(OTf)2] and 1r, which was expected to afford [Mn(dtbpy)2(1r-H)(OTf)], was followed by UV-Vis spectroscopy with or without blue light irradiation. As shown in Fig. 3, an obvious absorption at ca. 685 nm was observed, when the mixture was irradiated by blue light under O2 (Fig. 3, green dash line). Notably, a similar absorption band was also found in the mixture of [Mn(dtbpy)2(OTf)2] and 9 without blue light irradiation under N2, which increased over time and then reached saturation before beginning to decrease (Fig. S4 in ESI†). Finally, the product 2r was formed in 60% yield after 60 min (Scheme S1†). These observations indicate that a peroxide intermediate may be involved and complexed with the catalyst in the process of decarboxylative oxidation. Comparing with the previous literature, the absorption at 685 nm is likely to arise from a Mn-peroxide species.68–70
 | ||
| Fig. 3 In situ UV-Vis monitoring of the mixture of [Mn(dtbpy)2(OTf)2] and 9 and the UV-vis spectra of the mixture of [Mn(dtbpy)2(OTf)2] and 1r with blue light irradiation for 2 h (concentration of each compound: 25 μM in CH3CN). | ||
Based on the observations above and previous literature,25,36 a plausible mechanism is proposed by taking the oxidation of phenylacetic acid (1j) as an example. As shown in Scheme 7, 1j firstly reacts with the Mn(II) precatalyst to form the intermediate A, which is subsequently oxidized by O2 to afford the key species B under blue light irradiation.44,71–73 The unstable superoxide radical attacks the benzylic carbon of the coordinated acid driven by the release of CO2, resulting in the formation of the Mn(II)-peroxide C. Further decomposition of the unstable species C produces the carbonyl product and a Mn(II)-OH species D, which reacts with phenylacetic acid, regenerating A.
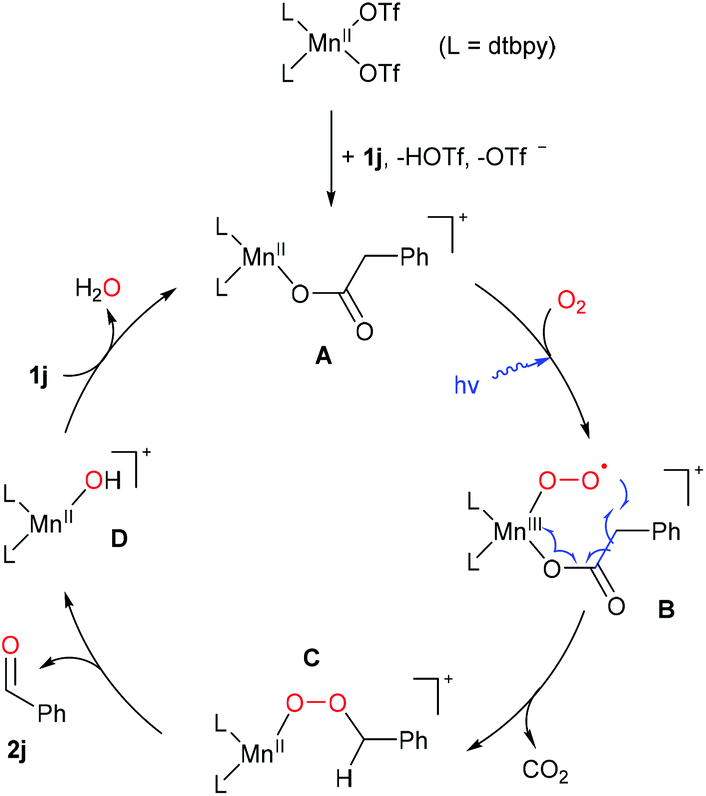 | ||
| Scheme 7 A plausible mechanism of decarboxylative oxygenation of carboxylic acids (possible coordination of solvent or other molecules is omitted). | ||
Conclusions
In conclusion, a selective, practical decarboxylative oxygenation protocol has been developed, which makes use of a non-heme Mn(II) complex as the catalyst and molecular oxygen as the oxidant under blue light irradiation. With this protocol, readily available carboxylic acids, including benzylic acids, amino acids, and pharmaceutical molecules, can be easily converted to a wide range of valuable aldehydes, ketones, and amides in good yields under mild conditions. Further mechanistic studies and applications of this catalytic system are underway in our laboratory and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the China Scholarship Council and University of Liverpool for a PhD studentship (RPG), the EPSRC (EP/R009694/1) for funding, and the Analytical Services of the Department of Chemistry of the University of Liverpool for product analysis.Notes and references
- M. Simonetta, S. Carra, L. Eberson, J. Koskikallio, R. Sneeden, V. Kucherov, L. Yanovskaya, L. Bergelson, M. Shemyakin and H. Kwart, Carboxylic acids and esters, Wiley, 1969 Search PubMed.
- M. Večeřa and J. Gasparič, Detection and Identification of Organic Compounds, Springer, 1971, pp. 247–293 Search PubMed.
- L. J. Goossen, N. Rodriguez and K. Goossen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef CAS PubMed.
- J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- F. Julia-Hernandez, M. Gaydou, E. Serrano, M. van Gemmeren and R. Martin, Top. Curr. Chem., 2016, 374, 45 CrossRef PubMed.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- A. Tortajada, M. Borjesson and R. Martin, Acc. Chem. Res., 2021, 54, 3941–3952 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522–6526 CrossRef CAS PubMed.
- N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- M. Pichette Drapeau and L. J. Goossen, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef CAS PubMed.
- S. Bhadra, W. I. Dzik and L. J. Goossen, J. Am. Chem. Soc., 2012, 134, 9938–9941 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- S. Ventre, F. R. Petronijevic and D. W. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS PubMed.
- A. Noble, S. J. McCarver and D. W. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- Z. Zuo and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- F. Bu, L. Lu, X. Hu, S. Wang, H. Zhang and A. Lei, Chem. Sci., 2020, 11, 10000–10004 RSC.
- Y. Yuan, Z. Zhou, L. Zhang, L. S. Li and A. Lei, Org. Lett., 2021, 23, 5932–5936 CrossRef CAS PubMed.
- Y. Liang, F. Strieth-Kalthoff, P. Bellotti and F. Glorius, Chem. Catal., 2021, 1, 1427–1436 CrossRef.
- C. Zheng, Y. Wang, Y. Xu, Z. Chen, G. Chen and S. H. Liang, Org. Lett., 2018, 20, 4824–4827 CrossRef CAS PubMed.
- L. Wang, T. Wang, G.-J. Cheng, X. Li, J.-J. Wei, B. Guo, C. Zheng, G. Chen, C. Ran and C. Zheng, ACS Catal., 2020, 10, 7543–7551 CrossRef CAS.
- Q. Feng and Q. Song, J. Org. Chem., 2014, 79, 1867–1871 CrossRef CAS PubMed.
- M. Liu, H. Wang, H. Zeng and C. J. Li, Sci. Adv., 2015, 1, e1500020 CrossRef PubMed.
- J. Li, H. Wang, Z. Qiu, C. Y. Huang and C. J. Li, J. Am. Chem. Soc., 2020, 142, 13011–13020 CrossRef CAS PubMed.
- L. Lv, D. Zhu and C. J. Li, Nat. Commun., 2019, 10, 715 CrossRef PubMed.
- L. Lv, D. Zhu, J. Tang, Z. Qiu, C.-C. Li, J. Gao and C.-J. Li, ACS Catal., 2018, 8, 4622–4627 CrossRef CAS.
- H. Wang, X. J. Dai and C. J. Li, Nat. Chem., 2017, 9, 374–378 CrossRef CAS PubMed.
- B. Cornils, W. A. Herrmann and M. Rasch, Angew. Chem., Int. Ed. Engl., 1994, 33, 2144–2163 CrossRef.
- R. Bruckner, Advanced organic chemistry: reaction mechanisms, Elsevier, 2001 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial organic chemistry, John Wiley & Sons, 2008 Search PubMed.
- Y. Sakakibara, P. Cooper, K. Murakami and K. Itami, Chem. – Asian J., 2018, 13, 2410–2413 CrossRef CAS PubMed.
- T. M. Faraggi, W. Li and D. W. C. MacMillan, Isr. J. Chem., 2019, 60, 410–415 CrossRef.
- S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 CrossRef CAS PubMed.
- M. Araghi and F. Bokaei, Polyhedron, 2013, 53, 15–19 CrossRef CAS.
- S. Tangestaninejad and V. Mirkhani, J. Chem. Res., 1998, 32, 820–821 RSC.
- S. Farhadi, P. Zaringhadam and R. Z. Sahamieh, Tetrahedron Lett., 2006, 47, 1965–1968 CrossRef CAS.
- Y. Pocker and B. C. Davis, J. Am. Chem. Soc., 1973, 95, 6216–6223 CrossRef CAS PubMed.
- K. Gholamreza and A. Roxana, Chem. Res. Chin. Univ., 2008, 24, 464–468 CrossRef.
- T. B. Mete, T. M. Khopade and R. G. Bhat, Tetrahedron Lett., 2017, 58, 2822–2825 CrossRef CAS.
- T. Rahman, G. Borah and P. K. Gogoi, Catal. Lett., 2020, 150, 2267–2272 CrossRef CAS.
- S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS PubMed.
- B. Zhu, T. Shen, X. Huang, Y. Zhu, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 11028–11032 CrossRef CAS PubMed.
- Y. F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed.
- S. Paria, L. Que Jr. and T. K. Paine, Angew. Chem., Int. Ed., 2011, 50, 11129–11132 CrossRef CAS PubMed.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS PubMed.
- K. A. Prokop and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 8014–8017 CrossRef CAS PubMed.
- K. A. Prokop, H. M. Neu, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2011, 133, 15874–15877 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. A. Prokop and D. P. Goldberg, J. Am. Chem. Soc., 2011, 133, 1859–1869 CrossRef CAS PubMed.
- S. Riaño, D. Fernández and L. Fadini, Catal. Commun., 2008, 9, 1282–1285 CrossRef.
- S. Barroso, G. Blay, I. Fernández, J. R. Pedro, R. Ruiz-García, E. Pardo, F. Lloret and M. C. Muñoz, J. Mol. Catal. A: Chem., 2006, 243, 214–220 CrossRef CAS.
- M. Guo, J. Zhang, L. Zhang, Y. M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2021, 143, 18559–18570 CrossRef CAS PubMed.
- M. Guo, Y. M. Lee, S. Fukuzumi and W. Nam, Coord. Chem. Rev., 2021, 435, 213807 CrossRef CAS.
- G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513–9517 CrossRef CAS.
- W. Liu, X. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard 3rd and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005–10013 CrossRef CAS PubMed.
- A. Uttry and M. van Gemmeren, Synthesis, 2019, 479–488 Search PubMed.
- K. Kiyokawa, S. Yahata, T. Kojima and S. Minakata, Org. Lett., 2014, 16, 4646–4649 CrossRef CAS PubMed.
- N. A. Herman and W. Zhang, Curr. Opin. Chem. Biol., 2016, 35, 22–28 CrossRef CAS PubMed.
- D. L. Boger and C. M. Baldino, J. Org. Chem., 2002, 56, 6942–6944 CrossRef.
- M. Scholz, R. Dedic, T. Breitenbach and J. Hala, Photochem. Photobiol. Sci., 2013, 12, 1873–1884 CrossRef CAS PubMed.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044–4060 RSC.
- D. Sheet and T. K. Paine, Chem. Sci., 2016, 7, 5322–5331 RSC.
- M. L. Neidig, A. Decker, O. W. Choroba, F. Huang, M. Kavana, G. R. Moran, J. B. Spencer and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12966–12973 CrossRef CAS PubMed.
- O. W. Choroba, D. H. Williams and J. B. Spencer, J. Am. Chem. Soc., 2000, 122, 5389–5390 CrossRef CAS.
- N. Kitajima, H. Komatsuzaki, S. Hikichi, M. Osawa and Y. Moro-oka, J. Am. Chem. Soc., 2002, 116, 11596–11597 CrossRef.
- T. A. Jackson, A. Karapetian, A. F. Miller and T. C. Brunold, Biochemistry, 2005, 44, 1504–1520 CrossRef CAS PubMed.
- S. Groni, G. Blain, R. Guillot, C. Policar and E. Anxolabehere-Mallart, Inorg. Chem., 2007, 46, 1951–1953 CrossRef CAS PubMed.
- J. A. Kovacs, Acc. Chem. Res., 2015, 48, 2744–2753 CrossRef CAS PubMed.
- M. K. Coggins, X. Sun, Y. Kwak, E. I. Solomon, E. Rybak-Akimova and J. A. Kovacs, J. Am. Chem. Soc., 2013, 135, 5631–5640 CrossRef CAS PubMed.
- M. Sankaralingam, Y. M. Lee, S. H. Jeon, M. S. Seo, K. B. Cho and W. Nam, Chem. Commun., 2018, 54, 1209–1212 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General information, preparation of substrates, optimization of reaction conditions, mechanistic studies, and characterization data. See DOI: 10.1039/d1gc04603a |
| This journal is © The Royal Society of Chemistry 2022 |