
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrodeoxygenation of acetophenone derivatives using a Fe25Ru75@SILP catalyst: a practical approach to the synthesis of alkyl phenols and anilines†
Lisa
Goclik
ab,
Henrik
Walschus
a,
Alexis
Bordet
 *a and
Walter
Leitner
*ab
*a and
Walter
Leitner
*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: alexis.bordet@cec.mpg.de; walter.leitner@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 11th March 2022
Abstract
A versatile synthetic pathway for the production of valuable alkyl phenols and anilines has been developed based on the selective hydrodeoxygenation of a wide range of hydroxy-, amino-, and nitro-acetophenone derivatives as readily available substrates. Bimetallic iron ruthenium nanoparticles immobilized on an imidazolium-based supported ionic liquid phase (Fe25Ru75@SILP) act as highly active and selective catalysts for the deoxygenation of the side-chain without hydrogenation of the aromatic ring. The catalytic system allows operation under continuous flow conditions with high robustness and flexibility as demonstrated for the alternating conversion of 3′,5′-dimethoxy-4′-hydroxyacetophenone and 4′-hydroxynonanophenone as model substrates.
Introduction
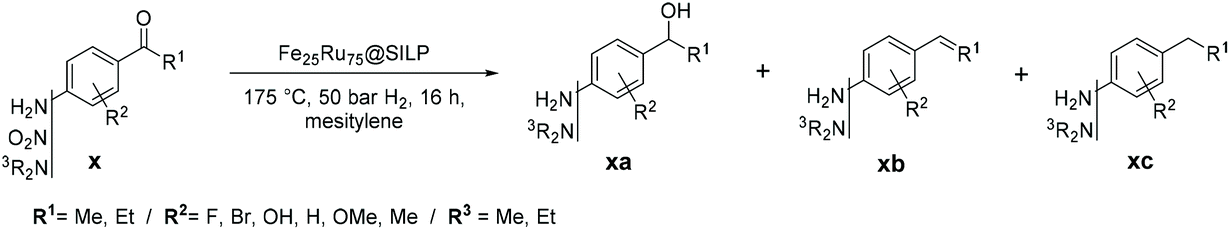
The development of synthetic pathways for the efficient access to alkyl phenols and anilines is attracting a lot of attention owing to the importance of these motifs as building blocks for the production of fine chemicals,1 polymers,2 and pharmaceuticals.3 Long chain alkyl phenols play a key role in the industrial production of non-ionic surfactants (e.g. alkyl phenol ethoxylates).4 Nowadays, long chain alkyl phenols are typically synthesized by direct alkylation of phenols with alkenes.4 However, the selectivity of this method is poor due to various side-reactions, including for example over-alkylation and isomerisation.4 Alkylated anilines, and in particular ethyl anilines, represent important building blocks for the production of agrochemicals5 and pharmaceuticals.3,6 While the Buchwald–Hartwig amination is generally the method of choice for the preparation of aromatic amine derivatives,7 it presents severe limitations including low tolerance toward functional groups (especially halides) and poor efficiency of the amination using NH3 in the case of primary amines.7,8 As a result, multistep approaches involving stoichiometric amounts of toxic reagents and waste are still commonly used.7dThus, the development of alternative methods providing easy access to these key motifs while being at the same time potentially more sustainable and respectful of the green chemistry principles9 is important. In this context, we propose to access alkyl phenols and anilines through the selective hydrodeoxygenation of hydroxy-, amino-, and nitro-acetophenone derivatives (Fig. 1). These substrates are in many cases commercially available at low to moderate costs. They can be produced via Friedel–Crafts (FC) acylation and Fries rearrangement of phenols,10 FC acylation of nitrobenzene and aniline derivatives,11 or nitration of acetophenones (Fig. 1).12
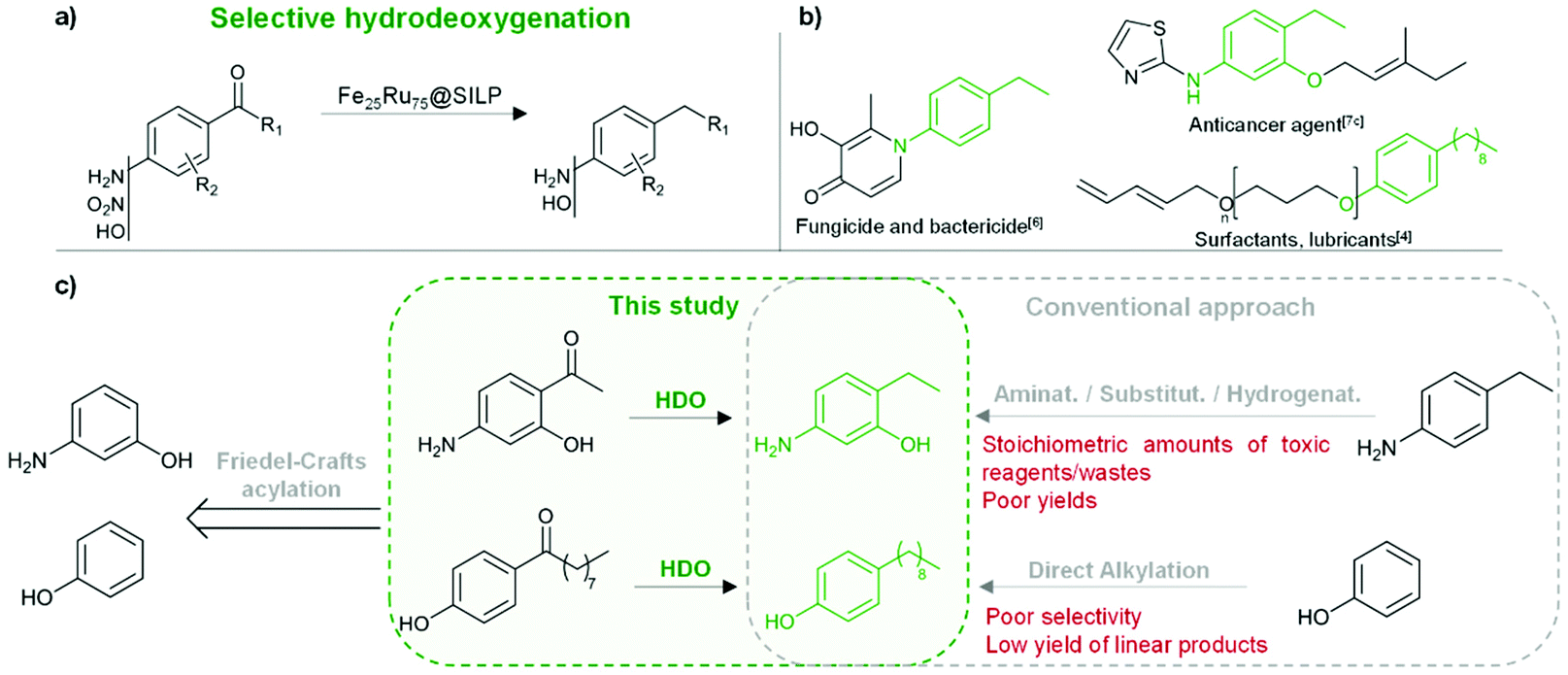 | ||
| Fig. 1 Production of alkyl anilines and alkyl phenols through Fe25Ru75@SILP-catalyzed hydrodeoxygenation of acetophenone derivatives. (a) Synthetic pathway; (b) examples of potential applications;4,6,7c (c) comparison with conventional approach4,7d for two selected examples. | ||
Unlocking the potential of this pathway requires a catalyst capable of effectively hydrodeoxygenating ketones, while leaving the aromatic ring and other functionalities untouched. Recently, promising catalysts have been reported for the selective hydrodeoxygenation of aromatic ketones to aromatic alkanes.13 However, catalytic systems capable of selectively hydrodeoxygenating hydroxy-, amino-, or nitro-acetophenones with H2 are scarce due to the tendency of these substrates to undergo various competing side reactions especially when acid sites are present in the catalyst materials.14 Consequently, the development of a catalytic system possessing excellent activity, selectivity, stability, and versatility for the production of alkyl phenols and anilines from acetophenone derivatives is highly desirable, but remains a challenge.
The immobilization of metal nanoparticles on molecularly modified surfaces, and in particular supported ionic liquid phases, is attracting increasing attention to generate catalytic systems with tailor-made reactivity.15,16 Careful tuning of the NPs composition and molecular modifier structure enables a large degree of control over H2 activation (homolytic versus heterolytic) providing hydrogenation16a–h and hydrodeoxygenation14b,16h–m catalysts with tunable activity and selectivity. Recently, we have shown that bimetallic Fe–Ru nanoparticles immobilized on an imidazolium-based SILP possess excellent activity and selectivity for the hydrodeoxygenation of substituted hydroxyacetophenones to ethyl phenol derivatives without hydrogenation of the aromatic ring.16m The acid-free Fe25Ru75@SILP catalyst prevented the acid-catalysed side-reactions typically observed when using conventional hydrodeoxygenation catalysts. Mesomeric stabilization of the intermediates was found essential to observe high hydrodeoxygenation activity in the absence of an acidic co-catalyst.
Based on these initial findings, we decided to explore the potential of the Fe25Ru75@SILP catalytic system for the conversion of a broad scope of long chain hydroxy-acetophenones as well as substituted amino- and nitro-acetophenones. For the first time, we report herein the remarkable versatility of the Fe25Ru75@SILP catalyst for their selective hydrodeoxygenation and demonstrate its robustness and flexibility under continuous flow conditions.
Results and discussion
Catalyst synthesis and characterization
The Fe25Ru75@SILP catalyst (Fig. 2) was synthesized following a recently reported procedure.16m In brief, the preparation of the SILP involved the condensation of [1-butyl-3-(3-triethoxy-silylpropyl)-imidazolium]NTf2 with dehydroxylated SiO2. The NPs were prepared through an organometallic approach, with the in situ reduction of a mesitylene solution of {Fe[N(Si(CH3)3)2]2}2 and [Ru(cod)(cot)] (cod = cyclooctadiene; cot = cyclooctatriene) in the presence of the SILP material under an atmosphere of H2 (3 bar) at 150 °C (for detailed procedure see ESI†). Scanning transmission electron microscopy with high angle annular dark field (STEM-HAADF) analysis confirmed the formation of small and well-dispersed nanoparticles with an average size of 3.3 nm (Fig. 2b). The BET surface area of 240.7 m2 g−1 is significantly lower than for the starting SiO2 (342.3 m2 g−1) due to the chemisorption of the ionic liquid. A metal loading of 0.4 mmol g−1 and a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru ratio of 25:75 were determined by scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX), well in agreement with the theoretical values (Table S1†). The bimetallic structure of the Fe25Ru75 nanoparticles as well as the zerovalent state of Fe and Ru atoms were previously confirmed by XANES and EXAFS characterization.16c
:Ru ratio of 25:75 were determined by scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX), well in agreement with the theoretical values (Table S1†). The bimetallic structure of the Fe25Ru75 nanoparticles as well as the zerovalent state of Fe and Ru atoms were previously confirmed by XANES and EXAFS characterization.16c
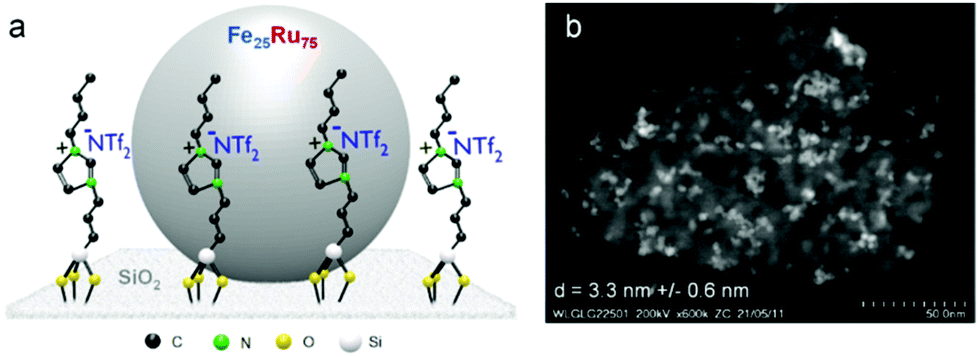 | ||
| Fig. 2 (a) Schematic illustration and (b) STEM-HAADF image of the Fe25Ru75@SILP catalyst. | ||
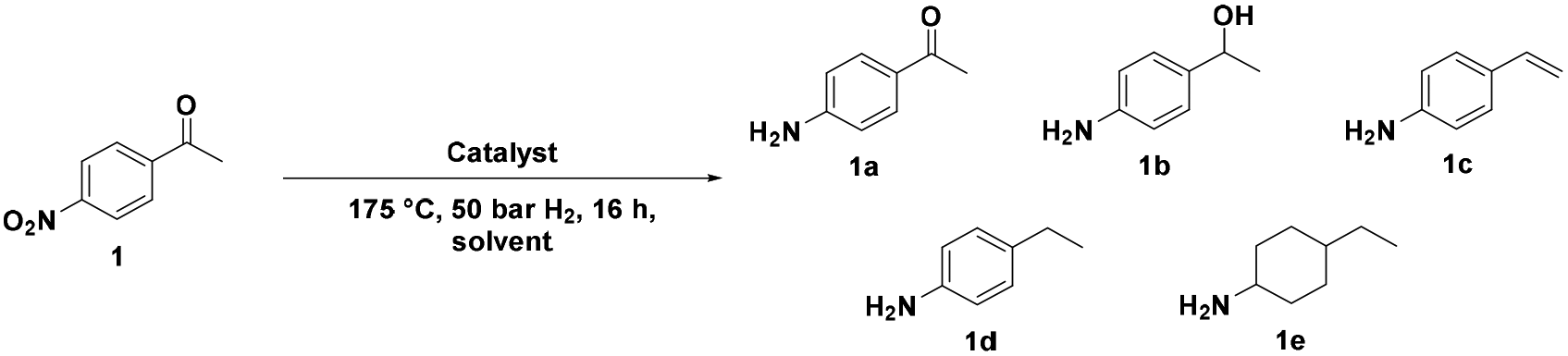
Catalytic study
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrodeoxygenation activity, conserving only activity for the hydrogenation of the nitro functionality (entries 5 and 6).
O hydrodeoxygenation activity, conserving only activity for the hydrogenation of the nitro functionality (entries 5 and 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | X [%] | Y (1a) [%] | Y (1d) [%] | Y (1e) [%] |
| Reaction conditions: Catalyst (10 mg, metal content: 0.004 mmol), substrate (25 eq.), solvent (0.5 mL), 175 °C, H2 (50 bar), 16 h, 500 rpm. Conversion and product yields determined by GD-FID using decane as internal standard. X = conversion, Y = yield. | ||||||
| 1 | Fe100@SILP | Mesitylene | 0 | 0 | 0 | 0 |
| 2 | Ru100@SILP | Heptane | >99 | 0 | 0 | >99 |
| 3 | Fe25Ru75@SILP | Mesitylene | >99 | 0 | >99 | 0 |
| 4 | Fe25Ru75@SiO2 | Mesitylene | >99 | 0 | 0 | >99 |
| 5 | Fe40Ru60@SILP | Mesitylene | >99 | 85 | 15 | 0 |
| 6 | Fe60Ru40@SILP | Mesitylene | >99 | >99 | 0 | 0 |
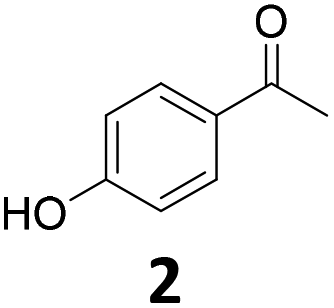
Long chain hydroxyacetophenones
The activity of the Fe25Ru75@SILP catalyst was investigated for a scope of hydroxyacetophenones with various alkyl chain lengths (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | X [%] | S [%] | Y [%] |
| Reaction conditions: Catalyst (37.5 mg, metal content: 0.015 mmol), substrate (0.375 mmol, 25 eq.), solvent (0.5 mL), 175 °C, H2 (50 bar), 16 h, 500 rpm.a By-product: 4′-octenylphenol.b 24 h. Conversion and product yields determined by GD-FID using decane as internal standard. X = conversion, S = selectivity, Y = yield. Isolated yields in brackets. | |||||
| 1 |
|
|
>99 | >99 | >99 (88) |
| 2 |
|
|
>99 | >99 | >99 (91) |
| 3 |
|
|
>99 | 99 | 99 (90) |
| 4 |
|
|
>99 | >99 | >99 (80) |
| 5 |
|
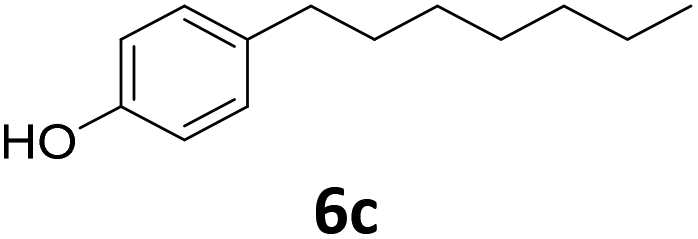
|
>99 | >99 | >99 (82) |
| 6a |
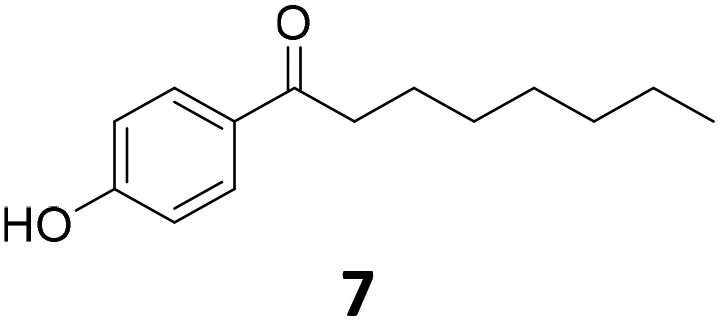
|
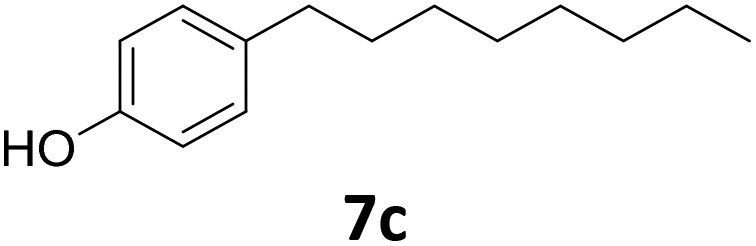
|
51 | 76 | 39 |
| 92b | 91 | 84 | |||
| 7 |
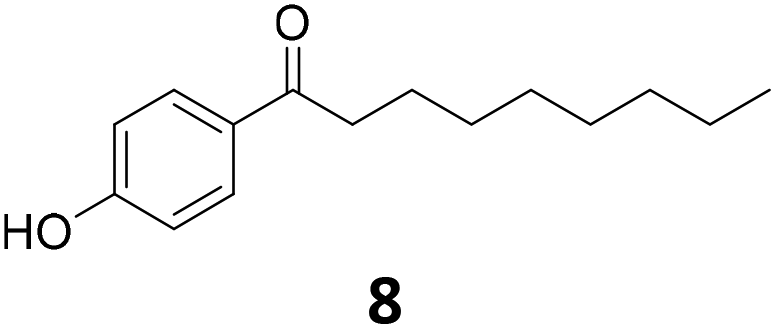
|
|
>99 | >99 | >99 (94) |
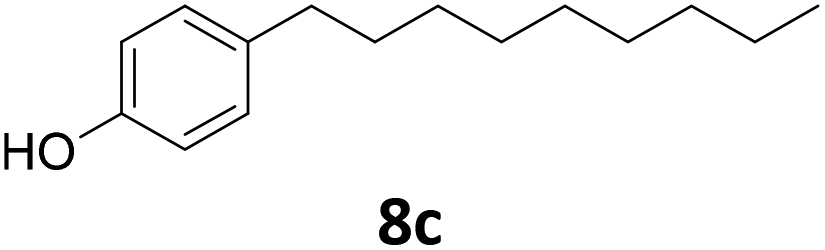
Potential main products for this transformation include the aromatic alcohols arising from the hydrogenation of the substrates, styrene derivatives resulting from alcohol dehydration, and the desired alkyl phenols obtained after hydrogenation of the styrene-type intermediates. Standard conditions for these reactions were set to 175 °C, 50 bar and 16 h reaction time. Substrates 2–8 with increasing side chain length (from R = Me to Oct) were readily hydrodeoxygenated under these conditions, giving the corresponding alkyl phenols 2c–8c in excellent yields and selectivities. No aromatic ring hydrogenation was observed with the bimetallic Fe25Ru75 NPs, in agreement with previous reports.14b,16m A reaction time of 24 h was necessary to fully convert substrate 6, presumably due to the presence of some impurities in the starting substrate. Isolation of the resulting alkyl phenols was readily accomplished by filtration of the reaction mixtures and evaporation of the solvent, providing the isolated compounds 2c–6c and 8c in excellent yields (80–94%). To the best of our knowledge, this is the first time that products 4c–8c are obtained through this pathway using molecular hydrogen. Interestingly, 4′-nonyl phenol (8c) is used industrially in combination with ethane or ethylene oxide to produce surfactants, technical non-ionic surfactants, wetting agents, emulsifiers and foam-stabilizers.4,17
Amino- and nitro-acetophenones
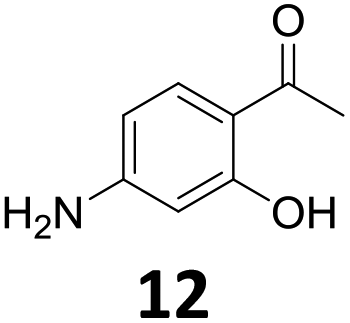
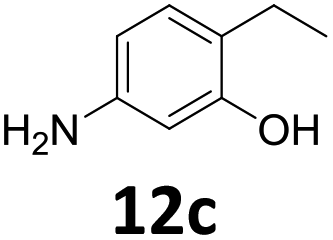
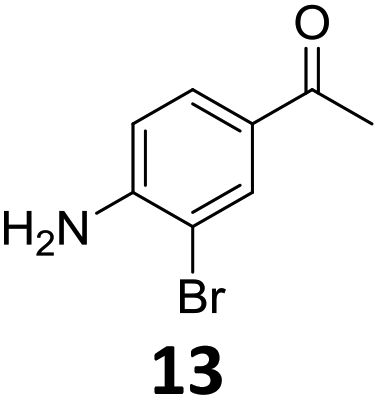
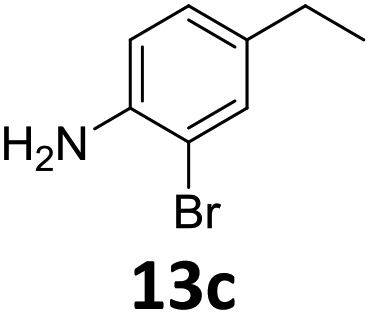
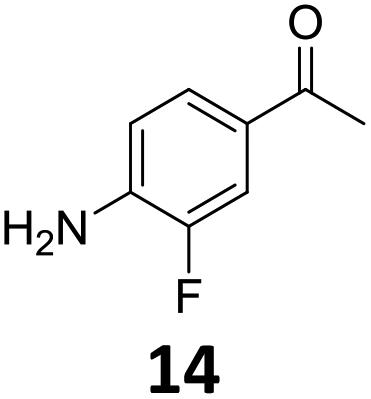
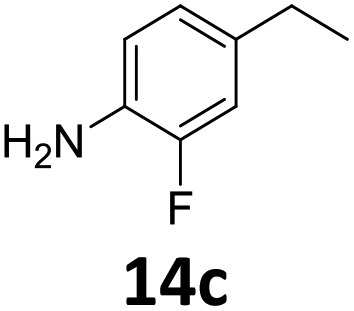
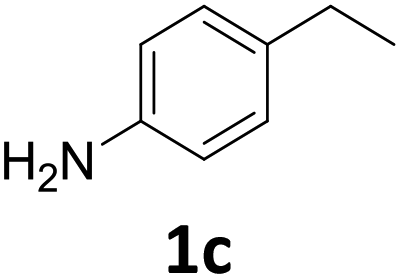
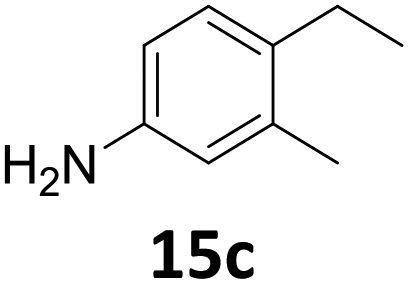
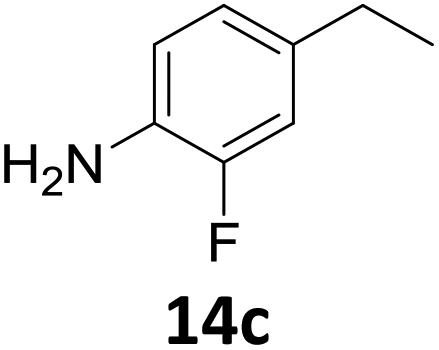
The versatility of the Fe25Ru75@SILP catalyst was then explored for a wide range of substituted amino- and nitroacetophenones (Table 3). Under standard conditions, aminoacetophenone (9) and 1-(4-aminophenyl)-ethanone (10) were hydrodeoxygenated to the corresponding 4′-ethylaniline (1c) and 4′-propylaniline (10c) in excellent yields (92% and 97%, respectively). Interestingly, 1c and 10c are important building blocks for the synthesis of fungicides and bactericides.5 Significant amounts of 3′-methoxyanilin were observed when using 2′-methoxy-4′-aminoacetophenone (11) as substrate (entry 3). Increasing the reaction temperature to 200 °C and using dioxane as solvent was beneficial to avoid deacylation, and the desired product 11c could be obtained in high yield (85%, entry 4). 2′-Hydroxy-4′-aminoacetophenone (12) was hydrodeoxygenated with excellent selectivity and yield (both >99%, entry 5), producing 5-amino-2-ethyl-phenol (12c), a building block entering in the preparation of promising anti-cancer agents.6c The production of 2-bromo-4-ethyl-benzenamine (13c) and 2-fluoro-4-ethyl-benzenamine (14c) in fair to high yields under optimized conditions (57% and 89%, respectively) is particularly interesting since these products are valuable building blocks18 that cannot be obtained via Buchwald–Hartwig amination7 and that typically require multistep synthetic approaches with limited selectivity.19|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | T [°C] | Product | X [%] | S [%] | Y [%] |
| Conditions: Fe25Ru75@SILP (10 mg catalyst containing 0.004 mmol total metal), 0.1 mmol substrate (25 equivalents), 0.5 mL mesitylene, 50 bar H2, 16 h, 500 rpm.a 24 h.b By-product: propylbenzene.c By-product: 3′-methoxyanilin.d Solvent: dioxane.e By-product: 2′-bromoanilin.f By-product: 4′-ethylanilin. X = conversion, S = selectivity, Y = yield, determined by GD-FID using decane as internal standard. Isolated yields in parentheses. | ||||||
| 1a |
|
175 |
|
92 | >99 | 92 |
| 2 |
|
175 |
|
>99 | 97b | 97 (94) |
| 3 |
|
175 |
|
>99 | 55c | 55 |
| 200 | >99 | 85c | 85 | |||
| 4 |
|
175 |
|
>99 | >99 | >99 (79) |
| 5d |
|
150 |
|
86 | 66e | 57 |
| 6d |
|
200 |
|
>99 | 89f | 89 |
| 7 |
|
175 |
|
>99 | >99 | >99 (93) |
| 8 |
|
175 |
|
>99 | >99 | >99 (94) |
| 9d |
|
150 |
|
>99 | >99 | >99 |
| 10 |
|
175 |
|
>99 | >99 | >99 |
| 11 |
|
175 |
|
>99 | >99 | >99 (80) |
| 12a |
|
175 |
|
>99 | >99 | >99 (87) |
A similar approach was adopted for the conversion of various nitro-acetophenones. First, a time profile was recorded using 4′-nitroacetophenone (1) as model substrate (Fig. 3). The conditions were adapted (120 °C, 50 bar H2) to slow down the reaction and allow observing intermediates.
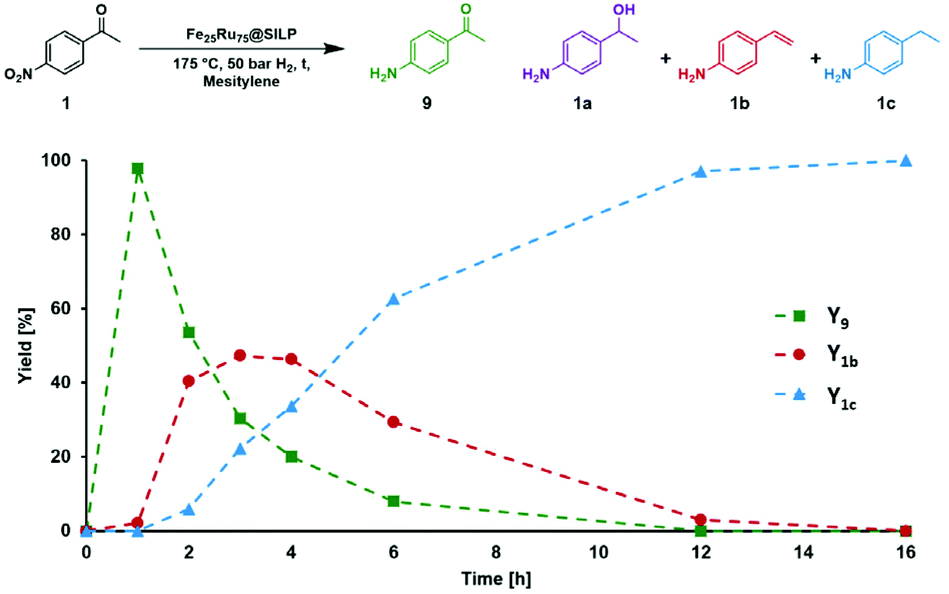 | ||
| Fig. 3 Reaction/time-profile for the hydrodeoxygenation of 4′-nitroacetophenone (1) using Fe25Ru75@SILP. Reaction conditions: Fe25Ru75@SILP (10 mg catalyst containing 0.004 mmol total metal), 0.1 mmol 1 (25 eq.), 120 °C, 0.5 mL mesitylene, 50 bar H2, 500 rpm. Y = yield. | ||
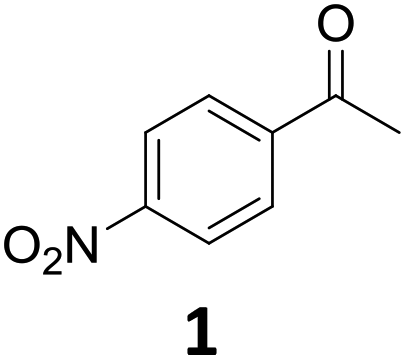
1 was very quickly (∼1 h) converted to the 4′-amino-acetophenone (1c), which was then hydrodeoxygenated to give 4′-vinylaniline (1b). 1b was then consumed over time to give a quantitative yield of 1c after 12 h in a typical kinetic profile for sequential reaction steps. These results suggest that the Fe25Ru75@SILP catalyst can be used to perform HDO reactions at relatively mild temperature. The observation of 1b as major intermediate indicates that the deoxygenation proceeds through the cleavage of the C–O bond and alcohol elimination via a E2-type mechanism resulting in an olefin intermediate. In addition, neither the alcohol 1a nor its nitro-analogue could be observed, suggesting that this elimination step is considerably faster than the hydrogenation of 9 to 1a (CO hydrogenation) and of 1b to 1c (CC hydrogenation).
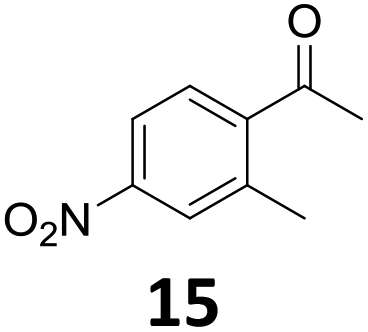
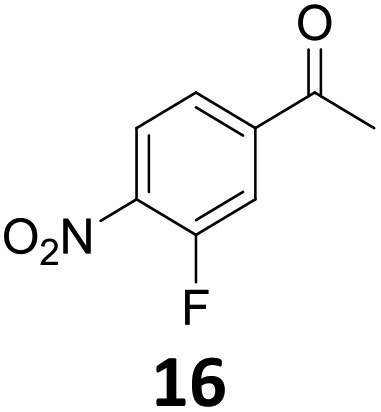
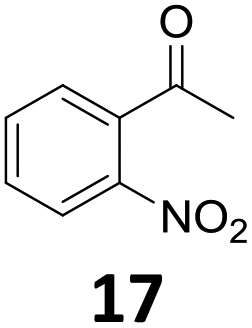
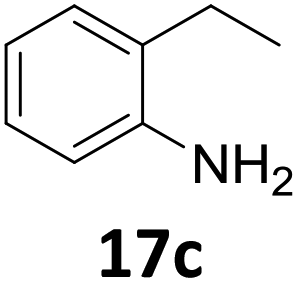
Starting from substituted nitroacetophenones (1, 15–17) was similarly successful, leading to the corresponding alkyl aniline derivatives in excellent yields (>99%). Notably, the conversion of 1-(3-fluoro-4-nitrophenyl)-ethanone (16) allowed obtaining 14c in higher yield and milder conditions (entry 9) that when starting directly from 1. Having the nitro functionality in ortho position (17) did not affect the catalyst's performances and gave quantitative yield of 2′-ethylanilin (17c) which can be used for the synthesis of enzyme inhibitors.18 To finish, two dialkylaminoacetophenones were investigated (1-[4-(dimethylamino)phenyl]-ethanone (18) and 1-[4-(diethylamino)phenyl]-ethanone (19). 18 and 19 were readily hydrodeoxygenated to the corresponding ethyl dialkylbenzeneamines 18c and 19c (>99% yield for both), highlighting the versatility of the Fe25Ru75@SILP catalysts as well as it tolerance toward various functional groups. In addition, product isolation was performed for several of the entries presented, giving 1c, 10c, 12c, 15c, 18c and 19c in good to excellent yields (79–94%) and thus confirming the good practicability of Fe25Ru75@SILP. To the best of our knowledge, this is also the first time that products 10c–14c and 15c–16c are accessed via the selective hydrodeoxygenation of substrates 10–14 and 15–16.
Satisfyingly, replacing mesitylene by anisole – a solvent recommended by the CHEM21 green chemistry selection guide20 – resulted also in excellent yields and selectivities, as evidenced for some selected examples (Table S2†).
Continuous flow operation
The performance of the Fe25Ru75@SILP catalyst was further investigated under continuous flow conditions. A solution of 4′-hydroxyacetophenone (2) (0.025 mol L−1 in anisole) was passed over a cartridge packed with the Fe25Ru75@SILP catalyst (433.5 mg, 0.173 mmol metal) at a flow rate of 0.6 mL min−1 under standard conditions (175 °C, 50 bar H2) using an H-Cube Pro reactor system from ThalesNano (Fig. S1†). Anisole was chosen as solvent for the continuous flow experiments to ensure good solubility of 2 at room temperature. The reaction conditions were selected to allow the observation of intermediates, evidencing any potential changes in the catalyst reactivity over time. The evolution of the substrate conversion and products yields is given in Fig. 4. Full conversion of 2 to a mixture of 4′-ethenylphenol (2b) and 4′-ethylphenol (2c) was observed for the complete duration of the experiment, i.e. 6 hours time-on-stream. After a short equilibration period (0–60 min), excellent yields of the hydrodeoxygenated product 4′-ethylphenol (2c, 90–95%) were maintained over the remaining 5 hours without any noticeable decay. TEM characterization of the Fe25Ru75@SILP material after 6 hours on stream did not show any change in the size and dispersion of the FeRu NPs (3.1 nm ± 0.6 nm) (Fig. S2 and Table S1†). In addition, slight variations in metal loading and Fe:Ru ratio evidenced by SEM-EDX mapping were within the measurement error and thus insignificant (Table S1†). BET analysis showed a minor decrease in surface area after catalysis (from 240.7 to 232.1 m2 g−1) again within measurement error (Table S1†). These results demonstrate that Fe25Ru75@SILP is active, selective, and stable for the hydrodeoxygenation of 2 under continuous flow operation.
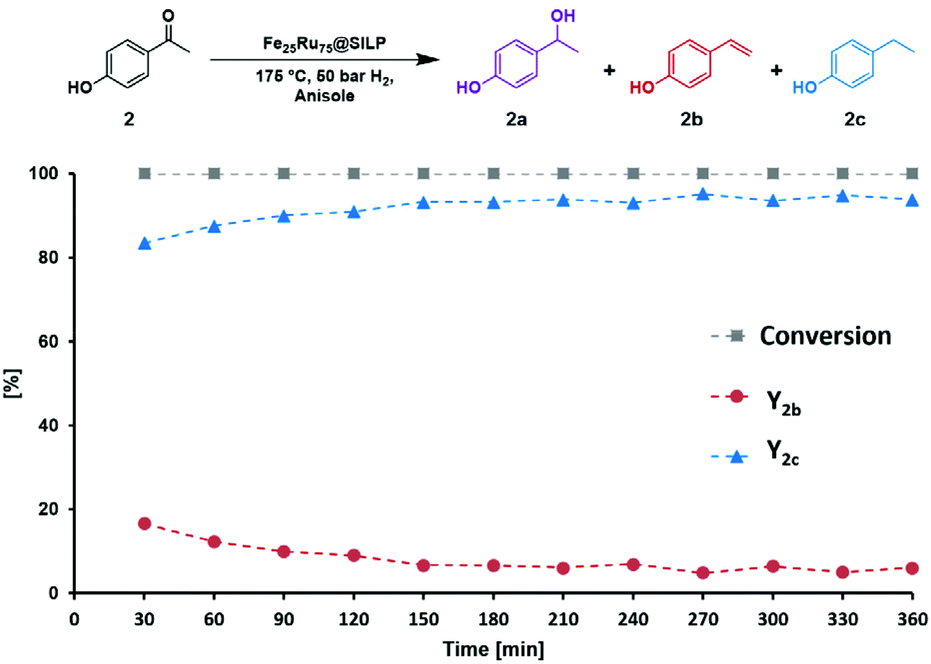 | ||
| Fig. 4 Stability test of Fe25Ru75@SILP under continuous flow conditions. Conditions: 175 °C, 50 bar H2, 60 NmL min−1 H2 flow, 0.6 mL min−1 substrate flow, substrate solution: 0.025 mol L−1 in anisole, residence time: 17 s, Fe25Ru75@SILP (433.5 mg, 0.173 mmol metal). Product yields determined by GD-FID using decane as internal standard. Y = yield. | ||
Based on these encouraging results, the versatility of Fe25Ru75@SILP was further explored under continuous flow conditions. Solutions containing biomass-derived 3′,5′-dimethoxy-4′-hydroxyaceto-phenone (20)21 in anisole and 4′-hydroxynonanophenone (8) in mesitylene were alternatingly passed over the catalyst bed using the continuous flow set-up described previously. Each substrate solution was applied with a flow rate of 0.5 mL min−1 under 50 bar H2 at 175 °C for 45 minutes without any catalyst make-up or changes in the set-up between the reactions. The substrate switch was performed three times in a row to study the impact of real-time repeated substrate changes on the catalyst's performances (Fig. 5). Under optimized reaction conditions (Table S3†), the substrate feed could be switched back and forth between substrate 20 and 8 to alternatively produce 20c and 8c in more than 90% yield. For both substrates, the styrene-type compounds 20b and 8b were observed as only intermediates, outlining once again that the deoxygenation step (C–O bond cleavage) is significantly faster than hydrogenation of the CO and CC bonds.
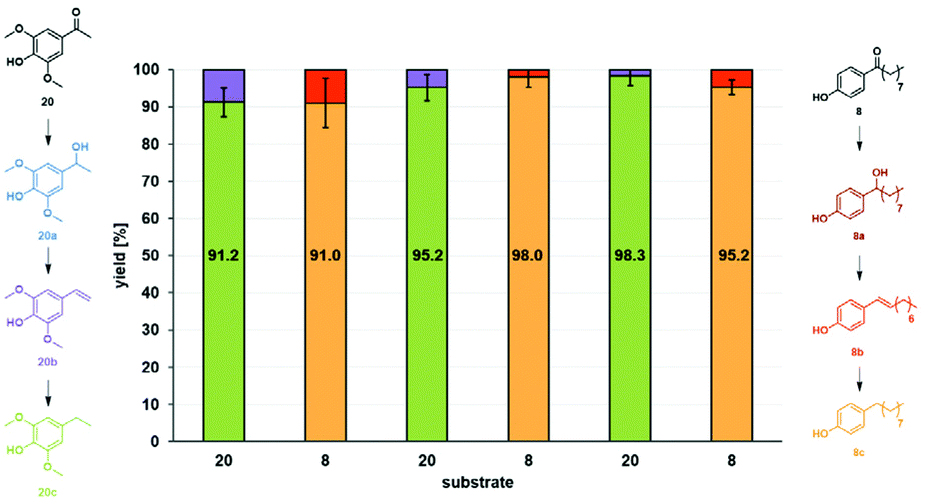 | ||
| Fig. 5 Substrate switch under continuous flow conditions. Conditions: 175 °C, 50 bar H2, 60 NmL min−1 H2 flow, 0.5 mL min−1 substrate flow, substrate solutions: 0.015 mol L−1 3′,5′-dimethoxy-4′-hydroxyacetophenone (20) in anisole and 0.015 mol L−1 4′-hydroxynonanophenone (8) in mesitylene, residence time: 18 s, Fe25Ru75@SILP (455.9 mg, 0.182 mmol metal). Between each substrate switch, the system was washed with anisole, mesitylene or heptane (for details see ESI†). Product yields determined by GD-FID using decane as internal standard. | ||
The experiment represents a total of 4.5 h time-on-stream without any change in activity, selectivity, or stability of the catalytic system for either of the substrates, highlighting its robustness and versatility in the selective hydrodeoxygenation of hydroxy-acetophenone derivatives.
Synthetic approach evaluation
To finish, the synthetic approach we propose here (FC acylation + hydrodeoxygenation) was systematically compared to conventional methods for the preparation of 2′-hydroxy-4-ethylaniline (12) and 4′-nonylphenol (8), the two products highlighted in Fig. 1b and c. Five parameters related to the green chemistry principles were taken into consideration to rank the pathways, i.e. the overall reaction yield (Y), the atom economy (AE), the number of steps (steps), the hazardous nature of the reagents (safety), and the economical aspect (difference of value between the product and the starting substrate, eco), giving the spider web-type graphs shown in Fig. 6 (details concerning the different synthetic pathways and corresponding rankings are available in the ESI, Fig. S3–S6 and Table S4†). While the hydrodeoxygenation-based approach necessitates in both cases an additional step as compared to the conventional pathways, the other parameters are either equal (AE), or clearly in favour (Y, eco, safety) of the alternative we propose, highlighting the potential benefits associated to the production of alkyl phenols and anilines via selective hydrodeoxygenation of acetophenone derivatives.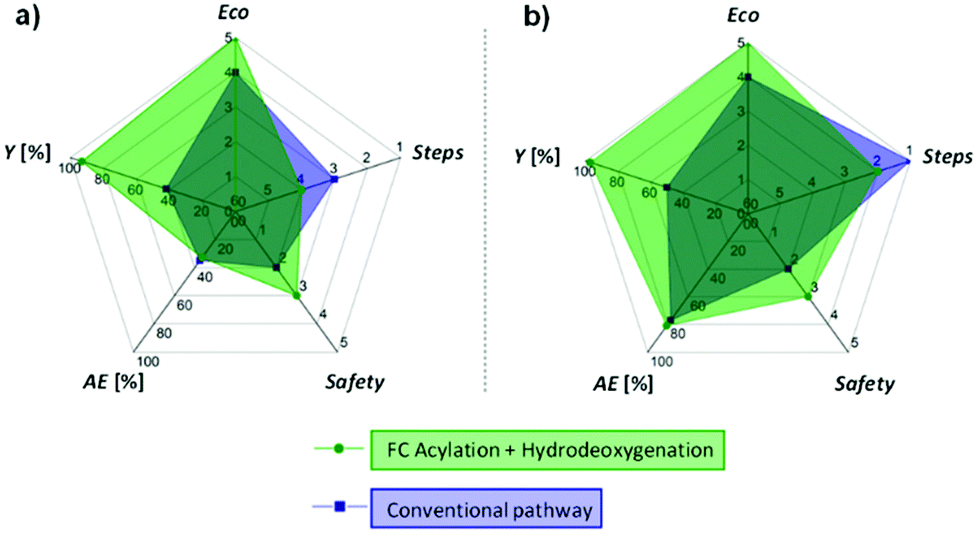 | ||
| Fig. 6 Comparison between our approach and conventional synthetic pathways for the production of (a) 2′-hydroxy-4-ethylaniline (12), and (b) 4′-nonylphenol (8). Area proportional to the green chemistry metrics (large area = good metrics). Details concerning the different synthetic pathways and corresponding rankings are available in the ESI.† | ||
Conclusion
In conclusion, the catalytic hydrodeoxygenation of hydroxy-, amino-, and nitro-acetophenone derivatives was established as a broadly applicable synthetic pathway for the synthesis of alkyl phenols and anilines. The bimetallic Fe25Ru75@SILP catalytic system proved highly effective for the selective hydrodeoxygenation of a broad range of substrates comprising various functionalities while conserving the aromaticity in the products. The activity, selectivity and stability of Fe25Ru75@SILP were demonstrated for the continuous flow hydrodeoxygenation of 4-hydroxyacetophenone (1) for over 6 h time-on-stream. In addition, the alternating hydrodeoxygenation of 3′,5′-dimethoxy-4′-hydroxyacetophenone (20) and 4′-hydroxynonanophenone (7) under continuous flow conditions was achieved in real-time over a single catalyst cartridge, producing the corresponding phenols alternatively in excellent yield and outlining the versatility of the Fe25Ru75@SILP catalytic system. The potential benefits of the proposed pathway with regards to sustainability were evidenced for two selected examples. This approach provides a promising alternative strategy for the effective synthesis of long chain alkyl phenols and ethyl anilines that are key building blocks for the production of important fine chemicals, agrochemicals, and pharmaceuticals.Experimental section
Synthesis of Fe25Ru75@SILP
A solution of {Fe[N(Si(CH3)3)2]2}2 (18.0 mg, 0.05 mmol Fe) in mesitylene (2 mL) was combined with a solution of [Ru(cod)(cot)] (47.0 mg, 0.15 mmol Ru) in mesitylene (2 mL) in a Fischer–Porter bottle (70 mL). The SILP (500 mg) was added to the solution of the metal precursors along with mesitylene (1 mL) and the reaction mixture was stirred under argon at room temperature for 30 min. The Fischer–Porter bottle was evacuated and backfilled with H2 and the suspension was stirred under H2 (3 bar) at 150 °C for 18 h. Under this reducing environment a black powder was obtained indicating the immobilization of the NPs onto the SILP. Mesitylene was decanted and Fe25Ru75@SILP were washed with fresh toluene (3 × 3 mL) and dried in vacuo at rt for 1 h.Autoclave reactions
The substrate (25 eq., 0.1 mmol), catalyst (10 mg, containing 0.004 mmol metal) and solvent (mesitylene, 0.5 mL) were weighed inside of a glovebox into the autoclave. The autoclave was closed, transferred out of the glovebox and pressurized with 50 bar hydrogen. After heating the autoclave under stirring (500 rpm) to 175 °C for 16 h, it was cooled down in a water bath and depressurized. The GC samples were prepared by adding acetone (250 mg) to the reaction mixture and taking a sample through a syringe with a filter. To the sample, decane (internal standard, 20 mg) and acetone (200 mg) were added. For all amines, methanol/dioxane (1/1) was used as solvent for the GC samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support by the Max Planck Society and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID:390919832. Furthermore, the authors thank Alina Jakubowski, Annika Gurowski, Justus Werkmeister and Norbert Pfänder (MPI-CEC, Mülheim/Ruhr) for their support with the analytics. Open Access funding provided by the Max Planck Society.References
- R. O. Hutchins, M. K. Hutchins and I. Fleming, in Comprehensive Organic Synthesis, Elsevier, Oxford, 1991, vol. 8, p. 327 Search PubMed.
- (a) F. Yang, G. Li, J. Qi, S.-M. Zhang and R. Liu, Appl. Surf. Sci., 2010, 257, 312 CrossRef CAS; (b) F. Yang, G. Li, N. Xu, R. Liu, S.-M. Zhang and Z.-J. Wu, J. Surfactants Deterg., 2011, 14, 339 CrossRef CAS; (c) D. Pan, L. Zhang, F. Li, Y. Chen, N. Tao, G. Zhang and Y. Dai, Faming Zhuanli Shenqing, CN102719820, 2012 Search PubMed; (d) Y. Nakai and N. Haruyama, PCT Int. Appl, WO2019130722, 2019 Search PubMed.
- (a) J. F. Lorenc, G. Lambeth and W. Scheffer, in Alkylphenols, John Wiley & Sons, Inc., 2003 Search PubMed; (b) J. L. Wang, K. Aston, D. Limburg, C. Ludwig, A. E. Hallinan, F. Koszyk, B. Hamper, D. Brown, M. Graneto and J. Talley, Bioorg. Med. Chem. Lett., 2010, 20(23), 7164 CrossRef CAS PubMed; (c) S. Bhattacharya and V. Kashaw, J. Drug Delivery Ther., 2019, 9, 591 CrossRef CAS.
- H. Fiege, H.-W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H.-J. Buysch, D. Garbe and W. Paulus, Phenol Derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- X. Yu, X. Zhu, Y. Zhou, Q. Li, Z. Hu, T. Li, J. Tao, M. Dou, M. Zhang, Y. Shao and R. Sun, J. Agric. Food Chem., 2019, 67(50), 13904 CrossRef CAS PubMed.
- (a) S. Bilginer, B. Gonder, H. I. Gul, R. Kaya, I. Gulcin, B. Anil and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2020, 35, 325 CrossRef CAS PubMed; (b) L. Xiu-jie, Z. Zhi-hao, D. Qing-song, C. Xin and W. Chao-qing, Med. Chem. Res., 2019, 28, 1864 CrossRef; (c) W. L. Jorgensen, J. Ruiz-Caro and A. D. Hamilton, PCT Int. Appl, WO2007038387, 2007 Search PubMed.
- (a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969 CrossRef CAS; (b) A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901 CrossRef CAS; (c) P. A. Forero-Cortés and A. M. Haydl, Org. Process Res. Dev., 2019, 23, 1478 CrossRef; (d) J. Ruiz-Caro, A. Basavapathruni, J. T. Kim, C. M. Baily, L. Wang, K. S. Anderson, A. D. Hamilton and W. L. Jorgensen, Bioorg. Med. Chem. Lett., 2006, 16, 688 CrossRef PubMed.
- (a) Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028 CrossRef CAS PubMed; (b) D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354 CrossRef CAS PubMed; (c) G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049 CrossRef CAS PubMed.
- (a) J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed; (b) P. T. Anastas, D. Constable and C. Jimenez-Gonzalez, Green Metrics, Wiley-VCH, Weinhiem, Germany, 2018, vol. 11, p. 400 Search PubMed.
- (a) H. Heaney and B. M. Trost and I. Fleming, in Comprehensive Organic Synthesis, Pergamon Press, Oxford, UK, 1991, vol. 2, p. 733 Search PubMed; (b) R. Faust, U.S. Pat. Appl. Publ, US 20170001936, 2017, 20pp. Search PubMed; (c) R. W. Stoughton, R. Baltzly and A. Bass, J. Am. Chem. Soc., 1934, 56, 2007 CrossRef CAS; (d) I. Jeon and I. K. Mangion, Synlett, 2012, 23, 1927 CrossRef CAS; (e) H. Paghandeh, H. Saeidian and M. Ghaffarzadeh, Lett. Org. Chem., 2018, 15, 809 CrossRef CAS; (f) R. Murashige, Y. Hayashi, S. Ohmori, A. Torii, Y. Aizu, Y. Muto, Y. Murai, Y. Oda and M. Hashimoto, Tetrahedron, 2011, 67, 641 CrossRef CAS.
- (a) M. Gopalakrischnan, P. Surechkumar, V. Kanagarajan and J. Thanusu, Catal. Commun., 2005, 6(12), 753 CrossRef; (b) S. Paul, P. Nanda, R. Gupta and A. Loupy, Synthesis, 2003, 2877 CAS; (c) G. Guerrini, M. Taddei and F. Ponticelli, J. Org. Chem., 2011, 76(18), 7597 CrossRef CAS PubMed; (d) M. Koura, T. Matsuda, A. Okuda, Y. Watanabe, Y. Yamaguchi, S. Kurobuchi, Y. Matsumoto and K. Shibuya, Bioorg. Med. Chem. Lett., 2015, 25(13), 2668 CrossRef CAS PubMed; (e) M. C. Bestwick, M. J. Drysdale, B. W. Dymock and E. McDonald, PTC int. Appl, WO2004/056782A1, 2004 Search PubMed; (f) C. Schultze and B. Schmidt, J. Org. Chem., 2018, 83, 5210 CrossRef CAS PubMed; (g) N. Hauel, H. Nar, H. Priepke, U. Ries, J. M. Stassen and W. Wienen, PCT Int. Appl, WO2000050419A120000831, 2000 Search PubMed.
- (a) A. H. Ford-Moore and H. N. Rydon, J. Chem. Soc., 1946, 679 RSC; (b) W. F. Hödlderich and H. van Bekkum, Stud. Surf. Sci. Catal., 1991, 58, 677 Search PubMed; (c) L. E. Bertea, H. W. Kouwnhoven and R. Prins, Stud. Surf. Sci. Catal., 1994, 84, 1973 CrossRef CAS; (d) S. Sana, K. C. Rajanna, K. R. Reddy, M. Bhoosha, M. Venkateswarlu, M. S. Kumar and K. Uppalaiah, Green Sustainable Chem., 2012, 2, 97 CrossRef.
- (a) A. Volkov, K. P. J. Gustafson, C.-W. Tai, O. Verho, J.-E. Bäckvall and H. Adolfsson, Angew. Chem., Int. Ed., 2015, 54, 5122 CrossRef CAS PubMed; (b) C. González, P. Marín, F. V. Díez and S. Ordóñez, Ind. Eng. Chem. Res., 2016, 55, 2319 CrossRef; (c) M. Li, J. Deng, Y. Lan and Y. Wang, ChemistrySelect, 2017, 2, 8486 CrossRef CAS; (d) L. Petitjean, R. Gagne, E. S. Beach, D. Xiao and P. T. Anastas, Green Chem., 2016, 18, 150 RSC; (e) S. C. Shit, R. Singuru, S. Pollastri, B. Joseph, B. S. Rao, N. Lingaiah and J. Mondal, Catal. Sci. Technol., 2018, 8, 2195 RSC; (f) F. Zaccheria, N. Ravasio, M. Ercolicand and P. Allegrini, Tetrahedron Lett., 2005, 46, 7743 CrossRef CAS; (g) V. Pandarus, R. Ciriminna, G. Gingras, F. Béland, M. Pagliaro and S. Kaliaguine, ChemistryOpen, 2018, 7, 80 CrossRef CAS PubMed; (h) V. Ranaware, D. Verma, R. Insyani, A. Riaz, S. Min Kim and J. Kim, Green Chem., 2019, 21, 1021 RSC; (i) L. Zhang, N. Shang, S. Gao, J. Wang, T. Meng, C. Du, T. Shen, J. Huang, Q. Wu, H. Wang, Y. Qiao, C. Wang, Y. Gao and Z. Wang, ACS Catal., 2020, 10, 8672 CrossRef CAS; (j) C. Guyon, M. Baron, M. Lemaire, F. Popowycz and E. Métay, Tetrahedron, 2014, 70, 2088 CrossRef CAS.
- (a) M. Kozelj and A. Petric, Synlett, 2007, 1699 CAS; (b) L. Offner-Marko, A. Bordet, G. Moos, S. Tricard, S. Rengshausen, B. Chaudret, K. L. Luska and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 12721 CrossRef CAS PubMed.
- (a) A. Bordet and W. Leitner, Acc. Chem. Res., 2021, 54, 2144 CrossRef CAS PubMed; (b) L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683 CrossRef CAS PubMed; (c) T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955 CrossRef CAS.
- (a) M. A. Gelesky, S. S. X. Chiaro, F. A. Pavan, J. H. Z. dos Santos and J. Dupont, Dalton Trans., 2007, 5549 RSC; (b) X. Yuan, N. Yan, C. Xiao, C. Li, Z. Fei, Z. Cai, Y. Kou and P. J. Dyson, Green Chem., 2010, 12, 228 RSC; (c) K. L. Luska, A. Bordet, S. Tricard, I. Sinev, W. Grünert, B. Chaudret and W. Leitner, ACS Catal., 2016, 6, 3719 CrossRef CAS; (d) A. Bordet, S. El Sayed, M. Sanger, K. L. Boniface, D. Kalsi, K. L. Luska, P. G. Jessop and W. Leitner, Nat. Chem., 2021, 13, 916–922 CrossRef CAS PubMed; (e) S. El Sayed, A. Bordet, C. Weidenthaler, W. Hetaba, K. L. Luska and W. Leitner, ACS Catal., 2020, 10, 2124 CrossRef CAS; (f) S. Kacem, M. Edmonds, A. Bordet and W. Leitner, Catal. Sci. Technol., 2020, 10, 8120 RSC; (g) A. Bordet, G. Moos, C. Welsh, P. Lience, K. L. Luska and W. Leitner, ACS Catal., 2020, 10(23), 13904 CrossRef CAS PubMed; (h) S. Rengshausen, C. Van Stappen, N. Levin, S. Tricard, K. L. Luska, S. DeBeer, B. Chaudret, A. Bordet and W. Leitner, Small, 2021, 2006683 CrossRef CAS PubMed; (i) Y. Wang, Z. Rong, Y. Wang, T. Wang, Q. Du, Y. Wang and J. Qu, ACS Sustainable Chem. Eng., 2017, 5, 1538 CrossRef CAS; (j) R. Insyani, D. Verma, S. M. Kim and J. Kim, Green Chem., 2017, 19, 2482 RSC; (k) G. Moos, M. Edmonds, A. Bordet and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 11977 CrossRef CAS PubMed; (l) S. Rengshausen, F. Etscheidt, J. Großkurth, K. L. Luska, A. Bordet and W. Leitner, Synlett, 2019, 30, A–H Search PubMed; (m) L. Goclik, L. Offner-Marko, A. Bordet and W. Leitner, Chem. Commun., 2020, 56, 9509 RSC.
- (a) F. Yang, G. Li, J. Qi, S.-M. Zhang and R. Liu, Appl. Surf. Sci., 2010, 257, 312 CrossRef CAS; (b) F. Yang, G. Li, N. Xu, R. Liu, S.-M. Zhang and Z.-J. Wu, J. Surfactants Deterg., 2011, 14, 339 CrossRef CAS; (c) M. H. Todd and C. Abell, J. Comb. Chem., 2001, 3, 319 CrossRef CAS PubMed.
- F. M. Khan, M. A. Abbasi, A. ur-Rehman, S. Z. Siddiqui, A. R. S. Butt, H. Raza, A. Zafar, S. A. A. Shah, M. Shahid and S.-Y. Seo, J. Heterocycl. Chem., 2021, 58, 1089 CrossRef CAS.
- (a) A. Aoyama, K. Endo-Umeda, K. Kishida, K. Ohgane, T. Noguchi-Yachide, H. Aoyama, M. Ishikawa, H. Miyachi, M. Makishima and Y. Hashimoto, J. Med. Chem., 2012, 55(17), 7360 CrossRef CAS PubMed; (b) Y.-J. Mao, G. Luo, H.-Y. Hao, Z.-H. Hu, S.-J. Lou and D.-Q. Xu, Chem. Commun., 2019, 55, 14458 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of ionic liquid synthesis, continuous flow experiments and analytics. See DOI: 10.1039/d1gc04189d |
| This journal is © The Royal Society of Chemistry 2022 |