
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cleavage of C–C double bond in cinnamic acids with hydrogen peroxide catalysed by vanadium(V) oxide†
Monika
Horvat
and
Jernej
Iskra
 *
*
University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: jernej.iskra@fkkt.uni-lj.si
First published on 31st January 2022
Abstract
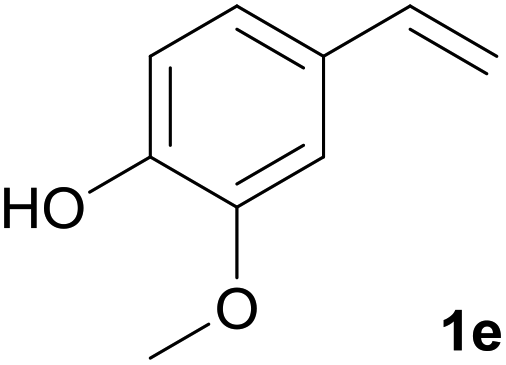
We have developed a cheap, green, mild and environmentally friendly method for the selective cleavage of carbon–carbon double bonds with a 30% aqueous solution of hydrogen peroxide as the oxidant and vanadium(V) oxide as the catalyst. The selectivity of the oxidative cleavage of cinnamic acid derivatives 1 depends on the substituents and the solvent used (DME – MeOCH2CH2OMe, TFE – 2,2,2-trifluoroethanol or MeCN). In DME, p-hydroxy derivatives were selectively converted to benzaldehyde derivatives 2, in TFE, oxidative cleavage led to the formation of benzoquinone derivatives 4, while in MeCN, cinnamic acid derivatives were selectively converted to benzoic acid derivatives 3. Ferulic acid 1a was quantitatively and selectively converted to vanillin 2a in a 91% isolated yield on a gram scale. Dimeric difurandione 1a′ was isolated as an intermediate, which was confirmed by in situ ATR-IR spectroscopy, while the formation of diols or epoxides was not observed. The analogous styrene derivative, 4-vinylguaiacol 1e was also selectively converted to either vanillin 2a or 2-methoxyquinone 4a in a high yield. The green metric for the conversion of ferulic acid to vanillin by different methods was calculated and compared to our method, and showed that our method has better environmental parameters.
Introduction
The selective oxidative cleavage of carbon–carbon double bonds is a synthetically important reaction to introduce oxygen functionality into molecules or to degrade complex compounds, especially those from natural sources and biomass, i.e. oils, terpenes, or cinnamic acids. It is important to prepare aromatic compounds from lignin, a sustainable alternative to conventional crude oil based methods.1–3 This is also a fundamental reaction in the industrial organic synthesis and valorisation of biomass i.e. oils and terpenes.4In recent decades, researchers have devoted much attention to this topic, and many protocols have been proposed. The standard method for the oxidative cleavage of olefins into aldehydes, ketones, and carboxylic acids is ozonolysis.5–7 However, the ozone is a highly reactive, ground-level air pollutant and a dangerous, toxic, corrosive, unstable and very potent oxidation reagent. Alternatively, many other oxidants have been investigated to achieve the selective oxidative cleavage of double bonds, e.g. OsO4/NaIO4 (Lemieux–Johnson reaction),8,9 Oxone,10,11m-CPBA,12t-BuOOH,13–15 KMnO4,16,17etc. These methods also have some disadvantages, such as toxicity, high price, and the generation of a stoichiometric oxidant as waste. Using biobased molecules as feedstock demands the use of environmentally friendly oxidants such as oxygen and hydrogen peroxide. Oxygen is used because of its lower cost, atom efficiency and environmental friendliness. The aerobic oxidative cleavage of carbon–carbon double bonds is very promising; however, improvements are still needed to compete with conventional oxidants.18–24 Hydrogen peroxide is similar to oxygen in that water is the only by-product, while the reactions can be carried out in a homogeneous phase.25–27 The use of H2O2 for the oxidative cleavage of olefins has been described in several articles, however there is a lack of simple and cost-effective methods.28–34
Cinnamic and ferulic acid derivatives are readily available and found in many plants, fruits, vegetables, etc.35,36 Ferulic acid, the most abundant natural aromatic compound, is found in the lignocellulosic biomass in the cell walls of plants, grasses, vegetables, flowers, leaves, seeds and nuts.37–39 Therefore, this class of compounds is interesting feedstock for various aromatic molecules and environmentally friendly processes are being sought for the production of various aromatic molecules. Some methods are known for converting the cinnamic acid model compounds into corresponding benzaldehydes or benzoic acids using H2O2, but the choice of substrates is extremely limited.40,41 The conversion of ferulic acid to vanillin is one of the best-known examples. Biotechnological processes proceed selectively, but many auxiliary components are required in a process that yields a dilute aqueous solution of vanillin.42 Synthetic processes with green oxidants are limited to hydrogen peroxide using catalysts, such as a metal–organic framework HKUST-1,43 metal–organic polyhedron Cu(II),44 Cu-MOF-7445 or photocatalytic Cu/TiO2.46 The selectivity of ferulic acid to vanillin conversion poses a serious problem due to further oxidation to vanillic acid.
We present a study on the development of a simple and cost-effective method for the oxidative cleavage of the C–C double bond in cinnamic acid derivatives with the use of vanadium(V) oxide and hydrogen peroxide, where the choice of solvent determines the selectivity towards benzaldehydes, benzoic acids, and benzoquinones. The selectivity and efficiency of the process was demonstrated on the ferulic acid to vanillin conversion. The green metric was used to evaluate the conversion of ferulic acid to vanillin with other similar methods.
Results and discussion
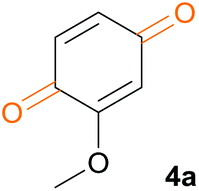
Ferulic acid 1a was used as a model substrate for the oxidative cleavage of the carbon–carbon double bond with a 30% aqueous solution of hydrogen peroxide. Various simple transition metal salts were tested as catalysts (Fe2O3, ZrBr2, (NH4)2MoO4, RuCl3, NiCl2, Co2(CO)8, Na2MoO4, CuBr, CuBr2, Na3VO4, NH4VO3, VO(acac)2, VOSO4·H2O, VCl2 and V2O5) (Table 1). The reaction was carried out in acetonitrile using 7 equiv. of H2O2 and 0.1 equiv. of catalyst at room temperature for 2 h. In the absence of a catalyst, the reaction did not take place. The same was observed in the presence of Fe2O3, ZrBr2 and (NH4)2MoO4 (Table 1, entries 1–3). When RuCl3, NiCl2, Co2(CO)8 and Na2MoO4 were used, some conversion occurred but the formation of vanillin 2a was not observed. Instead, a small amount (2–17%) of dimeric product 1a′ (vide supra) was formed (Table 1, entries 4–7, Scheme 1). Copper catalysts, CuBr and CuBr2, were found to be moderately effective in catalysing the conversion of 1a to 2a (Table 1, entries 8 and 9). Vanadium catalysts were the most efficient for the conversion to 2a, but further oxidation to vanillic acid 3a and 3-methoxy-1,2-benzoquinone 4a was also observed. The best result was obtained with V2O5 (68% of 2a) (Table 1, entry 15). The yield of 2a did not change when the amount of V2O5 was reduced to 5 mol% and further reactions were carried out with 0.05 equiv. of V2O5. | ||
| Scheme 1 Transformation of ferulic acid 1a to vanillin 2avia intermediate 1a′. | ||
| Entry | Catalyst | 1a [%] | 1a′ [%] | 2a [%] | 3a [%] | 4a [%] |
|---|---|---|---|---|---|---|
| Reaction conditions: 1a (0.1 mmol), 30%aq H2O2 (0.7 mmol), catalyst (0.01 mmol), MeCN (1 mL), rt, 2 h.a Conversion was determined by 1H NMR.b V2O5 (0.005 mmol).c V2O5 (0.001 mmol). | ||||||
| 1 | Fe2O3 | 100 | 0 | 0 | 0 | 0 |
| 2 | ZrBr2 | 100 | 0 | 0 | 0 | 0 |
| 3 | (NH4)2MoO4 | 100 | 0 | 0 | 0 | 0 |
| 4 | RuCl3 | 98 | 2 | 0 | 0 | 0 |
| 5 | NiCl2 | 97 | 3 | 0 | 0 | 0 |
| 6 | Co2(CO)8 | 92 | 8 | 0 | 0 | 0 |
| 7 | Na2MoO4 | 83 | 17 | 0 | 0 | 0 |
| 8 | CuBr | 79 | 10 | 11 | 0 | 0 |
| 9 | CuBr2 | 32 | 21 | 39 | 8 | 0 |
| 10 | Na3VO4 | 87 | 9 | 4 | 0 | 0 |
| 11 | NH4VO3 | 75 | 11 | 14 | 0 | 0 |
| 12 | VO(acac)2 | 0 | 6 | 66 | 9 | 19 |
| 13 | VOSO4·H2O | 1 | 15 | 57 | 15 | 12 |
| 14 | VCl2 | 0 | 0 | 57 | 13 | 30 |
| 15 | V2O5 | 0 | 0 | 68 | 8 | 24 |
| 16 | V2O5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
0 | 0 | 68 | 10 | 22 |
| 17 | V2O5c |
76 | 13 | 11 | 0 | 0 |
Next, we tested different solvents for the V2O5-catalysed C–C bond cleavage of 1a with H2O2. As can be seen from the results in Table 2, the solvent has an important effect on the transformation.
| Entry | Solvent | 1a [%] | 1a′ [%] | 2a [%] | 3a [%] | 4a [%] |
|---|---|---|---|---|---|---|
| Reaction conditions: 1a (0.1 mmol), 30%aq H2O2 (0.7 mmol), V2O5 (0.005 mmol), solvent (1 mL), rt, 2 h.a Conversion was determined by 1H NMR.b 8% of ethyl vannilate was formed. | ||||||
| 1 | H2O | 100 | 0 | 0 | 0 | 0 |
| 2 | TFA | 100 | 0 | 0 | 0 | 0 |
| 3 | DMSO | 100 | 0 | 0 | 0 | 0 |
| 4 | TFE | 0 | 0 | 0 | 0 | 100 |
| 5 | DMC | 64 | 5 | 16 | 0 | 15 |
| 6 | Isopropanol | 29 | 10 | 45 | 6 | 10 |
| 7 | Diethyl ether | 80 | 5 | 11 | 0 | 4 |
| 8 | EtOAc | 59 | 3 | 8 | 0 | 30 |
| 9 | Hexane | 58 | 8 | 24 | 5 | 5 |
| 10 | DCM | 0 | 40 | 32 | 24 | 4 |
| 11 | MeOH | 19 | 22 | 28 | 22 | 9 |
| 12 | THF | 53 | 1 | 39 | 5 | 2 |
| 13 | EtOH | 0b | 0 | 59 | 11 | 22 |
| 14 | MeCN | 0 | 0 | 68 | 8 | 24 |
| 15 | DME | 0 | 5 | 95 | 0 | 0 |
No reaction took place in water, TFA or DMSO (Table 2, entries 1–3). Reactions in other solvents were nonselective for 2a, with the notable exception of DME, where no overoxidation to 3a and 4a was observed (Table 2, entry 15), 95% of vanillin 2a was formed, and 5% of intermediate 1a′ remained. The oxidative cleavage of the C–C double bond occurred quantitatively in DCM, however the reaction was not selective (Table 2, entry 10). An interesting result was observed in THF where the selectivity was good but other products were formed (Table 2, entry 12). The NMR spectra show that THF reacted during the reaction and was oxidized to 2-hydroxytetrahydrofuran and γ-butyrolactone (ESI Scheme S2.1 and Fig. S2.1†). In ethanol and acetonitrile, the reaction was quantitative (Table 2, entries 13 and 14), the selectivity for 2a was better in acetonitrile than in ethanol, and 8% of ethyl vanillate was also formed in EtOH. The selectivity of the reaction in MeCN was not improved when the reaction was carried out at a lower temperature (0 °C) or when H2O2 was added in four portions. Only when DME was used as a solvent was the further oxidation of vanillin 2a to vanillic acid 3a sufficiently slow to allow for the selective formation of vanillin 2a. Fluorinated alcohols are activating solvents for oxidations,47,48 and the use of TFE resulted in the complete conversion of 1a to benzoquinone 4a, which was isolated in an 84% yield (Table 2, entry 4).
The reaction was further optimized by changing the amount of hydrogen peroxide (Table 3). Decreasing the amount of hydrogen peroxide to 5 equiv. resulted in a lower conversion, while a higher temperature only slightly improved the conversion. The quantitative conversion of 1a to 2a occurred with 7 equiv. of H2O2 for 3 h or 30 min at 60 °C (Table 3, entries 5 and 6). Further increasing H2O2 to 10 equiv. resulted in a partial overoxidation to 3a. The concentration of hydrogen peroxide (3% vs. 60% aq. solution) is also important. A higher concentration leads to the formation of 3a in a shorter time, while more dilute H2O2 is not strong enough.
| Entry | Equiv. of H2O2 | 1a [%] | 1a′ [%] | 2a [%] | 3a [%] |
|---|---|---|---|---|---|
| Reaction conditions: ferulic acid 1a (0.1 mmol), 30%aq H2O2 (0.2–1.0 mmol), V2O5 (0.005 mmol), DME (1 mL), rt, 3 h. Conversion was determined by 1H NMR.a 2 h, 60 °C.b 0.5 h, 60 °C.c 3%aq H2O2.d 3%aq H2O2, 24 h.e 60%aq H2O2. | |||||
| 1 | 2 | 70 | 8 | 22 | 0 |
| 2 | 3 | 69 | 8 | 23 | 0 |
| 3 | 5 | 37 | 9 | 54 | 0 |
| 4 | 5a | 36 | 2 | 62 | 0 |
| 5 | 7 | 0 | 0 | 100 | 0 |
| 6 | 7b | 0 | 0 | 100 | 0 |
| 7 | 7c | 91 | 4 | 5 | 0 |
| 8 | 7d | 86 | 4 | 10 | 0 |
| 9 | 7e | 0 | 0 | 85 | 15 |
| 10 | 10 | 0 | 0 | 86 | 14 |
We followed the conversion of 1a to 2a with 30%aq H2O2 (7 equiv.) and 5 mol% of V2O5 by NMR spectroscopy (Fig. 1). After 2 h of reaction, the conversion of 1a was 100% complete, but a small amount of the intermediate 1a′ was still present (5%). The latter was completely converted to product 2a after 3 h, while further conversion to vanillic acid 3a started after 4 h of reaction. Despite the longer reaction time, no quinone 4a was detected in DME.
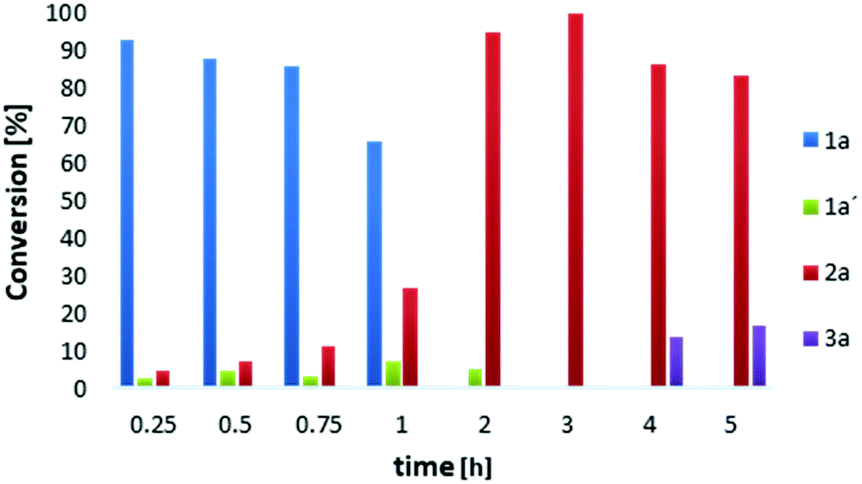 | ||
| Fig. 1 Reaction profile for vanillin 2a formation in DME. Reaction conditions: 1a (0.1 mmol), 30%aq H2O2 (0.7 mmol), V2O5 (0.005 mmol), DME (1 mL), rt. | ||
The reaction in DME was followed by in situ ATR-IR spectroscopy, but we had to use 100% H2O2 to avoid the peak of water in the IR spectrum (Fig. 2). Consequently, some acid 3a was also formed. The ATR-IR experiment confirmed that the product 1a′ with a characteristic peak at 1704 cm−1 is an intermediate product. When 100% H2O2 was used, the selectivity was lost and 3a (1521 cm−1) started to form after 2 hours, which slowly increased in the last phase of the reaction (Fig. 2 and ESI Fig. S2.2†). The same reaction course was observed when this reaction was followed by NMR spectroscopy (ESI Fig. S2.3†). Similarly, an in situ ATR-IR experiment was also performed for the reaction in TFE. The reaction proceeded too quickly to observe the formation of 1a′ and the signals of benzoquinone 4a predominated (ESI Fig. S2.4†).
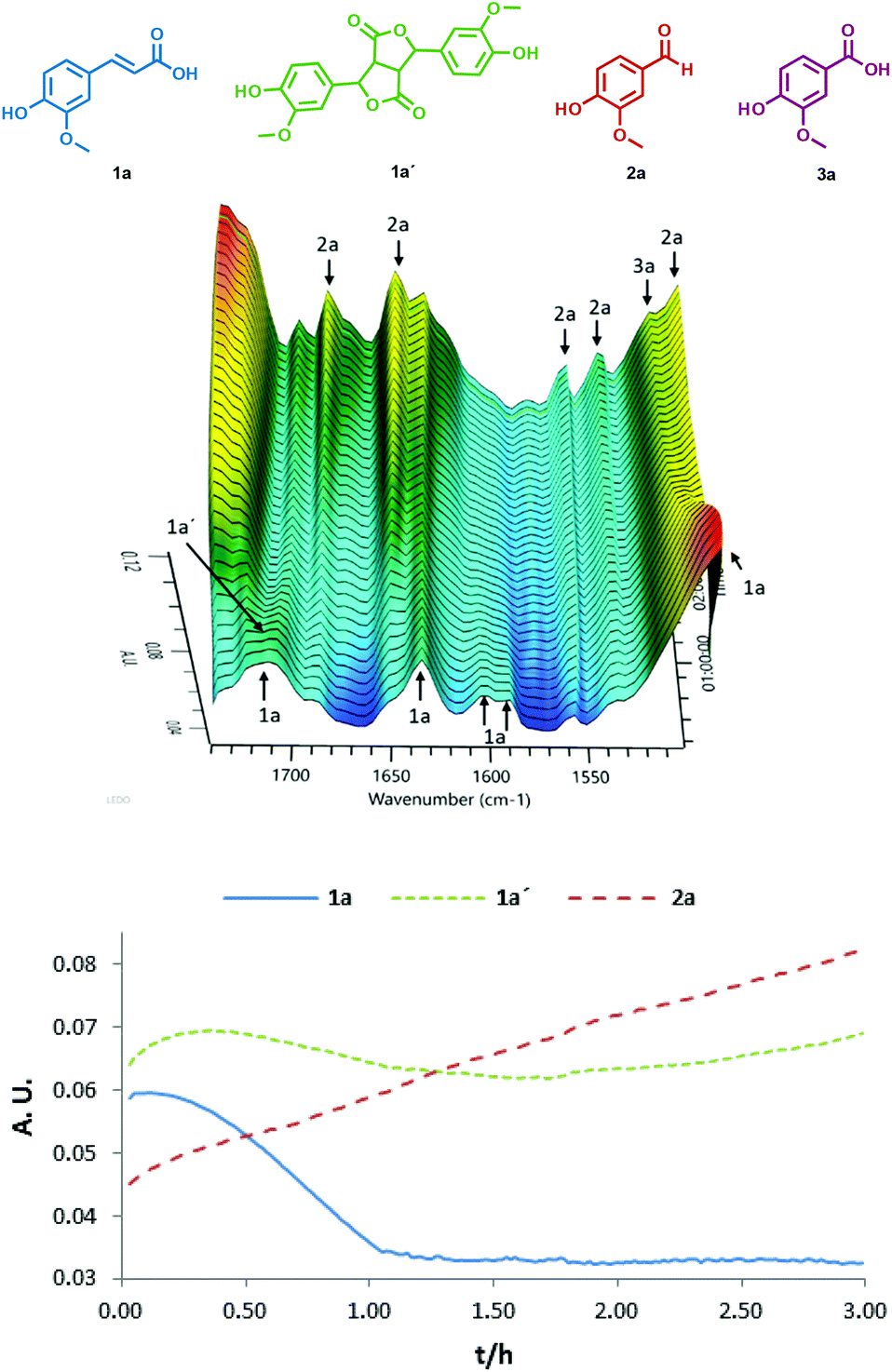 | ||
| Fig. 2 ATR-IR of the reaction of ferulic acid 1a (1517 cm−1) with 100% H2O2. Reaction conditions: 1a (6.2 mmol), 100% H2O2 (30.0 mmol), V2O5 (0.3 mmol), DME (25 mL), rt. | ||
To investigate the mechanism of the reaction, the experiments were performed with the addition of TEMPO, which is used as a radical quencher, and AIBN as a radical initiator. Upon the addition of TEMPO, the reaction was inhibited, indicating that the reaction proceeds via a radical pathway. On the other hand, the addition of the initiator AIBN resulted in the faster formation of vanillin 2a (Table 4).
Based on the experimental results, the oxidative cleavage of the C–C double bond occurs via a radical mechanism. The formation of the intermediate 1a′ indicates an initial reaction at the double bond, in which the OH group at the para position could play an important role in stabilizing the intermediate. The formation of epoxides and diols was not observed using NMR spectroscopy.
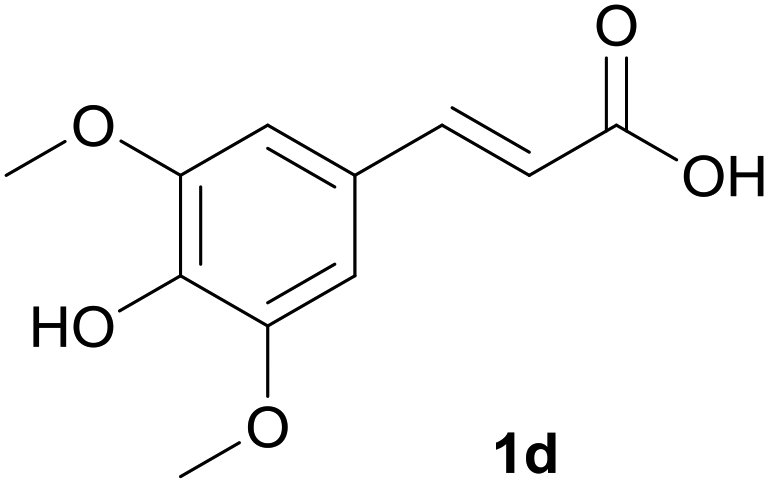
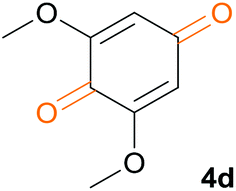
We tested this procedure on other derivatives containing an aromatic ring from the lignin core (Table 5). Ferulic acid 1a, its ethyl ester 1b, and p-coumaric acid 1c were converted to their corresponding aldehydes in excellent yields, however caffeic acid 1d was very reactive and benzoquinone 4d was always the major product, regardless of the reaction conditions, amount of oxidant, and time. The best yield of an aldehyde was only 15%. The reaction was also effective with a styrene derivative 1e.










Under the same reaction conditions, but replacing DME with a more activating solvent 2,2,2-trifluoroethanol (TFE), benzoquinones 4 were selectively prepared in good yields (79–91%), while the reaction proceeded at room temperature for 2 h (Table 6).





The reaction was also tested on various substrates without the presence of the p-hydroxy substituent. In all of the cases, the conversion of the starting substrates 1 did not occur in the presence of the solvent DME. In acetonitrile, the reaction was unselective for vanillin 2a, while the conversion to vanillic acid 3a was very high. Hence, a synthesis of aromatic benzoic acids 3 by the oxidative cleavage of cinnamic acid derivatives 1 was investigated.
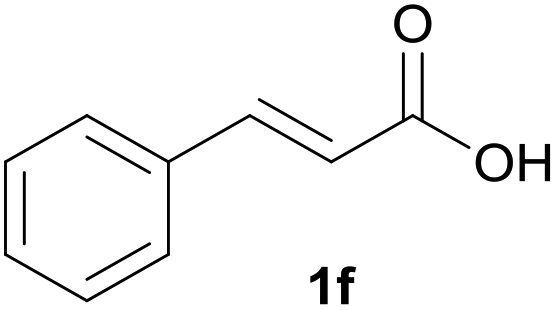
Numerous reaction conditions were carried out to find the optimum reaction conditions for the selective synthesis of benzoic acid 3f from cinnamic acid 1f (Fig. 3). Decreasing the amount of catalyst from 1 mol% to 0.5 mol% decreased the yield by 3–22%, depending on the amount of hydrogen peroxide. In contrast, the reaction yield was not improved when the amount of catalyst was increased to 0.3 equiv. The results show that 1 mol% of the vanadium catalyst gave the best result. Screening the amount of hydrogen peroxide showed that it had a very large effect on the reaction. Increasing the amount of oxidant led to more oxidative cleavage of the C–C double bond in 1f, resulting in the synthesis of benzaldehyde 2f and benzoic acid 3f. The complete conversion was only achieved with 30 equiv. of hydrogen peroxide. In these reactions, temperature did not have much of an influence. Comparing the results at room and reflux temperature (green columns and blue columns, respectively, in Fig. 3) with the same equivalence of catalyst and oxidant, the difference in conversion is only 3–24%.
 | ||
| Fig. 3 Oxidative cleavage of the C–C double bond of cinnamic acid 1f under different reaction conditions: cinnamic acid 1f (0.1 mmol), 30%aq H2O2 (5–30 equiv.), catalyst (0.005–0.3 equiv.), MeCN (1 mL), rt/Δ (60 °C), 24 h. | ||
Having found the optimal reaction conditions, we tested the general validity of the protocol for the oxidative cleavage of the carbon–carbon double bond in the cinnamic acid derivatives to the corresponding benzoic acid derivatives with 30%aq H2O2 catalyzed by V2O5 in acetonitrile (Table 7). All of the reactions resulted in a complete substrate conversion. The C–C double bond of the cinnamic acid derivatives 1 was effectively cleaved under optimized reaction conditions to afford benzoic acids 3 in excellent yields (84–94%). The presented protocol tolerates the presence of several functional groups on the aromatic ring. Compounds 1k, 1l, 1m and 1o were not sufficiently soluble, and a combination of acetonitrile and TFA (1:1) had to be used to achieve quantitative conversion.
|
|
|||
|---|---|---|---|
| Substrate | Product | Cond. | Yielda |
| Reaction conditions A: substrate 1 (0.5 mmol), 30%aq H2O2 (14.0 mmol), V2O5 (1 mol%), MeCN (5 mL), rt. Reaction conditions B: substrate 1 (0.5 mmol), 30%aq H2O2 (20.0 mmol), V2O5 (0.005 mmol), MeCN/TFA (1/1, 5 mL), rt.a Isolated yield. | |||
|
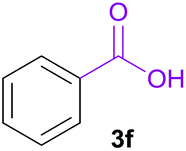
|
A | 94% |

|

|
A | 88% |

|

|
A | 91% |

|
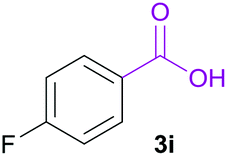
|
A | 86% |

|

|
A | 90% |
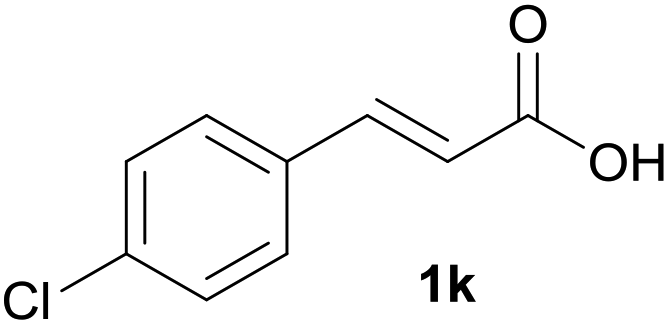
|

|
B | 87% |
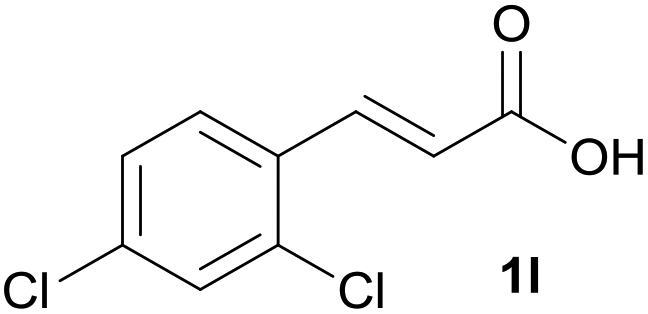
|

|
B | 93% |

|

|
B | 85% |
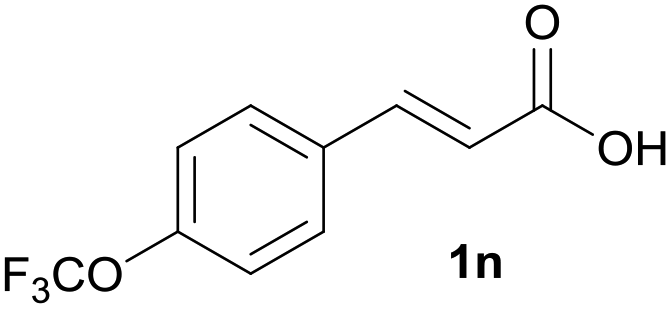
|

|
A | 91% |
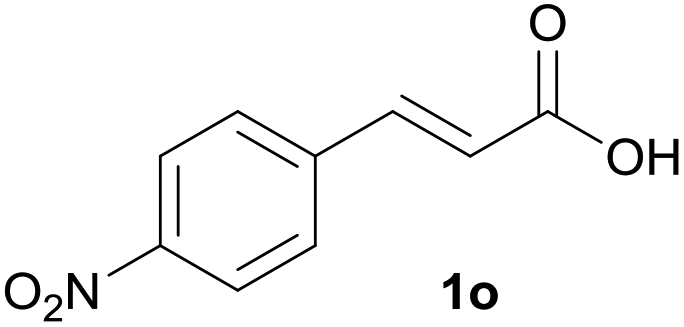
|
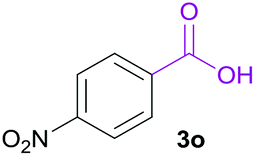
|
B | 84% |

|
|
A | 90% |

|

|
A | 89% |

To confirm the synthetic value of this method, the oxidation of ferulic acid to the important molecule vanillin was performed on a scale of 6.2 mmol (1.2 g) and after 3 h at 22 °C, 91% pure fragrant vanillin was isolated (Scheme 2).
 | ||
| Scheme 2 The gram-scale synthesis of vanillin 2a from ferulic acid 1a. | ||
We evaluated the green metrics for the conversion of ferulic acid 1a to vanillin 2a, focusing on the use of the green oxidants—H2O2 and O2—using a template proposed by Andraos.49 There is no research reporting on the aerobic oxidation of ferulic acid with useful data for calculating the green metrics. We chose a biotechnological method using the immobilized enzymes decarboxylase Fdc and oxygenase CsO2.50 The authors reported obtaining 2.5 mg of vanillin per 1 mL after ten reaction cycles, however, no isolation was reported. Three suitable methods were found with H2O2 using the catalysts HKUST-1,43 Cu(II)-MOP44 and Cu-MOF-74.45 Unfortunately, all of the data needed to calculate the green metrics are not available (the amount of solvent and additives in the work-up and the amount of silica gel and solvents in the isolation step). Furthermore, the isolation of vanillin 2a from the reaction mixture on an industrial scale would be quite different and the method used on a smaller scale is not as relevant. Hence, we made a three-level comparison – synthesis with workup, synthesis with workup and isolation and the synthesis with potential reuse of components. Where data in the original procedure was missing, we assumed that they used the same amounts of materials for the workup and isolation as we did since the reactions were similar. The parameters of the green metrics for the first level comparison are listed in Table 8 and on a pentagram in Fig. S2–5,† while the analysis of the whole process can be found in the ESI (xls file†). The atom economy was based on the reaction equation and is the same for all of the studied processes with hydrogen peroxide, while the biotechnological process has a better one. A major drawback is in the MRP and RME parameters, where the V2O5 method performs much better, but there is still room for improvement, especially in solvent recovery (ESI†). The E-factor defines the amount of waste produced per mass of product. It is obvious that there are significant differences in the E-factor (Table 8). The biotechnological process has a higher E-factor due to the highly diluted reaction media and additives. On the other hand, the catalytic processes have significant differences in the E-factor due to the differences in the amount of solvent and selectivity of the reaction as well as an excess of reagents. Again, the V2O5 catalytic process produces the least amount of waste.
| This article | HKUST-143 | Cu(II)-MOP44 | Cu-MOF-7445 | Biotech.50 | |
|---|---|---|---|---|---|
| AE (atom economy), SF (stoichiometric factor), MRP (materials recovery parameter), RME (reaction mass efficiency), E-factor (mass of waste/mass of 2a). | |||||
| Yield | 91% | 95% | 60% | 71% | 17% |
| AE | 58% | 58% | 58% | 58% | 78% |
| 1/SF | 0.623 | 0.307 | 0.294 | 0.193 | 1.00 |
| MRP | 0.107 | 0.0052 | 0.049 | 0.081 | 0.005 |
| RME | 3.5% | 0.2% | 0.5% | 0.6% | 0.1% |
| E-Factor | 28 | 430 | 198 | 154 | 1542 |
Experimental
Materials and methods
All of the chemicals and reagents were obtained from commercial suppliers (Sigma-Aldrich, Fluorochem) and were used as received without further purification. The TLC was performed on Merck-60-F254 plates using mixtures of EtOAc/heptane (2/1). The crude products were purified by column chromatography on a silica gel (63–200 μm, 70–230 mesh ASTM; Fluka). The products were characterized by 1H and 13C NMR spectroscopy, IR spectroscopy and HRMS. The 1H and 13C spectra were recorded on Bruker Avance III 500 instruments using TMS as the internal standard. The IR spectra were measured on a PerkinElmer 2000 Fourier transform infrared spectroscope. The HRMS data were recorded with an Agilent 6224 Accurate Mass TOF LC/MS System.FTIR: The spectra were recorded with a ReactIR™ 45 instrument with a resolution of 4 cm−1, averaging 128 scans. An EasyMax 102 controller was used to control the reaction conditions during the reaction. The instruments were controlled by iControl EasyMax 4.2 and iC IR 4.2 software.
General procedure for aldehyde synthesis
In a 10 mL volumetric flask, substrate 1 (0.5 mmol) and the V2O5 catalyst (0.05 equiv.) were added to a 5 mL solution of the solvent DME. The 30%aq H2O2 (0.36 mL, 7 equiv.) was first purged with argon and slowly added to the reaction mixture in three portions. The mixture was stirred at room temperature and monitored with TLC. After completion of the reaction, the reaction mixture was extracted with EtOAc (2 × 5 mL). The organic layer was evaporated under vacuum. The crude reaction was subjected to column chromatography with the mobile phase EtOAc/heptane (2/1). The solvent was evaporated in vacuo to provide the product.General procedure for benzoquinone synthesis
In a 5 mL volumetric flask, substrate 1 (0.2 mmol) and the V2O5 catalyst (0.05 equiv.) were added to a 2 mL solution of the solvent TFE (2,2,2-trifluoroethanol). The 30%aq H2O2 (0.144 mL, 7 equiv.) was first purged with argon and slowly added to the reaction mixture. The mixture was stirred at room temperature for 2 h. After completion of the reaction, the reaction mixture was extracted with EtOAc (2 × 3 mL). The organic layer was evaporated under vacuum. The crude reaction was subjected to column chromatography with the mobile phase EtOAc/heptane (2/1). The solvent was evaporated in vacuo to provide the product.General procedure for aromatic benzoic acid synthesis (conditions A)
In a 10 mL volumetric flask, substrate 1 (0.5 mmol) and the V2O5 catalyst (0.01 equiv.) were added to a 5 mL solution of the solvent MeCN. The 30%aq H2O2 (1.4 mL, 28 equiv., 14.0 mmol) was first purged with argon and slowly added to the reaction mixture in three portions. The mixture was stirred for 24 h at room temperature. After completion of the reaction, the reaction mixture was extracted with EtOAc (2 × 5 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The crude reaction was subjected to column chromatography with the mobile phase EtOAc/heptane (2/1). The solvent was evaporated in vacuo to provide the product.General procedure for acid synthesis (conditions B)
In a 10 mL volumetric flask, substrate 1 (0.5 mmol) and the V2O5 catalyst (0.01 equiv.) were added to a 5 mL solution of the solvent mixture MeCN/TFA (1/1). The 30%aq H2O2 (2 mL, 40 equiv., 20.0 mmol) was first purged with argon and slowly added to the reaction mixture in three portions. The mixture was stirred for 24 h at room temperature. After completion of the reaction, the reaction mixture was extracted with EtOAc (2 × 5 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The crude reaction was subjected to column chromatography with the mobile phase EtOAc/heptane (2/1). The solvent was evaporated in vacuo to provide the product.Scale-up procedure for the preparation of vanillin from ferulic acid
In a 50 mL volumetric flask, ferulic acid (6.2 mmol, 1.2 g) and the V2O5 catalyst (0.03 equiv., 0.2 mmol, 37.5 mg) were added to a 25 mL solution of the solvent DME. The 30%aq H2O2 (7 equiv., 4.2 mL) was first purged with argon and slowly added to the reaction mixture at 22 °C. The mixture was stirred at 24 °C and monitored with TLC. After completion of the reaction (3 h), the reaction mixture was extracted with EtOAc (2 × 20 mL). The organic layer was evaporated under vacuum. The product vanillin (858 mg, 91%) was obtained after column chromatography purification (EtOAc/heptane (2/1)) as a white solid.Conclusions
We have developed a method for the oxidative cleavage of the carbon–carbon double bond in cinnamic acid and related compounds under mild reaction conditions using the green and environmentally friendly oxidant hydrogen peroxide and the cheap catalyst vanadium(V) oxide. The choice of solvent (DME, TFE, MeCN) determines the selectivity, and benzaldehydes, aromatic carboxylic acids or benzoquinones were selectively prepared in DME, MeCN or TFE, respectively. The hydroxy substituent in the para position of the aromatic ring is necessary for the selective synthesis of the benzaldehyde derivatives. The process also provides a simple and cost-effective alternative for the conversion of ferulic acid to vanillin in a high yield. Calculation of the green metrics for similar processes showed that the V2O5 catalysed process has much better green parameters than other methods.Author contributions
Conceptualization: J. I.; formal analysis: M. H.; funding acquisition: J. I.; investigation: M. H.; methodology: M. H., J. I.; project administration: J. I.; supervision: J. I.; visualization: M. H., J. I.; writing – original draft: M. H., J. I.; writing – review & editing: M. H., J. I.Conflicts of interest
Patent pending (LU102332).Acknowledgements
We thank the Slovenian Research Agency (P1-0134) and the APPLAUSE project for financial support. The APPLAUSE project is co-financed by the European Regional Development Fund through the Urban Innovative Actions (UIA) initiative. The authors are grateful to the staff of The Centre for Research Infrastructure, Faculty of Chemistry and Chemical Technology and the Department of Physical and Organic Chemistry, Jožef Stefan Institute (ATR-IR spectroscopy).Notes and references
- C. J. Allpress and L. M. Berreau, Coord. Chem. Rev., 2013, 257, 3005–3029 CrossRef CAS.
- A. P. Y. Chan and A. G. Sergeev, Coord. Chem. Rev., 2020, 413, 213213 CrossRef CAS.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- S. P. Teong, X. Li and Y. Zhang, Green Chem., 2019, 21, 5753–5780 RSC.
- K. Koike, G. Inoue and T. Fukuda, J. Chem. Eng. Jpn., 1999, 32, 295–299 CrossRef CAS.
- G. Audran, S. R. A. Marque and M. Santelli, Tetrahedron, 2018, 74, 6221–6261 CrossRef CAS.
- Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Angew. Chem., Int. Ed., 2021, 60, 15138–15152 CrossRef CAS PubMed.
- W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217–3219 CrossRef CAS PubMed.
- S. Kim, J. Chung and B. M. Kim, Tetrahedron Lett., 2011, 52, 1363–1367 CrossRef CAS.
- B. R. Travis, R. S. Narayan and B. Borhan, J. Am. Chem. Soc., 2002, 124, 3824–3825 CrossRef CAS PubMed.
- J. N. Moorthy and K. N. Parida, J. Org. Chem., 2014, 79, 11431–11439 CrossRef CAS PubMed.
- F. V. Singh, H. M. S. Milagre, M. N. Eberlin and H. A. Stefani, Tetrahedron Lett., 2009, 50, 2312–2316 CrossRef CAS.
- M. M. Hossain, W.-K. Huang, H.-J. Chen, P.-H. Wang and S.-G. Shyu, Green Chem., 2014, 16, 3013–3017 RSC.
- T. M. Shaikh and F.-E. Hong, Adv. Synth. Catal., 2011, 353, 1491–1496 CrossRef CAS.
- Y.-L. Su, L. De Angelis, L. Tram, Y. Yu and M. P. Doyle, J. Org. Chem., 2020, 85, 3728–3741 CrossRef CAS PubMed.
- D. G. Lee, T. Chen and Z. Wang, J. Org. Chem., 1993, 58, 2918–2919 CrossRef CAS.
- S. Lai and D. G. Lee, Synthesis, 2001, 1645–1648 CrossRef CAS.
- G. Urgoitia, R. SanMartin, M. T. Herrero and E. Domínguez, ACS Catal., 2017, 7, 3050–3060 CrossRef CAS.
- W. Liu, G. Wu, W. Gao, J. Ding, X. Huang, M. Liu and H. Wu, Org. Chem. Front., 2018, 5, 2734–2738 RSC.
- A. Bhowmik and R. A. Fernandes, Org. Lett., 2019, 21, 9203–9207 CrossRef CAS PubMed.
- L.-F. Jia, H.-Z. Li, Z.-H. Li, R.-J. Li and G.-Y. Yang, Results Chem., 2021, 3, 100137 CrossRef CAS.
- J. Ou, S. He, W. Wang, H. Tan and K. Liu, Org. Chem. Front., 2021, 8, 3102–3109 RSC.
- J. Xu, Y. Zhang, X. Yue, J. Huo, D. Xiong and P. Zhang, Green Chem., 2021, 23, 5549–5555 RSC.
- Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005–10013 CrossRef CAS PubMed.
- A. Goti and F. Cardona, Peroxide in Green Oxidation Reactions: Recent Catalytic Processes, in Green Chemical Reactions, ed. P. Tundo and V. Esposito Hydrogen, Springer, Dordrecht, 2008, pp. 191–212 Search PubMed.
- A. S. Rao, H. R. Mohan and J. Iskra, in Encyclopedia of Reagents for Organic Synthesis, Wiley, Hydrogen Peroxide, 2013. DOI:10.1002/047084289X.rh040.pub2.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- C. Mi, L. Li, X.-G. Meng, R.-Q. Yang and X.-H. Liao, Tetrahedron, 2016, 72, 6705–6710 CrossRef CAS.
- D. Pyszny, T. Piotrowski and B. Orlińska, Org. Process Res. Dev., 2019, 23, 309–319 CrossRef CAS.
- C. Liu, J. Mao, X. Zhang and L. Yu, Catal. Commun., 2020, 133, 105828 CrossRef CAS.
- Y. Yang, B. Xu, J. He, J. Shi, L. Yu and Y. Fan, Appl. Catal., A, 2020, 590, 117353 CrossRef CAS.
- K. Peckh, D. Lisicki, G. Talik and B. Orlińska, Materials, 2020, 13, 4545 CrossRef CAS PubMed.
- L. Yu, H. Cao, X. Zhang, Y. Chen and L. Yu, Sustainable Energy Fuels, 2020, 4, 730–736 RSC.
- D. Yun, E. Z. Ayla, D. T. Bregante and D. W. Flaherty, ACS Catal., 2021, 11, 3137–3152 CrossRef CAS.
- N. Ruwizhi and B. A. Aderibigbe, Int. J. Mol. Sci., 2020, 21, 5712 CrossRef CAS PubMed.
- L. Chen, L. Zhang, G. Yan and D. Huang, Asian J. Org. Chem., 2020, 9, 842–862 CrossRef CAS.
- N. Kumar and V. Pruthi, Biotechnol. Rep., 2014, 4, 86–93 CrossRef PubMed.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- G. Banerjee and P. Chattopadhyay, J. Sci. Food Agric., 2019, 99, 499–506 CrossRef CAS PubMed.
- N. Anand, K. H. P. Reddy, K. S. R. Rao and D. R. Burri, Catal. Lett., 2011, 141, 1355 CrossRef CAS.
- S. R. Hart, D. C. Whitehead, B. R. Travis and B. Borhan, Org. Biomol. Chem., 2011, 9, 4741–4744 RSC.
- L. Yan, P. Chen, S. Zhang, S. Li, X. Yan, N. Wang, N. Liang and H. Li, Sci. Rep., 2016, 6, 34644 CrossRef CAS PubMed.
- R. Yepez, S. Garcia, P. Schachat, M. Sanchez-Sanchez, J. H. Gonzalez-Estefan, E. Gonzalez-Zamora, I. A. Ibarra and J. Aguilar-Pliego, New J. Chem., 2015, 39, 5112–5115 RSC.
- E. Sánchez-González, A. López-Olvera, O. Monroy, J. Aguilar-Pliego, J. Gabriel Flores, A. Islas-Jácome, M. A. Rincón-Guevara, E. González-Zamora, B. Rodríguez-Molina and I. A. Ibarra, CrystEngComm, 2017, 19, 4142–4146 RSC.
- J. G. Flores, E. Sanchez-Gonzalez, A. Gutierrez-Alejandre, J. Aguilar-Pliego, A. Martinez, T. Jurado-Vazquez, E. Lima, E. Gonzalez-Zamora, M. Diaz-Garcia, M. Sanchez-Sanchez and I. A. Ibarra, Dalton Trans., 2018, 47, 4639–4645 RSC.
- P. Gómez-López, N. Lázaro, G. C. Alvarado-Beltrán, A. Pineda, M. A. Balu and R. Luque, Molecules, 2019, 24, 3985 CrossRef PubMed.
- Š. Možina and J. Iskra, J. Org. Chem., 2019, 84, 14579–14586 CrossRef PubMed.
- Š. Možina, S. Stavber and J. Iskra, Eur. J. Org. Chem., 2017, 448–452 CrossRef.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
- T. Furuya, M. Kuroiwa and K. Kino, J. Biotechnol., 2017, 243, 25–28 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc04416h |
| This journal is © The Royal Society of Chemistry 2022 |