
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-step chemo-enzymatic synthesis of azelaic and pelargonic acids from the soapstock of high-oleic sunflower oil refinement†
Beatrice
Casali
,
Elisabetta
Brenna
 *,
Fabio
Parmeggiani
,
Francesca
Tentori
* and
Davide
Tessaro
*,
Fabio
Parmeggiani
,
Francesca
Tentori
* and
Davide
Tessaro
Dipartimento di Chimica, Materiali ed Ingegneria Chimica “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133, Milano, Italy. E-mail: mariaelisabetta.brenna@polimi.it; francesca.tentori@polimi.it
First published on 7th February 2022
Abstract
Soapstock recovery and manipulation represent one of the most burdensome aspects of the vegetable seed oil refining industry. In particular, soapstock splitting requires high amounts of concentrated acid and produces a low-value mixture of triglycerides and free fatty acids (oleins or acid oil), as well as huge volumes of acidulated wastewater. Oleins are currently converted into biodiesel or supplied to biodigesters, but alternative procedures to afford high-value products are sought after by the vegetable seed oil industry. In this paper, the valorization of soapstock from high-oleic sunflower oil refinement is investigated by biocatalytic methods. First, lipases are used to catalyze an efficient soapstock splitting, and reduce the environmental load of the procedure. Then, the high content of oleic acid (60–80%) is exploited by promoting its oxidative cleavage. Self-epoxidation of oleic acid by lipase-mediated perhydrolysis in the presence of H2O2 affords the corresponding epoxide, which is subsequently hydrolyzed to the diol derivative and oxidized to commercially valuable azelaic and pelargonic acids. The cleavage is performed using only sodium hypochlorite as an inexpensive and efficient oxidant. Epoxidation and glycol cleavage are optimized by a statistical approach and implemented under continuous-flow conditions to increase yields and productivity.
Introduction
Global production of vegetable oilseeds is forecast at nearly 600 million metric tons (Mt) for the year 2020–21, with a corresponding production of vegetable seed oil of 209 Mt.1 The refinement of such a high amount of oil is characterized by huge environmental footprint. Indeed, after extraction from seeds (mainly soybean and sunflower seeds), raw vegetable oil would not be suitable for human consumption because of the presence of different compounds (such as lecithin gums, free fatty acids, tocopherols, sterols, and squalene),2 hampering oil quality in terms of both organoleptic and nutritional qualities, and lowering its shelf life. Their removal is achieved through a process, producing a significant amount of waste. One of the most abundant by-products of vegetable oil refinement is the so-called soapstock, obtained by neutralization of raw oil with an alkaline solution (typically aqueous NaOH) to remove free fatty acids (FFAs). It consists of an alkaline emulsion containing approximately 50% water, 10% FFA sodium salts, 10% triglycerides and small percentages of partially hydrolyzed lipids (diacylglycerols and monoacylglycerols).2,3 Since about 6% of total refined oil volume is discharged as soapstock,4 its fate is a critical issue in vegetable oil refining, especially for its alkaline pH. In the refinement plant, soapstock, recovered by decantation, is generally treated with a concentrated acid solution (usually sulfuric or hydrochloric acid), and the resulting organic fraction is removed from water by settling and/or centrifugation.5 This effluent is a dark-coloured mixture of triglycerides, partially hydrolyzed derivatives and FFAs, containing also small amounts of mineral acids, phospholipids, and sterols.6 It is called high-acid oil or acidulated soapstock or oleins.7 Acid splitting of soapstock has several limitations: concentrated acid is required, the resulting acidic aqueous phase must be neutralized before disposal and the treatment of such wastewater is critical because of the high concentration of sulphate or chloride ions.6Being soapstock characterized by a wide variability in composition, it is considered a low-value source of FFAs, and it is currently employed in the production of soaps,8 biodiesel9 or for methane production in anaerobic biodigesters.10 It can also be added to animal feeding in limited and controlled amount.11 Increasing the sustainability of the vegetable seed oil refining process is an urging industrial call to be achieved by both reducing the environmental burden of the process and increasing the recycle of the related by-products.9
In this context, biocatalysis can offer interesting strategies. The enzymatic class of lipases is a valuable and broad source of effective catalysts for esterification or hydrolysis reactions, showing high efficiency, selectivity, stability and flexibility in a wide range of operative conditions. Lipases, many of which are produced industrially and marketed at a reasonable cost, can be employed in soapstock splitting as a convenient alternative to acid treatment, affording complete hydrolysis to the corresponding mixture of fatty acids.
Soapstock recovered from the refinement of high-oleic sunflower oil is characterized by 60–80% oleic acid (1, Fig. 1), which is of interest to the fine chemical industry, in particular as starting material for the production of pelargonic (2) and azelaic acids (3) by oxidative cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, currently carried out by ozonolysis.12 Pelargonic acid is used as a natural herbicide, a weed killer, a blossom thinner and for the synthesis of plasticizers, and flavours;13 azelaic acid is employed as an additive in anti-acne preparations and hair growth stimulators, as well as for the production of bio-based polyesters.14
C double bond, currently carried out by ozonolysis.12 Pelargonic acid is used as a natural herbicide, a weed killer, a blossom thinner and for the synthesis of plasticizers, and flavours;13 azelaic acid is employed as an additive in anti-acne preparations and hair growth stimulators, as well as for the production of bio-based polyesters.14
 | ||
| Fig. 1 Oleic acid and the products of its oxidative cleavage. | ||
A few years ago, our research group started a collaboration with Oleificio Zucchi, an Italian enterprise for vegetable seed oil refining settled in Cremona, aiming at the valorization of soapstock by biocatalytic methods. We started our investigation on high-oleic acid soapstock, and the first studies were performed on commercial oleic acid. In a recent paper,15 we discussed the use of a lipase-mediated Prilezhaev epoxidation of oleic acid with an immobilized form of Candida Antarctica lipase B (Novozym® 435) to obtain, after subsequent oxirane hydrolysis, threo-9,10-dihydroxystearic acid (4, Scheme 1), which represents the key intermediate towards the synthesis of azelaic and pelargonic acids.
 | ||
| Scheme 1 Synthesis of diol 4 from oleic acid. | ||
Oxidation to 9,10-dioxostearic acid (5) by means of catalytic quantities of Fe(NO3)3·9H2O, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), and NaCl in presence of atmospheric oxygen as stoichiometric oxidant (Scheme 2a) followed by cleavage with 35% aq. H2O2 completed the procedure to final products 2 and 3. In a further development of this work,16 we investigated the enzymatic oxidation of diol 4 by the recombinant alcohol dehydrogenase (ADH) from Micrococcus luteus NCTC2665, affording quantitative conversion into a mixture of the two regioisomeric hydroxyketones 6a,b, as an alternative to chemical oxidation (Scheme 2b). The final clevage to azelaic and pelargonic acids could be achieved by treatment with NaClO 10% aq. solution.
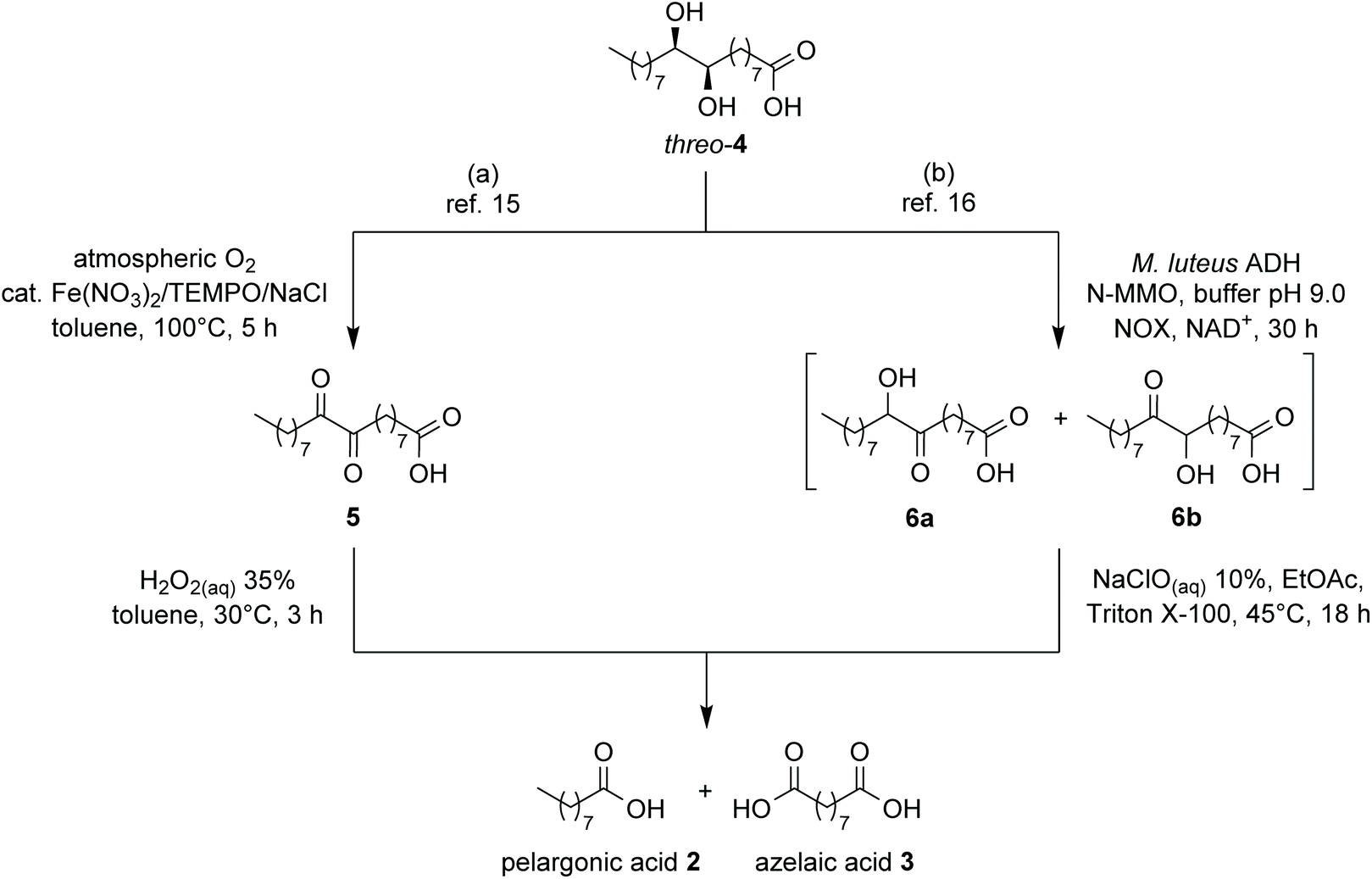 | ||
| Scheme 2 Conversion of diol 4 into pelargonic and azelaic acids, already described in our previous works. | ||
We herein report on the results we attained when high-oleic soapstock, provided by Oleificio Zucchi, was used as starting material. First, the lipase-mediated hydrolysis of soapstock was investigated, to obtain the corresponding mixture of FFAs according to a more sustainable procedure than the acid splitting employed at industrial level. The FFA mixture, enriched in oleic acid, was then submitted to Novozym® 435 mediated epoxidation to derivative 7, that was hydrolysed in situ to afford diol 4, recovered as a crystalline compound directly from the reaction mixture (Scheme 3). We studied the direct oxidation of diol 4 to acids 2 and 3 with aq. NaClO solution, thus avoiding the intermediate step to dioxo derivative 5 or hydroxyketones 6, described in our previous works. In order to increase the efficiency of the proposed synthesis, a statistical approach was applied to optimize both epoxidation and diol cleavage steps, and perform them in continuous-flow reactors.
 | ||
| Scheme 3 Multistep process for the conversion of soapstock into pelargonic and azelaic acids described in this work. | ||
Results and discussion
Enzymatic splitting of high-oleic sunflower oil soapstock
Enzymatic hydrolysis can represent a more effective and sustainable alternative to acid splitting, increasing the efficiency and reducing the associated waste. Several examples of soapstock treatment with lipases are described in the literature,17,18 although no reference could be found to any industrial large-scale application. Most of the published papers refer to lab-scale results: the lack of scale-up projects was mainly ascribed to high enzyme load and cost, long reaction times and low efficiency.17,19 Moreover, the high pH value of soapstock (9–10) usually has to be lowered in order to ensure the stability of most lipases, adding a further step to the process. Recently, Novozymes® has developed commercial enzymes working in a wide range of pH values,20 that are particularly suited to implement industrial-scale enzymatic splitting of soapstock. To achieve both the conversion of fatty acid sodium salts into the corresponding free acids, and the complete hydrolysis of residual triglycerides and phospholipids, a combination of the following preparations at pH 5 is suggested: lipase from Thermomyces lanuginosus (Eversa® Transform 2.0, aqueous solution) and two phospholipases, Novozym 40121 and Lecitase Ultra (aqueous solution).We received from Oleificio Zucchi samples of high-oleic sunflower oil soapstock showing pH ∼10. The molar ratio between triglycerides and free fatty acids was approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1H NMR analysis, see ESI†). No phospholipids were found in the samples (31P NMR analysis), thus only lipase Eversa® Transform 2.0 was employed. Soapstock was submitted to enzymatic hydrolysis without any acidification or pretreatment, by simply dispersing the material in water, and adding lipase Eversa® Transform 2.0 directly to the suspension. The reaction mixture was mechanically stirred at 25 °C. Conversion was monitored by 1H NMR (see ESI†) and complete hydrolysis was obtained after 12 h. After centrifugation of the final mixture, the organic phase (showing the following molar distribution of fatty acids by 1H NMR analysis: 80–87% oleic acid, 9% linoleic acid, 4–11% saturated acids, see ESI†) was recovered and submitted to the epoxidation step. Enzymatic soapstock hydrolysis proved to be a very advantageous and convenient alternative to acid splitting, occurring in mild reaction conditions, with high efficiency, producing smaller amounts of wastewater, that could be neutralized with phosphoric acid and disposed of with less drawbacks than that from acid splitting. Furthermore, since complete triglyceride conversion is accomplished, the corresponding product is richer in FFAs than commonly produced oleins.
:1 (1H NMR analysis, see ESI†). No phospholipids were found in the samples (31P NMR analysis), thus only lipase Eversa® Transform 2.0 was employed. Soapstock was submitted to enzymatic hydrolysis without any acidification or pretreatment, by simply dispersing the material in water, and adding lipase Eversa® Transform 2.0 directly to the suspension. The reaction mixture was mechanically stirred at 25 °C. Conversion was monitored by 1H NMR (see ESI†) and complete hydrolysis was obtained after 12 h. After centrifugation of the final mixture, the organic phase (showing the following molar distribution of fatty acids by 1H NMR analysis: 80–87% oleic acid, 9% linoleic acid, 4–11% saturated acids, see ESI†) was recovered and submitted to the epoxidation step. Enzymatic soapstock hydrolysis proved to be a very advantageous and convenient alternative to acid splitting, occurring in mild reaction conditions, with high efficiency, producing smaller amounts of wastewater, that could be neutralized with phosphoric acid and disposed of with less drawbacks than that from acid splitting. Furthermore, since complete triglyceride conversion is accomplished, the corresponding product is richer in FFAs than commonly produced oleins.
Chemo-enzymatic soapstock epoxidation (batch)
The most discussed example of soapstock valorization is the conversion into biodiesel,9 but no systematic evaluation of the economical applicability of the process has been reported in literature so far.2 Other added-value products, such as lipases21–23 and surfactants,24–26 have been obtained by using soapstock as medium for fermentation processes. The functionalization of the CC double bond of unsaturated FFAs as a strategy for soapstock valorization is scarcely documented. To the best of our knowledge, only Mashhadi et al.27 employed soapstock samples containing 16.7% by weight of total fatty acids to obtain natural epoxides to be studied as biodegradable plasticizers for PVC. The epoxidation reaction was promoted by producing peroxycarboxylic acids in situ by lipase-mediated perhydrolysis of the free fatty acids contained in the soapstock sample in the presence of H2O2.28Candida rugosa lipase was dispersed directly in soapstock and combined in a microchannel reactor with the aqueous phase containing H2O2. When the reaction was performed at 36 °C and pH 6.5, with a H2O2/CC molar ratio of 1.6:1 and a feed flow rate of 42 mL h−1, 85% epoxidation yield (with respect to the starting FFA content) was achieved.
In a previous paper,15 we used the same strategy for the self-epoxidation of commercial oleic acid, using an immobilized form of lipase B from Candida antarctica (Novozym® 435) as a catalyst. The epoxidation was carried out in acetonitrile as a solvent, because of its ability to solubilize both oleic acid and aqueous H2O2 35%. Final quantitative conversion was achieved in 5 h, at 50 °C with 0.15 M oleic acid and 0.27 M H2O2 with an enzyme loading of 5 g L−1. The reaction mixture was treated first with Na2SO3 sat. solution, then with H2SO4 2 M to promote oxirane hydrolysis. Compound 9,10-dihydroxystearic acid (4) was recovered directly by filtration (72%), after spontaneous crystallization from the reaction mixture. On the basis of these results, we planned the application of the same procedure to the mixture of FFAs produced by enzymatic hydrolysis of a sample of soapstock (from the refinement of high-oleic sunflower oil at Oleificio Zucchi) as described in the previous paragraph.
First, optimization of the epoxidation conditions in batch mode was carried out on the fatty acid mixture recovered from the enzymatic splitting of high-oleic sunflower soapstock (83% oleic acid), using a factorial design (elaborated through Minitab) according to the design of experiments (DOE) approach. This method allowed us to gain a better understanding of the system response to the reaction variables, with the final aim of finding the optimal conditions for continuous-flow implementation. A four-variables (oleic acid and H2O2 concentrations, temperature, and enzyme amount) factorial design, with a replicate for each point, was designed and analyzed through the software Minitab. We considered a range of 10–50 g L−1 of oleic acid concentration, that corresponded to 12–60 g L−1 of hydrolyzed mixture. As for the other conditions, we chose to evaluate system response to temperature in the range 30–50 °C, hydrogen peroxide concentration in the range 1–4% v/v (referred to 35% w/w aq. solution), Novozym® 435 in the range 1–2 g L−1. We observed that oleic acid concentration was by far the most influential parameter. In this transformation, oleic acid acts as both the substrate to be epoxidized, and the oxidant performing the epoxidation, after conversion into peroxy oleic acid. The best conversions (percentages evaluated by GC-MS analysis after treating samples with MeOH and trimethylsilyldiazomethane) were reached with the highest oleic acid concentration, likely for a positive effect on both equilibrium (shift towards the products) and reaction rate. The increase of enzyme load and temperature showed as well a beneficial effect on conversion, while H2O2 variation appeared almost irrelevant. The final optimized conditions were the following: 50 g L−1 oleic acid, 2 g L−1 Novozym® 435, 50 °C and 1% v/v H2O2 35% aq. solution.
When the reaction was performed under these conditions starting from 60 g L−1 of hydrolyzed soapstock (83% oleic acid by 1H NMR analysis), diol 4 was obtained in 40% yield in batch mode after 5 h. The presence of 9% linoleic acid (1H NMR analysis) in the starting mixture of fatty acids, had no influence on the quality of diol 4, that crystallized selectively from the reaction medium in pure form (35% isolated yield). In a separate experiment, we observed that when commercial linoleic acid 8 was submitted to epoxidation and hydrolysis in the same reaction conditions, no tetrahydroxy derivatives of stearic acid were formed, rather a complex mixture of stereoisomers of the two dihydroxy tetrahydrofuran derivatives 9 and 10 was obtained (Scheme 4). These compounds were identified by Li et al. (2018)29 as sea lamprey migratory pheromones, and fully characterized. Their formation was attributed to an intramolecular dehydration of dihydroxy derivatives formed by partial hydrolysis of the diepoxy stearic acid stereoisomers 11a and 11b. These compounds were also detected in the GC/MS analysis of the mother liquors after the filtration of diol 4.
 | ||
| Scheme 4 Products of epoxidation and oxirane hydrolysis of linoleic acid 8. | ||
Chemo-enzymatic soapstock epoxidation (continuous-flow)
Over the past decades, flow chemistry has gained exceptional interest in the field of organic synthesis for its advantages over conventional batch chemistry: due to the small size of flow reactors, the high surface to volume ratio affords a fast and efficient heat and mass transfer.30 The advantages are mainly related to an improvement of productivity, yield and safety, as well as a considerably easier scalability.In the paper by Mashhadi et al.,27 a biphasic aqueous–organic continuous system could be used for the lipase-mediated epoxidation of soapstock in a micro-channel reactor, since an aqueous solution of C. rugosa lipase was employed. In our work, Novozym® 435 was the catalyst of choice and, being an immobilized enzyme, a different configuration was devised.
A stirred-tank reactor was employed by Meyer et al.31,32 employed, working according to a CSTR mode, to promote the perhydrolysis of ethyl acetate by using a commercial formulation of immobilized C. antarctica lipase B dispersed in the liquid phase. We adopted a similar configuration, transferring into continuous-flow mode the best batch conditions found in our factorial design analysis. Although statistical analysis suggested to adopt the highest amount of oleic acid (50 g L−1, corresponding to 60 g L−1 of hydrolyzed soapstock), the maximum concentration of hydrolyzed soapstock we could obtain in MeCN was 52 g L−1 (corresponding to 43 g L−1 of oleic acid). This limitation was due to the fact that the solubility of oleic acid was lowered by the presence of other fatty acids and components in the starting mixture, and it had not been observed when lower amounts were used for DOE experiments in batch conditions.
To the final MeCN solution (160 mL), H2O2 35% (1.6 mL, 1% v/v) was added. The reactor volume was set to 45 mL and the flowrate at 150 μL min−1 to ensure a residence time of 5 h (equal to the reaction time in batch conditions). The reactor was kept at 50 °C and loaded with 90 mg of Novozym® 435 (corresponding to 2 g L−1 concentration). The outlet conversion was monitored every hour, for 12 h (Fig. 2): stationary state conversion was reached after 4 h and maintained for 4 h. From 8 h to 12 h a gradual decrease of conversion was observed, up to a final conversion of 45% in the last sample, probably due to a slow degradation of either the immobilized enzyme or its polymeric matrix caused by the oxidative reaction medium. The remaining solution inside the reactor was also analyzed, showing a similar conversion value (44%).
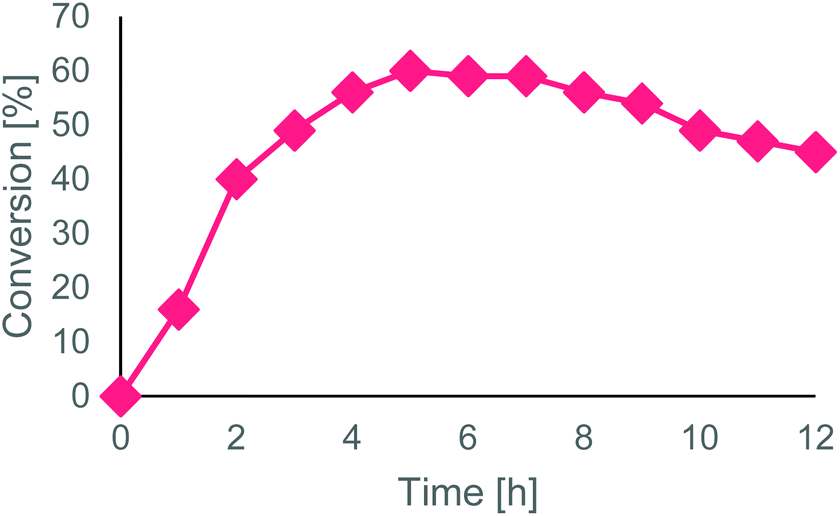 | ||
| Fig. 2 Enzymatic conversion of oleic acid from hydrolyzed soapstock (by GC/MS) in the CSTR during 12 h of continuous operating time (samples progressively collected from the output of the reactor during the reaction course). | ||
The fractions from 2 to 12 h, corresponding to a conversion value in the range of 40–60% (100 mL) and the solution inside the reactor (45 mL) were collected (total volume of 145 mL) and quenched by adding a saturated solution of Na2SO3 (6 mL). In the end, the solution was treated with 0.25 equiv. of H2SO4 2 M, to recover diol 4 as a pure and white solid after filtration. Starting from 7.3 g of hydrolyzed soapstock (corresponding to 6.2 g of oleic acid) we could recover 3.3 g of diol 4 (47% yield).
In order to compare the efficiency of batch and continuous-flow approaches, the space–time yield values (STY, amount of generated product per unit catalyst weight per unit time)33 were calculated in mmol gcat−1 h−1 according to the following formula.

nreagent (mmol) = amount of reagent; C (%) = conversion of the reagent into the desired product determined by GC/MS analysis; mN 435 (g) = Novozym® 435 load; Δt (h) = reaction time (for batch); τ (h) = residence time (for continuous-flow).
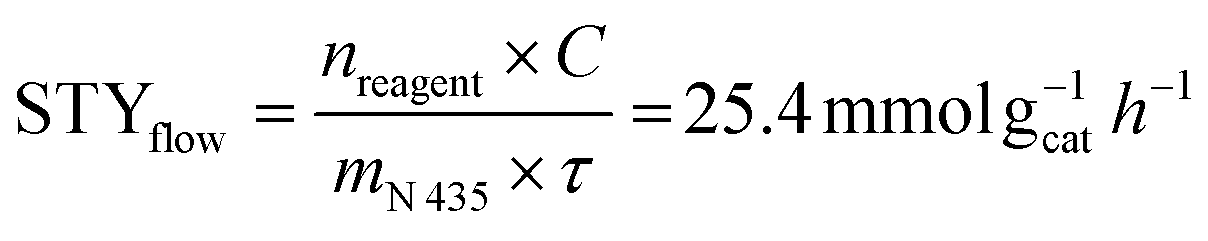
(i) Batch mode: 0.030 g Novozym® 435, 2.7 mmol oleic acid from hydrolyzed soapstock, 5 h reaction time, 40% conversion.
(ii) Continuous-flow mode: 0.090 g Novozym® 435, 22.0 mmol oleic acid from hydrolyzed soapstock, 5 h residence time, 52% conversion.
For the batch conditions, we considered the best conversion obtained using the factorial design. For the flow reaction, we took into account all the fractions collected from 2 to 12 h and the volume inside the reactor (145 mL) with a total epoxidation yield of nearly 52%.
As it can be observed, the reaction performed in a continuous-flow reactor led to an approximately three-fold higher STY with respect to the batch process, which accounts for the better exploitation of the same catalyst for a longer operation time.
Oxidative cleavage of diol 4 (batch)
Vicinal diol oxidative cleavage is a widely explored reaction, especially in carbohydrate chemistry.34 Typically, this transformation is carried out using either hypervalent iodine reagents, i.e., NaIO4, HIO4, Pb(OAc)4,34 or H2CrO4 and KMnO4 when a highly oxidant environment is required for the preparation of the corresponding (di)carboxylic acid. When milder reaction conditions are preferable, transition metal-based catalysts have to be employed, as ions (especially W35 and Mo36 derivatives) or nanoparticles.37 Unfortunately, these methods are often plagued by several disadvantages like the toxicity of the reagents, the harsh conditions required or the troublesome workups. Some alternative reagents for diol cleavage to (di)carboxylic acids have been recently proposed, such as tetrapropylammonium perruthenate38 or nitroxyl radicals like TEMPO.39A much more available and inexpensive oxidant, sodium hypochlorite, was tested also for glycol cleavage with appreciable results, favoring carboxylic acids instead of aldehydes as final products. In 2007, Khurana et al.40 successfully tested NaClO on various 1,2-diaryl- and dialkyl-1,2-diols, and in 2019 Kirihara et al.41 employed NaOCl·5H2O on sterically hindered trans-cyclic vicinal diols. The wide availability of NaClO attracted us as an interesting and economically viable replacement of traditional catalysts for the preparation of azelaic and pelargonic acids from diol 4. NaClO has already been reported to oxidize 4 into acids 2 and 3 in a patent by Lemaire et al. 2013:42 starting from 120 g L−1 of 4 in 1.1 M NaClO complete conversion into azelaic and pelargonic acids was achieved after 5 h. Unfortunately, in our hands this methodology did not provide the desired products with significant conversion. By diluting the system and increasing the reaction time, we obtained a 55% conversion of diol 4 after 3 days, with a concentration of 25 g L−1 in a 10% NaClO solution. Beyond 2 and 3, a small amount of the two isomers of hydroxyoxostearic acid (6a,b, Scheme 1) were formed, suggesting these are the intermediates in the cleavage of the glycol C–C bond. The addition of acetonitrile as a cosolvent to enhance the solubility of diol 4 in aqueous NaClO solution, as described in Khurana et al. (2007),40 had no significant effect to reduce reaction time and increase concentration.
The use of biphasic aqueous–organic systems to improve NaClO oxidation of poorly soluble molecules has been already studied,43 using tetrabutylammonium bromide to favor phase transfer. We tested a biphasic system EtOAc/aq. NaClO for the oxidative cleavage of diol 4 using either a quaternary ammonium salt (tetraethylammonium bromide), or a non-ionic surfactant (Triton-X 100) as a phase transfer catalyst. While the former prevented the reaction from happening and did not provide any conversion, the latter showed an impressive increase of conversion with respect to the previous monophasic experiments, affording complete conversion in 48 h. A further yield improvement was obtained by performing the reaction at 45 °C. Under these conditions, the biphasic approach showed a significant advantage in the isolation of products 2 and 3. We observed that pelargonic acid 2 was extracted completely in the still warm organic phase at the end of the reaction, while azelaic acid 3 was retained in the alkaline aqueous phase and could be isolated after treatment with a saturated solution of Na2SO3, acidification to pH 6 and extraction with EtOAc.
After these promising results, we attempted to optimize reaction efficiency by implementing a statistical analysis through a factorial DOE approach, already employed for the first step of this procedure. After some preliminary experiments that showed that surfactant concentration was not a strongly determining variable, we chose to keep substrate and surfactant concentrations constant In DOE investigation. For the application of the factorial design (see ESI†), the conversion of diol 4 (50 mg, 0.16 mmol) in EtOAc (5 mL, containing 1% Triton-X 100) into products 2 and 3 (determined by GC/MS analysis) was taken as the system response, choosing as variable parameters the total volume of water phase (in the range 3–5 mL), the molar ratio NaClO/diol (in the range 5.0–7.5), and temperature (in the range 45°–55 °C). The molar excess of NaClO and the total volume of the aqueous phase were found to be more determinant than temperature on the oxidative cleavage of diol, with positive effects due to the use of a high molar excess of the oxidant and a low volume of the aqueous phase. Starting from 50 mg diol 4 in 5 mL EtOAc, 38% of acid 2 (and of the corresponding co-product 3) and 19% of hydroxyketones 6 were obtained (GC/MS analysis) as mean values of three runs, performed at 45 °C, with 3 mL total aqueous phase and 7.5 mol of NaClO per mol of diol 4. Under these conditions, the effect of substrate concentration was investigated by performing batch experiments (in triplicates) with 25, 50, 75 and 100 mg of starting diol 4 in 5 mL EtOAc. A progressive increase of the percentage of acid 2 (and of the corresponding co-product 3) determined by GC/MS analysis was observed (16%, 38%, 55% and 58%, respectively), further improving process productivity. The best conversion was achieved starting from 20 mg mL−1 diol solution.
Oxidative cleavage of diol 4 (continuous-flow conditions)
Multiphase reactions constitute one of the main research areas in flow chemistry. When two immiscible solvents with high interfacial tension are introduced in the small-diameter channels of a flow system through two different inlets connected by a T-junction, a segmented flow is formed,44 in which the two liquids form short alternating segments with a regular periodic pattern depending on the flow rates. The application of phase-transfer catalysis is known to increase the interfacial area for the exchange of chemical species, thanks to the transfer of reactants from one phase to the other, where the main reaction occurs. Comparing phase-transfer catalyzed reactions in flow and batch reactors, the flow reactions result more efficient and sustainable.45,46 Nevertheless, the presence of the liquid–liquid interface limits overall kinetics, and flow chemistry has been more and more explored to push the yields. A biphasic segmented liquid–liquid flow system was thus considered the most suitable solution for the oxidative cleavage of diol 4. The best conditions obtained by DOE analysis were applied to a 10 mL tubular coil, using a 20 mg mL−1 solution of diol 4 in EtOAc containing 1% Triton-X 100. The optimal ratio between the organic and water phase was obtained by setting the flowrates as follows: 60 μL min−1 for aq. solution (containing the suitable amount of NaClO) and 100 μL min−1 for the organic solution. The two flows joined in a T-junction and entered the 10 mL tubular coil, thermostated at 45 °C, with a 62.5 min residence time. The solution collected at the outlet was quenched with a stoichiometric quantity of Na2SO3, the organic layer was separated and dried over Na2SO4. Its composition (by GC/MS) was 44% acid 2 (and the corresponding co-product 3), 26% 6a,b isomers, and 30% diol 4. To complete the reaction, the organic phase was flowed in a second 10 mL tubular coil with fresh aq. NaClO solution. At the outlet of the second coil, the conversion into final products was nearly complete (99%). Since the reaction does not depend on the presence of a catalyst, the STY is evaluated per unit of reaction volume: 8 mL for the batch reaction (5 mL EtOAc + 3 mL aqueous phase) and 20 mL for the two runs of the flow oxidation.

(i) Batch mode: 8 mL biphasic system, 0.316 mmol diol 4, 24 h reaction time, 58% conversion (i.e., the results of DOE optimization).
(ii) Continuous flow mode: 20 mL volume (considering the two subsequent flow reactions), 0.316 mmol diol 4, 2 h 5 min residence time, 99% conversion.
These results show that the reaction performed in a continuous flow reactor led to a 7.8 times higher STY when than in a batch process. The properties and reaction conditions in such microreactors are different to large-scale systems. The application of a biphasic reaction in microreactors, especially when phase-transfer catalysis is combined with segmentation, led to a significant increase the reaction rate, due to a high surface-to-volume ratio, short diffusion distances, fast and efficient heat dissipation and mass transfer.
Evaluation of the process efficiency and green metrics
In a recent paper, Kaspar47 suggested the following parameters as key tools for efficiency evaluation: (i) cost of reagents; (ii) reaction time and duration of work-up and purification procedures; (iii) material usage (as expressed by E-factor or process mass intensity).We adopted this approach to discuss the overall efficiency of our chemo-enzymatic procedure, comparing it to known reference synthetic protocols, i.e. (a) the commercial synthesis of azelaic acid by ozonolysis of oleic acid, (b) another approach employing the same diol 4 as intermediate,42 (c)–(e) three approaches representative of those using H2O2 as an oxidant in the presence of a suitable catalyst48 (Scheme 5).
 | ||
| Scheme 5 Known processes of oxidative cleavage of oleic acid (1) for the production of pelargonic (2) and azelaic acids (3) compared to that described in this work. | ||
(i) Cost of reagents. The procedure herein presented (Fig. 3) exploits soapstock (a side-product of vegetal seed oil refinement) as starting material, instead of commercial oleic acid like all the other known methods, including the processes used for comparison in this discussion. High-oleic sunflower oil soapstock is completely hydrolyzed in 12 h without preliminary acidification using a lipase as catalyst, and the mixture of FFAs (with ∼83% oleic acid content) is simply recovered by centrifugation. Specific economic advantages are connected not only to the use of a free and abundant renewable feedstock, but also to the beneficial effect on the environmental impact of seed oil refining due to switching from acidic to enzymatic splitting, and reducing the overall waste disposal costs. Continuous flow reaction mode allowed the use of the same enzyme loading for at least ten hours, producing diol 4 with STY = 25.4 mmol gcat−1 h−1. In the catalytic procedures herein considered for a comparison, the catalyst is recovered (but not re-used) in process (d), recovered and re-used four times in process (e).
 | ||
| Fig. 3 Overall scheme of the optimized process for the conversion of high-oleic sunflower oil soapstock into azelaic and pelargonic acids. | ||
(ii) Reaction time and duration of work-up and purification procedures. For both the preparation of diol 4 from hydrolyzed soapstock and its subsequent oxidative cleavage into acids 2 and 3, the use of continuous-flow modality led to short residence time, i.e. 5 h and 2 h 5 min, respectively. Using acetonitrile as solvent for the chemo—enzymatic epoxidation, the addition of diluted sulfuric acid caused precipitation of diol 4 as a pure solid, thus leaving in solution all impurities, including side-products from linoleic acid epoxidation, and avoiding time-consuming chromatographic purifications. No concentrated HCl is needed in the work-up as in process (b), and a crystalline pure compound instead of an oily crude mixture is obtained.
The biphasic reaction medium employed for NaOCl oxidation improves the yield and decreases the operation time of final isolation of acids 2 and 3, enabling one single extraction procedure from the still warm organic phase of the reaction mixture, instead of the multiple hot EtOAc/hot water extractions described in process (c). In procedures (b), (d) and (e) no separation of the two acids is described.
One of the main issues of the commercial production of azelaic acid12b is the purification of the final product, to eliminate oxidized impurities, dicarboxylic acids obtained by ozonolysis of other unsaturated fatty acids contaminating starting oleic acid, and residual saturated carboxylic acids inevitably present in the starting material. The recovery of intermediate diol 4 as a crystalline compound by filtration removes any acid contaminant, reduces the problems connected to final purifications, with a positive effect on the quality of final products.
(iii) Material usage (green metrics). Materials usage related to the procedure described in this work was compared to that characterizing the processes shown in Scheme 5, by using the values of the simplified environmental factor (sE-factor), calculated under the hypothesis of complete recycling of reaction and post-reaction solvents and water, according to the formula suggested by Roschangar et al.49 (see ESI†). The quantities of additives for reaction work-up (acid, base, and reductants) had not been reported in the corresponding literature for processes (b)–(e), so the amount of sulfuric acid and sodium bisulfite (the only work-up additives of our procedure) were not considered. No sE-factor was obtained for oleic acid ozonolysis, because it was difficult to retrieve all the information regarding the amounts of reagents and products employed during the process from patent literature (Table 1).
Although the process described in this work is not superior in terms of sE-factor, it is the only one that (i) affords directly the final products 2 and 3 as isolated compounds during the work-up of final NaClO oxidation, and (ii) produces high purity acids 2 and 3, due to the intermediate isolation of crystalline diol 4. For processes (b)–(e) only analytical yields of acids 2 and 3 obtained by GC of the final mixture are given, and the quality of final products, in terms of presence of acid by-products, is not discussed.
Conclusion
Soapstock splitting and disposal are critical issues in the vegetable oil refining industry. The potential of lipases in the manipulation of FFAs is well known but, at industrial scale, the complex composition and the highly alkaline pH of soapstock hinder lipase application for large scale enzymatic splitting. The three-step process described in this paper and summarized in Fig. 3 offers a possible solution apt to process-scale development.The collaboration with Oleificio Zucchi allowed us to test a new lipase (Eversa Transform 2.0) from Novozymes directly on a sunflower soapstock with a high content of oleic acid (∼83%) as it was provided from the refining plant. In order to valorize the hydrolyzed product, we aimed to prepare pelargonic and azelaic acids by oxidative CC bond cleavage of oleic acid. The FFAs (mainly composed of oleic acid with ∼9% of linoleic) was submitted to epoxidation promoted by the immobilized Candida Antarctica lipase B (Novozym® 435) in the presence of hydrogen peroxide according to the Prilezhaev reaction mechanism.
Intermediate crystalline diol 4, recovered by filtration, was easily submitted to oxidative cleavage to the final products in a biphasic EtOAc/aq. NaClO system using Triton-X 100 as phase-transfer catalyst. For both reaction steps, a statistical approach was applied to optimize the conversions, through a factorial DOE analysis. The optimal conditions obtained through the analysis were implemented on different reaction configurations, demonstrating and exploiting the advantages of continuous-flow chemistry. The epoxidation was run in a stirred-tank reactor in a CSTR mode, while for the oxidative cleavage a tubular coil in PFR mode was chosen. Both configurations proved to be stable and efficient, determining a substantial increase of the reaction yields with respect to the batch conditions, clearly demonstrating potential for further development and scale-up to industrial application in seed and vegetable oil refining plants.
Experimental
General methods
Chemicals and solvents were purchased from Merck Life Science s.r.l. (Milan, Italy) and used without further purification. Trimethylsilyldiazomethane 10% solution in hexane (TCI Europe N.V.) was purchased from Zentek s.r.l. (Milan, Italy). TLC analyses were performed on Macherey Nagel pre-coated TLC sheets Polygram® SIL G/UV254 purchased from Chimikart s.r.l. (Naples, Italy). Soapstock was provided by Oleificio Zucchi (Cremona, Italy) as obtained from caustic refining plant unit of high-oleic sunflower oil without further treatment. The continuous flow reactions were performed using an E-Series Integrated Flow Chemistry system from Vapourtec (Alfatech s.p.a., Genoa, Italy) equipped with Omnifit glass columns (6.6 mm i.d. × 150 mm length) and a 10 mL standard coiled tubular reactor (ambient to 150 °C temperature). For continuous enzymatic reaction, a flat bottom, 3-necked, jacketed reactor (50 mL) was purchased from Colaver s.r.l. (Milan, Italy). The mechanical stirrer was inserted in the central neck, while the two side necks were connected to the peristaltic pumps of the Vapourtec system (one pump for flowing the solution from the reservoir into the reactor, the other for moving the liquid out) through tubes. At the outlet of the suction pump, an HPLC solvent filter frit was fitted to prevent tube blockage because of the enzyme. 1H and 13C NMR spectra were recorded on a 400 or 500 MHz spectrometer in CDCl3 or CD3OD solution at r.t. unless otherwise specified. The chemical shift scale was based on internal tetramethylsilane. GC/MS analyses were performed using an HP-5MS column (30 m × 0.25 mm × 0.25 μm, Agilent Technologies Italia s.p.a., Cernusco sul Naviglio, Italy). The following temperature program was employed: 50 °C/10 °C min−1/250 °C (5 min)/50 °C min−1/300 °C (10 min). The samples for GC/MS were treated with MeOH and trimethylsilyldiazomethane 10% in hexane, to derivatize carboxylic acids by transformation into the respective methyl esters.General procedure for lipase-mediated hydrolysis of soapstock
Soapstock (10 g) was suspended in distilled water (30 mL). The initial pH was found to be 10.0. Lipase Eversa Transform 2.0, a liquid lipase from Thermomyces lanuginosus (100 mg, 1% w/w) was then added to the suspension. The reaction was kept under mechanical stirring for 12 h at r.t. in a two-neck round-bottom flask. Conversion was monitored by 1H NMR. At the end of the reaction, the pH value was 8.0. A diluted solution of H3PO4 (8% v/v) was added to the suspension to a final pH 4.5–5.0. The organic phase was recovered by centrifugation (5000 rpm, 25 °C, 20 min), yielding a mixture of free fatty acids (4.50 g, final yield 45% w/w), showing the following composition, obtained by 1H NMR analysis and confirmed by GC/MS analysis of the corresponding methyl esters (see ESI†): 80–87% oleic acid, 9% linoleic acid, 4–11% saturated acids (mainly palmitic and stearic acids).Epoxidation of hydrolyzed soapstock (factorial design analysis)
A 24 full factorial experimental design was applied to the epoxidation of the mixture of FFAs, containing 83% oleic acid derived from soapstock hydrolysis to study the influence of temperature, enzyme load, oleic acid and hydrogen peroxide concentration on final conversion. The conversion percentage of oleic acid into the corresponding epoxide (Y) was taken as response, while the four parameters (X1–X4) and their levels were: oleic acid concentration, X1 (10–50 g L−1, corresponding to 12–60 g L−1 of hydrolyzed mixture); Novozym® 435 load, X2 (1–2 g L−1); hydrogen peroxide 35% w/w concentration, X3 (1–4% v/v), temperature X4 (30–50 °C). All the experiments were conducted in duplicates, for a total of 32 reactions. The final reaction volume was 15 mL. All the 32 reactions were left in a thermoshaker at the controlled temperature for 5 h. The reactions were quenched with a saturated solution of Na2SO3, extracted with EtOAc and dried over Na2SO4. Conversion of oleic acid into the corresponding epoxide was analyzed as system response and was evaluated by GC/MS analysis after treating samples with MeOH and trimethylsilyldiazomethane. Data were processed using Minitab and the following multivariate regression model was obtained: Y = 21.7 + 8.2 × X1 + 5.4 × X2 − 0.53 × X3 + 4.4 × X4 + 2.34 × X1 × X2 − 0.15 × X2 × X3. The table of factorial plan and response and the factorial design analysis graphs are reported in the ESI.†Epoxidation of hydrolyzed soapstock followed by diol isolation (batch conditions)
The mixture of fatty acids recovered from the enzymatic splitting of soapstock (0.9 g) was dispersed in acetonitrile (15 mL). Then, aq. H2O2 35% (150 μL, 1% v/v) and Novozym® 435 (30 mg, 2 g L−1) were added. The reaction was left in a thermoshaker at 50 °C for 5 h. The enzyme was removed by filtration and the reaction quenched with a saturated solution of saturated Na2SO3 (1 mL), extracted with EtOAc and dried over Na2SO4. Oleic acid conversion to epoxide corresponded to 40% (GC/MS) A diluted solution of sulfuric acid 2 M (100 μL) was added to the mixture causing diol 4 precipitation as a white solid that was recovered by filtration (293 mg, yield 35%).Epoxidation of hydrolyzed soapstock followed by diol isolation (continuous-flow conditions)
The mixture of fatty acids recovered from the enzymatic splitting of soapstock (9.6 g) was dispersed in acetonitrile (160 mL), stirred in a shaker overnight at 50 °C, then cooled. The insoluble residue was filtered (1.3 g) and the solution obtained had a final concentration of 52 g L−1, corresponding to 43 g L−1 of oleic acid (83% of total FAAs). Aqueous H2O2 35% (1.6 mL, 1% v/v) was added. A portion of this solution (45 mL) was introduced into a 50 mL jacketed stirred tank reactor, containing Novozym® 435 (90 mg, 2 g L−1) and kept at the constant temperature (50 °C) by means of a thermostat. The remaining solution was continuously pumped into the reactor at a constant flowrate (150 μL min−1) by two peristaltic pumps, corresponding to a residence time of 5 h. A suction solvent filter for HPLC was placed at the suction tube to avoid tube blocking caused by enzyme beads. The system continuously operated for 12 h, and the outlet composition was periodically sampled and analysed by GC/MS. The fractions corresponding to a conversion in the range of 40–60% (from 2 to 12 h, 100 mL) and the volume inside the reactor (44% conversion, 45 mL) were collected (total volume 145 mL) and quenched by adding a saturated solution of Na2SO3 (6 mL). A diluted solution of sulfuric acid 2 M (2.5 mL, 0.25 eq.) was added to the mixture causing diol 4 precipitation as a white solid that was recovered by filtration (3.3 g, yield 47%): 1H NMR (CD3OD, 400 MHz):50δ = 3.45–3.35 (2H, m, 2CHOH), 2.29 (2H, t with J = 7.4 Hz, CH2COOH), 1.70–1.15 (26H, m, 13 CH2), 0.97–0.82 (3H, m, CH3). 13C NMR (CD3OD, 100.6 MHz):50δ = 177.6, 75.29, 75.26, 35.0, 34.0, 33.9, 33.0, 30.8, 30.7, 30.6, 30.42, 30.37, 30.2, 27.04, 26.96, 26.1, 23.7, 14.4. GC/MS (EI) as a methyl ester, obtained by treatment with MeOH and trimethylsilyldiazomethane 10% in hexane, tr = 23.85 min: m/z (%) = 294 (M+ −36, 1), 187 (48), 155 (100), 138 (30).Oxidative cleavage of threo-9,10-dihydroxystearic acid (4) to azelaic and pelargonic acid (factorial design)
Oxidative cleavage of diol 4 (50 mg) to azelaic and pelargonic acids was performed in a biphasic EtOAc/aq. NaClO solution using Triton X-100 (1 mg) as phase transfer catalyst. The influence of temperature (X1, in the range 45°–55 °C), the molar ratio NaClO/diol (X2, in the range 5.0–7.5), and the total volume of water phase (X3, in the range 3–5 mL) on the conversion was analyzed through a 23 full factorial experimental design. The amount of diol 4 converted into pelargonic acid (and the corresponding co-product azelaic acid) was taken as system response (Y). All the experiments were conducted in triplicates, for a total of 24 reactions. The volume of EtOAc (5 mL) was kept constant. The reactions were run at the suitable temperature for 24 h. The organic layer was then quenched with a saturated solution of Na2SO3 and dried over Na2SO4. Conversion of diol 4 into pelargonic acid (and the corresponding co-product azelaic acid) was determined by GC/MS after treating samples with MeOH and trimethylsilyldiazomethane. Data were processed using Minitab and the following multivariate regression model was obtained: Y = 20.17 − 0.50 × X1 + 5.67 × X2 − 7.83 × X3 − 0.83 × X1 × X2 − 0.17 × X1 × X3 − 1.67 × X2 × X3 + 0.83 × X1 × X2 × X3. The table of factorial plan and response and the factorial design analysis graphs are reported in the ESI.†Oxidative cleavage of threo-9,10-dihydroxystearic acid (4) to azelaic and pelargonic acid (continuous-flow conditions)
Diol 4 (100 mg, 0.316 mmol) and Triton-X 100 (1%) were added to EtOAc (5 mL). Diol solubilization was ensured by keeping the solution in a thermoshaker (170 rpm) at 50 °C for 1 h. This organic solution was pumped at 100 μL min−1 and was mixed in a T-junction with a solution of NaClO 10% (3 mL) pumped at a flowrate of 60 μL min−1 for a total flowrate of 160 μL min−1 corresponding to a residence time of 62.5 min. The segmented biphasic flow entered a 10 mL heated tubular coil kept at 45 °C. The mixture collected at the outlet was quenched by adding a stoichiometric quantity of Na2SO3, and the composition of the organic phase was evaluated by GC/MS analysis after derivatization with MeOH and trimethylsilyldiazomethane: 44% of acid 2, 26% of 6a,b isomers, 30% diol 4. The organic phase was flowed again through a 10 mL tubular coil at the same flowrate and temperature with fresh aq. NaClO solution. The mixture at the outlet was recovered and the still warm organic phase was separated, cooled to r.t. washed first with aq. Na2SO3, then with water, dried (Na2SO4), and concentrated under reduced pressure to afford pelargonic acid (44.9 mg, 90%): 1H NMR (CDCl3, 400 MHz, ppm):51δ = 9.81 (1H, s, COOH), 2.37 (2H, t with J = 7.5 Hz, CH2COOH), 1.75–1.2 (12H, m, 6CH2), 0.80–0.95 (3H, m, CH3). 13C NMR (CDCl3, 100.6 MHz, ppm):51δ = 180.3, 34.2, 31.9, 29.3, 29.23, 29.21, 24.8, 22.8, 14.2; GC/MS (EI) as a methyl ester, obtained by treatment with MeOH and trimethylsilyldiazomethane 10% in hexane, tr = 9.33 min: m/z (%) = 172 (M+, 0.5), 141 (15), 129 (18), 87 (45), 74 (100). The aqueous phase was treated with aq. Na2SO3, acidified with HCl 10% and extracted with EtOAc. The combined organic phases were dried (Na2SO4), and concentrated under reduced pressure, to obtain azelaic acid (46.3 mg, 78%): 1H NMR (CD3OD, 400 MHz, ppm):52δ = 2.28 (4H, t with J = 7.4 Hz, 2CH2COOH), 1.75–1.55 (4H, m, 2CH2), 1.4–1.2 (6H, m, 3CH2). 13C NMR (CD3OD, 100.6 MHz, ppm):52δ = 177.5, 34.9, 29.97, 29.93, 26.0. GC/MS (EI) as a methyl ester, obtained by treatment with MeOH and trimethylsilyldiazomethane 10% in hexane, tr = 13.9 min: m/z (%) = 185 (M+ −31, 55), 152 (100), 143 (47), 111 (63).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thomas Balle (Novozymes) is acknowledged for the kind gift of samples of Eversa® Transform 2.0, Novozym 40121, Lecitase Ultra and Novozym® 435. Fondazione Cariplo – INNOVHUB, project SOAVE (Seed and vegetable Oils Active Valorization through Enzymes) is acknowledged for funding (grant number 2017-1015).Notes and references
- United States Department of Agriculture Foreign Agricultural Service Oilseeds: World Markets and Trade, April 2021 (Accessed on 9th July 2021) https://apps.fas.usda.gov/psdonline/circulars/oilseeds.pdf.
- M.-J. Dumont and S. S. Narine, Int. Food Res. J., 2007, 40, 957–974 CrossRef CAS.
- M. J. Haas, D. J. Cichowicz, W. Jun and K. Scott, J. Am. Oil Chem. Soc., 1995, 72, 519–525 CrossRef CAS.
- M. J. Haas, S. Bloomer and K. Scott, J. Am. Oil Chem. Soc., 2000, 77, 373–379 CrossRef CAS.
- M. J. Haas, P. J. Michalski, S. Runyon, A. Nunez and K. M. Scott, J. Am. Oil Chem. Soc., 2003, 80, 97–102 CrossRef CAS.
- Z.-M. Wang, J.-S. Lee, J.-Y. Park, C.-Z. Wu and Z.-H. Yuan, Korean J. Chem. Eng., 2007, 24, 1027–1030 CrossRef CAS.
- C.-Y. Lin and Y.-W. Lin, Energies, 2012, 5, 2370–2380 CrossRef CAS.
- R. A. Pandey, P. B. Sanyal, N. Chattopadhyay and S. N. Kaul, Resour., Conserv. Recycl., 2003, 37, 101–117 CrossRef.
- B. Casali, E. Brenna, F. Parmeggiani, D. Tessaro and F. Tentori, Sustainable Chem., 2021, 2, 74–91 CrossRef.
- S. Cherif, F. Aloui, F. Carrière and S. Sayadi, J. Oleo Sci., 2014, 63, 109–114 CrossRef CAS PubMed.
- R. E. Beal, L. L. Lauderback and J. R. Ford, J. Am. Oil Chem. Soc., 1975, 52, 400–403 CrossRef CAS.
- (a) C. G. Goebel, A. C. Brown, H. F. Oehlschlaeger and R. P. Rolfes, Method of making azelaic acid, U.S. Patent2813113, 1957 Search PubMed; (b) H. F. Oehlschlaeger and H. G. Rodenberg, Process for the production of a purified grade of azelaic acid by treatment with ozone during purification, U.S. Patent3402108, 1968 Search PubMed.
- R. P. Pohanish, in Sittig's Handbook of Pesticides and Agricultural Chemicals, William Andrew, Norwich, New York, 2nd edn, 2015 Search PubMed.
- (a) Azelaic acid topical (Accessed on 9th July 2021) https://www.drugs.com/mtm/azelaic-acid-topical.html; (b) S. Sasmaz and O. Arican, Am. J. Clin. Dermatol., 2005, 6, 403–406 CrossRef PubMed; (c) J. A. Brydson, in Plastics Materials, Butterworth Heinemann, Oxford, 7th edn, 1999, ch. 18, pp. 478–530 Search PubMed.
- E. Brenna, D. Colombo, G. Di Lecce, F. G. Gatti, M. C. Ghezzi, F. Tentori, D. Tessaro and M. Viola, Molecules, 2020, 25, 1882 CrossRef CAS PubMed.
- D. Colombo, E. Brenna, B. Casali, M. C. Ghezzi, F. Parmeggiani, F. Tentori, D. Tessaro and F. Boratyński, ChemCatChem, 2021, 13, 1–9 CrossRef.
- K. Brunner, R. Frische and D. Kilian, Method for Enzymatic Splitting of Oils and Fats, U.S. Patent2002/0197687A1, 2002 Search PubMed.
- P. Kempers, U. Schörken, T. Wolf, S. Sato, W. Bueno de Almdeida, P. S. Bizzarri and A. S. Araujo, Process for Production of Fatty Acids, Fatty Acid Esters and Sterolesters from Soapstock, U.S. Patent8426622B2, 2013 Search PubMed.
- M. J. Haas and K. M. Scott, J. Am. Oil Chem. Soc., 1996, 73, 1393–1401 CrossRef CAS.
- Feedstock flexibility for your biodiesel plant – with Eversa® Transform. (Accessed on 9th July 2021) https://www.novozymes.com/en/advance-your-business/food-and-beverage/vegetable-oils-processing/biodiesel.
- K. D. Davranov, K. A. Gulyamova, B. K. Alimova and N. M. Turapova, Appl. Biochem. Microbiol., 2000, 36, 19–22 CrossRef.
- M. C. T. Damaso, M. A. Passianoto, S. C. de Freitas, D. M. G. de Freire, R. C. A. Lago and S. Couri, Braz. J. Microbiol., 2008, 39, 676–681 CrossRef PubMed.
- E. A. Silveira, P. W. Tardioli and C. S. Farinas, Appl. Biochem. Biotechnol., 2016, 179, 558–571 CrossRef CAS PubMed.
- M. Benincasa, J. Contiero, M. A. Manresa and I. O. Moraes, J. Food Eng., 2002, 54, 283–288 CrossRef.
- M. Benincasa and F. R. Accorsini, Bioresour. Technol., 2008, 99, 3843–3849 CrossRef CAS PubMed.
- Y. Shabtai, Int. J. Biol. Macromol., 1990, 12, 145–152 CrossRef CAS PubMed.
- F. Mashhadi, A. Habibi and K. Varmira, Ind. Crops Prod., 2018, 113, 324–334 CrossRef CAS.
- F. Björkling, H. Frykman, S. E. Godtfredsen and O. Kirk, Tetrahedron, 1992, 48, 4587–4592 CrossRef.
- K. Li, C. O. Brant, M. Huertas, E. J. Hessler, G. Mezei, A. M. Scott, T. R. Hoye and W. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8603–8608 CrossRef CAS PubMed.
- D. De Zani and M. Colombo, J. Flow Chem., 2012, 1, 5–7 CrossRef.
- J. Meyer, A. E. W. Horst, M. Steinhagen, D. Holtmann, M. B. Ansorge-Schumacher, M. Kraume and A. Drews, Eng. Life Sci., 2017, 17, 759–767 CrossRef CAS PubMed.
- J. Meyer-Waßewitz, D. Holtmann, M. B. Ansorge-Schumacher, M. Kraume and A. Drews, Biochem. Eng. J., 2017, 126, 68–77 CrossRef.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- A. S. Perlin, Adv. Carbohydr. Chem. Biochem., 2006, 60, 183–250 CrossRef CAS PubMed.
- C. Venturello and M. Ricci, J. Org. Chem., 1986, 51, 1599–1602 CrossRef CAS.
- E. P. Pultinas Jr., Preparation of Carboxylic Acids by the Oxidation of Vicinal Glycols, U.S. Patent3855257, 1974 Search PubMed.
- S. Solmi, E. Rozhko, A. Malmusi, T. Tabanelli, S. Albonetti, F. Basile, S. Agnoli and F. Cavani, Appl. Catal., A, 2018, 557, 89–98 CrossRef CAS.
- A.-K. C. Schmidt and C. B. W. Stark, Org. Lett., 2011, 13, 5788–5791 CrossRef CAS PubMed.
- M. Shibuya, T. Shibuta, H. Fukuda and Y. Iwabuchi, Org. Lett., 2012, 14, 5010–5013 CrossRef CAS PubMed.
- J. M. Khurana, P. Sharma, A. Gogia and B. M. Kandpal, Org. Prep. Proced. Int., 2007, 39, 185–202 CrossRef CAS.
- M. Kirihara, R. Osugi, K. Saito, K. Adachi, K. Yamazaki, R. Matsushima and Y. Kimura, J. Org. Chem., 2019, 84, 8330–8336 CrossRef CAS PubMed.
- M. Lemaire, A. Favre-Reguillon, B. Paquit, S. Claude and Y. Raoul, Method for Preparing Carboxylic Acids by Oxidative Cleavage of a Vicinal Diol, U.S. Patent2013/0131379 A1, 2013 Search PubMed.
- G. A. Mirafzal and A. M. Lozeva, Tetrahedron Lett., 1998, 39, 7263–7266 CrossRef CAS.
- B. Ahmed-Omer, D. Barrow and T. Wirth, Chem. Eng. J., 2008, 135, 280–283 CrossRef.
- B. Reichart, C. O. Kappe and T. N. Glasnov, Synlett, 2013, 24, 2393–2396 CrossRef CAS.
- S. K. Teoh, K. Sa-ei, M. S. Noorulameen, Q. Y. Toh, Y. L. Ng and P. N. Sharratt, Chem. Eng. Res. Des., 2015, 100, 467–480 CrossRef CAS.
- F. Kaspar, ChemRxiv, 2020. This content is a preprint and has not been peer-reviewed. 10.26434/chemrxiv.13251344.v1.
- (a) V. Benessere, M. E. Cucciolito, A. De Santis, M. Di Serio, R. Esposito, F. Ruffo and R. Turco, J. Am. Oil Chem. Soc., 2015, 9, 1701–1707 CrossRef; (b) A. Godard, P. de Caro, S. Thiebaud-Roux, E. Vedrenne and Z. Mouloungui, J. Am. Oil Chem. Soc., 2013, 90, 133–140 CrossRef CAS; (c) A. S. Ello, A. Enferadi-kerenkan, A. Trokourey and T.-O. Do, J. Am. Oil Chem. Soc., 2017, 94, 1451–1461 CrossRef CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- R. Awang, S. Ahmad, Y. B. Kang and R. Ismail, J. Am. Oil Chem. Soc., 2001, 78, 1249–1252 CrossRef CAS.
- Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212–11215 CrossRef CAS PubMed.
- E. Schievano, E. Morelato, C. Facchin and S. Mammi, J. Agric. Food Chem., 2013, 61, 1747–1755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR analysis of soapstock and of the corresponding mixtures of fatty acids obtained upon enzymatic hydrolysis; data of the factorial design study of enzymatic epoxidation and NaOCl oxidation. See DOI: 10.1039/d1gc03553c |
| This journal is © The Royal Society of Chemistry 2022 |