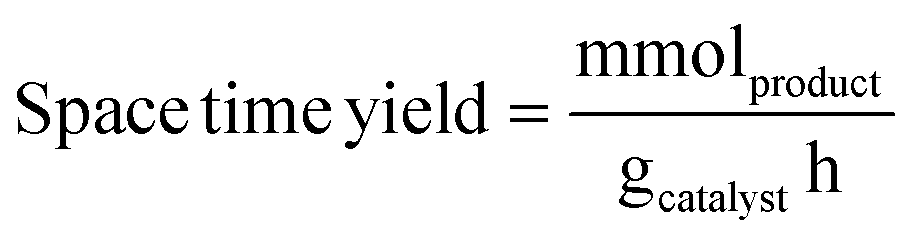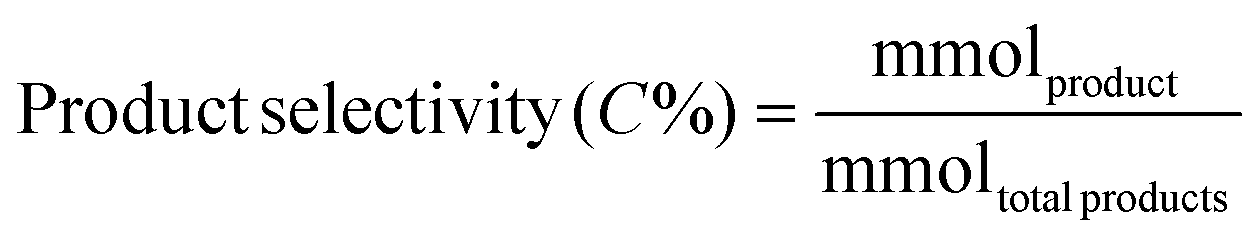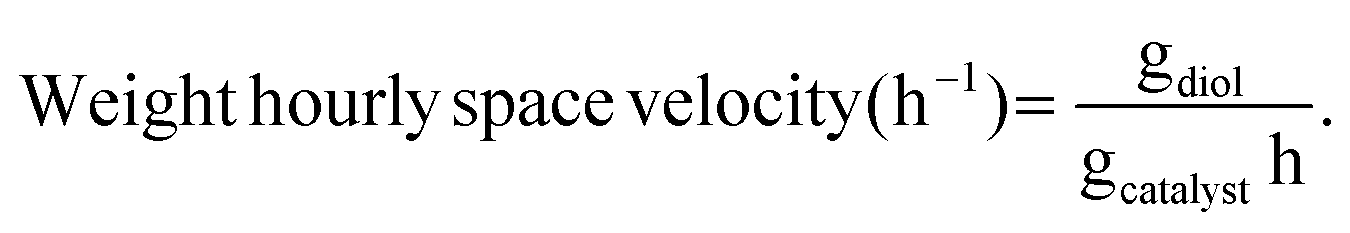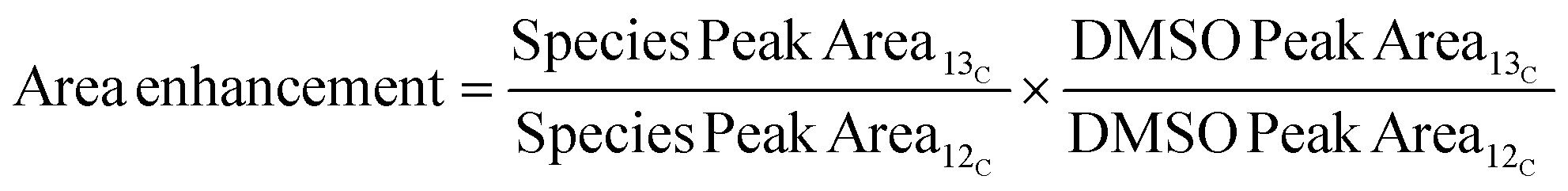DOI:
10.1039/D1GC03650E
(Paper)
Green Chem., 2022,
24, 350-364
Elucidation of reaction network and kinetics between cellulose-derived 1,2-propanediol and methanol for one-pot biofuel production†
Received
4th October 2021
, Accepted 8th December 2021
First published on 9th December 2021
Abstract
Cellulose upgrading with methanol (MeOH) to produce fuel-range alcohols involves the formation of α,β-diols like 1,2-propanediol (1,2-PDO) that can undergo C–C scission, C–O scission, and C–C coupling with MeOH. In this work, the reaction network for conversion of 1,2-PDO and MeOH in H2 flow over CuMgAlOx has been elucidated. Isotopic 13C-MeOH studies show that C–C coupling with 1,2-PDO produces 2,3-butanediol (14% selectivity) and 1,2-butanediol (5.3%), while 1,2-ethanediol (3.3%) forms from retro aldol condensation of a C–C coupling intermediate. Products observed from dehydrogenation, direct C–C scission, and C–O scission of 1,2-PDO include hydroxyacetone (44%), ethanol (15.2%), 1-propanol (5.5%) and 2-propanol (6%). C–C coupling is zero-order with respect to PHydrogen and P1,2-PDO, but has a 1.3 reaction order with respect to PMeOH. C–O scission is 1st order in PHydrogen and has low/near-zero reaction orders with respect to P1,2-PDO and PMeOH. Direct C–C scission has a 0.8–0.7 reaction order with respect to PHydrogen, P1,2-PDO and PMeOH. Dehydrogenation and C–C coupling have the lowest apparent activation energies of 45.6 and 47.2 kJ mol−1, respectively, while all other pathways have apparent activation energies at least 15 kJ mol−1 higher. Cofeed experiments with formaldehyde show that the rate determining step in C–C coupling is associated with the nucleophilic attack by 1,2-PDO. C–C coupling, direct C–C scission and esterification rates increase with PHydroxyacetone, while C–O scission and retro aldol condensation rates do not. C–C coupling is observed to occur with other α,β-diols and primary monoalcohols, but not with α,ω-diols or secondary monoalcohols.
Introduction
A major challenge with biofuel research has been the development of reaction conditions that can upgrade all fractions of lignocellulosic biomass in a single-stage reactor.1–3 Lignocellulose is composed of cellulose (40%), hemicellulose (25%), and lignin (20%).4 Matson et al. demonstrated that whole biomass can be converted to aliphatic alcohols in a one-pot process using supercritical methanol depolymerization and hydrodeoxygenation (SCM-DHDO) with a CuMgAlOx catalyst at 300 °C and 160 bar.5 The overall conversion achieved was greater than 98% and overall carbon yield was greater than 121 wt% due to methanol (MeOH) incorporation. Other one-pot processes use expensive precious metal catalysts but have lower yields than SCM-DHDO, such as: Ni-W2C in water (76% diol yield),6 LiTaMoO6 and Ru/C in aqueous phosphoric acid (82.4% gasoline yield),7 and Pt/NbOPO4 in cyclohexane (28.1% C5–6 alkane yield).8
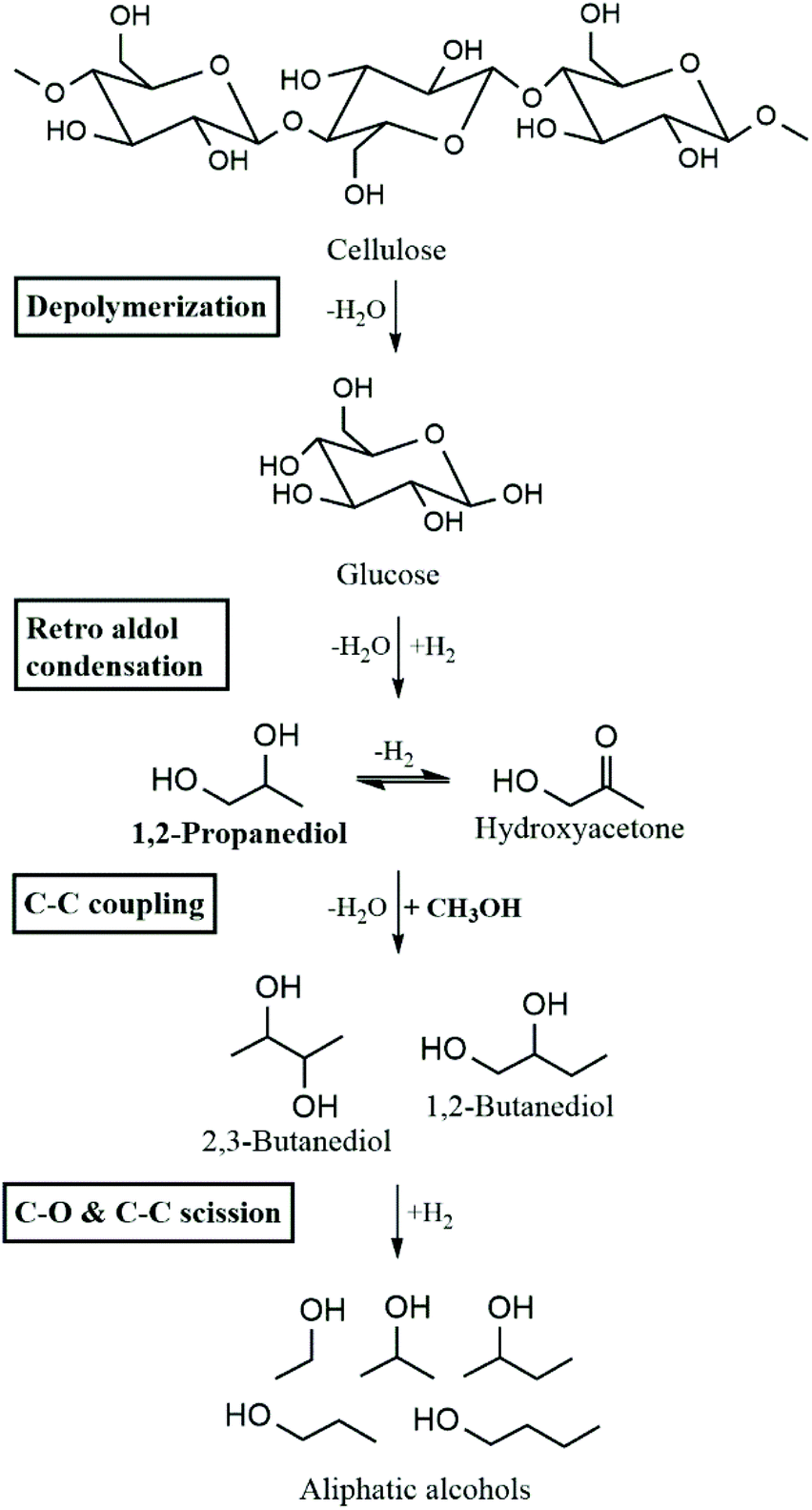
In previous work, Galebach et al. demonstrated the SCM-DHDO of cellulose to aliphatic alcohols over CuMgAlOx as shown in Scheme 1.9 In the first step, cellulose is solubilized in supercritical MeOH and depolymerized to monomeric sugars. Retro aldol condensation then produces C3 intermediates such as 1,2-propanediol (1,2-PDO) and the corresponding ketone, hydroxyacetone. C–C coupling of MeOH with 1,2-PDO and other α,β-diols leads to the formation of higher diols. Subsequent C–C scission and C–O scission of diols results in aliphatic monoalcohols as the stable final products of SCM-DHDO. The C–C coupling rate between MeOH and α,β-diols has been observed to be 100 times higher than the C–C coupling rate between MeOH and monoalcohols.10 MeOH incorporation was detected in all liquid products, accounting for 30–40% of carbon in the products. The importance of MeOH is three-fold: MeOH acts as the reaction medium to solubilize biomass, MeOH provides H2via MeOH reforming, and MeOH forms the electrophile during C–C coupling.11 Moreover, MeOH can be produced from sustainable sources like biomass, or CO2 and water.12 The CuMgAlOx catalyst has been characterized before and after reaction, and shown to be stable for over 100 h time on stream in a continuous flow reactor without leaching or coke formation.13 The reaction rates increased when the catalyst was reduced in situ before starting the reaction, indicating that metallic Cu is the active site for SCM-DHDO.
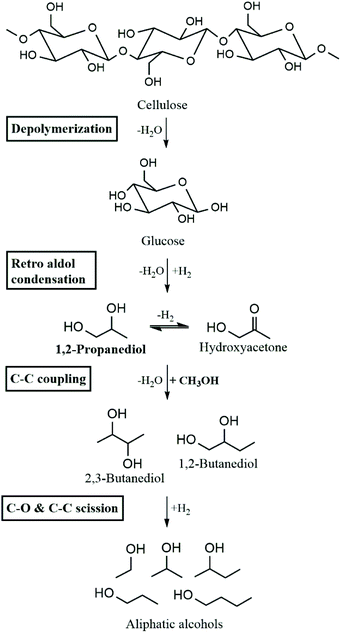 |
| | Scheme 1 Overall reaction pathway for SCM-DHDO of cellulose to aliphatic alcohols. Adapted from Galebach et al.9 | |
The objective of the present work is to better understand the fundamental reaction pathways and kinetics involved between 1,2-PDO and MeOH in order to increase the selectivity to C–C coupling reactions, while minimizing the scission of C–C and C–O bonds early in the reaction pathway. C–C coupling is proposed to occur via dehydrogenation of 1,2-PDO and MeOH to hydroxyacetone and formaldehyde, respectively, followed by nucleophilic attack by hydroxyacetone-derived enolate on the carbonyl carbon of formaldehyde, and hydrogenation to the product diol.10 This is similar to the Guerbet coupling of ethanol to produce n-butanol via acetaldehyde formation.14,15 The cross-coupling of MeOH has also been observed with ethanol16 and propanol.17 However, the reaction pathways involved in the formation of 2,3-butanediol and 1,2-butanediol from 1,2-PDO remain unclear as 1,2-PDO (and other α,β-diols) have two carbons with β-hydrogens that can carry out the nucleophilic attack on MeOH. The reaction orders with respect to MeOH and 1,2-PDO are also unknown, and the effects of varying the monoalcohol chain and diol type (α,β-diol versus α,ω-diols) are yet to be determined. In this study, we used isotopic 13C MeOH with quantitative 13C NMR to quantify the extent of MeOH incorporation. The reaction orders with respect to the partial pressures of H2, MeOH, 1,2-PDO and water have also been determined, along with the apparent activation energies. Cofeed experiments with hydroxyacetone and formaldehyde were carried out to determine the effect of proposed intermediates on reaction rates. Experiments with other α,β-diols (1,2-butanediol and 2,3-butanediol) and α,ω-diols (1,3-propanediol and 1,4-butanediol) in MeOH were carried out to determine how C–C coupling and C–C scission rates vary with diol position. Lastly, we also examined the effect of varying monoalcohol chain length and functionality from experiments with 1,2-PDO and other monoalcohols (ethanol, 1-propanol, and 2-propanol).
Experimental
Experimental setup
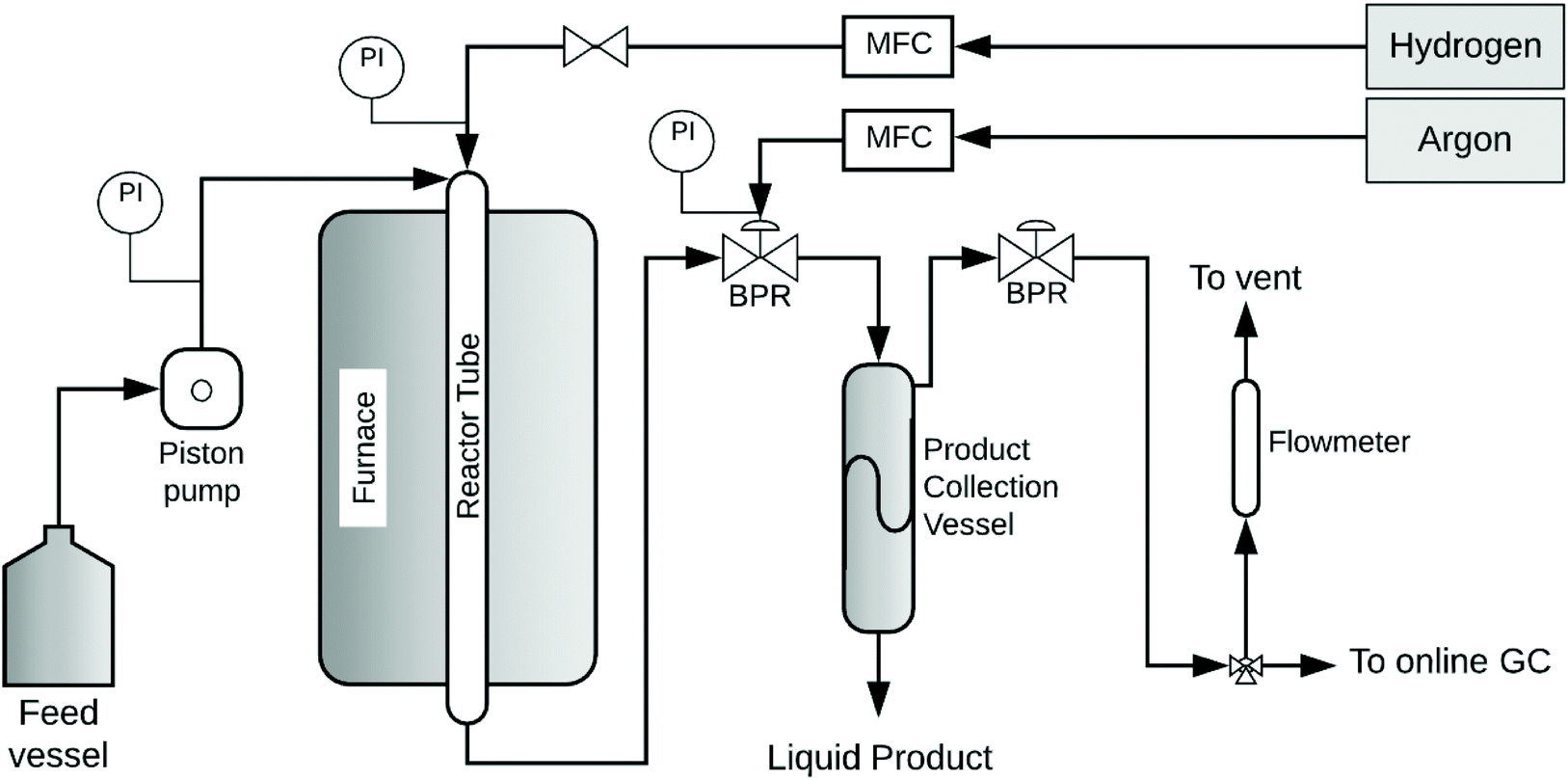
SCM-DHDO reactions of MeOH and 1,2-PDO in H2 flow were studied in a downflow packed-bed reactor, as shown in Fig. 1. The reactor is composed of a stainless-steel tube of length 15′′ and diameter ¼′′ with a packed bed of calcined CuMgAlOx powder in the middle, held in place with quartz wool and glass beads at both ends. The CuMgAlOx catalyst was synthesized using co-precipitation and characterized in previous work, as summarized in Table S1.†9,18 The reactor is enclosed in a cylindrical aluminum heating block and placed inside a clamshell furnace (Thermo Scientific), which ensures effective heat transfer and isothermal operation of the reactor. System pressure is maintained by an Equilibar dome-loaded back pressure regulator (BPR) with a polyimide diaphragm and Argon (UHP grade, Airgas) as the pilot fluid. The liquid feed consists of 10 mol% 1,2-PDO (99%, Acros Organics) in MeOH (HPLC grade, Thermofisher) and is pumped out of a sealed 1 L bottle in a downflow configuration using an ISM Eldex HPLC pump at 0.1 mL min−1. H2 gas (UHP grade, Airgas) is co-fed through a Brooks mass flow controller (MFC). As the liquid feed encounters the heated reactor walls above the catalyst bed, the diol-monoalcohol solution is vaporized. At the start of each experiment, the catalyst is reduced in situ with H2 flow of 102.5 mL min−1 at 350 °C with a temperature ramp of 1 °C min−1 for 5.5 h with a 4 h hold. The H2 feed is maintained at the same flow rate till the end of the experiment. To begin the reaction, the pressure of Ar in the BPR is increased to 1000 psig, the reactor temperature is changed to 300 °C, and the liquid feed pump is switched on. When the pressure in the reactor exceeds 1000 psig, products begin to collect via a dip-tube into a 100 mL stainless steel vessel, which is chilled using a circulating ethylene glycol/water bath and maintained at 45 psig with a spring-loaded pressure regulator. The time on stream (TOS) was measured from the moment the reactor temperature and pressure reached 300 °C and 1000 psig, respectively. The gas products are analyzed with an on-line GC and the gas flow rate is measured with a bubble flowmeter. The liquid products are sampled by draining the collection vessel periodically. The feed bottle is weighed before and after the reaction to determine the inlet flow rate, and the collected product volume is used to determine the outlet flow rate. Product yields for each set of reaction conditions are reported from the sample obtained at 24 h time on stream to ensure the catalyst system has reached steady state.
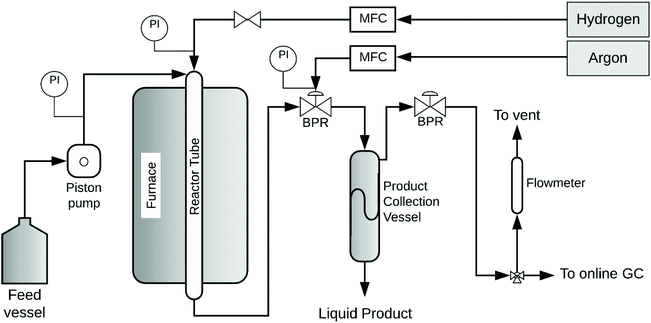 |
| | Fig. 1 Schematic of continuously operating, packed bed reactor for SCM-DHDO. Feed mixture of 1,2-PDO and MeOH is fed using an HPLC pump. H2 gas is cofed with a mass flow controller. Pressure is maintained by a dome loaded back pressure regulator with Argon as the pilot fluid. | |
Product analysis
Liquid products were analyzed with a Shimadzu GC-2010 equipped with an RTX-VMS column and FID detector, and with a Shimadzu GCMS-QP2010 mass spectrometer equipped with an RTX-VMS column. The GC-FID oven was held at 40 °C for 5 min, ramped to 240 °C at 7.5 °C min−1, and held for 15 min. Products were quantified using the GC-FID via external calibrations. Gas products were analyzed using an on-line Shimadzu GC-2014 with a 30 m RT-Q-Bond column, equipped with flame ionization (FID) and thermal conductivity (TCD) detectors. The GC oven was held at 40 °C for 5 min, ramped to 150 °C at 5 °C min−1, and held for 11 min. Products were quantified via calibrations with Scott Specialty Gas mixtures (Air Liquide). The equations used to calculate the product space time yield (STY), selectivity, and weight hourly space velocity (WHSV) are defined in eqn (1)–(3).| | 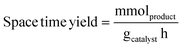 | (1) |
| | 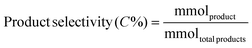 | (2) |
| | 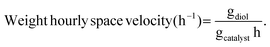 | (3) |
For the quantitative 13C nuclear magnetic resonance (NMR) analysis, the liquid product samples were dissolved in deuterated dimethyl sulfoxide (DMSO-d6). The experiments were completed on a Bruker Biospin (Billerica, MA) AVANCE III 500 MHz spectrometer fitted with a DCH (13C optimized) cryoprobe with the standard “zgig30” Bruker pulse sequence. The experiment was performed with a sweep width of 240 ppm centered at 110 ppm per 800 scans of 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 520 data points using a 1 s acquisition time and a 12 s interscan relaxation delay. The spectra were processed with Mestrelab Research's MestreNova software and were referenced to the residual DMSO solvent peak at 39.5 ppm. The area enhancement of product peaks was calculated using eqn (4), where the subscripts ‘12C’ and ‘13C’ indicate the sample was taken before and after the feed was changed to 5 wt% 13C MeOH, respectively.
520 data points using a 1 s acquisition time and a 12 s interscan relaxation delay. The spectra were processed with Mestrelab Research's MestreNova software and were referenced to the residual DMSO solvent peak at 39.5 ppm. The area enhancement of product peaks was calculated using eqn (4), where the subscripts ‘12C’ and ‘13C’ indicate the sample was taken before and after the feed was changed to 5 wt% 13C MeOH, respectively.
| | 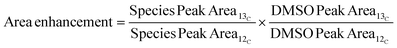 | (4) |
Results and discussion
Identification of reaction network from isotopic study of 1,2-PDO and 13C MeOH
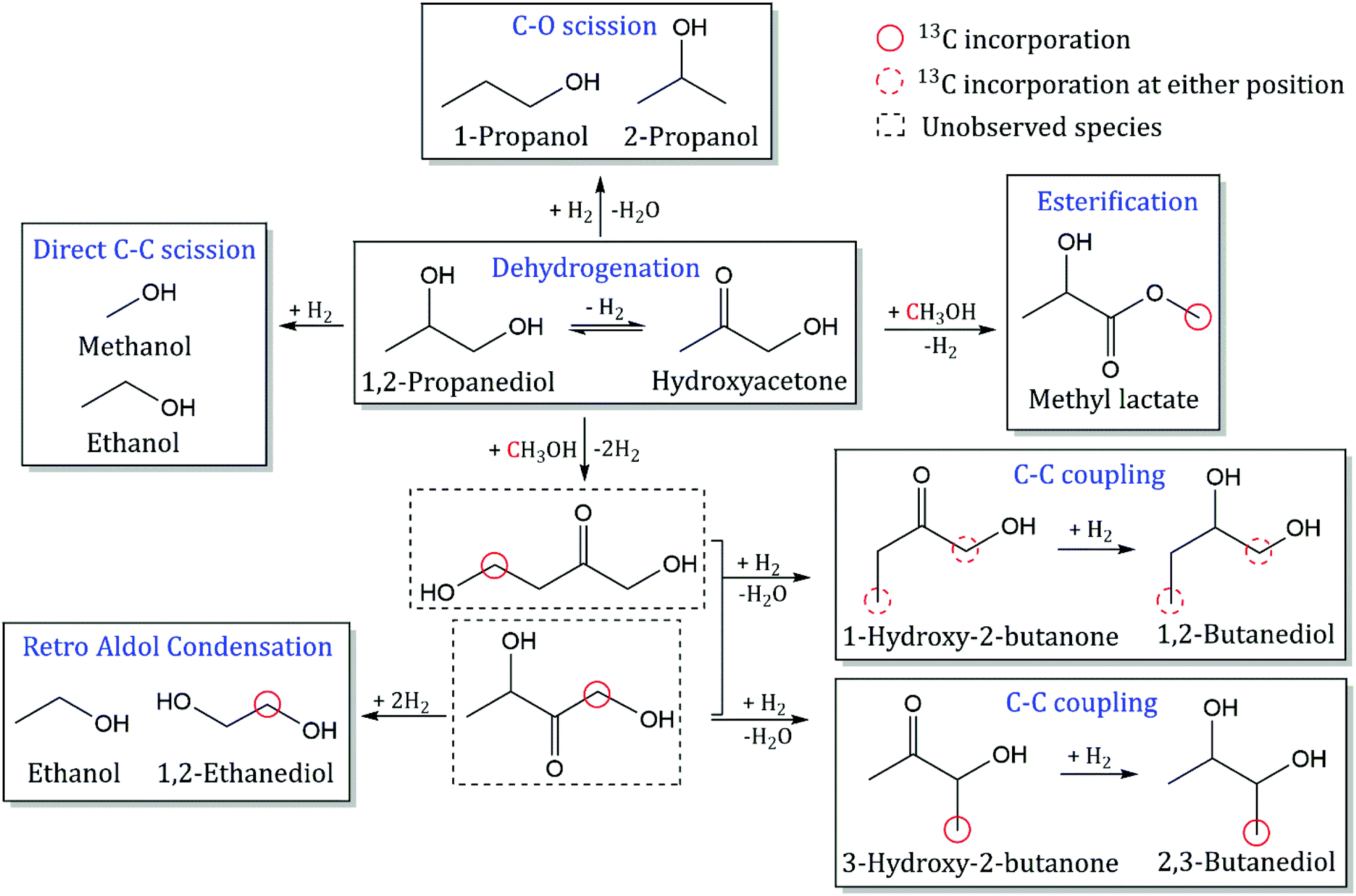
Reactions were carried out with 10 mol% 1,2-PDO in MeOH over a packed bed of CuMgAlOx at a WHSV of 40 h−1 to determine the reaction pathways involved between α,β-diols and MeOH. The corresponding reactant partial pressures of 1,2-PDO, MeOH, and H2 are 31.8, 318.2, and 650 psig, respectively. After the first 24 h of TOS, 5 wt% of MeOH in the liquid feed was replaced with 13C MeOH (99 atom%, Sigma-Aldrich) without disturbing the steady state of the reactor. The evolution of STY with TOS is shown in Fig. S1.† The product STYs remained nearly constant from 6 h to 30 h of TOS. The quantitative 13C NMR spectra of the products before and after the feed swap are shown in Fig. S2,† and the enhanced spectra for the major species are shown in Fig. S3.† The STY, selectivity, and area enhancement of liquid products were calculated using eqn (1), (2) and (4), as shown in Table 1. The carbon atom of MeOH was observed to have an area enhancement of 5.4, since MeOH has a 13C content of 1.1% (natural abundance) before the feed change and 5% after the feed change. The methyl carbon of 1,2-PDO has an area enhancement of 1. Any products with an area enhancement greater than 1 indicate 13C incorporation from MeOH. The main products observed were (in order of decreasing selectivity): hydroxyacetone, ethanol, 2,3-butanediol, 2-propanol, 1-propanol, 1,2-butanediol, 1,2-ethanediol, and methyl lactate. The 1,2-PDO conversion was 15% over the course of the experiment. The carbon balance was greater than 100% due to MeOH incorporation into products. Small amounts of ethyl acetate, 1-butanol, isobutanol, and 1,2-pentanediol were also observed but accounted for less than 1% of product yield. Ethers, aldehydes, alkanes, or products with unsaturated carbon bonds were not observed. CO and CO2 were detected at <0.5 mmol h−1 gcat−1, while CH4 was produced at an STY of 5 mmol h−1 gcat−1. This indicates that MeOH reforming was minimal under these reaction conditions. An experiment with a pure MeOH feed at the same reaction conditions was carried out and CH4 formation was observed at a lower STY of ∼1 mmol h−1 gcat−1. Amada et al. observed CH4 production from hydrogenolysis of 1,2-PDO at high H2 pressures (1160 psig) over Rh/SiO2 catalysts.19 CH4 produced during the experiment could be from either hydrogenolysis of 1,2-PDO and/or from methanation of MeOH-derived CO.20 The proposed reaction network based on the findings from GC-FID and quantitative 13C NMR has been elucidated in Scheme 2 and will be described in the following sections. When the reaction was carried out at 20 h−1 with 50 mg of catalyst, the conversion of 1,2-PDO was >95% and the selectivity to fuel-range C4+ monoalcohols (including butanol and hexanol) was >80%, while the amount of hydroxyacetone detected was negligible. This confirms that at higher conversions, SCM-DHDO is a useful technique for complete valorization of biomass to monoalcohols.
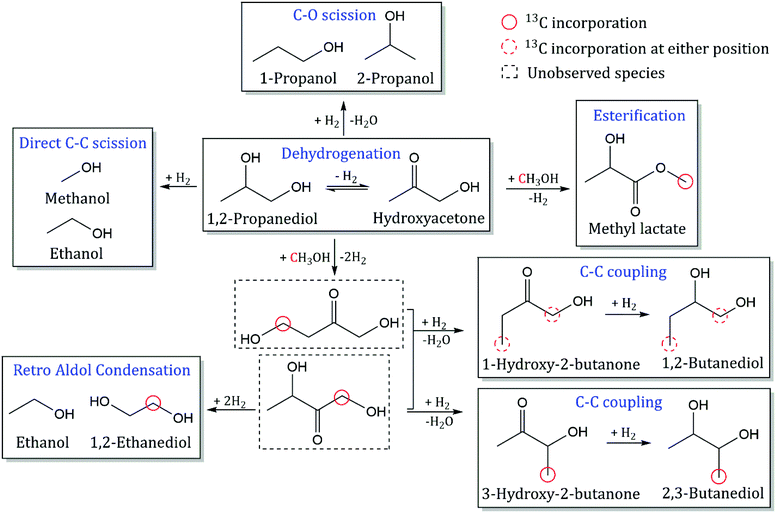 |
| | Scheme 2 Reaction pathway for catalytic conversion of 1,2-PDO with MeOH over CuMgAlOx. 13C incorporation from MeOH is indicated in red circles. Observed species are shown in solid rectangles while unobserved intermediates are shown in dotted rectangles. Reaction conditions: 300 °C, 1000 psig, 40 h−1 WHSV, 24 mg CuMgAlOx, 0.1 mL min−1 feed with 10 mol% 1,2-PDO in MeOH, 102.5 mL min−1 H2, 30 h, feed switched to 5% 13C MeOH after 24 h. | |
Table 1 STY, selectivity and 13C NMR area enhancement of major species from isotopic study of 1,2-PDO and 13C MeOHa
| |
STY (mmol h−1 gcat−1) |
Selectivity (%) |
Area enhancement (C atom) |
|
Reaction conditions: 300 °C, 1000 psig, 40 h−1 WHSV, 24 mg CuMgAlOx, 0.1 mL min−1 feed with 10 mol% 1,2-PDO in 5% 13C MeOH, 102.5 mL min−1 H2, 30 h.
Stereoisomers observed.
NMR-equivalent carbon present.
STY and selectivity from direct C–C scission only.
|
|
Reactants
|
| MeOH |
N/A |
N/A |
5.4 |
| 1,2-PDO |
N/A |
N/A |
1 (methyl) |
|
|
|
Products
|
| Hydroxyacetone |
47.1 |
44 |
1 (methyl) |
| 2-Propanol |
6.4 |
6 |
1 (primary methyl) |
| 1-Propanol |
5.8 |
5.5 |
1 (primary methyl) |
| Methyl lactate |
2.6 |
2.5 |
4.3 (alkoxy) |
| 2,3-Butanediol |
14.8 |
14 |
2.9b,c (methyl) |
| 1,2-Butanediol |
6.4 |
5.3 |
4.1 (primary hydroxyl) & 1.4 (primary methyl) |
| 1,2-Ethanediol |
3.5 |
3.3 |
3c (hydroxyl) |
| Ethanol |
12.1d |
11.5d |
1 (hydroxyl) |
Dehydrogenation and esterification of 1,2-PDO with MeOH.
Dehydrogenation of 1,2-PDO at the secondary hydroxyl group leads to the formation of hydroxyacetone with an area enhancement of 1 (methyl carbon) and selectivity of 44%. Esterification of MeOH and 1,2-PDO is observed to form methyl lactate with an area enhancement of 4.3 at the alkoxy carbon (circled in red in Scheme 2) and selectivity of 2.5%. Yfanti et al. observed hydroxyacetone formation from 1,2-PDO over a CuZnAl mixed metal oxide catalyst with 45% selectivity and demonstrated that dehydrogenation of 1,2-PDO is a reversible equilibrium-limited reaction.21 For a reaction to be at equilibrium, the ratio of Qreaction (mole fraction of products divided by the mole fraction of reactants) to Kequilibrium (equilibrium constant) should be 1. Dehydrogenation and esterification have Qreaction/Kequilibrium values of 0.1 and 0.01 respectively, indicating that both reactions are far from equilibrium at these reaction conditions. Methyl lactate forms from dehydrogenation of 1,2-PDO and nucleophilic attack by the oxygen of methoxide on the primary carbonyl carbon of lactaldehyde and subsequent deprotonation of the resulting hemiacetal. This dehydrogenative coupling of alcohols via hemiacetal formation to produce esters has been observed over other Cu-doped mixed oxide catalysts.22 Lactaldehyde formation from dehydrogenation of 1,2-PDO at the primary hydroxyl group is not observed, suggesting that it is either a rapidly consumed intermediate in the formation of methyl lactate, or the aldehyde-ketone tautomerization equilibrium is shifted far towards hydroxyacetone.23
C–C coupling of 1,2-PDO with MeOH.
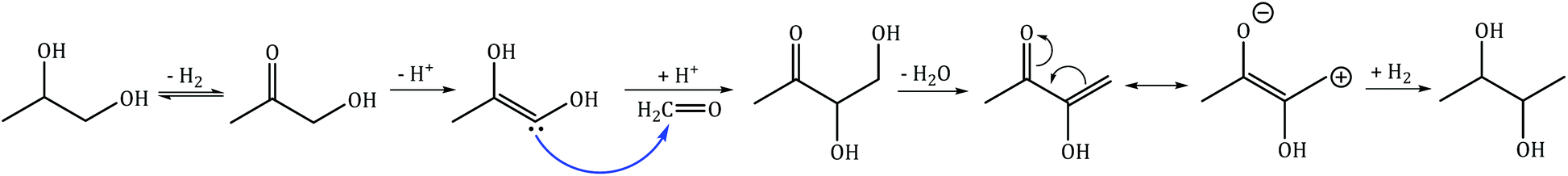
C–C coupling of 1,2-PDO with MeOH results in the formation of 2,3-butanediol and 1,2-butanediol with selectivities of 14% and 5.3% respectively. 2,3-Butanediol was observed as two stereoisomers (R,R and S,S) at nearly the same selectivities (7% each), and both molecules have area enhancements of 3 at the methyl carbons. For molecules with two NMR-equivalent carbons (such as the methyl carbons of 2,3-butanediol), if one of the carbons is 13C with area enhancement of 5 and the other is 12C with an area enhancement of 1, the average area enhancement of the molecule would be 3. The position of the 13C carbon indicates that 2,3-butanediol is produced from C–C bond formation at the primary hydroxyl carbon of 1,2-PDO. 1,2-Butanediol was observed to have an area enhancement of 4.1 at the primary hydroxyl carbon (from C–C bond formation at the primary hydroxyl carbon of 1,2-PDO) and an area enhancement of 1.4 at the primary methyl carbon (from C–C bond formation at the methyl carbon of 1,2-PDO). The formation of 1,2-butanediol with 13C incorporation at two different carbon positions reveals that C–C coupling of 1,2-PDO with MeOH is occurring at both the primary hydroxyl carbon and methyl carbon of 1,2-PDO at these reaction conditions. The selectivity to 1,2-butanediol is lower than the selectivity to 2,3-butanediol, indicating that the reactivity of the primary hydroxyl carbon of 1,2-PDO is much higher than that of the methyl carbon. C–C coupling at the primary hydroxyl carbon leads to the formation of 2,3-butanediol or 1,2-butanediol, while C–C coupling at the methyl carbon occurs at much lower rates and leads to the formation of only 1,2-butanediol. The proposed mechanism for formation of 2,3-butanediol is shown in Scheme 3.24 This includes the following steps: (1) dehydrogenation of 1,2-PDO and MeOH to hydroxyacetone and formaldehyde respectively; (2) deprotonation of hydroxyacetone to form an enol; (3) nucleophilic attack by the enol carbon on formaldehyde carbon to form a new C–C bond; (4) dehydration to form resonance-stabilized α,β-unsaturated ketone; and (5) hydrogenation to a saturated diol. Since formaldehyde could not be detected by GC-FID, a formaldehyde cofeed experiment will be carried out in the next section.
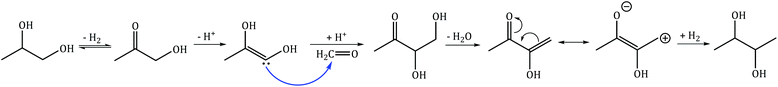 |
| | Scheme 3 Proposed mechanism for C–C coupling of 1,2-PDO with MeOH to form 2,3-butanediol via hydroxyacetone-derived enol formation and nucleophilic attack on MeOH-derived formaldehyde. | |
Retro aldol condensation and direct C–C scission of 1,2-PDO.
Quantitative 13C NMR results for C–C scission products reveal a previously unconsidered reaction pathway for the formation of 1,2-ethanediol. Ethanol was observed to have an area enhancement of 1 at both carbons, while 1,2-ethanediol has an area enhancement of 3. This indicates that after C–C bond formation between MeOH and the primary hydroxyl carbon of 1,2-PDO, an unobserved intermediate (1,3-dihydroxy-2-butanone) undergoes retro aldol condensation between the second and third carbons to form 1,2-ethanediol (with 13C MeOH incorporation) and ethanol (with no 13C MeOH incorporation). This intermediate (1,3-dihydroxy-2-butanone) can also undergo C–O hydrogenolysis to form 2,3-butanediol or 1,2-butanediol, as described in the previous section. Retro aldol condensation of the other proposed C–C coupling intermediate (1,4-dihydroxy-2-butanone), produced from C–C bond formation at the methyl carbon of 1,2-PDO, would lead to 13C incorporation in ethanol, which was not observed from the area enhancements. The experimental stoichiometric ratio from retro aldol condensation of 1,2-ethanediol to ethanol formation is 1:1 (by moles) but the STY of ethanol (16.1 mmol h−1 gcat−1) is much higher than the STY of 1,2-ethanediol (3.5 mmol h−1 gcat−1). This indicates that ethanol formation also occurs from direct C–C scission of 1,2-PDO. The C–C scission of 1,2-PDO has been split into 2 reaction pathways: retro aldol condensation of a C–C coupling intermediate (1,3-dihydroxy-2-butanone) to form 1,2-ethanediol and ethanol, and direct C–C scission to form ethanol and MeOH. The STY of retro aldol condensation is calculated as the STY of 1,2-ethanediol, while the STY of direct C–C scission is calculated by subtracting the STY of 1,2-ethanediol from the STY of ethanol.
C–O scission of 1,2-PDO.
C–O scission of 1,2-PDO is observed to occur at both the primary and secondary hydroxyl groups of 1,2-PDO, leading to the formation of 2-propanol and 1-propanol respectively. The difference in selectivity to 2-propanol (6%) and 1-propanol (5.5%) suggests that C–O scission rates are slightly higher at the primary hydroxyl group of 1,2-PDO. The area enhancement for both 1-propanol and 2-propanol at the primary alkyl carbon is 1, showing that 13C incorporation from MeOH is not involved. Peng et al. demonstrated that 1,2-PDO forms 1-propanol over metal sites via dehydration to propionaldehyde and consecutive hydride transfers from adjacent surface alkoxide species.25 Since propionaldehyde was not detected in the isotopic study, a co-feed experiment was carried out by replacing 5 mol% of 1,2-PDO with propionaldehyde in the feed. The STY of 1-propanol was observed to increase by an order of magnitude and very little propionaldehyde was detected in the product stream (>98% conversion), showing that CuMgAlOx can rapidly hydrogenate aldehydes to their corresponding alcohols at these reaction conditions.
Thermodynamics of reaction network
Overall, the products observed in the catalytic conversion of 1,2-PDO with MeOH can be classified into 6 types: dehydrogenation, esterification, C–C coupling, direct C–C scission, retro aldol condensation, and C–O scission. The thermodynamic parameters for these reactions were determined using the Aspen Plus software package at 300 °C and 1000 psig and can be found in Table S2.† All reactions have negative ΔG values, and all reactions except direct C–C scission and retro aldol condensation have negative ΔH values. The ΔG value for C–C coupling of 1,2-PDO with MeOH to form 2,3-butanediol is −148.4 kJ mol−1, which is higher in magnitude than the ΔG value for 1,2-butanediol formation (−99.6 kJ mol−1), suggesting that thermodynamic differences between the reaction pathways may explain the higher selectivity to 2,3-butanediol formation. Similarly, the ΔG value for C–O scission of 1,2-PDO to form 2-propanol (−132.8 kJ mol−1) is higher in magnitude than the ΔG value for 1-propanol formation (−119.4 kJ mol−1), indicating that thermodynamic differences are likely contributing to the higher selectivity to 2-propanol formation as well.
Determining reaction orders and apparent activation energies
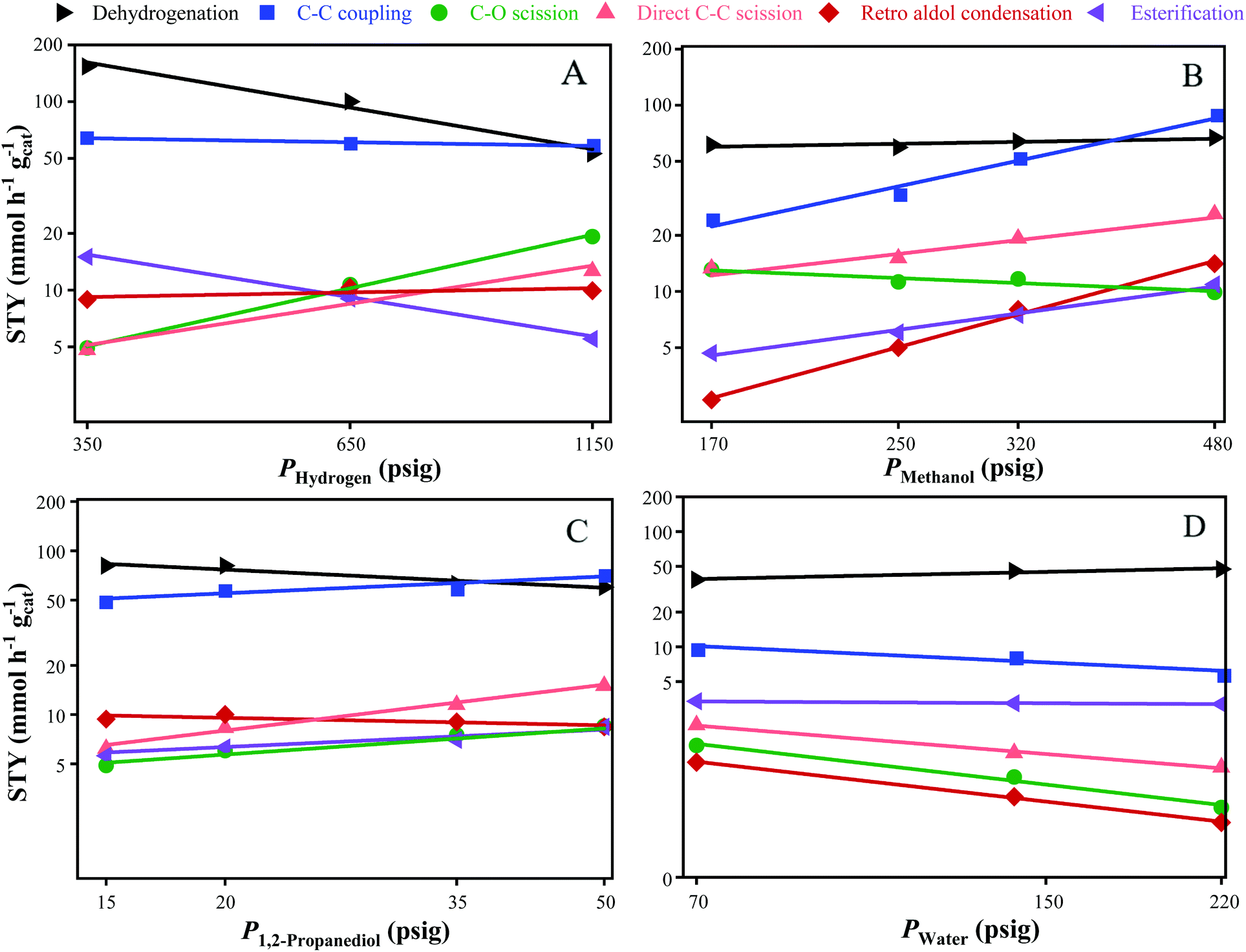
The reaction orders for dehydrogenation, C–C coupling, direct C–C scission, C–O scission, retro aldol condensation, and esterification with respect to the partial pressure of H2, MeOH, 1,2-PDO and water were determined by varying the partial pressure of one reactant while the keeping the others constant. The effect of replacing varying fractions of the feed MeOH with formaldehyde, and the feed 1,2-PDO with hydroxyacetone was also studied. The apparent activation energies were determined from the Arrhenius equation by varying the reactor temperature. Table 2 lists the range of parameters studied, with experiments carried out for 24 h of TOS at each condition. The variation in product STYs as a function of reactant partial pressures is shown in Fig. 2. Reaction orders for dehydrogenation, esterification, C–C coupling, C–O scission, direct C–C scission, and retro aldol condensation are listed in Table 3.
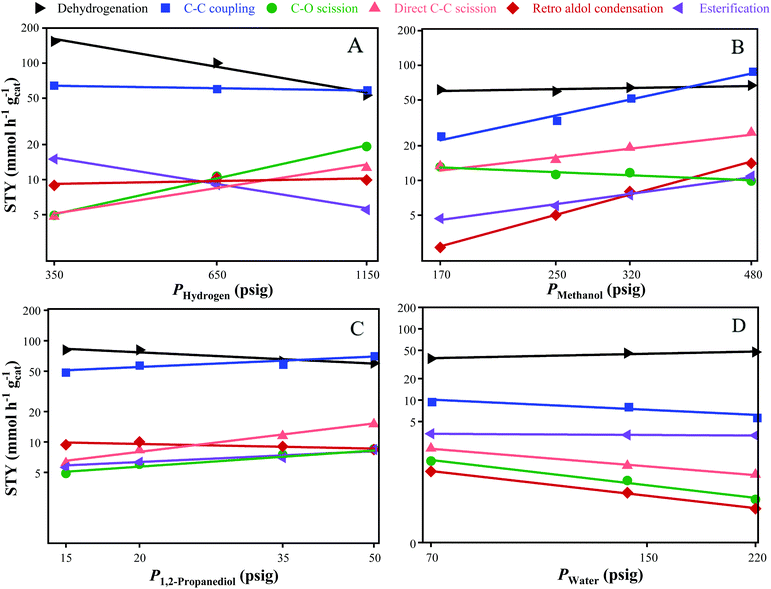 |
| | Fig. 2 Variation in product STYs with respect to the partial pressure of: (A) H2; (B) MeOH; (C) 1,2-PDO; and (D) water. Axes have been scaled to the natural log values. Standard reaction conditions: 300 °C, 8 mg CuMgAlOx, 120 h−1 WHSV, 24 h at each condition. | |
Table 2 Range of experimental conditions for the kinetic study of 1,2-PDO and MeOH in H2 flow
| Parameter |
Range |
| Standard reaction conditions: 300 °C, 8 mg CuMgAlOx, 120 h−1 WHSV, 24 h at each condition. |
| H2 partial pressure (psig) |
350–1150 |
| MeOH partial pressure (psig) |
170–480 |
| 1,2-PDO partial pressure (psig) |
15–50 |
| Water partial pressure (psig) |
415–650 |
| Temperature (°C) |
280–320 |
| % MeOH replaced with formaldehyde |
0–40 |
| % 1,2-PDO replaced with hydroxyacetone |
20–60 |
Table 3 Reaction orders with respect to the partial pressure of H2, MeOH, 1,2-PDO and water
| Reaction |
P
Hydrogen
|
P
MeOH
|
P
1,2-PDO
|
P
Water
|
| Standard reaction conditions: 300 °C, 8 mg CuMgAlOx, 120 h−1 WHSV, 24 h at each condition; partial pressures of H2, MeOH, 1,2-PDO and water are varied as needed. |
| Dehydrogenation |
−1 ± 0.1 |
0.1 ± 0.1 |
−0.3 ± 0.1 |
0.2 ± 0.1 |
| Esterification |
−0.8 ± 0.2 |
0.8 ± 0.0 |
0.3 ± 0.1 |
0 ± 0.1 |
| C–C coupling |
−0.1 ± 0.0 |
1.3 ± 0.1 |
0.3 ± 0.1 |
−0.4 ± 0.0 |
| C–O scission |
1.1 ± 0.1 |
−0.2 ± 0.1 |
0.4 ± 0.1 |
−1.1 ± 0.1 |
| Direct C–C scission |
0.8 ± 0.1 |
0.7 ± 0.1 |
0.7 ± 0.1 |
−0.7 ± 0.1 |
| Retro aldol condensation |
0.1 ± 0.1 |
1.6 ± 0.1 |
−0.1 ± 0.1 |
−0.7 ± 0.1 |
Reaction orders with respect to H2 partial pressure.
H2 is required for the removal of adsorbed atomic oxygen from water dissociation to create empty sites on the catalyst, and for the hydrogenation of unsaturated intermediates.26 The variation in product STYs with the partial pressure of hydrogen (PHydrogen) is shown in Fig. 2A. As PHydrogen is increased from 350 to 1150 psig, the STYs of hydroxyacetone and methyl lactate decrease three-fold from 150 to 50 mmol h−1 gcat−1, and from 15 to 5 mmol h−1 gcat−1, respectively. Dehydrogenation and esterification both produce 1 mole of H2 per mole of reactant consumed, which is in agreement with the observed reaction orders of −1 and −0.8 with respect to PHydrogen for dehydrogenation and esterification, respectively. As PHydrogen increases, the STYs of C–C coupling and retro aldol condensation remain nearly constant at 60 and 9 mmol h−1 gcat−1, respectively. C–C coupling and retro aldol condensation have a net-zero consumption of H2, and are observed to have near-zero reaction orders with respect to PHydrogen. This indicates that dehydrogenation of 1,2-PDO or subsequent hydrogenation of the unsaturated intermediates to form the product diols are not the rate-limiting steps in C–C coupling or retro aldol condensation. In contrast, the rate-limiting step during Guerbet coupling of ethanol over hydroxyapatite catalysts was observed to be the enolate formation from ethanol.27 As PHydrogen is increased from 350 to 1150 psig, direct C–C scission increases from 5 to 13 mmol h−1 gcat−1 while C–O scission increases from 5 to 20 mmol h−1 gcat−1. The corresponding reaction orders for direct C–C scission and C–O scission with respect to PHydrogen are 0.8 and 1.1 respectively, which are expected due to the stoichiometric consumption of 1 mole of H2 during both reactions. The near 1st order rate dependencies suggest that the STY of degradation products from direct C–C scission and C–O scission can be decreased by operating at lower PHydrogen without affecting C–C coupling rates.
Reaction orders with respect to MeOH partial pressure.
The variation of product STYs with PMeOH is shown in Fig. 2B. As PMeOH is increased from 170 to 480 psig, the STY of hydroxyacetone remains nearly constant at 60 mmol h−1 gcat−1, showing that dehydrogenation of 1,2-PDO is nearly zero-order with respect to PMeOH. All other reactions have positive or negative reaction orders with respect to PMeOH. As PMeOH is increased, C–C coupling increases three-fold from 24 to 88 mmol h−1 gcat−1, retro aldol condensation increases by an order of magnitude from 2 to 14 from mmol h−1 gcat−1, and esterification doubles from 5 to 10 mmol h−1 gcat−1. The corresponding reaction orders for C–C coupling, retro aldol condensation, and esterification with respect to PMeOH are 1.3, 1.6, and 0.8 respectively. The >1 reaction orders for C–C coupling and retro aldol condensation suggest that the rate-limiting step during C–C bond formation is related to the adsorption of MeOH and the subsequent nucleophilic attack on the carbonyl carbon of formaldehyde during aldol condensation. The 0.8 reaction order dependence of esterification on PMeOH indicates that the rate limiting step during methyl lactate formation is also associated with MeOH adsorption or the subsequent nucleophilic attack by the oxygen of methoxide species. Since direct C–C scission of 1,2-PDO leads to the formation of MeOH, it was expected that increasing PMeOH would lead to a decrease in direct C–C scission rates. However, the STY of direct C–C scission doubles from 13 to 26 mmol h−1 gcat−1, corresponding to a 0.7 reaction order with respect to PMeOH. As PMeOH is increased, C–O scission decreases from 13 to 9.5 mmol h−1 gcat−1, corresponding to a reaction order of −0.3 with respect to PMeOH.
Reaction orders with respect to 1,2-PDO partial pressure.
As seen in Fig. 2C, all reaction orders with respect to P1,2-PDO are lower than 1, suggesting high surface coverage of 1,2-PDO. As P1,2-PDO increases from 15 to 50 psig, the STY of dehydrogenation decreases from 80 to 60 mmol h−1 gcat−1 while the STY of retro aldol condensation remains nearly constant at 9 mmol h−1 gcat−1. The corresponding reaction orders with respect to 1,2 PDO for dehydrogenation and retro aldol condensation are −0.3 and −0.1 respectively. STYs for the other reactions increase with P1,2-PDO. C–C coupling and esterification increase from 50 to 72 mmol h−1 gcat−1, and from 5.6 to 8.4 mmol h−1 gcat−1, respectively. The reaction orders for C–C coupling and esterification with respect to P1,2-PDO is 0.3 for both. This is much lower than the reaction orders of C–C coupling and esterification with respect to PMeOH, suggesting that MeOH adsorption on the catalyst is weaker than 1,2-PDO adsorption. As P1,2-PDO is increased, C–O scission and direct C–C scission also increase from 5 to 8.5 mmol h−1 gcat−1, and from 6 to 15 mmol h−1 gcat−1, respectively. The corresponding reaction orders for C–O scission and direct C–C scission with respect to P1,2-PDO are 0.4 and 0.7 respectively. The similarity in reaction orders between C–C coupling and retro aldol condensation with respect to PHydrogen, PMeOH, and P1,2-PDO suggests that they involve the formation of a common C4O3Hx intermediate preceding a branch point, with the selectivity to final C–C coupling products being 6 times higher.28 This hypothesis is consistent with the isotopic 13C MeOH experiments discussed in the previous section.
Reaction orders with respect to water partial pressure.
As seen in Fig. 2D, the addition of water to the feed inhibits C–C coupling, C–O scission, direct C–C scission, and retro aldol condensation. As PWater increases from 70 to 140 psig, the STY of hydroxyacetone increases from 38 to 47 mmol h−1 gcat−1, corresponding to a reaction order of 0.2. Esterification remains nearly constant at 3.2 mmol h−1 gcat−1, indicating a zero-order dependence on PWater. As PWater increases from 70 to 140 psig, C–C coupling decreases from 9.8 to 5.9 mmol h−1 gcat−1 while C–O scission decreases from 1.4 to 0.4 mmol h−1 gcat−1. The stoichiometric reactions for C–C coupling and C–O scission involve the loss of 1 mole of water for each mole of product formed, which explains the decrease in rates with increasing PWater. The reaction orders for C–C coupling and C–O scission are −0.4 and −1.1 respectively. Direct C–C scission and retro aldol condensation also decrease from 2.1 to 0.9 mmol h−1 gcat−1, and from 1 to 0.3 mmol h−1 gcat−1, respectively, and both reactions have a reaction order of −0.7 with respect to PWater. The increase in PWater is observed to have the largest inhibitive effect on formation of 1-propanol and 2-propanol via C–O scission. Hanspal et al. observed the irreversible adsorption of water over MgO catalyst during Guerbet coupling of ethanol leads to inhibition of butanol formation, with the OH− from water coordinating to Mg2+ Lewis acid sites and the H+ coordinating to O2− Brønsted basic sites.29
Effect of formaldehyde cofeed.
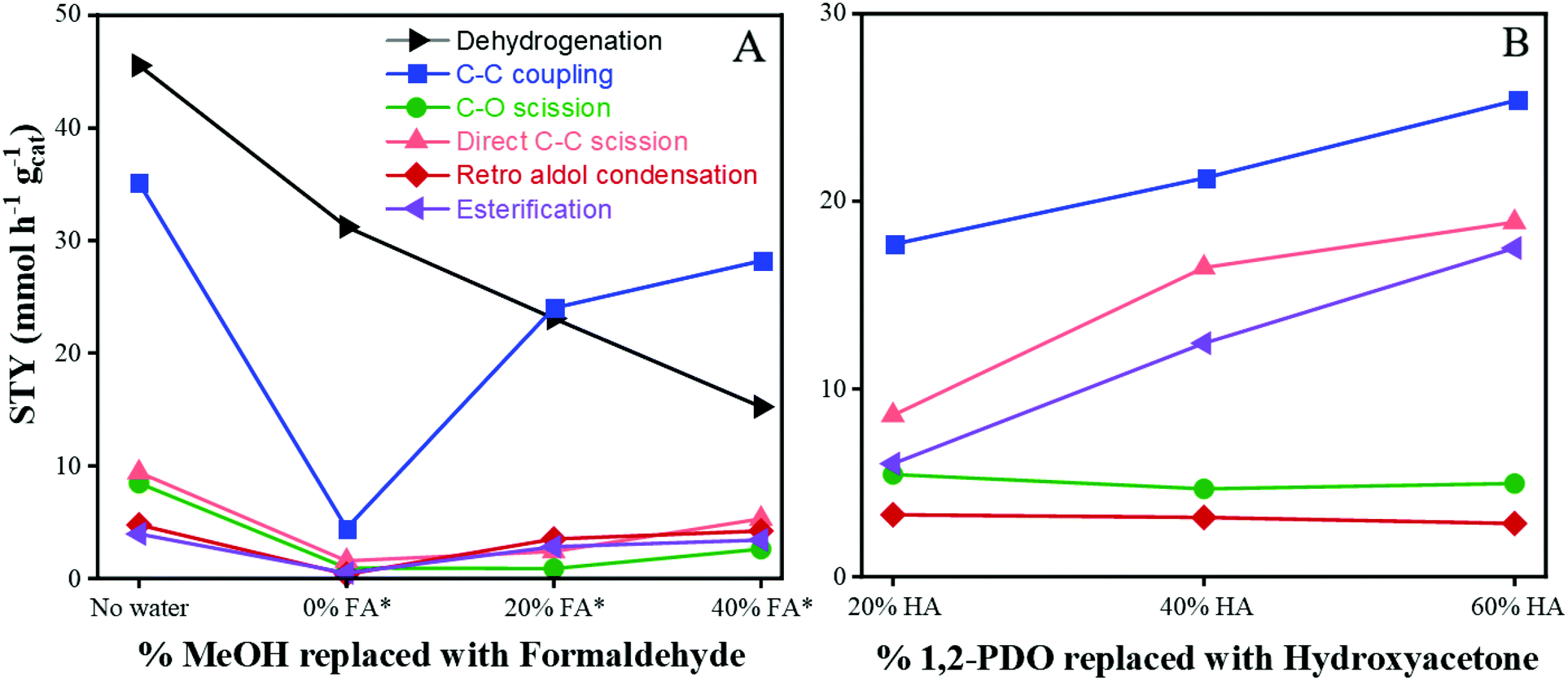
The effect of replacing MeOH with formaldehyde is shown in Fig. 3A. Since C–C coupling of 1,2-PDO and MeOH is proposed to occur via nucleophilic attack of formaldehyde by hydroxyacetone, the effect of replacing 0–40% of feed MeOH with formaldehyde, and 20–60% of feed 1,2-PDO with hydroxyacetone was studied. Formaldehyde (Sigma-Aldrich) was purchased as a 12 wt% aqueous solution so the impact of PWater should be considered when analyzing results from the formaldehyde cofeed experiments. As shown in Fig. 3A, an experiment with 10 mol% 1,2-PDO in MeOH with no added water or formaldehyde was carried out, labelled “No water”. PWater was maintained at 290 psig for the next 3 experiments (0, 20 and 40% MeOH replaced by formaldehyde) by adding water to the feed as needed. The corresponding partial pressures of formaldehyde (PFormaldehyde) for the 0, 20 and 40% replacement runs are 0, 60, and 130 psig respectively, while PMeOH is 320, 250, and 190 psig respectively. When water was added to the feed without any formaldehyde (labelled “0% FA”), all product STYs decreased from the “No water” run. Dehydrogenation and esterification decreased from 45 to 32 mmol h−1 gcat−1, and from 4 to 0.5 mmol h−1 gcat−1, respectively. C–O scission and direct C–C scission also decreased from 8 to 0.9 mmol h−1 gcat−1, and from 9.4 to 1.5 mmol h−1 gcat−1, respectively. C–C coupling and retro aldol condensation decreased by an order of magnitude from 35 to 4 mmol h−1 gcat−1, and from 5 to 0.3 mmol h−1 gcat−1, respectively. This is similar to the decrease in rates observed with the PWater experiments discussed in the previous section.
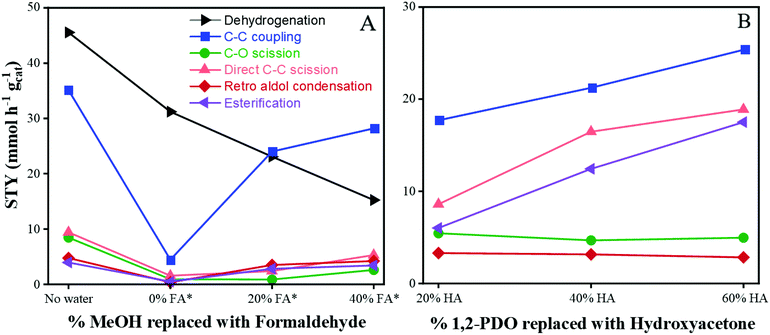 |
| | Fig. 3 Variation in product STYs with changes in feed. (A) MeOH replaced with formaldehyde (FA). *Indicates addition of water to maintain constant PWater of 290 psig. (B) 1,2-PDO replaced with hydroxyacetone (HA). Standard reaction conditions: 300 °C, 650 psig H2, 318.2 psig MeOH, 31.8 psig 1,2-PDO, CuMgAlOx, 120 h−1 WHSV, 24 h at each condition. | |
When the formaldehyde concentration was increased from 0 to 20% of MeOH replaced, the STYs of C–C coupling, retro aldol condensation, and esterification increased from 4.5 to 24 mmol h−1 gcat−1, from 0.3 to 3.5 mmol h−1 gcat−1, and from 0.5 to 3 mmol h−1 gcat−1, respectively. The opposite trend was observed for dehydrogenation – the STY of hydroxyacetone decreased from 32 to 23 mmol h−1 gcat−1. The corresponding STY of C–O scission remained nearly constant at 0.9 mmol h−1 gcat−1 and the STY of direct C–C scission increased slightly from 1.5 to 2.4 mmol h−1 gcat−1. This shows that C–C coupling, retro aldol condensation, and esterification rates increase by an order of magnitude when 20% of MeOH is replaced with formaldehyde, while C–O scission and direct C–C scission are relatively constant. With further increase in formaldehyde concentration to 40% of feed MeOH, the STYs of C–C coupling, C–O scission, direct C–C scission and retro aldol condensation increase to 28, 2.6, 5.3, and 4.2 mmol h−1 gcat−1 respectively. These results further elucidate that the rate determining step in C–C coupling is associated with the adsorption of MeOH, dehydrogenation of MeOH to formaldehyde, or the subsequent nucleophilic attack by the 1,2-PDO derived species.
Effect of hydroxyacetone cofeed.
The effect of replacing 1,2-PDO with hydroxyacetone is shown in Fig. 3B. When the hydroxyacetone concentration was increased from 20 to 60% of 1,2-PDO, the STY of C–C coupling increased linearly from 17 to 25 mmol h−1 gcat−1, while the STY of direct C–C scission nearly doubled from 9 to 19 mmol h−1 gcat−1. The STY of esterification nearly tripled from 6 to 17.5 mmol h−1 gcat−1. The corresponding STYs of C–O scission and retro aldol condensation remained nearly constant at 5 and 3 mmol h−1 gcat−1 respectively. This suggests that C–C coupling, direct C–C scission and esterification involve hydroxyacetone-mediated pathways, while C–O scission and retro aldol condensation rates do not vary with changes in PHydroxyacetone.
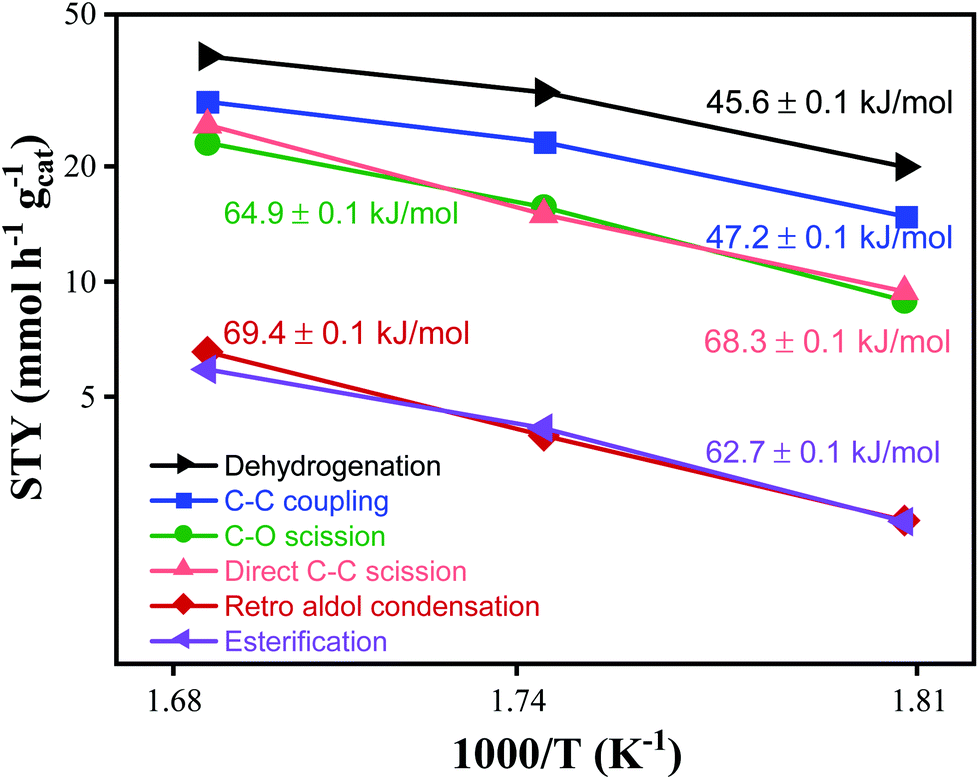
Apparent activation energies.
The apparent activation energies were calculated from the Arrhenius plot shown in Fig. 4. As the temperature was increased from 280 to 320 °C (i.e. from 1.81 to 1.68 × 103 K−1), the STY for dehydrogenation, C–C coupling, and esterification nearly doubled from 19 to 38 mmol h−1 gcat−1, from 15 to 30 mmol h−1 gcat−1, and from 2 to 5 mmol h−1 gcat−1 respectively. The STY for C–O scission, direct C–C scission and retro aldol condensation increased three-fold from 8 to 24 mmol h−1 gcat−1, from 9 to 26 mmol h−1 gcat−1, and from 2 to 6 mmol h−1 gcat−1, respectively. Dehydrogenation is observed to have the lowest apparent activation energy of 45.6 kJ mol−1, while C–C coupling is slightly higher at 47 kJ mol−1. The apparent activation energies of esterification and C–O scission are 64.9 and 62.7 kJ mol−1 respectively, while the apparent activation energies of direct C–C scission and retro aldol condensation are 68.3 and 69.4 kJ mol−1 respectively. The similarity in apparent activation energies for direct C–C scission and retro aldol condensation suggests the C–C hydrogenolysis mechanism involved in both pathways is likely very similar, or proceeds through pathways that happen to have similar thermodynamic barriers. The lower apparent activation energy for C–C coupling indicates that the selectivity to C–C coupling can be increased by operating at lower temperatures.
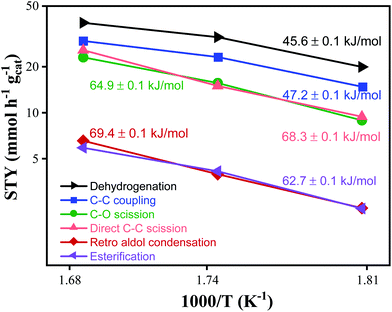 |
| | Fig. 4 Arrhenius plot of product STYs with apparent activation energies listed. Reaction conditions: 1000 psig, 8 mg CuMgAlOx, 120 h−1 WHSV, 24 h at each temperature. | |
Overall, the results from the partial pressure and apparent activation energy experiments indicate how to change the selectivity between C–C coupling, C–C scission, C–O scission and the retro aldol condensation reactions. The selectivity to C–C coupling can be increased by decreasing the reactor temperature and PH2, while increasing PMeOH. Increasing PHydrogen will increase the selectivity to direct C–C scission and C–O scission products.
Reaction pathways between alternative diols and monoalcohols
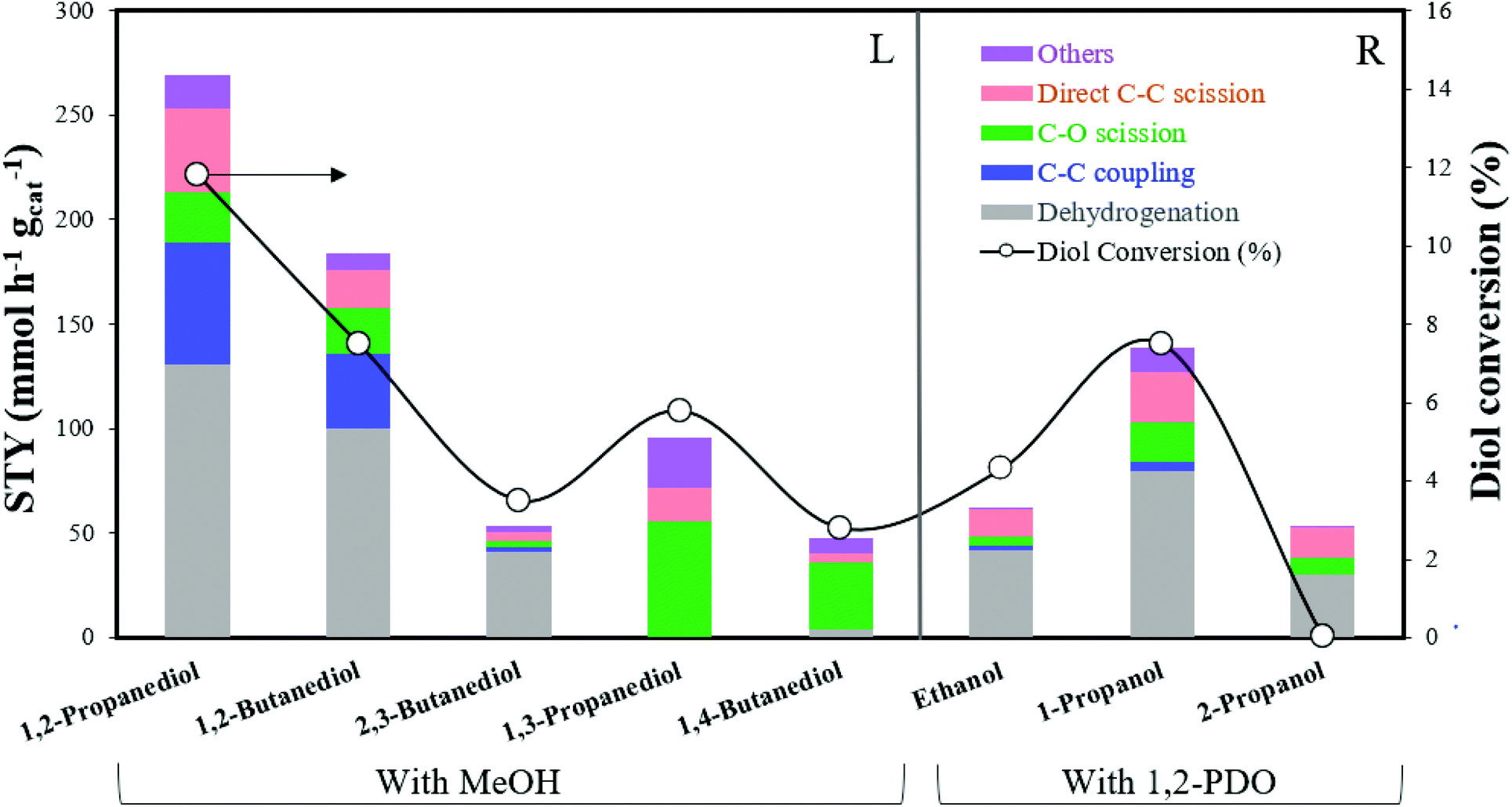
At higher conversions during SCM-DHDO of whole biomass, the chemistry between intermediate diols and monoalcohols besides MeOH becomes important. Monoalcohols with 2 or more carbons can also self-couple via the Guerbet reaction, which is not observed with MeOH due to the absence of β-hydrogens.30 In this section, experiments with MeOH and other α,β-diols (1,2-butanediol and 2,3-butanediol) and α,ω-diols (1,3-propanediol and 1,4-butanediol) have been carried out to examine the effect of diol carbon size and proximity of hydroxyl groups on product selectivities. Experiments with 1,2-PDO and other monoalcohols (ethanol, 1-propanol, 2-propanol) have also been carried out to examine the effect of monoalcohol carbon size and functionality of the hydroxyl carbon on product rates. The feed composition was maintained at 10 mol% diol in monoalcohol and the catalyst loading was adjusted accordingly to maintain a WHSV of 120 h−1 for each run. Fig. 5 summarizes the product STYs for C–C coupling, C–O scission, direct C–C scission, and the diol conversion for each diol/monoalcohol combination tested.
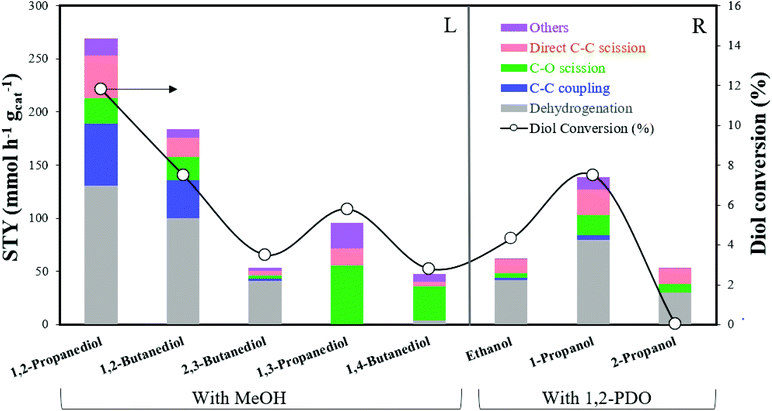 |
| | Fig. 5 STY of C–C coupling, C–O scission, direct C–C scission, and diol conversion for reaction between: (L) Diols with MeOH; (R) monoalcohols with 1,2-PDO. Reaction conditions: 300 °C, 1000 psig, CuMgAlOx, 120 h−1 WHSV, 0.1 mL min−1 feed with 10 mol% diol in monoalcohol, 102.5 mL min−1 H2, 24 h. | |
Reaction pathways of MeOH with other diols.
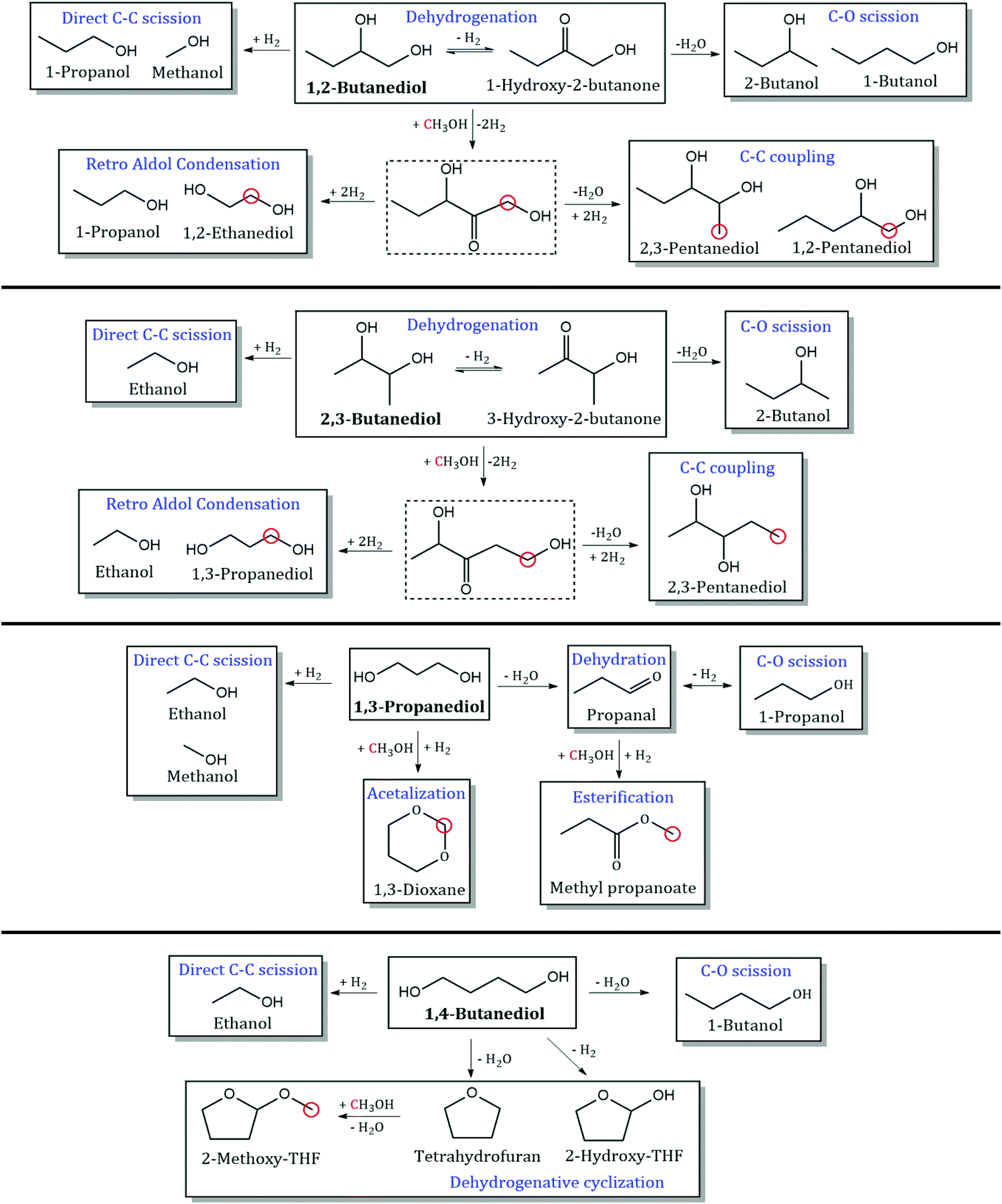
Detailed product STYs and diol conversion rates from the reaction of MeOH with other diols are shown in Table S3.† The reaction network for the major products has been shown in Scheme 4. C–C coupling is observed with all vicinal diols studied (1,2-PDO, 1,2-butanediol and 2,3-butanediol). 1,2-PDO has the highest conversion (11.8%), while 1,2-butanediol and 2,3-butanediol have lower conversions (7.5 and 3.5% respectively). 1,2-PDO also has the highest C–C coupling rate with MeOH, followed by 1,2-butanediol. The reaction pathways and selectivity trends of 1,2-butanediol are very similar to those of 1,2-PDO. 1,2-Butanediol dehydrogenates to form 1-hydroxy-2-butanone with a selectivity of 41%. C–C coupling at the primary hydroxyl carbon of 1,2-butanediol with MeOH leads to the formation of 1,2-pentanediol (5%) and 2,3-pentanediol (9%). C–C coupling at the secondary alkyl carbon with MeOH would lead to the formation of 2-methyl-1,2-butanediol, which is not observed at these reaction conditions, likely due to steric hindrance. Retro aldol condensation of the C–C coupling intermediate from 1,2-butanediol forms 1,2-ethanediol (3%) and 1-propanol, while direct C–C scission of 1,2-butanediol forms 1-propanol (7.2%) and MeOH. 1,2-Butanediol also undergoes C–O scission to form 1-butanol (4%) and 2-butanol (5%).
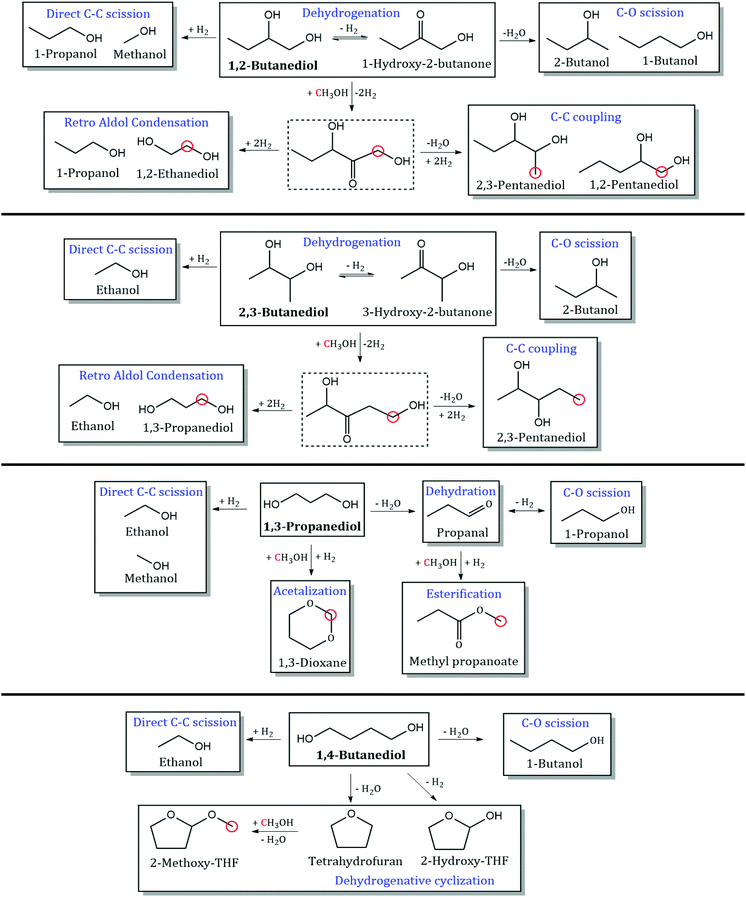 |
| | Scheme 4 Products observed from reaction of MeOH with 1,2-butanediol, 2,3-butanediol, 1,3-propanediol, and 1,4-butanediol. Dotted lines indicate unobserved intermediates. Red circles indicate incorporation of carbon from MeOH. Reaction conditions: 300 °C, 1000 psig, CuMgAlOx, 120 h−1 WHSV, 0.1 mL min−1 feed with 10 mol% diol in MeOH, 102.5 mL min−1 H2, 24 h. | |
The trends with an internal vicinal diol like 2,3-butanediol are different. 2,3-Butanediol dehydrogenates to form 3-hydroxy-2-butanone with a selectivity of 44%. C–C coupling of 2,3-butanediol with MeOH forms 2,3-pentanediol (3%) but the C–C coupling STY of 2,3-butanediol (2.6 mmol h−1 gcat−1) is an order of magnitude lower than that of 1,2-butanediol (36 mmol h−1 gcat−1). Retro aldol condensation is observed to form ethanol and 1,3-propanediol (4%), which is a terminal diol. As seen from the structure of the proposed C–C coupling intermediate (1,4-dihydroxy-3-pentanone), C–O scission of 2,3-butanediol forms 2-butanol (3%) and direct C–C scission forms ethanol (4%). Based on the observed reaction network from vicinal diols, some generalizations can be made. C–C coupling of MeOH with a 1,2-diol with n carbons leads to the formation of 1,2- and 2,3-diols with n + 1 carbons, with higher selectivity to the 2,3-diol. Retro aldol condensation occurs between the 2nd and 3rd carbons of the C–C coupling intermediate (with the 1st carbon originating from MeOH) to form 1,2-ethanediol and a monoalcohol with n − 1 carbons. C–O scission occurs at both hydroxyl groups of the 1,2-diol, with slightly higher selectivity to the secondary monoalcohol formed. 2,3-diols cannot form 1,2-diols via C–C coupling or retro aldol condensation. Instead, 2,3-diols with n carbons undergo C–C coupling with MeOH to form 2,3-diols with n + 1 carbons. Retro aldol condensation forms a terminal diol and a lower monoalcohol.
C–C coupling between MeOH and terminal diols (1,3-propanediol and 1,4-butanediol) is not observed at these reaction conditions. Nucleophilic attack by the β-carbon of α,ω-diols would lead to the formation of methyl-branched α,ω-diols, such as 2-methyl-1,3-propanediol, which are not detected. Unlike α,β-diols that form resonance stabilized α,β-unsaturated ketones during C–C coupling, the absence of C–C coupling products with α,ω-diols could be due to steric hindrance at the nucleophilic center of the diol or instability of the α,ω-unsaturated intermediate. 1,3-PDO was observed to undergo C–O scission to form 1-propanol (46%), and C–C scission to form ethanol (15%). Acetalization of 1,3-propanediol with formaldehyde produces 1,3-dioxane (23%), a reaction that is carried out commercially to extract diols from complex mixtures.31 1,4-Butanediol was observed to undergo C–O scission to form 1-butanol (19%), and dehydration to produce tetrahydrofuran (15%). Dehydrogenation of 1,4-butanediol leads to the formation of 2-hydroxy-THF (16%), which is proposed to occur via hemiacetalization of an unobserved intermediate, 4-hydroxybutanal. Ichikawa et al. observed similar reaction pathways in their work on dehydrogenative cyclization of 1,4-butanediol to produce γ-butyrolactone via 2-hydroxy-THF over Cu catalysts.32
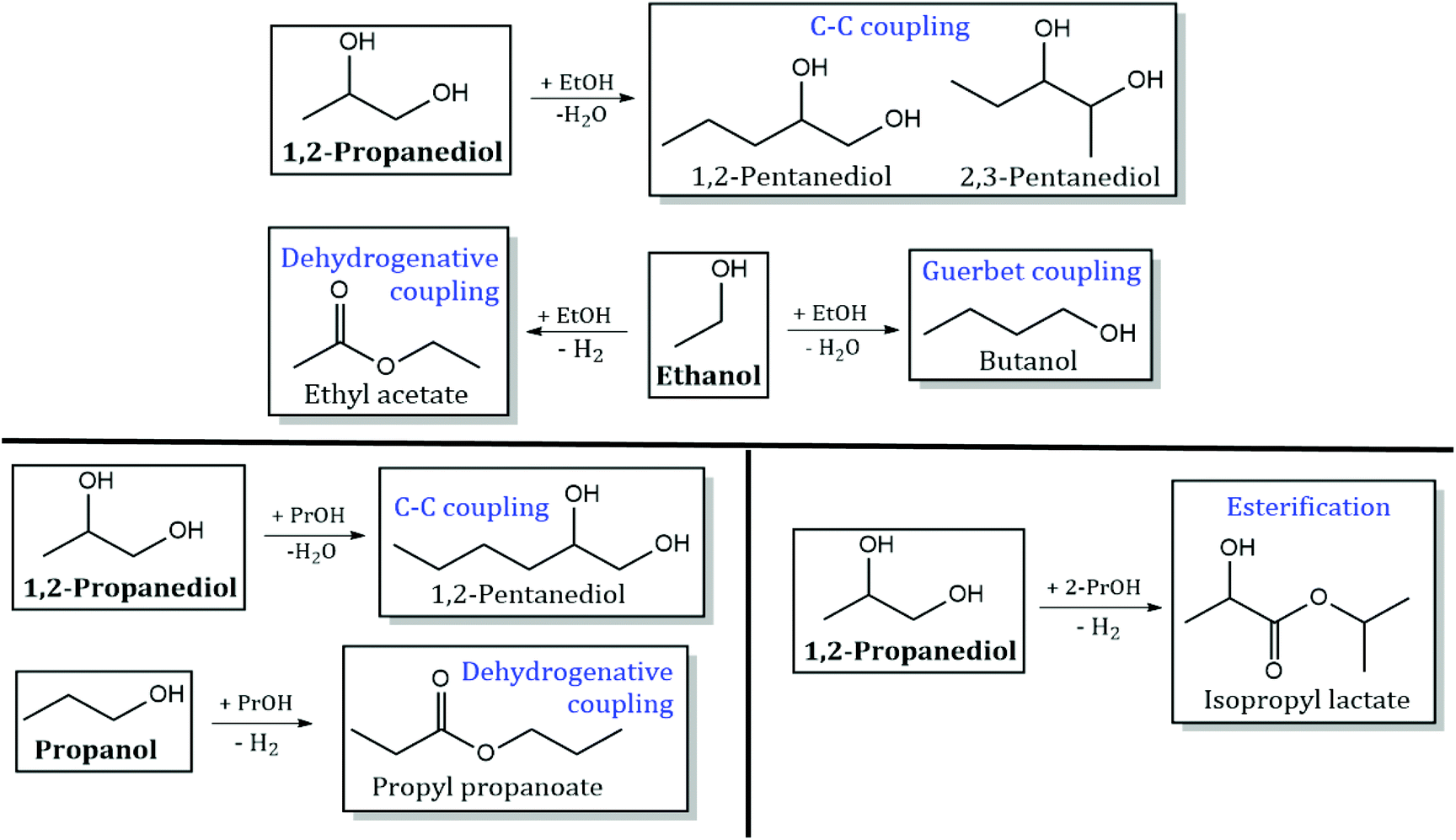
Reaction pathways of 1,2-PDO with other monoalcohols.
Detailed product STYs and 1,2-PDO conversion for experiments with 10 mol% 1,2-PDO in varying monoalcohols (ethanol, 1-propanol and 2-propanol) are shown in Table S4.† The reaction network for products formed from C–C coupling of 1,2-PDO with the monoalcohol, and self-coupling/esterification of the monoalcohol are shown in Scheme 5. The main products observed are from dehydrogenation of 1,2-PDO (hydroxyacetone), direct C–C scission of 1,2-PDO (ethanol and MeOH) and C–O scission of 1,2-PDO (1-propanol and 2-propanol).
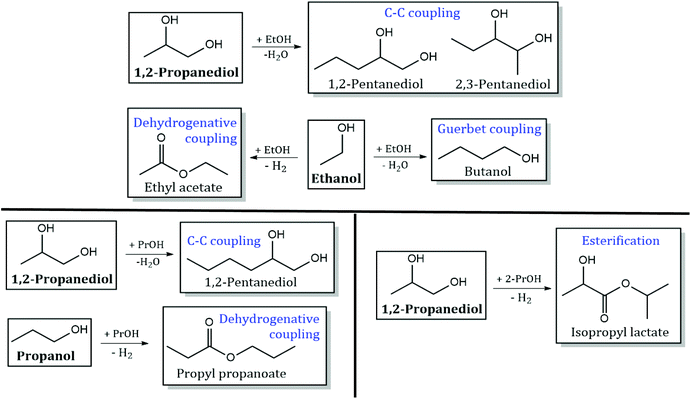 |
| | Scheme 5 Products observed from C–C coupling/esterification of 1,2-PDO with ethanol (EtOH), 1-propanol (1-PrOH), and 2-propanol (2-PrOH), and self-coupling of the monoalcohol. Products from direct C–C scission and C–O scission of 1,2-PDO are not shown. Reaction conditions: 300 °C, 1000 psig, CuMgAlOx, 120 h−1 WHSV, 0.1 mL min−1 feed with 10 mol% 1,2-PDO in monoalcohol, 102.5 mL min−1 H2, 24 h. | |
1,2-PDO conversion with ethanol is 4.3%, nearly half of that with MeOH (11.8%). Hydroxyacetone was observed at a selectivity of 66%, followed by MeOH at 23%. C–O scission of 1,2-PDO produces 1-propanol (3%) and 2-propanol (1.5%). A small amount of C–C coupling of 1,2-PDO with ethanol is observed to occur at both terminal carbons of 1,2-PDO to form 2,3-pentanediol (1.5% selectivity) and 1,2-pentanediol (1%), but the total selectivity to C–C coupling of 1,2-PDO with ethanol (2.5%) is 10 times lower than that with MeOH (24%). No retro aldol condensation products are observed when 1,2-PDO is reacted with C2+ monoalcohols at these reaction conditions. Acetaldehyde is observed to form from dehydrogenation of ethanol at an STY of 33 mmol h−1 gcat−1. 1-Butanol (0.1 mmol h−1 gcat−1) and ethyl acetate (1 mmol h−1 gcat−1) are also detected from Guerbet coupling and dehydrogenative coupling of ethanol, respectively.15 1,2-PDO conversion with 1-propanol is 7.5%, which is lower than 1,2-PDO conversion with MeOH but higher than that with ethanol. Hydroxyacetone is observed with a selectivity of 44%, followed by MeOH and ethanol (each with a selectivity of 14%). 2-Propanol is produced at a selectivity of 5%, but 1-propanol produced from C–O scission of 1,2-PDO could not quantified as 1-propanol is the solvent in this reaction. C–C coupling of 1,2-PDO with 1-propanol is observed to form 1,2-hexanediol at a selectivity of 2%. Retro aldol condensation products are not seen. Propyl propanoate is observed to form at an STY of 12 mmol h−1 gcat−1via dehydrogenative coupling of 1-propanol with itself, similar to the pathway involved in ethyl acetate formation. With 2-propanol, 1,2-PDO conversion drops to 0.05% and C–C coupling is not observed. Shimura et al. observed the self-coupling of secondary alcohols like isopropanol to 4-methyl-2-pentanone at low temperatures (80–144 °C) over Ni/CeO2 catalysts.33 This was not observed over CuMgAlOx at the given reaction conditions. Hydroxyacetone is observed to form with a selectivity of 67%, followed by MeOH (22%) and 1-propanol (11%). Esterification of 1,2-PDO with 2-propanol to form isopropyl lactate was observed at a very low selectivity of 0.5%.
Conclusions
α,β-Diols like 1,2-PDO have been identified as important cellulose-derived intermediates during biomass upgrading with SCM-DHDO. In this work, the reaction network between 1,2-PDO and MeOH was elucidated at 300 °C and 1000 psig over a CuMgAlOx catalyst in a continuous flow reactor. An isotopic study with 13C MeOH demonstrated that C–C coupling occurs at both terminal carbons of 1,2-PDO. C–C coupling at the primary hydroxyl carbon occurs at higher rates and leads to the formation of 2,3-butanediol or 1,2-butanediol, while retro aldol condensation of the C–C coupling intermediate forms 1,2-ethanediol. C–C coupling at the primary alkyl carbon of 1,2-PDO only forms 1,2-butanediol. 13C incorporation from MeOH was only observed in 2,3-butanediol, 1,2-butanediol, 1,2-ethanediol, and methyl lactate. Direct C–C scission of 1,2-PDO produces ethanol and MeOH, while C–O scission produces 1-propanol and 2-propanol.
C–C coupling has a 1.3 reaction order dependence on PMeOH partial pressure but is zero-order with respect to PH2 and P1,2-PDO. C–O scission is 1st order in PH2 but has low/near-zero reaction orders with respect to PMeOH and P1,2-PDO. Direct C–C scission has 0.8–0.7 reaction orders with respect to PH2, PMeOH and P1,2-PDO. Water inhibits all reactions, with C–O scission having a negative 1st order dependence on PH2O. Cofeed experiments were also carried out by replacing 0–40% of MeOH with formaldehyde, and 20–60% of 1,2-PDO with hydroxyacetone. As formaldehyde concentration was increased, C–C coupling yields increased by an order of magnitude, while dehydrogenation decreased. As hydroxyacetone concentration was increased, the yields of C–C coupling, direct C–C scission and esterification increased, while C–O scission and retro aldol condensation remained nearly the same. These results reveal that the rate determining step in C–C coupling is associated with the adsorption of MeOH, dehydrogenation of MeOH to formaldehyde, or the subsequent nucleophilic attack by the 1,2-PDO derived species.
The apparent activation energy of dehydrogenation and C–C coupling were found to be 45.6 and 47.2 kJ mol−1 respectively, the lowest among all reactions observed. The apparent activation energies for C–O scission (64.9 kJ mol−1), direct C–C scission (68.3 kJ mol−1), retro aldol condensation (69.4 kJ mol−1) and esterification (62.7 kJ mol−1) were found to be at least 15 kJ mol−1 higher. We also carried out experiments with MeOH and other α,β-diols (1,2-butanediol and 2,3-butanediol), and α,ω-diols (1,3-propanediol and 1,4-butanediol). C–C coupling was only observed between MeOH and α,β-diols, while the major products from α,ω-diols were C–O scission and C–C scission. Experiments carried out with 1,2-PDO and other monoalcohols (ethanol, 1-propanol and 2-propanol) showed C–C coupling occurring between 1,2-PDO and primary alcohols, but not with secondary alcohols like 2-propanol. Moreover, products from self-coupling/esterification of the monoalcohols, such as 1-butanol and ethyl acetate from ethanol, were observed with C2+ monoalcohols.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was funded by ExxonMobil Research and Engineering Company. The authors would like to thank the Magnetic Resonance Facility in the Chemistry Department at the University of Wisconsin–Madison for the use of the Bruker Avance III 500 MHz spectrometer gifted by Paul J. Bender. The authors gratefully acknowledge the use of facilities and intrumentation supported by NSF through the University of Wisconsin Materials Research Science and Engineering Center (DMR-1121288).
References
- M. G. Alriols, A. García, R. Llano-Ponte and J. Labidi, Combined organosolv and ultrafiltration lignocellulosic biorefinery process, Chem. Eng. J., 2010, 157(1), 113–120 CrossRef CAS.
- G. De Bhowmick, A. K. Sarmah and R. Sen, Lignocellulosic biorefinery as a model for sustainable development of biofuels and value added products, Bioresour. Technol., 2018, 247, 1144–1154 CrossRef CAS PubMed.
- A. K. Chandel, V. K. Garlapati, A. K. Singh, F. A. F. Antunes and S. S. da Silva, The path forward for lignocellulose biorefineries: bottlenecks, solutions, and perspective on commercialization, Bioresour. Technol., 2018, 264, 370–381 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, The Path Forward for Biofuels and Biomaterials, Science, 2006, 311(5760), 484 CrossRef CAS PubMed.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, One-pot catalytic conversion of cellulose and of woody biomass solids to liquid fuels, J. Am. Chem. Soc., 2011, 133(35), 14090–14097 CrossRef CAS PubMed.
- C. Li, M. Zheng, A. Wang and T. Zhang, One-pot catalytic hydrocracking of raw woody biomass into chemicals over supported carbide catalysts: simultaneous conversion of cellulose, hemicellulose and lignin, Energy Environ. Sci., 2012, 5(4), 6383–6390 RSC.
- Y. Liu, L. Chen, T. Wang, Q. Zhang, C. Wang, J. Yan and L. Ma, One-Pot Catalytic Conversion of Raw Lignocellulosic Biomass into Gasoline Alkanes and Chemicals over LiTaMoO6 and Ru/C in Aqueous Phosphoric Acid, ACS Sustainable Chem. Eng., 2015, 3(8), 1745–1755 CrossRef CAS.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Direct hydrodeoxygenation of raw woody biomass into liquid alkanes, Nat. Commun., 2016, 7(1), 11162 CrossRef PubMed.
- P. H. Galebach, D. J. McClelland, N. M. Eagan, A. M. Wittrig, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Production of Alcohols from Cellulose by Supercritical Methanol Depolymerization and Hydrodeoxygenation, ACS Sustainable Chem. Eng., 2018, 6(3), 4330–4344 CrossRef CAS.
- P. H. Galebach, S. Thompson, A. M. Wittrig, J. S. Buchanan and G. W. Huber, Investigation of the Reaction Pathways of Biomass-Derived Oxygenate Conversion into Monoalcohols in Supercritical Methanol with CuMgAl-Mixed-Metal Oxide, ChemSusChem, 2018, 11(23), 4007–4017 CrossRef CAS PubMed.
- Z. Sun and K. Barta, Cleave and couple: toward fully sustainable catalytic conversion of lignocellulose to value added building blocks and fuels, Chem. Commun., 2018, 54(56), 7725–7745 RSC.
- F. Dalena, A. Senatore, A. Marino, A. Gordano, M. Basile and A. Basile, Methanol production and applications: An overview, Methanol, 2018, 3–28 Search PubMed.
- P. H. Galebach, J. K. Soeherman, A. M. Wittrig, M. P. Lanci and G. W. Huber, Supercritical Methanol Depolymerization and Hydrodeoxygenation of Maple Wood and Biomass-Derived Oxygenates into Renewable Alcohols in a Continuous Flow Reactor, ACS Sustainable Chem. Eng., 2019, 7(18), 15361–15372 CrossRef CAS.
- Z. Sun, A. S. Couto Vasconcelos, G. Bottari, M. C. Stuart, G. Bonura, C. Cannilla, F. Frusteri and K. Barta, Efficient catalytic conversion of ethanol to 1-butanol via the Guerbet reaction over copper-and nickel-doped porous, ACS Sustainable Chem. Eng., 2017, 5(2), 1738–1746 CrossRef CAS.
- J. T. Kozlowski and R. J. Davis, Heterogeneous Catalysts for the Guerbet Coupling of Alcohols, ACS Catal., 2013, 3(7), 1588–1600 CrossRef CAS.
- J. J. Bravo-Suárez, B. Subramaniam and R. V. Chaudhari, Vapor-phase methanol and ethanol coupling reactions on CuMgAl mixed metal oxides, Appl. Catal., A, 2013, 455, 234–246 CrossRef.
- C. Carlini, C. Flego, M. Marchionna, M. Noviello, A. M. Raspolli Galletti, G. Sbrana, F. Basile and A. Vaccari, Guerbet condensation of methanol with n-propanol to isobutyl alcohol over heterogeneous copper chromite/Mg–Al mixed oxides catalysts, J. Mol. Catal. A: Chem., 2004, 220(2), 215–220 CrossRef CAS.
- P. H. Galebach, J. K. Soeherman, E. Gilcher, A. M. Wittrig, J. Johnson, T. Fredriksen, C. Wang, M. P. Lanci, J. A. Dumesic and G. W. Huber, Production of renewable alcohols from maple wood using supercritical methanol hydrodeoxygenation in a semi-continuous flowthrough reactor, Green Chem., 2020, 22(23), 8462–8477 RSC.
- Y. Amada, S. Koso, Y. Nakagawa and K. Tomishige, Hydrogenolysis of 1,2-propanediol for the production of biopropanols from glycerol, ChemSusChem, 2010, 3(6), 728–736 CrossRef CAS PubMed.
- M. Araki and V. Ponec, Methanation of carbon monoxide on nickel and nickel-copper alloys, J. Catal., 1976, 44(3), 439–448 CrossRef CAS.
- V. L. Yfanti and A. A. Lemonidou, Mechanistic study of liquid phase glycerol hydrodeoxygenation with in-situ generated hydrogen, J. Catal., 2018, 368, 98–111 CrossRef CAS.
- F. Zaccheria, N. Scotti and N. Ravasio, The role of copper in the upgrading of bioalcohols, ChemCatChem, 2018, 10(7), 1526–1535 CrossRef CAS.
- E. Taarning, A. T. Madsen, J. M. Marchetti, K. Egeblad and C. H. Christensen, Oxidation of glycerol and propanediols in methanol over heterogeneous gold catalysts, Green Chem., 2008, 10(4), 408–414 RSC.
- N. M. Eagan, B. M. Moore, D. J. McClelland, A. M. Wittrig, E. Canales, M. P. Lanci and G. W. Huber, Catalytic synthesis of distillate-range ethers and olefins from ethanol through Guerbet coupling and etherification, Green Chem., 2019, 21(12), 3300–3318 RSC.
- G. Peng, X. Wang, X. Chen, Y. Jiang and X. Mu, Zirconia-supported niobia catalyzed formation of propanol from 1, 2-propanediol via dehydration and consecutive hydrogen transfer, J. Ind. Eng. Chem., 2014, 20(5), 2641–2645 CrossRef CAS.
- T. Gabrysch, B. Peng, S. Bunea, G. Dyker and M. Muhler, The Role of Metallic Copper in the Selective Hydrodeoxygenation of Glycerol to 1,2-Propanediol over Cu/ZrO2, ChemCatChem, 2018, 10(6), 1344–1350 CrossRef CAS.
- C. R. Ho, S. Shylesh and A. T. Bell, Mechanism and Kinetics of Ethanol Coupling to Butanol over Hydroxyapatite, ACS Catal., 2016, 6(2), 939–948 CrossRef CAS.
- M. Neurock, Z. Tao, A. Chemburkar, D. D. Hibbitts and E. Iglesia, Theoretical insights into the sites and mechanisms for base catalyzed esterification and aldol condensation reactions over Cu, Faraday Discuss., 2017, 197, 59–86 RSC.
- S. Hanspal, Z. D. Young, J. T. Prillaman and R. J. Davis, Influence of surface acid and base sites on the Guerbet coupling of ethanol to butanol over metal phosphate catalysts, J. Catal., 2017, 352, 182–190 CrossRef CAS.
- D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization, Catal. Sci. Technol., 2015, 5(8), 3876–3902 RSC.
- J. Hao, H. Liu and D. Liu, Novel route of reactive extraction to recover 1, 3-propanediol from a dilute aqueous solution, Ind. Eng. Chem. Res., 2005, 44(12), 4380–4385 CrossRef CAS.
- N. Ichikawa, S. Sato, R. Takahashi, T. Sodesawa and K. Inui, Dehydrogenative cyclization of 1,4-butanediol over copper-based catalyst, J. Mol. Catal. A: Chem., 2004, 212(1–2), 197–203 CrossRef CAS.
- K. Shimura, K. Kon, S. M. A. Hakim Siddiki and K.-I. Shimizu, Self-coupling of secondary alcohols by Ni/CeO2 catalyst, Appl. Catal., A, 2013, 462–463, 137–142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03650e |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Michael P.
Lanci
b,
Chengrong
Wang
b,
Yi
Du
a,
Michael P.
Lanci
b,
Chengrong
Wang
b,
Yi
Du