
Dihydrolevoglucosenone (Cyrene™) as a versatile biobased solvent for lignin fractionation, processing, and chemistry†
Antoine
Duval
 * and
Luc
Avérous
*
* and
Luc
Avérous
*
BioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: antoine.duval@unistra.fr; luc.averous@unistra.fr
First published on 10th December 2021
Abstract
The solubility of technical lignins is a complex issue that depends on many parameters, such as the lignin structure governed by the botanical origin and the extraction process. Only polar aprotic solvents, such as dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAc), are able to fully dissolve a large range of technical lignins. However, DMF and DMAc are fossil-based and present serious toxicity issues. In this study, we evaluated the potential of a biobased, non-cytotoxic and non-mutagenic solvent, dihydrolevoglucosenone (Cyrene™) as an alternative polar aprotic solvent for lignins. The solubility of Kraft (KL), soda and organosolv lignins was first evaluated. It appeared that full solubility could be achieved in Cyrene–water mixtures, between 60 and 80–90 vol% Cyrene, probably because of the peculiar property of Cyrene to form a geminal diol upon water addition. Cyrene and water could then be used as solvent and non-solvent to refine KL by fractional precipitation, leading to the recovery of fractions of controlled molar masses. Finally, after ensuring that it was inert to lignin, Cyrene was shown to be a suitable solvent for the chemical modification of different lignins. Three common reactions were tested as proof-of-concept: esterification with an acyl chloride or a cyclic anhydride, and formation of urethane linkages with an isocyanate. This study thus shows that Cyrene is a promising and versatile green solvent for lignin fractionation, processing and chemistry, to ensure greener processes when solvents are mandatory.
Introduction
Lignin is the second most abundant component of biomass, and the first source of renewable aromatic structures. The interest in lignin is broad and fast-growing, involving academic and industrial researchers from different disciplines. Therefore, a lot of work has been dedicated in the past decade to the development of new processes to isolate lignin from lignocellulosic biomass. Many biorefinery processes have been proposed, based on different biomass pretreatments, solvents or catalysts,1–3 with some of them scaling-up towards pilot or industrial facilities.4 On the other hand, pulp and paper processes are industrially mature, with an annual production of paper pulp of more than 100 Mt per year.5 They generate about 50 Mt per year of lignin as by-product of cellulose pulp, mainly resulting from the Kraft process. Although the large majority of Kraft lignin (KL) is still burnt to generate energy, different processes to recover KL from the black liquor have been developed,6,7 leading to a rapid increase in its commercial availability.8Driven by the worldwide depletion of some petrol fractions and the growing demand for more sustainability, an increasing amount of research is focusing on lignin depolymerization to replace the conventional BTX (benzene–toluene–xylene) fractions or to produce aromatic platform molecules.9,10 Our group, among others, is rather focusing on the direct use of lignin macromolecules in polymer materials applications.11,12 Efficient use of lignin in polymer materials requires addressing a set of challenges according to the type and the origin of the resource. Technical lignins usually have important variability and large dispersity, which complicate their use. Among the various techniques that have been employed to refine lignins into controlled fractions of low dispersity, those relying on lignin partial solubility in organic solvents or on the controlled precipitation using a non-solvent afforded good results.13,14 Applications of lignin in polymer science also require chemical modifications to achieve the right architectures and better properties. They can aim at modifying the lignin polarity to increase its compatibility with apolar polymer matrices,15,16 or introducing new chemical functions that can be used in further polymerization reactions,17–19 for instance.
All these objectives require most of the time to solubilize the lignin. The solubility of lignin is a complex issue that depends on many parameters, including its chemical structure, molar mass and dispersity, which all result from both the botanical origin and the process used for its isolation from the lignocellulosic biomass. Very few organic solvents are indeed able to dissolve a large range of technical lignins, such as Kraft (KL), soda (SL) or organosolv (OSL) lignins. Polar aprotic solvents, such as dimethylsulfoxide (DMSO), dimethylformamide (DMF) and dimethylacetamide (DMAc) are among them. DMSO, which can be obtained as by-product of the Kraft process, is often used as solvent for lignin NMR (in its deuterated form) and for size-exclusion chromatography (SEC),20,21 but more occasionally for chemical reactions22–27 because of its limited chemical stability.28 On the other hand, DMF and DMAc are widely used as solvents for lignin chemical modifications,29–37 especially for esterification reactions with carboxylic acids,38–41 acyl chlorides42–48 or anhydrides.49 However, they are fossil-based and present serious health issues (reprotoxicity). They are classified as hazardous by the most recent solvent selection guides,50,51 therefore encouraging to look for safer alternatives.
The need to ban toxic solvents has led us to look for new strategies for lignin chemical modification in the past few years. Reactions in solvent-free conditions were found to be possible when the reagent has indeed a polar aprotic character, which allows it to dissolve the lignin, as in the case of cyclic carbonates52–55 or trimethyl phosphate.16 However, this strategy requires using the reagent in large excess, and is not applicable to all kinds of chemical modifications of lignins. Looking for safer and greener polar aprotic solvents is another option to consider. Recently, dihydrolevoglucosenone has appeared as a non-cytotoxic, non-mutagenic, and biobased alternative to common polar aprotic solvents.56 It is obtained from the acid-catalyzed pyrolysis of cellulose forming levoglucosenone, followed by hydrogenation.57 It is now produced industrially on 50 t per year scale by the Australian company Circa,57 under the trade name Cyrene™, with a scale-up to 1000 t per year under development.58 Cyrene has been shown to be a potential substitute for DMF in various chemical reactions, including palladium-catalyzed Sonogashira and Suzuki–Miyaura cross-coupling reactions,59,60 Menschutkin reaction,56 fluorination,56 ureas61 and amides syntheses.62,63 Its solvent parameters (Hansen and Kamlett–Taft) are relatively close to those of DMSO, DMF and DMAc (Table 1), thus presaging good potential for lignin dissolution.
| Solvent | Hansen solubility parameters (MPa1/2) | Kamlett–Taft parameters | bp (°C) | ρ (g mL−1) | η 25 °C (mPa s) | ||||
|---|---|---|---|---|---|---|---|---|---|
| δ D | δ P | δ H | α | β | π* | ||||
| a 0.61 was obtained by the solvatochromic method using 4-nitroaniline/N,N-diethyl-4-nitroaniline dyes.56 Recent results obtained by the 19F solvatomagnetic method rather suggest a lower value of 0.40.67 | |||||||||
| Cyrene56 | 18.8 | 10.6 | 6.9 | 0.00 | 0.61 (0.40) a | 0.93 | 203 | 1.25 | 14.5 |
| DMSO65,66 | 18.4 | 16.4 | 10.2 | 0.00 | 0.76 | 1.00 | 189 | 1.10 | 1.97 |
| DMF65,66 | 17.4 | 13.7 | 11.3 | 0.00 | 0.69 | 0.88 | 153 | 0.95 | 0.93 |
| DMAc66 | 16.8 | 11.5 | 10.2 | 0.00 | 0.76 | 0.88 | 165 | 0.94 | 1.0 |
Recently, Ragauskas and co-workers used Cyrene–water mixtures as pretreatment of poplar wood to extract lignin.64 Delignification was achieved in mild conditions, thus preserving the lignin structure, with a low level of condensation and a high content in β-O-4 linkages. Preliminary tests in their study also showed a good solubility of ethanol OSL in Cyrene–water mixtures from 60 to 100 vol% Cyrene.
The aim of this study is to further examine the potential of Cyrene as biobased and safe solvent of technical lignins. We first studied in detail the solubility of the most common technical lignins (KL, SL and OSL) in pure Cyrene and in Cyrene–water mixtures. Then, we used Cyrene to refine KL into low dispersity fractions using fractional precipitation in water. Finally, we examined the possibility to use Cyrene as solvent for the chemical modification of lignin, focusing on three different reactions as a global proof of concept.
Materials and methods
Materials
Softwood Kraft lignin (KL, Indulin AT) was obtained from Mead Westvaco, soda lignin from a mixture of wheat straw and Sarkanda grass (SL, Protobind 1000) was obtained from GreenValue and acetone organosolv lignin from beech wood (OSL) was extracted in a pilot plant at Fraunhofer CBP (Leuna, Germany) using the Fabiola™ process.68 Lignins were dried overnight in a vacuum oven at 40 °C, then stored in a desiccator prior to use.Dihydrolevoglucosenone (Cyrene™), heptanoyl chloride (HC, 99%) and succinic anhydride (SA, ≥99%) were purchased from Sigma-Aldrich. N,N-Dimethylformamide (DMF, 99.8%, extra dry, AcroSeal®) and hexyl isocyanate (HI, 99%) were purchased from Acros Organics. All chemicals were used as received without further purification.
Solubility tests in Cyrene–water mixtures
50 mg of lignin (mlig) were introduced into pre-weighed Eppendorf tubes (mtube), to which 1 mL of Cyrene–water mixture was added. The tubes were then shaken for 16 h at 200 rpm on a platform shaker at room temperature (about 25 °C). The soluble and insoluble fractions were separated by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rpm for 3 min), and the insoluble fractions were washed 3 times with deionized water to ensure complete removal of Cyrene, then dried in a vacuum oven at 40 °C for 24 h, before the final mass mf was measured.
800 rpm for 3 min), and the insoluble fractions were washed 3 times with deionized water to ensure complete removal of Cyrene, then dried in a vacuum oven at 40 °C for 24 h, before the final mass mf was measured.
The solubility was then calculated according to eqn (1):
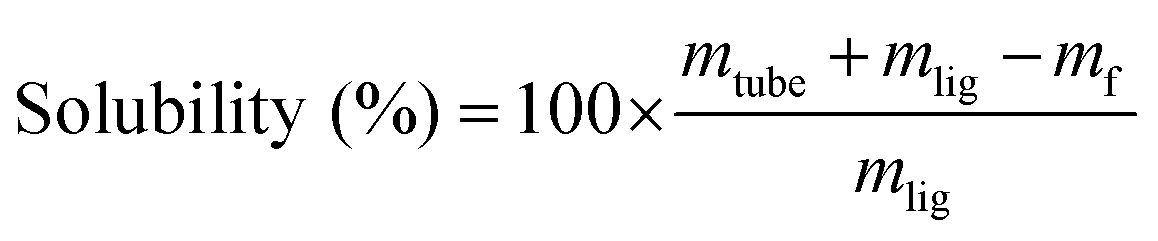 | (1) |
Thermal treatment of lignin in Cyrene
250 mg KL and 2 mL Cyrene were introduced in a glass vial and placed under stirring in an oil bath regulated at 50, 75 or 100 °C for 16 h. After cooling to room temperature, the solution was added dropwise to an aqueous HCl solution (pH 2) to precipitate the lignin, which was recovered by filtration over 0.45 μm PVDF membranes and dried under vacuum at 40 °C overnight.Fractional precipitation of KL
1 g of KL was dissolved in 20 mL of 60 vol% Cyrene solution in water. Water was then added to obtain a 50 vol% Cyrene solution, causing the precipitation of part of the lignin, which was recovered by centrifugation, washed with deionized water to remove all Cyrene, and dried under vacuum at 40 °C overnight. Cyrene concentration in the solution was then successively reduced to 40, 30 and 5 vol% by addition of water, and the precipitated lignin was recovered as before. Finally, the 5 vol% Cyrene solution was acidified to pH 2 by addition of a 2 M HCl solution, causing the precipitation of some lignin, which was recovered by filtration over 0.45 μm PVDF membranes and dried under vacuum at 40 °C overnight.Chemical modification of lignin with acyl chloride
The protocol for lignin modification with acyl chloride was adapted from Koivu et al.47 Lignin (0.25 g), triethylamine (TEA, 1 molar equivalent to lignin OH groups) and Cyrene or DMF (2 mL) were added to a round bottom flask, which was flushed with argon for 5 min under stirring. Heptanoyl chloride (HC, 1.3 molar equivalents to lignin OH groups) was then added dropwise to the mixture with a syringe, and the mixture was placed in an oil bath regulated at 50 °C. The reaction was conducted for 16 h. At the end of the reaction, a homogeneous black solution was obtained. It was poured into about 150 mL of cold water, leading to the precipitation of the product, which was recovered by filtration and dried overnight at 40 °C under vacuum.Chemical modification of lignin with acid anhydride
Lignin (0.25 g), 1-methylimidazole (1-MIM, 0.1 molar equivalent to lignin OH groups) and Cyrene or DMF (2 mL) were added to a round bottom flask, which was flushed with argon for 5 min under stirring. Succinic anhydride (SA, 1 molar equivalent to lignin OH groups) was then added to the mixture, which was then immersed in an oil bath regulated at 50 °C. The reaction was conducted for 16 h. At the end of the reaction, a homogeneous black solution was obtained. It was poured into about 150 mL of cold water, leading to the precipitation of the product, which was recovered by filtration and dried overnight at 40 °C under vacuum.Chemical modification of lignin with isocyanate
Lignin (0.25 g), N,N′-dimethylcyclohexylamine (DMCHA, 0.1 molar equivalent to lignin OH groups) and Cyrene or DMF (2 mL) were added to a round bottom flask, which was flushed with argon for 5 min under stirring. Hexyl isocyanate (HI, 1 molar equivalent to lignin OH groups) was then added dropwise to the mixture, which was then immersed in an oil bath regulated at 50 °C. The reaction was conducted for 16 h. At the end of the reaction, a homogeneous black solution was obtained. It was poured into about 150 mL of cold water, leading to the precipitation of the product, which was recovered by filtration and dried overnight at 40 °C under vacuum.Lignins characterizations
1D NMR spectra were recorded on a Bruker 400 MHz spectrometer at 25 °C. For 1H NMR spectroscopy, about 20 mg of sample were dissolved in DMSO-d6 or CDCl3, depending on the solubility, and 16 scans were collected. Quantitative 31P NMR spectroscopy was performed following the standard literature protocol.69,70 The lignin samples were derivatized with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, Sigma-Aldrich, 95%) in pyridine/CDCl3 (1.6:1) in the presence of cholesterol as internal standard, and 128 scans were recorded using an inverse-gated decoupling and 15 s relaxation delay. 2D HSQC NMR spectra were recorded on a Bruker 500 MHz spectrometer at 25 °C. About 100 mg of sample were dissolved in DMSO-d6, and 32 scans were recorded (1024 × 256 increments, 1.5 s relaxation delay, 0.2 s acquisition time).
Size-exclusion chromatography (SEC) was performed on an Acquity Advanced Polymer Chromatography (APC) system (Waters, USA), equipped with three 150 mm APC XT columns (a 45 Å, 1.7 μm column; a 200 Å, 2.5 μm column; and a 450 Å, 2.5 μm column) thermostated at 40 °C. Tetrahydrofuran (THF, HPLC grade, Fisher Scientific) was used as the eluent at a flow rate of 0.6 mL min−1. Detection was performed with a refractive index (RI) detector and a tunable UV (TUV) detector operating at 280 nm. The samples were dissolved in THF at 5 mg mL−1 and filtered through 0.2 μm PTFE syringe filters prior to injection. To ensure their full solubility in THF, the neat lignins (KL, SL and OSL) were acetylated. Lignins modified with HC and HI were fully soluble in THF and were analyzed as such. Lignins modified with SA were methylated following a protocol described elsewhere.16 The average molar masses and the dispersity were calculated from a calibration with polystyrene standards.
Results and discussion
Solubility of lignins in pure Cyrene and mixtures with water
The only data on lignin solubility in Cyrene were reported by Ragauskas and co-workers for the development of a new pretreatment of poplar biomass.64 They report a near complete solubility of organosolv poplar lignins in Cyrene at a concentration of 10 g L−1. The solubility remained high (>90%) up to 40% water addition, and then decreased sharply.Based on these preliminary results showing that Cyrene is a potential green solvent for lignins, we decided to expand the study by examining the solubility of the three main kinds of technical lignins (KL, SL and OSL) in Cyrene–water mixtures at a concentration of 50 g L−1. The results are shown in Fig. 1. In good agreement with the previous results, OSL was found to be almost fully soluble in pure Cyrene (95% solubility), but KL and SL were only partially soluble (61–63%). It confirms that organosolv lignins have better solubility in organic solvents than Kraft or soda lignins. However, the solubility of all the tested lignins increased when water was added to Cyrene, and full solubility was achieved between 60 and 80 to 90 vol% Cyrene. Similar results were reported for the solubilization of various aromatic molecules, which showed best solubilities in Cyrene–water mixtures between 60 and 95 vol% Cyrene.71 The addition of water to Cyrene leads to the reversible formation of a geminal diol by hydration of the ketone group (Scheme 1), thus introducing additional polarity and enhancing the hydrogen bonding capacity of the mixture. Cyrene–water mixtures are thus in fact ternary mixtures of Cyrene, geminal diol and water, whose compositions over the whole range have been determined by De bruyn et al. based on 1H and 13C NMR.71 Using their data, we calculated the molar fraction of each constituents in the mixture depending on the amount of Cyrene (Fig. 2a, the details of the calculation are given in the ESI†). The results show that the region of maximum lignin solubility coincides with the maximum molar fraction of the geminal diol, suggesting that it plays a substantial role in lignin solubility. Further addition of water led to a sharp decrease of the solubility, which was more brutal for OSL than for KL and SL.
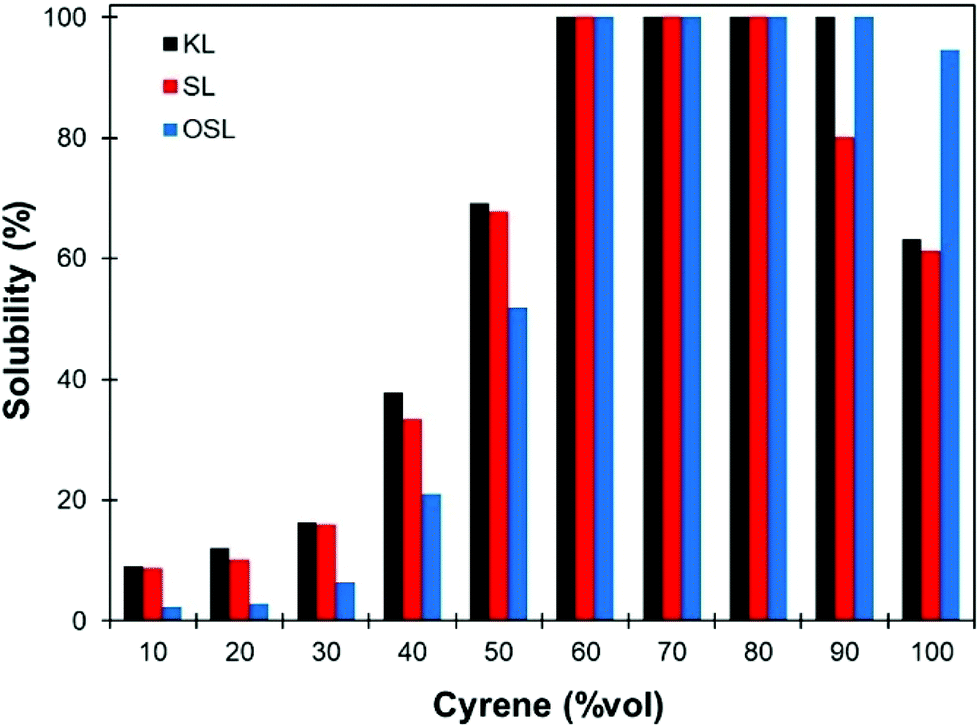 | ||
| Fig. 1 Solubility of different technical lignins (KL, SL and OSL) in Cyrene–water mixtures at a concentration of 50 g L−1, depending on the proportion of Cyrene. | ||
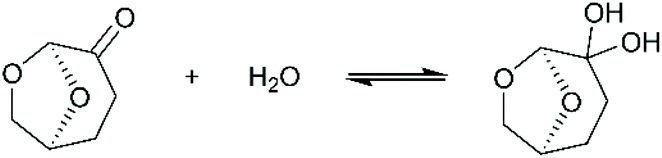 | ||
| Scheme 1 Reversible formation of geminal diol upon reaction of Cyrene with water. | ||
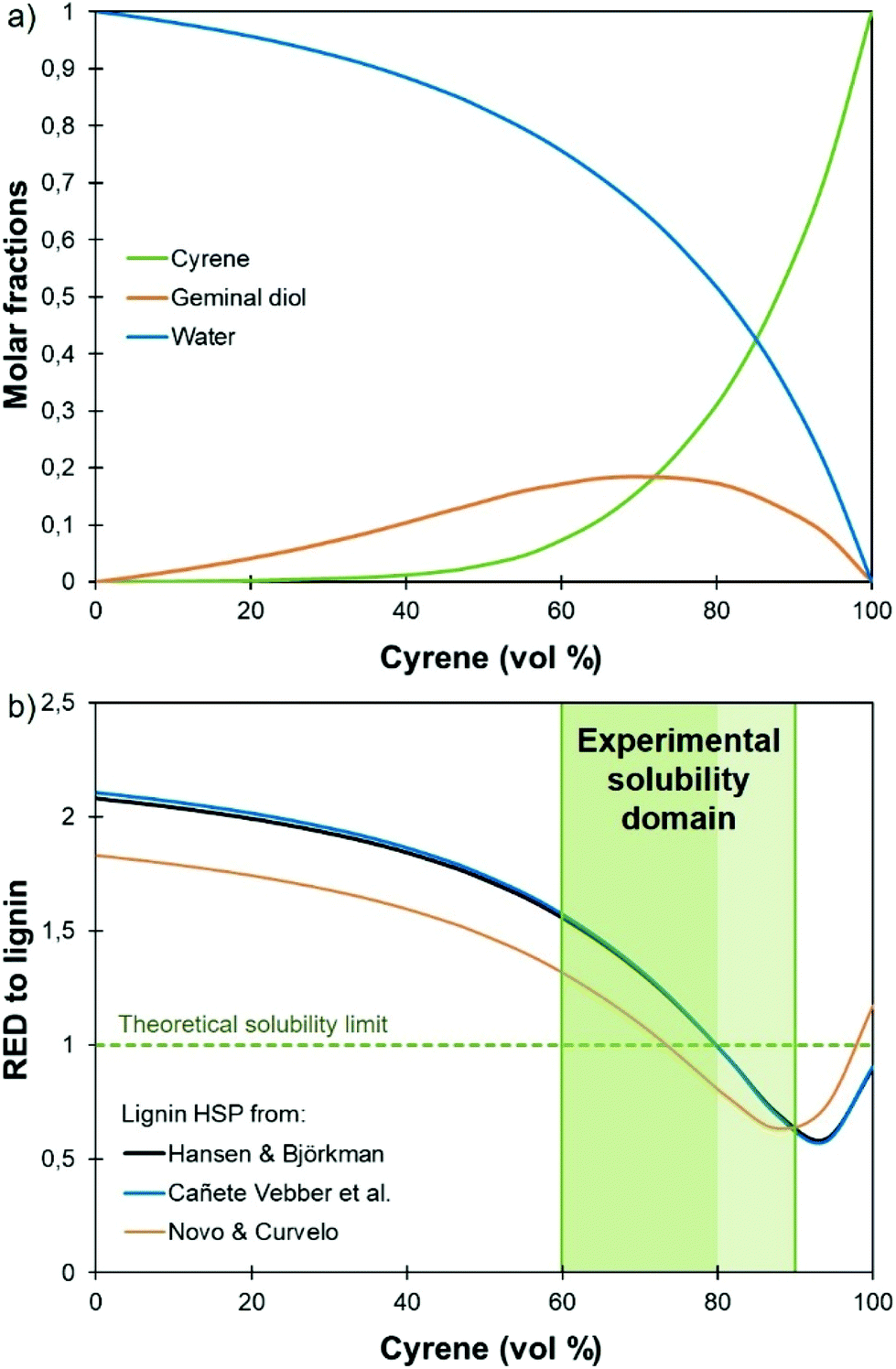 | ||
| Fig. 2 Molar fraction of the different constituents in Cyrene–water mixtures (a) and relative energy difference (RED) to lignin (b), depending on the amount of Cyrene vol%. The calculations have been done based on the HSP of the Cyrene–water ternary mixture calculated in this study (details are given in the ESI†) and literature values of the HSP of lignin.74–76 | ||
The results were further analyzed with Hansen solubility parameters (HSP),72 which were already used to describe the solubility of different lignins in organic solvents.21,55 HSP are three parameters related to the solvent or solute dispersity (δD), polarity (δP) and hydrogen bonding ability (δH). A solvent is commonly represented as a point in a three-dimensional space whose axis are the individual HSP, called Hansen's space. A solute is represented by a sphere whose center has for coordinates the compound HSP (δD, δP, δH), and radius Ra. All the good solvents of a solute are supposed to lie within its solubility sphere, whereas the non-solvents should be outside. The “distance” between a solvent and a solute in Hansen's space, Ra, can be calculated according to eqn (2):
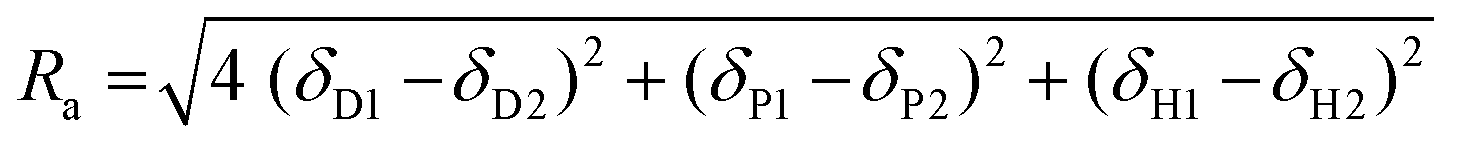 | (2) |
The good solvents lie within the solubility sphere of the solute, meaning that Ra < R0, or
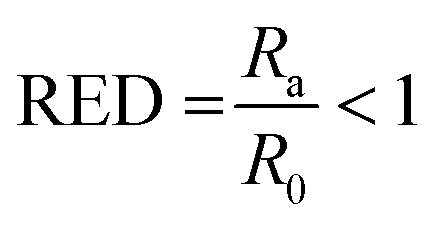 | (3) |
As discussed above, the Cyrene–water mixture is indeed a ternary mixture of Cyrene, geminal diol and water. The HSP of Cyrene56 and its geminal diol73 were both estimated by Clark's group, giving the opportunity to calculate the HSP of the Cyrene–water ternary mixture depending on the Cyrene concentration. The details of the calculation are given in the ESI.† The results confirm that the hydrogen bonding ability of the solvent mixture (δH) increases rapidly when water is added to Cyrene as a result of the formation of the geminal diol.71
Several approaches exist to estimate a solute HSP, and different values have been calculated in the literature for lignin.74–76Fig. 2b presents the RED between the Cyrene–water mixture and lignin, depending on the concentration of Cyrene, calculated with the different lignin HSP proposed in the literature. Depending on the chosen values, the predicted solubility range of lignin is between 78 and 100 vol% Cyrene74,75 or 73 and 98 vol% Cyrene,76 and only partially matches the experimental solubility domain. Indeed, the reported HSP of lignin were calculated for softwood74 and sugarcane bagasse lignin,76 respectively. They cannot accurately describe all technical lignins, which show significant structural variations depending on the botanical origin and the extraction process. Nevertheless, the fact that the addition of water improves the solubility of lignin in Cyrene is well reflected here, since the RED decreases significantly and presents a minimum at about 90 vol% Cyrene.
Influence of the temperature on the solubility and stability of KL in pure Cyrene
The use of Cyrene as organic solvent for chemical modifications of lignin is however often incompatible with the addition of water, which would prevent many chemical reactions. The influence of the temperature on KL solubility was then evaluated. After overnight stirring in pure Cyrene at 50, 75 or 100 °C, homogeneous solutions were obtained. To evaluate whether the thermal treatment in Cyrene could affect the lignin structure, KL was recovered from the Cyrene solutions by precipitation in acidic water (pH 2) and analyzed by SEC, 31P, 1H and HSQC NMR.The SEC distributions are presented in Fig. 3. They are nearly identical for KL treated in Cyrene at 50 to 100 °C and do not show significant differences with the neat KL, meaning that crosslinking or major degradation reactions did not occur. Some low molar mass fractions have however been lost, but this is most likely related to the loss of acid-soluble low molar mass fractions during the precipitation process (the recovered yields are between 92 and 94%). It also causes a slight increase in Mn (Table 2).
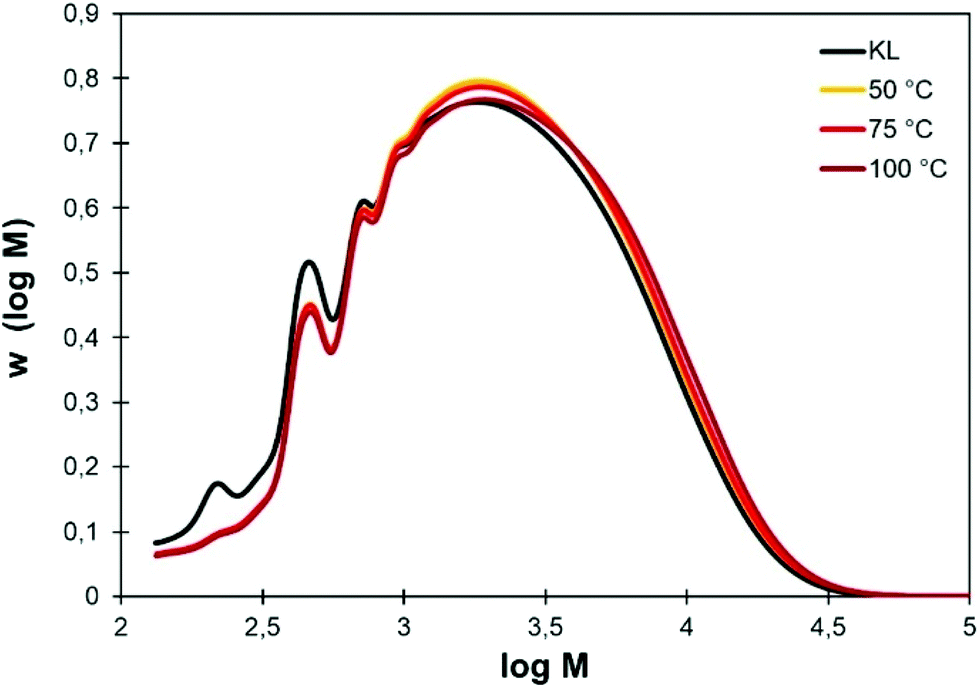 | ||
| Fig. 3 Molar mass distributions of KL before and after overnight stirring in pure Cyrene at different temperatures. | ||
| Sample name | Functional groups by 31P NMR (mmol g−1) | SEC results | Yield (%) | |||
|---|---|---|---|---|---|---|
| Al-OH | Ph-OH | COOH | M n (g mol−1) | Đ | ||
| KL | 2.42 | 4.18 | 0.36 | 1340 | 3.0 | — |
| KL – 50 °C | 2.12 | 3.54 | 0.24 | 1510 | 2.9 | 92 |
| KL – 75 °C | 2.09 | 3.48 | 0.21 | 1510 | 2.9 | 94 |
| KL – 100 °C | 1.95 | 3.36 | 0.20 | 1510 | 2.9 | 92 |
31P and 1H NMR spectra of KL treated in Cyrene are available in the ESI.† Surprisingly, two new peaks are visible on the 31P NMR spectra, at respectively 142.3 and 141.3 ppm (Fig. S6†). These peaks were also observed when Cyrene was treated with the phosphorylating reagent (TMDP) and analyzed by 31P NMR (Fig. S5†). It means that Cyrene reacts with TMDP, yielding at least two distinct products. Indeed, TMDP or its non-methylated analogue 2-chloro-1,3,2-dioxaphospholane were already found to react with compounds containing aldehydes or ketone groups.77,78 Residual Cyrene was thus present in the KL samples, despite the work-up and vacuum drying of the samples. It was also detectable on the 1H NMR spectra (Fig. S20†).
The content in functional groups calculated from the 31P NMR data are given in Table 2. There is a slight decrease in functional groups content after the treatment in Cyrene (−0.30 to 0.47 mmol g−1 of aliphatic OH, −0.64 to 0.82 mmol g−1 of phenolic OH groups and −0.12 to 0.16 mmol g−1 of COOH groups). It probably originates from the loss of low molar mass fractions during the precipitation, as they have the highest content in phenolic OH and COOH groups.79–82 Indeed, similar decrease was observed when lignin was dissolved in basic solutions and recovered by acidic precipitation.17,83
HSQC NMR was then used to analyze the lignin structure and detect if side reactions occurred between Cyrene and lignin. Fig. 4 shows the aliphatic side chain region of the HSQC spectra. The aromatic region is available in the ESI (Fig. S26†). The main inter-unit linkages detected in KL have been assigned according to literature data.84,85 Cyrene traces are visible on the spectra of KL treated in Cyrene at 50 °C (Fig. 4b) or 100 °C (Fig. 4c), confirming the 31P and 1H NMR results. They were assigned based on the comparison with the HSQC NMR spectrum of Cyrene (ESI, Fig. S27†). When Cyrene was used as solvent for the enzymatic polymerization of galactaric acid, it was found to be partially included in the polymer chain by formation of acetal linkages.86 Cyrene can also form acetals with various diols.87,88 The β-O-4 linkage of KL contains an α,γ-diol that could potentially react with Cyrene to form an acetal. However, signals from the β-O-4 linkage are still present on the HSQC spectra of KL after treatment in Cyrene. The HSQC NMR spectrum of the hypothetical structure that would result from the acetal formation between Cyrene and the β-O-4 motif was simulated (ESI, Fig. S28†). None of the expected signals appear on the spectra of KL after treatment in Cyrene, thus excluding acetal formation as potential side reaction. Signals corresponding to the potential formation of aldol adducts59 were also not detected. More generally, the inter-unit linkages present in KL seem to be unaffected by the thermal treatment in Cyrene (Fig. 4), and no reactions between lignin and Cyrene could be detected. Cyrene can thus be considered as an inert solvent for lignin.
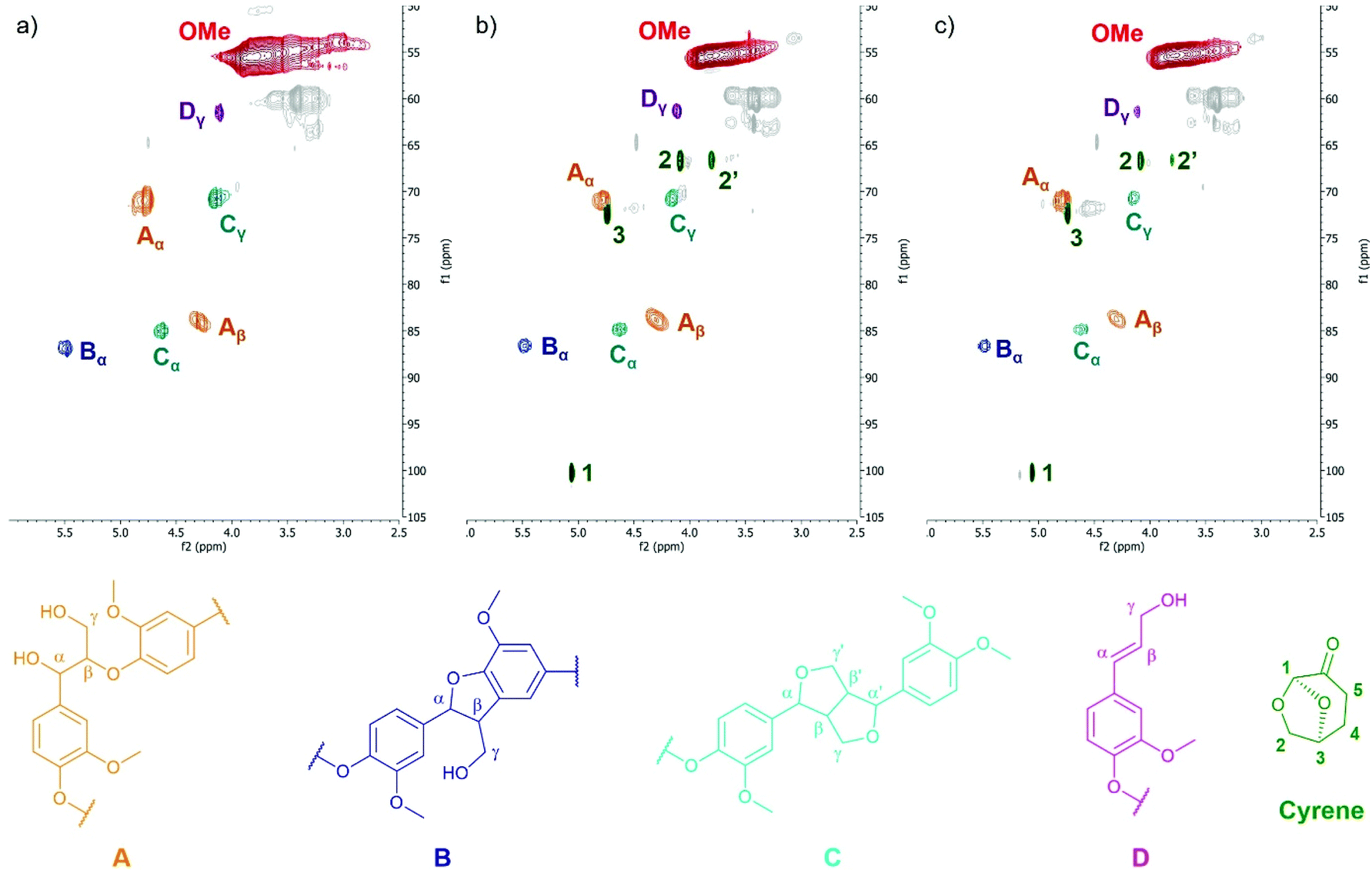 | ||
| Fig. 4 Oxygenated aliphatic side chain regions of the HSQC NMR spectra of KL (a) and KL treated in Cyrene at 50 °C (b) or 100 °C (c). Assignation of the main lignin structural motifs was performed according to recent literature.84,85 | ||
Lignin refining by fractional precipitation from Cyrene–water mixtures
Lignin refining into fractions of low dispersity has been the topic of numerous researches, as it allows to improve the properties in many applications. Recently, the different protocols available and the properties of the obtained fractions have been thoroughly reviewed.13,14 Fractional precipitation involves dissolving the lignin in a solvent, followed by the addition of a non-solvent to precipitate part of the lignin. After recovering the precipitate, non-solvent is added again, leading to additional precipitation and to the progressive recovery of successive fractions of decreasing molar mass. The first example used acetone as solvent and hexane as non-solvent.89 Later on, different couples of solvent/non-solvent were proposed, especially looking for safer and greener solvents. Most interestingly, water can be used as the non-solvent for the fractional precipitation of lignin previously dissolved in THF,90 aqueous ethanol, acetone or propyleneglycol monomethyl ether,91 or the biobased solvent γ-valerolactone (GVL).92The results of the solubility tests show that Cyrene and water can be a good solvent/non-solvent couple for lignin fractionation. We have thus studied the fractional precipitation of KL, starting from a solution in 60 vol% Cyrene, with successive additions of water to reach 50, 40, 30 and 5 vol% Cyrene (Fig. 5). The yields of the different fractions are reported in Table 3, together with the results of the characterization of the fractions by 31P NMR and SEC. The yields of the different fractions are between 11.8 and 36.9%. The total recovery is however relatively low, only 78.7%, meaning that 21.3% of KL was still dissolved in the 5 vol% Cyrene solution. Acidification of the solution to pH 2 allows to recover an additional 7.2%, reducing the global loss to 14.1%. It is still higher than in the fractionation of lignin from THF–water (2.8%) or GVL–water (5.4%). However, fractionation was only performed in small scale as a proof-of-concept, and we can expect to reduce the mass loss upon scaling up or working with more concentrated KL solutions.
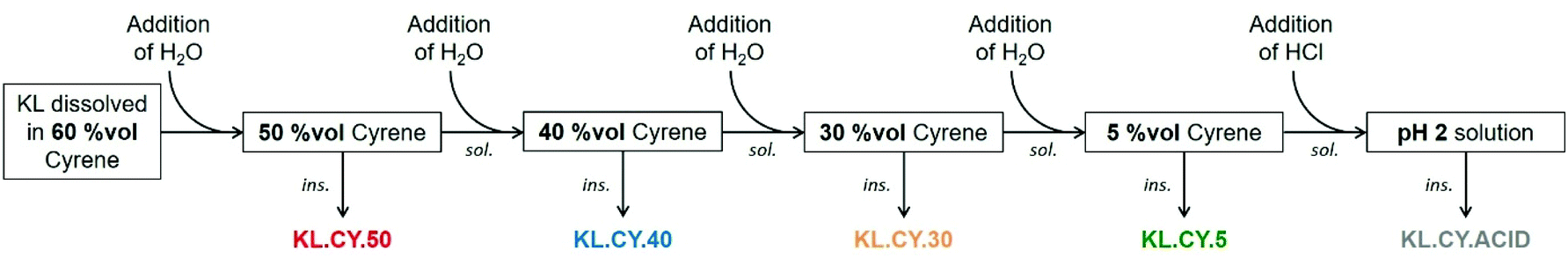 | ||
| Fig. 5 Scheme of KL fractionation in Cyrene–water mixtures. | ||
| Sample name | Functional groups by 31P NMR (mmol g−1) | SEC results | Yield (%) | |||
|---|---|---|---|---|---|---|
| Al-OH | Ph-OH | COOH | M n (g mol−1) | Đ | ||
| KL | 2.42 | 4.18 | 0.36 | 1340 | 3.04 | — |
| KL.CY.50 | 2.12 | 3.07 | 0.28 | 2250 | 3.48 | 15.2 |
| KL.CY.40 | 2.06 | 3.21 | 0.28 | 1840 | 2.93 | 36.9 |
| KL.CY.30 | 2.08 | 3.47 | 0.31 | 1140 | 3.32 | 11.8 |
| KL.CY.5 | 1.95 | 3.41 | 0.33 | 1160 | 2.41 | 14.8 |
| KL.CY.ACID | 1.84 | 3.20 | 0.25 | 1160 | 2.80 | 7.2 |
Fig. 6 shows the SEC distributions and average molar masses of the different fractions. The molar mass is reduced when the amount of water for precipitation increased, showing that fractional precipitation from Cyrene–water mixtures is an efficient protocol for lignin refining into fractions of controlled molar mass. The last two fractions (KL.CY.5 and KL.CY.ACID) have similar molar mass distributions, meaning that they can be combined to obtain a low molar mass fraction in higher yield.
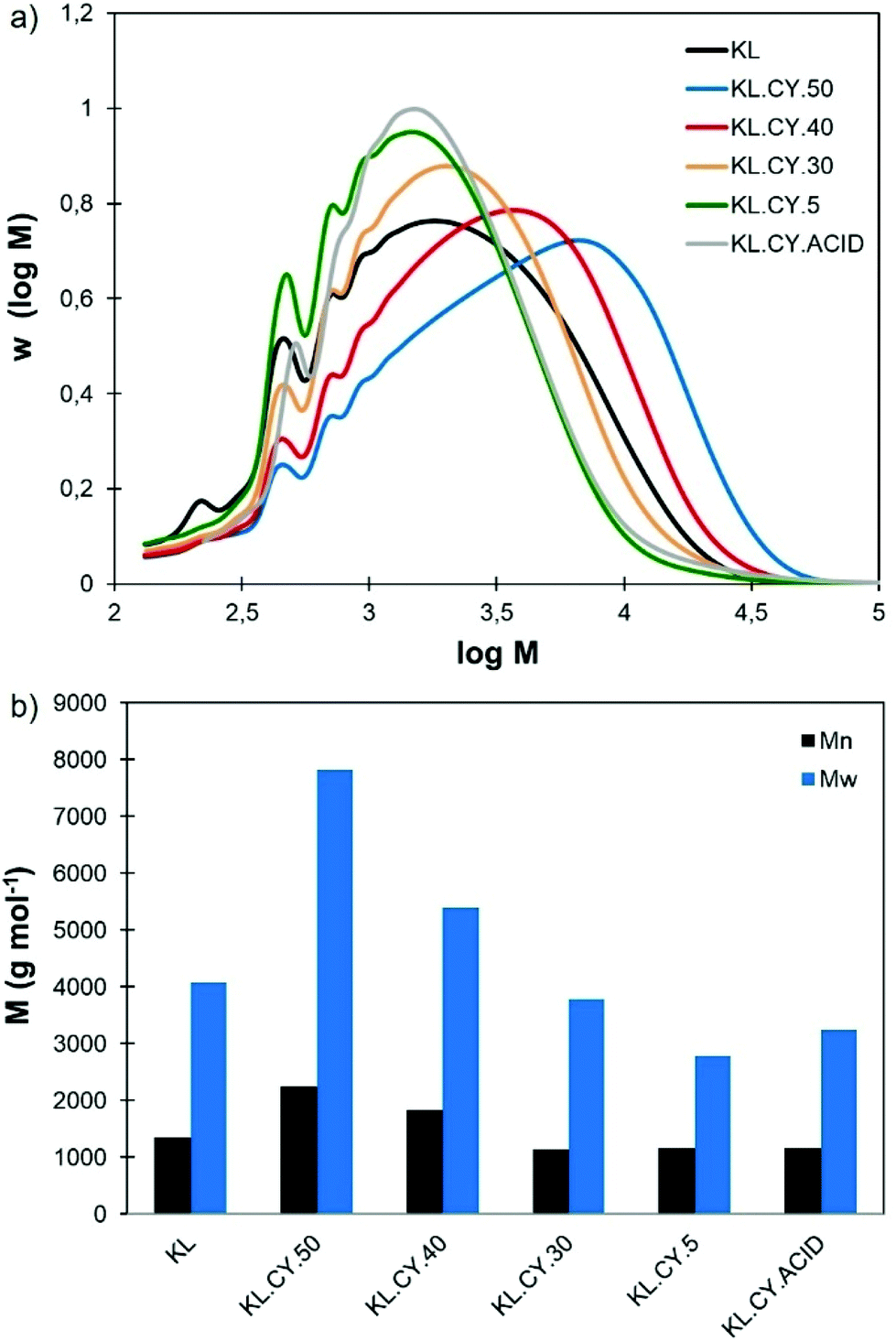 | ||
| Fig. 6 Molar mass distributions (a) and average molar masses (b) of KL fractions obtained by sequential precipitation from Cyrene–water mixtures. | ||
The content in functional groups of the fractions has been evaluated by 31P NMR (Table 3). The content in phenolic OH groups increases when the molar mass is reduced, as previously reported.89–92 There is no clear trend regarding the aliphatic OH groups. All the fractions present a lower content in functional groups (Al-OH, Ph-OH and COOH) than the parent KL. This trend, already observed during fractionation of lignin in GVL–water,92 is probably due to the loss of lignin that remain in solution. Indeed, it is probably the fraction of lowest molar mass, and hence highest content in functional groups, which is not recovered.
Use of Cyrene as solvent for the chemical modification of lignins
The potential use of Cyrene as green solvent for lignin chemical modifications was finally evaluated for three different types of chemical reactions commonly applied to lignin: (i) esterification with an acyl chloride, (ii) esterification with an acid anhydride and (iii) formation of an urethane linkage with an isocyanate. Three different lignins (KL, SL and OSL) were thus modified in Cyrene and characterized by SEC, 1H and 31P NMR. All the NMR spectra and SEC chromatograms are available in the ESI.† For each reaction, the reactivity of KL in Cyrene was also compared to the reactivity in DMF for similar reaction conditions.Lignins were first esterified with heptanoyl chloride (HC, Scheme 2). Acyl chlorides were already used for reactions in Cyrene to prepare various amides,63 but not yet to prepare esters. HC was used in slight excess (1.3 molar equivalents to OH groups in lignin), based on the results of Koivu et al.47 They reported the complete esterification of KL with C2, C6 and C12 acyl chlorides in a mixture of THF and DMF as solvent at 60 °C, with pyridine as catalyst. However, Cyrene is not stable in the presence of pyridine above 25 °C.59 Triethylamine (TEA, 1 equivalent) was thus used instead, as it is compatible with Cyrene for at least 24 h at 50 °C.59
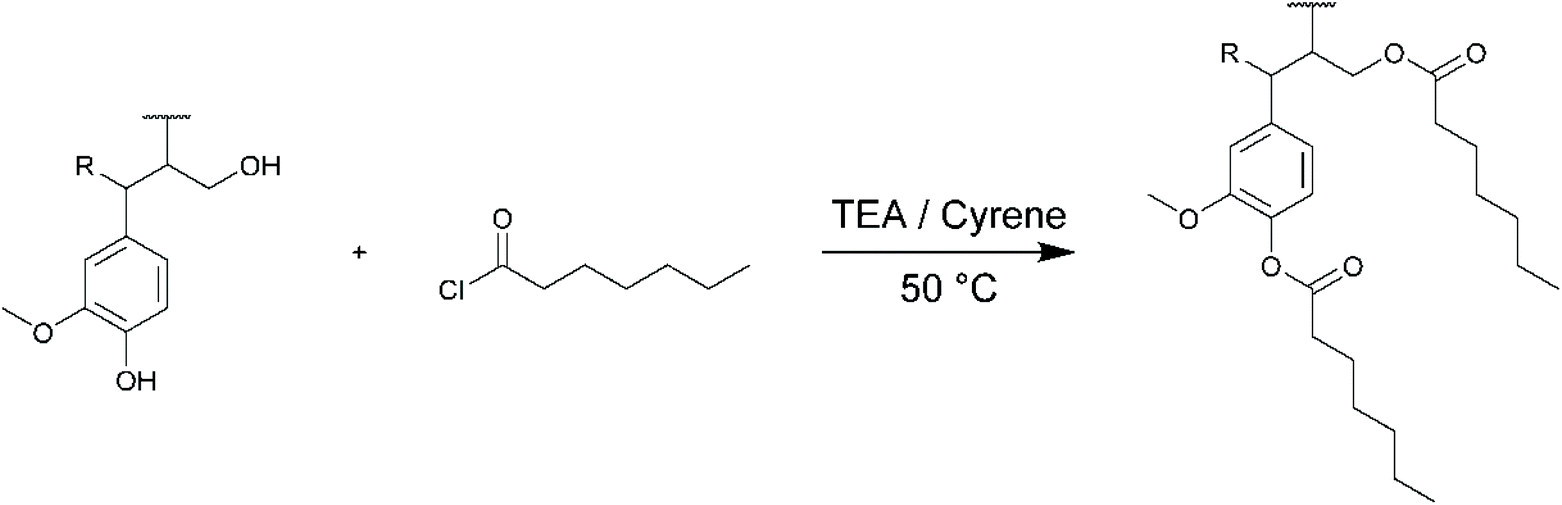 | ||
| Scheme 2 Esterification of lignin with heptanoyl chloride (HC). | ||
The main characterization results are summarized in Table 4. For KL, the conversion of the aliphatic OH groups is nearly quantitative in both Cyrene and DMF (86 to 89% conversion). As expected, the phenolic OH groups are much less reactive than the aliphatic OH groups towards esterification. Their reactivity is similar in Cyrene and DMF. Koivu et al.47 achieved full conversion of phenolic OH, but for longer reaction time (48 h vs. 16 h), higher temperature (65 °C vs. 50 °C) and higher catalyst loading (1.4 eq. pyridine vs. 1 eq. TEA). The reaction conditions in Cyrene could thus probably be optimized to improve the conversion of phenolic OH, but KL esterified in Cyrene already presents interesting properties, such as a full solubility in various organic solvents (acetone, THF, chloroform and ethyl acetate). SEC shows that KL modified with HC has a molar mass relatively close to that of acetylated KL (Table 4). One could expect a higher molar mass because of the higher length of the grafts, but the conversion is also lower than in the case of acetylation. SL and OSL were also successfully esterified with HC in Cyrene. The conversion of their aliphatic OH is similar to KL, but their phenolic OH were found to be more reactive (62 to 64% conversion against only 37% for KL).
| Lignin type | Solvent | Reagent | Functional groups by 31P NMR (mmol g−1) | Conversion (%) | SEC results | Yield (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Al-OH | Ph-OH | COOH | Al-OH | Ph-OH | Total OH | M n (g mol−1) | Đ | ||||
| a SEC measured after acetylation. b SEC measured after methylation. | |||||||||||
| KL | — | — | 2.42 | 4.18 | 0.36 | — | — | — | 1340a | 3.04a | — |
| DMF | HC | 0.27 | 2.64 | 0.32 | 89 | 37 | 56 | 1420 | 2.81 | 78 | |
| SA | 0.20 | 1.84 | 3.05 | 92 | 56 | 69 | 2160b | 3.47b | 82 | ||
| HI | 0.90 | 0.19 | 0.11 | 63 | 95 | 83 | 1680 | 2.60 | 98 | ||
| Cyrene | HC | 0.34 | 2.23 | 0.19 | 86 | 47 | 61 | 1250 | 2.73 | 92 | |
| SA | 0.24 | 2.72 | 2.58 | 90 | 35 | 55 | 2160b | 3.44b | 85 | ||
| HI | 1.10 | 0.65 | 0.13 | 55 | 84 | 74 | 1380 | 2.48 | 92 | ||
| SL | — | — | 2.06 | 3.34 | 1.28 | — | — | — | 940a | 2.80 | — |
| Cyrene | HC | 0.34 | 1.26 | 0.41 | 84 | 62 | 70 | 900 | 2.91 | 87 | |
| SA | 0.49 | 1.75 | 2.47 | 76 | 48 | 59 | 1760b | 3.01 | 79 | ||
| HI | 0.84 | 0.70 | 0.36 | 59 | 79 | 72 | 1090 | 2.57 | 81 | ||
| OSL | — | — | 1.84 | 2.86 | 0.08 | — | — | — | 1470a | 2.73 | — |
| Cyrene | HC | 0.27 | 1.02 | 0.08 | 86 | 64 | 73 | 1590 | 2.70 | 89 | |
| SA | 0.31 | 2.16 | 1.69 | 83 | 24 | 47 | 1920b | 2.91 | 73 | ||
| HI | 0.62 | 0.38 | 0.01 | 54 | 80 | 70 | 1620 | 2.38 | 105 | ||
The esterification of lignins with succinic anhydride (SA) in Cyrene was then studied (Scheme 3). The main results are reported in Table 4. The esterification of lignin with anhydrides can efficiently proceed without additional solvent if the anhydride is able to dissolve lignin, as usually observed with linear anhydrides,93 but in this case they have to be used in large excess to play the role of reactive solvents. However, the modification of lignin with cyclic anhydrides of higher melting points, such as maleic (mp 53 °C) or succinic anhydrides (mp 120 °C), requires the dissolution of lignin in an appropriate solvent, usually 1,4-dioxane93,94 or DMF.49 1-Methylimidazole (1-MIM) was used as catalyst,93 after confirming by 1H NMR that Cyrene was stable in its presence at 50 °C (Fig. S2 in the ESI†).
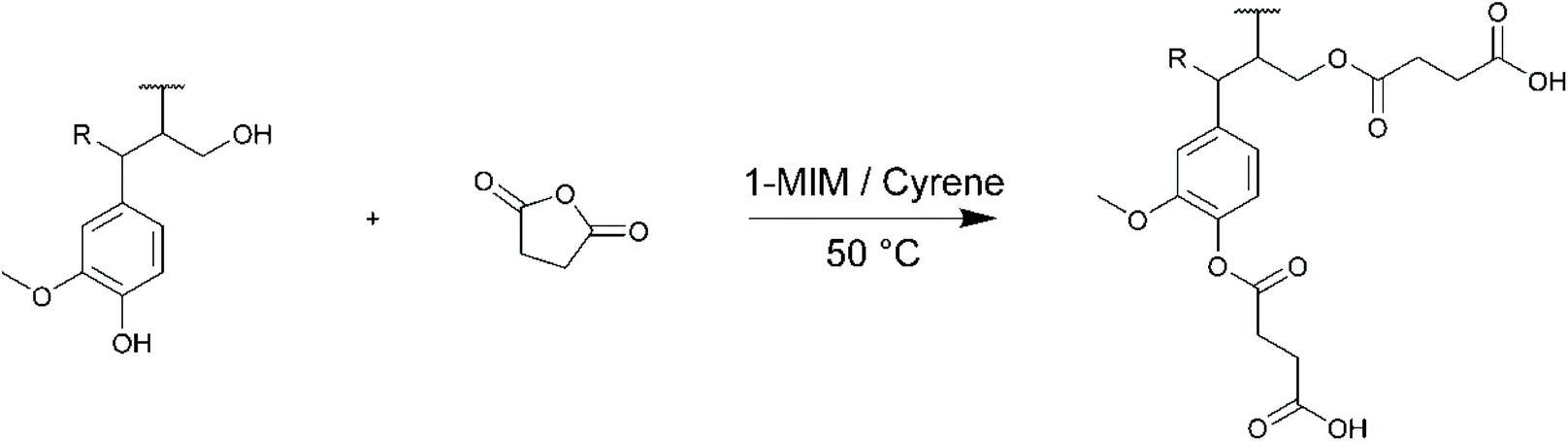 | ||
| Scheme 3 Esterification of lignin with succinic anhydride (SA). | ||
For KL, the results show a near quantitative conversion of aliphatic OH groups in both DMF and Cyrene (90–92%, Table 4). The reactivity of the phenolic OH groups is much lower, as already reported for this kind of esterification. The conversion of phenolic OH is lower in Cyrene than in DMF (35% against 56%), probably because of the higher basicity of the latter. However, it is higher than previously reported for similar reactions in 1,4-dioxane94 or THF.95 KL modified with SA was fully soluble in THF and was thus directly analyzed by SEC (Fig. S29 in the ESI†). Surprisingly, the product eluted at very long retention times, even higher than the toluene used as flow marker, leading to unexpectedly low molar mass. KL modified with SA indeed possess a lot of polar groups (COOH formed after the reaction and unreacted Ph-OH, Scheme 3), which can cause undesired association with the column,96 and delay the elution. We thus performed a methylation of KL modified with SA, to convert in a single step the carboxyl groups into methyl esters and the phenolic OH into methoxy groups, following a protocol previously developed in our laboratory (Scheme 4).16 The elution profile was drastically modified after methylation, confirming that polar interactions were responsible for these results (Fig. S29 in the ESI†). KL modified in DMF or Cyrene present similar molar masses, notably higher than that of neat KL or KL modified with HC. In addition to the grafting which should induce an increase in molar mass, potential crosslinking by esterification cannot be completely excluded, although the relatively low temperature used for the reaction should limit this phenomenon. The modification of SL and OSL with SA in Cyrene was also successful, although the conversion of the aliphatic OH groups was slightly lower than for KL (76 and 83% conversion, respectively). The molar masses, measured after methylation, were also found to be significantly higher than for the neat lignins.
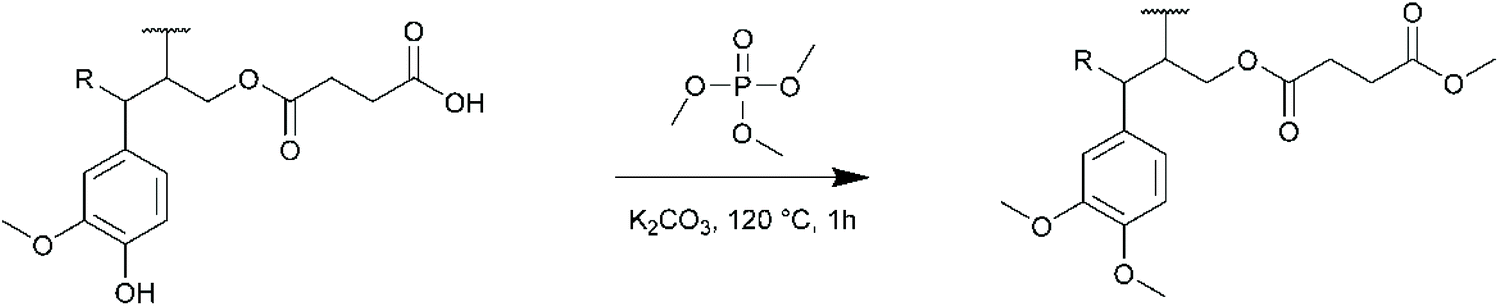 | ||
| Scheme 4 Methylation of KL modified with SA prior to SEC analysis, to convert the carboxylic acid and phenolic OH groups into methyl esters and methoxy groups, respectively.16 | ||
Finally, the modification of lignins with hexyl isocyanate (HI) was performed (Scheme 5). HI was chosen as a model isocyanate to study the formation of urethane linkages with the OH groups of KL. First attempts were made with dibutyltin dilaurate (DBTL) as catalyst, after ensuring that it was compatible with Cyrene (Fig. S3 in the ESI†). It yielded satisfactory results, but the work-up by precipitation in water was not efficient to remove the catalyst from the precipitated lignin. N,N′-Dimethylcyclohexylamine (DMCHA) was then used instead. The results show that Cyrene is a suitable solvent for the reaction of lignin with isocyanates. The formation of urea in Cyrene was already reported,61 but it is to the best of our knowledge the first time that Cyrene is used to form urethanes, thus opening potential applications in polyurethane synthesis. In this case also, the reactivity was found to be slightly lower in Cyrene than in DMF, especially for the phenolic OH groups (84% conversion against 95%). As a result, the molar mass of KL modified with HI was slightly lower after reaction in Cyrene than in DMF. SL and OSL were also successfully modified, with conversions almost identical to KL. For OSL, an aberrant yield of 105% was measured. Replicate of the reaction gave the same result. However, the 31P NMR spectrum does not show the presence of residual Cyrene (Fig. S19†). The extra mass could then come from the presence of residual hexylamine (formed from HI during the aqueous work-up) or from the second addition of HI onto the urethane group forming allophanate structures, although this cannot be clearly evidenced from the 1H NMR spectra (Fig. S25†).
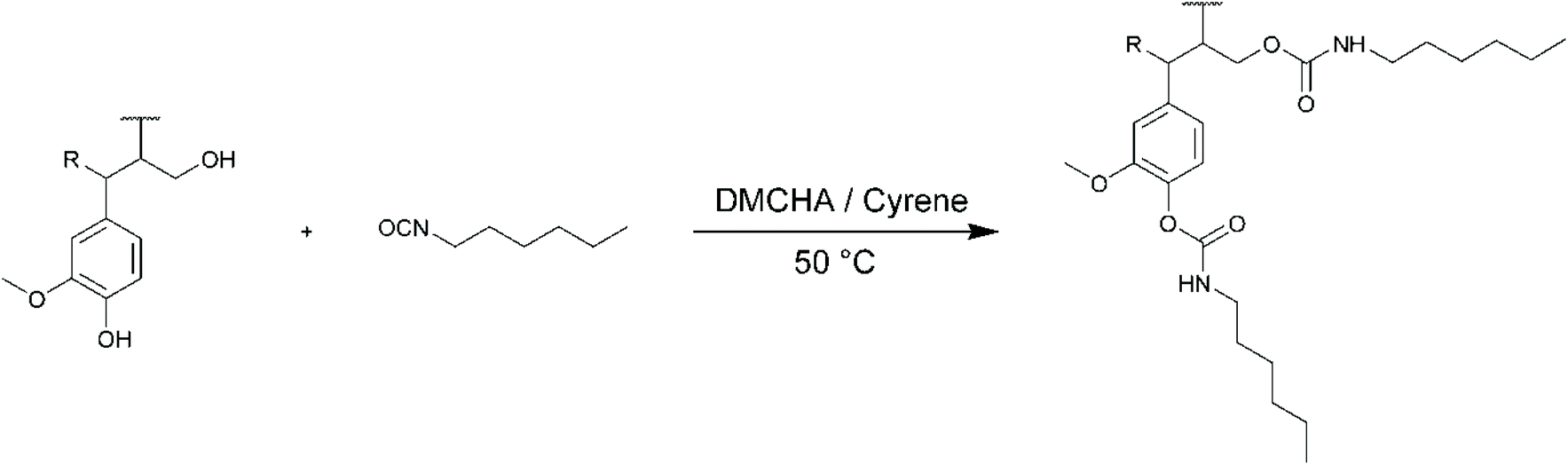 | ||
| Scheme 5 Reaction of lignin with hexyl isocyanate (HI). | ||
Globally, Cyrene appears as a suitable alternative to DMF or other undesired solvents for lignin chemical modifications. Although none of the tested reactions were optimized, the conversions were always satisfactory and comparable to literature data, when available. The modified lignins were easily recovered by precipitation in high yields, almost always higher than 80%. Traces of Cyrene could sometimes be detected on the 31P NMR spectra after reaction (Fig. S8 and S19 in the ESI†), despite careful washing of the products with deionized water and vacuum drying. In this case, an additional wash of the products allowed to remove Cyrene traces, confirming that it did not react with lignin. However, it shows that Cyrene can be complex to remove from the products, which can be problematic for reactions at higher scale.
Conclusion
This study evaluated the potential of the biobased solvent dihydrolevoglucosenone (Cyrene) as a green substitute for toxic polar aprotic solvents like DMF in lignin processing and chemistry. The solubility of different technical lignins, KL, SL and OSL, was first evaluated. None of the tested lignins were fully soluble in Cyrene, but it was greatly enhanced by the addition of water, to reach full solubility in 60 to 80–90 vol% Cyrene solutions. The addition of water to Cyrene leads to the formation of a geminal diol by hydration of the ketone group, which increases the hydrogen bonding capacity and probably plays a significant role in the solubilization of lignin. Further increasing the water content leads to a decrease in solubility, which gives the opportunity to use Cyrene and water as a solvent/non-solvent couple for the fractional precipitation of lignin. As proof-of-concept, KL was thus fractionated into 5 different fractions of decreasing molar mass, thus proving the efficiency of the method. More detailed analyses of the isolated fractions should now be conducted to fully assess the potential of the method for lignin fractionation.After confirming that it was inert to lignin, Cyrene was successfully used as solvent for the chemical modification of lignins with an acyl chloride, a cyclic anhydride, and an isocyanate. To the best of our knowledge, this is the first time that Cyrene is used as solvent for esterification and urethane formation. The reactivity of KL was compared for reactions in Cyrene and in DMF. The reactivity of the aliphatic OH groups was similar, whereas the conversion of the phenolic OH was slightly lower in Cyrene than in DMF, probably because of the higher basicity of the latter. Although further optimization would be needed depending on the reaction type and lignin feedstock, this study proves that Cyrene is a valid alternative for greener lignin chemistry when solvents are mandatory.
This study clearly showed the high potential of Cyrene as green solvent for lignin, but also evidenced some difficulties. For instance, the high viscosity of Cyrene (about 7 times that of DMSO and 15 times that of DMF, Table 1) can lead to practical issues, for instance when it comes to filtration or centrifugation processes. Lignin solutions in Cyrene can reach high viscosities (about 40 mPa s for a solution of KL in 90 vol% Cyrene at 100 g L−1, against 5 mPa s for an equivalent concentration in DMSO), thus complicating handling. In addition, traces of Cyrene could sometimes be observed in the products after reaction, despite careful washing. As its boiling point is very high (203 °C), it cannot be easily removed by vacuum drying. However, the advantages of using Cyrene over e.g., DMF should lead to consider it as an additional efficient tool towards greener lignin chemistry and fractionation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Arjan T. Smit (TNO-Energy Transition, Petten, Netherlands) is acknowledged for providing the organosolv beech lignin. We also thank Céline Piras (ICPEES) for performing the SEC measurements.Notes and references
- Z. Jiang, P. Zhao and C. Hu, Bioresour. Technol., 2018, 256, 466–477 CrossRef CAS.
- M. Galbe and O. Wallberg, Biotechnol. Biofuels, 2019, 12, 294 CrossRef PubMed.
- P. P. Thoresen, L. Matsakas, U. Rova and P. Christakopoulos, Bioresour. Technol., 2020, 306, 123189 CrossRef CAS PubMed.
- S. S. Hassan, G. A. Williams and A. K. Jaiswal, Trends Biotechnol., 2019, 37, 231–234 CrossRef CAS.
- D. Lachenal, in Lignocellulosic Fibers and Wood Handbook, John Wiley & Sons, Ltd, 2016, pp. 207–223 Search PubMed.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- L. Kouisni, A. Gagné, K. Maki, P. Holt-Hindle and M. Paleologou, ACS Sustainable Chem. Eng., 2016, 4, 5152–5159 CrossRef CAS.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- W. Schutyser, T. Renders, S. V. den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- H. Sadeghifar and A. Ragauskas, ACS Sustainable Chem. Eng., 2020, 8, 8086–8101 CrossRef CAS.
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustainable Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS.
- A. Duval and L. Avérous, Green Chem., 2020, 22, 1671–1680 RSC.
- A. Duval, H. Lange, M. Lawoko and C. Crestini, Green Chem., 2015, 17, 4991–5000 RSC.
- P. Buono, A. Duval, L. Averous and Y. Habibi, ChemSusChem, 2017, 10, 984–992 CrossRef CAS.
- P. Buono, A. Duval, L. Averous and Y. Habibi, Polymer, 2017, 133, 78–88 CrossRef CAS.
- O. Ringena, S. Lebioda, R. Lehnen and B. Saake, J. Chromatogr. A, 2006, 1102, 154–163 CrossRef CAS.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2016, 70, 11–20 CAS.
- D. S. Argyropoulos, H. Sadeghifar, C. Cui and S. Sen, ACS Sustainable Chem. Eng., 2014, 2, 264–271 CrossRef CAS.
- J.-Y. Kim, P. Hafezi-Sefat, S. Cady, R. G. Smith and R. C. Brown, Energy Fuels, 2019, 33, 1248–1255 CrossRef CAS.
- J. J. Meister and M. J. Chen, Macromolecules, 1991, 24, 6843–6848 CrossRef CAS.
- J. J. Meister and C. T. Li, Macromolecules, 1992, 25, 611–616 CrossRef.
- S. Sen, H. Sadeghifar and D. S. Argyropoulost, Biomacromolecules, 2013, 14, 3399–3408 CrossRef CAS.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- H. Chung and N. R. Washburn, ACS Appl. Mater. Interfaces, 2012, 4, 2840–2846 CrossRef CAS PubMed.
- L. Mu, Y. Shi, X. Guo, J. Wu, T. Ji, L. Chen, X. Feng, X. Lu, J. Hua and J. Zhu, ACS Sustainable Chem. Eng., 2016, 4, 3877–3887 CrossRef CAS.
- I. Panovic, J. R. D. Montgomery, C. S. Lancefield, D. Puri, T. Lebl and N. J. Westwood, ACS Sustainable Chem. Eng., 2017, 5, 10640–10648 CrossRef CAS.
- J. Wang, B. Wu, S. Li, G. Sinawang, X. Wang and Y. He, ACS Sustainable Chem. Eng., 2016, 4, 4036–4042 CrossRef CAS.
- W. Liu, C. Fang, S. Wang, J. Huang and X. Qiu, Macromolecules, 2019, 52, 6474–6484 CrossRef CAS.
- Y. Yu, S. Fu, P. Song, X. Luo, Y. Jin, F. Lu, Q. Wu and J. Ye, Polym. Degrad. Stab., 2012, 97, 541–546 CrossRef CAS.
- Y. Zhang, J. Liao, X. Fang, F. Bai, K. Qiao and L. Wang, ACS Sustainable Chem. Eng., 2017, 5, 4276–4284 CrossRef CAS.
- L. Zoia, A. Salanti, P. Frigerio and M. Orlandi, BioResources, 2014, 9, 6540–6561 CrossRef.
- Y. Han, L. Yuan, G. Li, L. Huang, T. Qin, F. Chu and C. Tang, Polymer, 2016, 83, 92–100 CrossRef CAS.
- A. Chandran, S. Kuriakose and T. Mathew, Polym. Adv. Technol., 2013, 24, 437–440 CrossRef CAS.
- A. Chandran, S. Kuriakose and T. Mathew, Polym. Adv. Technol., 2014, 25, 589–594 CrossRef CAS.
- K. Huang, S. Ma, S. Wang, Q. Li, Z. Wu, J. Liu, R. Liu and J. Zhu, Green Chem., 2019, 21, 4964–4970 RSC.
- H. Liu and H. Chung, Macromolecules, 2016, 49, 7246–7256 CrossRef CAS.
- L. N. Dang, T. B. N. Thi, L. D. Duong, D. O. Kim, D.-S. Kim, S. H. Lee, B. J. Kim, Y. S. Lee and J.-D. Nam, Polym. Bull., 2012, 68, 879–890 CrossRef.
- D. V. Evtugin and A. Gandini, Acta Polym., 1996, 47, 344–350 CrossRef CAS.
- Z. X. Guo, A. Gandini and F. Pla, Polym. Int., 1992, 27, 17–22 CrossRef CAS.
- X. Liu, J. Wang, J. Yu, M. Zhang, C. Wang, Y. Xu and F. Chu, Int. J. Biol. Macromol., 2013, 60, 309–315 CrossRef CAS PubMed.
- N. T. T. Binh, N. D. Luong, D. O. Kim, S. H. Lee, B. J. Kim, Y. S. Lee and J.-D. Nam, Compos. Interfaces, 2009, 16, 923–935 CrossRef.
- K. A. Y. Koivu, H. Sadeghifar, P. A. Nousiainen, D. S. Argyropoulos and J. Sipilä, ACS Sustainable Chem. Eng., 2016, 4, 5238–5247 CrossRef CAS.
- X. Zhang, S. Keck, Y. Qi, S. Baudis and Y. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 10959–10970 CAS.
- D. E. García, N. Delgado, F. L. Aranda, M. A. Toledo, G. Cabrera-Barjas, E. M. Sintjago, D. Escobar-Avello and S. Paczkowski, Ind. Crops Prod., 2018, 123, 154–163 CrossRef.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2016, 4, 3103–3112 CrossRef CAS.
- A. Duval and L. Avérous, ChemSusChem, 2017, 10, 1813–1822 CrossRef CAS PubMed.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- A. Duval, G. Layrac, A. van Zomeren, A. T. Smit, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 387–397 CrossRef CAS PubMed.
- J. Sherwood, M. D. Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- Resolute Project, https://www.resolute-project.eu/, (accessed August 18, 2021).
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Synlett, 2018, 29, 650–654 CrossRef CAS.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC.
- X. Meng, Y. Pu, M. Li and A. J. Ragauskas, Green Chem., 2020, 22, 2862–2872 RSC.
- Md. S. Alam, B. Ashokkumar and A. M. Siddiq, J. Mol. Liq., 2019, 281, 584–597 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- C. Laurence, S. Mansour, D. Vuluga and J. Legros, Green Chem., 2021, 23, 1816–1822 RSC.
- A. Smit and W. Huijgen, Green Chem., 2017, 19, 5505–5514 RSC.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS.
- M. De bruyn, V. L. Budarin, A. Misefari, S. Shimizu, H. Fish, M. Cockett, A. J. Hunt, H. Hofstetter, B. M. Weckhuysen, J. H. Clark and D. J. Macquarrie, ACS Sustainable Chem. Eng., 2019, 7, 7878–7883 CrossRef CAS PubMed.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- R. A. Milescu, M. L. Segatto, A. Stahl, C. R. McElroy, T. J. Farmer, J. H. Clark and V. G. Zuin, ACS Sustainable Chem. Eng., 2020, 8, 18245–18257 CrossRef CAS.
- C. M. Hansen and A. Björkman, Holzforschung, 1998, 52, 335–344 CrossRef CAS.
- G. C. Vebber, P. Pranke and C. N. Pereira, J. Appl. Polym. Sci., 2014, 131, 39696 CrossRef.
- L. P. Novo and A. A. S. Curvelo, Ind. Eng. Chem. Res., 2019, 58, 14520–14527 CrossRef CAS.
- J. Heine and G.-V. Röschenthaler, Chem. Ber., 1988, 121, 379–381 CrossRef CAS.
- Y. Archipov, D. S. Argyropoulos, H. I. Bolker and C. Heitner, J. Wood Chem. Technol., 1991, 11, 137–157 CrossRef.
- A. P. Dodd, J. F. Kadla and S. K. Straus, ACS Sustainable Chem. Eng., 2015, 3, 103–110 CrossRef CAS.
- D. Barana, M. Orlandi, L. Zoia, L. Castellani, T. Hanel, C. Bolck and R. Gosselink, ACS Sustainable Chem. Eng., 2018, 6, 11843–11852 CrossRef CAS.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- A. Tagami, C. Gioia, M. Lauberts, T. Budnyak, R. Moriana, M. E. Lindström and O. Sevastyanova, Ind. Crops Prod., 2019, 129, 123–134 CrossRef CAS.
- A. Duval, H. Lange, M. Lawoko and C. Crestini, Biomacromolecules, 2015, 16, 2979–2989 CrossRef CAS PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- M. Vastano, A. Pellis, C. B. Machado, R. Simister, S. J. McQueen-Mason, T. J. Farmer and L. D. Gomez, Macromol. Rapid Commun., 2019, 40, 1900361 CrossRef CAS PubMed.
- A. A. C. Pacheco, J. Sherwood, A. Zhenova, C. R. McElroy, A. J. Hunt, H. L. Parker, T. J. Farmer, A. Constantinou, M. De bruyn, A. C. Whitwood, W. Raverty and J. H. Clark, ChemSusChem, 2016, 9, 3503–3512 CrossRef CAS PubMed.
- R. A. Milescu, A. Zhenova, M. Vastano, R. Gammons, S. Lin, C. H. Lau, J. H. Clark, C. R. McElroy and A. Pellis, ChemSusChem, 2021, 14, 3367–3381 CrossRef CAS PubMed.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS.
- X. Meng, A. Parikh, B. Seemala, R. Kumar, Y. Pu, C. E. Wyman, C. M. Cai and A. J. Ragauskas, Bioresour. Technol., 2019, 272, 202–208 CrossRef CAS PubMed.
- A.-S. Jääskeläinen, T. Liitiä, A. Mikkelson and T. Tamminen, Ind. Crops Prod., 2017, 103, 51–58 CrossRef.
- G. Wang, X. Liu, B. Yang, C. Si, A. M. Parvez, J. Jang and Y. Ni, ACS Sustainable Chem. Eng., 2019, 7, 10112–10120 CrossRef CAS.
- W. Thielemans and R. P. Wool, Biomacromolecules, 2005, 6, 1895–1905 CrossRef CAS PubMed.
- D. Schorr, G. Komba Yoya, P. Rodrigue, P. Niokhor Diouf and T. Stevanovic, Int. Wood Prod. J., 2015, 6, 112–122 CrossRef.
- C. Scarica, R. Suriano, M. Levi, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2018, 6, 3392–3401 CrossRef CAS.
- A. A. Andrianova, N. A. Yeudakimenka, S. L. Lilak, E. I. Kozliak, A. Ugrinov, M. P. Sibi and A. Kubátová, J. Chromatogr., A, 2018, 1534, 101–110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03395f |
| This journal is © The Royal Society of Chemistry 2022 |