
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upcycling agricultural waste into membranes: from date seed biomass to oil and solvent-resistant nanofiltration†
Abdulaziz
Alammar
 a,
Rifan
Hardian
b and
Gyorgy
Szekely
*ab
a,
Rifan
Hardian
b and
Gyorgy
Szekely
*ab
aDepartment of Chemical Engineering & Analytical Science, School of Engineering, The University of Manchester, The Mill, Sackville Street, Manchester, M1 3BB, UK. E-mail: gyorgy.szekely@manchester.ac.uk
bAdvanced Membranes and Porous Materials Center, Physical Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: gyorgy.szekely@kaust.edu.sa; Web: http://www.szekelygoup.com
First published on 3rd December 2021
Abstract
Membranes hold a great promise for replacing energy-intensive separations across various industrial sectors. However, membrane production heavily relies on petrochemical-based raw materials; the need for greener membranes is a challenge that is yet to be solved. In this work, we solubilized date seed biomass (abundantly available from the multimillion-metric-ton date industry) using ionic liquids and dimethyl sulfoxide (which are greener than traditional organic solvents) to fabricate biodegradable nanofiltration membranes. The resultant membranes were coated with mussel-inspired polydopamine (PDA) via a layer-by-layer deposition method. The obtained membranes demonstrated excellent performance for organic solvent nanofiltration (OSN) and oil-in-water separation. The deposition time and the number of PDA layers correlated with the molecular sieving performance of the membranes and allowed the fine-tuning of the molecular weight cutoff (MWCO). The best-performing membrane exhibited an acetonitrile permeance of 7.8 L m−2 h−1 bar−1 and a 96% rejection of acid fuchsin (585 g mol−1). Moreover, an oil-removal efficiency of up to 97% was achieved with a water permeance of 5.7 L m−2 h−1 bar−1. The prepared membranes showed excellent stability for over seven days in continuous nanofiltration tests. The biodegradability of the membranes was demonstrated in an aqueous cellulase solution. Our work offers a sustainable production of waste biomass-based membranes for liquid-separation applications.
1. Introduction
Conventional thermal separation processes are crucial in various industrial sectors, but they consume a substantial amount of energy. The alternative membrane-based separation technologies offer lower energy consumption; however, membrane production mainly relies on petrochemical-based materials. Energy conservation and its responsible consumption and production have become increasingly important, as stated in the United Nations’ Sustainable Development Goals. Therefore, alternative materials are required to reduce our dependency on petroleum-based polymers and eventually replace them with bio-based materials.1 Edible oils, fats, and sugar are limited sources for developing new materials because of their high demand in the food sector.2 Therefore, waste and residue-based lignocellulosic biomass sources are economically more favorable and sustainable raw materials for developing bio-based polymers. However, the environmental costs of the techniques and solvents used in the manufacturing process must be considered alongside the use of sustainable raw materials to achieve green membrane production.3Lignocellulosic biomass is an abundant natural material that comprises cellulose, lignin, hemicellulose, and other minor components called extractives. Recently, lignocellulosic biomass is getting considerable interest due to its application in biofuel development and the potential for replacing fossil-based chemicals and materials. However, their sustainable utilization for developing advanced materials has remained challenging because of the versatility and complexity of their structure (i.e., different biomass sources have unique characteristics of cellulose, lignin, and hemicellulose) as well as the low solubility of cellulose in both water and organic solvents.4 A common approach to isolate lignin involves mechanical grinding, extensive heat, and highly acidic and alkaline pretreatment. Utilizing lignocellulosic biomass is challenging without employing multiple physical and chemical treatment steps because common organic solvents cannot directly deconstruct lignocellulosic constituents. By contrast, the robust and insoluble nature of lignocellulosic biomass make them a promising candidate for fabricating advanced materials for application in harsh environments, such as for preparing organic solvent nanofiltration (OSN) membranes.
The seeds of date fruits (Phoenix dactylifera L.), which are abundant and nonedible by-products, can be the source of lignocellulosic biomass. Dates are popular for their high content of antioxidants and dietary fibers, and their seeds have antibacterial activity.5 The annual worldwide production of dates is ∼9 million metric tons with at least 2 million metric tons being wasted owing to their limited use of the seed.6,7 Approximately 90% of the cultivated date palm trees (i.e., 100 million palm trees) are grown in the Middle East and North Africa. Date seeds are rich in mannan fibers, a hemicellulose component, and commonly used as food additives in fodder for sheep and camels in the desert. However, they have not been extensively studied as a natural material to be used in different applications compared with other common natural materials such as bamboo, wheat straw, corncob, and sugarcane bagasse. In the field of separation science and technology, date seed has been used as adsorbent for pollutant removal.8 To the best of our knowledge, there is no report on the application of date seed to be used in membrane fabrication.
The direct utilization of biomass remains challenging owing to the involvement of multiple treatment stages. Ionic liquids (ILs) have been proposed as a viable option for lignocellulosic biomass's pretreatment.9,10 ILs consist of an organic cation and an organic or inorganic anion salt, with an intermediate melting temperature of up to 100 °C. The main advantages of ILs are their highly tunable polarity, ionic conductivity, nonflammability, thermal stability, and negligible vapor pressure.11,12 Direct dissolution of various lignocellulosic biomass in ILs enabled the production of advanced materials, such as hydrogels,13 fibers,14 and films.15
ILs containing butyl cations, such as (1-butyl-3-methylimidazolium) or [Bmim], with different anions have been widely studied for cellulose dissolution. The chloride in [Bmim]Cl is speculated to highly influence the dissociation of hydrogen bonding in cellulose. However, [Bmim]Cl is highly toxic and forms a crystalline solid at room temperature.16 Alternatively, [Bmim][OAc] has been considered effective in lignocellulosic biomass swelling, and its lignin solubility is not affected by moisture as much as that of [Bmim]Cl.17 However, [Bmim][OAc] is an expensive solvent and generates large amounts of wastewater during the membrane fabrication stage (e.g., long demixing time and multiple washing steps). Therefore, the use of organic solvent-assisted ILs has been proven a viable option for lignocellulosic biomass treatment. Herein, dimethyl sulfoxide (DMSO EVOL) has been used as a co-solvent with [Bmim][OAc] to make a dope solution for membrane casting. DMSO was selected because it is considered an inexpensive and green organic solvent.18,19
To improve the membranes performance, the nanoscale deposition of dopamine on a porous support has been demonstrated to produce nanofiltration-based membranes with dense separation layers, improving their long-term stability in acidic and alkaline environments.20,21 Mussel-inspired polydopamine (PDA) has attracted attention for improving membrane performance and antibacterial properties. The use of PDA catechol chemistry has been proved to enhance the long-term stability of NF membranes in strong alkaline and acidic conditions.22 In addition, the layer-by-layer dip-coating method to prepare PDA-coated membranes is a simple way to increase membrane rejection, hydrophilicity, solvent stability, antifouling, and antibacterial performance.20 Therefore, we postulated that coating biomass-based membranes with PDA could improve their separation performance.
In this work, we proposed a co-solvent system based on ILs to dissolve date seeds with minimum pretreatment steps and develop highly durable high-performance membranes with adequate mechanical and chemical stability for OSN and oil-in-water separation. The schematic presenting date seeds treatments and membrane fabrication processes is shown in Fig. 1. Our approach can be easily integrated with the current membrane manufacturing lines in the market and provide a low-cost, sustainable process for developing high-performance membranes.
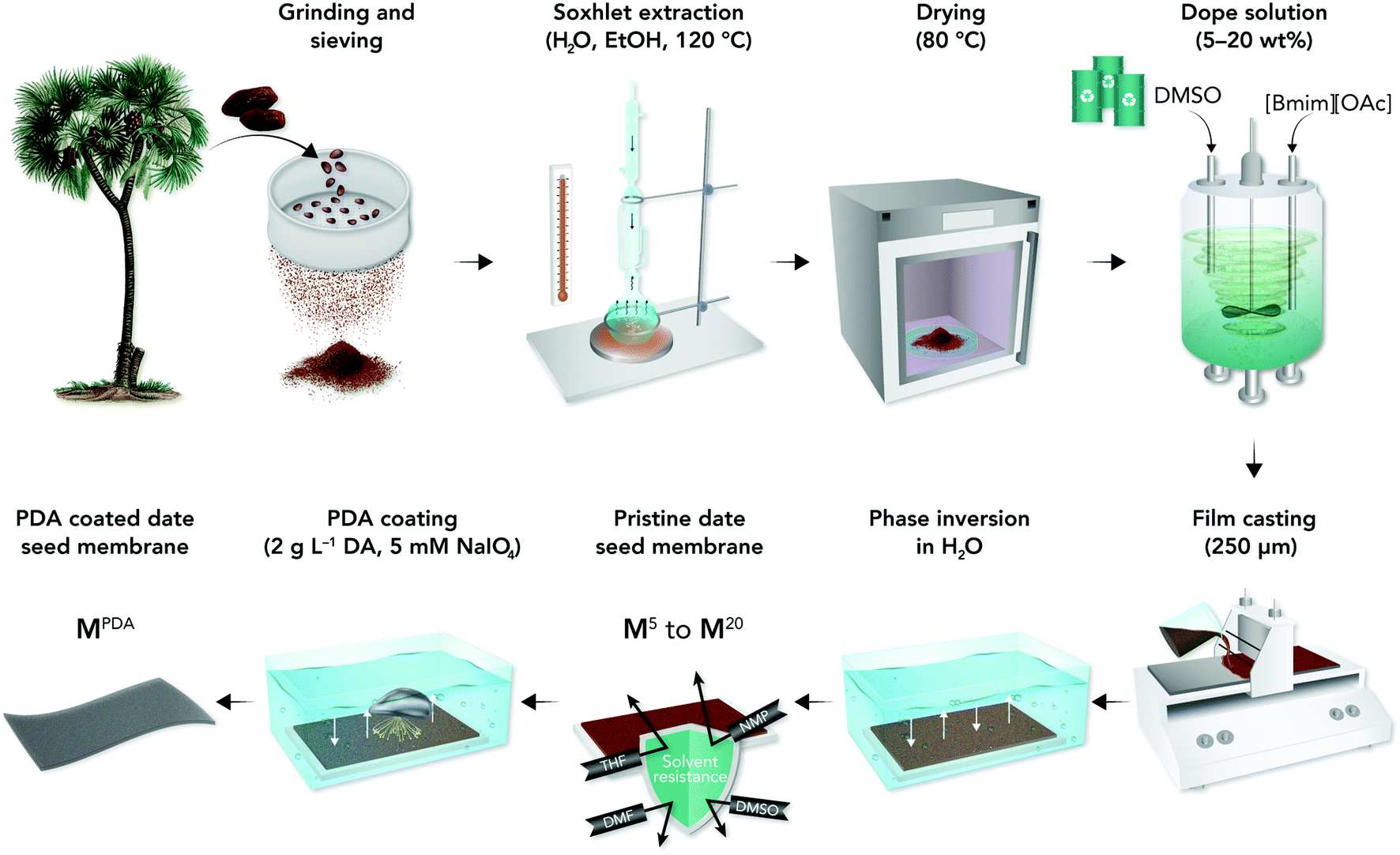 | ||
| Fig. 1 Schematic of the preparation of pure and PDA-coated date seed membranes. | ||
2. Results and discussion
A series of membranes with varying date seed concentrations, PDA deposition time, and numbers of PDA coating layers were prepared (Table 1). A mixture of 1-butyl-3-methylimidazolium acetate ([Bmim][OAc]) with dimethyl sulfoxide (DMSO) as a co-solvent was successfully used to dissolve the date seeds. We opted for a co-solvent system because it is a common approach to lower the viscosity and improve the miscibility in biomass processing.23 In particular, both experimental and in silico methods suggest that using DMSO as a co-solvent enhances the solubility of cellulose in [Bmim][OAc].24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (1:1) as a solvent
:DMSO (1:1) as a solvent
| Membrane | Date seed (wt%) | Solvent (wt%) | Number of PDA layers | PDA coating time (h per layer) |
|---|---|---|---|---|
| M5 | 5 | 95 | NA | NA |
| M10 | 10 | 90 | NA | NA |
| M15 | 15 | 85 | NA | NA |
| M1 PDA1 h | 10 | 90 | 1 | 1 |
| M2 PDA1 h | 10 | 90 | 2 | 1 |
| M3 PDA1 h | 10 | 90 | 3 | 1 |
| M4 PDA1 h | 10 | 90 | 4 | 1 |
| M5 PDA1 h | 10 | 90 | 5 | 1 |
| M6 PDA1 h | 10 | 90 | 6 | 1 |
| M7 PDA1 h | 10 | 90 | 7 | 1 |
| M8 PDA1 h | 10 | 90 | 8 | 1 |
| M9 PDA1 h | 10 | 90 | 9 | 1 |
| M10 PDA1 h | 10 | 90 | 10 | 1 |
| M11 PDA1 h | 10 | 90 | 11 | 1 |
| M1 PDA24 h | 10 | 90 | 1 | 24 |
| M2 PDA24 h | 10 | 90 | 2 | 24 |
| M3 PDA24 h | 10 | 90 | 3 | 24 |
| M4 PDA24 h | 10 | 90 | 4 | 24 |
2.1. Membrane characterization
The membrane with 5 wt% date seed concentration (M5) was too fragile, whereas high dope solution concentrations (15 wt%) produced wrinkled and nonuniform M15 membranes (Fig. S1†). The 10 wt% dope solution concentration was found to be optimal because it provided effective solubilization of date seed components to produce a membrane (M10) with acceptable mechanical flexibility, uniform thickness, and a smooth surface (Fig. 2a). Therefore, the membrane with 10 wt% date seed concentration was selected to be further treated by PDA to investigate the effects of coating time and the number of layers.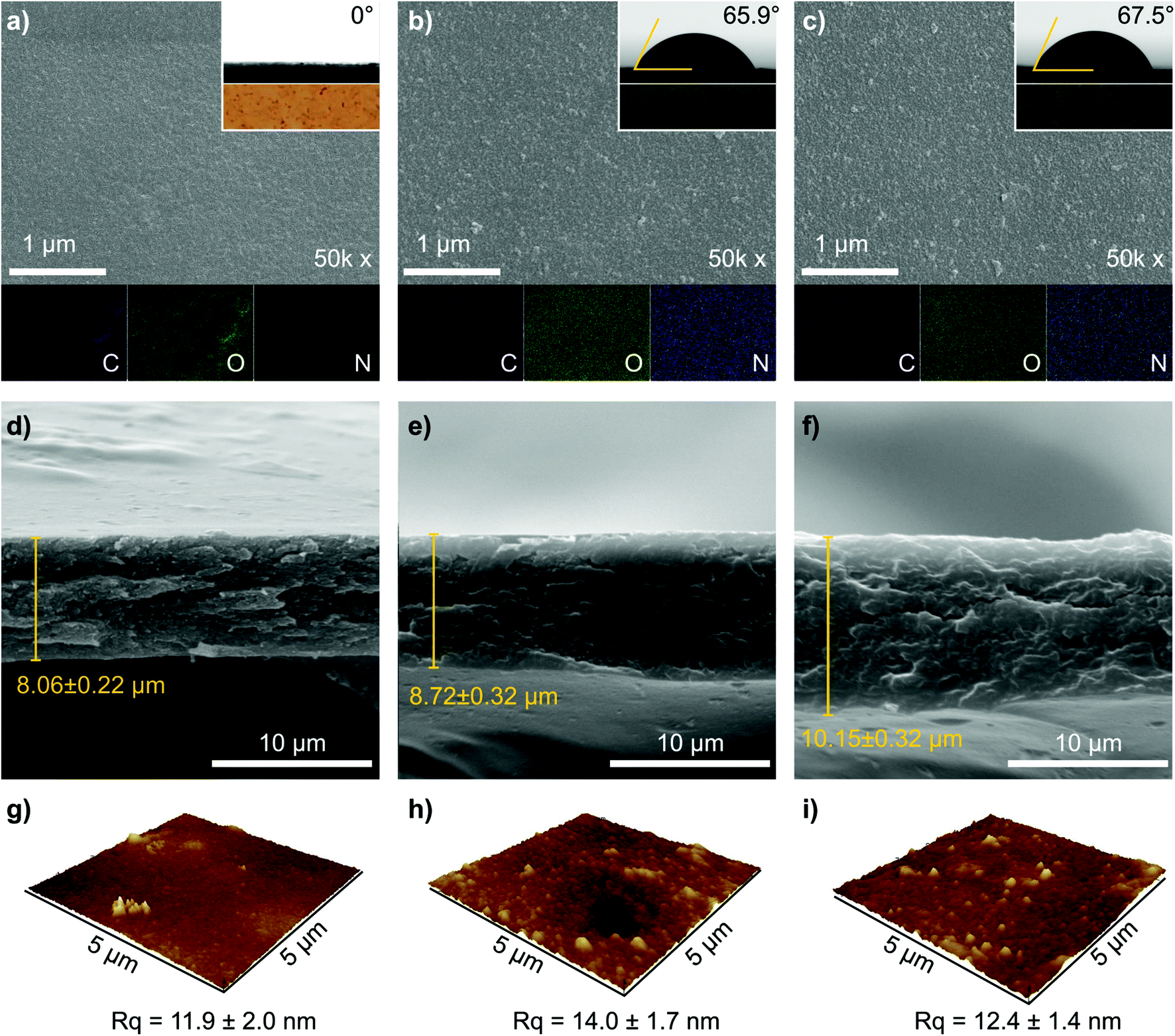 | ||
| Fig. 2 (a–c) Representative SEM images of the top surfaces, (d–f) cross sections, and (g–i) AFM images of M10, M11 PDA1 h, and M4 PDA24 h membranes. The insets in Fig. 2a–c are the water contact angles and the optical photos for each membrane. The EDX elemental mapping of carbon, oxygen, and nitrogen are indicated in Fig. 2a–c in purple, green, and blue, respectively. The AFM images for the complete membrane series are presented in Fig. S2, ESI.† | ||
We characterized representative membranes using scanning electron microscopy (SEM) and atomic force microscopy (AFM) to investigate the effects of the number of PDA layers and coating time on the membrane's morphology. The morphologies of the benchmark membrane (M10), the membrane that was coated 11 times (layers) for 1 h per layer (M11 PDA1 h), and the membrane that was coated 4 times (layers) for 24 h per layer (M4 PDA24 h) are shown in Fig. 2. SEM top surface images of M10, M11 PDA1 h, and M4 PDA24 h had a relatively homogenous surface and no defects were observed (Fig. 2a–c). A small number of particles can be observed on the surface of the PDA-coated membranes, which is assumed to originate from the precipitation route of the PDA polymerization.20,25 The optical photos of the membrane (inset in Fig. 2a–c) distinguish the brown color of the pristine membrane from the black color of the PDA-coated membranes. The SEM cross-sectional images exhibited a dense membrane profile with a thickness of approx. 8–10 μm (Fig. 2d–f). The increase in thickness correlates with the number of PDA coating layers as well as the coating time. However, other factors, such as slight variations in the membrane casting and phase inversion parameters, can also affect the thickness. The SEM observations indicated that the number of PDA layers and coating time did not affect both the membranes’ surface and cross-sectional morphologies. A quasi-similar surface topographies of M10, M11 PDA1 h, and M4 PDA24 h can also be seen with AFM height images (Fig. 2g–i), with comparable surface roughness (Rq of approx. 12–14 nm). The AFM images of the other prepared membranes are provided in the (ESI, Fig. S2†). The presence of PDA coating on the membrane surface was confirmed using energy-dispersive X-ray (EDX) elemental mapping (Fig. 2a–c), where carbon and oxygen were present in the benchmark and PDA-coated membranes and nitrogen originating from the deposited PDA was strongly observable in M11 PDA1 h and M4 PDA24 h.
The presence of the PDA layer was found to modify the membrane hydrophilicity, as evidenced by the water contact-angle measurements (insets of Fig. 2a–c and Fig. S3, ESI†). The uncoated pristine date seed membrane (M10) was highly hydrophilic, causing the water droplet to instantaneously penetrate the membrane surface. Varying the dope solution concentration for the pristine membranes (5–15 wt%) did not change the water contact-angle values (Fig. S3 and S4, ESI†). On the contrary, increasing the number of PDA layers (up to 9) with 1 h of coating time per layer gradually increased the water contact angle from 0° to 66°. Further deposition of up to 11 layers PDA resulted in virtually constant contact-angle values at approx. 66°, which is similar to that of the reported value for pure PDA films.26,27 These results indicate that the full surface coverage by PDA can be achieved at nine layers with 1 h of coating time per layer. PDA deposition with a coating time of 24 h per layer resulted in a contact-angle value of approx. 66° irrespective of the number of layers, indicating that the membrane surface was fully covered during the deposition of the first PDA layer. Interestingly, the PDA coating did not affect the membrane hardness, which was constant at around 0.2 GPa (Fig. S4†).
The chemical compositions of the membranes were investigated using X-ray photoelectron spectroscopy (XPS), and the wide spectra are shown in Fig. 3a. The XPS spectra revealed the presence of carbon, oxygen, and nitrogen in the membranes. A small nitrogen peak was also observed in the pristine membrane, which might be naturally included in the biomass source (date seed); however, its content was very low (0.54%). The atomic content of nitrogen showed enhancement with increase in the number of PDA layers and/or coating time (i.e., M1 PDA1 h, M11 PDA1 h, M1 PDA24 h, and M4 PDA24 h), evidencing the increased PDA content on the membrane surface (Table S1, ESI†). The XPS quantitative analysis was also in line with the EDX results where the nitrogen content slightly increased with the increase in the PDA deposition, as shown in Fig. S5 (ESI†).
 | ||
| Fig. 3 (a) XPS wide and narrow (b) C 1s, (c) O 1s, and (d) N 1s spectra and (e) FTIR spectra, (f) XRD patterns, and (g) thermogravimetric analysis curves of M10, M1 PDA1 h, M11 PDA1 h, M1 PDA24 h, and M4 PDA24 h. | ||
The C 1s high-resolution XPS spectrum (Fig. 3b and Fig. S6†) was deconvoluted into four peaks (i.e., C–C/C–H, C–O/C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and π → π*). The C–N peak at 286.5 eV was not observable because it overlapped with the C–O peak. The ratio of the C–O/C–C peak (at 286.5 and 285 eV, respectively) gradually decreased as the number of PDA layers and coating time increased, indicating the coverage of the membrane surface (containing C–O from cellulose) with PDA (containing C–C). The π → π* of the C 1s peak (at 289.1 eV) indicates a common energy loss feature for aromatic carbon species, and its intensity increased with PDA deposition. Consistent with the C 1s observation, the O–C peak of the XPS narrow O 1s spectra (Fig. 3c) decreased with the increasing number of PDA layers. By contrast, the R2NH peak in the N 1s spectra (Fig. 3d) increased as the number of PDA layers increased, supporting the argument that the cellulose (containing O–C) in the date seed membrane is covered by the PDA layer (containing R2NH). The XPS peaks reported here are in line with the previously reported literature on PDA thin film deposition.28
O, and π → π*). The C–N peak at 286.5 eV was not observable because it overlapped with the C–O peak. The ratio of the C–O/C–C peak (at 286.5 and 285 eV, respectively) gradually decreased as the number of PDA layers and coating time increased, indicating the coverage of the membrane surface (containing C–O from cellulose) with PDA (containing C–C). The π → π* of the C 1s peak (at 289.1 eV) indicates a common energy loss feature for aromatic carbon species, and its intensity increased with PDA deposition. Consistent with the C 1s observation, the O–C peak of the XPS narrow O 1s spectra (Fig. 3c) decreased with the increasing number of PDA layers. By contrast, the R2NH peak in the N 1s spectra (Fig. 3d) increased as the number of PDA layers increased, supporting the argument that the cellulose (containing O–C) in the date seed membrane is covered by the PDA layer (containing R2NH). The XPS peaks reported here are in line with the previously reported literature on PDA thin film deposition.28
The characteristic peaks of cellulose—the main constituent of the date seed—were observed in the Fourier transform infrared (FTIR) spectra at 3469, 1158, and 890 cm−1 (Fig. 3e), which were ascribed to the intermolecular hydrogen bonding (–OH), (C–O–C), and existence of β-D-glucopyranosyl groups, respectively.29 Additionally, the peak at 2870–2920 cm−1 can be attributed to the C–H stretching (–CH2−) in both lignin and cellulose.30,31 The presence of PDA is difficult to identify because of the overlapping of its peaks with those of the pristine date seed membrane. However, a small shift and increase in the peak intensity at 1595–1610 cm−1 (highlighted in yellow) with the increase in PDA deposition were observed, which could be attributed to α,β-unsaturated ketone, a characteristic peak of PDA. All the representative membranes exhibited broad X-ray diffraction (XRD) patterns (Fig. 3f) owing to their amorphous nature of the components, such as hemicellulose and lignin. Some XRD peaks (i.e., 16.3°, 18.4° and 21.7°) were observable, which can be attributed to cellulose.32 The thermal analyses of the pristine and coated membranes are presented in Fig. 3g and S8 (ESI†). It was found that the cellulose degradation temperature in PDA-coated membranes was lower (232 °C) compared with the pristine date seed membrane (278 °C). The mass loss above 500 °C can be associated with the CO and CO2 formation due to intense heat (burning of the carbonaceous residue).33
2.2. Membrane performance and biodegradability
Membrane's resistance toward organic solvents was determined by contacting the membrane with common organic solvents, as shown in Table S3 (ESI†). The pristine date seed membrane (M10) showed virtual stability in all the tested solvents, including harsh polar aprotic solvents (DMAc, DMF, and DMSO), whereas the PDA-coated membranes disintegrated and dissolved in DMF. Furthermore, M10 was stable in acidic solution and in basic solutions, it was stable only up to pH 9 (Table S4, ESI†).The prepared membranes were screened in a cross-flow nanofiltration setup using valsartan (VS) as an active pharmaceutical ingredient (API) in acetonitrile to evaluate the membranes’ rejection and permeance (Fig. 4a). The effects of 1 h versus 24 h coating time on the filtration performance were compared (highlighted in cyan and olive, respectively). Without PDA coating, the membrane exhibited a permeance value of 94.9 ± 2.4 L m−2 h−1 bar−1 and no rejection was observed. The permeance significantly decreased to approx. 60% for the PDA-coated membrane with a coating time of 1 h and remained constant until four layers of deposition. However, solute rejections were still not observed. These observations suggest that low-molecular-weight PDA was insufficient to narrow the large pore sizes in the pristine membrane in a short deposition time or with few deposition layers. Depositing more PDA layers resulted in additional mass transfer resistance due to the reduction of the large pore size and blockage of the smaller pores. The rejection increased monotonously from five (M5 PDA1 h) to eleven (M11 PDA1 h) PDA layers with the latter having a maximum rejection of 61.5% ± 3.3% and permeance of 38.8 ± 2.0 L m−2 h−1 bar−1.
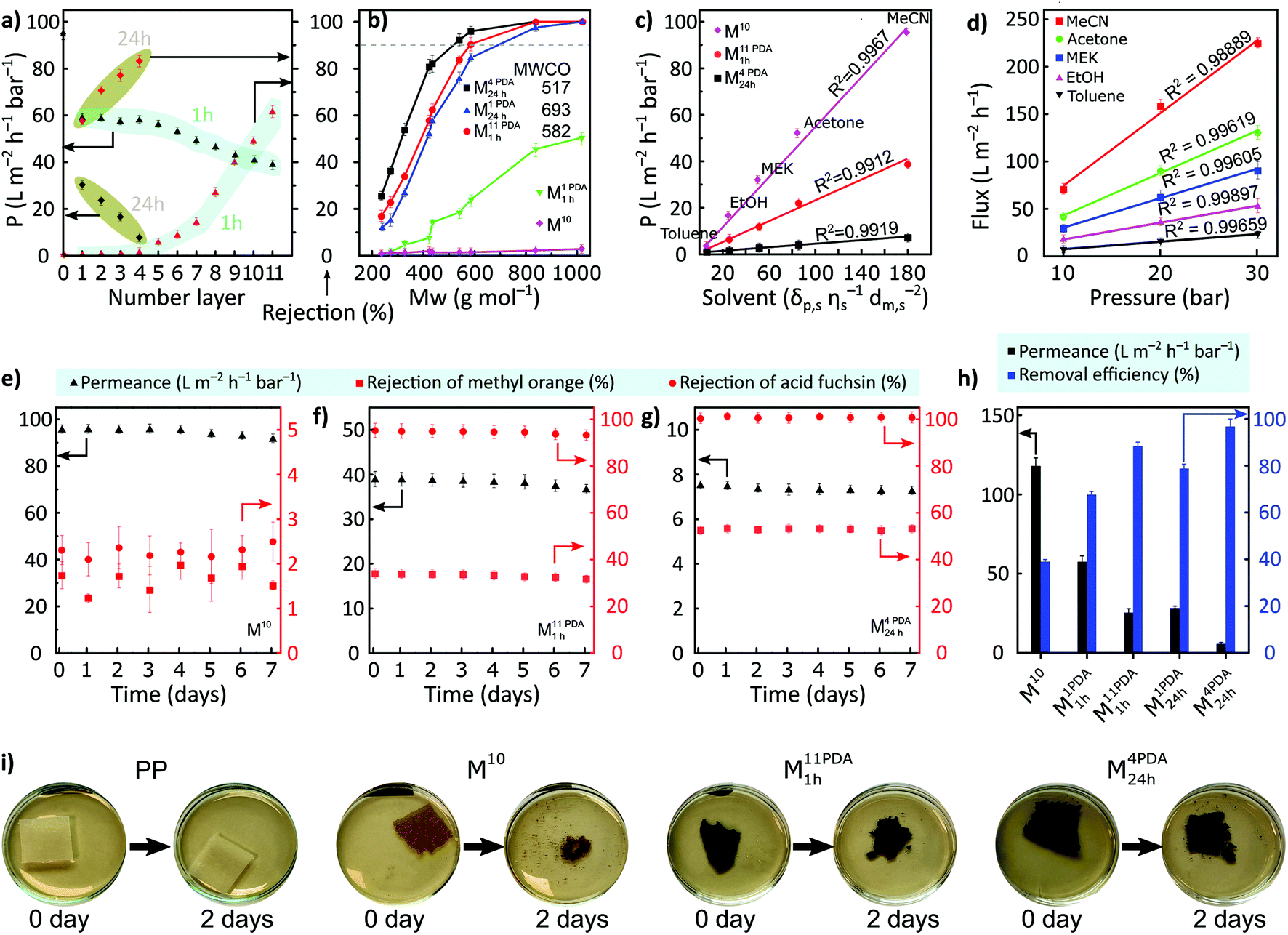 | ||
| Fig. 4 (a) Acetonitrile permeance and rejection of valsartan for all the membranes coated for 1 h (cyan) and 24 h (olive) per layer versus the number of PDA layers. (b) Rejection profiles of various solutes using pristine and PDA-coated date seed membranes in acetonitrile. (c) Permeance values for M10, M11 PDA1 h, and M4 PDA24 h as the functions of solubility parameters using various solvents. (d) Effect of pressure on the flux of various solvents for M4 PDA24 h. Nanofiltration testing of methyl orange and acid fuchsin for over 7 days for (e) M10, (f) M11 PDA1 h, and (g) M4 PDA24 h. (h) Oil-in-water emulsion separation performances of M10, M1 PDA1 h, M11 PDA1 h, M1 PDA24 h, and M4 PDA24 h. (i) Biodegradation of the prepared membranes using cellulase at room temperature. All the nanofiltration tests were performed at 30 bar. | ||
The permeance considerably declined with a longer PDA coating time (24 h) compared to that with multiple layers deposited for a shorter time (1 h). In particular, the permeance decreased from 94.9 ± 2.4 L m−2 h−1 bar−1 for M10 to 30.3 ± 0.7 L m−2 h−1 bar−1 for M1 PDA24 h, while the rejection of valsartan increased from 0% to 57.7% ± 1.70%. A sharp increase in membrane rejection (by approx. 44%) was realized with the increase in the PDA layers for a coating time of 24 h (M1 PDA24 h → M4 PDA24 h) at the expense of permeance, which further decreased to 7.8 ± 0.2 L m−2 h−1 bar−1 for M4 PDA24 h. M4 PDA24 h was the tightest membrane with a maximum obtained valsartan rejection of 83.1% ± 2.4%.
The separation performance of the representative membranes was further tested using different types of solutes including oligomers, APIs, and dyes having their molecular weight between approx. 200 and 1000 g mol−1 (Fig. 4b). M10 and M1 PDA1 h did not exhibit rejections above 90% in the nanofiltration molecular weight range, and therefore, the molecular weight cutoff (MWCO) value could not be determined. The molecular selectivity of the membrane can be successfully controlled by increasing the number of PDA layers and/or the coating time. Increasing the coating time to 24 h (M1 PDA24 h) resulted in an MWCO of approx. 693 g mol−1. The MWCO of the membrane exhibited a further decrease by 25% as the number of PDA layers increased with a coating time of 24 h per layer (M1 PDA24 h → M4 PDA24 h). Further, 16% reduction in the MWCO was obtained for the highest number of PDA layers with a coating time of 1 h per layer (M11 PDA1 h) compared with that of M1 PDA24 h.
The pristine date seed membrane and the membranes with the lowest MWCO (M11 PDA1 h and M4 PDA24 h) were tested with various organic solvents (Fig. 4c). The permeances of solvents and their solubility parameters (i.e., solubility, viscosity, and molar diameter) were linearly correlated. The PDA-coated M4 PDA24 h membrane exhibited a linear correlation between pressure and flux, which indirectly proves the structural stability of the membrane under the nanofiltration operating pressure range (Fig. 4d). A long-term stability test was performed over 7 days of continuous filtration of two dyes, methyl orange (MO) and acid fuchsin (AF), for the same membrane series (M10, M11 PDA1 h, and M4 PDA24 h). The rejections of both dyes and the permeance were found to be stable during the filtration (Fig. 4e–g). The separation performances obtained were compared with those reported in the literature in terms of solute rejection, MWCO, and permeance (Table S13, ESI†). The tightest membrane with a longer PDA coating time (M4 PDA24 h) showed better results by exhibiting lower MWCO. The rejection of methyl orange is lower than that of acid fuchsin because the molecular weight of methyl orange (327 g mol−1) is lower compared to acid fuchsin (586 g mol−1). According to the surface zeta potential measurements, the PDA-coated membranes were more negatively charged compared to the pristine membrane, with the values of −23 ± 2.1 mV and −2 ± 1.2 mV, respectively. Therefore, we propose that in the pristine membrane, the separation mechanism is dominated by size-exclusion. However, in the PDA-coated membranes, the separation mechanism is likely to be governed by both size exclusion and Donnan exclusion.
Moreover, oil-in-water filtration experiments were performed using M10, M1 PDA1 h, M11 PDA1 h, M1 PDA24 h, and M4 PDA24 h (Fig. 4h). The pristine membrane (M10) exhibited a high water permeance of 117.9 ± 5.1 L m−2 h−1 bar−1 and 39% of oil-removal efficiency owing to its high hydrophilicity and MWCO to channel water molecules. PDA coating for 1 h per layer from 1 to 11 layers decreased the water permeance from 117.9 ± 5.1 to 25.3 L m−2 h−1 bar−1 and increased the oil-removal efficiency from 67.6 ± 1.4% to 88.5 ± 1.6%, respectively. As the number of PDA layers increased from 1 to 4 layers, with the 24 h coating time per layer, the permeance decreased from 28.3 ± 1.2 to 5.7 ± 0.9 L m−2 h−1 bar−1, while the oil-removal efficiency increased from 78.8 ± 1.9% to 96.8 ± 3.2%, respectively. Finally, the biodegradability of the membranes using cellulase was tested because the membrane's decomposition is crucial considering the environmental aspects. We used polypropylene (PP) support as a control and observed that this support did not disintegrate even after 30 days of biodegradation testing (Fig. 4i and S10, ESI†). By contrast, the pristine and PDA-coated membranes possessed excellent biodegradability as their disintegration was observed after 2 days of biodegradation testing (Fig. 4h). Microorganisms started to cultivate after 5 days (Fig. S9 and S10, ESI†), and the pristine date seed membrane (M10) was completely degraded within 7 days of biodegradation testing. The degradation was relatively slower for the PDA-coated membranes compared with M10. Based on previous reports,34,35 we infer that the increased nitrogen content in PDA-coated membranes delayed the biodegradation of M11 PDA1 h and M4 PDA24 h compared with M10.
3. Conclusions
We demonstrated the upcycling of waste biomass, i.e., date seeds, with minimum processing steps using a greener solvent system ([Bmim][OAc]:DMSO; 1:1) to prepare biodegradable membranes. The pristine date seed membranes can be tailored via the facile layer-by-layer PDA deposition method to fine-tune their molecular sieving properties. The gradual increase in the number of PDA layers from 0 to 11 and the increase in the coating time from 1 to 24 h enabled the control of the separation performance and afforded proportionally tighter membranes. Having screened 25 organic solvents covering eight solvent classes, we found that the pristine date seed membranes were stable in all the solvents, including DMF at 100 °C. Moreover, the PDA-coated membranes exhibited excellent stability, except in DMF. The best-performing membrane (M4 PDA24 h) achieved an MWCO value as low as 517 g mol−1 at the expense of lowering acetonitrile permeance from 95 (pristine membrane) to 8 L m−2 h−1 bar−1. The membranes exhibited stable performance for over seven days of continuous nanofiltration in acetonitrile. Further, the pristine and PDA-coated membranes were biodegraded within few days using cellulase. Our work demonstrates that lignocellulosic biomass can be used to fabricate solvent-resistant biodegradable membranes via a sustainable cradle-to-grave approach. The robustness and control over the porosity of the membranes make them excellent candidates for demanding applications such as organic solvent nanofiltration and water treatment.
4. Experimental
4.1. Materials
1-Butyl-3-methylimidazolium acetate ([Bmim][OAc], ≥95%), 1-ethyl-3-methylimidazolium acetate ([Emim][OAc]), N,N-dimethylacetamide (DMAc, 99.8%), dimethyl sulfoxide (DMSO, ≥99.9%), N,N-dimethylformamide (DMF, 99.8%), dimethyl carbonate (DMC, 99%), 1-methyl-2-pyrrolidinone (NMP, ≥99%), tetrahydrofuran (THF, ≥99.9%), dichloromethane (DCM, ≥99.8%), propylene carbonate (PC, 99.7%), γ-valerolactone (99%), glycerol (≥99.5%), 1-butanol (99.8%), 1,4-dioxane (99.8%), hexane (≥95%), and acetone (≥99.5%) were obtained from Sigma Aldrich. Ethanol (≥99.5%), methanol (≥99.9%), toluene (≥99%), and acetonitrile (ACN, ≥99.9%) were purchased from Fisher. Isopropanol (99.5%) and cyclopentyl methyl ether (CPME, 99.5%) were obtained from Acros Organics. Heptane (≥99%) was purchased from Honeywell. Cellulase from Trichoderma species (10 units per mg solid) and sodium periodate (NaIO4) were purchased from Sigma Aldrich. Dopamine hydrochloride was obtained from Alfa. Date seed powder was obtained from a local distributor in Saudi Arabia (Siafa International Mfg. CO., Saudi Arabia). Vegetable oil was purchased from a local market in the United Kingdom. Hexadecyltrimethylammonium bromide (CTAB) was purchased from Acros Organics. Novatexx 2471 (polypropylene nonwoven support) was obtained from Freudenberg Filtration Technologies SE & Co. KG. Deionized water (DI) with a resistivity of 18.2 MΩ cm at 25 °C (Milli-Q) was employed in all the experiments. All the materials were used as received without further modification.4.2. Treatment of date seeds
The raw date seeds were sieved through 150 μm filter and dried in an oven at 80 °C for 24 h before treatment. Then, the sieved date seeds (5 g) were treated with hot DI water (250 mL) at 120 °C for at least 24 h via a Soxhlet apparatus to remove water-soluble extractives until the filtered water was clear, followed by ethanol (250 mL) Soxhlet extraction at 120 °C for 24 h. The hot water and ethanol treatment (at 120 °C) is a critical prestep to solubilize date seeds in [Bmim][OAc]:DMSO solvent as it reduces recalcitrance and the disturbance of the biomass network, making cellulose and other components such as lignin and hemicellulose more accessible.36 The treated date seeds were further dried at 80 °C until a constant weight of dry seeds was obtained.
4.3. Fabrication of membranes
Dope solutions with varying amounts of date seeds (5–20 wt%) were developed using a co-solvent of [Bmim][OAc] and DMSO with a mass ratio of 1:1. The dope solutions were mixed using an overhead stirrer at 35 °C at 100 rpm for 24 h. Then, they were placed in an incubator for 6 h at 40 °C with a shaking speed of 400 rpm, and finally, the temperature was set to 25 °C for 2 h before casting. The dope solutions were cast on polypropylene nonwoven (PP) support using a film applicator (Elcometer 4340) at a casting speed of 6 cm s−1 and a blade gap of 150 and 250 μm for 5–10 and 15–20 wt% dope solutions, respectively. The resulting film was immediately immersed into a 10 L DI water bath (15 MΩ cm) at 22 °C for 24 h and then immersed in a 2 L water bath for 24 h. The resulting films were rolled and stored in a measuring cylinder containing 1 v/v% acetonitrile–water mixture.
4.4. PDA coating
The common dip-coating method was used to prepare the PDA-coated membranes.20 In a typical experiment, a membrane was cut into a disk (8.5 cm in diameter) and soaked in a dopamine monomer solution (2 mg mL−1 concentration in water, 140 mL) at room temperature. Then, 5 mM NaIO4 was added (as oxidant to accelerate the formation of a homogenous polydopamine film37) and shaken for 1- or 24 h deposition time, followed by washing the membrane with water to terminate the coating process for one PDA layer. The same procedure was repeated for coating each PDA layer.4.5. Biodegradation of membranes
The pristine and best-performing membranes (M10, M11 PDA1 h, and M4 PDA24 h) were cast on glass without the PP support following the same procedure of PDA coating (section 4.4). The membranes were dried using an oven at 80 °C and a vacuum desiccator and cut into small pieces (10 × 10 mm2). The membranes were soaked in a Petri dish containing an aqueous solution of cellulase (i.e., 1.5 wt% cellulase in 10 mL of phosphate buffer solution, pH 7) at 25 °C, as reported elsewhere.38 The membranes including the PP support were also cut into larger pieces (30 × 30 mm2) and soaked in a Petri dish containing approx. 20 mL of 1.5 wt% cellulase at room temperature without the phosphate buffer solution, mimicking natural biodegradation conditions. After 30 d, the samples were skimmed carefully to remove the top layer and diluted with DI water to show the remaining pieces.4.6. Characterization
SEM images for surface and cross-sectional analysis were acquired using an FEI Quanta 200 instrument with an acceleration voltage of 15 kV. For improving the conductivity, the samples were spin-coated with a 6 nm platinum layer using Quorum Q150TES under an Ar atmosphere. Membrane thickness was estimated using ImageJ software; at least 10 measurements across the SEM cross-sectional images were taken for each membrane. AFM (BioAFM Bruker Multimode 8) with a standard tapping mode was used under air at 25 °C. A scanning area of 5 × 5 μm2 was used with a scanning rate of 1 Hz. The results were analyzed using NanoScope Analysis software. The root-mean-square average roughness (Rq) was selected to quantify the surface roughness of the prepared membranes. The FTIR spectra of the prepared membranes were obtained via an Alpha-P instrument (Bruker Instruments). The spectra were generated using an average of 32 scans under air over the range of 600–4000 cm−1. Thermogravimetric analysis (TGA) curves were obtained using TGA-550 (TA Instruments) with a temperature ramp rate of 20 °C min−1 from 25 °C to 800 °C under a N2 atmosphere. The XRD patterns were acquired via a PANalytical X'Pert Pro X'Celerator (XRD5) diffractometer at room temperature using Cu Kα radiation (1.541 Å) over 5°–50° angular range (2θ) with a step size of 0.03°. The dried membranes were peeled off from the PP support and flattened on an XRD sample holder. XPS was conducted using an Axis Ultra Hybrid spectrometer (Kratos Analytical, Manchester, United Kingdom) equipped with a monochromatic Al Kα X-ray source with a base vacuum pressure of ∼5 × 10−9 mbar (hν = 1486.6 eV, spot size = 300 × 700 μm2, and 10 mA emission at 150 W). Charge neutralization was achieved using a filament. Binding energy scale calibration was performed using C–C in the C 1s photoelectron peak at 285 eV for each sample. Data analysis and curve fitting was performed using CasaXP 2.3.24 software.39Viscosity was determined at 25 °C at 5 rpm using Elcometer 2300 RV (Elcometer Inc., USA) with TL6 and TL7 connections for solvents and dope solutions, respectively. The nanoindentation technique was used to evaluate the mechanical hardness of the membranes using a NanoTest Vantage instrument with a pyramidal diamond indenter. 1 cm2 membrane samples were cut and then adhered to a silicon wafer using superglue. At least four indentions were obtained per specimen. A Kruss EasyDrop instrument was used to record the water contact angle of the membranes; the sessile drop method was employed, and the Young–Laplace fitting model was used. Prior to the measurement, the membranes were adhered to a glass plate using a double-sided tape, and five measurements were taken at different locations on a membrane to obtain the average contact-angle value for each sample. 2 μL of water was dropped on each membrane, and contact-angle reading was taken after 5 s. The surface zeta potential measurements were caried out by using a Zetasizer-Nano ZS. The membrane samples were peeled off from the support, and attached to the holder using double-sided tape. The measurements were performed using deionized water with pH of 7.2.
4.7. Nanofiltration
Nanofiltration experiments were performed using a cross-flow setup, which comprised a high-pressure pump, a recirculation gear pump, a backpressure regulator, and multiple membrane cells with an effective area (A) of 53 cm2. The operating parameters were kept constant as the following. A pressure difference (ΔP) of 10–30 bar, a feed flow rate of 3 L h−1, and a cross-flow volume rate of 100 L h−1 was used to reduce the concentration polarization. The permeate volume (V) was collected over time (t) until constant flow was realized for permeance measurement (eqn (1)). Samples from the feed and permeate were collected to analyze the feed concentration (Cf) and permeate concentration (Cp) using high-performance liquid chromatography. Membrane rejection or removal efficiency (%) was determined using eqn (2). The MWCO was defined as the molecular weight of solute to be 90% retained by the membrane. The prepared membranes were tested with nine solutes in the feed stream, namely, styrene dimer (236 g mol−1), estradiol (272.38 g mol−1), MO (327 g mol−1), losartan (423 g mol−1), VS (435 g mol−1), oleuropein (541 g mol−1), AF (586 g mol−1), roxithromycin (837 g mol−1), and rose bengal (1017 g mol−1). A long-term test of continuous filtration for over 7 days was performed using two different solutes (MO and AF) in acetonitrile. For the separation of the oil-in-water emulsion, the concentrations were analyzed using InfraCal 2 (AMETK, Inc.). The reported filtration results are the average values of two membranes prepared independently.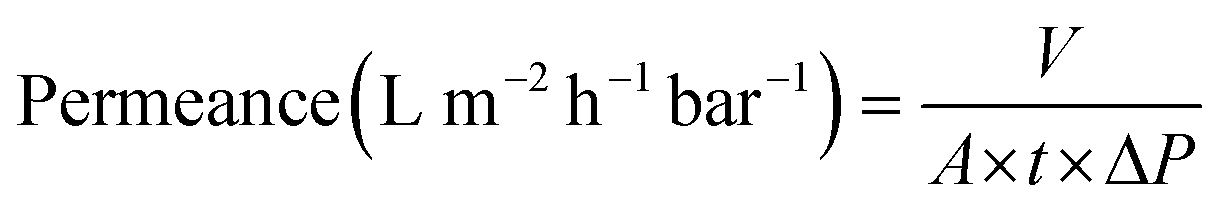 | (1) |
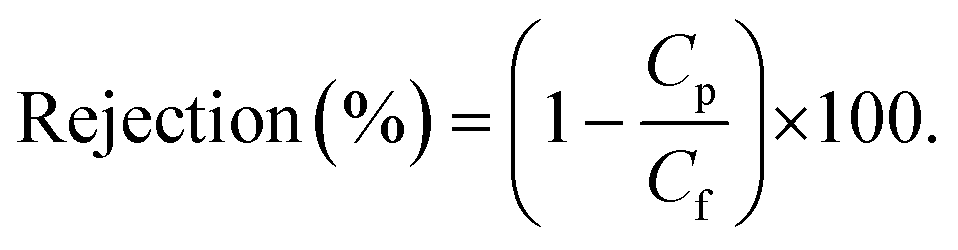 | (2) |
Oil-in-water emulsions were prepared via high mechanical shearing using CTAB as an emulsifier. The feed solution was prepared by mixing 0.5 g of vegetable oil (0.5 g L−1) with 50 mg of CTAB surfactant in 1 L of water using T-18 ULTRA-TURRAX® (IKA England Ltd) at 15000 rpm for 2 min at 25 °C, as reported elsewhere.40 The stability of the pristine membrane has been evaluated by placing square membrane pieces (0.5 × 0.5 cm2) in 4 mL of various solvents and aqueous solutions with different pH levels at room temperature in closed-top cap vials for up to 30 days.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the King Abdullah University of Science and Technology (KAUST). RH acknowledges the postdoctoral fellowship from the Advanced Membranes and Porous Materials Center at KAUST. AA acknowledges the PhD scholarship awarded by Saudi Aramco.References
- W. Xie, T. Li, A. Tiraferri, E. Drioli, A. Figoli, J. C. Crittenden and B. Liu, ACS Sustainable Chem. Eng., 2021, 9, 50–75 CrossRef CAS.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- D. Zou, S. P. Nunes, I. F. J. Vankelecom, A. Figoli and Y. M. Lee, Recent advances in polymer membranes employing non-toxic solvents and materials, Green Chem., 2021 10.1039/D1GC03318B.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499–3535 RSC.
- M. ALrajhi, M. AL-Rasheedi, S. E. M. Eltom, Y. Alhazmi, M. M. Mustafa, A. M. Ali and F. Yildiz, Cogent Food Agric., 2019, 5, 1625479 CrossRef.
- C. de la Cruz-Lovera, F. Manzano-Agugliaro, E. Salmerón-Manzano, J. L. de la Cruz-Fernández and A. J. Perea-Moreno, Sustainability, 2019, 11, 711 CrossRef CAS.
- S. Ghnimi, S. Umer, A. Karim and A. Kamal-Eldin, NFS J., 2017, 6, 1–10 CrossRef.
- H. Al Subhi, M. S. Adeeb, M. Pandey, H. Al Sadeq, D. Kumar and S. K. Shukla, Appl. Water Sci., 2020, 10, 166 CrossRef CAS.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- N. Sathitsuksanoh, K. M. Holtman, D. J. Yelle, T. Morgan, V. Stavila, J. Pelton, H. Blanch, B. A. Simmons and A. George, Green Chem., 2014, 16, 1236–1247 RSC.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- A. A. C. Toledo Hijo, G. J. Maximo, M. C. Costa, E. A. C. Batista and A. J. A. Meirelles, ACS Sustainable Chem. Eng., 2016, 4, 5347–5369 CrossRef CAS.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63–69 RSC.
- N. Sun, W. Li, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 1158–1161 RSC.
- A. Abdulkhani, E. Hojati Marvast, A. Ashori and A. N. Karimi, Carbohydr. Polym., 2013, 95, 57–63 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- N. Sun, H. Rodríguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405–1421 RSC.
- S. Wang, W. Zhao, T. S. Lee, S. W. Singer, B. A. Simmons, S. Singh, Q. Yuan and G. Cheng, ACS Sustainable Chem. Eng., 2018, 6, 4354–4361 CrossRef CAS.
- T. Marino, F. Galiano, S. Simone and A. Figoli, Environ. Sci. Pollut. Res., 2019, 26, 14774–14785 CrossRef CAS PubMed.
- F. Fei, H. A. Le Phuong, C. F. Blanford and G. Szekely, ACS Appl. Polym. Mater., 2019, 1, 452–460 CrossRef CAS PubMed.
- D. Zhao, J. F. Kim, G. Ignacz, P. Pogany, Y. M. Lee and G. Szekely, ACS Nano, 2019, 13, 125–133 CrossRef CAS PubMed.
- F. Meng, F. Song, Y. Yao, G. Liu and S. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 10928–10938 CAS.
- A. S. Patri, B. Mostofian, Y. Pu, N. Ciaffone, M. Soliman, M. D. Smith, R. Kumar, X. Cheng, C. E. Wyman, L. Tetard, A. J. Ragauskas, J. C. Smith, L. Petridis and C. M. Cai, J. Am. Chem. Soc., 2019, 141, 12545–12557 CrossRef CAS PubMed.
- J. M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. H. Pádua and M. F. C. Gomes, Green Chem., 2014, 16, 2528–2538 RSC.
- Y. Ding, L.-T. Weng, M. Yang, Z. Yang, X. Lu, N. Huang and Y. Leng, Langmuir, 2014, 30, 12258–12269 CrossRef CAS PubMed.
- Q. Wei, F. Zhang, J. Li, B. Li and C. Zhao, Polym. Chem., 2010, 1, 1430–1433 RSC.
- J. Zhao, Y. Su, X. He, X. Zhao, Y. Li, R. Zhang and Z. Jiang, J. Membr. Sci., 2014, 465, 41–48 CrossRef CAS.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- S. Zhang, F. Zhang, Y. Pan, L. Jin, B. Liu, Y. Mao and J. Huang, RSC Adv., 2018, 8, 5678–5684 RSC.
- T. Zhang, G. Cai and S. Liu, J. Cleaner Prod., 2018, 172, 1788–1799 CrossRef CAS.
- X. Cui, X. Yang, K. Sheng, Z. He and G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16520–16528 CrossRef CAS.
- M. K. D. Rambo, F. L. Schmidt and M. M. C. Ferreira, Talanta, 2015, 144, 696–703 CrossRef CAS PubMed.
- M. Zhao, Z. Han, C. Sheng and H. Wu, Energy Fuels, 2013, 27, 898–907 CrossRef CAS.
- G. Fuchs, M. Boll and J. Heider, Nat. Rev. Microbiol., 2011, 9, 803–816 CrossRef CAS PubMed.
- C. C. Chen, L. Dai, L. Ma and R. T. Guo, Nat. Rev. Chem., 2020, 4, 114–126 CrossRef.
- J. Wang, R. Boy, N. A. Nguyen, J. K. Keum, D. A. Cullen, J. Chen, M. Soliman, K. C. Littrell, D. Harper, L. Tetard, T. G. Rials, A. K. Naskar and N. Labbé, ACS Sustainable Chem. Eng., 2017, 5, 8044–8052 CrossRef CAS.
- F. Ponzio, J. Barthès, J. Bour, M. Michel, P. Bertani, J. Hemmerlé, M. d'Ischia and V. Ball, Chem. Mater., 2016, 28, 4697–4705 CrossRef CAS.
- D. Beaton, P. Pelletier and R. R. Goulet, Frontiers in Microbiology, 2019, 10, 204 CrossRef PubMed.
- N. Fairley, V. Fernandez, M. Richard–Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- D. Kim, S. Livazovic, G. Falca and S. P. Nunes, ACS Sustainable Chem. Eng., 2019, 7, 5649–5659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental design, calculations, data analysis, structure and morphology analysis. See DOI: 10.1039/d1gc03410c |
| This journal is © The Royal Society of Chemistry 2022 |