
Manganese oxide as an alternative to vanadium-based catalysts for effective conversion of glucose to formic acid in water†
Jialu
Li
 a,
Richard Lee
Smith
Jr.
b,
Siyu
Xu
a,
De
Li
a,
Jirui
Yang
a,
Keqiang
Zhang
*a and
Feng
Shen
*a
a,
Richard Lee
Smith
Jr.
b,
Siyu
Xu
a,
De
Li
a,
Jirui
Yang
a,
Keqiang
Zhang
*a and
Feng
Shen
*a
aAgro-Environmental Protection Institute, Ministry of Agriculture and Rural Affairs, No. 31 Fukang Road, Nankai District, Tianjin 300191, China. E-mail: shenfeng@caas.cn; keqiangzhang68@163.com
bGraduate School of Environmental Studies, Research Center of Supercritical Fluid Technology, Tohoku University, Aramaki Aza Aoba 6-6-11, Aoba, Sendai 980-8579, Japan
First published on 26th November 2021
Abstract
MnOx catalysts were synthesized under hydrothermal conditions for conversion of glucose into formic acid (FA) in water with the objective to develop vanadium-free reaction systems. An FA yield of 81.1% was obtained with a MnOx catalyst and glucose substrate in water at 160 °C and is almost twice the value obtained with vanadium-based heterogeneous catalysts. MnOx materials prepared hydrothermally at 100 °C had higher Mn2+/Mn3+ ratios and adsorbed oxygen species than those prepared at higher temperatures and gave the highest FA yields among the catalysts evaluated. Mechanistic studies of glucose–MnOx–water reaction systems revealed that two parallel reactions existed with arabinose being the intermediate in the α-scission route and glyoxylic acid being the intermediate in the β-scission route where CO2 co-forms with FA. Small water-soluble carbohydrates (glucose, sucrose, cellobiose, maltose, xylose) afforded FA yields greater than 50%, while starch afforded FA yields greater than 20%, thus demonstrating the potential of MnOx catalysts for converting biomass into FA.
Introduction
The conversion of lignocellulosic biomass into chemicals and fuels is necessary for achieving sustainable society. Glucose has been examined as the starting material for producing organic acids (formic acid,1 lactic acid,2 gluconic acid3), alcohols (sorbitol,4 ethanol5), and furans (5-hydroxymethylfurfural,6 furfural7). Among the organic acids that can be derived from glucose, formic acid (FA) is a promising candidate for hydrogen storage since it has a volumetric density of 53 g of H2 per liter and low toxicity and exists as a liquid under ambient conditions facilitating transportation. FA can be used to produce hydrogen at room temperature8 elevating its importance in the textile, pharmaceutical, and food chemical sectors9 as well as in the leather and tanning industries which require high amounts of FA every year. Moreover, FA is widely used in animal feed as an antibacterial agent.10 About 1.137 million metric tons of FA are required every year worldwide, wherein the compound annual growth report estimates that there will be a 3.7% increase in FA consumption over the period from 2019 to 2024.11 Industrial production of FA is mainly based on the carbonylation of methanol followed by hydrolysis of methyl formate.12 However, more than 80% of the methanol used in FA production is from natural gas and coal.13 Production of FA directly from biomass would be desirable and would help in creating sustainable production routes.Wet oxidation and catalytic oxidation are two common routes for the synthesis of FA from biomass. Wet oxidation is a hydrothermal process (>200 °C) conducted in alkaline solutions (NaOH, KOH, LiOH)14 using H2O2 as the oxidant. For instance, 75% FA yield can be obtained from glucose using 1.2 mol L−1 KOH and 240% excess H2O2 at 250 °C.15 Although high FA yields are achieved from glucose via wet oxidation, other organic acids such as lactic acid, acetic acid, and propanoic acid are generated simultaneously making FA separation challenging,15 and furthermore, when the obtained product is formate instead of formic acid, an additional hydrolysis step is required. Compared with wet oxidation, catalytic oxidation can be performed at relatively low reaction temperatures (<180 °C) to obtain high FA selectivities. For instance, V-substituted heteropolyacid H5PV2Mo10O40 catalysts used for FA production from biomass afford FA yields of 45 to 65% with FA being found as the sole liquid phase product.16,17 However, most catalytic oxidation routes for FA synthesis use vanadium(V)-containing homogeneous catalysts like NaVO3,18 VOSO4,19 and V-substituted heteropolyacids (Hn+3PVnMo12−nO40),17 wherein V-based catalytic systems have biological toxicity20,21 and issues associated with recycling of homogeneous catalysts. Although vanadium-free metal oxides, such as ZrO2,22 CaO23 and Mg–Al hydrotalcite,24 have emerged as recyclable solid catalysts for FA synthesis from biomass, most of these provide moderate FA yields (ca. 50%).
As a typical metal oxide, manganese oxide (MnOx) has been widely used in the field of catalysis due to the multiple valences of manganese (Mn2+, Mn3+, and Mn4+),25 its oxidative properties, its narrow bandgap, its relatively low toxicity, its abundance in the Earth's crust and its low cost. For instance, Mn-based oxides have been used as substitutes for V2O5 in the selective catalytic reduction (SCR) of nitrogen oxide where they exhibit outstanding activity at low temperatures26 and show high activity for catalytic degradation of volatile organic compounds (VOCs).27 As one of the most important reactions for valorization of biomass, Mn-based oxides are yet to be applied to the synthesis of FA from biomass.
In this work, MnOx nanorods were synthesized by a hydrothermal method and applied to the synthesis of FA from glucose. The relationship between the MnOx structure and catalytic activity in glucose oxidation was elucidated. Key factors of glucose oxidation, such as reaction atmosphere, were studied and a reaction mechanism is proposed.
Experimental section
Materials
D(+)-Glucose (99.9%) and D-fructose (99%) were purchased from Macklin. Co., Ltd. D(+)-Maltose monohydrate (98%) and D(+)-cellobiose (98%) were supplied by Beijing J&K Co., Ltd. Sucrose (AR) was provided by Jiangtian Chemical Co., Ltd. The D(+)-Xylose (98%) was bought from Aladdin Biochemical Technology Co., Ltd. Starch was purchased from Beijing Qitexin Chemical Company. KMnO4 (GR) was provided by Tianjin Kemiou Chemical Reagent Co., Ltd. Hydrochloric acid HCl (GR, 37 wt%) was purchased from Yongfei Chemical Reagent Co., Ltd. Water used in the experiments was ultrapure grade.Preparation of MnOx samples
Manganese oxide catalysts were prepared by a hydrothermal method. Typically, 0.31 g of KMnO4 was dissolved in 29.3 mg of water. Then, 0.7 mL of HCl aqueous solution (37 wt%) was added to the KMnO4 solution and stirred for 30 min. The resulting mixture was transferred into a 50 mL autoclave and heated to specific temperatures (100, 120, 140) °C for 12 h. Next, the mixture was filtered and the solids were washed with water three times and ethanol three times, and dried at 80 °C for 12 h. After that, the resulting solids were calcined in a muffle furnace at a ramp of 1 °C min−1 from room temperature to 500 °C and kept for another 3 h. The obtained samples were marked as MnOx-100, MnOx-120 and MnOx-140, respectively, to denote their hydrothermal treatment temperature.Catalyst characterization
The morphologies of the samples were analyzed on a scanning electron microscope (SEM, SU8010, HITACHI, Japan) operated at 10 kV. X-ray diffraction patterns (XRD) were obtained on a Rigaku Ultima IV in the range of 5° to 90°, operating at 40 kV and 40 mA. X-ray photoelectron spectroscopy (XPS) was performed with a Thermo Fisher EscaLab 250xi spectrometer, which was used to determine the Mn 2p and O 1s binding energies of surface manganese and oxygen species, respectively, with mono Al Kα radiation. The specific surface area of catalysts was calculated by nitrogen adsorption–desorption analysis (Micromeritics ASAP 2020).Catalytic reactions
All reactions were conducted in a 30 mL stainless steel autoclave reactor equipped with a magnetic stirrer. Typically, the autoclave was loaded with 100 mg of glucose, 50 mg of catalyst and 5 mL of ultrapure water. After purging with O2 several times, the reactor was pressurized with O2 to 3 MPa and heated using an electrical heating jacket to the specified temperature (130 to 170) °C with stirring at 1000 rpm. Warning: adiabatic compression and appropriate system components must be considered when using oxygen. At the desired reaction time, the autoclave was removed immediately from the heating jacket, immersed in cold water and cooled down to room temperature. The liquid product was separated by filtration and filtrates were analyzed by ultraperformance liquid chromatography (Waters Acquity UPLC) with a sugar column (Shodex SH1011) and a refractive index (RI) detector. The temperature of the SH1011 column and RI detector was 50 °C and 35 °C, respectively. The mobile phase was 5 mmol L−1 H2SO4 aqueous solution with a flow rate of 0.5 mL min−1. Glucose conversion, FA yield, and FA selectivity were calculated according to the following equations: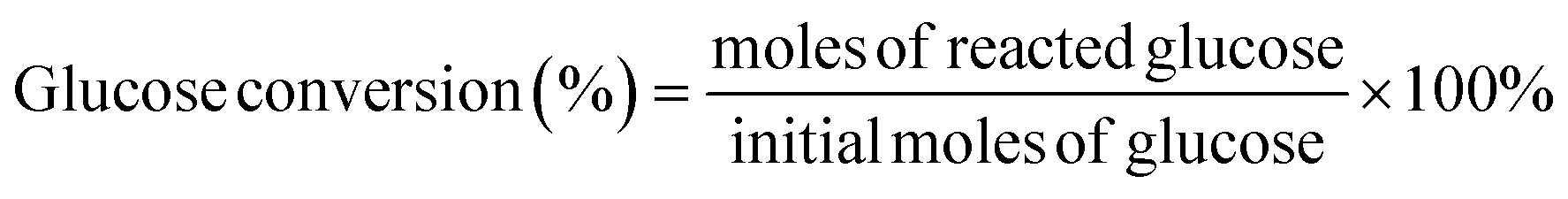 | (1) |
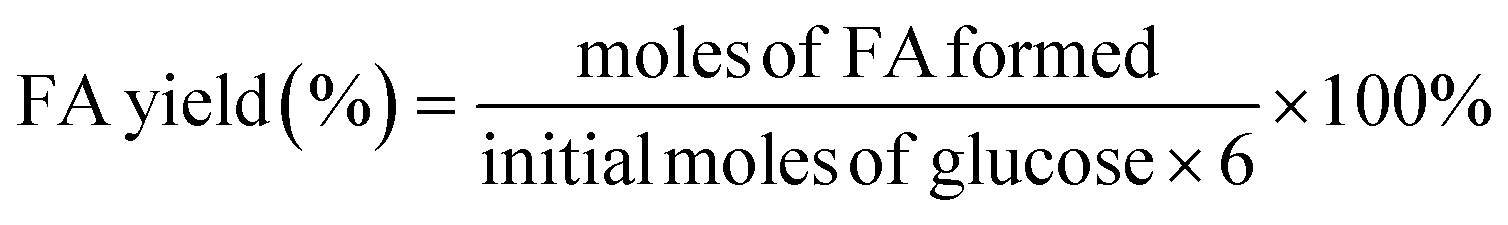 | (2) |
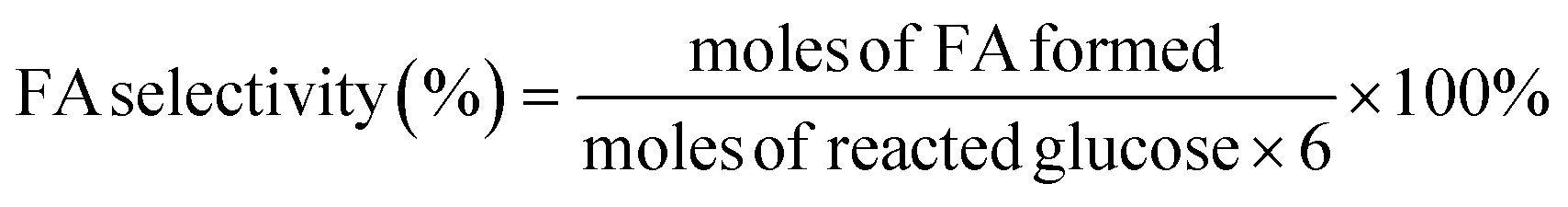 | (3) |
Results and discussion
Catalytic activity of MnOx materials
The catalytic performance of the as-prepared MnOx materials for conversion of glucose to formic acid (FA) in aqueous solution was evaluated (Fig. 1). An FA yield of 50.6% with 57.2% selectivity was afforded when MnOx-100 materials were applied, whereas MnOx-120 and MnOx-140 materials exhibited lower FA yields and selectivities. Based on these results, it was speculated that the hydrothermal reaction temperature for material preparation played an important role in the synthesis procedure and might be responsible for both physical (e.g. porosity, morphology) and chemical structure changes such as crystallinity including the metal valence state and oxygen vacancies. The structure–activity relationship of the reaction mechanism for conversion of glucose to FA in water with MnOx was studied as described next.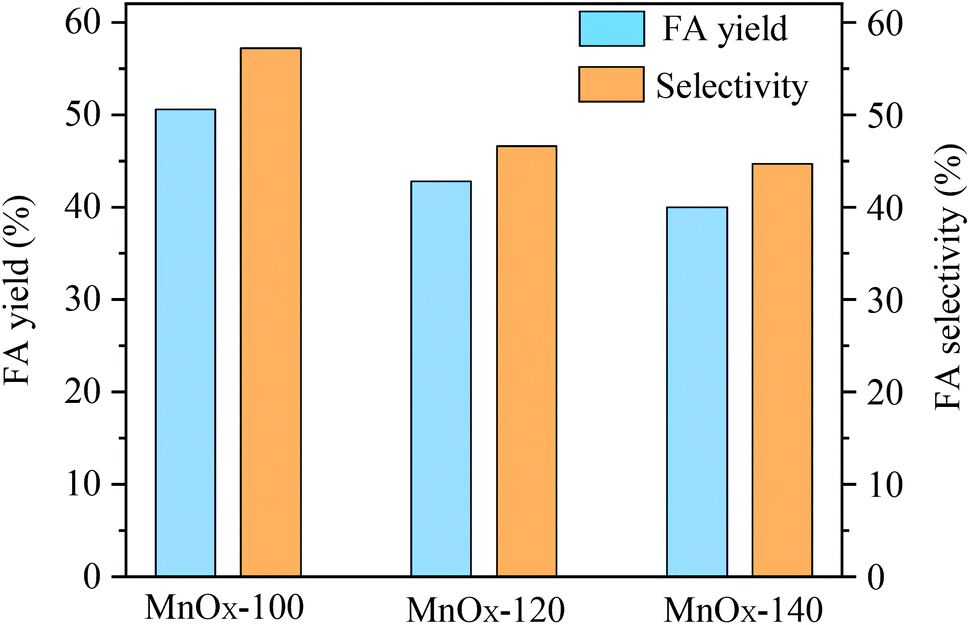 | ||
| Fig. 1 Catalytic oxidation of glucose to FA by various MnOx samples (reaction conditions: 50 mg catalyst, 100 mg glucose, 5 mL H2O, 150 °C, 3 MPa O2, 150 min). | ||
Morphology of MnOx materials
The surface morphology of the as-synthesized MnOx samples was observed by SEM (Fig. 2), where it was found that MnOx prepared at 100 °C exhibited well-grown nanorod structures having uniform diameters (∼84 nm). The formation of MnOx from KMnO4 and HCl by a hydrothermal method probably occurs in two steps:| 2KMnO4 + 16HCl → 2MnCl2 + 2KCl + 5Cl2 + 8H2O | (4) |
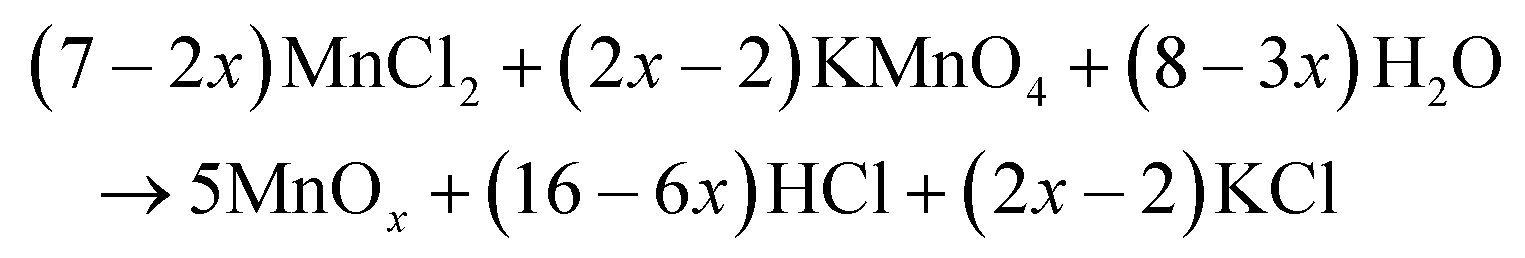 | (5) |
 | ||
| Fig. 2 SEM images of MnOx materials obtained by the hydrothermal treatment of KMnO4–HCl solutions for 12 h at (a) 100 °C (MnOx-100), (b) 120 °C (MnOx-120), and (c) 140 °C (MnOx-140). | ||
First, KMnO4 rapidly reacts with HCl to reduce Mn7+ to Mn2+ and form MnCl2. After that, the generated Mn2+ is oxidized by excess KMnO4 under hydrothermal conditions,28,29 to gradually form Mn3O4, Mn2O3, and MnO2 with increasing temperature. At low reaction temperatures (<120 °C) under hydrothermal conditions, MnOx mainly exists in the form of Mn3O4 and Mn2O3, whereas at high temperatures (>120 °C) MnOx mainly exists in the form of MnO2.
When the temperature in hydrothermal treatment was increased to 120 °C, some nanorods remained, but had increased diameters of around 121.8 nm (MnOx-120) indicating growth in the radial direction primarily (Fig. 2b). A further increase of the temperature used in hydrothermal treatment (140 °C) leads to both radial and longitudinal growth of the regular nanorod structures and aggregation and destruction of their regular structures (Fig. 2c). The SEM results confirmed that lower hydrothermal temperatures promoted the formation of uniform MnOx nanorods with small diameters; however, at the treatment temperature of 80 °C, a small amount of MnOx powders was collected indicating incomplete reduction of KMnO4 to MnOx. Porosity analysis (Table S1†) showed that MnOx-100 had a higher specific surface area (28.0 m2 g−1) than MnOx-120 (16.6 m2 g−1) or MnOx-140 (16.9 m2 g−1). Pores in MnOx are possibly formed by erosion and dissolution of solid surfaces by the generated Cl2.
XRD and XPS analyses of MnOx materials
The crystal phases of the synthesized MnOx catalysts were analyzed by XRD (Fig. 3), in which it was found that all samples had characteristic diffraction peaks at 2θ = 13.8°, 18.1°, 28.8°, 37.7°, 21.1°, and 49.6° attributed to the (110), (200), (310), (121), (301) and (411) planes of MnO2 (JCPDS PDF# 72-1982), respectively. Interestingly, the characteristic diffraction peaks with 2θ at 23.3°, 32.9°, 38.3°, 45.1°, 55.1°, and 64.1° are attributed to the Mn2O3 phase (JCPDS PDF# 41-1442) appearing only in MnOx-100, with a possible reason that crystalline Mn2O3 forms at low temperatures and then transforms into higher valence state MnO2 with an increase in hydrothermal treatment temperature.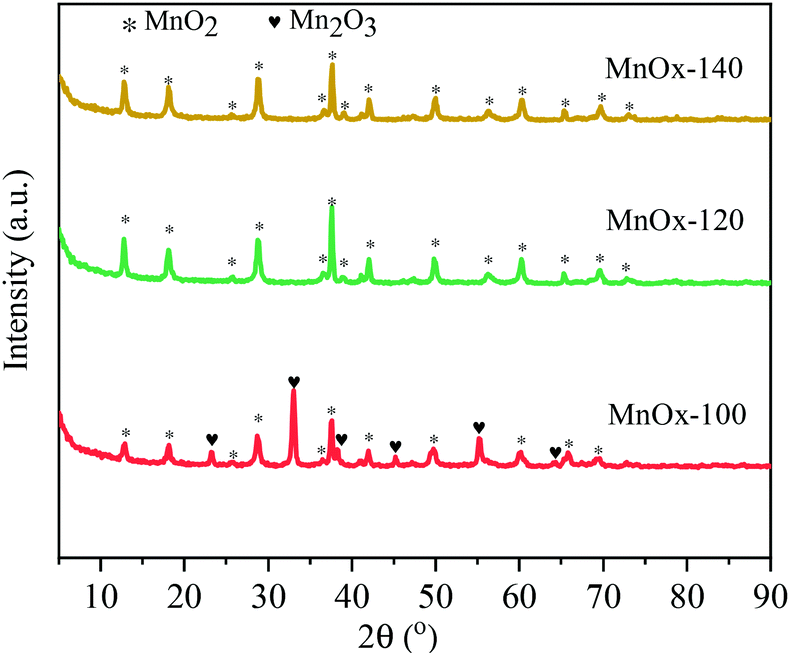 | ||
| Fig. 3 XRD spectra of MnOx solids obtained at 100 °C, 120 °C and 140 °C by hydrothermal treatment of solutions. | ||
The surface state of the MnOx catalysts was studied by XPS (Fig. 4), in which the Mn 2p3/2 spectra of MnOx-100 (Fig. 4a) could be divided into three components with binding energies of 640.2, 641.4, and 642.7 eV, corresponding to Mn2+, Mn3+, and Mn4+ species, respectively.30,31 Other samples prepared at higher hydrothermal temperatures (MnOx-120 and MnOx-140) can only be divided into two components, Mn3+ and Mn4+ as shown in Fig. 4a. Table 1 summarizes the proportion of surface Mn elements, where it can be seen that the ratio of the total low valence Mn (Mn2+ and Mn3+) in MnOx-100 is 69.9%, which is much higher than that of MnOx-120 (57.6%) or MnOx-140 (56.9%). Mixed valence states of Mn in catalysts are important in redox reactions. The mutual conversion among Mn2+, Mn3+, and Mn4+ facilitates electron transport between the substrates and the MnOx catalyst, which improves the catalytic performance.32 Yu et al.33 reported that an increase of low valence Mn ratio (Mn2+ and Mn3+) improves its catalytic activity due to redox reactions. In this work, MnOx-100 had the largest amount of low valence Mn (Mn2+ and Mn3+) and showed the best oxidative performance among the materials evaluated, which infers that the formation of low valence Mn (Mn2+ and Mn3+) in MnOx improves the reaction efficiency of glucose oxidation to FA.
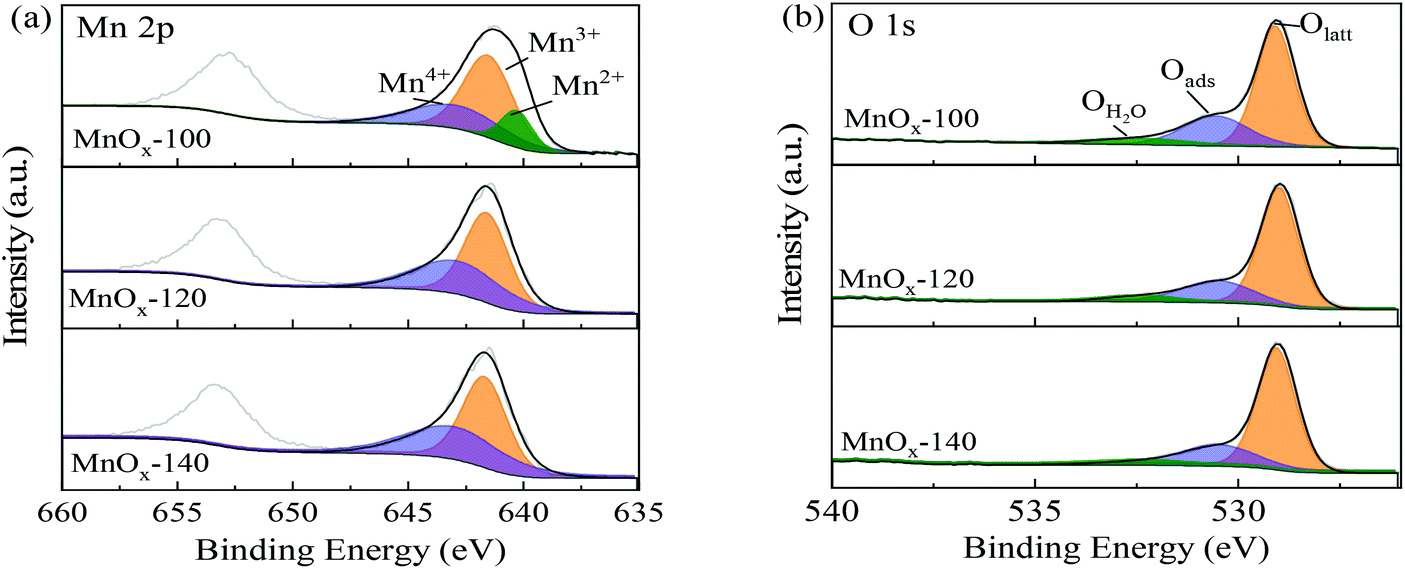 | ||
| Fig. 4 (a) Mn 2p and (b) O 1s XPS spectra of different MnOx samples. | ||
| Catalyst | Mn | O | |||
|---|---|---|---|---|---|
| (Mn2+ + Mn3+)/Mn (%) | Mn4+/Mn (%) | Olatt/Ototal (%) | Oads/Ototal (%) | OH2O/Ototal (%) | |
| MnOx-100 | 69.91 | 30.09 | 65.24 | 27.02 | 7.74 |
| MnOx-120 | 57.64 | 42.36 | 71.60 | 22.45 | 5.95 |
| MnOx-140 | 56.87 | 43.13 | 72.95 | 22.24 | 4.81 |
XPS spectra of O 1s of MnOx samples are shown in Fig. 4b. Peaks at (529.8 ± 0.2) eV, (531.0 ± 0.2) eV, and (532.4 ± 0.2) eV can be assigned to lattice oxygen (OLatt), surface adsorbed oxygen (Oads), and adsorbed molecular water (OH2O), respectively.34 It is widely considered that surface adsorbed oxygen species are more reactive than lattice oxygen in oxidation reactions because of their higher mobility.31,35 The Oads/Ototal value of MnOx-100 was 27%, which was the highest among the prepared MnOx samples, such that MnOx-100 was expected to have higher activity for glucose oxidation than the other materials.
Glucose oxidation with the MnOx-100 catalyst
Synthesis of formic acid (FA) from glucose in water using an MnOx-100 catalyst was performed at reaction temperatures of 130 to 170 °C. As shown in Fig. 5(a), both glucose conversion and FA yield gradually increased with an increase in reaction temperature when the temperature was below 160 °C. At 160 °C, the maximum FA yield (60.9%) was obtained in 150 min with 98.4% glucose conversion. A further increase in reaction temperature to 170 °C led to a slightly lower FA yield (57.4%). The MnOx-100 catalyst before and after the reaction at 170 °C had a similar weight loss ratio (∼14%) as measured by thermal gravimetric analyses (Fig. S1†), showing that the reaction conditions did not seem to affect the stability of the catalysts. Thus, the slightly lower FA yield at the reaction temperature of 170 °C could be ascribed to the thermal degradation of FA.36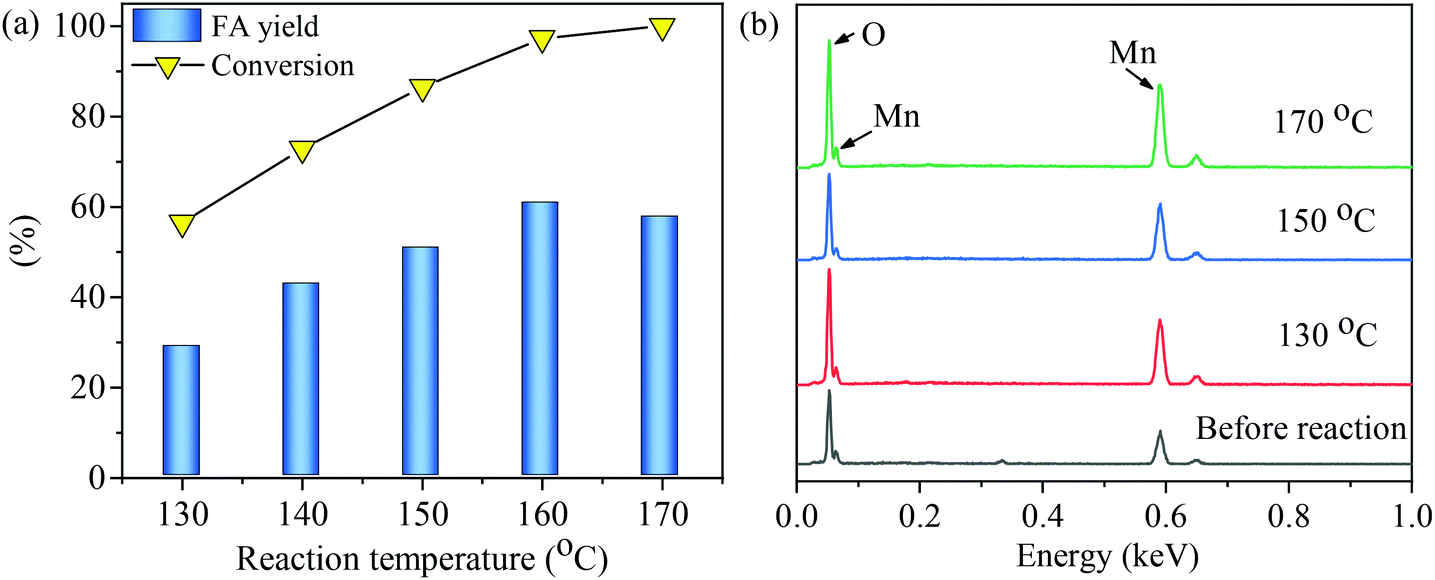 | ||
| Fig. 5 Conversion of glucose to formic acid (FA) with the MnOx-100 catalyst: (a) influence of reaction temperature on the FA yield, (b) EDS of MnOx-100 before and after the reaction at different temperatures (reaction conditions: 50 mg catalyst, 100 mg glucose, 5 mL H2O, 150 min, 3 MPa O2). | ||
The total carbon content of the aqueous solutions after the reaction was determined for reactions at 130 °C, 150 °C, and 170 °C and the carbon balance was determined to be 99%, 88%, and 75%, respectively (Fig. S2†). The carbon balance can be affected by the formation of insoluble carbon-containing by-products, such as humins, that deposit on the catalyst or by the formation of carbon-containing gaseous products, such as CO2. Each of these possibilities is discussed next. Surface elements of the MnOx-100 catalyst were analyzed by EDS after the reaction to investigate whether solid humins were deposited on the material. As shown in Fig. 5b, only Mn and O in the MnOx-100 catalyst could be detected for all solid substrates collected from solutions after the reaction, indicating that insoluble carbon-containing by-products were not adsorbed onto the catalytic materials in the reaction systems. Gaseous products were collected from the glucose–water–MnOx-100 catalytic system after the reaction at 150 °C for 150 min and analyzed by GC-MS, which showed that 11 mol% CO2 yield was obtained assuming that 1 mol glucose gives 6 mol CO2. CO2 was probably formed by decomposition of intermediates, such as glyoxylic acid.37 Thus, the lower carbon balance at higher reaction temperatures can be attributed to the formation of gaseous products rather than compounds adsorbing onto the catalyst active sites.
The influence of initial loading oxygen partial pressure of 0 to 3 MPa on the oxidation of glucose to FA by MnOx-100 at 160 °C was studied to investigate the role of the reaction atmosphere. As shown in Fig. 6(a), the gaseous atmosphere had a great influence on the product distribution. Although 99.3% of glucose was converted using the MnOx-100 catalyst under a nitrogen atmosphere (3 MPa), negligible FA was formed (<5%), whereas the FA yield increased to 54.6% under an oxygen atmosphere (1 MPa). When the reaction was performed under a pure oxygen atmosphere (3 MPa), an FA yield of 60.8% was obtained (Fig. 6).
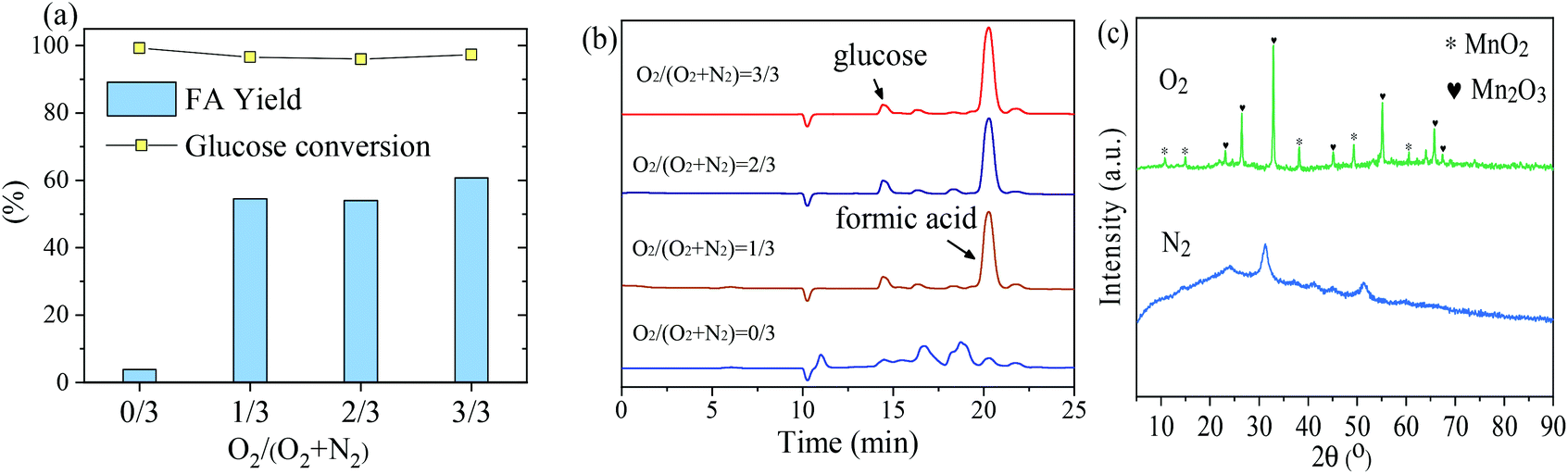 | ||
| Fig. 6 Conversion of glucose to formic acid (FA) with the MnOx-100 catalyst: (a) effect of initial oxygen partial pressure, (b) HPLC analysis of product solutions for oxygen partial pressures of 0 to 3 MPa, (c) XRD patterns of MnOx-100 after the reaction for a O2 or N2 partial pressure of 3 MPa. Reaction conditions: 50 mg catalyst, 100 mg glucose, 5 mL H2O, 160 °C, 150 min. | ||
According to HPLC analyses (Fig. 6b), by-products were formed in the absence of oxygen for conversion of glucose to FA under the given reaction conditions. After the introduction of oxygen into the catalytic system, signals of by-products were significantly reduced and at a retention time of 21.0 min, a peak assigned to FA appeared. The structure of the MnOx-100 catalyst after the reaction under different gaseous atmospheres was analyzed by XRD. MnOx-100 became amorphous (Fig. 6c) after the reaction under a nitrogen atmosphere, while it retained its crystalline structure under an oxygen atmosphere.
The effect of catalyst amount on the FA yield was studied at a reaction temperature of 160 °C and a reaction time of 150 min (Fig. 7a). Glucose conversion and FA yield were negligible (<2%) in the absence of a catalyst; however, when a small amount of MnOx-100 (12.5 mg) was added, the FA yield remarkably increased to 28.9% with 80.8% glucose conversion. As the MnOx-100 amount was increased from 12.5 mg to 75 mg, the FA yield gradually increased to 72.6% (Fig. 7a) and when the catalyst dosage was increased from 75 mg to 100 mg, the FA yield showed a small increase from 72.6% to 74.5%, which infers that the reaction system became saturated at a catalytic dosage of 100 mg.
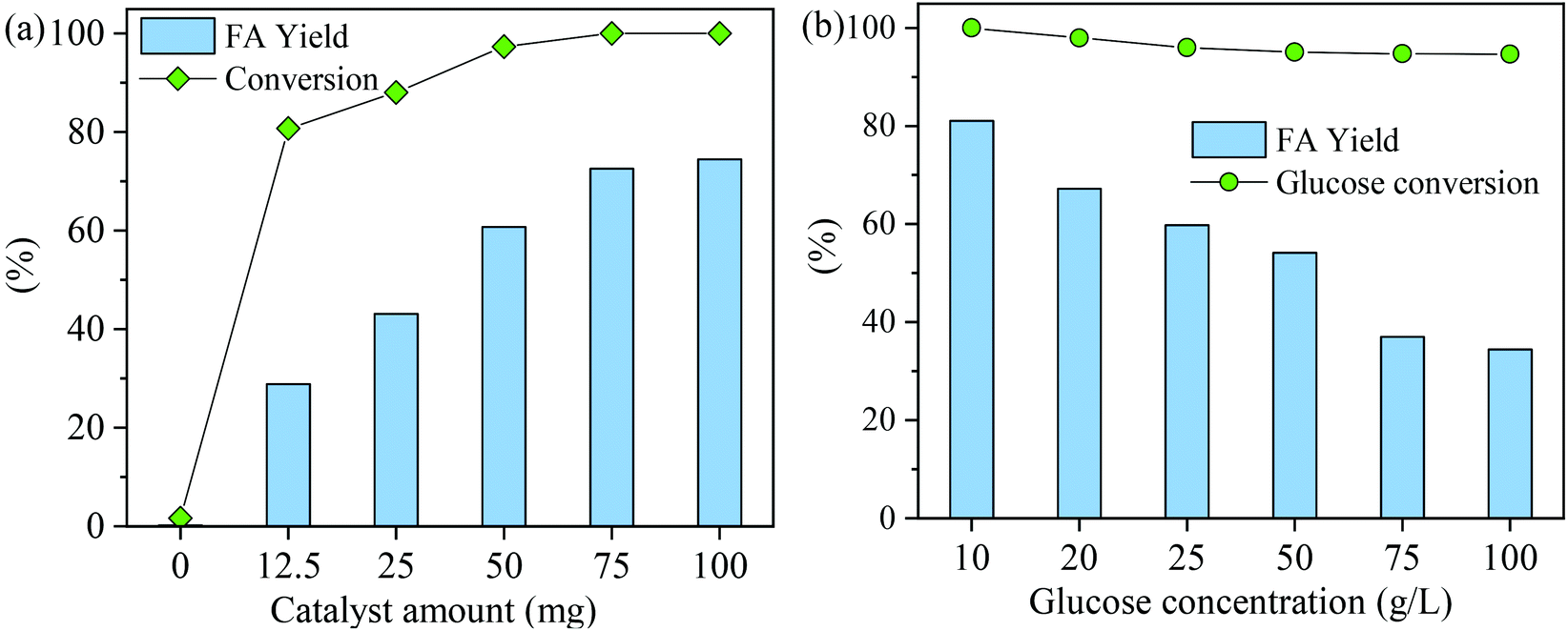 | ||
| Fig. 7 Conversion of glucose to formic acid (FA) with the MnOx-100 catalyst: (a) variation of glucose conversion and FA yield with the catalyst amount (reaction conditions: 100 mg glucose, 5 mL H2O, 160 °C, 3 MPa O2, 150 min) and (b) variation of glucose conversion and FA yield with glucose concentration (reaction conditions: 50 mg catalyst, 5 mL H2O, 160 °C, 3 MPa O2, 150 min). | ||
While considering the initial glucose concentration in the conversion of glucose to FA with the MnOx-100 catalyst (Fig. 7b), glucose could be converted almost completely (>95%) at all initial glucose concentrations, indicating the high activity of the MnOx-100 catalyst. When the glucose loading concentration was 10 g L−1, the FA yield reached 81.1% with 100% glucose conversion at 160 °C in 150 min reaction time. In the reaction system (Fig. 7b), FA yields of 62.1% and 59.8% were obtained when the initial glucose concentration was 20 g L−1 and 25 g L−1, respectively, and FA yields gradually decreased with increasing initial glucose concentration which can be attributed to an insufficient number of active sites for the substrate.
The maximum yields obtained in this work for glucose conversion to FA in aqueous solutions with MnOx-100 were compared with the results obtained with heterogeneous catalysts reported in the literature. As shown in Table 2, most heterogeneous catalysts afford low to moderate FA yields of 9.8 to 37.0%, with CaO23 and Cu-CTAB/MgO38 exhibiting the highest activity for the conversion of glucose to FA among literature studies. Thus, MnOx-100 obtained in this work has higher activity for promoting glucose to FA conversion than the reported heterogeneous catalysts. When the glucose concentration was as high as 50 g L−1, which is the same as that reported for a V2O5 catalyst (Table 2), FA yields obtained with MnOx-100 were much higher than those obtained using V2O5 (Table 2), indicating that the MnOx-100 catalyst has high selectivity for the glucose substrate and is tolerant to the substrate concentration. Production of FA from glucose in water using homogeneous catalysts was compared (Table S2†) in which it was found that all homogeneous catalysts afforded relatively high FA yields (45.0 to 91.3)% and that an FA yield of 91.3% could be obtained when LiOH was used as the catalyst.1 However, in the LiOH–water system, the product is formate (HCOOLi) using LiOH as the catalyst and additional acidulation is required to obtain FA (HCOOH). Furthermore, large amounts of H2O2 (2.5 mmol) are required in LiOH-based catalytic systems that require energy and development for the production of FA and reuse of the catalyst in the homogeneous system. The MnOx-100 developed in this work can be recycled by filtration and reused in subsequent catalytic cycles (Fig. S3†).
| Catalyst | Key reaction conditions | Yield (%) | Ref. | |||
|---|---|---|---|---|---|---|
| Glucose concentration (g L−1) | Oxidant | Temperature (°C) | Time (h) | |||
| a HT: hydrotalcite. b CTAB: the capping agent cetyltrimethylammonium bromide. | ||||||
| MnOx-100 | 10 | O2 (3 MPa) | 160 | 2.5 | 81.1 | This work |
| MnOx-100 | 20 | O2 (3 MPa) | 160 | 2.5 | 67.2 | This work |
| MnOx-100 | 50 | O2 (3 MPa) | 160 | 2.5 | 54.2 | This work |
| SrO | 10 | H2O2 (5.5 mmol) | 90 | 2.0 | 9.8 | 23 |
| BaO | 10 | H2O2 (5.5 mmol) | 90 | 2.0 | 15.0 | 23 |
| Mg–Al/HTa | 36 | H2O2 (2.8 mmol) | 90 | 5.0 | 30.0 | 24 |
| V2O5 | 50 | O2 (3 MPa) | 150 | 3.0 | 30.0 | 39 |
| MgO | 36 | H2O2 (2.8 mmol) | 90 | 5.0 | 37.0 | 24 |
| CaO | 10 | H2O2 (5.5 mmol) | 70 | 0.5 | 52.0 | 23 |
| CuCTAB/MgOb | 18 | H2O2 (2.0 mmol) | 120 | 12.0 | 65.0 | 38 |
The spent MnOx-100 catalyst after three cycles (Fig. S4†) had a similar crystalline structure to that of the fresh MnOx-100 catalyst (Fig. 3). XPS analyses (Fig. S5†) showed that the ratio of total low valence Mn (Mn2+ and Mn3+) slightly decreased from 69.9% in fresh MnOx-100 to 61.2% in the spent MnOx-100 catalyst, which may be caused by oxidation under hydrothermal conditions in an oxygen atmosphere. Correspondingly, the adsorbed oxygen (Oads) ratio of MnOx-100 decreased from 27.0% to 22.2% after three cycles. Adsorbed oxygen (Oads) is responsible for the catalytic activity as discussed in the above section on XRD and XPS analyses. Leaching of Mn species from the MnOx-100 catalyst by the liquid phase was detected by ICP-AES. The amount of Mn leached by the FA-containing aqueous phase between successive runs was 16%, 24% and 38% of Mn for recycling runs 1 to 3, respectively. The stability of the MnOx-100 catalyst in different solvents under reaction conditions (150 °C, 3 MPa O2, 100 min reaction time) was examined. In pure water solutions, negligible Mn losses were observed (96% recovery). However, only 62% of the MnOx-100 catalyst was collected in the FA–H2O mixed solvent (66 mg FA, 5 mL H2O) after hydrothermal treatment at 150 °C for 100 min. Therefore, it could be concluded that Mn was leached from the MnOx-100 catalyst by the formed FA–H2O mixtures which is an issue that can be addressed by anchoring the catalyst to a support.
Isolation of FA using ethyl acetate from the reaction aqueous solutions was carried out by two successive extractions with ethyl acetate (product solution![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 1:3 v/v, room temperature, 10 h extraction time), where it was found that more than 96.4% of FA could be isolated from the product solution.
:ethyl acetate = 1:3 v/v, room temperature, 10 h extraction time), where it was found that more than 96.4% of FA could be isolated from the product solution.
Mechanism of FA production from glucose
To explore the reaction pathway and conversion mechanism, intermediates during the reaction were analyzed (Fig. 8). HPLC analysis showed that the intensity of the glucose peak (14.5 min retention time) decreased with an increase in reaction time from 60 min to 420 min. At 420 min reaction time, the peak of glucose disappeared indicating the complete conversion of glucose. On the other hand, the peak area of FA (retention time, 21 min) gradually increased as the reaction time increased suggesting that the FA yield increased with reaction time. Except for the peaks of the reactant glucose and the product FA, a new peak located at a retention time of 16.2 min appeared at 60 min reaction time and became smaller and finally disappeared with prolonged reaction time. It can be speculated that the substance which appeared at a retention time of 16.2 min in the HPLC chromatogram was a stable intermediate in the conversion of glucose to FA. From the chromatogram of standard substances (Fig. 8a), the unknown peak appearing at 16.2 min retention time closely matched the chromatogram of arabinose. To confirm that arabinose was indeed the intermediate, product solutions obtained from the conversion of glucose catalyzed by MnOx-100 for 60 min reaction time were analyzed by 13C-NMR. As shown in Fig. 8(b), some characteristic peaks of glucose could be observed in the product solutions due to the unreacted substrate and characteristic peaks at 68.6 ppm and 72.1 ppm appeared which can be assigned to arabinose. Based on these analysis results, arabinose was concluded to be the main intermediate in the conversion of glucose to FA catalyzed by MnOx-100. Arabinose was thus used as a feedstock which gave 69.8% FA yield using the MnOx-100 catalyst in water, which is higher than the FA yield (55.7%) with glucose as the feedstock under the same reaction conditions (Table S3†) and consistent with the reaction pathway, since arabinose is an intermediate in the conversion of glucose to FA.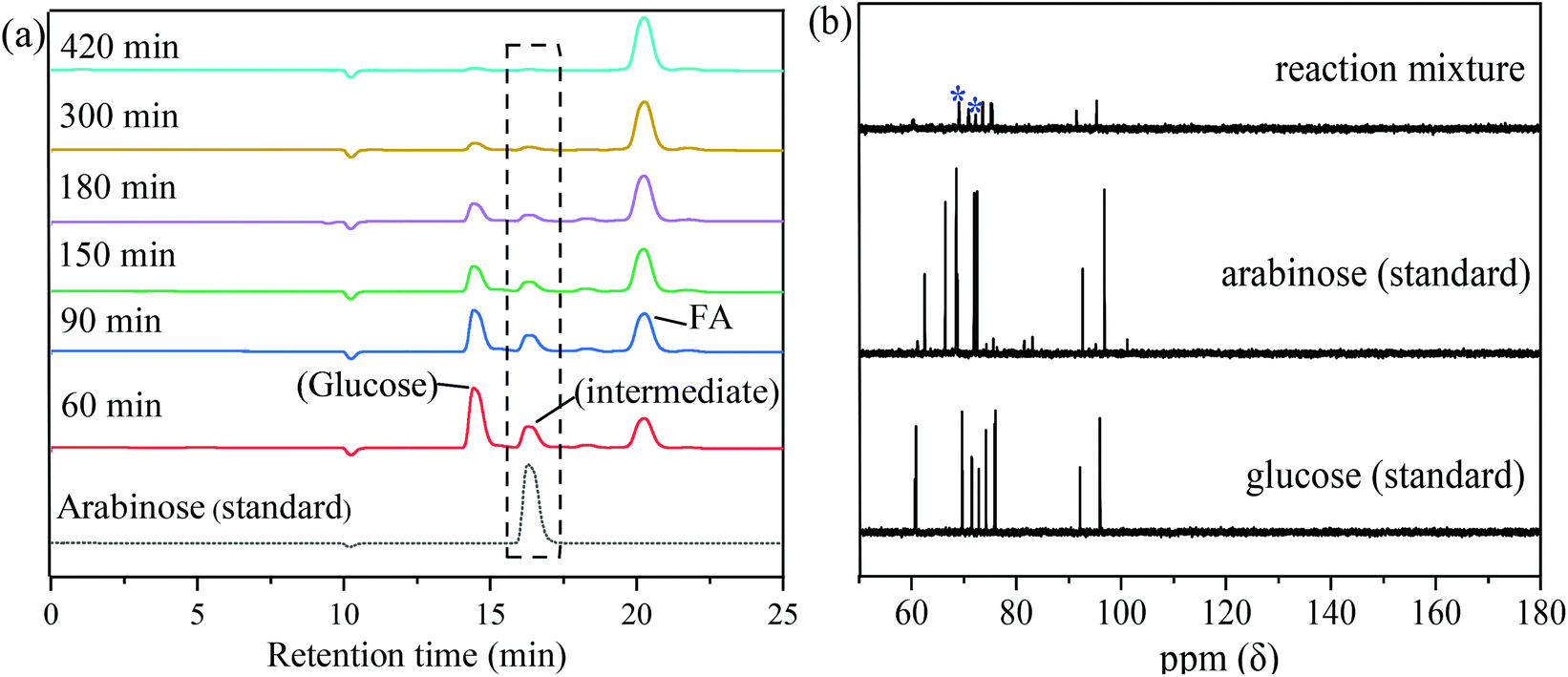 | ||
| Fig. 8 Intermediates identified in the conversion of glucose to formic acid (FA) in water with the MnOx-100 catalyst: (a) HPLC analysis of reaction solutions at different reaction times (reaction conditions: 50 mg catalyst, 100 mg glucose, 5 mL H2O, 150 °C, 3 MPa O2) and (b) 13C-NMR chemical shifts of reaction mixtures with * representing the characteristic peaks of arabinose (reaction conditions: 75 mg catalyst, 1500 mg glucose, 5 mL D2O, 150 °C, 60 min, 3 MPa O2). | ||
In the arabinose reaction system, the carbon balance of the reaction system decreased (Fig. 5b) which means that gaseous products are formed and although some arabinose may be gasified with the MnOx-100 catalyst, other parallel conversion pathways are likely to exist.
Oxidation pathways of glucose to FA promoted by the MnOx-100 catalyst are proposed in Fig. 9. Oxidation reactions over metal catalysts mainly begin via adsorbed O2 molecules on oxygen vacancies (Oads) as noted in the literature.40,41 Oxygen vacancies in MnOx are generated via a charge compensation mechanism and their concentration increases for low valence Mn (Mn2+ and Mn3+).42 Oxidation of glucose to FA undergoes either C1–C2 (α-scission) or C2–C3 (β-scission) bond cleavage as induced by the adsorbed oxygen species (Fig. 9). Since arabinose was detected as the main intermediate, the reaction may first undergo successive C1–C2 bond cleavage (Fig. 9a) in which the breakage of the C1–C2 bond (α-scission) in glucose occurs forming an equivalent of FA and arabinose, followed by repetition of the process until aldose is entirely converted into FA. However, small amounts of CO2 (11 mol% based on glucose) were detected in the reaction system (20 g L−1 glucose, 50 mg catalyst, 150 °C for 150 min), implying that another reaction occurred in parallel to Route I, since Route I does not generate appreciable CO2. In Route II, rupture of C2–C3 (β-scission) through a successive retro-aldol reaction yields three molecules of glycolaldehyde.16,39,43 Simultaneously, the cleavage of C2–C3 (β-scission) via oxidation gives two molecules of glyoxal and one molecule of glycolaldehyde.44 The resulting glycolaldehyde and glyoxal can be further oxidized to glyoxylic acid to afford FA and CO2 in about a 1:1 ratio via an electron-transfer and oxygen-transfer pathway.45 Glyoxylic acid was thus used as a feedstock which gave 73.4% FA yield using the MnOx-100 catalyst in water (Table S3†). Inhibition of reaction Route II could reduce the formation of CO2 and thus improve the FA yield. Namely, the use of mixed solvents could be a practical way to control the FA selectivity. For instance, Guo et al.37 reported that the addition of DMSO (1 vol%) in water greatly reduced the generation of intermediates such as glyoxylic acid that was attributed to the hydrogen bonds formed between –diol groups and DMSO.
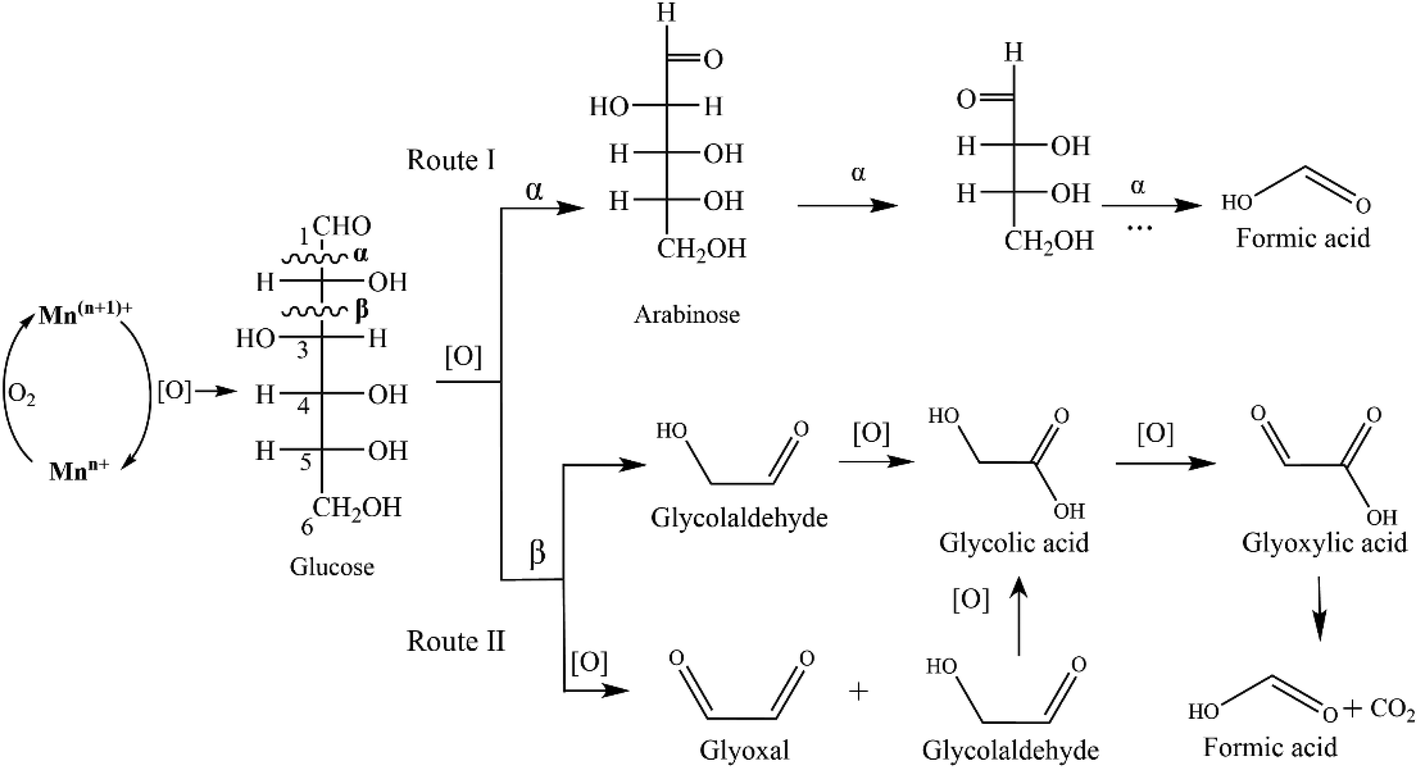 | ||
| Fig. 9 Proposed oxidation pathways of glucose to FA in water for the MnOx-100 catalyst. | ||
After the conversion of glucose to FA, oxidation states of Mn in the catalyst are reduced to their lower valence which can be returned to their initial high valence by oxygen and reused in the next reaction cycle. Based on the above results, it was found that glucose oxidation is more likely to be initiated at the aldehyde end of glucose (C1–C2) instead of at the C5–C6 position, since the –CHO group has much higher activity than the –OH group.1 As support for the proposed mechanism in Fig. 9, Wang et al. reported that the use of –CHO-containing intermediates (formaldehyde and glycolaldehyde) gave much higher FA yields (>90%) using LiOH catalysts than –COOH and –OH-containing intermediates (glycolic acid, sorbitol, and oxalic acid).1
Scope of the reaction system
The scope of the reaction system for FA production from other biomass-derived carbohydrates, including monosaccharides (xylose), disaccharides (cellobiose, sucrose, maltose) and polysaccharides (starch) was investigated. As shown in Fig. 10, small water-soluble carbohydrates including glucose, sucrose, cellobiose, maltose and xylose all gave relatively high FA yields (>50%) using MnOx-100 as the catalyst. It is worth noting that since xylose is the main unit of hemicellulose, primary carbohydrates (cellulose and hemicellulose) in waste biomass (e.g. straw) can potentially be converted into FA with the MnOx-100 catalyst. For a complex polysaccharide (starch), 20.8% FA yield was obtained using the MnOx-100 catalyst, which means that acid hydrolysis pretreatment is most likely needed for the conversion of complex polysaccharides (starch and cellulose) into FA. The combination of MnOx-100 with an acidic catalyst such as Nb2O5 may be another way for one-step conversion of complex polysaccharides to FA.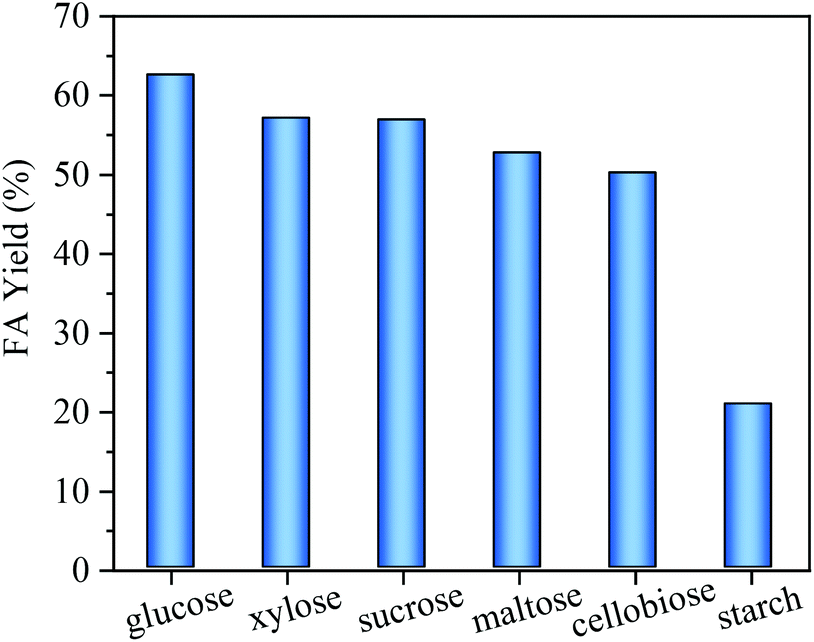 | ||
| Fig. 10 Formic acid (FA) yields from carbohydrates with the MnOx-100 catalyst in water (reaction conditions: 50 mg catalyst, 100 mg substrate, 5 mL H2O, 160 °C, 150 min, 3 MPa O2). | ||
Conclusions
In this work, a vanadium(V)-free heterogeneous catalyst, MnOx, was synthesized via a hydrothermal method and applied to the production of formic acid from glucose in water. The MnOx-100 material prepared under hydrothermal conditions at 100 °C exhibited the highest catalytic activity for conversion of glucose to formic acid in water (81.1% yield) among the catalyst preparation conditions examined, which can be attributed to higher ratios of lower valence Mn (Mn2+ and Mn3+) and more adsorbed oxygen species than MnOx catalysts prepared under other conditions. Mechanistic studies showed that MnOx-100 promoted the oxidation of glucose to formic acid in parallel pathways via an α-scission reaction route with arabinose being an intermediate and via a β-scission reaction route with glyoxylic acid being the main intermediate, while the rupture of α-scission (C1–C2 bond cleavage) was found to be the major pathway for the developed MnOx-100 catalyst.Author contributions
Jialu Li: methodology, data curation, and writing – original draft. Richard Lee Smith Jr.: methodology, investigation, and writing – reviewing and editing. Siyu Xu, De Li and Jirui Yang: data curation and methodology. Keqiang Zhang: conceptualization, supervision, and writing – reviewing and editing. Feng Shen: conceptualization, methodology, supervision, writing – reviewing, and funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, no. 21706139) and the Central Public-interest Scientific Institution Basal Research Fund (Agro-Environmental Protection Institute, Ministry of Agriculture and Rural Affairs, 2021-jbkyywf-sf).References
- C. Wang, X. Chen, M. Qi, J. Wu, G. Gözaydın, N. Yan, H. Zhong and F. Jin, Green Chem., 2019, 21, 6089–6096 RSC.
- L. Li, F. Shen, R. L. Smith and X. Qi, Green Chem., 2017, 19, 76–81 RSC.
- S. Guo, Q. Fang, Z. Li, J. Zhang, J. Zhang and G. Li, Nanoscale, 2019, 11, 1326–1334 RSC.
- S. Yamaguchi, S. Fujita, K. Nakajima, S. Yamazoe, J. Yamasaki, T. Mizugaki and T. Mitsudome, Green Chem., 2021, 23, 2010–2016 RSC.
- M. Negro, C. Alvarez, I. Ballesteros, I. Romero, M. Ballesteros, E. Castro, P. Manzanares, M. Moya and J. Oliva, Bioresour. Technol., 2014, 153, 101–107 CrossRef CAS PubMed.
- S. Sun, L. Zhao, J. Yang, X. Wang, X. Qin, X. Qi and F. Shen, ACS Sustainable Chem. Eng., 2020, 8, 7059–7067 CrossRef CAS.
- L. Zhang, G. Xi, Z. Chen, J. Dong, H. Yu and X. Wang, Chem. Eng. J., 2017, 307, 868–876 CrossRef CAS.
- Y. Luo, Q. Yang, W. Nie, Q. Yao, Z. Zhang and Z. H. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 8082–8090 CrossRef CAS PubMed.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, Ullmann's encyclopedia of industrial chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2016 Search PubMed.
- L. Pan, Z. Shen, L. Wu, Y. Zhang, X. Zhou and F. Jin, J. Zhejiang Univ., Sci., A, 2010, 11, 613–618 CrossRef CAS.
- https://www.mordorintelligence.com/industry-reports/formic-acid-market , accessed 1 September 2021.
- X. Chen, Y. Liu and J. Wu, Mol. Catal., 2020, 483, 110716 CrossRef CAS.
- C. Li, H. Bai, Y. Lu, J. Bian, Y. Dong and H. Xu, J. Cleaner Prod., 2018, 188, 1004–1017 CrossRef CAS.
- F. Shen, R. L. Smith Jr., J. Li, H. Guo, X. Zhang and X. Qi, Green Chem., 2021, 23, 1536–1561 RSC.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Green Chem., 2008, 10, 612–615 RSC.
- R. Wölfel, N. Taccardi, A. Bösmann and P. Wasserschei, Green Chem., 2011, 13, 2759–2763 RSC.
- J. Reichert and J. Albert, ACS Sustainable Chem. Eng., 2017, 5, 7383–7392 CrossRef CAS.
- M. Niu, Y. Hou, S. Ren, W. Wang, Q. Zheng and W. Wu, Green Chem., 2015, 17, 335–342 RSC.
- Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang and Y. Wang, ChemSusChem, 2014, 7, 1557–1567 CrossRef CAS.
- Y. Pu, X. Xie, W. Jiang, L. Yang, X. Jiang and L. Yao, Chin. Chem. Lett., 2020, 31, 2549–2555 CrossRef CAS.
- Z. Liu, Y. Liu, B. Chen, T. Zhu and L. Ma, Catal. Sci. Technol., 2016, 6, 6688–6696 RSC.
- L. Cheng, H. Liu, Y. Cui, N. Xue and W. Ding, J. Energy Chem., 2014, 23, 43–49 CrossRef CAS.
- A. Takagaki, W. Obata and T. Ishihara, ChemistryOpen, 2021, 10, 954–959 CrossRef CAS PubMed.
- R. Sato, H. Choudhary, S. Nishimura and K. Ebitani, Org. Process Res. Dev., 2015, 19, 449–453 CrossRef CAS.
- I. Kruk, P. Zajdel, W. van Beek, I. Bakaimi, A. Lappas, C. Stock and M. A. Green, J. Am. Chem. Soc., 2011, 133, 13950–13956 CrossRef CAS PubMed.
- G. He, M. Gao, Y. Peng, Y. Yu, W. Shan and H. He, Environ. Sci. Technol., 2021, 55, 6995–7003 CrossRef CAS.
- P. Wu, X. Jin, Y. Qiu and D. Ye, Environ. Sci. Technol., 2021, 55, 4268–4286 CrossRef CAS PubMed.
- X. Huang, D. Lv, H. Yue, A. Attia and Y. Yang, Nanotechnology, 2008, 19, 225606 CrossRef PubMed.
- K. Chen, Y. Dong Noh, K. Li, S. Komarneni and D. Xue, J. Phys. Chem. C, 2013, 117, 10770–10779 CrossRef CAS.
- Y. Wang, S. Aghamohammadi, D. Li, K. Li and R. Farrauto, Appl. Catal., B, 2019, 244, 438–447 CrossRef CAS.
- V. Santos, M. Pereira, J. Orfao and J. Figueiredo, Appl. Catal., B, 2010, 99, 353–363 CrossRef CAS.
- T. Wang, S. Chen, H. Wang, Z. Liu and Z. Wu, Chin. J. Catal., 2017, 38, 793–803 CrossRef CAS.
- D. Yu, Y. Liu and Z. Wu, Catal. Commun., 2010, 11, 788–791 CrossRef CAS.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 18642–18642 CrossRef CAS PubMed.
- J. Liu, H. Cheng, J. Tan, B. Liu, Z. Zhang, H. Xu, M. Zhao, W. Zhu, J. Liu and Z. Zhao, J. Mater. Chem. A, 2020, 8, 6717–6731 RSC.
- T. Lu, Y. Hou, W. Wu, M. Niu and Y. Wang, Fuel Process. Technol., 2018, 171, 133–139 CrossRef CAS.
- Y. Guo, S. Li, Y. Sun, L. Wang, W. Zhang, P. Zhang, Y. Lan and Y. Li, Green Chem., 2021, 23, 7041–7052 RSC.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., B, 2015, 162, 1–10 CrossRef CAS.
- J. Li, D. J. Ding, L. Deng, Q. X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS PubMed.
- L. Zhu, J. Wang, S. Rong, H. Wang and P. Zhang, Appl. Catal., B, 2017, 211, 212–221 CrossRef CAS.
- F. Wang, H. Dai, J. Deng, G. Bai, K. Ji and Y. Liu, Environ. Sci. Technol., 2012, 46, 4034–4041 CrossRef CAS PubMed.
- X. Yang, X. Yu, M. Jing, W. Song, J. Liu and M. Ge, ACS Appl. Mater. Interfaces, 2019, 11, 730–739 CrossRef CAS.
- J. Xu, H. Zhang, Y. Zhao, Z. Yang, B. Yu, H. Xu and Z. Liu, Green Chem., 2014, 16, 4931–4935 RSC.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- M. Niu, Y. Hou, S. Ren, W. Wu and K. N. Marsh, Green Chem., 2015, 17, 453–459 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03637h |
| This journal is © The Royal Society of Chemistry 2022 |