
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid-assisted grinding/compression: a facile mechanosynthetic route for the production of high-performing Co–N–C electrocatalyst materials†
Akmal
Kosimov
a,
Gulnara
Yusibova
a,
Jaan
Aruväli
b,
Päärn
Paiste
b,
Maike
Käärik
a,
Jaan
Leis
a,
Arvo
Kikas
c,
Vambola
Kisand
c,
Krišjānis
Šmits
d and
Nadezda
Kongi
 *a
*a
aInstitute of Chemistry, University of Tartu, Tartu 50411, Estonia. E-mail: nadezda.kongi@ut.ee
bInstitute of Ecology and Earth Sciences, University of Tartu, Tartu 50411, Estonia
cInstitute of Physics, University of Tartu, Tartu 50411, Estonia
dInstitute of Solid State Physics, University of Latvia, LV-1063 Riga, Latvia
First published on 10th December 2021
Abstract
Worldwide implementation of energy conversion devices such as metal–air batteries and fuel cells needs an innovative approach for the sustainable design of noble metal-free electrocatalysts. A key factor to be considered is the industry-scale production method, which should be cost and energy-effective, and environmentally friendly. A novel solid-phase-based methodology is introduced herein as a new approach for the mechanosynthesis of M–N–C-type catalysts. This method employs low-cost commercially available materials, is time and energy-efficient, results in no solvent/toxic waste and does not require a complex post-synthetic treatment. The liquid-assisted grinding/compression approach yielded a series of meso- and microporous Co–N–C catalysts, with excellent bifunctional activity towards oxygen evolution and reduction reactions. In-depth physical characterization confirmed that all NaCl-supported catalysts possess cross-linked sheet-like mesoporous carbon structures with high exposure of catalytically active sites. This study provides a new avenue for the large-scale production of high-performance and low-cost M–N–C materials via energy-effective and environmentally sustainable synthetic protocols.
Introduction
In recent years, fuel cells, metal–air batteries, and water electrolysers have been striking as excellent alternative energy conversion and storage devices.1 However, the performance of these systems depends on sluggish kinetics of the key electrochemical processes – oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER), which determine their overall performance and significantly impede the advancements of these energy systems. Nevertheless, despite the drawbacks, electrochemical energy devices, with their zero-emission operation, promise to be a good candidate for replacing fossil fuel-based energy processes.2,3One of the key limiting factors for the commercialization of fuel-cells and metal–air batteries is the relatively high cost caused by the use of platinum group metals (PGMs), which are applied to effectively overcome the ORR/OER overpotentials.4,5 Despite the advances in decreasing their cost, the PGM-based catalyst still constitutes the major part of the fuel-cell/metal–air battery price.6 Thus, it is necessary to find a replacement for noble-metal-based catalysts to make these energy systems more accessible for widespread applications.
Among the recently developed non-noble-metal-based materials, transition metal–nitrogen-doped carbon (M–N–C) materials demonstrate catalytic activity comparable with commercially available catalysts.7,8 Their low cost and a wide variety of sources make them excellent candidates for replacing Pt-based catalysts.9–13 ORR electroactivity of M–N–C materials is often attributed to various single-atom sites, where metal is coordinated to nitrogen (M–Nx), whereas OER activity is generally enhanced by a presence of nanoparticles in pure metallic form.14–18 Furthermore, to improve catalytic efficiency, access to the catalytically active sites should also be ensured by increasing the porosity of the carbon materials. Thus, recent advances in this field involved the application of templating agents, which, upon their removal, are expected to leave vacant pores and enhance the porosity.19–21 As the templated catalyst precursor materials usually object to carbonization, the templating agent should withstand elevated temperatures. Hence preference is given to cheap and environmentally sustainable inorganic sacrificial templates such as (SiO2, MgO, Mg(OH)2, NaCl).22–26 Efficiency of the template-assisted method was proven by recent studies, which show that the resulting catalysts exhibited greater catalytic activity compared to their non-templated analogs.23,26,27
Despite the extensive research in this field, fabrication of M–N–C materials heavily relies on solvent-based techniques (precipitation, deposition–precipitation, hydrothermal treatments, and impregnation), which are considered from the industrial perspective laborious and complex, and unsustainable to scale up.28 On the other hand, solid-phase synthetic protocols are gaining traction as green and facile alternatives to solution-based methods.29,30 Mechanochemistry, where kinetic force is utilized in the form of ball milling (grinding) or compression, is becoming a major method for fabricating various materials.31–33 Recent examples of mechanochemistry in the domain of materials include the synthesis of MOFs,31–33 nanoparticles,34 organic molecules,35etc. Moreover, mechanosynthesis possesses numerous attractive hallmarks such as more straightforward, easy-to-scale, and affordable techniques, reduced formation of hazardous substances, and minimized energy requirement, which is considered advantageous from an industrial point of view.36,37 Nevertheless, to the best of our knowledge, there were hardly any attempts to study mechanochemical reactions to produce M–N–C-type materials. Therefore, our research aimed to develop an optimal mechanosynthetic protocol to improve the fabrication efficiency of M–N–C materials.
Herein, we report a liquid-assisted grinding/compression strategy to produce a highly active and stable bifunctional Co–N–C catalyst using green and cost-effective template-assisted mechanochemistry techniques. The electrocatalytic behavior of produced materials is comparable with commercially available ones, and our original mechanosynthetic protocols have the potential to become a stepping stone to industrial manufacturing of PGM-free catalysts.
Experimental section
General
Tripyridyl triazine (TPTZ) (≥98%) and Co(NO3)2·6H2O were purchased from Aldrich and used as received. MilliQ water was used to prepare all solutions. NaCl crystals were pulverized in a ball mill to achieve an average crystallite size of 10–50 μm. Milling was conducted in a ball mill (Fritsch Planetary Mill Pulverisette 7) using 12 mL zirconium oxide grinding bowls and 5 mm diameter zirconium oxide milling balls. Furnace (Carbolite Gero EST 12/300B) was used for the carbonization of catalyst precursors.Template-assisted mechanosynthesis of Co–N–C catalysts
The green mechanosynthesis method for producing the series of Co–N–C catalysts is schematically depicted in Fig. 1 and described in details below. For the synthesis of catalyst precursors, liquid-assisted grinding and compression (LAG and LAC) methods and their combinations were applied.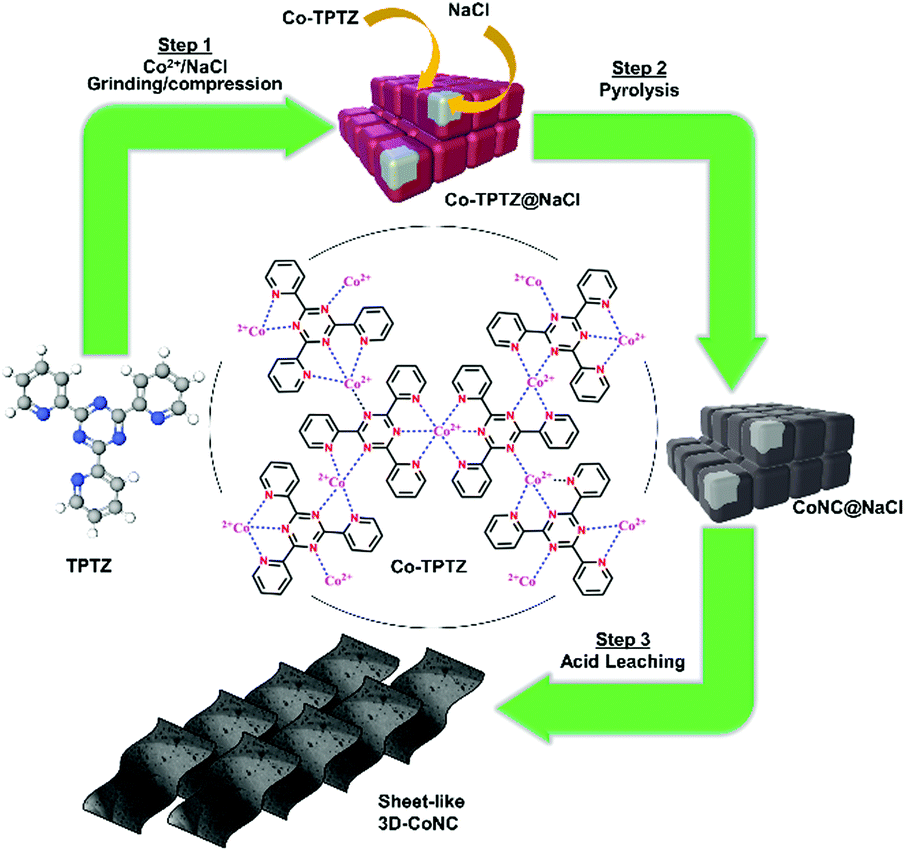 | ||
| Fig. 1 Schematic diagram of green mechanochemical approach toward synthesis of Co–N–C-type catalysts. | ||
Physical characterization
Electrochemical characterization
All electrochemical measurements were carried out in a five-neck electrochemical cell. A reversible hydrogen electrode (RHE) served as a reference electrode and glassy carbon (GC) rod as a counter electrode. Potentials were applied using Autolab PGSTAT128N potentiostat/galvanostat (Metrohm Autolab B.V., The Netherlands), controlled by Nova 2.1.4 software. Rotating disk electrode (RDE) measurements were carried out by OrigaTrod rotating disk electrode linked to OrigaBox speed controller. The electrolyte solution was prepared by dissolving KOH pellets (purity ≥85%, Sigma-Aldrich) in Milli-Q water, and obtained 0.1 M KOH solution was saturated with pure O2 (99.999%, Linde Gas) and deaerated with Ar gas (99.999%, Linde Gas) prior ORR/OER experiments.Before modification, GC disk electrodes (OrigaTip GC disks with a diameter of 5 mm) were polished to a mirror finish with alumina slurries (1 and 0.3 μm, Buehler). They were sonicated in both isopropanol and Milli-Q water for 5 min to clean polishing residue. The catalyst suspension was prepared by mixing 5 mg of catalyst powder, 5 μl of Nafion ionomer solution (5 wt%, Sigma-Aldrich), and 495 μl of 2-propanol and homogenized by sonication for 1 hour. The catalyst ink was deposited onto a clean GC surface to yield a catalyst loading of 0.5 mg cm−2. The ORR measurements were performed in O2 saturated KOH solution (0.1 M) at a scan rate of 10 mV s−1 and at various electrode rotation speeds. The RDE data was analyzed by the Koutecky–Levich (K–L) equation:32
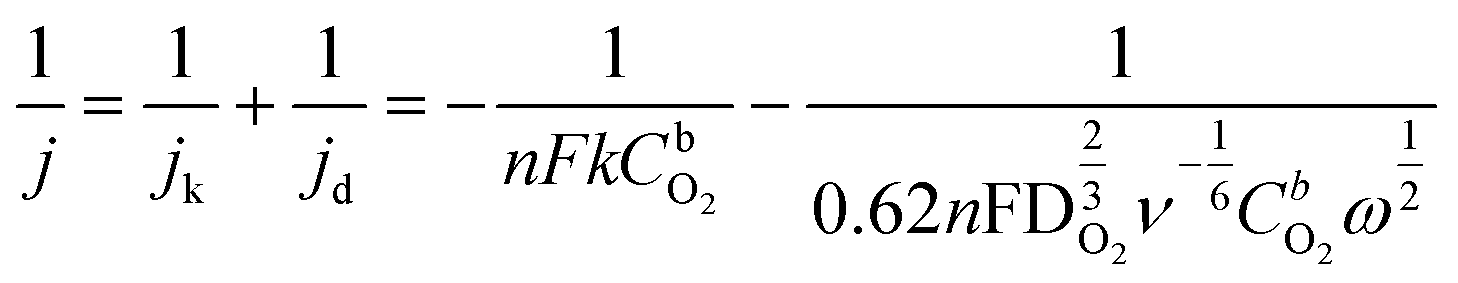
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and n is the number of electrons transferred per oxygen molecule. k is the electrochemical rate constant for O2 reduction, CbO2 is the concentration of O2 (1.22 × 10−6 mol cm−3 in 0.1 M KOH),33 while DO2 shows the diffusion coefficient of O2 (1.93 × 10−5 cm2 s−1 in 0.1 M KOH),33ω represents the rotation rate of the electrode (rad s−1), and ν is the kinematic viscosity of the solution (0.01 cm2 s−1).34
485 C mol−1), and n is the number of electrons transferred per oxygen molecule. k is the electrochemical rate constant for O2 reduction, CbO2 is the concentration of O2 (1.22 × 10−6 mol cm−3 in 0.1 M KOH),33 while DO2 shows the diffusion coefficient of O2 (1.93 × 10−5 cm2 s−1 in 0.1 M KOH),33ω represents the rotation rate of the electrode (rad s−1), and ν is the kinematic viscosity of the solution (0.01 cm2 s−1).34
The OER performances of the as-prepared Co–N–C materials were investigated in a 0.1 M KOH solution with a scan rate of 10 mV s−1. The ORR/OER durability was investigated by a chronoamperometry technique in 0.1 M O2/Ar-saturated KOH electrolyte at a constant potential of 0.6/1.6 V vs. RHE and the current response collected against time over a period of 30000 s was recorded at an electrode rotation speed of 1600 rpm. An iR correction was applied to all initial electrochemical data.
Results
The synthesis of M–N–C catalysts often employs organic molecules to ensure sufficient carbon and nitrogen content in the catalyst matrix.15,16,38,39 In this work, 2,4,6-tris(2-pyridyl)-1,3,5-triazine (TPTZ) was implemented both as C/N-source and complexation agent to coordinate towards Co2+ ions and form the Co-TPTZ complex. Recent reports show that using Co-TPTZ precursor is beneficial for good catalytic activity, which is assigned to the effective formation of Co–Nx species. Moreover, coordinative bonding between the organic molecules stabilizes Co-ions and inhibits their agglomeration during pyrolysis.24 Additionally, the NaCl-template-assisted approach was implemented in this work, which was reported to effectively stabilize Co-TPTZ-complex during heat treatment, increase the accessibility of catalytically active sites, and improve porosity.24 Moreover, NaCl crystallites act as a grinding aid, facilitating the milling, improving materials dispersion, and providing finer particles and narrower particle size distribution.40 Previously described template-assisted procedures provide highly active catalysts, however these methodologies implement solvent-based approach and rather complex workup. Thus, mechanochemistry was introduced to improve the sustainability and cost/energy-efficiency.23,24 Synthetic conditions and material/time-cost are summarized and compared in Table S1,† which indicates that the introduction of mechanochemical protocol and optimization of post-synthetic treatment yielded faster synthesis and less sophisticated operation, thus, three times less time and energy consumption.To assess the effect of various mechanosynthetic procedures on the catalytic activity, three different NaCl-templated (3D) Co–N–C catalysts were synthesized. Procedures involved liquid-assisted grinding (LAG) and compression (LAC) and a combination of both (LAG–LAC). The resulting catalyst materials were designated as 3D-CoNC-LAG, 3D-CoNC-LAC, and 3D-CoNC-LAG-LAC. Nontemplated catalyst CoNC-LAG was prepared via ball milling for comparative purposes. Upon mechanosynthesis, the reaction mixture changes color from yellow, and the Co-TPTZ complex appears as a pale-red powder, indicating that the TPTZ molecule is coordinated to Co2+. The complex formation was also confirmed by the comparative powder X-ray diffraction analysis, where the product pattern did not match any of the starting materials (Fig. S1†).
Microscope pictures (Fig. 2a) demonstrate even integration of micro-sized NaCl crystallites into the framework of the Co-TPTZ complex or the complex evenly surrounding the NaCl crystallites. After the carbonization of NaCl-templated precursors, materials show visible inclusion of NaCl crystallites into the framework of carbon catalysts (Fig. 2c). In contrast, Co-TPTZ-LAG demonstrates a large amount of metallic-Co upon carbonization and appears as dense bulky carbon blocks after acid leaching. In contrast, NaCl-supported catalysts after removing NaCl and acid treatment yielded homogeneous porous carbon materials. This difference after the heat treatment can be attributed to the stabilization effect of NaCl during the pyrolysis, which was previously reported in solution-based syntheses.23,24 Remarkably, our study indicates that the same effect is maintained in green solid-phase-based mechanosynthesis.
 | ||
| Fig. 2 Microscope pictures of (a) catalyst precursors, (b) non-washed/leached carbonized precursors; (c) CoNC catalysts. | ||
The PXRD analysis also confirms the above-mentioned stabilizing effect. The spectra shown in Fig. 3a are acquired after the carbonization of Co-TPTZ-LAG and Co-TPTZ@NaCl-LAG before the acid leaching. Spectra demonstrate the presence of larger amounts of various Co-particles in non-leached CoNC-LAG (Co-cubic and Co3O4) compared to the NaCl-supported carbon material – 3D-CoNC-LAG, which shows only the peaks attributed to Co-cubic.
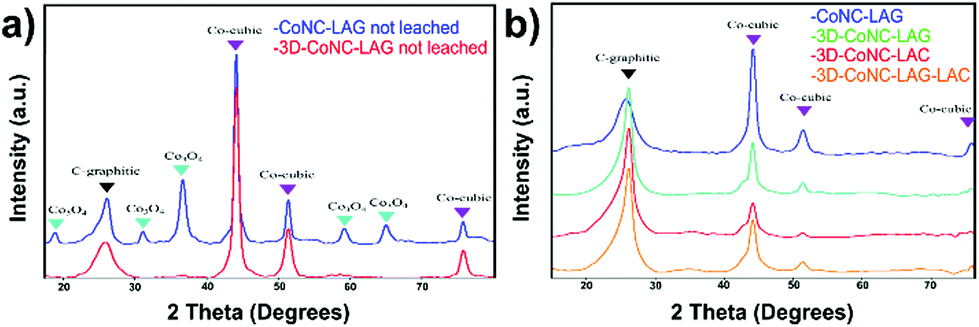 | ||
| Fig. 3 (a) PXRD pattern of CoNC-LAG and 3D-CoNC-LAG before leaching, (b) PXRD patterns of synthesized catalysts after leaching. | ||
Upon the leaching (Fig. 3b), all four products mainly exhibit four strong peaks at 26.5° (graphitic-C) and 44.1°, 51.7°, and 75.9° (Co-cubic), proving that the amount of agglomerated cobalt can be effectively decreased by acid treatment. Furthermore, the pattern of CoNC-LAG shows a distinctly higher intensity of metallic-Co, indicating that its amount is higher than in NaCl-supported catalysts. Overall, the decreased formation of Co-particles in NaCl-supported catalysts can be assigned to the template-induced complex stabilization effect during the pyrolysis, as it was described previously.23,24
The TEM morphology analyses of prepared materials represented in Fig. 4 support the results of XRD analysis, showing that CoNC-LAG visually accommodates more metallic nanoparticles. CoNC-LAG appears as a large bulky microscale particle, which serves as another evidence of the agglomeration of catalyst particles and packing of carbon material, resulting in decreased exposure of active sites when carbonized without NaCl crystallites.
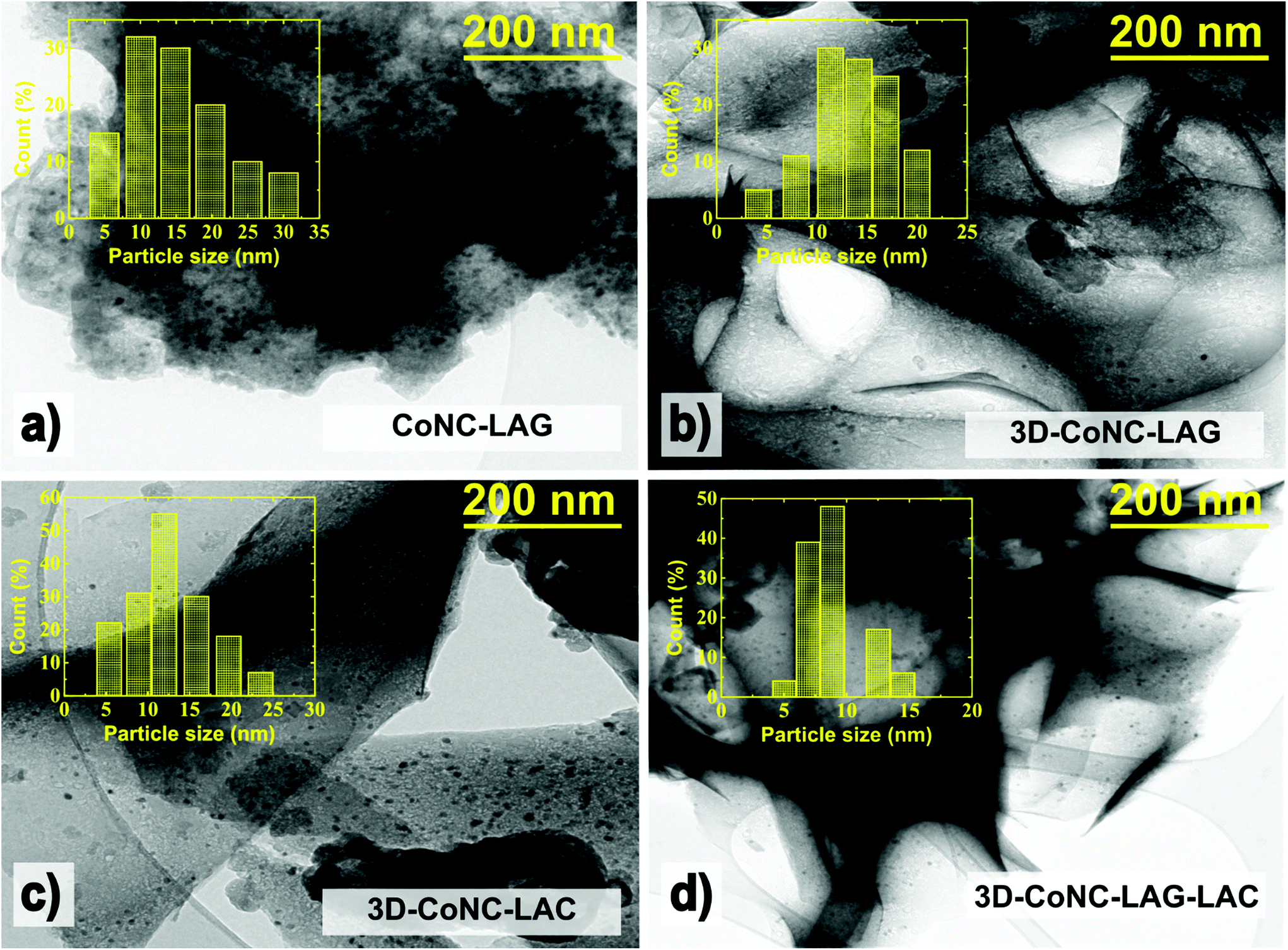 | ||
| Fig. 4 TEM images and particle size distribution of (a) CoNC-LAG, (b) 3D-CoNC-LAG, (c) 3D-CoNC-LAC, and (d) 3D-CoNC-LAG-LAC. | ||
On the other hand, the three catalysts, CoNC-LAG, 3D-CoNC-LAC, and 3D-CoNC-LAC, made by the NaCl-templating method, demonstrate a cross-linked sheet-like structure, which can be seen in their STEM and TEM images (Fig. S2† and Fig. 4). Moreover, the particle size distribution analysis suggests that the average size of the Co-nanoparticles is bigger for CoNC-LAG and 3D-CoNC-LAG (14–16 nm) in comparison to 3D-CoNC-LAC and 3D-CoNC-LAG-LAC (12 and 9 nm, respectively). Although small (4–10 nm) Co-nanoparticles can effectively empower OER activity in the produced materials,41 larger Co particles result in decreased catalytic activity by blocking the pores and hindering the mass transfer.23,24 HR-TEM images in Fig. S3† demonstrate that Co-nanoparticles are coated with nitrogen-doped carbon forming hollow nanocapsules. The nanocapsule structure of carbon sheets around metallic-Co in combination with meso-/macroporous architecture enhances the mass transport of reactants to the OER/ORR catalytically active sites. Furthermore, carbon sheet layers serve as protection of Co nanoparticles from the electrolyte and agglomeration.42
The porosity data collected from the N2 adsorption–desorption isotherms analysis are summarized in Table 1. The CoNC-LAG possesses a slightly higher specific surface area compared to 3D-CoNC-LAG, 286 and 263 m2 g−1, respectively. This difference may be attributed to the specifics of the grinding method. It was observed that NaCl-crystallites coverage by Co-TPTZ complex is rather “loose” compared to the compression method; thus, it was hypothesized that not tight enough contact of the complex with the templating agent might reduce the advantageous effects of support. The highest surface area (Sa) was observed with 3D-CoNC-LAC (490 m2 g−1), followed by 3D-CoNC-LAG-LAC (406 m2 g−1), which can be another indicator of the importance of tight contact of the template with complex. Results of total volume (Vtot) calculation correlate with Sa values among the products, with 3D-CoNC-LAC having the highest total volume (0.55 cm3 g−1) and 3D-CoNC-LAG – the lowest (cm3 g−1). Average pore sizes of 3D-CoNC-LAC and 3D-CoNC-LAC are 2.2 nm and 3D-CoNC-LAG and CoNC-LAG – 1.9 and 2.0, accordingly. The pore size distribution graph (Fig. S4†) indicates that all the materials possess a mixed meso- and microporous nature, with the highest peaks in the region of 1–30 nm. The presence of both micropores and mesopores is essential for facilitating the charge transfer and mass transport in the catalytic process.43
| Sample | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|
| CoNC-LAG | 286 | 0.29 | 2.0 |
| 3D-CoNC-LAG | 263 | 0.25 | 1.9 |
| 3D-CoNC-LAC | 490 | 0.55 | 2.2 |
| 3D-CoNC-LAG-LAC | 406 | 0.45 | 2.2 |
To analyze the surface elemental composition and presence of catalytically active sites, the XPS technique was utilized. As shown in the survey XPS spectra (Fig. S5†), all produced catalysts mainly contain cobalt, carbon, nitrogen, and oxygen species.
The total surface content of four elements (Co, N, C, and O) in all catalysts is summarized in Table 2. Notably, an examination of the bulk composition of metals in the catalyst materials by MP-AES shows the highest content of cobalt in CoNC-LAG (12.35 wt%). This can be explained by lower stability of Co-TPTZ-LAG towards pyrolysis, thus resulting in bulky Co-metal pieces, which are harder to remove by acid treatment; hence these results correlate with previously acquired data.
| Catalyst | Surface elemental composition (at%) | Bulk metal composition (wt%) | |||
|---|---|---|---|---|---|
| C | N | O | Co | Co | |
| CoNC-LAG | 85.9 | 4.1 | 7.6 | 0.8 | 12.35 ± 0.11 |
| 3D-CoNC-LAG | 87.0 | 7.2 | 5.0 | 0.5 | 5.55 ± 0.05 |
| 3D-CoNC-LAC | 88.8 | 7.0 | 4.1 | 0.5 | 4.69 ± 0.02 |
| 3D-CoNC-LAG-LAC | 84.3 | 9.0 | 5.0 | 0.8 | 5.70 ± 0.04 |
Deconvoluted high-resolution C 1s photoelectron spectra of all catalyst materials (Fig. S6†) reveal the characteristic peak of sp2 carbon at the binding energy (B.E.) of ∼284.3 eV, which can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonding and suggests an effective N-atoms doping.23 N 1s XPS spectra (Fig. 5a) of the obtained catalysts demonstrate the presence of pyridinic (∼398.4 eV), graphitic (∼400.5 eV), and Co–Nx-type nitrogens (∼399.4 eV), which are considered as main contributors to the catalytic activity of the M–N–C type catalysts.14–17 To check the chemical state of Co atoms, the Co2p spectra of all catalysts were thoroughly analyzed. (Fig. 5a) Spectra were deconvoluted into three peaks with binding energies of ∼779.8, ∼781.8, and ∼785.6, which can be assigned to the Co–C, Co–Nx, and Co–Nx satellite bonds, respectively.23
N bonding and suggests an effective N-atoms doping.23 N 1s XPS spectra (Fig. 5a) of the obtained catalysts demonstrate the presence of pyridinic (∼398.4 eV), graphitic (∼400.5 eV), and Co–Nx-type nitrogens (∼399.4 eV), which are considered as main contributors to the catalytic activity of the M–N–C type catalysts.14–17 To check the chemical state of Co atoms, the Co2p spectra of all catalysts were thoroughly analyzed. (Fig. 5a) Spectra were deconvoluted into three peaks with binding energies of ∼779.8, ∼781.8, and ∼785.6, which can be assigned to the Co–C, Co–Nx, and Co–Nx satellite bonds, respectively.23
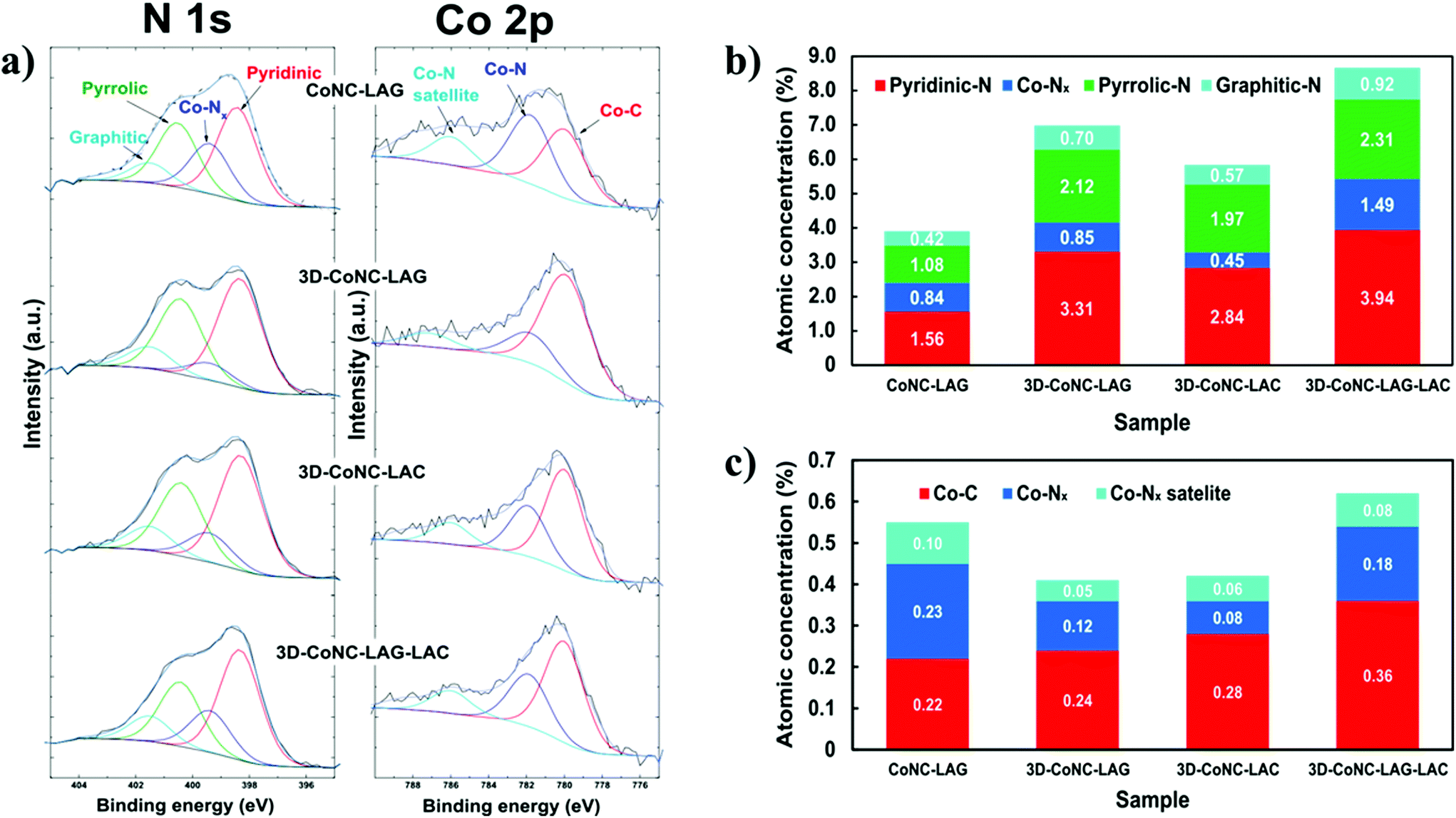 | ||
| Fig. 5 (a) Deconvoluted high-resolution N 1s and Co 2p photoelectron spectra of Co–N–C catalysts. (b) The content distribution of various nitrogen-rich groups in the prepared catalysts (c) The content distribution of single-atom Co-species in the prepared catalysts. | ||
Content analysis shows that the 3D-CoNC-LAG-LAC has the highest N/C ratio (10.7%) with a carbon content of 84.32 at% and nitrogen 9.05 at%. Furthermore, the comparison of catalytically active N- and Co-species concentrations (Fig. 5b and c) reveal that the 3D-CoNC-LAG-LAC has the highest load of pyridinic, pyrrolic, and graphitic-N sites and second-high Co-sites content. These results suggest the efficiency of the combinatorial mechanosynthetic method towards N-atom doping. Both CoNC-LAG and 3D-CoNC-LAG-LAC have the highest Co/C ratio (∼0.9%); however, the ratio of metallic-Co is almost twice as big for the NaCl-free catalyst. (7.8% and 4.0%, respectively). Contrarily, the lowest N/C ratio (4.77%) was observed for CoNC-LAG with C-content 85.87 at% and N-content – 4.1 at%, and the lowest concentration of active activity N-species, which further proves the positive effect of the NaCl-template method on the efficient N-atom doping. Nevertheless, the concentration of Co–Nx species – one of the major contributors to catalytic activity, in the non-templated catalyst is the highest in the series. (Fig. 5c) Thus, despite the findings, one could still expect a rather high activity towards ORR from this material. Notably, the 3D-CoNC-LAC demonstrates the lowest concentration of Co–Nx, indicating that the compression technique alone is unable to provide sufficient energy for the effective formation of the Co-TPTZ complex, which further results in Co–Nx species. It was assumed in our study that LAG method, in comparison to LAC, provides higher amount of active Co–Nx species, proving the better complexation reaction efficiency. However, upon the compression of LAG material we observe boost in the concentration of active sites, which can be attributed to an improved stability during the pyrolysis due to the better packing after the compression. The electrochemical assessment of materials is discussed below.
The oxygen electroactivity of prepared Co–N–C catalysts was evaluated by cyclic voltammetry (CV) and rotating disk electrode (RDE) techniques. RDE data (Fig. 6a) demonstrate that 3D-CoNC-LAG-LAC has the highest ORR activity with an ORR onset potential (Eon) value of 0.98 V and a half-wave potential (E1/2) value of 0.83 V, which is comparable to the recently reported results for Co–N–C materials (Table S2†). Enhanced ORR electroactivity can be attributed to a highly porous structure in combination with an abundance of pyridinic-N sites. Template-free catalyst – CoNC-LAG showed a second-best result in activity towards ORR, which can be explained by the highest concentration of Co–Nx sites (Fig. 6c). Tafel analysis was performed to get an insight into ORR mechanism (Fig. 6b). Tafel slope values were −61, −59, −37, and −47 mV dec−1 for 3D-Co-NC-LAC, Co-NC-LAG, 3D-Co-NC-LAG, and 3D-Co-NC-LAG–LAC, demonstrating that the rate determining step of ORR is the transfer of the first electron to the oxygen molecule.
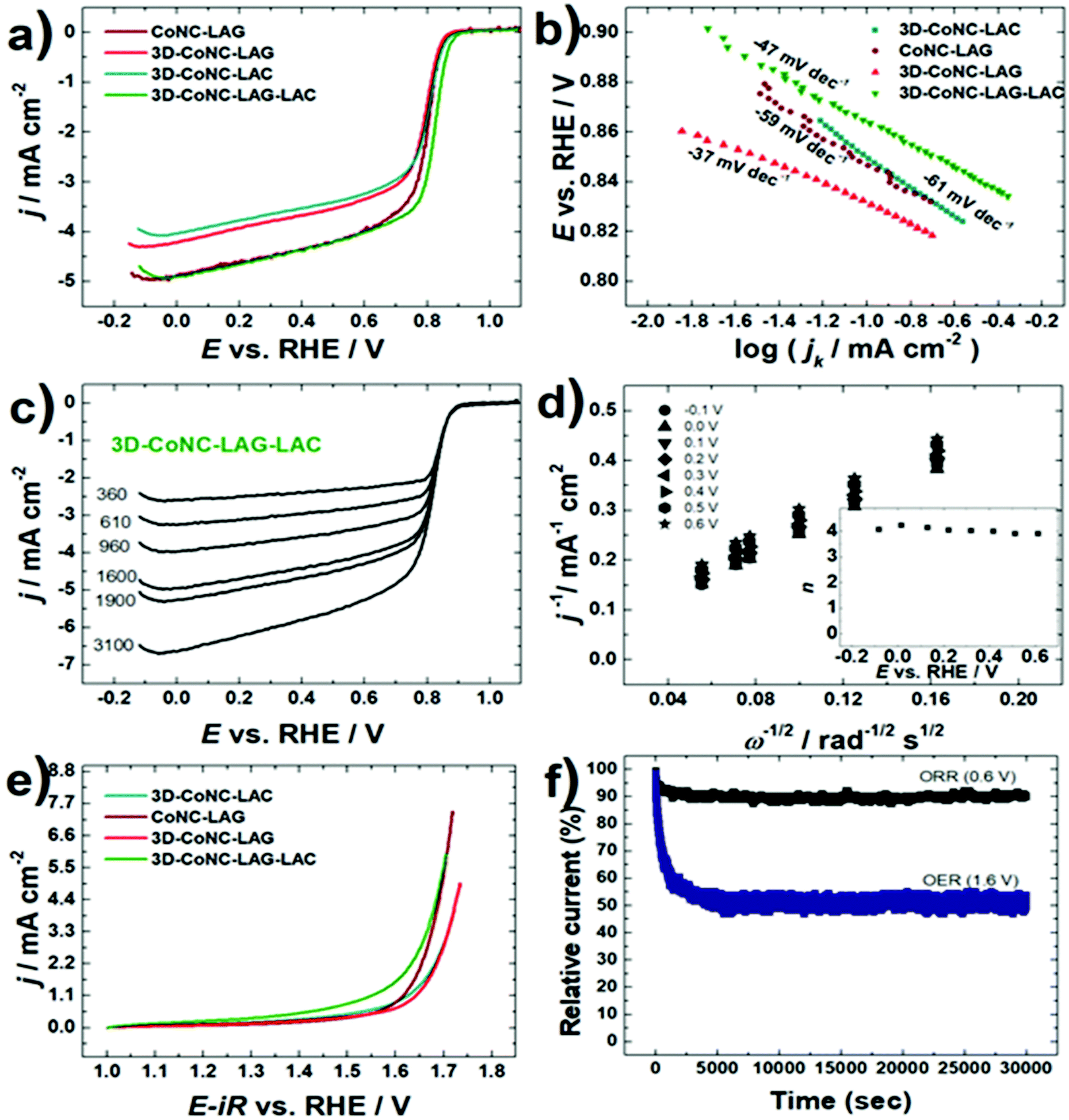 | ||
| Fig. 6 (a) ORR polarization curves obtained for all catalyst materials in O2-saturated 0.1 M KOH at 1600 rpm; (b) Tafel plots constructed from the RDE data; (c) ORR polarization curves recorded for 3D-CoNC-LAG-LAC at different electrode rotation rates; (d) Koutecky–Levich plots obtained for 3D-CoNC-LAG-LAC modified GC electrode (Inset: number of electrons transferred per O2 molecule); (e) OER polarization curves recorded in Ar-saturated 0.1 M KOH; ν = 10 mV s−1. (f) Chronoamperometric response for 3D-CoNC-LAG-LAC recorded at an applied potential of 0.6 V for ORR and 1.6 V for OER. | ||
The Koutecky–Levich (K–L) plots constructed from the RDE data in demonstrate good linearity and parallelism (Fig. 6d), indicating first-order reaction kinetics with respect to the dissolved O2 concentration. The electron transfer number calculated from the K–L equation was close to four, indicating that the ORR proceeds to water formation (inset in Fig. 6d).
To further investigate the OER behavior, Co–N–C modified GC electrodes were scanned at 1600 rpm in the potential range of 1–1.8 V vs. RHE (Fig. 6e). The benchmark OER current density of 10 mA cm−2 (Ej=10) was achieved at 1.70 V for 3D-CoNC-LAG-LAC and resulted in the lowest OER overpotential value (η = 0.46). Overall bifunctional ORR/OER activity (ΔE = Ej=10 − E1/2) was calculated, and the lowest ΔE value was acquired for 3D-Co-NC-LAG-LAC. Kinetic parameters for the obtained catalysts are summarized in Table 3. As discussed above, the OER activity is associated with the presence of Co nanoparticles in the structure of catalysts. The difference in the electrocatalytic efficiency of the materials is the influence of porosity and the size of Co nanoparticles (CoNPs), where the most electroactive 3D-CoNC-LAG-LAC has the smallest average size of CoNPs. The above-mentioned results indicate that 3D-CoNC-LAG-LAC has great bifunctional activity towards ORR and OER.
| Sample | E 1/2 (V) | E onset (V) | n | E j=10 (V) | η OER (V) | ΔE (V) |
|---|---|---|---|---|---|---|
| CoNC-LAG | 0.79 | 0.96 | 3.1 | 1.73 | 0.49 | 0.94 |
| 3D-CoNC-LAG | 0.78 | 0.92 | 3.4 | 1.80 | 0.56 | 1.04 |
| 3D-CoNC-LAC | 0.79 | 0.94 | 3.5 | 1.80 | 0.56 | 1.01 |
| 3D-CoNC-LAG-LAC | 0.83 | 0.98 | 4.0 | 1.70 | 0.46 | 0.87 |
The stability of the most active 3D-CONC-LAG-LAC catalyst was investigated employing the chronoamperometry technique, where the fixed potential of the working electrode was held at 0.6 V for ORR and at 1.6 V for OER (Fig. 6f). According to chronoamperometry test results, the ORR current was highly stable during ca. 8 hours of continuous operation. During the OER chronoamperometry measurement, a catalyst exhibited a current decay, which is caused by the production of gas bubbles that partially block the catalyst surface. Nevertheless, after ca. 3000 s the current density attained a stable state and the 3D-CONC-LAG-LAC catalyst exhibited stable electrocatalytic performance towards OER. The promising results of the proposed mechanosynthetic methodology show that high-performing PGM-free catalysts can be produced in an inexpensive and sustainable way. The data summarized in Table S2† shows that the catalysts produced in our study possess compatible activity, however the comparative data in Table S3† shows that the proposed methodology has several major advantages: minimal amount of non-toxic solvent, short total reaction time, and easy-to-conduct protocols. These benefits in turn result in lower-to-none toxic waste and considerably less time and energy consumption. This study is dedicated to bringing the global goal closer to reality: a cheap, completely PGM-free catalyst-based fuel cell system with high performance and long life.
Conclusions
In this work we present a novel green and cost-effective method for producing M–N–C-type catalysts with excellent bifunctional oxygen electroactivity. The suggested method is based on the application of mechanochemistry in the form of liquid-assisted grinding and compression. The current study yielded a series of meso/microporous Co and N doped carbon materials as a highly active and stable oxygen electrocatalyst. Porosity, electrocatalytic activity, and stability of materials were improved by applying the NaCl sacrificial template. This research confirms the efficiency of the mechanochemical approach towards catalyst production, indicating that the advantageous effect of NaCl-template is also maintained. The excellent ORR/OER performance can be attributed to the conventional active sites, including Co–Nx moieties, whose presence was confirmed by XPS analysis. A relatively large surface area is also suggested to be a contributor facilitating mass transport.A proposed method opens new avenues for environmentally sustainable large-scale implementation of high-performance M–N–C catalysts for clean energy systems. This low-cost and facile method may empower the switch to Pt-free electrocatalysts for ORR in fuel cells.
Author contributions
A. K. conceived the concept and experiments design, fabricated the catalysts, wrote the manuscript. G. Y. performed the material electrochemical characterization and analyzed the data. K. Š. performed the electron microscopy imaging. J. A. performed PXRD. J. L. and M. K. together performed N2 physisorption analysis. P. P. performed MP-AES experiments. A. K. and V. K. together performed XPS analysis. A. K and N. K. were the primary writers of the manuscript. N. K. supervised the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Estonian Research Council grant PSG250; and by the EU through the European Regional Development Fund (TK141, “Advanced materials and high-technology devices for energy recuperation systems” and TK143, “Molecular Cell Engineering”).References
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- V. Das, Renewable Sustainable Energy Rev., 2017, 9 Search PubMed.
- Z. Wu, X. F. Lu, S. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- T. J. Omasta, Y. Zhang, A. M. Park, X. Peng, B. Pivovar, J. R. Varcoe and W. E. Mustain, J. Electrochem. Soc., 2018, 165, F710–F717 CrossRef CAS.
- M. Li, X. Bi, R. Wang, Y. Li, G. Jiang, L. Li, C. Zhong, Z. Chen and J. Lu, Matter, 2020, 2, 32–49 CrossRef.
- H. A. Miller and F. Vizza, Anion Exchange Membrane Fuel Cells: Principles, Materials and Systems, Springer, 1st edn., 2018, vol. 63 Search PubMed.
- Z.-C. Yao, T. Tang, J.-S. Hu and L.-J. Wan, Energy Fuels, 2021, 35, 6380–6401 CrossRef CAS.
- M. D. Bhatt and J. Y. Lee, Energy Fuels, 2020, 34, 6634–6695 CrossRef.
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, 7970 CrossRef PubMed.
- X. Fu, P. Zamani, J.-Y. Choi, F. M. Hassan, G. Jiang, D. C. Higgins, Y. Zhang, M. A. Hoque and Z. Chen, Adv. Mater., 2017, 29, 1604456 CrossRef.
- X.-F. Lu, L.-F. Gu, J.-W. Wang, J.-X. Wu, P.-Q. Liao and G.-R. Li, Adv. Mater., 2017, 29, 1604437 CrossRef.
- P. Song, M. Luo, X. Liu, W. Xing, W. Xu, Z. Jiang and L. Gu, Adv. Funct. Mater., 2017, 27, 1700802 CrossRef.
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef.
- P. Song, Y. Wang, J. Pan, W. Xu and L. Zhuang, J. Power Sources, 2015, 300, 279–284 CrossRef CAS.
- L. Osmieri, A. H. A. Monteverde Videla and S. Specchia, J. Power Sources, 2015, 278, 296–307 CrossRef CAS.
- F. Roncaroli, E. S. Dal Molin, F. A. Viva, M. M. Bruno and E. B. Halac, Electrochim. Acta, 2015, 174, 66–77 CrossRef CAS.
- R. Othman, Md. S. Hossain and N. H. Jabarullah, Appl. Organomet. Chem., 2017, 31, 3738 CrossRef.
- K. Zhang and R. Zou, Small, 2021, 17, 2100129 CrossRef CAS.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- J. Pampel and T.-P. Fellinger, Adv. Energy Mater., 2016, 6, 1502389 CrossRef.
- J. Pampel, A. Mehmood, M. Antonietti and T.-P. Fellinger, Mater. Horiz., 2017, 4, 493–501 RSC.
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS.
- C. Guo, R. Zhou, Z. Li, Y. Si, W. Liao, W. Sun, S. Xiang, X. Luo, M. Luo and Z. Luo, Microporous Mesoporous Mater., 2020, 303, 110281 CrossRef CAS.
- C. Guo, Y. Li, W. Liao, Y. Liu, Z. Li, L. Sun, C. Chen, J. Zhang, Y. Si and L. Li, J. Mater. Chem. A, 2018, 6, 13050–13061 RSC.
- J. Cao, Z. Chen, J. Xu, W. Wang and Z. Chen, Electrochim. Acta, 2013, 88, 184–192 CrossRef CAS.
- R. Wu, J. Wang, K. Chen, S. Chen, J. Li, Q. Wang, Y. Nie, Y. Song, H. Chen and Z. Wei, Electrochim. Acta, 2017, 244, 47–53 CrossRef CAS.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Chem. – Eur. J., 2021, 27, 6819–6847 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- T. Friščić, Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2014, pp. 1–19 Search PubMed.
- D. Chen, J. Zhao, P. Zhang and S. Dai, Polyhedron, 2019, 162, 59–64 CrossRef CAS.
- T. Friščić and L. Fábián, CrystEngComm, 2009, 11, 743 RSC.
- L. Paseta, G. Potier, S. Sorribas and J. Coronas, ACS Sustainable Chem. Eng., 2016, 7, 3780–3785 CrossRef.
- B. G. Fiss, N.-N. Vu, G. Douglas, T.-O. Do, T. Friščić and A. Moores, ACS Sustainable Chem. Eng., 2020, 8, 12014–12024 CrossRef CAS.
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 19, 1507–1518 RSC.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571 RSC.
- R. A. Buyanov, V. V. Molchanov and V. V. Boldyrev, Catal. Today, 2009, 144, 212–218 CrossRef CAS.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefèvre, R. Chenitz, J.-P. Dodelet, D. Jones and F. Jaouen, Angew. Chem., Int. Ed., 2013, 52, 6867–6870 CrossRef CAS.
- L. Osmieri, A. H. A. Monteverde Videla and S. Specchia, J. Solid State Electrochem., 2016, 20, 3507–3523 CrossRef CAS.
- V. Chipakwe, P. Semsari, T. Karlkvist, J. Rosenkranz and S. C. Chelgani, J. Mater. Res. Technol., 2020, 9, 8148–8162 CrossRef CAS.
- E. Locke, S. Jiang and S. K. Beaumont, Top. Catal., 2018, 61, 977–985 CrossRef CAS PubMed.
- Y. Zhang, P. Wang, J. Yang, S. Lu, K. Li, G. Liu, Y. Duan and J. Qiu, Carbon, 2021, 177, 344–356 CrossRef CAS.
- H.-W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03433b |
| This journal is © The Royal Society of Chemistry 2022 |