
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ reactive extraction with oleic acid for process intensification in amine transaminase catalyzed reactions†
Moritz
Doeker‡
 ab,
Laura
Grabowski‡
bcd,
Dörte
Rother
bcd and
Andreas
Jupke
*ab
ab,
Laura
Grabowski‡
bcd,
Dörte
Rother
bcd and
Andreas
Jupke
*ab
aRWTH Aachen University AVT.FVT - Fluid Process Engineering, Forckenbeckstraße 51, 52074 Aachen, Germany. E-mail: andreas.jupke@avt.rwth-aachen.de; Tel: +49 241 80 95490
bBioeconomy Science Center (BioSC), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
cForschungszentrum Jülich GmbH IBG-1, Wilhelm-Johnen-Straße, 52425 Jülich, Germany
dRWTH Aachen University ABBt, Worringer Weg 1, 52074 Aachen, Germany
First published on 10th December 2021
Abstract
Amine transaminase catalyzed reaction systems allow the efficient production of high value active pharmaceutical ingredients via stereoselective amination of prochiral precursors. Metaraminol is a bioactive amino alcohol directly used in hypotension treatment and further acts as important precursor for the synthesis of more complex bioactive compounds. The amination reaction to such compounds makes use of an amine donor, which is usually derived from non-renewable resources despite other drawbacks. To allow a greener production route to metaraminol, L-alanine can be used as an alternative amine donor, although conversions are thermodynamically limited. To overcome this limitation, extraction-based in situ product removal (ISPR) can be applied to achieve higher conversions. Here, we describe a reactive extraction strategy to recover metaraminol under transaminase-favorable process conditions, achieving higher yields as for previously described physical extraction concepts. Suitable process parameters, such as pH and required reactant concentration were defined by extraction experiments and enzyme compatibility tests. An operational window was found to establish reactive ISPR within the biocatalytic production of metaraminol. Our approach makes use of oleic acid as a sustainable reactant and allows efficient in situ recovery of metaraminol, thus improving the enzymatic production.
1. Introduction
In recent years, enzyme-catalyzed reaction steps have become an important green tool in the production of pharmaceuticals and fine chemicals.1,2 The used biocatalysts are valued for their high (stereo)selectivity and their moderate reaction conditions, thereby enabling alternative reaction routes compared to standard organic chemistry. These alternative synthesis routes facilitate the integration of bio-based precursors and therefore play an important role in the development of more sustainable production processes.3,4 One important group of these enzymatic catalysts are amine transaminases, formerly known as ω-transaminases, which convert ketones or aldehydes towards chiral amines or amino alcohols.5 One example for these amino alcohols is metaraminol ((1R,2S)-2-amino-1-(3-hydroxyphenyl)propan-1-ol), a bioactive compound used for the treatment of hypotension and as high-value building block in the synthesis of tetrahydroisoquinolines.6,7 Amine transaminase catalyzed reaction systems suffer from inefficiencies, which are related to an often unfavorable reaction equilibrium,8 leading to low overall conversion.9 To cope with these inefficiencies, strategies such as enzyme engineering of transaminases,10 optimal choice of the amine donor11 and intelligent process design are required for efficient operation of the transaminase reaction system. Specifically, the choice of amine donor is limited to few possibilities, where the most commonly used compound is isopropylamine (IPA), which was shown to be a suitable amine donor for many amine transaminase reactions.12 When using IPA as amine donor, acetone is produced as side product, which can be removed from the reaction medium either by evaporation or gas stripping.13 This side product removal allows to shift the reaction equilibrium and thus yields high conversions. Nevertheless, the large-scale application of IPA exhibits some major drawbacks as it is derived from fossil feedstock, is often not accepted by wild type amine transaminases and is toxic.14 Moreover, IPA is often used in excess in the reaction medium to further push the reaction equilibrium. This can result in unwanted side reactions15 and product contamination, complicating the downstream processing. Therefore alternative amine donors have been investigated.11,16 One such amine donor is L-alanine which is non-toxic and was shown to be a favorable co-substrate in amine transaminase reactions.17,18 The use of bio-based L-alanine is advantageous as no polluting side products are released. Moreover, in the envisaged cascade the recycling of the co-product pyruvate is possible in the preliminary carboligation step with 3-OH-benzaldehyde to yield the intermediate (R)-3-OH-phenylacetylcarbinol (1-hydroxy-1-(3-hydroxyphenyl)propan-2-one), in the following referred to as (R)-3-OH-PAC. In this way, a high atom economy of the cascade can be achieved. The enzyme cascade towards metaraminol is depicted in Fig. 1.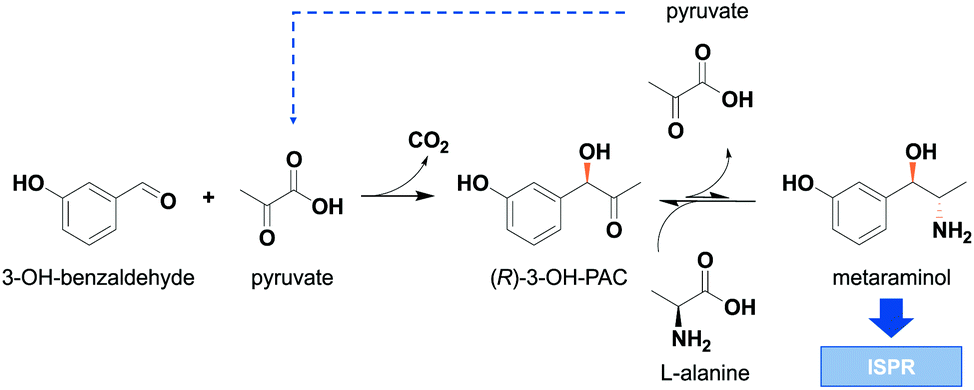 | ||
| Fig. 1 Two-step enzymatic cascade towards metaraminol consisting of a carboligation and transamination step. 3-OH-benzaldehyde and pyruvate are converted by a pyruvate decarboxylase variant from Acetobacter pasteurianus (ApPDC). The intermediate (R)-3-OH-phenylacetylcarbinol (–PAC) is converted by the amine transaminase Cv2025 from Chromobacterium violaceum with L-alanine as the amine donor. In situ product removal (ISPR) is needed to shift the reaction equilibrium of the transamination step. | ||
The major challenge in using L-alanine in transaminations are low conversions towards the amino alcohol due to an unfavorable thermodynamic reaction equilibrium. The thermodynamic equilibrium of transaminase reactions has already been extensively studied and it was shown that a combination of thermodynamic studies and prediction tools allow to successfully describe and optimize reaction conditions for improved conversions.19–21 Still to further increase conversions, an equilibrium shift is required. This equilibrium shift is predominantly realized by in situ product removal (ISPR) strategies such as reactive crystallization22 and supported liquid membrane product extraction.23 Also, a two-phase system for a similar case has already been studied, but with less success than the approach shown in this present work.20 In a recent publication, we investigated physical liquid–liquid extraction as another suitable ISPR method to increase conversion during enzymatic production of metaraminol using L-alanine as the amine donor.24 One major drawback we encountered during the physical extraction was the requirement of metaraminol being present in its neutral dissociation state, which is only present under alkaline reaction conditions between pH 9 and 10. However, these high pH values were not ideal for the biotransformation as they led to decreased initial enzyme activity and also caused unwanted side reactions, lowering the overall process selectivity. To overcome these restraints, an alternative ISPR strategy suitable to recover metaraminol at neutral pH conditions is required. Such an ISPR strategy is reactive liquid–liquid extraction, in which an additionally added reactant selectively forms complexes with the target product, allowing for the product removal from the aqueous reaction phase into an organic extraction phase. The applicability of reactive extraction studies has been presented for fermentative production of organic acids.25–27 Few examples, however, have been shown for the selective extraction of chiral amines and for equilibrium shifts in enzymatic amine synthesis, which were solely used in downstream operations independent of the reaction system.28 The suitability of the herein presented method was already shown for the recovery of various amines from aqueous streams.29–31 In this contribution, we show the successful integration of the reactive extraction with oleic acid as reactant within the transamination reaction. Oleic acid can be gained from renewables and it can be recycled after successful product removal, making it a sustainable target for in situ reactive extraction, allowing for process intensification of the enzymatic production of metaraminol.
2. Materials and methods
2.1. Chemicals and materials
The following chemicals were purchased as given: (1R,2S)-3-(2-amino-1-hydroxypropyl)phenol (metaraminol bitartrate, TRC, CA), thiamine pyrophosphate (ThDP; AppliChem, DE), 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris; AlfaAesar, DE), L(+)-tartaric acid (Merck, DE), orthophtalic aldehyde/mercaptoethanol reagent (Maisch, DE), phosphoric acid (Fluka, DE), magnesium chloride hexahydrate (MgCl2·6H2O, Fluka, DE), L-alanine (Fluka, DE), α-methylbenzylamine (α-MBA; Sigma-Aldrich, DE), isopropylamine (IPA, Sigma-Aldrich, DE), pyridoxal-5-phosphate (PLP, Sigma-Aldrich, DE), oleic acid (Sigma-Aldrich, DE), disodium tetraborate decahydrat (Merck, DE), di-sodium sulfate (VWR, DE), HCl (Carl Roth, DE), pyruvic acid sodium salt (Carl Roth, DE), potassium dihydrogenphosphate (KH2PO4, Carl Roth, DE), di-potassium hydrogen phosphate (K2HPO4, Carl Roth, DE), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES, Carl Roth, DE), sodium hydroxide (NaOH, Carl Roth, DE), 3-(trimethylsilyl)propanoic acid (TMSP, Carl Roth, DE), trifluoroacetic acid (TFA, Carl Roth, DE), diethylamine (DEA, Carl Roth, Germany), 1-octanol (Merck, DE).2.2. LC/DAD quantification
Metaraminol quantification in the enzymatic reaction system
To quantify metaraminol production, 20 μL sample from conversion experiments were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 in 50% (v/v) acetonitrile (ACN):water containing 0.1% diethylamine (DEA) and 0.075% trifluoroacetic acid (TFA) and centrifuged for 3 min at 14000 rpm. The supernatant was measured by HPLC (Agilent 1290 Infinity II, DAD 220 nm, Agilent Zorbax Eclipse plus C18 column, 2 × 100 mm, 1.8 μm, 20 °C, 5 μL injection volume). For product quantification an isocratic method was used (0.3 mL min−1, 15% ACN and 85% water containing 0.1% DEA, 0.075% TFA). The retention time of metaraminol was 0.9 min. The substrate depletion was quantified using the same mobile phase in a gradient method (0.5 mL min−1, 10–90% ACN in 5 min). The retention time of (R)-3-OH-PAC was 1.9 min.
:20 in 50% (v/v) acetonitrile (ACN):water containing 0.1% diethylamine (DEA) and 0.075% trifluoroacetic acid (TFA) and centrifuged for 3 min at 14000 rpm. The supernatant was measured by HPLC (Agilent 1290 Infinity II, DAD 220 nm, Agilent Zorbax Eclipse plus C18 column, 2 × 100 mm, 1.8 μm, 20 °C, 5 μL injection volume). For product quantification an isocratic method was used (0.3 mL min−1, 15% ACN and 85% water containing 0.1% DEA, 0.075% TFA). The retention time of metaraminol was 0.9 min. The substrate depletion was quantified using the same mobile phase in a gradient method (0.5 mL min−1, 10–90% ACN in 5 min). The retention time of (R)-3-OH-PAC was 1.9 min.
Quantification of metaraminol in artificial systems
Analysis of extraction samples of metaraminol was performed by HPLC analysis (Agilent 1200, DAD 334 nm, Machery Nagel Nucleodur C18ec-pre-column 2 × 4 mm, 40 °C, 5 μL injection volume). The method was derived in a previous publication and optimized for shorter method run times.24 Each standard and sample was diluted with 0.1 M HCl solution to achieve metaraminol concentrations below 0.01 mol L−1. Before measurement pre-column online derivatization using orthophthalic aldehyde solution (Maisch) was performed following the derivatization procedure given in ESI.† The derivatized samples were injected onto the HPLC. The eluents were (A) 2.5 mM acetic acid in water with 10% methanol at pH 6 and (B) methanol. The gradient elution is given in ESI.†2.3. Biocatalyst production and formulation
Competent E. coli BL21 (DE3) cells were transformed with the amine transaminase from Chromobacterium violaceum ATCC 12472 Cv2025. The enzyme was produced in shake-flask cultivations using auto-induction medium (specifics are given in the ESI†). The cells were harvested by centrifugation at 4 °C and 7000 rpm for 30 min. Cell disruption was performed by ultrasonification at 70% amplitude and 0.5 cycle time in a continuous flow cell at 4 °C. The enzyme was either purified or used as whole cell formulation. For protein purification, an immobilized-metal affinity chromatography (IMAC) via a C-terminal histidine-tag was used and the purified enzyme was obtained in 10 mM potassium phosphate buffer pH 7.5 supplemented with 0.2 mM PLP. After lyophilization at −80 °C and 0.2 mbar, the enzyme lyophilisate was stored at −20 °C. To obtain lyophilized, whole cells, the cells were cultivated as stated above, the frozen cell pellet was lyophilized and subsequently ground to a fine powder, which was stored at −20 °C.2.4. Enzymatic production and purification of (R)-3-OH-PAC
In 400 mL total volume, 70 mM 3-hydroxybenzaldehyde and 180 mM pyruvate were dissolved in 100 mM potassium phosphate buffer pH 6.5 + 1 mM thiamine pyrophosphate (ThDP) and 2.5 mM MgSO4. Lyophilized whole cells containing a variant of the pyruvate decarboxylase from Acetobacter pasteurianus (ApPDC var), were added to the reaction mixture (10 mg mL−1) and the reaction was stirred at 350 rpm in a 500 mL glass reactor at 25 °C for 24 h (EasyMax 402, Mettler Toledo, US). The reaction was performed in duplicates and reached >99% conversion (>98% ee). The conversion was analyzed via HPLC analysis as described before. For product purification, the cells were removed by centrifugation (6500 rpm, 20 min, 10 °C). The supernatant was acidified with 20% HCl to precipitate the protein and subsequently centrifuged again. The supernatant was filtered twice. To extract the target compound from the aqueous phase, three extraction steps with 100 mL ethyl acetate were performed. Afterwards, the aqueous phase was adjusted to pH 7 and two more extraction step were performed. The organic phase was washed with a saturated NaHCO3 solution and dried over MgSO4. The drying agent was removed by filtration and the organic solvent was evaporated at 40 °C and 100 mbar. The isolated yield and purity were determined via HPLC measurements.2.5. Activity and stability of Cv2025 under process relevant conditions
The enzyme activity of Cv2025 was determined using a standard photometric assay for transaminases (100 mM HEPES buffer, 0.1 mM PLP, 2.5 mM α-methylbenzylamine, 5 mM pyruvate).32 The reaction components were combined in a quarz-glass cuvette and equilibrated to a reaction temperature of 25 °C. The protein concentration of the enzyme stock solution was determined via Bradford assay using BSA for calibration.33 The enzyme was added at a final concentration of 0.1 mg mL−1 to the assay solution. The initial activity was derived from the linear increase in acetophenone absorbance at 300 nm within 10% conversion. One Unit is defined as the amount of enzyme producing 1 μmol of acetophenone per minute under the defined conditions. The stability in the presence of oleic acid was determined after the addition of 10% (v/v) 1-octanol with 0.1 M, 0.25 M or 0.5 M oleic acid to the enzyme stock solution after incubation rotating at 20 °C by measuring the enzyme activity after 1–7 days. The activity measurements were performed in technical triplicates from two biological replicates. The effect of different oleic acid concentrations was investigated in the target reaction to metaraminol using isopropylamine as the amine donor (20 mM (R)-3-OH-PAC, 100 mM isopropylamine, 100 mM HEPES buffer pH 7.5, 0.2 mM PLP, 10 mg mL−1 lyophilized, whole cells Cv2025 or 1 mg mL−1 purified Cv2025, 10% 1-octanol with 0.1 M, 0.25 M and 0.5 M oleic acid, 1 mL reaction volume in 1.5 mL glass reaction vial with sealed screw cap). The reaction was incubated at 30 °C and 750 rpm and the conversion was determined via endpoint measurements after 24 h. The measurements were performed in three technical replicates.2.6. Reactive extraction experiments in pure substance systems without enzyme addition
Experimental characterization of the extraction behavior
The initial aqueous solution consisted of 0.02 M metaraminol bitartrate, adjusted to pH 5. The solution was divided into individual samples and mixed with equal volumes of NaOH solutions. The NaOH solutions differed in the NaOH concentrations, which were calculated as a molar proportion of the metaraminol between 0 to 200% in steps of 10%. The obtained aqueous phase thus yielded 0.01 M of metaraminol. The organic phase consisted of 0 M, 0.1 M, 0.25 M and 0.5 M oleic acid in 1-octanol and was then saturated with water prior to the extraction experiments. 1 mL of the aqueous phase and 1 mL of the saturated organic phase were added into a 2 mL micro centrifuge tube and shaken for 16 hours at 20 °C. Subsequently phase separation was achieved via centrifugation at 2000 rpm for 5 minutes. Initial and equilibrium metaraminol concentrations were determined via HPLC measurements and the equilibrium pH after extraction was measured. All experiments were performed in duplicates.Influence of side components on the extraction efficiency
The reactive extractions with side components were performed following the procedure used for the characterization of the reactive extraction behavior. Additionally, 20 mM of either L-alanine, Na2SO4, or pyruvic acid were added to the aqueousphase prior to pH adjustment, which results in a final molar concentration of 10 mM of the side components. Furthermore, an oleic acid concentration of 0.25 M in 1-octanol was used.2.7. In situ reactive extraction within the biosynthesis of metaraminol
The in situ reactive extraction experiments within the vessel for enzymatic production of metaraminol from (R)-3-OH-PAC were conducted in a 100 mL stirred glass reactor with 40 mL aqueous reaction phase (EasyMax 402, Mettler Toledo; Columbus, USA). To enable reactive extraction of metaraminol, an organic extraction phase consisting of 1-octanol with different concentrations of oleic acid was added to the aqueous reaction phase in a 1:4 phase ratio. The organic phase was pre-saturated with water beforehand. The pH in the reaction phase was measured in the aqueous reaction phase with a pH electrode (InLab Semi-Micro L pH electrode, Mettler Toledo, Columbus, USA) and controlled via automated titration of 0.5 M NaOH. Before the start of the reaction, the biphasic system was equilibrated at 30 °C and 150 rpm for 1 h. Afterwards, the phase distribution of the substrate (R)-3-OH-PAC was determined via a sample from the aqueous reaction phase. The biotransformation was started by the addition of 10 mg mL−1 lyophilized, whole cells containing Cv2025 and the reaction system was stirred at 150–200 rpm and 30 °C for 48 h. For product recovery, the organic extraction phase was transferred to an acidic re-extraction phase (50 mM tartaric acid, pH 2–3) and incubated shaking at 20 °C for 1 h. To aid in phase separation, biphasic systems were centrifuged at 4000 rpm and 10 °C for 10 min in 50 mL Falcon reaction tubes. Afterwards, samples from both aqueous phases were analyzed via HPLC. Control reactions without ISPR were performed under identical conditions without oleic acid in the organic phase (40 mM (R)-3-OH-PAC, 250 mM L-alanine, 1 mM PLP, pH 7–7.5).
2.8. Repeated batch set-up for process intensification
The same setup and reaction concentrations as specified in section 2.7 were used. After 24 h, the biphasic reaction system was centrifuged at 4000 rpm and 10 °C for 10 minutes to separate both the organic extraction phase and aqueous reaction phase completely. The organic phase (10 mL) was transferred to 10 mL 50 mM tartaric acid in a shot flask and stirred at 600 rpm for 30 minutes at 20 °C. Afterwards, the same unloaded organic phase was transferred back to the aqueous reaction phase and the process was resumed for another 24 h.2.9. Calculation of performance indicators
The extraction experiments show the extraction yield plotted over the measured equilibrium pH, where the extraction yield is given in eqn (1).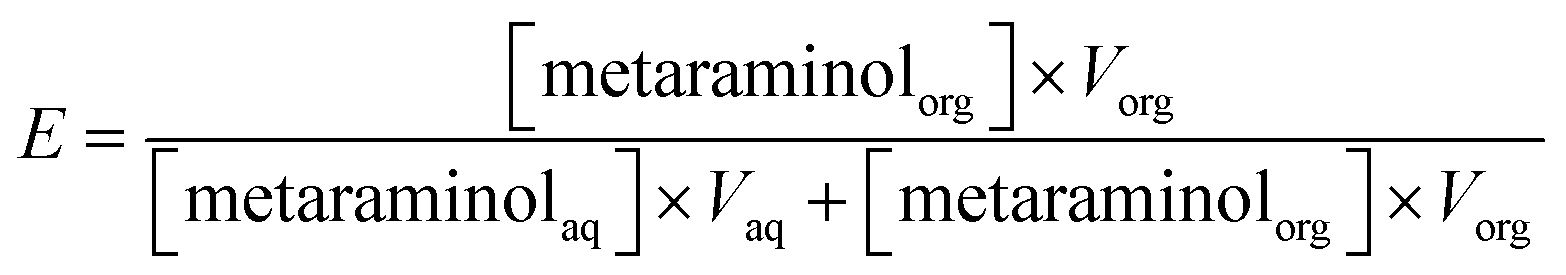 | (1) |
The enzymatic conversion was calculated as shown in eqn (2). [metaraminol,t] and [3OHPAC,t] are the aqueous concentration of product and substrate of the reaction at the time of sampling:
 | (2) |
The product yield was defined as the total amount of produced metaraminol from the initial amount of (R)-3-OH-PAC as follows:
 | (3) |
3. Results and discussion
3.1. Investigation of the liquid–liquid reactive extraction behavior in artificial medium without enzyme addition
In the following section, extraction experiments of metaraminol from an artificial aqueous phase, using oleic acid as reactant, are shown. The results allow to characterize the reactive extraction system, where special focus is laid on the concentration of the oleic acid, the equilibrium pH and possible side components. Following the results of this investigation, a general understanding of the extraction behavior is derived, which lays the basis to develop a process concept for metaraminol ISPR (refer to Fig. 4) and finally test its ability to shift the reaction equilibrium by reactive product removal in the enzymatic reaction system (section 3.3).Metaraminol extraction without addition of side components
To characterize the reactive extraction behavior, batch extractions of metaraminol without addition of further side components, from an aqueous to an organic phase (oleic acid and 1-octanol), were performed. Fig. 2 shows the pH dependent reactive extraction yield at different oleic acid concentrations plotted over the equilibrium pH. As shown in our earlier publication, physical extraction of metaraminol with pure organic solvent such as 1-octanol or 1-decanol is only feasible at high pH, with a maximum extraction yield at pH 9.6.24 The addition of oleic acid to the organic phase allows to extract metaraminol via complex formation with superior extraction yields compared to the physical extraction system and at a lower pH, in the range of pH 5 to 8. Furthermore, no precipitation of solids was observed in the experiments. Due to this, the reactive extraction system is ideal for the use in transaminase reaction systems. As can be observed, the reactive extraction shows strong dependence on the equilibrium pH of the aqueous phase. This pH dependence cannot be explained by changes of the dissociation states of metaraminol, which is mostly present in its cationic form at neutral pH and must therefore be due to the oleic acid present in the system. Here, the oleic acid acts as liquid cation exchanger, which will exchange the initially bound H+ ion with metaraminol forming metaraminol oleate ion pairs. As common for ion exchange in general, the exchange is governed by the different concentrations of the involved species, which includes H+ ions. As this H+ ion concentration is linked to the pH via the activity coefficients, a change in pH significantly influences the ion exchange and thus the efficiency of the reactive extraction.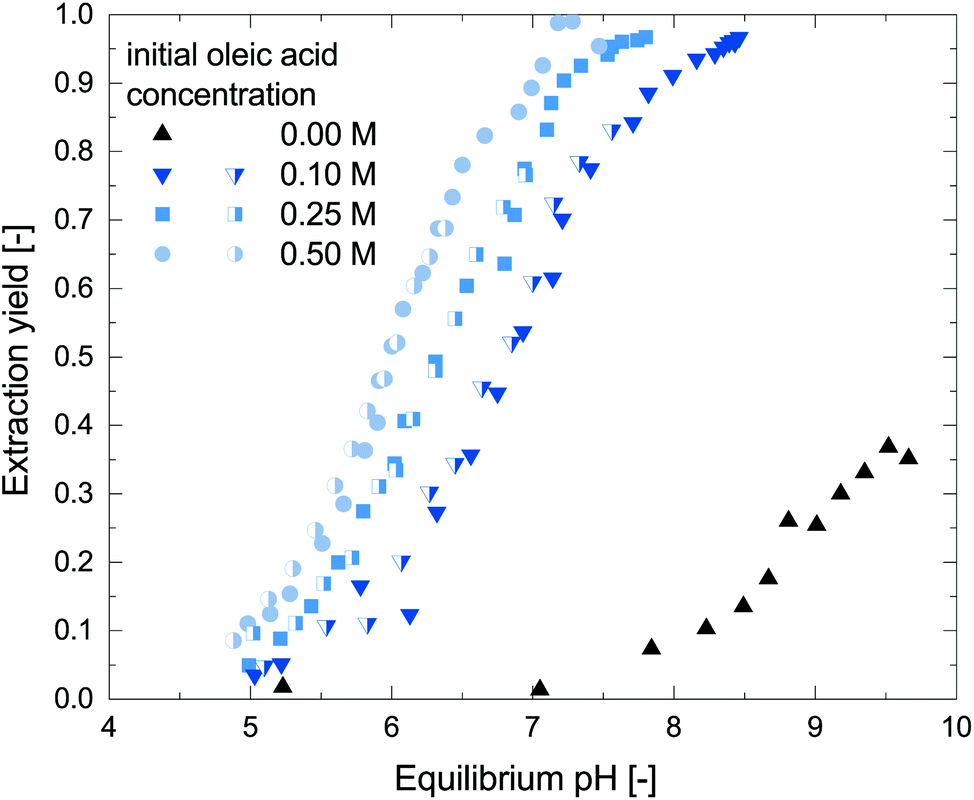 | ||
| Fig. 2 Duplicate determination of the pH dependant mole-related extraction yield of metaraminol at different oleic acid concentrations. The aqueous phase was prepared with pure metaraminol bitartrate. | ||
Furthermore, the concentration of oleic acid influences the liquid cation exchange, where increased extraction yields are achieved with increased oleic acid concentrations. A similar behavior was already shown for the reactive extraction of some primary amines, and is therefore expected to be observed in all reactive extractions of amines.29,30 Thus, for an efficient use of the reactive extraction in the transaminase reactions system, high oleic acid concentrations are favorable. Nevertheless, as oleic acid concentrations of 0.5 M were shown to strongly reduce the stability of the enzymatic catalyst (refer to section 3.2), we chose to further investigate oleic acid concentrations in the range of 0.1 M to 0.5 M for the following investigations.
Influence of side components
For an efficient integration of the reactive extraction into the enzymatic system, the influence of the reaction components on the reactive extraction has to be taken into account. Therefore, we investigated the influence of the salt Na2SO4, pyruvate and L-alanine, which are present in the investigated reaction setup, on the reactive extraction. The results are shown in Fig. 3. The investigated side components did not influence the reactive extraction of metaraminol and no further cationic side components are present in the reaction setup. Thus, we concluded that the reactive extraction of metaraminol is highly selective for metaraminol at the investigated pH. | ||
| Fig. 3 Influence of 10 mM Na2SO4, pyruvate and L-alanine on the mole-related extraction yield during the reactive extraction of metaraminol. The organic phase consisted of 0.25 M oleic acid in 1-octanol. | ||
Process concept for metaraminol in situ product removal
Following the results of the metaraminol extraction with and without side components, the process concept shown in Fig. 4 was developed. Oleic acid is used as reactant and 1-octanol as the diluent. In this concept, in situ reactive extraction of metaraminol is realized resulting in an organic phase loaded with metaraminol oleate and the acidification of the reaction phase due to proton release upon complex formation. This acidification is counteracted via addition of a base to ensure constant pH and optimal operating conditions for the transaminase catalyst. The loaded organic phase is removed and contacted with a second acidic aqueous phase (pH < 3), at which the complexation reaction is reversed. This facilitates the recovery and concentration of metaraminol in the second aqueous phase as was shown in previous investigations.30 | ||
| Fig. 4 Presented process concept for pH-driven in situ extraction and back extraction of metaraminol. | ||
3.2. Identification of suitable process parameters compatible with Cv2025
The amine transaminase from Chromobacterium violaceum Cv2025 was previously characterized as a robust biocatalyst capable of producing, amongst other valuable chiral amines, metaraminol in an enzymatic cascade with high stereoselectivity.6 Therefore, our investigations concentrated further on optimizing the transamination step towards metaraminol with this enzyme. After defining an operation window for metaraminol extraction in a system without enzymatic catalyst, we focused on determining a suitable operational window by assessing the optimal pH and non-inhibiting oleic acid concentration for Cv2025. In order to perform reactive in situ extractions of metaraminol, the product must be mostly present in cationic form (section 3.1). To ensure this, we focused on the neutral pH range more than one pH step below the lowest pKa value of metaraminol (pKa = 8.9). This suitable pH range of pH 7.5–8 has been reported before.34 The initial activity of Cv2025 was therefore tested between a relevant range of pH 6.7 to 7.6. The highest specific activity was determined at pH 7.6 (Fig. 5). | ||
| Fig. 5 Dependence of initial enzyme activities of Cv2025 on the reaction pH. Final enzyme concentration 0.1 mg mL−1. Error bars show standard deviation of three technical replicates. | ||
Since both, the enzyme activity and the extraction, is heavily dependent on the pH in the aqueous reaction phase, a pH of 7.6 was used for the following experiments, as it allowed highest enzyme activity while keeping the reaction pH far below the first pKa of metaraminol. In the next step, different oleic acid concentrations in the organic phase (1-octanol) were screened, as the reactive extraction studies indicated a strong influence of higher oleic acid concentrations on the achievable extraction yield. To assess any influence of the reactant on the biocatalyst, the activity of Cv2025 with oleic acid in the presence of an organic overlay with oleic acid concentration between 0.1 M and 0.5 M was tested in the target reaction with IPA as amine donor. Using IPA in this reaction setup allowed for high conversions permitting to evaluate any effects of the oleic acid on the enzyme (Fig. 6).
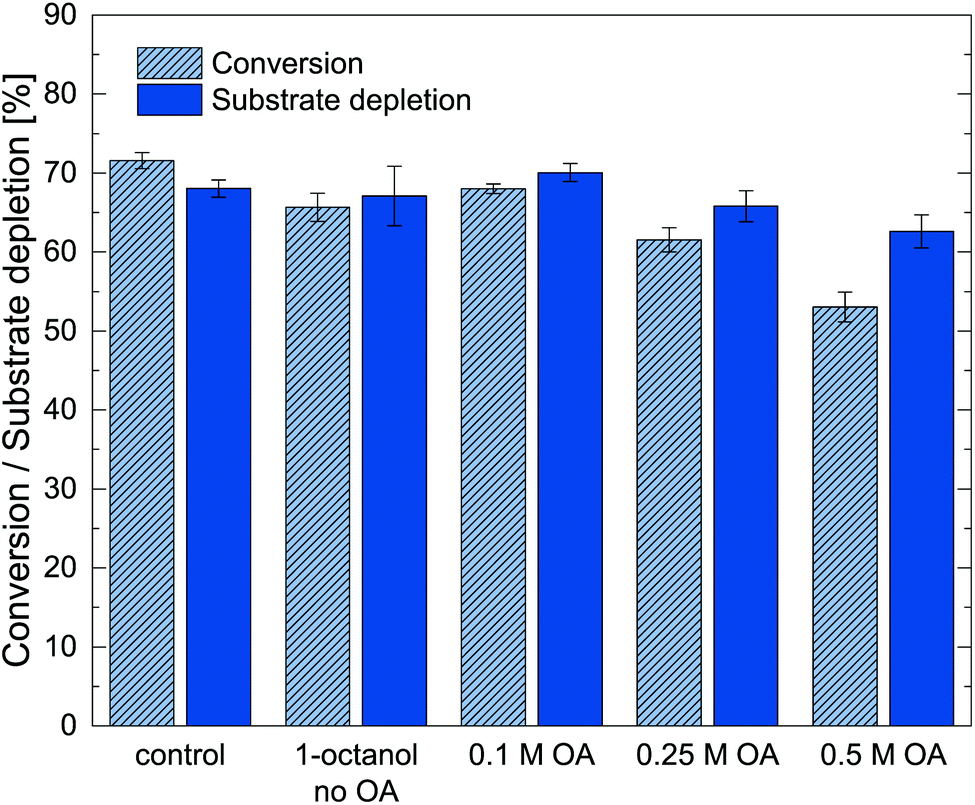 | ||
| Fig. 6 Determined mole-related conversion and substrate depletion in the transamination of (R)-3-OH-PAC to metaraminol in the presence of 10% 1-octanol containing different oleic acid (OA) concentrations after 24 h. IPA was used as amine donor to facilitate high conversions (20 mM (R)-3-OH-PAC, 100 mM IPA, 100 mM HEPES buffer pH 7.5, 0.2 mM PLP, 10 mg mL−1 lyophilized, whole cells containing Cv2025). Error bars show standard deviation of three technical replicates. | ||
The results show that upon the increase of oleic acid concentrations to 0.25 M and 0.5 M the conversion decreased, suggesting that an upper optimal concentration of the reactant was found. In general, the influence of the oleic acid in 1-octanol was small. One has to note that stronger reactive product extraction could have been present for increased oleic acid concentration. The addition of pure oleic acid, however, caused immediate inactivation of the biocatalyst (data not shown). As back extractions of the organic phase was not possible in the small-scale applied here, only samples from the aqueous reaction phase were used to calculate conversions. Consequently, extraction of metaraminol into the organic phase could also account for apparent losses in conversion for 0.5 M oleic acid, even though a complete assessment was not possible in this case. Conclusively, an oleic acid concentration in the range of 0.1 M to 0.25 M was chosen to maintain a high catalytic performance of Cv2025.
To further assess the influence of oleic acid on the enzyme activity, we additionally tested for long-term storage stability under such conditions. In our previous study, Cv2025 exhibited substantial stability in the presence of the polar-protic organic solvents over long time periods.24 To further assess the compatibility of oleic acid with the enzyme, stability assays with Cv2025 in the presence of 0.1 M, 0.25 M and 0.5 M oleic acid in 1-octanol were performed. An organic overlay with the respective reactant concentrations were added to buffered enzyme stock solutions forming a biphasic system. These enzyme solutions were incubated in an overhead shaker at 20 °C for 7 days. The initial activity of the incubated enzyme solution was measured after 1, 2 and 7 days. The results are shown in Fig. 7.
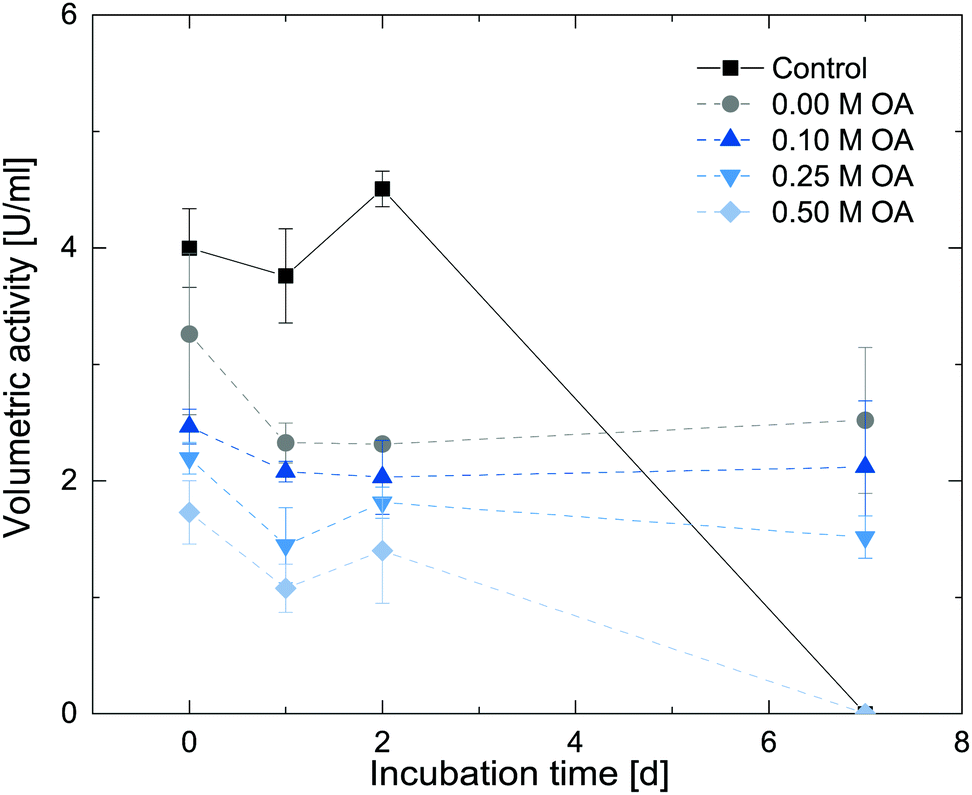 | ||
| Fig. 7 Storage stability of the amine transaminase Cv2025 in the presence of 10% 1-octanol containing different oleic acid concentrations at pH 7.6. The control contains no organic solvent. The initial activity was measured photometrically by the acetophenone production at 300 nm for 5 minutes (100 mM HEPES, 0.1 mM PLP, 2.5 mM α-MBA, 5 mM pyruvate, 0.1 mg mL−1Cv2025, 25 °C). Error bars show standard deviation of three technical replicates. | ||
Compared to the control reactions without addition of the organic solvent, the specific activity was lowered upon addition of the second phase to the enzyme stock. Lower initial rate were observed with increasing oleic acid concentrations in the organic phase. Upon storage under these conditions, the enzyme however retains this activity for 7 days for 1-octanol and with 0.1 M and 0.25 M oleic acid added to it. In contrast, the enzyme stocks without organic solvent or with 0.5 M oleic acid were inactive after 7 days. These results suggest a partly stabilizing effect of 10% 1-octanol on the enzyme as already reported previously. Based on these results, 0.1 M and 0.25 M oleic acid in 1-octanol can be suitable concentrations to test for in situ product removal in the final application.
3.3. Implementation of the reactive extraction coupled with the enzymatic production of metaraminol
Following the results of the investigation of the reactive extraction and the enzymatic reaction, a pH in the range of 7.3–7.6 and oleic acid concentrations of 0.1 M to 0.25 M were identified to be suitable for the production process towards metaraminol from (R)-3-OH-PAC and L-alanine. The (R)-3-OH-PAC, which was used as substrate in the transamination step, was produced in a carboligation reaction (400 mL-scale using 70 mM 3-hydroxybenzaldehyde and a 2.6-fold molar excess of pyruvate) and purified (2.6 g, 64% isolated yield) as given in the ESI.† For the transamination reaction, lyophilized, whole cells containing Cv2025 were used as a cost-efficient biocatalyst formulation.35 The transamination reaction was conducted in a stirred-reactor setup to ensure sufficient suspension of the whole-cell biocatalyst and to use automated pH control. In the biphasic reaction system, 10 mL overlay of 1-octanol containing oleic acid at varying concentrations (4:1 aq:org phase ratio) enabled the reactive in situ extraction of metaraminol. During the experiments no precipitated solids were observed. The product yields of different experiments with and without in situ product removal are shown in Fig. 8.
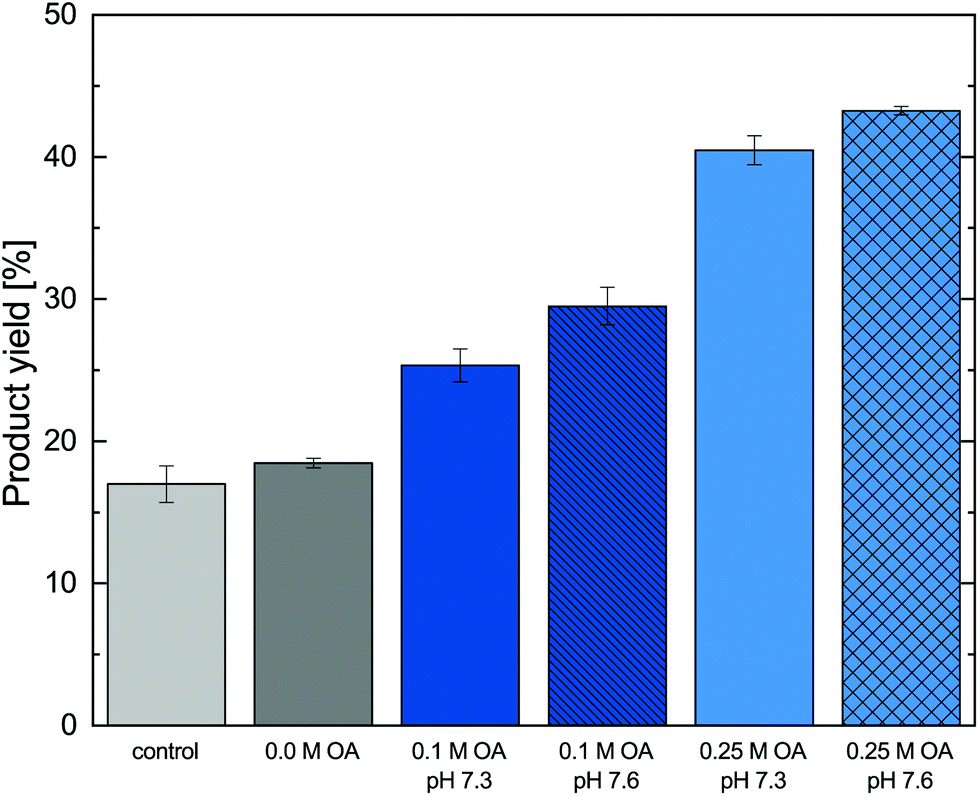 | ||
| Fig. 8 In situ product removal of metaraminol during the enzymatic transamination of (R)-3-OH-PAC using L-alanine as amine donor. The total mole-related product yield was calculated from the sum of metaraminol in the aqueous reaction and the organic extraction phase (after back extraction) after 48 h (40 mM (R)-3-OH-PAC, 250 mM L-alanine, 1 mM PLP, 10 mg mL−1 lyophilized, whole cells containing Cv2025). | ||
The experimental results show that for the control without organic phase and the system with 1-octanol, a reaction equilibrium of 18% was achieved. A maximal conversion in equilibrium of 15–20% was already determined in prior investigations.24 No physical extraction occurred at the investigated pH with solely 1-octanol added. In contrast, the addition of oleic acid to the organic phase led to a significant higher overall of product yields at both investigated pH. Here, slightly higher product yields at pH 7.6 were observed when compared to the same oleic acid concentration at pH 7.3, which can be explained by the pH dependence of the reactive extraction. The addition of oleic acid to the organic phase allows an increase in product yield of 10–15% for 0.1 M and 20–25% for 0.25 M oleic acid. This effect is due to the shift of the reaction equilibrium via in situ product removal, where an increase of the used oleic acid concentration leads to higher overall product yields. As the achievable extraction equilibrium (section 3.2) was also shown to be strongly dependent on the oleic acid concentration, this behavior was expected and leads to the conclusion that highest product yields could be achieved for pure oleic acid. Nevertheless, as higher oleic acid concentrations significantly reduced the transaminase stability, a simple increase in oleic acid concentration is not possible. Thus, alternative solutions such as increased organic phase ratios or repeated batch extractions were investigated.
3.4. Investigation of an increase in the organic to aqueous phase ratio
A higher organic phase ratio was investigated, while the amount of the used substrate was kept constant. Here, no significant increase in product yields could be observed after 48 hours. We suspect, that due to the increased phase ratio, higher amounts of (R)-3-OH-PAC were co-extracted to the organic phase resulting in lower aqueous concentrations and a slower conversion. This slower conversion counteracted the advantages of improved ISPR. Therefore, to increase overall conversions, also the total amount of used (R)-3-OH-PAC must be increased, which would result in higher losses of (R)-3-OH-PAC in batch operation.3.5. Implementation of repeated batch reactive extraction within the enzymatic production of metaraminol
As further alternative, repeated ISPR batch extractions were investigated, which allow to overcome the observed extraction equilibrium of the ISPR process and thus lead to higher total product yields. The biotransformation thereby remains one batch, while the in situ product extraction was broken up into three repeated extraction steps. For this approach, we used the same experimental setup as for the experiments shown in Fig. 8 and performed back extractions of the organic phase after 24 h, 48 h and 72 h hours with a tartaric acid aqueous phase at pH < 3. This allowed to unload the extracted metaraminol, yielding a recovered organic phase, which was then reused for the next ISPR extraction step. During back extractions, the (R)-3-OH-PAC remained largely in the organic phase and did not distribute significantly into the tartaric acid aqueous phase due to the low pH. Nevertheless, the volume of each back extraction phase was kept minimal (10 mL) to further reduce (R)-3-OH-PAC losses and to concentrate the product. The result of the repeated ISPR extraction steps are shown in Fig. 9, depicting both the product yield for three repeated extraction steps as well as the total product yield from three extracted fractions and the remaining aqueous reaction phase. Thereby, the efficiency of each additional extraction step can be assessed.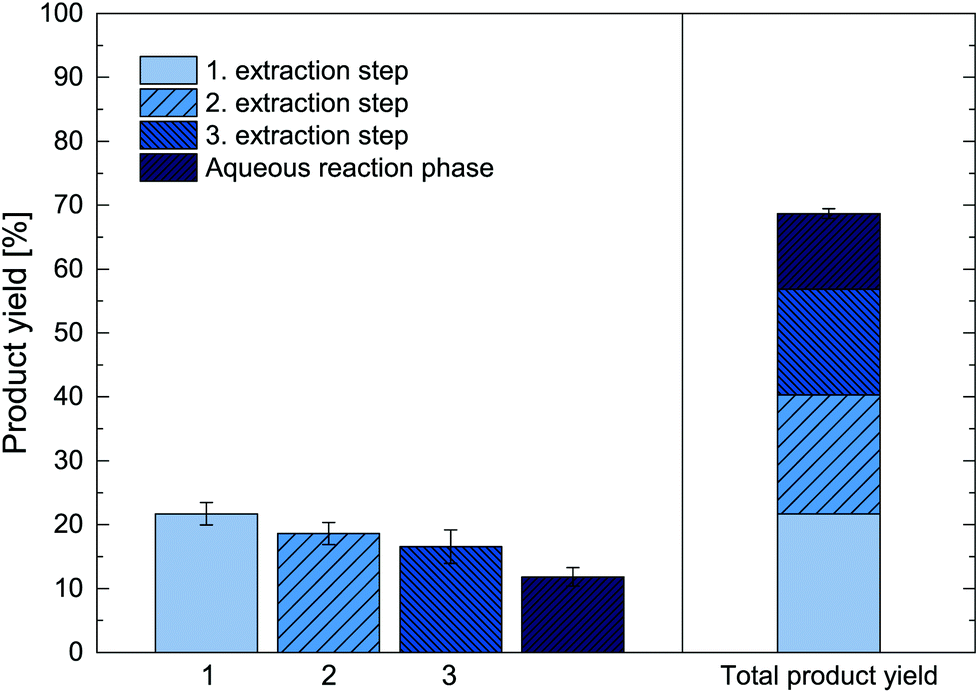 | ||
| Fig. 9 Mole-related product yield of in situ reactive extracted metaraminol during enzymatic transamination after three repeated ISPR extraction steps. The metaraminol amount in the back extraction phases for each of three repeated ISPR extraction steps (1–3) and the remaining product in the aqueous reaction phase (dark blue) are shown. 40 mM (R)-3-OH-PAC, 250 mM L-alanine, 1 mM PLP, 10 mg mL−1 lyophilized, whole cells containing Cv2025, 1-octanol + 0.25 M oleic acid, pH 7.6. The total product yield refers the total amount of produced metaraminol from the initial amount of (R)-3-OH-PAC. Error bars show the standard deviation of two independent experiments. | ||
The results show that in the first extraction step after 24 h, a product yield of 21% was achieved, referring only to the amount of product in the first extraction phase. After back extraction of the organic phase, it was reused for another two extraction step in the biphasic reaction system, each was incubated for another 24 h. In both extraction steps, an additional 19 and 17% product yield was achieved. The decrease in extraction yield in each repetition is expected, as in each extraction step a lower substrate concentrations and enzymatic rates and thus also lower product concentration is reached. The aqueous concentration of metaraminol remained stable at 6 mM, corresponding to 12% product yield that remained in the aqueous reaction phase at the end of the experiment. No solid precipitates were observed.
Thus, by applying a second and third extraction step, the total product yield was increased by additional 17% and 12%. After three repetitions, the repeated ISPR extraction steps achieved an increase of the total product yield of the enzymatic reaction to a total value of 69% (Fig. 9, right), corresponding to a 3.8-fold increase compared to the reaction without ISPR (Fig. 8). In total, 79% of the produced metaraminol were recovered in the acidic back extraction phase. As multiple repeated extractions allow to significantly boost the total achievable product yield, investigations into a continuous operation of the reactive extraction are required to further push the total product yield as well as the overall space–time-yield. Several aspects of the presented process are indicative for this. The results indicate that the organic extraction phase can be recycled and reused at least three times, which significantly reduces process waste streams and contributes to the overall greenness of the process. Furthermore, the biocatalyst remained active for up to 72 h in the repeated batch experiments, suggesting high operational stability in relevant process conditions, in addition to profound storage stability in the presence of 1-octanol and oleic acid as shown in section 7. This good operational stability allows for long-term application of the biocalatyst in multiple ISPR-enhanced production cycles, which significantly reduces catalyst costs and ecological impact.
Nevertheless, during operation slow phase separation behavior and the formation of a small third interphase between organic and aqueous phase could be observed. These were traced back to cell debris and degraded proteins and lipids, which accumulated at the organic–aqueous interface and may complicate large scale applications of the presented process. To overcome this, spatial separation of reaction and extraction via retention of the biocatalyst through whole cell immobilization techniques could be used. This might expand the reusability of the biocatalyst, substantially enhancing the specific productivity in the reactive extraction production process for metaraminol. Beneficial effects of continuous extraction regimes integrated in biocatalytic syntheses have been shown in previous studies.36,37 An elaborate review on the benefits and challenges of reactive ISPR-integrated biocatalytic processes was presented by Fellechner et al.38
4. Conclusion
The chiral amino alcohol metaraminol is an active pharmaceutical ingredient for hypotension treatment and can, apart from direct pharmaceutical applications, serve as a precursor molecule for other bioactive compounds. One large substance class relying on this precursor are tetrahydroisoquinoline derivatives. In contrast to lengthy and complex chemo-catalytic synthesis routes, metaraminol can be produced in an enzymatic cascade in two subsequent steps starting from 3-OH-benzaldehyde and (bio-based) pyruvate under mild process conditions. The transamination step can make use of bio-based L-alanine as the amine donor. Both, pyruvate and L-alanine, can be produced by microbial transformations from second generation feedstock, and the microbial production of 3-OH-benzoate was shown.39,40 In combination with the enzymatic reduction of benzoates, as recently described by Weber et al., the entire cascade to metaraminol can be established on solely bio-based precursor molecules.41 However, the low equilibrium constant of (enzymatic) transaminations with L-alanine as amine donor results in low conversions, requiring an integrated solution to shift the reaction equilibrium. Here, we showed a reactive extraction-based ISPR strategy that realizes the equilibrium shift and product recovery in a simple setup. We defined relevant process parameters for the extraction system and enzyme compatibility screenings to define an operational window for the in situ product extraction of metaraminol. Upon integration with the enzymatic production, the reactive ISPR of metaraminol increased the total product yield to 42–44% in batch mode and 69% after three repeated extractions steps. In comparison to the physical extraction of metaraminol, the reactive extraction principle allows increased maximal extraction efficiencies while also allowing operation in the neutral pH range preferable for the investigated transaminase reaction system. Furthermore no background reactions were observed as seen during physical extraction, allowing for very high selectivities (>95%). Thus, reactive extraction is more favorable for the transaminase reaction system. The product, metaraminol was recovered by a simple back extraction step using an acidic tartaric acid phase, providing the amino alcohol in an aqueous phase for further (chemo)enzymatic conversion or further work-up. A clear advantage of this approach is the sustainable extraction system consisting of 1-octanol as the diluent, which can be produced microbially from renewable resources,42 and oleic acid as the reactant, which can be derived from (waste) plant oil.43 The possibility to recycle the extraction phase, as partly shown in the repeated batch application, further adds to a sustainable process concept. While this approach presents an efficient method to make use of L-alanine as amine donor in enzymatic transaminations with substantially increased yields and efficient product recovery, some distinct points of optimization are clearly evident. The main limitation in the presented process concerns the maximal loading of the oleic acid and the consequently reduction in free oleic acid in the organic extraction phase. Since higher oleic acid concentrations and organic phase ratios did not succeed to increase conversions in batch mode further, the expansion of the repeated batch strategy to a continuous extraction mode can be advantageous. In conclusion, reactive extraction using oleic acid as reactant was shown to be a suitable strategy to improve enzymatic transaminations with L-alanine and might contribute to the integration of more efficient yet sustainable enzymatic processes in the future.Author contributions
Experimental contribution in chapter 3.1: MD. Experimental contribution in chapter 3.2 and 3.3: LG. Data analysis and interpretation: LG and MD. The manuscript was written by MD and LG. Project conception: AJ and DR. All authors revised and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The scientific activities of the Bioeconomy Science Center were financially supported by the Ministry of Culture and Science within the framework of the NRW Strategieprojekt BioSC (No. 313/323-400-002 13). We sincerely thank Helen C. Hailes and John M. Ward, University College London, for providing the plasmids for the expression of the amine transaminase from Chromobacterium violaceum (Cv2025).References
- P. de María, G. de Gonzalo and A. Alcántara, Catalysts, 2019, 9, 802 CrossRef.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- R. de Regil and G. Sandoval, Biomolecules, 2013, 3, 812–847 CrossRef PubMed.
- M. S. Malik, E.-S. Park and J.-S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- M. Nishihachijo, Y. Hirai, S. Kawano, A. Nishiyama, H. Minami, T. Katayama, Y. Yasohara, F. Sato and H. Kumagai, Biosci., Biotechnol., Biochem., 2014, 78, 701–707 CrossRef CAS.
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS.
- J.-S. Shin and B.-G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS PubMed.
- C. Xiang, S. Wu and U. T. Bornscheuer, Bioorg. Med. Chem., 2021, 43, 116271 CrossRef CAS.
- M. T. Gundersen, R. Abu, M. Schürmann and J. M. Woodley, Tetrahedron: Asymmetry, 2015, 26, 567–570 CrossRef CAS.
- A. W. H. Dawood, M. S. Weiß, C. Schulz, I. V. Pavlidis, H. Iding, R. O. de Souza and U. T. Bornscheuer, ChemCatChem, 2018, 10, 3943–3949 CrossRef CAS.
- P. Tufvesson, C. Bach and J. M. Woodley, Biotechnol. Bioeng., 2013, 111, 309–319 CrossRef.
- P. Kelefiotis-Stratidakis, T. Tyrikos-Ergas and I. V. Pavlidis, Org. Biomol. Chem., 2019, 17, 1634–1642 RSC.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2017, 118, 349–367 CrossRef.
- A. Gomm, W. Lewis, A. P. Green and E. O'Reilly, Chemistry, 2016, 22, 12692–12695 CrossRef CAS PubMed.
- S.-W. Han and J.-S. Shin, Biosci., Biotechnol., Biochem., 2014, 78, 1788–1790 CrossRef CAS PubMed.
- N. Richter, J. E. Farnberger, D. Pressnitz, H. Lechner, F. Zepeck and W. Kroutil, Green Chem., 2015, 17, 2952–2958 RSC.
- M. Voges, F. Schmidt, D. Wolff, G. Sadowski and C. Held, Fluid Phase Equilib., 2016, 422, 87–98 CrossRef CAS.
- M. Voges, C. Fischer, D. Wolff and C. Held, Org. Process Res. Dev., 2017, 21, 1059–1068 CrossRef.
- M. Voges, R. Abu, M. T. Gundersen, C. Held, J. M. Woodley and G. Sadowski, Org. Process Res. Dev., 2017, 21, 976–986 CrossRef CAS.
- D. Hülsewede, J.-N. Dohm and J. von Langermann, Adv. Synth. Catal., 2019, 361, 2727 Search PubMed.
- T. Börner, G. Rehn, C. Grey and P. Adlercreutz, Org. Process Res. Dev., 2015, 19, 793–799 CrossRef.
- K. Mack, M. Doeker, L. Grabowski, A. Jupke and D. Rother, Green Chem., 2021, 23, 4892–4901 RSC.
- A. Eggert, T. Maßmann, D. Kreyenschulte, M. Becker, B. Heyman, J. Büchs and A. Jupke, Sep. Purif. Technol., 2019, 215, 463–472 CrossRef CAS.
- J. Gorden, E. Geiser, N. Wierckx, L. M. Blank, T. Zeiner and C. Brandenbusch, Eng. Life Sci., 2017, 17, 809–816 CrossRef CAS.
- K. L. Wasewar, A. M. Heesink, G. F. Versteeg and V. G. Pangarkar, J. Biotechnol., 2002, 97, 59–68 CrossRef CAS.
- L. Tang, S. Choi, R. Nandhakumar, H. Park, H. Chung, J. Chin and K. M. Kim, J. Org. Chem., 2008, 73, 5996–5999 CrossRef CAS PubMed.
- A. Bednarz, A. C. Spieß and A. Pfennig, J. Chem. Technol. Biotechnol., 2017, 92, 1817–1824 CrossRef CAS.
- M. Doeker, V. Hüttche and A. Jupke, Sep. Purif. Technol., 2021, 118229 CrossRef CAS.
- R. N. C. Utomo, W.-J. Li, T. Tiso, C. Eberlein, M. Doeker, H. J. Heipieper, A. Jupke, N. Wierckx and L. M. Blank, ACS Sustainable Chem. Eng., 2020, 8, 17466–17474 CrossRef.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81, 8244–8248 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- U. Kaulmann, K. Smithies, M. E. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- P. Tufvesson, J. Lima-Ramos, N. A. Haque, K. V. Gernaey and J. M. Woodley, Org. Process Res. Dev., 2013, 17, 1233–1238 CrossRef CAS.
- E. Ritter and I. Smirnova, Chem. Ing. Tech., 2018, 90, 348–357 CrossRef.
- E.-L. Teo, G.-K. Chuah, A. R. Huguet, S. Jaenicke, G. Pande and Y. Zhu, Catal. Today, 2004, 97, 263–270 CrossRef CAS.
- O. Fellechner, M. Blatkiewicz and I. Smirnova, Chem. Ing. Tech., 2019, 91, 1522–1543 CrossRef CAS.
- Y. Zhou, Z. Li, X. Wang and H. Zhang, Eng. Life Sci., 2019, 19, 389–395 CrossRef CAS PubMed.
- S. Wang, C. Fu, K. Liu, J. Cui, H. Hu, W. Wang and X. Zhang, Front. Bioeng. Biotechnol., 2020, 8, 622226 CrossRef.
- D. Weber, D. Patsch, A. Neumann, M. Winkler and D. Rother, ChemBioChem, 2021, 22, 1823–1832 CrossRef CAS.
- M. K. Akhtar, H. Dandapani, K. Thiel and P. R. Jones, Metab. Eng. Commun., 2015, 2, 1–5 CrossRef PubMed.
- R. Elkacmi, N. Kamil, M. Bennajah and S. Kitane, BioMed Res. Int., 2016, 2016, 1–9 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03289e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |