
Lactation-dependent vertical transmission of natural probiotics from the mother to the infant gut through breast milk†
Ce
Qi
a,
Jingbo
Zhou
a,
Huayu
Tu
a,
Rundan
Tu
a,
Hong
Chang
b,
Jie
Chen
c,
Duo
Li
a,
Jin
Sun
 *a and
Renqiang
Yu
*d
*a and
Renqiang
Yu
*d
aInstitute of Nutrition and Health, Qingdao University, Qingdao 266071, PR China. E-mail: sunj@qdu.edu.cn; Fax: +86 18151559959; Tel: +86 18151559959
bDepartment of Pediatric Cardiology, Nephrology and Rheumatism, The Affiliated Hospital of Qingdao University Medical College, Qingdao 266003, PR China
cDepartment of Obstetrics, Affiliated Hospital of Qingdao University, Qingdao 266000, PR China
dDepartment of Neonatology, The Affiliated Wuxi Maternity and Child Health Care Hospital of Nanjing Medical University, Wuxi 214002, PR China. E-mail:. E-mail: yurenqiang@njmu.edu.cn
First published on 18th November 2021
Abstract
The transmission of certain bacteria from the mother's gut to the infant's gut via breast milk (BM) is critical for the offspring's immune system development. Dysbiosis of the BM microbiota can be caused by a variety of reasons, which can be influenced by probiotics delivered via the enteromammary route. The goal of this study was to investigate the bacteria that can be transmitted from the mother to the infant's intestine during various lactation periods in 19 mother–child dyads. Bacterial transmission is most common during the colostrum phase when bacteria with certain amplicon sequence variants (ASVs) enter the newborn intestine and inhabit it permanently. We have established that anaerobic gut-associated bacteria, such as Faecalibacterium, Blautia and Lachnoclostridium, transfer from the mother to the infant's gut with lactation dependence using the idea of weighted transfer ratios. Streptococcus salivarius, Bifidobacterium longum, and Lactobacillus gasseri are transferred from the maternal gut to the BM, as well as from the BM to the newborn gut, depending on different ASVs. These findings suggest that isolation of key microorganisms from breast milk could be utilized to modify the microbiota of BM or newborns by giving the mother a probiotic or adding it to artificial milk to promote neonatal health.
Introduction
Probiotics are live microorganisms that, when administered in adequate amounts, confer a health benefit to the host.1 Well-studied and commercially available species include Bifidobacterium (adolescentis, animalis, bifidum, breve, and longum) and Lactobacillus (acidophilus, casei, fermentum, gasseri, johnsonii, reuteri, paracasei, plantarum, rhamnosus, and salivarius). Probiotics are approved in many countries for administration to newborns, to prevent necrotising enterocolitis, to reduce the risk of eczema, to treat colic or to simply supplement formula. The benefits of these microbes on respiratory and skin health have been documented, but beyond their influence on the immune system and intestinal permeability, the mechanisms have not been fully explored.2Human milk (BM) contains a wide range of bacterial types, and with so many confounders it is challenging to correlate the species with distant side effects.2 The entero-mammary pathway3 was hypothesized to be responsible for the vertical transmission of gut-specific bacteria, such as Bacteroides, Bifidobacterium and Clostridium.4 BM microbiota is a more essential source of live bacteria for daily continuous inoculation in a full-term vaginally born baby.5 BM includes 102 to 104 live bacterial cells per ml.6 A range of these bacteria even in low counts per ml would lead to a high diversity infant gut, yet, it is dominated by bifidobacteria and therefore is defined as of low diversity and linked to reduced risk of certain chronic illnesses in adulthood.7 Breast milk-derived beneficial gut bacteria may help reduce the risk of chronic diseases such as asthma, obesity, allergies, dermatitis, inflammatory bowel disease, and neurodevelopmental disorders.8,9 They work together with breast milk oligosaccharides to create a symbiotic environment in which the infant's innate intestinal immunity functions and the intestine develops.10
Dysbiosis of the BM microbiota can be caused by a variety of conditions, which can harm the mother's and child's health. Antibiotic usage by mothers during pregnancy or nursing, for example, dramatically lowers the amounts of Lactobacillus and Bifidobacterium in BM.4 The BM microbiota diversity of obese mothers was considerably lower.11 Maternal mastitis was linked to reduced maternal milk microbiota diversity and raised relative levels of Staphylococcus aureus and Staphylococcus epidermidis.12
The enteromammary pathway for delivering probiotics may improve the gut microbiota and mammary gland health of the offspring. This also provides the opportunity to develop personalized probiotics for infants.13 A number of microorganisms isolated from BM have been tested as probiotics.14–17 When women are given BM-derived Lactobacillus as a probiotic, the same strain can be found in BM.18 Prenatal and postnatal intake of a multispecies probiotic could minimize the incidence of allergic illnesses in allergy-prone infants.19 When mothers consume non-BM probiotics, however, these bacteria are rarely transported to the BM, showing that BM-derived probiotics have specific features that allow for vertical transmission.20 The deliberate isolation and screening of BM-derived probiotics that are not restricted to Lactobacillus and Bifidobacterium will be facilitated by a full understanding of the commensal bacteria conveyed to the infant's gut via the enteromammary pathway. While some recent research studies have used paired samples to identify the co-occurring taxa in the mother's gut, BM, and infant's gut, statistical proof of vertical species transmission is absent. Weighted transfer ratios (WTRs) has been proposed a unique statistical technique for detecting bacterial transmission.20
Here, we propose the hypothesis that mastitis or infant-specific diseases can be prevented by personalized probiotics with the intestinal-mammary transmission property when administered to lactating mothers or infants. This study aims to elucidate potentially common commensal bacteria in healthy individuals that pass through the intestinal–lacteal transmission to the infant's gut. We investigated the bacterial composition of the mother's gut and BM at different phases of lactation, and the infant's gut in paired samples in this study. The WTR was used to assess lactation-specific bacterial transmission from the mother's gut to the BM and infant gut, as well as the genus colonizing the infant gut at the colostrum stage surviving to the mature milk stage.
Experimental section
Volunteers and sample collection
Nineteen mother and infant dyads were recruited after delivery at The Affiliated Wuxi Maternity and Child Health Care Hospital of Nanjing Medical University, China, from March 2018 to June 2020 (Fig. 1). The recruitment criteria for mother–infant dyads were as follows: (i) vaginal delivery at term (≥37 weeks gestation), (ii) exclusive breastfeeding during the sampling period, (iii) no exposure of either the mother or infant to an antibiotic/probiotic during the pregnancy, intrapartum or postnatal period. Clinical data, including maternal age and weight, delivery mode, neonatal weight, lactation stage, gestational time, blood biochemistry, and routine blood tests, were collected. The pregnancy duration was >38 weeks for all mothers. Maternal faeces, BM, and infant faeces samples were collected within 7 days (colostrum stage), 8–14 days (transitional milk stage) and the 42nd day (mature milk stage) after delivery. The protocol (registration number 2020-01-0302-03) was approved by the Ethics Committee of Wuxi Maternal and Child Health Care Hospital and all experiments were performed in compliance with the Chinese relevant laws and institutional guidelines. Written informed consent was obtained from all donors.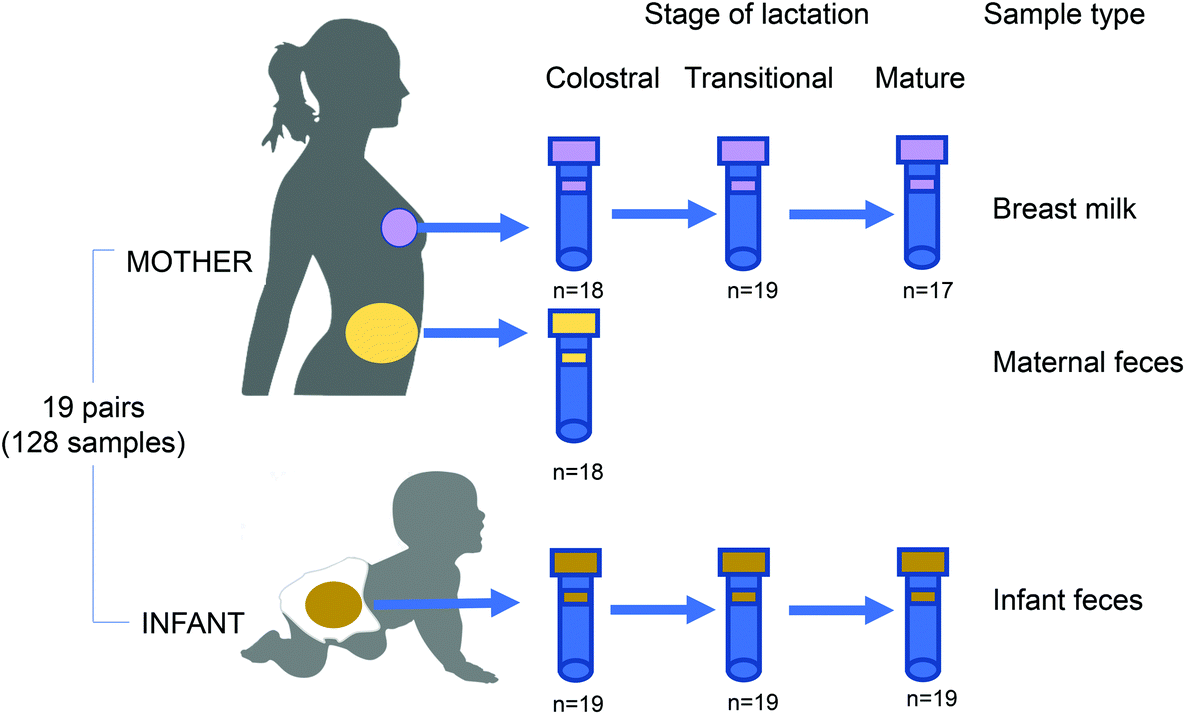 | ||
| Fig. 1 16S rRNA gene sequencing of the microbiome of mother–infant pairs. Samples were collected from 19 mother–infant pairs at colostrum (0–7 days), transitional (8–14 days) and mature (>15 days) stages of lactation. | ||
16S rDNA sequencing and processing
The procedures for total DNA extraction, amplification, and sequencing of the V3–V4 region of the 16S rRNA genes, and the construction of the sequencing library are illustrated in ESI. The raw paired-end reads were primer-trimmed using the Cutadapt software (version 2.3).21 The downstream amplicon bioinformatic analyses were performed using EasyAmplicon v1.0.22 Next, dereplication was performed using the –derep_fullength command of VSEARCH (v2.15).23 After that, nonredundant sequences were denoised into ASVs via the unoise3 command of USEARCH (v10.0).24 Taxonomy assignments for all ASVs were carried out using the sensitive and highly accurate Kraken2 (v2.0.9) using the MiniKraken2_v1_8GB Kraken 2 database built from the RefSeq bacteria and RDP databases.25 In the filtration and normalization module, both Kraken2 feature table outputs were filtered with the Kraken 2 filter command (confidence threshold: 0.95, indicated ∼95% precision to the lowest common ancestor). The USEARCH command cluster_agg was used to construct a phylogenetic tree. The sequences of all samples were rarefied to rarefaction before alpha and beta diversity analysis.Shotgun metagenomic sequencing
DNA was sequenced using an Illumina HiSeq 3000 at GENEWIZ Co. (Suzhou, China). Cluster generation, template hybridization, isothermal amplification, linearization, and blocking denaturing, and hybridization of sequencing primers were performed according to the workflow specified by the service provider. Libraries were constructed with an insert size of approximately 500 bp, followed by high-throughput sequencing to obtain paired-end reads with 150 bp in the forward and reverse directions. For data quality control, Prinseq26 was employed to (1) trim reads from the 3′ end until reaching the first nucleotide with a quality threshold of 20; (2) remove read pairs when either read was <60 bp or contained “N” bases, and (3) de-duplicate reads. In addition, reads that could be aligned to the human genome (Homo sapiens, UCSC hg19) were removed (aligned with Bowtie227 using –reorder–no-hd–no-contain–dovetail). High-quality paired-end reads from each sample were used for species profiles by MetaPhlAn3, with high accuracy for taxonomic profiling.28 The sequencing data supporting the results of this article are available in the NCBI Sequence Read Archive, BioProject PRJNA542027 (accession # SUB5578272).Statistical analysis
EasyAmplicon v1.0 was used for alpha diversity analysis of Shannon's index and the Chao1 index. Differences between groups were used as measures of intra-individual diversity and were evaluated using Wilcoxon rank-sum tests. The relative abundances at the genus level were compared between sample types using Student's T-test for paired samples, and differences in β-diversity were visualized with principal coordinates analysis (PCoA) plots.Vertical transfer of the maternal gut microbiota to BM, infant's gut, and that from BM to infant's gut were further analyzed for all ASVs without rarefaction according to a method developed by Mortensen et al.29 Briefly, univariate Fisher's exact test was carried out to compare the presence/absence of ASVs between dyads of maternal gut/BM, maternal gut/infant gut, and BM/infant gut, respectively, and an OR of 1 for transfer with a one-sided p-value toward the null hypothesis was recorded. Only ASVs showing the presence/absence in both the giving and receiving compartments were included in the analysis. WTR was used as an overall measure of transfer. It is defined as the ratio of the sum of the positive and negative associations from the volcano plot. It is described as follows:
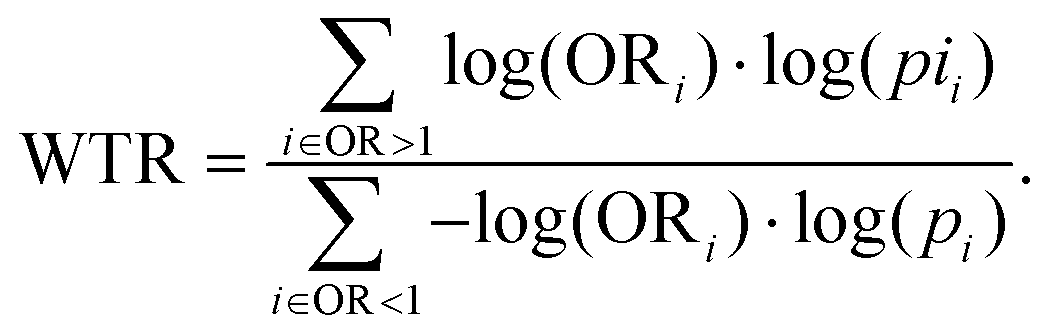
WTR should be around one in cases of no transfer and more significant when the transfer occurred, but due to the high sparsity, the null distribution is not always centred on 1 and ranges from 1.08 to 1.39.
Results
Shared amplicon sequence variants (ASVs) among samples
We first investigated the co-occurrence of high abundance ASV (>0.05%) among different samples (Fig. 2). The largest abundance of ASVs was found in the colostrum (339) and infant faeces of the same stage (313). Co-occurrence of ASV in infant faeces of the colostrum stage, mature milk, and infant faeces of the mature milk stage was the most common (72). The faeces of newborns in the colostrum and mature milk stages came next (31). Bifidobacterium (122), Fusicatenibacter (64) and Blautia (56), respectively, had the largest number of ASVs in the colostrum. As lactation progressed, the number of dominant ASVs in these three genera decreased, and in mature milk, the dominant ASVs in Bifidobacterium and Fusicatenibacter were only 51 and 45, respectively. Staphylococcus and Bifidobacterium are the first two genus with the most ASVs in the feces of infants at the colostrum stage, but the number of ASVs in Staphylococcus is reduced by half at the mature milk stage. Throughout the stage, two Blautia powerful ASVs were consistently present. At the mature milk stage, the number of prominent ASVs of different genera in the mother's and infant's faeces was rather evenly distributed. This shows that bacterial inoculation from the mother's milk to the infant's gut occurs mostly during the colostrum period, with a small number of variations that can adapt to the infant's intestinal environment colonizing later in the evolutionary process.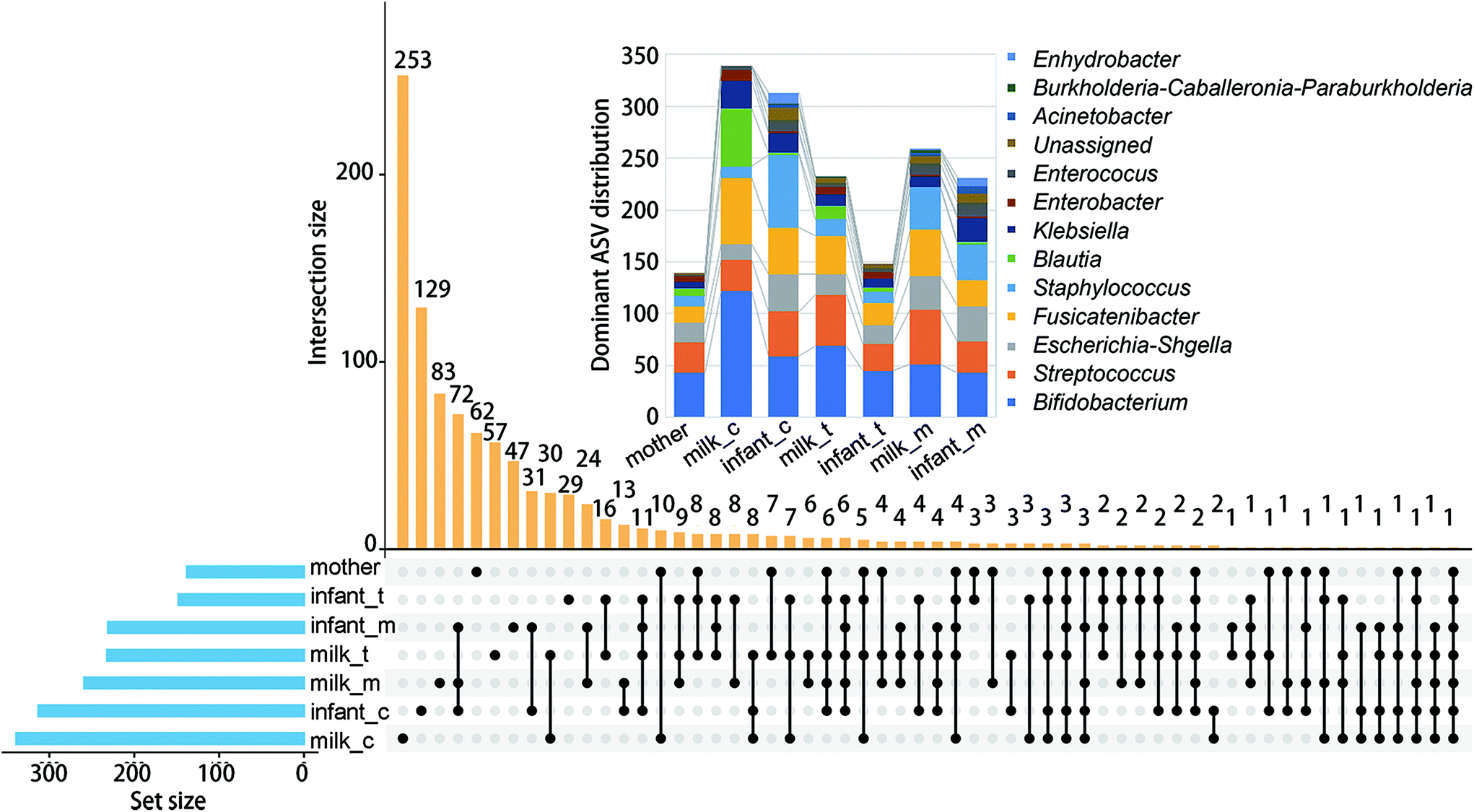 | ||
| Fig. 2 ASV Venn diagrams depicting overlapping taxa and distribution of ASV related to a specific genus (inset). Paired sequencing samples including maternal faeces (mother), BM at colostrum, transition and mature stages (milk_c, milk_t, milk_m) and infant faeces (infant_c, infant_t, infant_m). | ||
The alpha diversity of BM and newborn intestinal microbiota changes over time as the breastfeeding period progresses
The alpha diversity of microbiota from the mother's faeces was significantly different from all other samples (P < 0.01), except for the Shannon index of mature milk (Fig. 3). The richness of the mature stage of the infant's faeces differed greatly from that of the colostrum stage. The mature stage's Shannon index was significantly different from both the colostrum and transitional stages (P < 0.05). Only during the colostrum stage do both the richness and Shannon index demonstrate a substantial difference between BM and newborn faeces (P < 0.05). | ||
| Fig. 3 The richness and Shannon and weighted and unweighted UniFrac PCo2 distances of different sample microbiota. Paired sequencing samples included maternal faeces and breast milk and infant faeces from colostrum (C), transitional (T) and mature (M) lactation stages. &, significantly different from the mother (<0.01). *, significantly different between breast milk and infant faeces of the same lactation period; #, significantly different between the infant faeces of the mature milk stage; Student's t-test for paired samples was used. | ||
The beta diversity of BM and newborn gut microbiota changes over time as the breastfeeding period progresses
There were substantial changes between the intestinal microbiota of newborns in different lactations and the BM microbiota in the same period, according to unweighted and weighted UniFracPCo2 (P < 0.05) (Fig. 3). Fig. 4 shows that maternal intestine bacteria differed significantly from the BM and newborn intestinal microbiota, but heterogeneity was lower than the latter. Individual heterogeneity in both BM and newborn gut microbiota was higher in mature milk than in early BM. | ||
| Fig. 4 Compositional changes in the microbiota. Weighted and unweighted UniFrac ordination of microbiota coloured according to the sample type and joined within individuals. Paired sequencing samples included maternal faeces (mother), breast milk at colostrum, transition and mature stages (milk_c, milk_t, milk_m) and infant faeces (infant_c, infant_t, infant_m). Ellipses indicate the mean ± SD. | ||
Putative oxygen-related phenotypes and functional pathway of the microbiome
The maternal and newborn gut microbiomes have a low abundance of aerobic bacteria (Fig. 5). All BM samples were substantially higher in aerobic bacteria than in maternal faecal samples (p < 0.01) (Fig. 5). Because of the significant prevalence of Bifidobacterium in infant faeces (Fig. 6), the faecal anaerobic bacteria were greater than the BM microbiota in infants of different breastfeeding periods (P = 0.003). They also exhibited similar quantities of facultatively anaerobic bacteria, which were higher than those found in the mother's faeces. Small levels of oxidative stress-tolerant bacteria were found in mature BM and infant faeces of colostrum phases, which were much greater than the mother's faecal samples.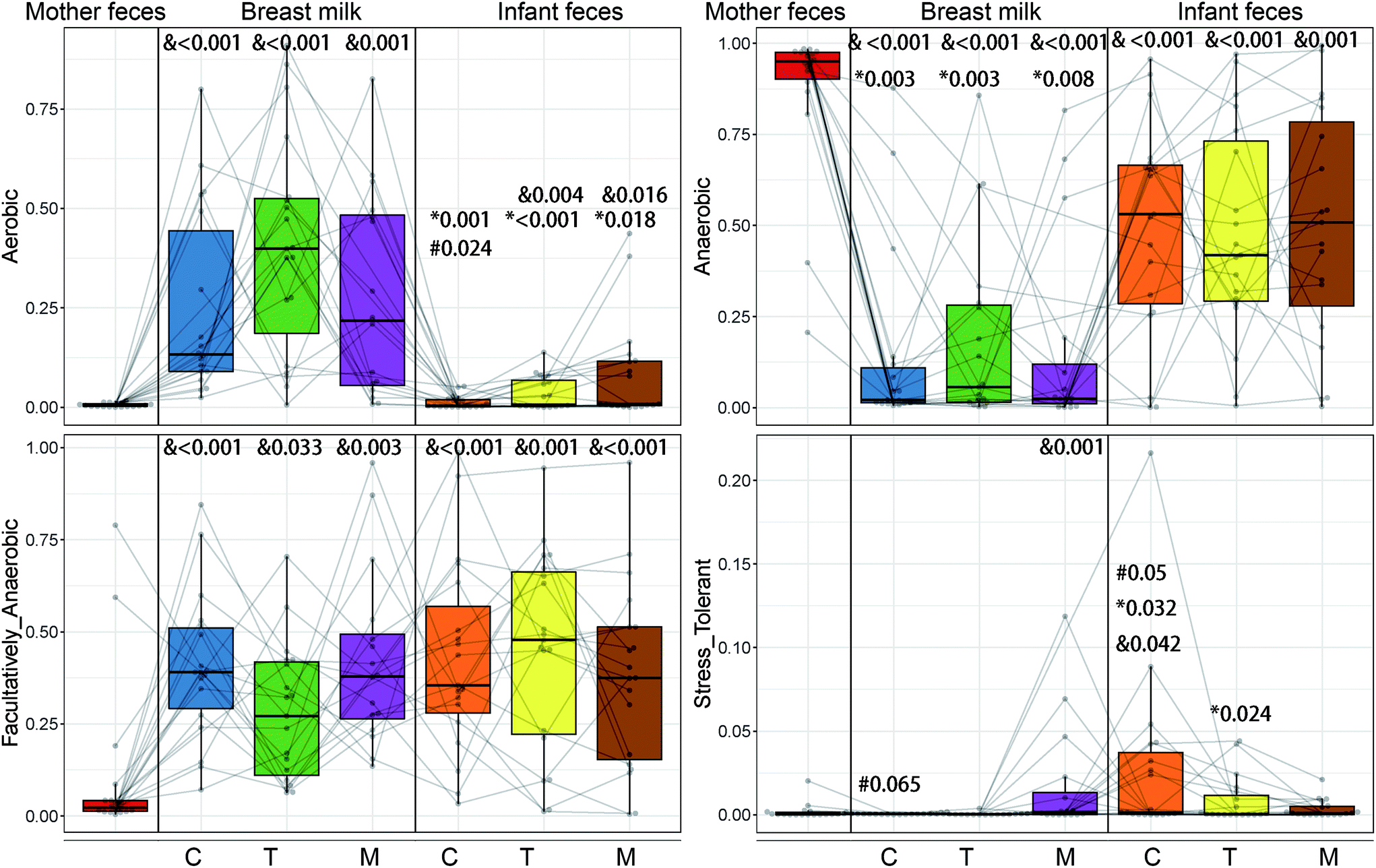 | ||
| Fig. 5 Bugbase predicted the proportion of the bacteria involved in the oxygen-related phenotypes. Paired sequencing samples included maternal faeces and breast milk and infant faeces from colostrum (C), transitional (T) and mature (M) lactation stages. &, significantly different from the mother (<0.01). *, significantly different between breast milk and infant faeces of the same lactation period; #, significantly different between the infant faeces of the mature milk stage, and adjusted P values are shown with each label. Student's t-test for paired samples was used. | ||
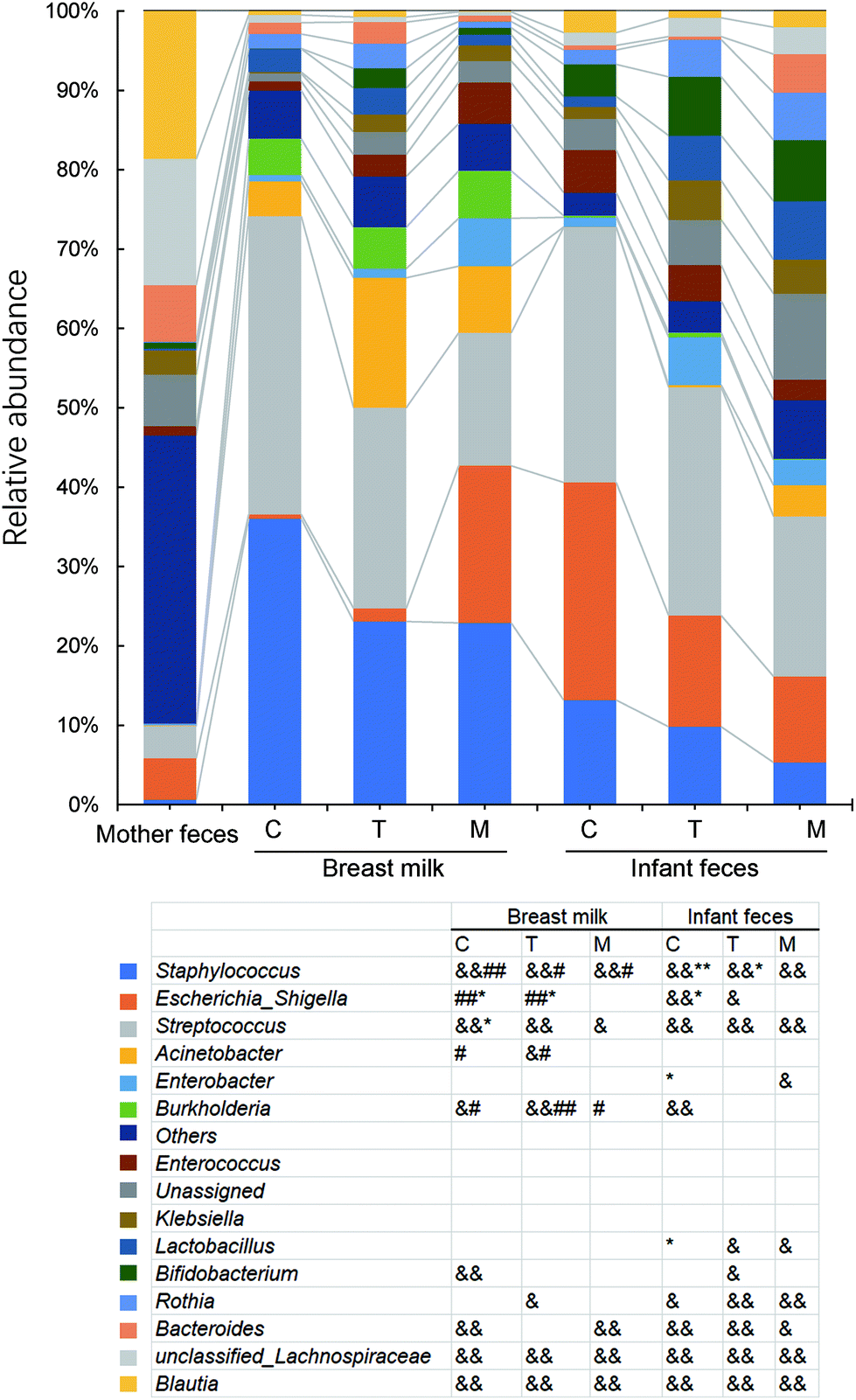 | ||
| Fig. 6 Relative abundance of the top ten genera in the different types of samples. Paired sequencing samples included maternal faeces and breast milk and infant faeces from colostrum (C), transitional (T) and mature (M) lactation stages. The colour order of the legend is based on the relative abundance of the genera in the mature milk in descending order. & (p < 0.05) and && (p < 0.01), significantly different from the mother. * (p < 0.05) and ** (p < 0.01), significantly different between breast milk and infant faeces of the same lactation period; # (p < 0.05) and ## (p < 0.01), significantly different between the infant faeces of the mature milk stage. Student's t-test for paired samples was used. | ||
Furthermore, based on the rarefied 16S rRNA data, functional pathway abundances were predicted using PICRUSt2 (Fig. 7). The functional difference between BM and newborn faeces grows from 9 to 16 pathways from colostrum to mature milk. Glucose1pmetab-PWY, P124-PWY, and PWY-5384 (sucrose degradation IV (sucrose phosphorylase)) were the most significant pathways enriched in newborn faeces compared to BM. In some strains of Escherichia coli, the glucose1pmetab-PWY pathway allows them to use glucose as their sole source of carbon and energy. For glucose metabolism, P124-PWY and PWY-5384 are found in the genus Bifidobacterium. PWY-3781 is a function involved in aerobic respiration that is unique to all stages of the BM microbiota, primarily found in Pseudomonas putida. The microbiota of the colostrum is particularly rich in two pathways. Lactose breakdown is linked to LACTOSECAT-PWY in Firmicutes such as Lactococcus lactis, Staphylococcus aureus, and Streptococcus mutans. In bacteria like Helicobacter pylori, the tricarboxylic acid cycle REDCITCYC is involved in aerobic respiration. The transition and mature milk microbiota were particularly enriched for the other two pathways. FASYN-ELONG-PWY is related to saturated fatty acid elongation in bacteria including E. coli and Streptococcus pneumoniae. PWY-7094 is related to fatty acid biosynthesis in Pseudomonas aeruginosa.
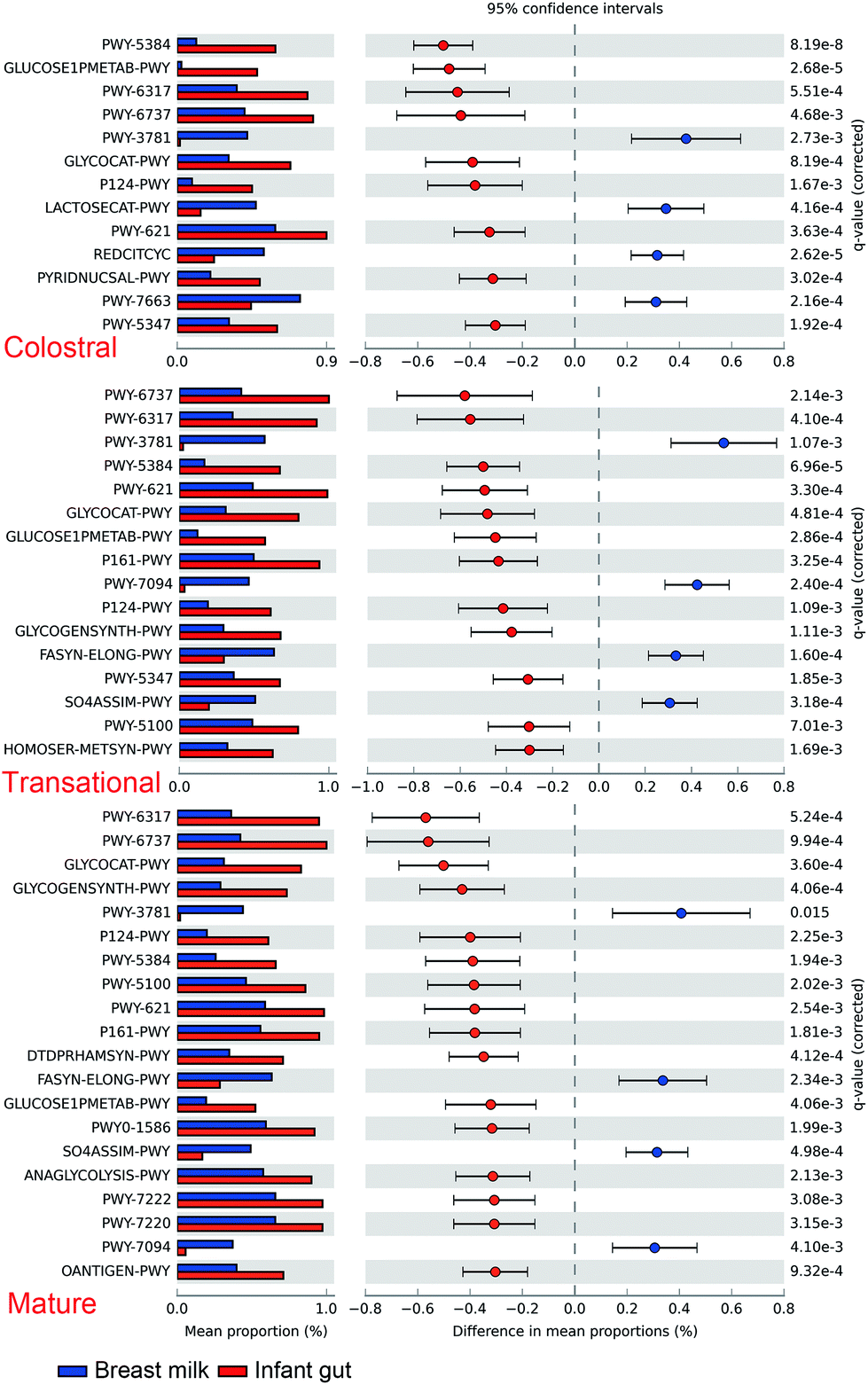 | ||
| Fig. 7 Difference of PICURSt2 function prediction of microbiota between breast milk and infant faeces. Data were analyzed by two-sided Welch's t-test and pathway with Q values less than 0.05 and affect less than 0.30 were listed. | ||
Vertical transfer of specific bacteria from the mother to the infant's gut via breastfeeding
The transfer was first analyzed as the presence/absence of a ASV by Fisher's exact test based on a method developed by Rasmussen et al. (2020).30Fig. 8 shows volcano plots of the odds ratio of observing a specific ASV in the receiving environment, BM or infant faecal samples, given the presence in the given environment, maternal faecal samples, or BM. An odds ratio >1 is in favour of positive transfer. The ASVs transmitted from the mother's intestine to the infant's intestine increased with lactation (Fig. 8A). In the colostrum, there were 23 ASVs, with a dramatic increase to 211 in mature lactation. The number of ASVs transmitted from the mother's intestine to the BM increased with lactation as well, although it was lower than the number transmitted to the infant's intestine, especially during full lactation when only 69 ASVs were transmitted (Fig. 8A). Bacterial transfer from the BM to the infant's gut is extremely common. In the colostrum and transition milk stages, it reaches 184 and 188, respectively, and then drops to 150 in the mature milk stage (Fig. 8A). From the colostrum to mature milk stage, the newborn intestinal microbiota contains 4328 ASVs, 83 per cent of which belong to Bifidobacterium (Fig. 8B). However, only 2.7 per cent of ASVs are found in the intestines of more than half of all infants.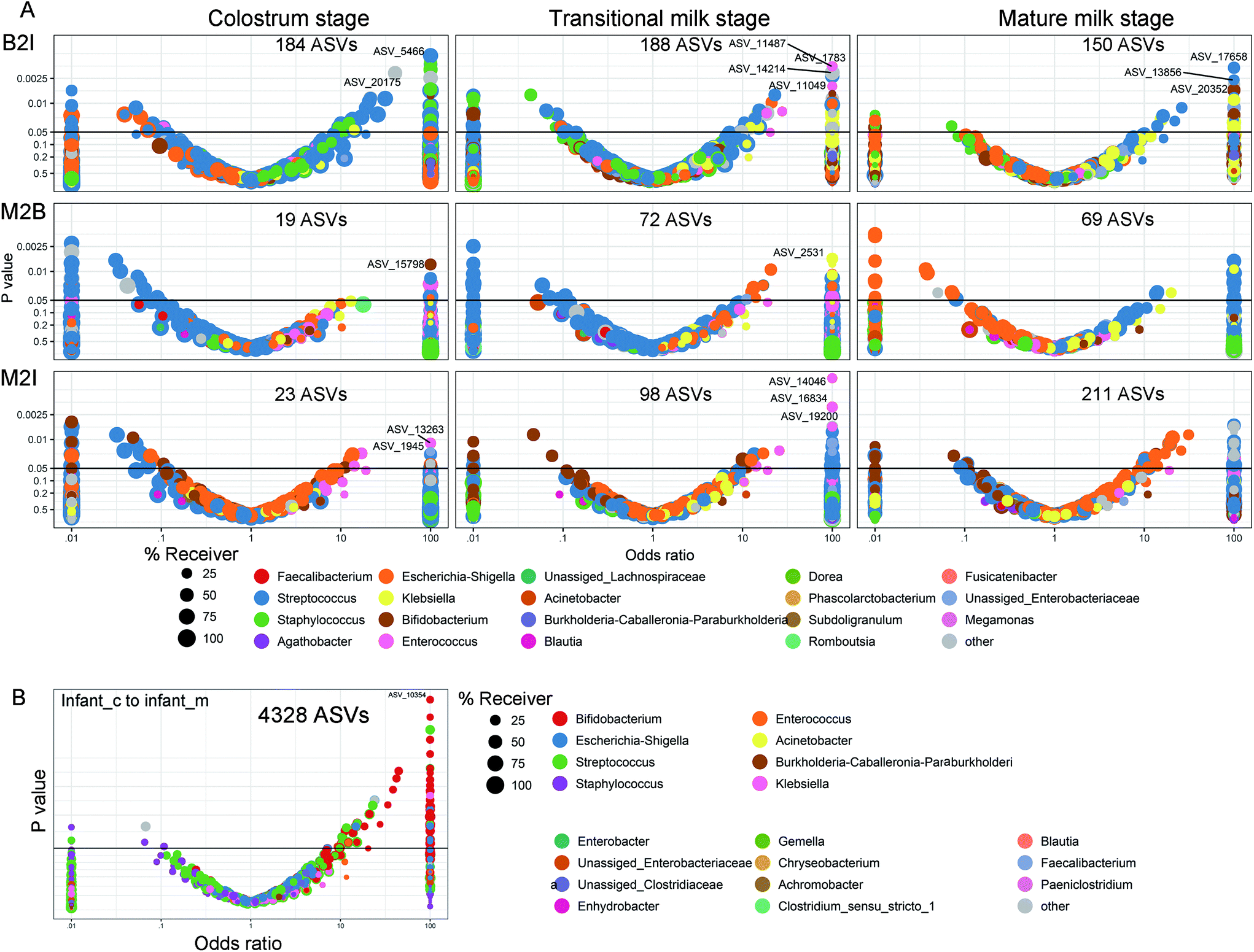 | ||
| Fig. 8 Volcano plot of odds ratios for the transmission of individual ASVs. (A) Transmission from the maternal gut to breast milk (M2B) or infant gut (M2I), and from breast milk to infant gut (B2I) at three lactation periods, respectively. (B) Transmission from the colostrum stage to the mature milk stage in the infant's gut. The significant (P < 0.05) ASVs above the horizontal line are labelled according to the lowest assignable phylogenetic rank. Odds ratios less than or more than 100 were truncated to these boundaries. The size of the dots indicates the number of receiving compartments related to the taxa. | ||
The WTR between positive and negative ORs was also calculated for all ASVs belonging to a single genus, and this was employed as a general measure of transfer. The WTR should be around one in the absence of transfer and bigger when a transfer is present, but because of the high sparsity, the null distribution is centred on 1.08–1.39, depending on sample dyads. The genera in which transmission was demonstrated to occur by WTR values can be classified into four groups based on the direction of bacterial transmission and genus in distinct lactations (Fig. 9). Bifidobacterium, Achromobacter, Burkholderia, and Staphylococcus are among the genera that transfer from BM to the infant's gut and from the mother's intestine to BM during the colostrum and transition milk stages. Cluster II contains Enterobacter, Lactobacillus, Gemella, and Rothia, which are all transmitted from the BM to the infant's gut. Acinetobacter, Faecalibacterium, Blautia, and Lachnoclostridium are among the shared genera that are transmitted from the mother's gut to the infant's gut. Cluster IV includes unique genera that are transmitted from the mother's intestine to BM and include Bacteroides, Loctococcushe and Clostridium sensu stricto 1, and also Bifidobacterium. During the transitional milk phase, Monoglobus, Enterococcus, Collinsella, and Klebsiella are also specifically transferred from the mother's intestine to the infant's intestine.
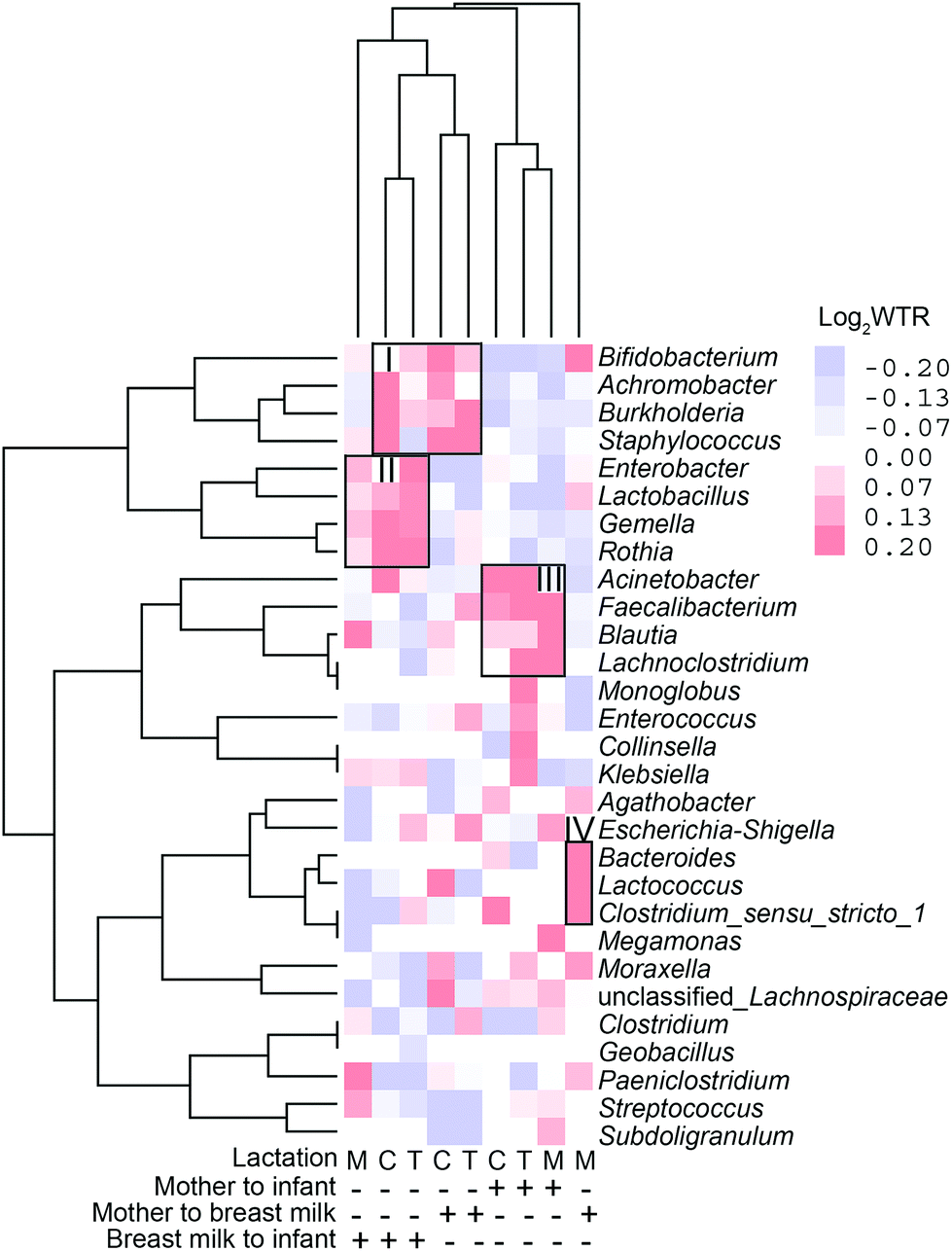 | ||
| Fig. 9 Clustering analysis of weighted transfer ratio values of genera transferred at colostrum (C), transitional (T) and mature (M) milk stages and in different transferring directions. | ||
Streptococcus salivarius is transmitted during all lactations, except for the transitional lactation from the mother's gut to the BM (Fig. 10). In all lactations, less than 30% of dyads of Bifidobacterium longum transferred from the mother's intestine to the infant's intestine. During the mature milk stage, Bifidobacterium longum is transmitted from BM to the infant's intestine at a rate of more than 30% dyads. Starting in the transitional milk stage and continuing through the mature milk stage, E. coli is transmitted from the mother's gut to the infant's intestine, and some ASVs are found in more than 30% of dyads.
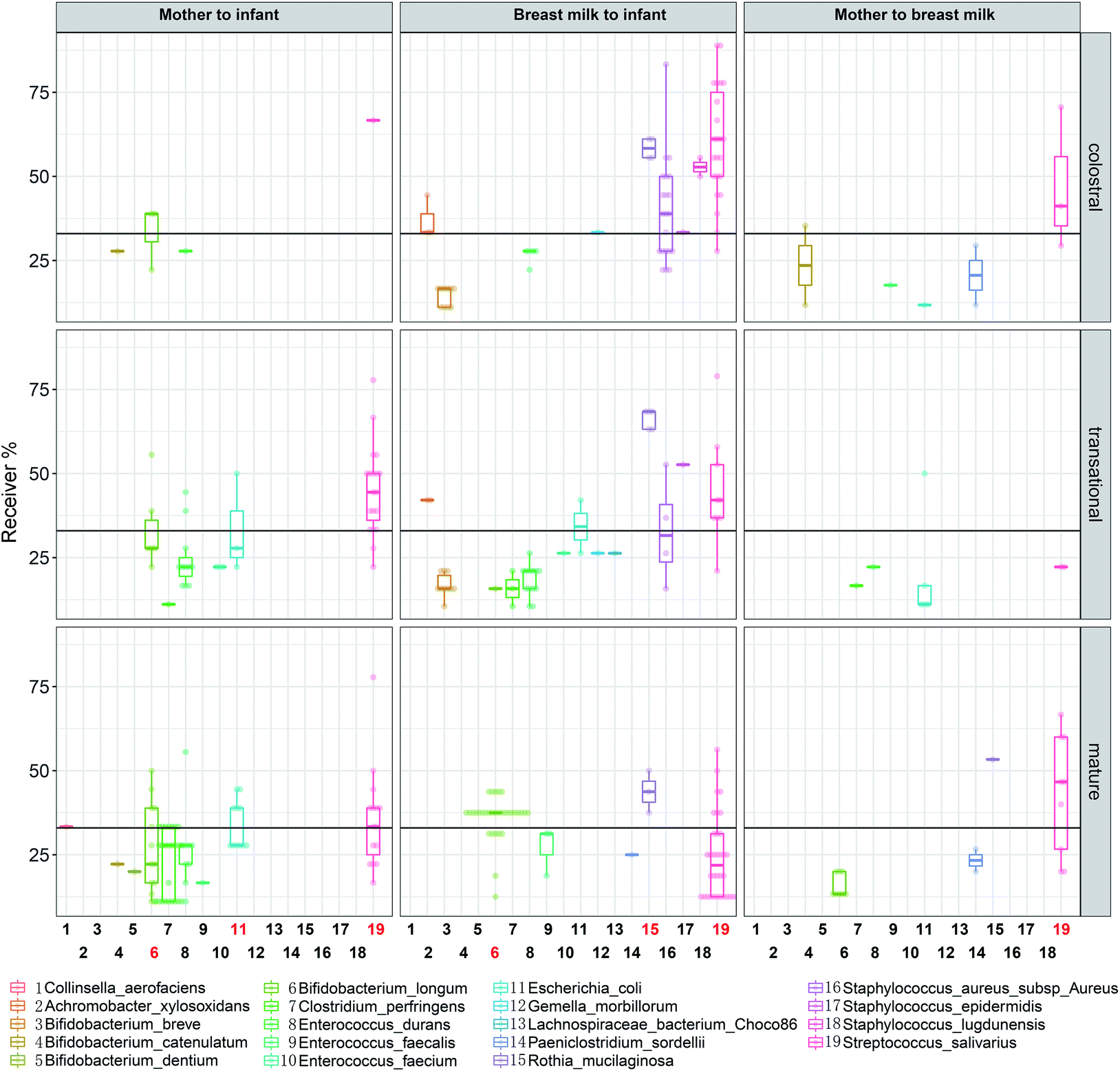 | ||
| Fig. 10 ASVs for significantly transmitted species according to odds ratios. | ||
Metagenomic sequencing analysis
Three pairs of BM–infant faecal samples from the mature milk period were also metagenomically sequenced (Table 1). Bifidobacterium longum was found in all three pairings of samples. L. gasseri, L. paragasseri, L. salivarius, S. epidermidis, Enterococcus faecium, Enterococcus faecium, Eubacterium rectale, and E. coli were all found in one pair of samples. Ruminococcus bromii and S. salivarius were found in the other two pairs of samples, respectively.| Infant1 | Milk1 | Infant2 | Milk2 | Infant3 | Milk3 | |
|---|---|---|---|---|---|---|
| Bifidobacterium longum | 1.121 | 0.108 | 0.816 | 0.421 | 1.753 | 0.676 |
| Lactobacillus gasseri | 0.000 | 0.022 | 0.000 | 0.010 | 0.023 | 33.171 |
| Lactobacillus paragasseri | 0.000 | 0.068 | 0.000 | 0.070 | 0.053 | 55.117 |
| Lactobacillus salivarius | 0.000 | 42.428 | 0.000 | 0.029 | 0.648 | 0.010 |
| Staphylococcus epidermidis | 0.000 | 34.587 | 0.000 | 92.290 | 0.100 | 0.028 |
| Enterococcus faecium | 0.000 | 22.454 | 0.000 | 0.396 | 0.008 | 0.002 |
| Enterococcus faecium | 0.000 | 22.454 | 0.000 | 0.396 | 0.008 | 0.002 |
| Eubacterium rectale | 32.208 | 0.000 | 0.833 | 0.000 | 6.853 | 0.001 |
| Escherichia coli | 0.526 | 0.000 | 1.167 | 0.000 | 0.008 | 0.057 |
| Ruminococcus bromii | 6.451 | 0.000 | 0.786 | 0.001 | 13.159 | 0.000 |
| Streptococcus salivarius | 3.113 | 0.000 | 0.382 | 0.040 | 0.146 | 0.000 |
Discussion
Here, we describe the lactation-dependent succession of newborn intestinal microbiota and transfer from the mother's gut and breast milk. Traditionally, bacteria were thought to colonize mainly the gastrointestinal tract, but recent studies have shown that some bacteria also colonize distal tissues different from the gastrointestinal tract,2 and the present study provides evidence that some of the bacteria in tissues such as the breast are of intestinal origin. At the colostrum stage, there is a considerable difference in microbiota diversity between BM and infant faeces, but as nursing develops, the infant's gut becomes enriched with anaerobic bacteria. The most commonly co-occurring ASVs are found in BM, newborn faeces at the mature milk stage, and colostrum. According to WTR, anaerobic gut-associated bacteria, such as Faecalibacterium, Blautia, and Lachnoclostridium, are transferred from the mother to the infant's gut in a lactation-dependent manner. For potential probiotics, Streptococcus, particularly S. salivarius, transmits during the mature milk stage. Bifidobacterium, particularly B. longum, is transmitted from the mother's gut to the BM and from the BM to the infant's gut, whereas Lactobacillus is transmitted at the mature milk stage. After colostrum transfer, Bifidobacterium continues to colonize the infant's gut.A recent study found the co-occurrence of bacterial ASVs in the paired sample of human milk and the infant faeces, including Veillonella, Rothia, Streptococcus, Bifidobacterium, Haemophilus, and Staphylococcus.31 We not only focused on the bacterial co-occurrence but also explored the bacterial genera and some species transferred between the mother's intestine–breast milk–infant intestine during different lactation periods based on WTR analysis. For the mother–infant specific transmission genus, Lachnoclostridium and Faecalibacterium are common members of the mammary gland specific microbiome,32 which may colonize the mammary gland via physiological translocation before lactation. They may colonize the gut of breastfeeding infants due to promotion by milk oligosaccharides, such as 6′-SL, and Bifidobacterium longum.33Blautia is a gut anaerobic bacterium with probiotic characteristics,34 which was detected in BM previously.35 In the present study, BM Blautia was present at levels higher than 0.1% in 50% of individuals in early lactation, along with a large number of different ASVs, while it was detectable in only 21% of individuals in mature lactation. Its vertical transmission may be related to physiological changes in the mother in the early postpartum period.
Staphylococcus and Streptococcus are the most dominant genera in the BM of different national and geographical origins.35,36 In the present study, BM Staphylococcus was mainly Staphylococcus aureus, S. epidermidis and S. lugdunensis, which are common causes of severe bacterial disease in neonates.37 They undergo strong breast-feeding-infant transmission during early lactation, but no longer thereafter, possibly related to antimicrobial and anticolonial components of BM.38 The present study provides evidence for frequent transmission of Streptococcus salivarius from the mother's intestine to the infant's intestine via BM (Fig. 10). S. salivarius is an oral lactic acid bacterium most closely related to S. thermophilus that is extensively used for yoghurt manufacture.39 It is among the earliest colonizers of the infant oral mucosal surfaces,39 and is an intestinal commensal inhabitant of adults40 and breastfed infants.41 The maternal gut is enriched with S. salivarius in late pregnancy, which may prepare for vertical transmission in the postpartum period.42S. salivarius may deplete the oxygen in the infant's gut and create a suitable environment with low redox potential for the growth of anaerobic bacteria.43 Therefore, it is a potential probiotic for infant formula milk.44
Bifidobacterium is one of the predominant genera in an infant's gut increasing with lactation (Fig. 6) and responsible for the enrichment of pathways in glucose metabolism (P124-PW and PWY-5384). One investigator found consistent phenotypes of Bifidobacterium longum subsp. longum isolates from paired mother–infant stool samples at different stages of lactation, demonstrating that the vertical transmission through each strain is unique to a particular mother–infant pair.45 Our study showed the presence of a large number of different bifidobacterial ASVs in the breast milk and infant gut (Fig. 2). Although significant bifidobacterial transmission was found from the mother's gut to BM and from BM to the infant's gut, the mother's gut and the infant's gut did not show significant transmission, indicating significant individual differences in the ASVs where transmission occurred. BM Lactobacillus is a popular source of probiotics. In the present study, Lactobacillus was transferred especially from BM to the infant's intestine. After metagenomic analysis, co-occurring species in one sample pairs were mainly L. gasseri, L. paragasseri and L. salivarius. It was reported that after oral administration of L. gasseri to mice, it can enter the dendritic cell-enriched region of small intestinal Peyer's patches,46 indicating a potential for transmission via the gut–mammary pathway.
There were significant individual differences in the vertical transmission of the strains. Individual differences in the adhesion of Lactobacillus and Bifidobacterium are common.47 Therefore, further understanding of the mechanisms of bacterial migration to the mammary gland could help isolate suitable strains for use in lactating mothers. It will be necessary to predict the potential breast milk microbiota by testing the maternal gut microbiota in different lactations. This could help develop potential personalized, preventive medicine or future food for preventing infant diseases by regulating bacterial transmission.
The main limitation of our study is that the sample size is small. This study only focused on a small group of healthy mother–infant pairs, to clarify the characteristics of mother–infant transmission of healthy individuals. It is necessary to investigate mother-to-child pairs with different diseases and dietary characteristics and also the persistence of beneficial bacteria in BM, by large-scale metagenomic or culturomic studies in the future. Also, the culture-independent method was not able to confirm the viability of bacteria. However, WTR-based analysis enhances the value of the 16S rRNA gene-based molecular approach for studying vertical transfer aspects.
In summary, our research identifies potential beneficial and commensal bacteria that are vertically transmitted by breast milk, including Faecalibacterium, Blautia, Lachnoclostridium, L. gasseri, L. paragasseri, and L. salivarius, as well as S. salivarius and B. longum, which needs to be validated with a larger sample size study. To improve newborn health, it will be important in the future to target the isolation of relevant strains from breast milk for modifying the microbiota of BM or infants by supplementing the mother or adding to infant formula. To fully exploit this possibility, more knowledge of the microbial composition that undergoes vertical transmission through BM is required. Some questions that should be addressed in the future include the following: (1) why these strains can be the most effective personalized probiotics in the future life? (2) Will these bacteria still colonize the intestines stably in adulthood?
Author contributions
J. S. and R. Y. designed the experiments with input from J. Z., and J. S. performed the overall data analysis; C. Q., H. C., J. C. and R. Y. contributed to volunteer recruitment and sample collection; J. S., D. L., H. T., R. T. and J. Z. contributed to data analysis and figure generation and wrote the manuscript with input from all co-authors who read and approved the manuscript.Data availability statement
The 16s sequencing data supporting the results of this article are available in the NCBI Sequence Read Archive, BioProject PRJNA542027 (accession # SUB5578272). The metagenome sequencing data are available in the NCBI Sequence Read Archiv PRJNA687137 (accession # SUB8771143).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Wuxi Municipal Science and Education Strengthening Health Engineering Medical Key Discipline Construction Program (ZDXK003), the Wuxi Young and Middle-aged Medical Talents Project (BJ2020075), the Jiangsu Maternal and Child Health Research Project (F201908), the Wuxi Commission of Health and Family Planning (Z202012 and M202171), the Nutrition and Care of Maternal & Child Research Fund Project of Biostime Institute of Nutrition & Care (2020BINCMCF056).References
- C. Hill, F. Guarner, G. Reid, G. R. Gibson, D. J. Merenstein, B. Pot, L. Morelli, R. B. Canani, H. J. Flint, S. Salminen, P. C. Calder and M. E. Sanders, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 506–514 CrossRef PubMed.
- G. Reid, T. Abrahamsson, M. Bailey, L. B. Bindels, R. Bubnov, K. Ganguli, C. Martoni, C. O'Neill, H. M. Savignac, C. Stanton, N. Ship, M. Surette, K. Tuohy and S. van Hemert, Benefic. Microbes, 2017, 8, 521–533 CrossRef CAS PubMed.
- J. M. Rodríguez, Adv. Nutr., 2014, 5, 779–784 CrossRef PubMed.
- S. Duranti, G. A. Lugli, L. Mancabelli, F. Armanini, F. Turroni, K. James, P. Ferretti, V. Gorfer, C. Ferrario and C. Milani, Microbiome, 2017, 5, 66 CrossRef PubMed.
- E. Biagi, S. Quercia, A. Aceti, I. Beghetti, S. Rampelli, S. Turroni, G. Faldella, M. Candela, P. Brigidi and L. Corvaglia, Front. Microbiol., 2017, 8, 1214 CrossRef PubMed.
- P. Ferretti, E. Pasolli, A. Tett, F. Asnicar, V. Gorfer, S. Fedi, F. Armanini, D. T. Truong, S. Manara and M. Zolfo, Cell Host Microbe, 2018, 24, 133–145 CrossRef CAS PubMed.
- F. Goldsmith, A. O'Sullivan, J. T. Smilowitz and S. L. Freeman, J. Mammary Gland Biol. Neoplasia, 2015, 20, 149–158 CrossRef PubMed.
- G. Bigman, Acta Paediatr., 2020, 109, 1627–1633 CrossRef PubMed.
- N. Soto-Ramírez, S. Kar, H. Zhang and W. Karmaus, Clin. Exp. Allergy, 2017, 47, 1285–1298 CrossRef PubMed.
- K. E. Lyons, C. A. Ryan, E. M. Dempsey, R. P. Ross and C. Stanton, Nutrition, 2020, 12 Search PubMed.
- R. Cabrera-Rubio, M. C. Collado, K. Laitinen, S. Salminen, E. Isolauri and A. Mira, Am. J. Clin. Nutr., 2012, 96, 544–551 CrossRef CAS PubMed.
- E. Jiménez, A. J. De, M. Manrique, P. Parejatobes, R. Tobes, J. F. Martínezblanch, F. M. Codoñer, D. Ramón, L. Fernández and J. M. Rodríguez, J. Hum. Lact, 2015, 31, 406–415 CrossRef PubMed.
- R. V. Bubnov, M. Y. Spivak, L. M. Lazarenko, A. Bomba and N. V. Boyko, EPMA J., 2015, 6, 14 CrossRef PubMed.
- N. A. Reis, M. A. F. Saraiva, E. A. A. Duarte, E. A. de Carvalho, B. B. Vieira and N. S. Evangelista-Barreto, J. Appl. Microbiol., 2016, 121, 811–820 CrossRef CAS PubMed.
- M. Jiang, F. Zhang, C. Wan, Y. Xiong, N. P. Shah, H. Wei and X. Tao, J. Dairy Sci., 2016, 99, 1736–1746 CrossRef CAS PubMed.
- S. Arboleya, P. Ruas-Madiedo, A. Margolles, G. Solís, S. Salminen, C. G. de Los Reyes-Gavilán and M. Gueimonde, Int. J. Food Microbiol., 2011, 149, 28–36 CrossRef CAS PubMed.
- K. Kozak, D. Charbonneau, R. Sanozky-Dawes and T. Klaenhammer, Gut Microbes, 2015, 6, 341–351 CrossRef PubMed.
- E. Jiménez, L. Fernández, A. Maldonado, R. Martín, M. Olivares, J. Xaus and J. M. Rodríguez, Appl. Environ. Microbiol., 2008, 74, 4650–4655 CrossRef PubMed.
- M. Kuitunen, K. Kukkonen, K. Juntunen-Backman, R. Korpela, T. Poussa, T. Tuure, T. Haahtela and E. Savilahti, J. Allergy Clin. Immunol., 2009, 123, 335–341 CrossRef PubMed.
- M. R. Simpson, E. Avershina, O. Storro, R. Johnsen, K. Rudi and T. Oien, J. Dairy Sci., 2018, 10, 889–899 CrossRef PubMed.
- A. Kechin, U. Boyarskikh, A. Kel and M. Filipenko, J. Comput. Biol., 2017, 24, 1138–1143 CrossRef CAS PubMed.
- Y.-X. Liu, Y. Qin, T. Chen, M. Lu, X. Qian, X. Guo and Y. Bai, Protein Cell, 2021, 12, 315–330 CrossRef PubMed.
- T. Rognes, T. Flouri, B. Nichols, C. Quince and F. Mahé, PeerJ, 2016, 4, e2584 CrossRef PubMed.
- R. C. Edgar, Bioinformatics, 2010, 26, 2460–2461 CrossRef CAS PubMed.
- J. Lu and S. L. Salzberg, Microbiome, 2020, 8, 124 CrossRef PubMed.
- R. Schmieder and R. Edwards, Bioinformatics, 2011, 27, 863–864 CrossRef CAS PubMed.
- B. Longmead and S. L. Salzberg, Nat. Methods, 2012, 9, 357–359 CrossRef PubMed.
- F. Beghini, L. J. McIver, A. Blanco-Míguez, L. Dubois, F. Asnicar, S. Maharjan, A. Mailyan, P. Manghi, M. Scholz, A. M. Thomas, M. Valles-Colomer, G. Weingart, Y. Zhang, M. Zolfo, C. Huttenhower, E. A. Franzosa and N. Segata, eLife, 2021, 10, e65088 CrossRef PubMed.
- M. S. Mortensen, M. A. Rasmussen, J. Stokholm, A. D. Brejnrod, C. Balle, J. Thorsen, K. A. Krogfelt, H. Bisgaard and S. J. Sørensen, eLife, 2021, 10, e57051 CrossRef CAS PubMed.
- M. A. Rasmussen, J. Thorsen, M. G. Dominguez-Bello, M. J. Blaser, M. S. Mortensen, A. D. Brejnrod, S. A. Shah, M. H. Hjelmsø, J. Lehtimäki, U. Trivedi, H. Bisgaard, S. J. Sørensen and J. Stokholm, ISME J., 2020, 14, 2325–2335 CrossRef CAS PubMed.
- K. Fehr, S. Moossavi, H. Sbihi, R. C. T. Boutin, L. Bode, B. Robertson, C. Yonemitsu, C. J. Field, A. B. Becker, P. J. Mandhane, M. R. Sears, E. Khafipour, T. J. Moraes, P. Subbarao, B. B. Finlay, S. E. Turvey and M. B. Azad, Cell Host Microbe, 2020, 28, 285–297 CrossRef CAS PubMed.
- C. A. Shively, T. C. Register, S. E. Appt, T. B. Clarkson, B. Uberseder, K. Y. J. Clear, A. S. Wilson, A. Chiba, J. A. Tooze and K. L. Cook, Cell Rep., 2018, 25, 47–56 CrossRef CAS PubMed.
- L. Cheng, M. B. G. Kiewiet, M. J. Logtenberg, A. Groeneveld, A. Nauta, H. A. Schols, M. T. C. Walvoort, H. J. M. Harmsen and P. de Vos, Front. Microbiol., 2020, 11, 569700 CrossRef PubMed.
- X. Liu, B. Mao, J. Gu, J. Wu, S. Cui, G. Wang, J. Zhao, H. Zhang and W. Chen, Gut Microbes, 2021, 13, 1–21 CrossRef CAS PubMed.
- T. Jost, C. Lacroix, C. Braegger and C. Chassard, Br. J. Nutr., 2013, 110, 1253 CrossRef CAS PubMed.
- K. A. Lackey, J. E. Williams, C. L. Meehan, J. A. Zachek, E. D. Benda, W. J. Price, J. A. Foster, D. W. Sellen, E. W. Kamau-Mbuthia and E. W. Kamundia, Front. Nutr., 2019, 6, 45 CrossRef PubMed.
- A. Roca, A. Bojang, B. Camara, C. Oluwalana, K. Lette, P. West, U. D'Alessandro and C. Bottomley, Clin. Microbiol. Infect., 2017, 23, 974–979 CrossRef CAS PubMed.
- S. D. Townsend, Breastfeed. Med., 2019, 14, S5–S6 CrossRef PubMed.
- P. A. Wescombe, N. C. K. Heng, J. P. Burton, C. N. Chilcott and J. R. Tagg, Future Microbiol., 2009, 4, 819–835 CrossRef CAS PubMed.
- B. Couvigny, S. Kulakauskas, N. Pons, B. Quinquis, A.-L. Abraham, T. Meylheuc, C. Delorme, P. Renault, R. Briandet, N. Lapaque and E. Guédon, Front. Microbiol., 2018, 9, 273 CrossRef PubMed.
- G. Solís, C. G. de los Reyes-Gavilan, N. Fernández, A. Margolles and M. Gueimonde, Anaerobe, 2010, 16, 307–310 CrossRef PubMed.
- O. Koren, J. K. Goodrich, T. C. Cullender, A. Spor, K. Laitinen, H. K. Backhed, A. Gonzalez, J. J. Werner, L. T. Angenent, R. Knight, F. Backhed, E. Isolauri, S. Salminen and R. E. Ley, Cell, 2012, 150, 470–480 CrossRef CAS PubMed.
- S. Wang, C. A. Ryan, P. Boyaval, E. M. Dempsey, R. P. Ross and C. Stanton, Trends Microbiol., 2020, 28, 28–45 CrossRef CAS PubMed.
- T. Kryuchko and O. Tkachenko, Int. J. Nutraceuticals, Funct. Foods Nov. Foods, 2021 DOI:10.17470/NF-021-0033.
- H. Makino, A. Kushiro, E. Ishikawa, D. Muylaert, H. Kubota, T. Sakai, K. Oishi, R. Martin, K. Ben Amor, R. Oozeer, J. Knol and R. Tanaka, Appl. Environ. Microbiol., 2011, 77, 6788–6793 CrossRef CAS PubMed.
- G. Harata, F. He, M. Kawase, A. Hosono, K. Takahashi and S. Kaminogawa, Microbiol. Immunol., 2009, 53, 475–480 CrossRef CAS PubMed.
- R. V. Bubnov, L. P. Babenko, L. M. Lazarenko, V. V. Mokrozub and M. Y. Spivak, EPMA J., 2018, 9, 205–223 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fo03131g |
| This journal is © The Royal Society of Chemistry 2022 |