
Steatosis induced by nonylphenol in HepG2 cells and the intervention effect of curcumin
Dandan
Shan
,
Jinming
Wang
,
Qiannan
Di
,
Qianqian
Jiang
and
Qian
Xu
 *
*
Key Laboratory of Environmental Medicine Engineering, Ministry of Education, School of Public Health, Southeast University, Nanjing, China. E-mail: q_xu68@163.com; q-xu@seu.edu.cn; Tel: +86 025 83272563
First published on 6th December 2021
Abstract
Non-alcoholic fatty liver disease (NAFLD) has increasingly become a serious public health problem. There is growing evidence that nonylphenol (NP) exposure may cause steatosis, but the underlying mechanism is not fully understood. Curcumin (CUR) improves NAFLD-related lipid metabolism disorders and oxidative stress, but its preventive and therapeutic effects on NP-induced steatosis have not been reported. The objective of this investigation was to determine the capability and potential mechanism of NP to induce steatosis in vitro and the intervention of curcumin. HepG2 cells were treated with 0 μM, 20 μM, 30 μM, 40 μM NP for 24 h. Lipid droplets accumulated significantly in HepG2 cells after NP treatment, and the concentration of triglyceride (TG) and total cholesterol (T-CHO) increased significantly. Simultaneously, lipogenesis gene expression was up-regulated significantly, fatty acid oxidation (FAO) gene expression was significantly down-regulated, and reactive oxygen species (ROS) were overproduced. Meanwhile, the expression of p-AMPK/AMPK in the AMPK/mTOR signaling pathway was significantly down-regulated and the expression of p-mTOR/mTOR was markedly up-regulated. However, blocking ROS production with N-acetyl-L-cysteine (NAC) can reverse these phenomena. In addition, our study found that curcumin effectively ameliorated the effects of NP-induced steatosis. Our study indicates that NP can induce steatosis in HepG2 cells, and may be implicated in inhibiting the ROS-dependent AMPK/mTOR pathway, and that curcumin ameliorates the NAFLD-like changes induced by NP in HepG2 cells.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) refers to a group of liver-acquired metabolic diseases in which the pathological changes of lipid deposition dominated by triglycerides occur in hepatocytes without excessive alcohol consumption and other factors that can cause liver damage.1–3 The global prevalence of NAFLD is about 25%, affecting the health of 1.7 billion people. The overall prevalence of NAFLD in China is as high as 29.6%, and the annual incidence is still increasing.4,5 NAFLD has become a serious public health problem of worldwide concern. The pathogenesis and progression of NAFLD are complex. The ‘two-hit’ hypothesis and the ‘Multiple-Hit’ theory propose the important risk factors lipid metabolism disturbances, insulin resistance, oxidative stress, inflammation, along with diet, genetics and environment. However, the pathogenesis of NAFLD remains inconclusive.6,7 Recent studies indicate that the development and progression of NAFLD may be related to exposure to EDCs (endocrine disrupting chemicals).8–12As a consequence of industrialization and the modernization of cities, EDCs are widely present in the environment. Among them, Nonylphenol ethoxylate (NPEO), an important nonionic surfactant, has become a major EDC common in both industrial and residential detergents, agricultural products and plastic products.13 NP (Nonylphenol) a degradation product of Nonylphenol ethoxylate (NPEO) and an intermediate in the NPEO manufacturing process, can be detected in water, soil and air and can ultimately reach the food chain.14 After enrichment and concentration by the food chain, NP can affect animal or human health directly or indirectly. It has been shown that NP can be detected in almost all foodstuffs in different countries and regions, ranging from 30 ng kg−1 to 2101 μg kg−1.15–17 In addition to NP presence in food as mentioned above, NP can also contaminate food through other pathways, including contaminated packaging materials or soil bioaccumulation processes.18–20 Therefore, the main source of NP for an organism is dietary intake. Although the daily intake of NP in China (520 ng per kg-bw per day)15 is lower than the tolerable daily intake (5 μg per kg-bw per day, the Danish Institute of Safety and Toxicology),21 the level is 4 times higher than those in Germany (125 ng per kg-bw per day).22 And the average daily intake of alkylphenols for males over 25 years old in New Zealand was reported to be 3.6 μg day−1.23 Attention should be paid to health risks from NP chronic exposure even at a relatively low dietary dosage.
Previous studies on rodent models and avian and aquatic organisms have focused on the estrogenic effects of NP, and its toxic effects on reproduction, development, immunity, and nerves.24–27 Due to its lipophilicity, the NP can also concentrate in adipose, brain, liver and other systems to exert its toxic effects.28 The liver appears to be the major organ for NP aggregation, biotransformation and degradation.29 The main feature of NAFLD is accumulation of lipids in the liver. Prior studies have observed that tissue damage, including steatosis, occurs after NP exposure in different species.30–36 A study of 4-nonylphenol impact on the oxidant/antioxidant balance system inducing hepatic steatosis in male rat showed that 4-NP might promote hepatic steatosis in male rat, activate the expression of reactive oxygen species, reduce the expression of antioxidant enzymes and cause apoptosis.34 In addition, lipid accumulation was observed in hepatocytes of adult male zebrafish exposed to NP resulting in hepatic steatosis.37 Therefore, NP can cause NAFLD-like changes. However, the effect and mechanism of NP-induced steatosis of hepatocytes have not been determined, and further investigation is needed.
A recent study showed that changing diet, increasing physical exercise, and some antioxidants such as Vitamin E, Betaine, Cysteamine, are the main strategies for prevention and treatment of NAFLD.38 In recent years, a growing number of scientific studies have explored the possibility of targeted treatment of NAFLD with active substances from Chinese herbs.39 Among them, curcumin has become a research hotspot because of its enormous potential in the prevention and treatment of NAFLD.40–42 Curcumin, a polyphenolic compound with high biological activity extracted from the rhizome of the culinary and medicinal perennial herb Curcuma longa L, is anti-inflammatory, anti-oxidant, anti-viral, immunomodulatory, hypolipidemic, hepatoprotective, cancer preventive and anti-cancer.43–47 The US Food and Drug Administration (FDA) reported that curcumin as a dietary supplement is a generally safe alternative medicine.48 Curcumin has promising applications in the protection and treatment of NAFLD.49,50 In a clinical population study, the serum levels of total cholesterol (T-CHO), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG) and uric acid (UA) in 80 NAFLD patients were reduced by curcumin.51 An NAFLD animal model experiment indicated that compared with the placebo control group, liver TG and T-CHO concentrations of male SD rats on a curcumin diet were significantly reduced.52 In addition, studies have shown that curcumin can also improve steatosis by regulating expression of liver lipid metabolism-related genes and affecting oxidative stress response.53,54 Although curcumin can reduce lipid accumulation and alleviate NAFLD symptoms in vivo, there is currently no evidence that curcumin is a protective agent against NP-induced hepatocyte steatosis.
The purpose of this study was to investigate the ability and potential mechanism of NP-induced steatosis in HepG2 cells, and to explore the interventional effects of curcumin on it. Therefore, the first in vitro model of NP-induced steatosis in HepG2 cells was established, then the molecular mechanism was examined, and further verified by oxidative stress scavenger N-acetyl-L-cysteine (NAC). In addition, the effect and possible mechanism of curcumin intervention on NP-induced steatosis in HepG2 cells were explored. The structures of nonylphenol, N-acetyl-L-cysteine (NAC) and curcumin are shown in (Fig. 1).
 | ||
| Fig. 1 Structures of nonylphenol, N-acetyl-L-cysteine (NAC), curcumin. | ||
2. Materials and methods
2.1. Reagents and antibodies
Nonylphenol (Linear isomer, CAS: 84852-15-3, >99.9%) was purchased from Sigma-Aldrich (Shanghai, China). Curcumin (CAS: 458-37-7, >98%) was purchased from Alfa Biotechnology (Chengdu, China). Ultrapure water (resistance 18.2 MΩ cm) used throughout the experiment was obtained by using a Milli-Q system from Millipore, Bedford, Massachusetts U.S.A. NAC (≥99%) and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St Louis, MO, USA). Fetal bovine serum (FBS) was obtained from Biological Industries (Beit HaEmek, Israel). The medium and Phosphate buffered saline (PBS) were obtained from Gibco (New York City, USA). 0.25% trypsin was purchased from KGI Bio (Nanjing, China). Cell counting kit-8 (CCK-8) was purchased from APExBIO (Boston, MA, USA). Nile red was purchased from Shanghai Yuanye Biotechnology (Shanghai, China). TG and T-CHO assay kits were purchased from Jiancheng Bioengineering Institute (Nanjing, China). RNA isolater Total RNA Extraction Reagent was purchased from Vazyme (Nanjing, China). HiScript® III RT SuperMix for qPCR (+gDNA wiper) was purchased from Vazyme (Nanjing, China). ChamQ Universal SYBR qPCR Master Mix was obtained from Vazyme (Nanjing, China). The reactive oxygen species (ROS) assay kit was purchased from Beyotime Biotechnology (Shanghai, China). Acetyl–CoA carboxylase (C83B10), fatty acid synthase (C20G5), phospho-mTOR (Ser2481), mTOR (7C10) were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-AMPK alpha phosphor (anti-AMPK alpha 1 (phospho T183) + AMPK alpha 2 (phospho T172) antibody [EPR5683]), anti-AMPK alpha (alpha 1 and alpha 2), and anti-PPAR gamma were purchased from Abcam (Cambridge, UK). CPT1A, PPARA, CEBPA, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Proteintech (Wuhan, China). SREBP1c and SCD were purchased from ABclonal (Wuhan, China). Goat anti-rabbit IgG-HRP was purchased from Absin Bioscience (Shanghai, China). All antibodies were used at the dilutions recommended by the manufacturer.2.2. Cell culture and treatment
The human hepatocellular carcinoma cell line (HepG2) was obtained from the Institute of Oncology of Fudan University-affiliated Oncology Hospital (Shanghai, China). Cells were grown in a carbon dioxide cell incubator at 37 °C, carbon dioxide concentration 5%, and Dulbecco's modified Eagle's medium (DMEM) basic (1×) medium containing 10% fetal bovine serum (FBS) was added to provide nutrients required for the cells. Logarithmically grown cells were used for the experiments. The stock solution was prepared by dissolving NP and curcumin in DMSO. Cells in the control group were treated with medium, HepG2 cells were treated with NP and/or curcumin (the control group, the NP group, and the curcumin group) for 24 h.2.3. Cell viability
To evaluate the impact of NP and curcumin on HepG2 cell viability, a CCK-8 test was performed. The procedure was executed according to the instructions. Briefly, HepG2 cells were seeded in 96-well plates at a density of 0.8 × 104 cells per well and grown to 80%. 24 h later, the medium was replaced with 100 μL medium containing 10% CCK-8, then the cells were treated with different concentrations of NP and/or curcumin (the concentrations of NP are 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 μM and the concentrations of CUR are 5, 10, 20, 30, 40, 50, and 60 μM), then the plates were further incubated at 37 °C in the dark for 2 h. Lastly, the optical density (OD) was measured at 450 nm wavelength with an Infinite F50 enzyme-labeled instrument (TECAN, Switzerland). All measurements were repeated in triplicate.2.4. Nile red staining
To investigate the Lipid accumulation in HepG2 cells, Nile red staining was used. First, HepG2 cells were inoculated at a density of 2–4 × 104 cells per well on 24 mm × 24 mm cell slide and grown to 80%. Next, the cells were divided into four groups and treated separately. Control group (normal medium treatment for 24 h), NP alone (40 μM NP treatment for 24 h), NAC alone (10 mM NAC pretreatment for 2 h followed by normal medium change for 24 h) and combined NP and NAC treatment (10 mM NAC pretreatment for 2 h followed by 40 μM NP treatment for 24 h). Then, cells were stained with 0.5 mg L−1 Nile Red staining working solution and DAPI solution and sealed with anti-fluorescence quenching sealing solution. Finally, the fluorescence microscope was used to visualize the HepG2 cells steatosis and take photos. Three fields of view were selected for each sample, each group of experiments was repeated three times independently. After selecting images of the same number of cells, Image J software was applied to quantify the fluorescence intensity.2.5. Measurement of TG, T-CHO levels
Lipid accumulation in HepG2 cells was evaluated by TG, T-CHO. After treatment with different configurations of NP and/or curcumin for 24 h, or pretreatment with 10 mM NAC for 2 h. The TG and T-CHO content were performed by TG and T-CHO assay kits and BCA kits. The optical density (OD) at the corresponding wavelengths was determined by an Infinite F50 enzyme marker (TECAN, Männedorf, Switzerland).2.6. Measurement of ROS level
The level of ROS in HepG2 cells was detected by flow cytometry. The procedure was performed in accordance with the manufacturer's instructions of Beyotime ROS assay kits. In brief, the cells were treated with different concentrations of NP and/or curcumin for 24 h, or pretreated with 10 mM NAC for 2 h. The ROS levels in HepG2 cells were measured by quantifying the average cellular fluorescence intensity of 2,7-dichloro-fluorescein diacetate (DCFH-DA), and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample were collected using a BD FACSCaliburTM flow cytometer. Lastly, the results were analyzed by Flow JO software.
000 cells per sample were collected using a BD FACSCaliburTM flow cytometer. Lastly, the results were analyzed by Flow JO software.
2.7. Real-time quantitative PCR (qPCR)
After being treated with different concentrations of NP and/or curcumin for 24 h, cells were collected and total RNA was extracted with Trizol reagent. RNA purity was measured by spectrophotometry at the ratio of absorbance at 260 and 280 nm (ND 1000 ultra-micro ultraviolet spectrophotometer, ThermoFisher, Waltham, Massachusetts USA). Total RNA (1 μg) was reverse transcribed into cDNA with a reverse transcriptase kit (Mastercycler pro gradient PCR instrument, Eppendorf, Germany). And cDNA was amplified using SYBR Green real-time fluorescence qPCR kit. qPCR was performed using a CFX ConnectTM real-time fluorescence qPCR instrument (BIO-RAD, Hercules, California USA) with the following conditions: after pre-denaturation at 95 °C for 5 min, denaturation at 95 °C for 5 s, and annealing temperature of 60 °C for 30 s, the fluorescence signal was collected at an extension of 30 s at 72 °C for 40 cycles; and the melting curve analysis was performed by increasing the temperature from 65 °C to 95 °C. The 2-ΔΔCt formula was used to analyze the calculation results, the singularity of PCR products was examined according to the melting curve, and the relative content expression levels of individual gene mRNAs were determined using GAPDH as the internal reference gene. The primer sequences used in this work are listed in Table 1.| Primer | Sequence (5′ to 3′) | Sequence (5′ to 3′) |
|---|---|---|
| ACC | GCCTCAGGAGGATTTGCTGT | AGGATCTACCCAGGCCACAT |
| FAS | GGCCCCTCTGTTAATTGGCT | GGATCTCAGGGTTGGGGTTG |
| Srebp-1c | CAGACTCACTGCTGCTGACA | GATGGTCCCTCCACTCACCA |
| PPARγ | CCTTCACCACCGTTGACTTCT | GATACAGGCTCCACTTTGATTGC |
| C/EBPα | GAGCCCGGCAACTCTAGTAT | TGACAAGGCACGATTTGCTC |
| SCD-1 | GTACCGCTGGCACATCAACT | AACTCAGAAGCCCAAAGCTCA |
| CPT1α | GGACTCCGCTCGCTCATT | GAGATCGATGCCATCAGGGG |
| PPARα | TGCCTTCCCTGTGAACTGAC | TGGGGAGAGAGGACAGATGG |
| GAPDH | ATCACTGCCACCCAGAAGAC | AGATCCACGACGGACACATT |
2.8. Western blotting analysis
After NP, CUR or NAC treatment, the cells were placed in RIPA lysis buffer containing PMSF and phosphatase inhibitors and lysed on ice for 15 minutes. After centrifugation at 4 °C, 14000 rpm for 15 minutes, the total protein from HepG2 cells was obtained by collecting the supernatant. Protein quantification was determined in accordance with the instructions of Beyotime BCA Protein Concentration Assay Kit (Beyotime, China). 10–20 μg of total protein from HepG2 cells was separated by 7.5% PAGE Gel Rapid Preparation Kit and transferred to 0.45 μM PVDF membranes (DYCZ-24DN electrophoresis instrument, DYCZ-40D membrane transfer instrument, Liuyi, China). The membranes were blocked with 5% nonfat skim milk or 5% BSA for 2 h at room temperature. Then primary antibodies were incubated overnight at 4 °C and then rinsed three times with 1× TBST buffer for 10 min once. Secondary antibodies were incubated for 2 h at room temperature and rinsed four times with 1× TBST buffer for 10 min each time. The immunoblot signals were examined by ECL chemiluminescence kit (Tanon 5200 luminescence imaging workstation, Tanon, China). The expression levels of target proteins were assessed using GAPDH as an internal reference. The grayscale values of the western blot bands were analyzed and counted using Image J software.
2.9. Statistical analysis
Statistical graphs were drawn using GraphPad Prism 6.0 software, and the data were statistically analyzed with SPSS 22.0 software. The experimental data was expressed as mean ± standard deviation (S.D.) of three independent experiments. Each group of experiments was repeated three times independently. The statistical significance among the groups was examined using the analysis of variance (ANOVA), and the LSD test was used to analyze the differences between two comparisons of multiple groups. A p-value of less than 0.05 was considered statistically significant.3. Results
3.1. Establishment of NP-induced HepG2 cell steatosis model
To establish NP-induced HepG2 cell steatosis model, CCK8 kit was used to investigate the effect of NP on the viability of HepG2 cells, Nile red staining and TG, T-CHO kits were used to explore the impact of NP on HepG2 cell steatosis. As shown in Fig. 2, the results indicated that when treated with NP at concentrations of 10–40 μM, no effect was observed on HepG2 cells (P > 0.05), while NP at concentrations above 40 μM dramatically decreased the cell viability of HepG2 in a dose-dependent manner (P < 0.001), and the viability of HepG2 cells treated with 70 μM NP was only 38.1%. Then, HepG2 cells were incubated with 20 μM, 30 μM and 40 μM NP for 24 h, and compared with the control group, enhanced Nile red fluorescence intensity and lipid droplet accumulation were found in all HepG2 cells of the NP-exposed group, and the expression levels of TG and T-CHO were remarkably increased (P < 0.001). The above results indicated that the NP-induced steatosis model of HepG2 cells was successfully established in this study.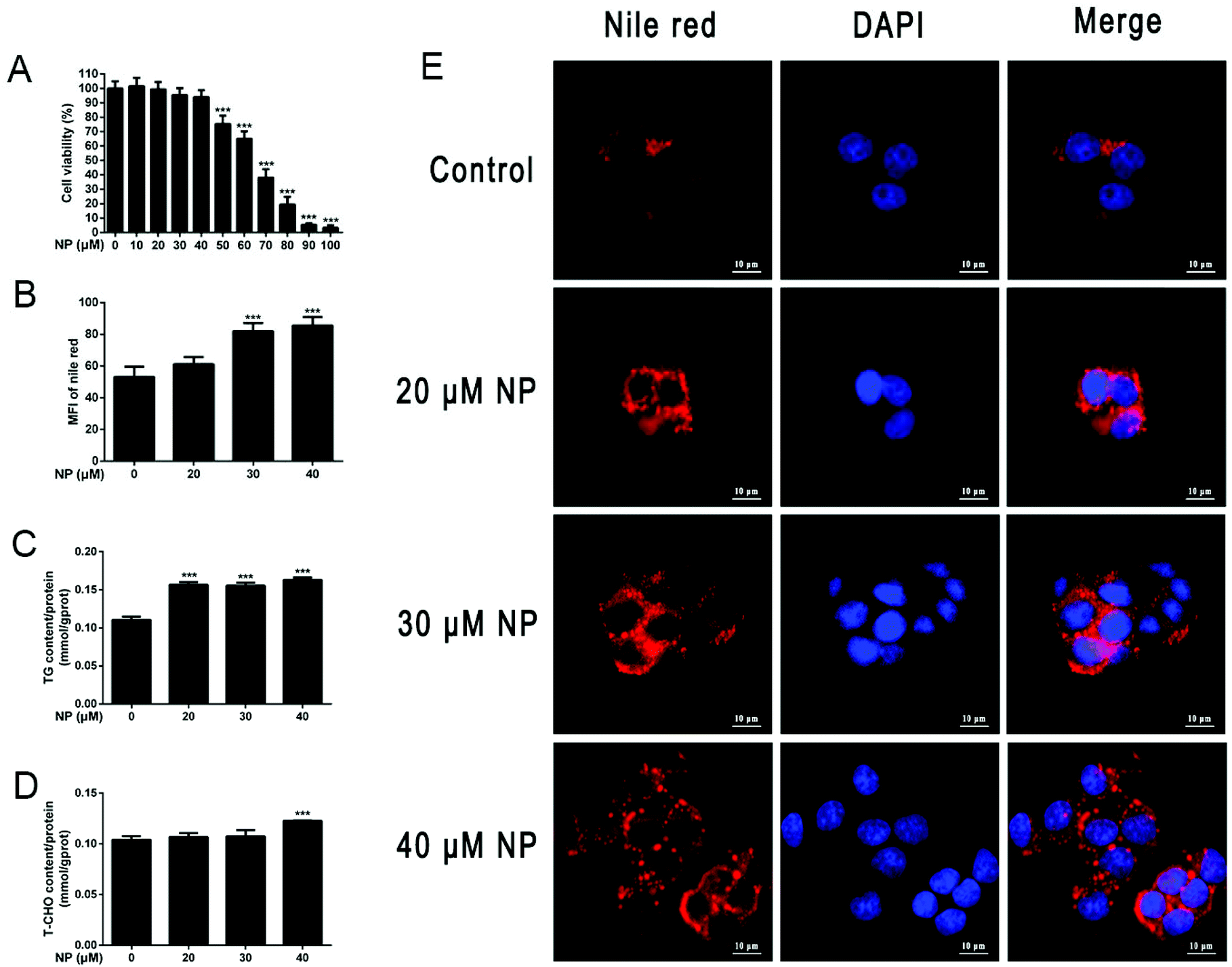 | ||
| Fig. 2 Establishment of NP-induced HepG2 cell steatosis model. (A) HepG2 cells were treated with different concentration gradients of NP (0–100 μM) for 24 h. The effect of NP on cell viability was analyzed by CCK-8 assay. (B–E) HepG2 cells were treated with 20 μM, 30 μM, and 40 μM NP for 24 h. (B and E) Nile red staining was used to investigate the changes of lipid droplets in HepG2 cells and the statistical results. (C) The expression levels of triglycerides in HepG2 cells were determined. (D) The expression levels of total cholesterol in HepG2 cells were measured. *** P < 0.001, significantly different as compared with the control group. Note: CCK-8: cell counting kit-8; NP: nonylphenol. | ||
3.2. NP induces disruption of lipid metabolism in HepG2 cells
To further determine the effect of NP exposure on lipid metabolism of HepG2 cells, the expression of mRNA and protein by lipogenesis-related genes acetyl–CoA carboxylase (ACC), fatty acid synthase (FAS), sterol regulatory element-binding protein factor 1c (Srebp-1c), CCAAT-enhancer-binding proteins (C/EBPα), peroxisome proliferator-activated receptor gamma (PPARγ), stearoyl–CoA desaturase-1 (SCD-1), and fatty acid oxidation (FAO)-related genes carnitine palmitoyl transferase I (CPT1α) and Peroxisome proliferator-activated receptor alpha (PPARα) in HepG2 cells after NP treatment was investigated by qPCR and western blot. The results of real-time fluorescence quantitative PCR showed that the mRNA expression levels of the lipogenesis-related genes were all up-regulated after NP exposure. The mRNA expression level of SCD-1 was statistically upregulated after 40 μM NP treatment (Fig. 3A1; P < 0.05). The results of qPCR demonstrated that the mRNA expression levels of CPT1α were significantly decreased with 20–40 μM NP treatment (Fig. 3B1; P < 0.01) and PPARα were significantly decreased with 30 μM and 40 μM NP treatment (Fig. 3B1; P < 0.01) compared with the control group. Western blot was used to detect the expression level of the corresponding protein, which was consistent with the mRNA level. These results indicated that NP can cause disruption of lipid metabolism in HepG2 cells.3.3. NP induces oxidative stress in HepG2 cells
To determine the role of oxidative stress in the development of cellular steatosis. The production of ROS in HepG2 cells treated with NP for 24 h was observed by flow cytometry. The results displayed that ROS content was significantly increased in HepG2 cells with 20–40 μM NP treatment compared to the control group (Fig. 3C; P < 0.001). The results indicated that NP treatment increased the expression of intracellular ROS levels in a dose-dependent manner.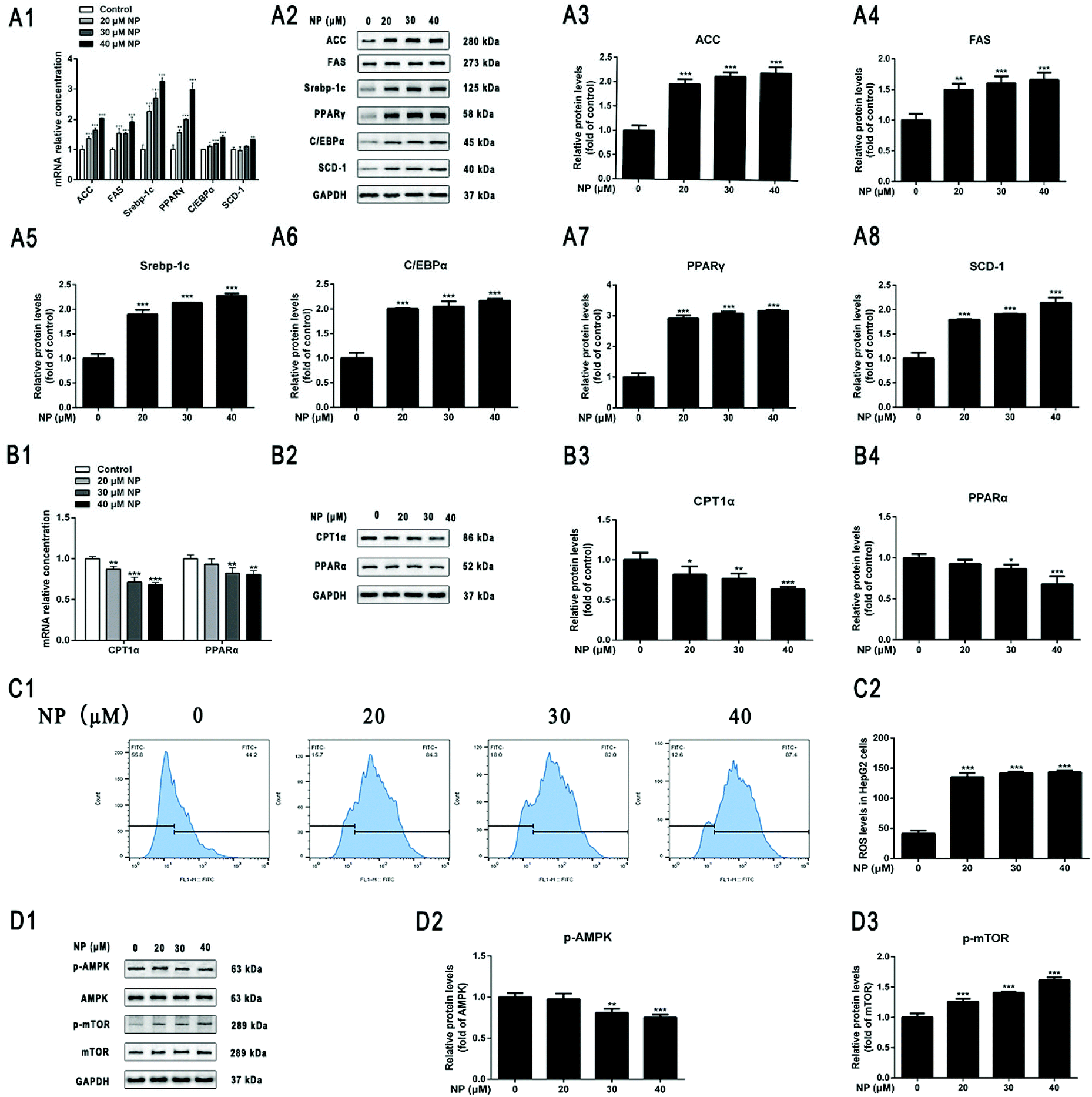 | ||
| Fig. 3 Effect of NP exposure on lipid metabolism and ROS levels and AMPK/mTOR signaling pathway in HepG2 cells. HepG2 cells were treated with 20 μM, 30 μM, 40 μM NP for 24 h. (A) The effect of NP exposure on lipogenic in HepG2 cells. (A1) RT-qPCR was applied to explore the expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1 mRNA levels. (A2–A8) Western blot was used to determine the expression of protein levels of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1. (B) Effect of NP exposure on lipid acid oxidation in HepG2 cells. (B1) RT-qPCR was applied to explore the expression of CPT1α and PPARα mRNA levels. (B2–B4) Western blot was performed to determine the expression of CPT1α and PPARα protein levels. (C) Intracellular ROS levels were quantified by flow cytometry, and 10000 cells were analyzed for each sample. (D) Effect of NP exposure on AMPK/mTOR signaling pathway in HepG2 cells. The expression of p-AMPK, AMPK, p-mTOR, and mTOR protein levels were examined by western blot. * P < 0.05, ** P < 0.01, *** P < 0.001, significantly different as compared with the control group. Note: ACC: acetyl–CoA carboxylase; AMPK: AMP-dependent protein kinase; C/EBPα: CCAAT-enhancer-binding proteins; CPT1α: carnitine palmitoyl transferase I; FAS: fatty acid synthase; mTOR: target of rapamycin protein; p-AMPK: phosphorylated AMP-dependent protein kinase; p-mTOR: phosphorylated target of rapamycin protein; PPARα: peroxisome proliferator-activated receptor alpha; PPARγ: peroxisome proliferator-activated receptor gamma; RT-qPCR: real-time quantitative PCR; ROS: reactive oxygen species; SCD-1: stearoyl–CoA desaturase-1; Srebp-1c: sterol regulatory element-binding protein factor 1c. | ||
3.4. NP inhibits AMPK/mTOR signaling pathway in HepG2 cells
Current studies have confirmed that the regulation of AMPK/mTOR signaling pathway is extremely important in mediating cell growth and proliferation and energy homeostasis.55,56 The role of AMPK/mTOR signaling pathway in the process of NP exposure-induced steatosis in HepG2 cells was investigated by detecting the expression of signaling pathway-related protein levels in cells by western blot. As can be seen in Fig. 3D, the ratio of p-AMPK/AMPK was significantly lower at NP exposure concentrations of 30 μM–40 μM compared with the control (Fig. 3D; P < 0.01), whereas the ratio of p-mTOR/mTOR was significantly higher at NP exposure concentrations of 20 μM–40 μM (Fig. 3D; P < 0.001). These results indicated that NP may induce HepG2 cell steatosis by suppressing AMPK phosphorylation in HepG2 cells and significantly promoting mTOR signaling.3.5. NP induces HepG2 cells steatosis through ROS via AMPK/mTOR signaling pathway
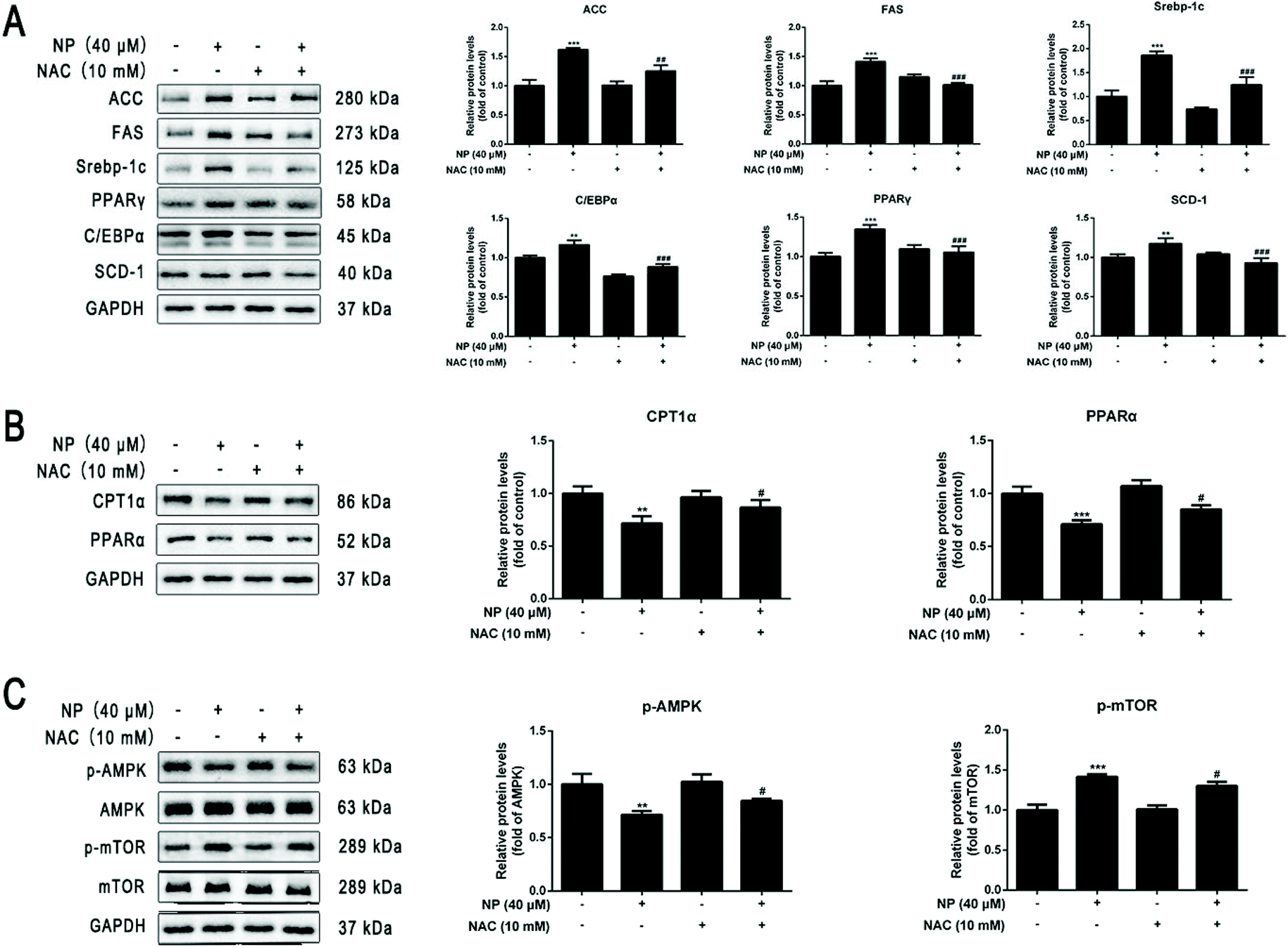
To further investigate the mechanism of the role of ROS in NP-induced steatosis in HepG2 cells. The ROS scavenger NAC was used to inhibit the NP-induced oxidative stress response in HepG2 cells. Experiments were performed by comparing the control group (normal medium treatment for 24 h), NP alone (40 μM NP treatment for 24 h), NAC alone (10 mM NAC pretreatment for 2 h followed by normal medium change for 24 h) and combined NP and NAC treatment (10 mM NAC pretreatment for 2 h followed by 40 μM NP treatment for 24 h). The results were used to analyze the role of ROS in NP-induced steatosis in HepG2 cells.The effect of NAC on NP-induced ROS production in HepG2 cells was measured by detecting the expression of ROS content in each group of HepG2 cells by flow cytometry. The results demonstrated that the expression of ROS levels in HepG2 cells was significantly inhibited in the combined NP and NAC-treated group compared with the NP-treated group alone (Fig. 4A; P < 0.001).
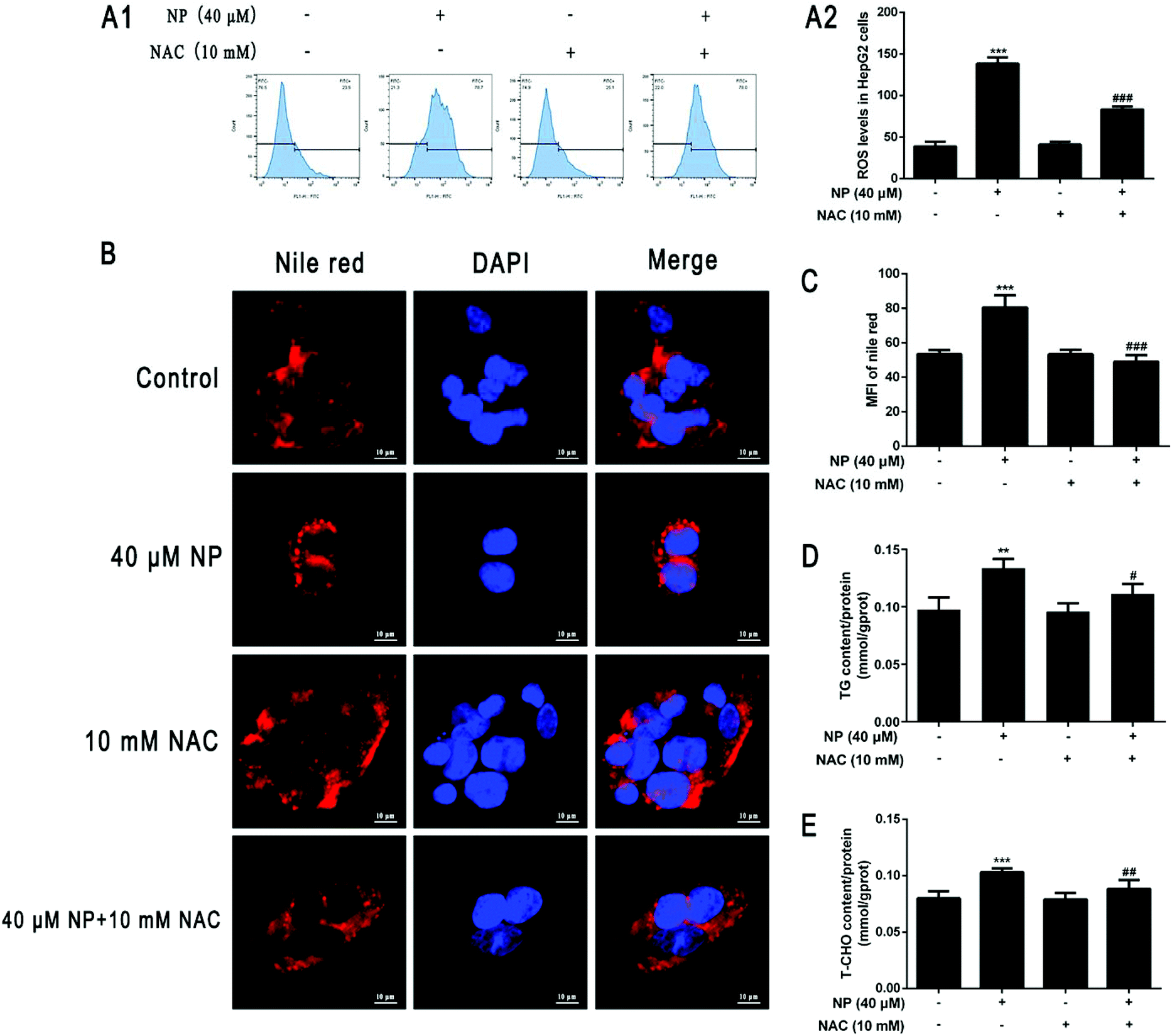 | ||
| Fig. 4 Effect of ROS in NP-induced steatosis in HepG2 cells. HepG2 cells were treated with 0 μM and 40 μM NP for 24 h, 10 mM NAC pretreated for 2 h and then changed to normal medium for 24 h and 10 mM NAC pretreated for 2 h followed by 40 μM NP for 24 h. (A) Effect of NAC on NP-induced ROS generation in HepG2 cells. The expression of ROS levels in HepG2 cells was quantified by flow cytometry using the DCFH-DA staining method. (B–C) Nile red staining was used to observe the changes of lipid droplets in HepG2 cells and statistical results, (D) determination of intracellular triglyceride expression level in HepG2 cells, (E) determination of total cholesterol expression level in HepG2 cells. ** P < 0.01, *** P < 0.001, significantly different as compared with the control group. # P < 0.05, ## P < 0.01, #### P < 0.001, significantly different as compared with the NP group. | ||
The impact of ROS on NP-induced steatosis in HepG2 cells was assessed by Nile Red staining and kits to detect intracellular lipid droplet production and determine TG and T-CHO content in each treatment group. Nile red staining fluorescence results showed that NAC pretreatment significantly reduced the number and shape size of lipid droplets compared with NP treatment alone (Fig. 4B and C; P < 0.001). The results of further detection of TG and T-CHO contents revealed that the combined treatment of NP and NAC significantly reduced the expression of TG and T-CHO levels in HepG2 cells (Fig. 4D; P < 0.05) compared with NP treatment alone (Fig. 4E; P < 0.01). Taken together, it can be concluded that ROS may be one of the factors of NP-induced steatosis and induction of NAFLD-like changes in HepG2 cells.
The effects of ROS on NP-induced lipogenesis in HepG2 cells were investigated by western blot analysis of the expression of lipogenesis-related key protein levels in each group. The results showed that the expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, and SCD-1 protein levels were significantly reversed in HepG2 cells in the combined NP and NAC-treated group compared with the NP-treated group alone (Fig. 5A; P < 0.01), indicating that ROS may indispensable in the promotion of lipogenesis in NP treated HepG2 cells.
 | ||
| Fig. 5 Role of ROS in NP-induced lipid metabolism and AMPK/mTOR signaling pathway in HepG2 cells. HepG2 cells were treated with 0 μM and 40 μM NP for 24 h, 10 mM NAC pretreated for 2 h and then changed to normal medium for 24 h and 10 mM NAC pretreated for 2 h followed by 40 μM NP for 24 h. (A) Role of ROS in NP-induced lipogenesis in HepG2 cells. The ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1 were measured by western blot. (B) Role of ROS in fatty acid oxidation in HepG2 cells inhibited by NP. The expression of CPT1α and PPARα protein levels were determined by western blot. (C) Role of ROS in AMPK/mTOR signaling pathway in NP-inhibited HepG2 cells. The expression of p-AMPK, AMPK, p-mTOR, and mTOR protein levels were examined by western blot after treatment. ** P < 0.01, *** P < 0.001, significantly different as compared with the control group. # P < 0.05, ## P < 0.01, #### P < 0.001, significantly different as compared with the NP group. | ||
The effects of ROS on FAO in NP-inhibited HepG2 cells were investigated by western blot analyses of the expression of key protein levels associated with FAO in each group. The results showed that NAC pretreatment significantly reversed (decreased) the protein expression levels of CPT1α and PPARα in control and NP treated HepG2 cells (Fig. 5B; P < 0.05), demonstrating that ROS may be one of the important triggers of FAO inhibition by NP in HepG2 cells.
Based on the above experimental results, it was further investigated whether NP affects the AMPK/mTOR signaling pathway via ROS and thus has an impact in the induction of HepG2 cell steatosis. As shown in Fig. 5C, the results of western blot experiments displayed that the ratio of p-AMPK/AMPK was significantly higher (Fig. 5C; P < 0.05) and the ratio of p-mTOR/mTOR was significantly lower (Fig. 5C; P < 0.05) in HepG2 cells in the combined NP and NAC treatment group compared with the NP treatment group alone. These results indicate that NP may inhibit AMPK phosphorylation in HepG2 cells through ROS action and significantly promote mTOR signaling, thus inducing NAFLD-like changes in HepG2 cells.
3.6. Curcumin ameliorates NP-induced steatosis in HepG2 cells
HepG2 cells were co-incubated with different concentration gradients of curcumin (5–60 μM) combined with 40 μM NP for 24 h. The effect of NP on HepG2 cell viability was previously determined by CCK-8 kit. The results demonstrated that curcumin had no significant effect on HepG2 cell viability when treated with 5–20 μM concentrations (P > 0.05), while HepG2 cell viability decreased significantly (P < 0.001) at concentrations above 20 μM, and approximately 57.3% at 40 μM curcumin treatment (Fig. 6A). Furthermore, HepG2 cells were incubated with curcumin at concentrations of 5 μM, 10 μM, and 20 μM in combination with 40 μM NP for 24 h. The results showed that curcumin reduced the lipid accumulation of HepG2 cells caused by NP (Fig. 6B and E). In comparison, the curcumin significantly inhibited the expression of TG and T-CHO levels in HepG2 cells, and the TG content for co-treatment with 10 μM curcumin was cleared by about 46.2% (Fig. 6C; P < 0.001). The TCHO content under the co-treatment with 20 μM curcumin was cleared by about 45.1% (Fig. 6D; P < 0.001). The above results indicate that curcumin can ameliorate NP-induced steatosis in HepG2 cells.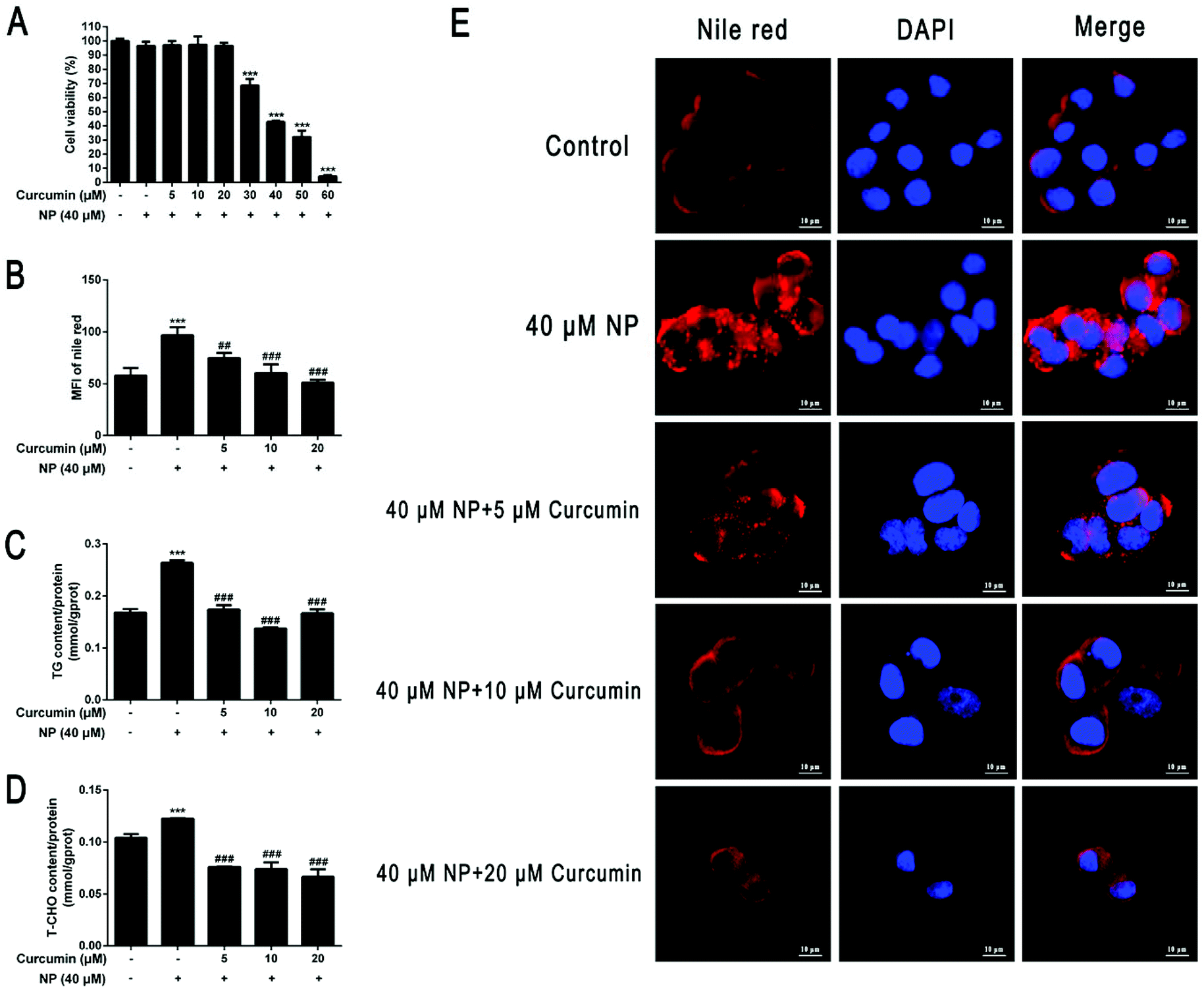 | ||
| Fig. 6 (A) Effect of curcumin on the viability of HepG2 cells. HepG2 cells were co-treated with different concentration gradients of curcumin (5–60 μM) combined with 40 μM NP for 24 h. The effect of curcumin on cell viability was analyzed by CCK-8 kit. (B–E) Effect of curcumin on NP-induced HepG2 cell steatosis. HepG2 cells were treated with 40 μM NP for 24 h. 5 μM, 10 μM, 20 μM curcumin and 40 μM NP were co-treated for 24 h. (B and E) Nile red staining was used to observe the changes of lipid droplets in HepG2 cells and the statistical results. (C) The determination of the expression levels of triglycerides in HepG2 cells. (D) The determination of the expression levels of total cholesterol in HepG2 cells. *** P < 0.001, significantly different as compared with the control group. ## P < 0.01, ## P < 0.001, significantly different as compared with the NP group. | ||
3.7. Curcumin ameliorates NP-induced disruption of lipid metabolism in HepG2 cells
Whether the curcumin mechanism ameliorating NP-induced steatosis in HepG2 cells was associated with the inhibition of lipogenesis and promotion of fatty acid oxidation was investigated further. The results of real-time fluorescence quantitative PCR and western blot analysis showed that compared with NP treatment alone, curcumin co-treatment significantly down-regulated the expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, and SCD-1 mRNA levels and protein levels in HepG2 cells (Fig. 7A), improving the NP-exposed HepG2 cell mRNA and protein expression levels of CPT1α and PPARα that were down-regulated in HepG2 cells after NP exposure (Fig. 7B; P < 0.001). The above results indicate that curcumin may facilitate the expression of key genes related to fatty acid oxidation, suppress key genes related to lipogenesis and, thus, play an essential role in ameliorating NP-induced disruption of lipid metabolism in HepG2 cells.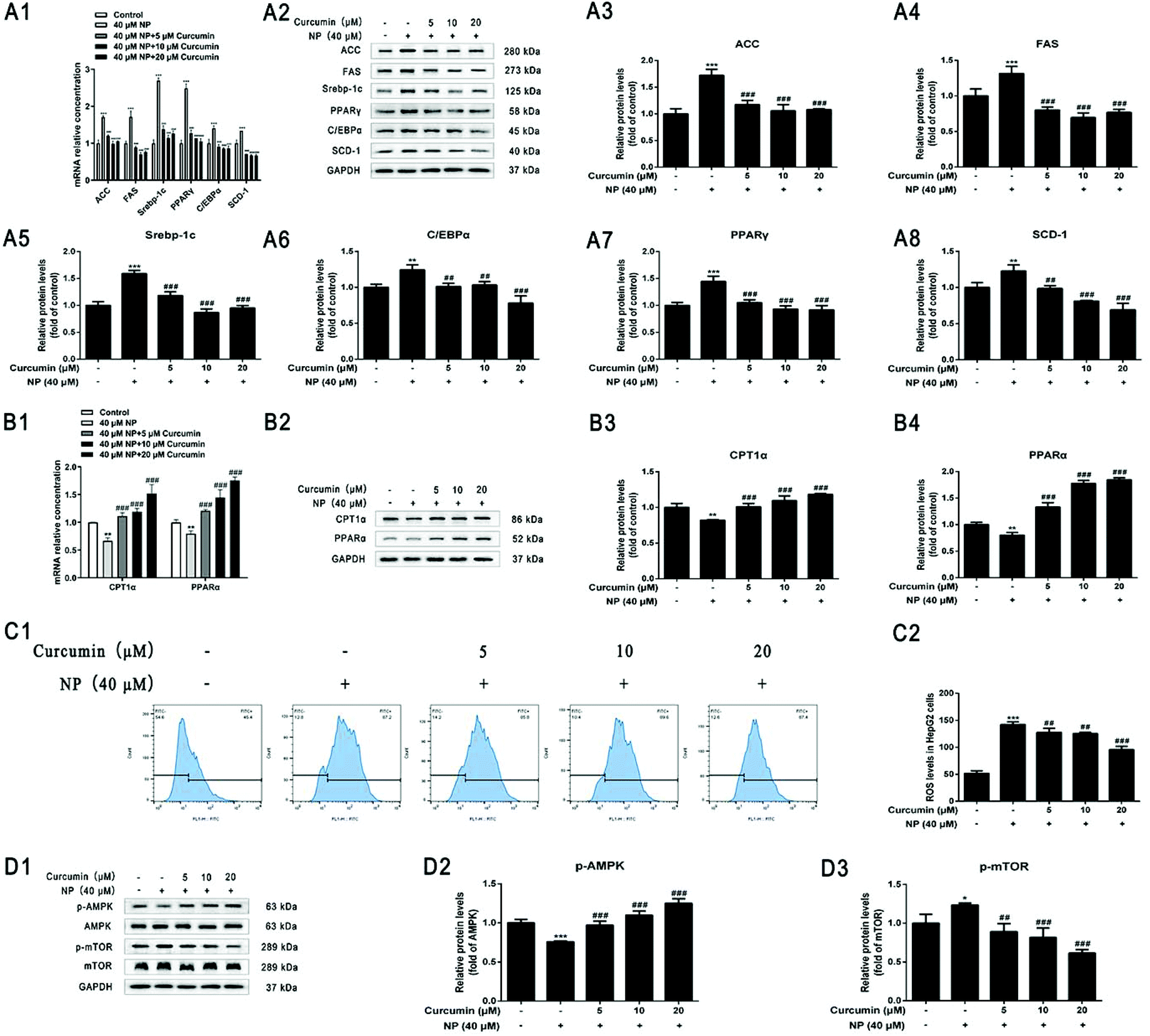 | ||
| Fig. 7 Effect of curcumin on NP-induced lipid metabolism and ROS levels and AMPK/mTOR signaling pathway in HepG2 cells. HepG2 cells were treated with 40 μM NP for 24 h. 5 μM, 10 μM, 20 μM curcumin and 40 μM NP were co-treated for 24 h. (A) Role of curcumin in NP-induced lipogenesis in HepG2 cells. (A1) RT-qPCR was performed to determine the expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1 mRNA levels, (A2–A8) western blot was applied to determine the protein level expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1. (B) Role of curcumin in fatty acid oxidation in NP-inhibited HepG2 cells. (B1) RT-qPCR was applied to determine the expression of CPT1α and PPARα mRNA levels, and (B2–B4) western blot was performed to determine the expression of CPT1α and PPARα protein levels. (C) The expression of ROS levels in HepG2 cells was quantified by flow cytometry. (D) The expression of p-AMPK, AMPK, p-mTOR, and mTOR protein levels were explored by western blot. * P < 0.05, ** P < 0.01, *** P < 0.001, significantly different as compared with the control group. ## P < 0.01, #### P < 0.001, significantly different as compared with the NP group. | ||
3.8. Curcumin ameliorates NP-promoted oxidative stress in HepG2 cells
The impact of curcumin on ameliorating oxidative stress response was further examined in HepG2 cells with NP-induced steatosis. Flow cytometry results showed a significant reduction in the level of ROS expression in HepG2 cells after curcumin co-treatment compared with NP alone (Fig. 7C; P < 0.01), indicating that the mechanism of curcumin ameliorating NP-induced steatosis in HepG2 cells may be related to the inhibition of ROS production.3.9. Curcumin activates the NP-inhibited ROS/AMPK/mTOR signaling pathway
The impact of curcumin to ameliorate NP-inhibited ROS/AMPK/mTOR pathway was further determined in HepG2 cells with NP-induced steatosis. As shown in Fig. 7D, western blot results indicated that co-treatment with curcumin significantly elevated the p-AMPK/AMPK ratio (Fig. 7D; P < 0.001) and significantly decreased the p-mTOR/mTOR ratio (Fig. 7D; P < 0.01) in HepG2 cells compared to NP alone. These results demonstrate that curcumin may ameliorate NAFLD-like alterations in HepG2 cells by activating AMPK phosphorylation and significantly interfering with mTOR signaling. In other words, the mechanism by which curcumin ameliorates NP-induced steatosis in HepG2 cells may be associated with the activation of the AMPK/mTOR signaling pathway.4. Discussion
NAFLD as a chronic liver disease has complex pathological manifestations. The accumulation of substantial fat in hepatocytes causes hepatic steatosis, which in turn triggers inflammation and fibrosis resulting in severe liver damage.57 NAFLD has captured the attention of various countries around the world due to its high global prevalence, diversity of consequences, severity, and tremendous economic burden.4.1. NP exposure causes HepG2 cell steatosis through lipid metabolism disorders
Lipid metabolism regulates the balance between TG production and clearance to maintain intrahepatic lipid homeostasis, and disruption of lipid metabolism is an important feature in the development of NAFLD.58Recent studies have indicated that NP, a widespread EDC, can induce hepatic steatosis and may be one of the important risk factors for the occurrence and development of NAFLD.30–35 In this study, we attempted to investigate whether HepG2 cells undergo NAFLD-like changes after NP exposure. The effect of NP on the viability of HepG2 cells was first examined by CCK-8, and then its effect and potential mechanism of steatosis in HepG2 cells was investigated using 20 μM, 30 μM, and 40 μM NP exposure to induce steatosis. In the steatosis initiation experiments, lipid droplets accumulated significantly in HepG2 cells after NP exposure (P < 0.001), and the expression levels of TG and T-CHO increased significantly (P < 0.001). Thus, a model of NP exposure-induced steatosis in HepG2 cells was successfully established.
To investigate the mechanism of lipid accumulation in HepG2 cells due to NP exposure, gene and protein expression levels of important transcription factors and key enzymes in the process of lipogenesis and fatty acid oxidation were examined. We observed that the expression of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, SCD-1 lipogenesis-related key gene mRNA levels and protein levels were significantly upregulated in HepG2 cells treated with NP for 24 h (P < 0.05). In contrast, the expression of fatty acid oxidation-related key genes CPT1α, and PPARα mRNA levels and protein levels were significantly decreased in HepG2 cells (P < 0.05). Excessive accumulation of fatty acids occurs in NAFLD, and fatty acid synthesis is mediated mainly by ACC and FAS. ACC generates malonyl mono-acyl coenzyme A through the condensation of adenosine triphosphate-dependent acetyl coenzyme A with carbonate, whereas FAS catalyzes the synthesis of palmitic acid using acetyl coenzyme A or malonyl mono-acyl coenzyme A.59 The essential transcription factors Srepb-1c, PPARγ, C/EBPα can individually or synergistically regulate the expression of key lipid-producing genes to activate lipogenesis. And perinatal 4-NP exposure increased the expression of hepatic FAS, Srebp-1 and PPARγ in F1 and F2 generation rats.60 SCD-1 in fatty acid synthesis is responsible for the formation of stearoyl coenzyme A double bond that converts saturated fatty acids to monounsaturated fatty acids.61 In NAFLD, free fatty acids (FFA) accomplish lipid degradation mainly through the β-oxidation pathway.62 β-Oxidation occurs mainly in the mitochondria. Long-chain FFA enters the mitochondria to form lipoyl coenzyme A, which is then transported to the mitochondrial matrix for β-oxidation via CPT1 on the inner membrane.38 The transcription factor PPARα plays an essential regulatory role in the 4-step β-oxidation sequential reaction of dehydrogenation, water addition, rehydrogenation and thiolysis.63 Previous population and animal experimental studies have shown that impaired FAO occurs in NAFLD.64,65 The results of the present study are consistent with those described in the literature above, indicating that NP exposure can lead to changes in gene and protein expression levels of transcription factors and key enzymes important in lipogenesis and fatty acid oxidation in HepG2 cells, promoting hepatic fatty acid synthesis and excessive accumulation, leading to steatosis in HepG2 cells.
4.2. Study on the mechanism of NP induced HepG2 cells steatosis through ROS
In the development of NAFLD, lipid metabolism, oxidative stress, and inflammation are the three pillars of the pathogenesis.6 An epidemiological theory suggests that an excessive increase in hepatic FFA hinders the efficiency of β-oxidation and in the process leads to an excessive production of ROS, which increases oxidative stress, resulting in abnormal lipogenesis and fatty acid oxidation, massive formation of lipid droplets, altering the course of lipid metabolism, causing hepatic steatosis and NAFLD-like changes.66–68 Our study found that NP may cause HepG2 cell steatosis through the ROS-mediated AMPK/mTOR signaling pathway.First, by detecting the ROS content in HepG2 cells after 24 h of NP exposure, we found that NP promoted an oxidative stress response in HepG2 cells. Second, to explore the mechanism of ROS action in HepG2 cell steatosis caused by NP exposure, the ROS scavenger N-acetyl-L-cysteine NAC was used to play a role in improving liver function by supplementing glutathione (GSH) levels to downregulate ROS levels.69 In addition, NAC as a ROS scavenger, may attenuate NAFLD lipid accumulation and reverse the severity of liver injury by regulating the transcription factors of FFA and signaling molecules Srebp-1c/-2, PPARγ, and CD36.70–72 This suggests that the excessive accumulation of ROS in NAFLD may regulate the reprogramming of hepatic lipid metabolism.73 In our experiments, the number of intracellular lipid droplets decreased and became smaller in shape after NAC pretreatment, and the TG and T-CHO contents were significantly lower compared with the NP exposure alone group (P < 0.05), indicating that inhibition of ROS can reduce lipid accumulation in HepG2 cells and attenuate the effect of NP exposure-induced steatosis. Since ROS signaling may cause alterations in lipid metabolism homeostasis.73 The results of western blot experiments in this study illustrated that, compared with NP treatment alone, the combined treatment of NP and NAC significantly down-regulated the expression of key genes for lipogenesis ACC, FAS, Srebp-1c, C/EBPα, PPARγ, and SCD-1 protein levels (P < 0.01) and significantly up-regulated the expression of key genes for fatty acid oxidation CPT1α, PPARα protein levels (P < 0.05), and these results suggest that, by affecting the expression of key transcription factors and key enzymes of lipid metabolism (lipogenesis/fatty acid oxidation), ROS may regulate HepG2 cell steatosis caused by NP exposure.
Last, the activity of Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) is controlled by nutritional metabolic diseases such as NAFLD. For instance, a high-fat diet rich in palmitic acid can reverse the activation of hepatic AMPK.74 AMPK is a serine/threonine protein kinase composed of three subunits: α catalytic subunit, β and γ regulatory subunits.75 The function of AMPK as an energy sensor is influenced by the ratio of AMP/ATP. When AMP/ATP is high, it is activated and phosphorylated, it increases lipid oxidation and mitochondrial biogenesis while reducing lipogenesis and gluconeogenesis, thereby increasing intracellular ATP levels to maintain the homeostasis of intracellular energy.76,77 In contrast, in the hepatic lipid metabolism, mTOR can form the catalytic center of two multiprotein complexes (mTORC1 and mTORC2) in response to upstream signals (AMPK).78,79
In present study, western blot detection of p-AMPK, p-mTOR, AMPK, mTOR related protein expression results illustrated that p-AMPK/AMPK was markedly decreased (P < 0.01), p-mTOR/mTOR was dramatically increased (P < 0.001, Fig. 3), indicating that the effect of NP exposure on the AMPK/mTOR signaling pathway may inhibit AMPK phosphorylation and promote the activation of mTOR signaling, thereby inducing steatosis of HepG2 cells. Based on the above experiments, the effects of ROS on AMPK/mTOR signaling pathway were further explored. The results revealed that NAC pretreatment significantly reversed the expression of p-AMPK/AMPK and p-mTOR/mTOR in HepG2 cells, resulting in elevated AMPK phosphorylation and decreased mTOR phosphorylation (P < 0.05, Fig. 5). These results suggest that NP may cause HepG2 cell steatosis through the ROS-mediated AMPK/mTOR signaling pathway.
4.3. Study on the effect mechanism of curcumin intervention on nonylphenol-induced steatosis in HepG2 cells
NAFLD-related therapeutic agents have been developed and are mostly in clinical phase II and phase III trials, but there are still significant challenges to overcome for these agents to enter clinical use.80 Curcumin, a polyphenolic bioactive compound, has antioxidant and anti-inflammatory properties, and existing studies have described the therapeutic potential of curcumin in the prevention and treatment of human diseases, especially NAFLD.47,81–83 By establishing a model of NP-induced steatosis in HepG2 cells for the first time, we found that curcumin could play a protective role against NP-induced NAFLD-like changes in HepG2 cells by ameliorating oxidative stress and possibly through the ROS-mediated AMPK/mTOR signaling pathway.In previous studies, curcumin could improve NAFLD by regulating lipid metabolic pathways, such as downregulating TG expression in NAFLD.51,84 High-fat diet supplementation with curcumin reduces circulating lipids and hepatic lipid levels.85 The results of in vivo and in vitro experiments indicated that curcumin can significantly decrease the expression of intracellular lipid metabolism-related genes FAS, ACC, CD36, Srebp1, and PPARγ to inhibit hepatic lipogenesis and increase PPARα and CPT1 levels to promote related β-oxidation and thus improve NAFLD symptoms.86,87 Fewer studies have been conducted on whether curcumin ameliorates NP-induced steatosis in HepG2 cells. In our experiments, after establishing an NP-induced steatosis model in HepG2 cells, by the detection of indicators related to lipid metabolism disorders, lipid droplet particles were smaller or even disappeared under curcumin co-treatment compared to NP alone, and the TG and T-CHO contents in HepG2 cells were significantly cleared (P < 0.001). mRNA levels and protein levels of ACC, FAS, Srebp-1c, C/EBPα, PPARγ, and SCD-1 lipogenesis-related key genes were significantly reduced (P < 0.01), while the expression of mRNA levels and protein levels of key genes related to fatty acid oxidation in CPT1α and PPARα were significantly increased (P < 0.001). The above results suggest that curcumin intervention could reverse the disorder of lipid metabolism caused by NP and could decrease lipid degeneration in HepG2 cells by affecting the expression of key transcription factors and key enzymes of lipid metabolism.
Existing studies in methionine and choline-deficient (MCD) diet models have shown that curcumin can reduce hepatocyte lipid accumulation by upregulating AMPK mRNA expression and decreasing Srebp-1c, ACC, and FAS expression in mouse liver, while inhibiting the inflammatory NF-κB pathway, thereby ameliorating the severity of steatosis.88,89 In addition, curcumin exerts antioxidant properties by inhibiting the expression of ROS and malondialdehyde levels and promoting the expression of antioxidants such as SOD, CAT and GSH Px to improve hepatic lipid metabolism and reduce lipid accumulation.90 Therefore, it was further explored whether curcumin could ameliorate NP-induced HepG2 cell steatosis by ameliorating oxidative stress in HepG2 cells after NP exposure and possibly through the ROS-mediated AMPK/mTOR signaling pathway. Curcumin was found to reverse the inhibition of AMPK/mTOR signaling pathway by NP when combined with NP (40 μM) treatment of HepG2 cells, and exerted a protective effect against NP-induced NAFLD-like alterations in HepG2 cells by activating the AMPK/mTOR signaling pathway (Fig. 7).
Despite the robust ability of curcumin to improve NAFLD, pharmacokinetic and bioavailability studies have revealed that the clinical promotion of curcumin as a therapeutic agent for NAFLD is extremely restricted by its low bioavailability and rapid metabolic elimination profile.46,91–93 One study found that human serum concentrations peaked at 1 to 2 hours after taking 4000 mg, 6000 mg and 8000 mg of curcumin, with mean peak serum concentrations of 0.51 ± 0.11 μM, 0.63 ± 0.06 μM and 1.77 ± 1.87 μM, respectively.94 Although its plasma concentration is low, curcumin has become a research hotspot because of its great potential for NAFLD prevention and treatment.40–42 It has been shown that curcumin can be detected as curcumin glucuronide and curcumin sulfate in plasma after oral administration.95,96 However, the data to date do not support the utilization of curcumin as a prodrug for the treatment of diseases.97,98 For example, it was found that tetrahydrocurcumin, a major reducing metabolite of curcumin, was a less effective inhibitor of NF-κB, cyclooxygenase-1,5-lipoxygenase and proliferation at the same concentrations.98,99 In our study, curcumin was found to have a significant ameliorative effect on NP-induced NAFLD-like changes in HepG2 cells. Therefore, the bioavailability of curcumin must be improved to achieve reasonable and effective therapeutic benefits in humans.
On the one hand, the application of modern technologies has considerably addressed the bioavailability of curcumin. The bioavailability of curcumin can be substantially improved by modern formulation technologies, including nanoparticles, nanoemulsions, phospholipid complexes, liposome encapsulation, capsules, tablets, and powders.100–103 A study of recent clinical trials has demonstrated that the bioavailability of curcumin in different formulations is 100 times higher than that of curcumin alone.104 On the other hand, the chemical structure of curcumin is a crucial part in its biological activity. The bioavailability of curcumin can be significantly improved by modifying the structure of curcumin, such as novel polyethylene glycolized curcumin derivatives that can improve blood concentration and tissue distribution.105 In summary, many approaches have been taken in existing studies to improve the bioavailability of curcumin. As formulation techniques and structural modification methods are developed and matured, curcumin will have higher bioavailability and will most likely be used at the forefront of NAFLD treatment.
5. Conclusion
In conclusion, our study demonstrated that NP exposure could induce HepG2 cell steatosis, cause lipid metabolism disorders, and promote ROS production, possibly involving inhibition of the AMPK/mTOR signaling pathway. In contrast, curcumin intervention may reverse the effects caused by NP, thereby ameliorating NP-induced NAFLD-like alterations in HepG2 cells.Abbreviations
| ACC | Acetyl–CoA carboxylase |
| AMPK | AMP-dependent protein kinase |
| CCK-8 | Cell counting kit-8 |
| C/EBPα | CCAAT-enhancer-binding proteins |
| CPT1α | Carnitine palmitoyl transferase I |
| EDCs | Endocrine disrupting chemicals |
| FAO | Fatty acid oxidation |
| FAS | Fatty acid synthase |
| FFA | Free fat acid |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| mTOR | Target of rapamycin protein |
| NAC | N-Acetyl-L-cysteine |
| NAFLD | Non-alcoholic fatty liver disease |
| NP | Nonylphenol |
| p-AMPK | Phosphorylated AMP-dependent protein kinase |
| p-mTOR | Phosphorylated target of rapamycin protein |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| RT-qPCR | Real-time quantitative PCR |
| ROS | Reactive oxygen species |
| SCD-1 | Stearoyl–CoA desaturase-1 |
| Srebp-1c | Sterol regulatory element-binding protein factor 1c |
| T-CHO | Total cholesterol |
| TG | Triglyceride |
Author contributions
The corresponding author is responsible for ensuring that the descriptions are accurate and agreed by all authors. Dandan Shan: Formal analysis; writing – original draft; writing – review & editing. Jinming Wang: Conceptualization; methodology; investigation. Qiannan Di: Methodology; investigation; visualization. Qianqian Jiang: Validation; data curation. Qian Xu: Resources; supervision; project administration; funding acquisition.Conflicts of interest
The author declare that this report content has no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project No. 81673171) and Social Development Research Program of Jiangsu Province Science and Technology Department (Project No. BE 2015646).References
- S. Gitto, G. Vitale, E. Villa and P. Andreone, Treatment of nonalcoholic steatohepatitis in adults: present and future, Gastroenterol. Res. Pract., 2015, 732870, DOI:10.1155/2015/732870.
- Q. M. Anstee, G. Targher and C. P. Day, Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 330–344 CrossRef CAS PubMed.
- Q. M. Anstee, S. McPherson and C. P. Day, How big a problem is non-alcoholic fatty liver disease?, Br. Med. J., 2011, 343, d3897 CrossRef PubMed.
- J. Zhou, F. Zhou, W. Wang, X. J. Zhang, Y. X. Ji, P. Zhang, Z. G. She, L. Zhu, J. Cai and H. Li, Epidemiological Features of NAFLD From 1999 to 2018 in China, Hepatology, 2020, 71, 1851–1864 CrossRef PubMed.
- Y. Fazel, A. B. Koenig, M. Sayiner, Z. D. Goodman and Z. M. Younossi, Epidemiology and natural history of non-alcoholic fatty liver disease, Metabolism, 2016, 65, 1017–1025 CrossRef CAS PubMed.
- E. Buzzetti, M. Pinzani and E. A. Tsochatzis, The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD), Metabolism, 2016, 65, 1038–1048 CrossRef CAS PubMed.
- C. P. Day and O. F. James, Steatohepatitis: a tale of two “hits”?, Gastroenterology, 1998, 114, 842–845 CrossRef CAS.
- J. J. Heindel, B. Blumberg, M. Cave, R. Machtinger, A. Mantovani, M. A. Mendez, A. Nadal, P. Palanza, G. Panzica, R. Sargis, L. N. Vandenberg and F. Vom Saal, Metabolism disrupting chemicals and metabolic disorders, Reprod. Toxicol., 2017, 68, 3–33 CrossRef CAS PubMed.
- A. Marmugi, S. Ducheix, F. Lasserre, A. Polizzi, A. Paris, N. Priymenko, J. Bertrand-Michel, T. Pineau, H. Guillou, P. G. Martin and L. Mselli-Lakhal, Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver, Hepatology, 2012, 55, 395–407 CrossRef CAS PubMed.
- C. E. Foulds, L. S. Treviño, B. York and C. L. Walker, Endocrine-disrupting chemicals and fatty liver disease, Nat. Rev. Endocrinol., 2017, 13, 445–457 CrossRef CAS PubMed.
- L. S. Treviño and T. A. Katz, Endocrine Disruptors and Developmental Origins of Nonalcoholic Fatty Liver Disease, Endocrinology, 2018, 159, 20–31 CrossRef PubMed.
- R. Cano, J. L. Pérez, L. A. Dávila, Á. Ortega, Y. Gómez, N. J. Valero-Cedeño, H. Parra, A. Manzano, T. I. Véliz Castro, M. P. D. Albornoz, G. Cano, J. Rojas-Quintero, M. Chacín and V. Bermúdez, Role of Endocrine-Disrupting Chemicals in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: A Comprehensive Review, Int. J. Mol. Sci., 2021, 22, 4807 CrossRef CAS PubMed.
- A. Ekdal, Fate of nonylphenol ethoxylate (NPEO) and its inhibitory impact on the biodegradation of acetate under aerobic conditions, Environ. Technol., 2014, 35, 741–748 CrossRef CAS PubMed.
- A. Soares, B. Guieysse, B. Jefferson, E. Cartmell and J. N. Lester, Nonylphenol in the environment: a critical review on occurrence, fate, toxicity and treatment in wastewaters, Environ. Int., 2008, 34, 1033–1049 CrossRef CAS PubMed.
- Y. Niu, J. Zhang, H. Duan, Y. Wu and B. Shao, Bisphenol A and nonylphenol in foodstuffs: Chinese dietary exposure from the 2007 total diet study and infant health risk from formulas, Food Chem., 2015, 167, 320–325 CrossRef CAS PubMed.
- Y. Y. Lu, M. L. Chen, F. C. Sung, P. S. Wang and I. F. Mao, Daily intake of 4-nonylphenol in Taiwanese, Environ. Int., 2007, 33, 903–910 CrossRef CAS PubMed.
- K. Guenther, E. Kleist and B. Thiele, Estrogen-active nonylphenols from an isomer-specific viewpoint: a systematic numbering system and future trends, Anal. Bioanal. Chem., 2006, 384, 542–546 CrossRef CAS PubMed.
- I. Aparicio, J. Martín, C. Abril, J. L. Santos and E. Alonso, Determination of household and industrial chemicals, personal care products and hormones in leafy and root vegetables by liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 2018, 1533, 49–56 CrossRef CAS PubMed.
- Y. Kawamura, Y. Ogawa and M. Mutsuga, Migration of nonylphenol and plasticizers from polyvinyl chloride stretch film into food stimulants, rapeseed oil, and foods, Food Sci. Nutr., 2017, 5, 390–398 CrossRef CAS PubMed.
- A. Careghini, A. F. Mastorgio, S. Saponaro and E. Sezenna, Bisphenol A, nonylphenols, benzophenones, and benzotriazoles in soils, groundwater, surface water, sediments, and food: a review, Environ. Sci. Pollut. Res. Int., 2015, 22, 5711–5741 CrossRef CAS PubMed.
- E. Nielsen, G. Østergaard, I. Thorup, O. Ladefoged, O. Jelnes and J. E. Jelnes, Toxicological evaluation and limit values for nonylphenol, nonylphenol ethoxylates, tricresyl, phosphates and benzoic acid, Danish Environmental Protection Agency, Copenaghen, DK, Environmental Project, 2000, vol. 512, p. 43 Search PubMed.
- K. Guenther, V. Heinke, B. Thiele, E. Kleist, H. Prast and T. Raecker, Endocrine disrupting nonylphenols are ubiquitous in food, Environ. Sci. Technol., 2002, 36, 1676–1680 CrossRef CAS PubMed.
- B. M. Thomson, P. J. Cressey and I. C. Shaw, Dietary exposure to xenoestrogens in New Zealand, J. Environ. Monit., 2003, 5, 229–235 RSC.
- B. Roig, A. Cadiere, S. Bressieux, S. Biau, S. Faure and P. de Santa Barbara, Environmental concentration of nonylphenol alters the development of urogenital and visceral organs in avian model, Environ. Int., 2014, 62, 78–85 CrossRef CAS PubMed.
- S. Pal, P. Nath, S. Biswas, U. Mukherjee and S. Maitra, Nonylphenol attenuates SOCS3 expression and M1 polarization in lipopolysaccharide-treated rat splenic macrophages, Ecotoxicol. Environ. Saf., 2019, 174, 574–583 CrossRef CAS PubMed.
- W. Xu, J. Yu, S. Li and J. Xu, Depressive behavior induced by nonylphenol and its effect on the expression of ER-α and ER-β in nerve cells of rats, J. Affective Disord., 2020, 263, 373–381 CrossRef CAS PubMed.
- R. L. Spehar, L. T. Brooke, T. P. Markee and M. D. Kahl, Comparative toxicity and bioconcentration of nonylphenol in freshwater organisms, Environ. Toxicol. Chem., 2010, 29, 2104–2111 CAS.
- Z. Noorimotlagh, N. J. Haghighi, M. Ahmadimoghadam and F. Rahim, An updated systematic review on the possible effect of nonylphenol on male fertility, Environ. Sci. Pollut. Res. Int., 2017, 24, 3298–3314 CrossRef CAS PubMed.
- S. Kazemi, S. N. Mousavi Kani, M. Ghasemi-Kasman, F. Aghapour, H. Khorasani and A. A. Moghadamnia, Nonylphenol induces liver toxicity and oxidative stress in rat, Biochem. Biophys. Res. Commun., 2016, 479, 17–21 CrossRef CAS PubMed.
- R. Jubendradass, S. C. D'Cruz and P. P. Mathur, Long-term exposure to nonylphenol affects insulin signaling in the liver of adult male rats, Hum. Exp. Toxicol., 2012, 31, 868–876 CrossRef CAS PubMed.
- V. Meucci and A. Arukwe, Transcriptional modulation of brain and hepatic estrogen receptor and P450arom isotypes in juvenile Atlantic salmon (Salmo salar) after waterborne exposure to the xenoestrogen, 4-nonylphenol, Aquat. Toxicol., 2006, 77, 167–177 CrossRef CAS PubMed.
- J. Yu, X. Yang, X. Yang, M. Yang, P. Wang, Y. Yang, J. Yang, W. Li and J. Xu, Nonylphenol aggravates non-alcoholic fatty liver disease in high sucrose-high fat diet-treated rats, Sci. Rep., 2018, 8, 3232 CrossRef PubMed.
- F. Maradonna, V. Nozzi, S. Santangeli, I. Traversi, P. Gallo, E. Fattore, D. G. Mita, A. Mandich and O. Carnevali, Xenobiotic-contaminated diets affect hepatic lipid metabolism: Implications for liver steatosis in Sparus aurata juveniles, Aquat. Toxicol., 2015, 167, 257–264 CrossRef CAS PubMed.
- A. Kourouma, H. Keita, P. Duan, C. Quan, K. K. Bilivogui, S. Qi, N. A. Christiane, A. Osamuyimen and K. Yang, Effects of 4-nonylphenol on oxidant/antioxidant balance system inducing hepatic steatosis in male rat, Toxicol. Rep., 2015, 2, 1423–1433 CrossRef CAS PubMed.
- I. Bernabò, P. Biasone, R. Macirella, S. Tripepi and E. Brunelli, Liver histology and ultrastructure of the Italian newt (Lissotriton italicus): normal structure and modifications after acute exposure to nonylphenol ethoxylates, Exp. Toxicol. Pathol., 2014, 66, 455–468 CrossRef PubMed.
- J. Zha, Z. Wang, N. Wang and C. Ingersoll, Histological alternation and vitellogenin induction in adult rare minnow (Gobiocypris rarus) after exposure to ethynylestradiol and nonylphenol, Chemosphere, 2007, 66, 488–495 CrossRef CAS PubMed.
- M. Huff, W. da Silveira, E. Starr Hazard, S. M. Courtney, L. Renaud and G. Hardiman, Systems analysis of the liver transcriptome in adult male zebrafish exposed to the non-ionic surfactant nonylphenol, Gen. Comp. Endocrinol., 2019, 271, 1–14 CrossRef CAS PubMed.
- Z. Li, Y. Li, H. X. Zhang, J. R. Guo, C. W. K. Lam, C. Y. Wang and W. Zhang, Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease, Mol. Nutr. Food Res., 2019, 63, e1900043 CrossRef PubMed.
- Y. Xu, W. Guo, C. Zhang, F. Chen, H. Y. Tan, S. Li, N. Wang and Y. Feng, Herbal Medicine in the Treatment of Non-Alcoholic Fatty Liver Diseases-Efficacy, Action Mechanism, and Clinical Application, Front. Pharmacol., 2020, 11, 601 CrossRef CAS PubMed.
- S. Saadati, A. Sadeghi, A. Mansour, Z. Yari, H. Poustchi, M. Hedayati, B. Hatami and A. Hekmatdoost, Curcumin and inflammation in non-alcoholic fatty liver disease: a randomized, placebo controlled clinical trial, BMC Gastroenterol., 2019, 19, 133 CrossRef PubMed.
- Y. Panahi, P. Kianpour, R. Mohtashami, R. Jafari, L. E. Simental-Mendía and A. Sahebkar, Efficacy and Safety of Phytosomal Curcumin in Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial, Drug Res., 2017, 67, 244–251 CrossRef CAS PubMed.
- S. Rahmani, S. Asgary, G. Askari, M. Keshvari, M. Hatamipour, A. Feizi and A. Sahebkar, Treatment of Non-alcoholic Fatty Liver Disease with Curcumin: A Randomized Placebo-controlled Trial, Phytother. Res., 2016, 30, 1540–1548 CrossRef CAS PubMed.
- D. Jiao, J. Wang, W. Lu, X. Tang, J. Chen, H. Mou and Q. Y. Chen, Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3 K/Akt/mTOR signaling pathways in lung cancer, Mol. Ther.–Oncolytics, 2016, 3, 16018 CAS.
- L. M. Ailioaie and G. Litscher, Curcumin and Photobiomodulation in Chronic Viral Hepatitis and Hepatocellular Carcinoma, Int. J. Mol. Sci., 2020, 21, 7150 CrossRef CAS PubMed.
- S. Qin, L. Huang, J. Gong, S. Shen, J. Huang, H. Ren and H. Hu, Efficacy and safety of turmeric and curcumin in lowering blood lipid levels in patients with cardiovascular risk factors: a meta-analysis of randomized controlled trials, Nutr. J., 2017, 16, 68 CrossRef PubMed.
- M. Dei Cas and R. Ghidoni, Dietary Curcumin: Correlation between Bioavailability and Health Potential, Nutrients, 2019, 11, 2147 CrossRef PubMed.
- A. B. Kunnumakkara, D. Bordoloi, G. Padmavathi, J. Monisha, N. K. Roy, S. Prasad and B. B. Aggarwal, Curcumin, the golden nutraceutical: multitargeting for multiple chronic diseases, Br. J. Pharmacol., 2017, 174, 1325–1348 CrossRef CAS PubMed.
- U.S. Food & Drug Administration, GRN No. 686 Curcumin from turmeric (Curcuma longa L.), Available at: https://www.fda.gov/media/104050/download (accessed September 7, 2021).
- F. Mansour-Ghanaei, M. Pourmasoumi, A. Hadi and F. Joukar, Efficacy of curcumin/turmeric on liver enzymes in patients with non-alcoholic fatty liver disease: A systematic review of randomized controlled trials, Integr. Med. Res., 2019, 8, 57–61 CrossRef PubMed.
- N. Ghosh, R. Ghosh, V. Mandal and S. C. Mandal, Recent advances in herbal medicine for treatment of liver diseases, Pharm. Biol., 2011, 49, 970–988 CrossRef CAS PubMed.
- Y. Panahi, P. Kianpour, R. Mohtashami, R. Jafari, L. E. Simental-Mendía and A. Sahebkar, Curcumin Lowers Serum Lipids and Uric Acid in Subjects With Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial, J. Cardiovasc. Pharmacol., 2016, 68, 223–229 CrossRef CAS PubMed.
- R. Li, Y. Yao, P. Gao and S. Bu, The Therapeutic Efficacy of Curcumin vs. Metformin in Modulating the Gut Microbiota in NAFLD Rats: A Comparative Study, Front Microbiol., 2020, 11, 555293 CrossRef PubMed.
- F. Salomone, J. Godos and S. Zelber-Sagi, Natural antioxidants for non-alcoholic fatty liver disease: molecular targets and clinical perspectives, Liver Int., 2016, 36, 5–20 CrossRef CAS PubMed.
- K. Egashira, H. Sasaki, S. Higuchi and I. Ieiri, Food-drug interaction of tacrolimus with pomelo, ginger, and turmeric juice in rats, Drug Metab. Pharmacokinet., 2012, 27, 242–247 CrossRef CAS PubMed.
- X. Wu, Y. Song, S. Li, X. Liu, W. Hua, K. Wang, W. Liu, S. Li, Y. Zhang, Z. Shao and C. Yang, Pramlintide regulation of extracellular matrix (ECM) and apoptosis through mitochondrial-dependent pathways in human nucleus pulposus cells, Int. J. Immunopathol. Pharmacol., 2018, 31, 394632017747500 Search PubMed.
- T. Sanli, K. Linher-Melville, T. Tsakiridis and G. Singh, Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells, PLoS One, 2012, 7, e32035 CrossRef CAS PubMed.
- R. J. Schulze and M. A. McNiven, Lipid Droplet Formation and Lipophagy in Fatty Liver Disease, Semin. Liver Dis., 2019, 39, 283–290 CrossRef CAS PubMed.
- J. C. Cohen, J. D. Horton and H. H. Hobbs, Human fatty liver disease: old questions and new insights, Science, 2011, 332, 1519–1523 CrossRef CAS PubMed.
- C. S. Heil, S. S. Wehrheim, K. S. Paithankar and M. Grininger, Fatty Acid Biosynthesis: Chain-Length Regulation and Control, ChemBioChem, 2019, 20, 2298–2321 CrossRef CAS PubMed.
- H. Y. Zhang, W. Y. Xue, Y. S. Zhu, W. Q. Huo, B. Xu and S. Q. Xu, Perinatal exposure to 4-nonylphenol can affect fatty acid synthesis in the livers of F1 and F2 generation rats, Toxicol. Res., 2018, 7, 283–292 CrossRef CAS PubMed.
- J. Kim, E. J. Kang, M. N. Park, J. E. Kim, S. C. Kim, E. B. Jeung, G. S. Lee, D. Y. Hwang and B. S. An, The adverse effect of 4-tert-octylphenol on fat metabolism in pregnant rats via regulation of lipogenic proteins, Environ. Toxicol. Pharmacol., 2015, 40, 284–291 CrossRef CAS PubMed.
- J. P. Arab, M. Arrese and M. Trauner, Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease, Annu. Rev. Pathol.: Mech. Dis., 2018, 13, 321–350 CrossRef CAS PubMed.
- D. Moseti, A. Regassa and W. K. Kim, Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules, Int. J. Mol. Sci., 2016, 17, 124 CrossRef PubMed.
- S. Tanaka, K. Miyanishi, M. Kobune, Y. Kawano, T. Hoki, T. Kubo, T. Hayashi, T. Sato, Y. Sato, R. Takimoto and J. Kato, Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma, J. Gastroenterol., 2013, 48, 1249–1258 CrossRef CAS PubMed.
- S. Seki, T. Kitada, T. Yamada, H. Sakaguchi, K. Nakatani and K. Wakasa, In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases, J. Hepatol., 2002, 37, 56–62 CrossRef CAS PubMed.
- F. Nassir, R. S. Rector, G. M. Hammoud and J. A. Ibdah, Pathogenesis and Prevention of Hepatic Steatosis, Gastroenterol. Hepatol., 2015, 11, 167–175 Search PubMed.
- R. Vuppalanchi and N. Chalasani, Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management, Hepatology, 2009, 49, 306–317 CrossRef PubMed.
- Y. Jin, Y. Tan, L. Chen, Y. Liu and Z. Ren, Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression, Int. J. Mol. Sci., 2018, 19, 3445 CrossRef PubMed.
- G. F. Rushworth and I. L. Megson, Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits, Pharmacol. Ther., 2014, 141, 150–159 CrossRef CAS PubMed.
- Y. Ma, M. Gao and D. Liu, N-acetylcysteine Protects Mice from High Fat Diet-induced Metabolic Disorders, Pharm. Res., 2016, 33, 2033–2042 CrossRef CAS PubMed.
- I. K. Lai, K. Dhakal, G. S. Gadupudi, M. Li, G. Ludewig, L. W. Robertson and A. K. Olivier, N-acetylcysteine (NAC) diminishes the severity of PCB 126-induced fatty liver in male rodents, Toxicology, 2012, 302, 25–33 CrossRef CAS PubMed.
- C. C. Lin and M. C. Yin, Effects of cysteine-containing compounds on biosynthesis of triacylglycerol and cholesterol and anti-oxidative protection in liver from mice consuming a high-fat diet, Br. J. Nutr., 2008, 99, 37–43 CrossRef CAS PubMed.
- Z. Chen, R. Tian, Z. She, J. Cai and H. Li, Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, Free Radicals Biol. Med., 2020, 152, 116–141 CrossRef CAS PubMed.
- Y. Wu, P. Song, J. Xu, M. Zhang and M. H. Zou, Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase, J. Biol. Chem., 2007, 282, 9777–9788 CrossRef CAS PubMed.
- Q. Wang, S. Liu, A. Zhai, B. Zhang and G. Tian, AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation, Biol. Pharm. Bull., 2018, 41, 985–993 CrossRef CAS PubMed.
- D. G. Hardie, AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function, Genes Dev., 2011, 25, 1895–1908 CrossRef CAS PubMed.
- M. Daval, F. Foufelle and P. Ferré, Functions of AMP-activated protein kinase in adipose tissue, J. Physiol., 2006, 574, 55–62 CrossRef CAS PubMed.
- D. M. Sabatini, Twenty-five years of mTOR: Uncovering the link from nutrients to growth, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11818–11825 CrossRef CAS PubMed.
- A. Caron, D. Richard and M. Laplante, The Roles of mTOR Complexes in Lipid Metabolism, Annu. Rev. Nutr., 2015, 35, 321–348 CrossRef CAS PubMed.
- M. A. Konerman, J. C. Jones and S. A. Harrison, Pharmacotherapy for NASH: Current and emerging, J. Hepatol., 2018, 68, 362–375 CrossRef CAS PubMed.
- G. Li, J. B. Chen, C. Wang, Z. Xu, H. Nie, X. Y. Qin, X. M. Chen and Q. Gong, Curcumin protects against acetaminophen-induced apoptosis in hepatic injury, World J. Gastroenterol., 2013, 19, 7440–7446 CrossRef PubMed.
- H. Jaeschke, M. R. McGill, C. D. Williams and A. Ramachandran, Current issues with acetaminophen hepatotoxicity–a clinically relevant model to test the efficacy of natural products, Life Sci., 2011, 88, 737–745 CrossRef CAS PubMed.
- C. Yan, Y. Zhang, X. Zhang, J. Aa, G. Wang and Y. Xie, Curcumin regulates endogenous and exogenous metabolism via Nrf2-FXR-LXR pathway in NAFLD mice, Biomed. Pharmacother., 2018, 105, 274–281 CrossRef CAS PubMed.
- A. Asai and T. Miyazawa, Dietary curcuminoids prevent high-fat diet-induced lipid accumulation in rat liver and epididymal adipose tissue, J. Nutr., 2001, 131, 2932–2935 CrossRef CAS PubMed.
- E. M. Jang, M. S. Choi, U. J. Jung, M. J. Kim, H. J. Kim, S. M. Jeon, S. K. Shin, C. N. Seong and M. K. Lee, Beneficial effects of curcumin on hyperlipidemia and insulin resistance in high-fat-fed hamsters, Metabolism, 2008, 57, 1576–1583 CrossRef CAS PubMed.
- H. G. Kim, S. B. Lee, J. S. Lee, W. Y. Kim, S. H. Choi and C. G. Son, Artemisia iwayomogi plus Curcuma longa Synergistically Ameliorates Nonalcoholic Steatohepatitis in HepG2 Cells, J. Evidence-Based Complementary Altern. Med., 2017, 2017, 4390636 Search PubMed.
- J. Lin, Y. Tang, Q. Kang, Y. Feng and A. Chen, Curcumin inhibits gene expression of receptor for advanced glycation end-products (RAGE) in hepatic stellate cells in vitro by elevating PPARγ activity and attenuating oxidative stress, Br. J. Pharmacol., 2012, 166, 2212–2227 CrossRef CAS PubMed.
- M. Y. Um, K. H. Hwang, J. Ahn and T. Y. Ha, Curcumin attenuates diet-induced hepatic steatosis by activating AMP-activated protein kinase, Basic Clin. Pharmacol. Toxicol., 2013, 113, 152–157 CrossRef CAS PubMed.
- J. D. Lee, M. A. Choi, S. W. Ro, W. I. Yang, A. E. Cho, H. L. Ju, S. Baek, S. I. Chung, W. J. Kang, M. Yun and J. H. Park, Synergic chemoprevention with dietary carbohydrate restriction and supplementation of AMPK-activating phytochemicals: the role of SIRT1, Eur. J. Cancer Prev., 2016, 25, 54–64 CrossRef CAS PubMed.
- X. Lin, D. Bai, Z. Wei, Y. Zhang, Y. Huang, H. Deng and X. Huang, Curcumin attenuates oxidative stress in RAW264.7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2-Keap1 pathway, PLoS One, 2019, 14, e0216711 CrossRef CAS PubMed.
- A. Siviero, E. Gallo, V. Maggini, L. Gori, A. Mugelli, F. Firenzuoli and A. Vannacci, Curcumin, a golden spice with a low bioavailability, J. Herb. Med., 2015, 5, 57–70 CrossRef.
- S. Prasad, S. C. Gupta, A. K. Tyagi and B. B. Aggarwal, Curcumin, a component of golden spice: from bedside to bench and back, Biotechnol. Adv., 2014, 32, 1053–1064 CrossRef CAS PubMed.
- V. O. Gutierres, M. L. Campos, C. A. Arcaro, R. P. Assis, H. M. Baldan-Cimatti, R. G. Peccinini, S. Paula-Gomes, I. C. Kettelhut, A. M. Baviera and I. L. Brunetti, Curcumin Pharmacokinetic and Pharmacodynamic Evidences in Streptozotocin-Diabetic Rats Support the Antidiabetic Activity to Be via Metabolite(s), J. Evidence-Based Complementary Altern. Med., 2015, 2015, 678218 Search PubMed.
- A. L. Cheng, C. H. Hsu, J. K. Lin, M. M. Hsu, Y. F. Ho, T. S. Shen, J. Y. Ko, J. T. Lin, B. R. Lin, W. Ming-Shiang, H. S. Yu, S. H. Jee, G. S. Chen, T. M. Chen, C. A. Chen, M. K. Lai, Y. S. Pu, M. H. Pan, Y. J. Wang, C. C. Tsai and C. Y. Hsieh, Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions, Anticancer Res., 2001, 21, 2895–2900 CAS.
- S. K. Vareed, M. Kakarala, M. T. Ruffin, J. A. Crowell, D. P. Normolle, Z. Djuric and D. E. Brenner, Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects, Cancer Epidemiol. Biomarkers Prev., 2008, 17, 1411–1417 CrossRef CAS PubMed.
- R. A. Sharma, S. A. Euden, S. L. Platton, D. N. Cooke, A. Shafayat, H. R. Hewitt, T. H. Marczylo, B. Morgan, D. Hemingway, S. M. Plummer, M. Pirmohamed, A. J. Gescher and W. P. Steward, Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance, Clin. Cancer Res., 2004, 10, 6847–6854 CrossRef CAS PubMed.
- C. Ireson, S. Orr, D. J. Jones, R. Verschoyle, C. K. Lim, J. L. Luo, L. Howells, S. Plummer, R. Jukes, M. Williams, W. P. Steward and A. Gescher, Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production, Cancer Res., 2001, 61, 1058–1064 CAS.
- S. K. Sandur, M. K. Pandey, B. Sung, K. S. Ahn, A. Murakami, G. Sethi, P. Limtrakul, V. Badmaev and B. B. Aggarwal, Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism, Carcinogenesis, 2007, 28, 1765–1773 CrossRef CAS PubMed.
- J. Hong, M. Bose, J. Ju, J. H. Ryu, X. Chen, S. Sang, M. J. Lee and C. S. Yang, Modulation of arachidonic acid metabolism by curcumin and related beta-diketone derivatives: effects on cytosolic phospholipase A(2), cyclooxygenases and 5-lipoxygenase, Carcinogenesis, 2004, 25, 1671–1679 CrossRef CAS PubMed.
- T. Feng, Y. Wei, R. J. Lee and L. Zhao, Liposomal curcumin and its application in cancer, Int. J. Nanomed., 2017, 12, 6027–6044 CrossRef CAS PubMed.
- B. J. Douglass and D. L. Clouatre, Beyond Yellow Curry: Assessing Commercial Curcumin Absorption Technologies, J. Am. Coll. Nutr., 2015, 34, 347–358 CrossRef CAS PubMed.
- R. Jäger, R. P. Lowery, A. V. Calvanese, J. M. Joy, M. Purpura and J. M. Wilson, Comparative absorption of curcumin formulations, Nutr. J., 2014, 13, 11 CrossRef PubMed.
- C. Schiborr, A. Kocher, D. Behnam, J. Jandasek, S. Toelstede and J. Frank, The oral bioavailability of curcumin from micronized powder and liquid micelles is significantly increased in healthy humans and differs between sexes, Mol. Nutr. Food Res., 2014, 58, 516–527 CrossRef CAS PubMed.
- R. Jamwal, Bioavailable curcumin formulations: A review of pharmacokinetic studies in healthy volunteers, J. Integr. Med., 2018, 16, 367–374 CrossRef PubMed.
- Y. Liu, F. Cheng, Y. Luo, Z. Zhan, P. Hu, H. Ren, H. Tang and M. Peng, PEGylated Curcumin Derivative Attenuates Hepatic Steatosis via CREB/PPAR-γ/CD36 Pathway, BioMed Res. Int., 2017, 2017, 8234507, DOI:10.1155/2017/8234507.
| This journal is © The Royal Society of Chemistry 2022 |