DOI:
10.1039/D2DT02681C
(Paper)
Dalton Trans., 2022,
51, 18136-18142
Palladium(II) ortho-cyano-aminothiophenolate (ocap) complexes†
Received
16th August 2022
, Accepted 10th November 2022
First published on 11th November 2022
Abstract
A series of Pd(II) complexes containing ortho-cyano-aminothiophenolate (ocap) ligands have been prepared and their molecular structures elucidated. Hg(II) ocap complexes, [Hg{SC6H3XN(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)}]n (X = H, Me) (1), react with Na2S to afford HgS and Na2[ocap] which reacts in situ with K2[PdCl4] to afford palladium ocap complexes [Pd{SC6H3XN(CN)}]n (2). A second route to these coordination polymers has also been developed from reactions of 2-aminobenzothiazole (abt) complexes, trans-PdCl2(abt)2 (3), with NaOH. We have not been able to crystallographically characterise coordination polymers 2, but addition of PPh3, a range of phosphines and cyclic diamines affords mono and binuclear complexes in which the ocap ligand adopts different coordination geometries. With PPh3, binuclear [Pd(μ-κ2,κ1-ocap)(PPh3)]2 (4) results, in which the ocap bridges the Pd2 centre acting as an S,N-chelate to one metal centre and binding the second via coordination of the cyanide nitrogen. In contrast, with diphosphines, Ph2P(CH2)nPPh2 (n = 1–4), mononuclear species predominate as shown in the molecular structures of Pd(κ2-ocap){κ2-Ph2P(CH2)nPPh2} (5–7; n = 1–3). With 2,2′-bipy and 1,10-phen we propose that related monomeric chelates Pd(κ2-ocap)(κ2-bipy) (9) and Pd(κ2-ocap)(κ2-phen) (10) result but we have been unable to substantiate this crystallographically. Addition of HgCl2(phen) to 9a (generated in situ) affords heterobimetallic Pd(κ2-phen)(μ-κ2,κ1-ocap)HgCl2(κ2-phen) (11), in which Hg(II) is coordinated through the ring sulfur.
N)}]n (X = H, Me) (1), react with Na2S to afford HgS and Na2[ocap] which reacts in situ with K2[PdCl4] to afford palladium ocap complexes [Pd{SC6H3XN(CN)}]n (2). A second route to these coordination polymers has also been developed from reactions of 2-aminobenzothiazole (abt) complexes, trans-PdCl2(abt)2 (3), with NaOH. We have not been able to crystallographically characterise coordination polymers 2, but addition of PPh3, a range of phosphines and cyclic diamines affords mono and binuclear complexes in which the ocap ligand adopts different coordination geometries. With PPh3, binuclear [Pd(μ-κ2,κ1-ocap)(PPh3)]2 (4) results, in which the ocap bridges the Pd2 centre acting as an S,N-chelate to one metal centre and binding the second via coordination of the cyanide nitrogen. In contrast, with diphosphines, Ph2P(CH2)nPPh2 (n = 1–4), mononuclear species predominate as shown in the molecular structures of Pd(κ2-ocap){κ2-Ph2P(CH2)nPPh2} (5–7; n = 1–3). With 2,2′-bipy and 1,10-phen we propose that related monomeric chelates Pd(κ2-ocap)(κ2-bipy) (9) and Pd(κ2-ocap)(κ2-phen) (10) result but we have been unable to substantiate this crystallographically. Addition of HgCl2(phen) to 9a (generated in situ) affords heterobimetallic Pd(κ2-phen)(μ-κ2,κ1-ocap)HgCl2(κ2-phen) (11), in which Hg(II) is coordinated through the ring sulfur.
1. Introduction
Amides are widely used ligands in coordination chemistry, being able to stabilise both high and low-valent metal centres by varying the degree of metal–ligand interaction by virtue of being able to participate in metal–ligand π-bonding.1–3 In general they are relatively easily prepared via the deprotonation of secondary amines and in this way a large range of metal–amide complexes have been developed especially at high-valent metal centres via metathesis reactions. However, closer inspection reveals that these are almost exclusively those with alkyl or trimethylsilyl-substituents and examples with electron-withdrawing groups are, in comparison, rare.4 The cyanide group has a unique set of chemical and physical properties. Thus, along with being electron-withdrawing, its linear nature makes it sterically non-demanding, it has the ability to take part in π-bonding in either a donor or acceptor capacity and is also able to coordinate to secondary metal centres. As far as we are aware, cyanide-substituted amines and amides remain virtually unexplored.5
In contrast, ortho-aminothiophenolate complexes (SC6H4NH) are relatively common,6–12 being accessible from the double deprotonation of 2-aminothiophenol, N-substituted variants remain virtually unknown. In extensive studies, Wieghardt has shown that the ortho-aminothiophenolate ligand is redox-active being able to stabilise coordinated metals in a range of oxidation and spin states,6 while related ortho-phenylenediamido (RNC6H4NR) ligands are also capable of redox behaviour.13–15 A key component of this redox activity is the ability of the ligand to delocalise charge, being able to do this even with the non-participating proton on nitrogen. Thus, the ability to delocalise developing negative charge onto a cyanide substituent suggests that these species will show a rich redox chemistry, potentially providing low energy pathways for oxidation state changes during catalysis.
We recently reported the high yield synthesis of a series of Hg(II) ortho-cyano-aminothiophenolate (ocap) complexes, [Hg{SC6H3XN(CN)}]n (1a–e) (Scheme 1) resulting from the simple addition of 2-aminobenzothiazole and substituted derivatives to mercuric acetate in warm EtOH.16,17 These contain the previously unreported ocap ligand and result from loss of hydrogen and sulfur–carbon bond scission. While they have limited solubility in common organic solvents, addition of phosphines affords a series of soluble derivatives in which the ocap ligand was shown to be highly versatile, three different binding modes (A–C) being shown crystallographically,16,17 while a fourth (D) was proposed for 1a–e (Chart 1).
 |
| | Scheme 1 Synthesis of [Hg(ocap)]n (1a–e) upon dehydrogenative ring-opening of 2-aminothiazoles upon addition of Hg(OAc)2. | |
 |
| | Chart 1 Binding modes (A–D) of the ortho-cyano-aminothiophenolate (ocap) ligand. | |
In seeking to develop the chemistry of highly delocalised redox-active ocap ligands we sought a route to late transition metal derivatives, especially Pd(II), as we reasoned that such complexes were potential oxidation catalysts. Herein we provide details of the successful application of this strategy which allows access to Pd(II)-ocap complexes, which in analogy with the mercury complexes can adopt different binding modes.
2. Results and discussion
2.1 Synthesis of coordination polymers [Pd{SC6H3XN(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N)}]n (2)
N)}]n (2)
Stirring [Hg{SC6H3XN(CN)}]n (1a–b)16,17 with a slight excess of Na2S in a water–EtOH mixture for ca. 1 h resulted in formation of a black precipitate, assumed to be HgS. This was removed by filtration and to the resulting yellow solution, which we assume to contain Na2[ocap] but have made no attempt to confirm, we added K2[PdCl4] in EtOH and refluxed this mixture for ca. 3 h. This resulted in formation of a red-brown solid which was isolated by filtration to give [Pd{SC6H3XN(CN)}]n (2a–b) (Scheme 2). We have also developed a second route to 2a–b which avoids the use of mercury. Thus, reaction of K2[PdCl4] with 2-aminothiazoles affords simple substitution complexes, trans-PdCl2(2-abt)2 (3a–b), 3a being previously prepared and crystallographically characterised.16 Refluxing basic solutions of 3a–b resulted in the slow formation of 2a–b. Thus, after heating 3a for 4 h, 2a was isolated in ca. 71% yield, and while slightly lower than the mercury route (95%) it does negate the use of toxic mercury salts.
 |
| | Scheme 2 Synthetic routes to [Pd(ocap)]n (2a–b). | |
Complexes 2a–b have poor solubility in common organic solvents suggesting they are coordination polymers (or oligomers) in a similar fashion to 1a–e.16 Unfortunately, we have been unable to obtain suitable crystalline forms of either 1a–e or 2a–b to unequivocally confirm this. Complexes 2a–b are sparingly soluble in dmso, which we associate with the partial breakdown of the polymeric structure to afford dmso adducts [Pd{SC6H3XN(CN)}(dmso)x], and this allowed us to record 1H and 13C{1H} NMR data for these adducts.
We have previously investigated the formation of 1a from reaction of 2-aminobenzothiazole (abt) and Hg(OAc)2,18 a transformation which occurs without added base. Thus, upon coordination, neutral enamine and zwitterionic enamide forms (Chart 2a) are accessible and at the Lewis acidic Hg(II) centre (Chart 2b) there is an obvious shortening of the C–NH2 bond suggesting a significant degree of zwitterionic enamide form. In contrast in 3a (Chart 2c) the two crystallographically inequivalent abt ligands have longer C–NH2 bonds suggestive of the enamine form. Nevertheless, these protons must still be acidic such that upon addition of base deprotonation results in activation leading to eventual C–N bond scission and ring-opening, the mechanism of which has been probed at Hg(II).17
 |
| | Chart 2 (a) Enamine and zwitterionic resonance hybrids of coordinated abt, (b) important bond lengths in Hg(sac)2(abt)L (L = MeOH, dmso (brackets)),20 (c) important bond lengths in trans-PdCl2(2-abt)2 (3a) which has two crystallographically inequivalent abt ligands.19 | |
2.2 Reaction with PPh3: synthesis of [Pd(μ-κ2,κ1-ocap)(κ1-PPh3)]2 (4)
Given the relative insolubility of coordination polymers 2a–b and our previous success in breaking down the analogous Hg(II) complexes 1a–b upon addition of phosphines,16,17 we attempted to prepare mononuclear square planar complexes of the type Pd(κ2-ocap)(PPh3)2via addition of 2 equivalents of PPh3 to 2a–b in refluxing CHCl3. However, in no instance did we generate such mononuclear complexes, but rather dimeric [Pd(μ-κ2,κ1-ocap)(PPh3)]2 (4a–b) resulted, being isolated as red-brown crystalline solids in good yields (Scheme 3).
 |
| | Scheme 3 Reactions of 2 with PPh3. | |
Formation of a dimeric product was confirmed through the X-ray structure of 4a details of which are given in Fig. 1 and Table 1. The two crystallographically inequivalent Pd(II) centres are square-planar, each being bound by a single PPh3 ligand and bridged by two ocap ligands. Each of the latter act as an N,S-chelate to one Pd centre coordinating with bite angles of 84.75(10) and 85.27(10)° at Pd(1) and Pd(2) respectively. The final coordination site is taken up by the nitrogen of the cyanide group, which lies trans to sulfur. Thus each ocap ligand acts in a tridentate μ-κ2,κ1 manner, donating a total of 6 electrons, four to one, and two to the second, Pd(II) centre. The central 8-membered Pd2N4C2 ring is not flat but rather is bowl-like, with the phenyl rings of the ortho-aminothiophenolate moieties lying on the same side and extending the bowl-like structure. Spectroscopic data are in accord with the solid-state structure. Each shows a singlet resonance in the 31P{1H} NMR spectrum and the cyanide is seen as a strong peak between 2162–2169 cm−1 in the IR spectrum. As mentioned above, heating 4a–b with excess PPh3 did not result in scission of the binuclear structure. Thus, we suggest that in coordination polymers 2a–b, the substructure seen in 4a likely is adopted the polymeric network being completed by further coordination of sulfur, which is lost upon addition of PPh3.
 |
| | Fig. 1 Molecular structure of 4a with thermal ellipsoids at the 50% level and hydrogen atoms omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for palladium ocap complexes
| |
4a
|
5a
|
6a
|
7b
|
11
|
| Pd–S |
2.2221(14) & 2.2233(14) |
2.2818(8) |
2.2949(7) |
2.3141(18) |
2.2334(13) |
| Pd–N |
2.047(4) & 2.058(4) |
2.041(2) |
2.0655(19) |
2.084(6) |
2.018(4) |
| Pd–NC |
2.039(4) & 2.047(4) |
|
|
|
|
| Pd–P (trans S) |
|
2.2744(8) |
2.2969(6) |
2.2976(19) |
|
| Pd–P (trans N) |
2.2118(14) & 2.2157(14) |
2.2217(9) |
2.2358(6) |
2.255(2) |
|
| Pd–N (bipy) |
|
|
|
|
2.082(4) & 2.017(4) |
| N–C |
1.275(6) & 1.282(6) |
1.299(4) |
1.288(3) |
1.341(10) |
1.304(6) |
| CN |
1.160(6) & 1.165(6) |
1.147(4) |
1.185(3) |
1.152(10) |
1.157(6) |
| S–Pd–N |
84.71(11) & 85.25(11) |
84.79(8) |
84.96(6) |
84.87(15) |
84.31(12) |
| P–Pd–P |
|
72.96(3) |
85.36(2) |
91.20(7) |
|
| N–Pd–N |
91.19(15) & 91.19(15) |
|
|
|
80.83(17) |
2.3 Reactions with diphosphines, Ph2P(CH2)nPPh2 (n = 1–4): synthesis of Pd(κ2-ocap){κ2-Ph2P(CH2)nPPh2} (5–8)
Given the dimeric structure of 4a–b we reasoned that a chelating diphosphine may be capable of displacing the metal-bound cyanide group with concomitant formation of a mononuclear chelate. We first considered the small bite angle diphosphine, 1,1′-bis(diphenylphosphino)methane (dppm), which while often found to bridge two metal centres, is also a good chelate ligand.20 Indeed, heating a solution of dppm and a red suspension 1a–b in CHCl3 for 2 h led to the slow formation of a clear red solution, which after cooling to room temperature and partial evaporation of solvent gave 5a–b as orange and brown crystalline solids respectively in good yields (Scheme 4).
 |
| | Scheme 4 Synthesis of Pd(κ2-ocap)(κ2-dppm) (5). | |
The structure of 5a was elucidated using X-ray crystallography, the details of which are given in Fig. 2 and Table 1. The complex contains a single square planar Pd(II) centre which is coordinated to a dppm-chelate and a κ2-ocap ligand, forming bite angles of 72.98(3) and 84.79(7)° respectively. The latter is in accord with those found in 4a showing that de-coordination of the cyanide does not affect ligand binding. Complexes 5a–b could also be prepared in similar yields upon heating [Pd(κ2-dppm)2]Cl2 and the yellow filtrate, believed to be Na2[ocap] formed upon addition of Na2S to 1a–b. Solution NMR data support the solid-state structure two phosphorus centres are inequivalent, and this is confirmed in solution as seen by a pair of doublets (JPP = 99 Hz) at −45.5 and −28.6 (d, J 99 Hz) in the 31P{1H} NMR spectrum. The two Pd–P bond lengths are very similar, that lying trans to sulfur being slightly longer [by ca. 0.05 Å].
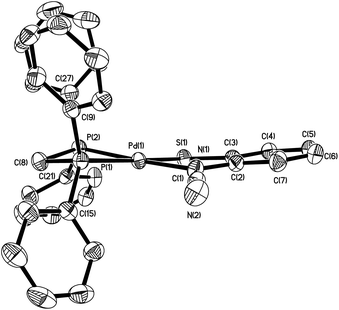 |
| | Fig. 2 Molecular structure of 5a with thermal ellipsoids at the 50% level and hydrogen atoms omitted for clarity. | |
Following on from the unexpected isolation of dppm-chelate complexes 5, we next explored reactions of 1 with the more flexible diphosphines, Ph2P(CH2)nPPh2 (n = 2–4) anticipating that related chelate complexes would result. This is indeed the case with red-brown dppe, dppp and dppb complexes 6–8 respectively being isolated in good yields (ca. 60–90%) upon addition of the diphosphines to 2 in EtOH (Scheme 5). The dppe complexes, Pd(κ2-ocap)(κ2-dppe) (6a–b), are also accessible in comparable yields following reaction of Na2[ocap] with [Pd(dppe)2]Cl2. Spectroscopic data are in full accord with the proposed structures, the chelating nature of the diphosphine being confirmed by the observation of a pair of doublets in the 31P{1H} NMR spectra with JPP couplings of ca. 29 Hz (dppe) and 53 Hz (dppp), which do not vary significantly with substituent X. More unexpected were the JPP values of 44.5 and 29.0 Hz for the dppb complexes 8a and 8b respectively. It is not clear why these vary to such a large extent but suggests that the precise geometry, especially the P–Pd–P bond angle is flexible for this large diphosphine.
 |
| | Scheme 5 Synthesis of Pd(κ2-ocap){κ2-Ph2(CH2)nPPh2} (6–8). | |
Molecular structures of 6a and 7b (Fig. 3) were confirmed by crystallographic studies. While bond lengths and angles are generally very similar to those in 5a (Table 1), the bite angle of the diphosphine varies. Thus, in 6a and 7b the P–Pd–P bond angles of 85.36(2)° and 91.20(7)° respectively are ca. 12–19° larger than that in 5a, highlighting the flexibility of the Pd(κ2-ocap) moiety to support a range of diphosphine ligands. While we have been unable to crystallographically characterise 8a–b, the phosphorus–phosphorus coupling constants are indicative of chelate formation.
 |
| | Fig. 3 Molecular structures of (a) 6a and (b) 7b with hydrogen atoms omitted for clarity. | |
2.4 Reactions with 2,2′-bipy and 1,10-phen: synthesis of Pd(κ2-ocap)(κ2-bipy) (9) and Pd(κ2-ocap)(κ2-phen) (10)
Reactions of 2 with cyclic diamines, 2,2′-bipyridine (bipy) and 1,10-phenanthroline (phen) also lead to formation of mononuclear chelate complexes, namely Pd(κ2-ocap)(κ2-bipy) (9a–b) and Pd(κ2-ocap)(κ2-phen) (10a–b) upon heating in CHCl3 (Scheme 6). Products were isolated as solids in good yields following simple filtration and washing, and NMR and analytical data are in full accord with the proposed formulations. Unfortunately, we have been unable to grow X-ray quality crystals of any of these complexes to confirm their mononuclear nature.
 |
| | Scheme 6 Synthesis of Pd(κ2-ocap)(κ2-bipy) (9) and Pd(κ2-ocap)(κ2-phen) (10). | |
2.5 Synthesis of bimetallic Pd(κ2-phen)(μ-κ2,κ1-ocap)HgCl2(κ2-phen) (11)
Following our inability to fully characterise 9–10 in the solid state, we added one equivalent HgCl2(κ2-phen)21 to 9a prepared in situ. This gave a red-brown solid and a red filtrate, slow evaporation of which led to the isolation of well-formed crystals suitable for X-ray diffraction, the results of which are shown in Fig. 4 and Table 1. The complex is heterobimetallic Pd(κ2-phen)(μ-κ2,κ1-ocap)HgCl2(κ2-phen) (11), which co-crystallises with three molecules of CHCl3. The Pd(κ2-phen)(μ-κ2,κ1-ocap) sub-unit is as expected and contains a square planar Pd(II) centre ligated by phenanthroline and ocap chelates with bite angles of 80.83(17) and 84.31(12)° respectively. The sulfur of the ocap ligand is also bound to the Hg(II) centre [Hg(1)–S(1) 2.6113(15) Å] which adopts a distorted square-based pyramidal coordination geometry. Thus, the two chlorides remain cis [Cl(1)–Hg(1)–Cl(2) 100.63(6)°] and subtend angles of 99.64(5) and 113.03(5)° with the S(1)–Hg(1) vector, while mercury-bound phenanthroline is also distorted out of the plane [S(1)–Hg(1)–N(5) 94.33(5), S(1)–Hg(1)–N(6) 106.38(5)°] and the bridging sulfur brings the two metal ions into relatively close contact [Hg(1)⋯Pd(1) 3.482(2) Å]. As might be anticipated, the two phenanthrolines are approximately parallel to one another and there is also a stacking of individual molecules with the chloroform molecules weakly binding to either side of the stack.
 |
| | Fig. 4 Molecular structure of 11 with thermal ellipsoids at the 50% level and hydrogen atoms omitted for clarity. | |
We have been unable to collect NMR data for 11 due to its insolubility in all common solvents and this suggests that the bulk material is also comprised of 11 since 10a is soluble in dmso. This (in part) supports our view that coordination polymers 1–2 are held together by M–S–M interactions in the solid-state. It is also noteworthy that while in 11 the cyanide remains uncoordinated, in PPh3 derivatives 4a–b, sulfur is bound to a single metal centre, while the cyanide is metal-bound. Possibly the relatively soft Hg(II) centre favours sulfur coordination, although the π-stacking of the phenanthrolines may also lead to this form being favoured.
3. Summary and conclusions
We have reported here the synthesis of the first examples of Pd(II) ortho-cyano-aminothiophenolate (ocap) complexes. In many respects the chemistry is similar to that found at Hg(II) centres.16,17 Thus, both [Hg{SC6H3XN(CN)}]n (1) and [Pd{SC6H3XN(CN)}]n (2) are coordination polymers, showing low solubility in common organic solvents. They are soluble in dmso, but likely this leads to a breakdown of the polymeric structure to afford M(ocap)(dmso)n. The polymeric structure is broken down upon addition of PPh3. For palladium this leads to formation of dimeric complexes in which the Pd2 centre is held together by secondary coordination of the cyanide. Thus, the ocap ligand acts as binds via mode B (Chart 1) and we have found that even addition of excess PPh3 does not lead to scission of this bond. With the small bite-angle diphosphine, dppm, cyanide binding is released, and simple mononuclear square planar complexes are formed, as is the case for the more flexible diphosphines, while similar behaviour is also believed to occur in 2,2′-bipyridine and 1,10-phenanthroline adducts. Such small Pd(II) complexes may have potential as oxidation catalysts and we are actively investigating this and also their electrochemical properties to understand the nature of the electronic delocalisation. While in the phosphine adducts, sulfur is bound only to a single metal centre, addition of HgCl2(κ2-phen) to 10a affords an unexpected bimetallic complex in which the two metal centres are bridged by sulfur. Thus, in the crystallographically characterised complexes reported herein, the ocap ligand displays three coordination modes, something previously noted at the Hg(II) centre.16,17 These results show that this ligand type is coordinatively flexible and given its expected electronic delocalisation, a rich chemistry is anticipated. Aspects of the redox chemistry of palladium and other ocap complexes is currently under study in our laboratories and will be reported in due course.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We thank the University of Tikrit and King's College London for partial support of this work. S. B.-M. thanks Erasmus Mundus for a Postdoctoral Fellowship, S. G. thanks the Commonwealth Scholarship Commission for the award of a Commonwealth Scholarship and G. R. F. O. thanks King's College London for PhD funding.
References
- D. C. Bradley and M. H. Chisholm, Acc. Chem. Res., 1976, 9, 273–280 CrossRef CAS.
- M. D. Fryzuk and C. D. Montgomery, Coord. Chem. Rev., 1989, 95, 1–40 CrossRef CAS.
- O. Clement, B. M. Rapko and B. P. Hay, Coord. Chem. Rev., 1998, 170, 203–243 CrossRef CAS.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232–8245 CrossRef CAS.
-
(a) R. J. Crutchly and M. L. Naklicki, Inorg. Chem., 1989, 28, 1955–1958 CrossRef;
(b) R. J. Crutchly, Coord. Chem. Rev., 2001, 219–221, 125–155 CrossRef;
(c) T. Ramana, P. Saha, M. Das and T. Punniyamurthy, Org. Lett., 2010, 12, 84–87 CrossRef CAS PubMed.
-
(a) S. R. Presow, M. Ghosh, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chim. Acta, 2011, 374, 226–239 CrossRef CAS;
(b) P. Ghosh, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 3967–3969 CrossRef CAS PubMed;
(c) P. Ghosh, A. Begum, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2003, 42, 3208–3215 CrossRef CAS PubMed;
(d) S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2010, 254, 1358–1382 CrossRef CAS.
- J. Vincente, M. T. Chicote, M. D. Bermudez, P. G. Jones, C. Fittschen and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1986, 2361–2366 RSC.
-
(a) D. Sellman, S. Emig, F. W. Heinemann and F. Knoch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1201–1203 CrossRef;
(b) D. Sellman, S. Emig and F. W. Heinemann, Angew. Chem., Int. Ed. Engl., 1997, 36, 1734–1736 CrossRef.
- K. Sorasaenee, J. R. Galán-Mascarós and K. R. Dunbar, Inorg. Chem., 2002, 41, 433–436 CrossRef CAS PubMed.
- J. C. Noveron, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1998, 37, 1138–1139 CrossRef CAS PubMed.
-
(a) W.-F. Liaw, N.-H. Lee, C.-M. Chen, G.-H. Lee and S.-M. Peng, J. Am. Chem. Soc., 2000, 122, 488–494 CrossRef CAS;
(b) W.-F. Liaw, C.-M. Lee, G.-H. Lee and S.-M. Peng, Inorg. Chem., 1998, 37, 6396–6398 CrossRef CAS;
(c) W.-F. Liaw, C.-K. Hsieh, G.-Y. Lin and G.-H. Lee, Inorg. Chem., 2001, 40, 4468–3475 CrossRef PubMed.
- A. Majumber and S. Sarkar, Inorg. Chim. Acta, 2009, 362, 3493–3501 CrossRef.
- M. M. Khusniyarov, K. Harms, O. Burghaus, J. Sundermeyer, B. Sarkar, W. Kaim, J. van Slageren, C. Duboc and J. Fiedler, Dalton Trans., 2008, 1355–1365 RSC.
- M. M. Khusniyarov, T. Weyhermueller, E. Bill and K. Wieghardt, J. Am. Chem. Soc., 2009, 131, 1208–1221 CrossRef CAS PubMed.
- R. Zhang, Y. Wang, Y. Zhao, C. Redshaw, I. L. Fedushkin, B. Wu and X.-J. Yang, Dalton Trans., 2021, 50, 13634–13650 RSC.
- S. A. Al-Jibori, A. A. Irzoqi, E. G. H. Al-Saraj, A. S. Al-Janabi, S. Basak-Modi, S. Ghosh, K. Merzweiler, C. Wagner, H. Schmidt and G. Hogarth, Dalton Trans., 2015, 44, 14217–14219 RSC.
- S. A. Al-Jibori, A. A. Irzoqi, A. S. M. Al-Janabi, A. I. A. Al-Nassiry, S. Basak-Modi, S. Ghosh, C. Wagner and G. Hogarth, Dalton Trans., 2022, 51, 7889–7898 RSC.
-
(a) E. J. Gao, L. Liu, M. C. Zhu, Y. Huang, F. Guan, X. N. Gao, M. Zhang, L. Wang, W. Z. Zhang and Y. G. Sun, Inorg. Chem., 2011, 50, 4732–4741 CrossRef CAS PubMed;
(b) S. A. Al-Jibori, A. T. Habeeb, G. H. H. Al-Jibori, N. A. Dayaaf, K. Merzweiler, C. Wagner, H. Schmidt and G. Hogarth, Polyhedron, 2014, 67, 338–343 CrossRef CAS.
- S. A. Al-Jibori, Z. S. Afandi, K. Merzweiler, C. Wagner, H. Schmidt, S. Basak-Modi and G. Hogarth, Polyhedron, 2014, 81, 442–449 CrossRef CAS.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- G. J. Sutton, Aust. J. Chem., 1959, 12, 637–642 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental methods and characterising data, along with details of the X-ray crystallographic structure determinations. CCDC 2114230, 2114233, 2114234, 2181464 and 2201775. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02681c |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ahmed A.
Irzoqi
b,
Ali I. A.
Abdullah
c,
Sucharita
Basak-Modi
d,
Georgia R. F.
Orton
e,
Shishir
Ghosh
de,
Christof
Wagner
f and
Graeme
Hogarth
a,
Ahmed A.
Irzoqi
b,
Ali I. A.
Abdullah
c,
Sucharita
Basak-Modi
d,
Georgia R. F.
Orton
e,
Shishir
Ghosh
de,
Christof
Wagner
f and
Graeme
Hogarth