
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
To chelate thallium(I) – synthesis and evaluation of Kryptofix-based chelators for 201Tl†
Angelo
Frei
 *ab,
Alex
Rigby
b,
Thomas T. C.
Yue
ab,
George
Firth
b,
Michelle T.
Ma
b and
Nicholas J.
Long
*a
*ab,
Alex
Rigby
b,
Thomas T. C.
Yue
ab,
George
Firth
b,
Michelle T.
Ma
b and
Nicholas J.
Long
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: a.frei@imperial.ac.uk; n.long@imperial.ac.uk
bSchool of Biomedical Engineering and Imaging Sciences, King's College London, 4th Floor Lambeth Wing, St Thomas’ Hospital, London, SE1 7EH, UK
First published on 23rd May 2022
Abstract
While best known for its toxic properties, thallium has also been explored for applications in nuclear diagnostics and medicine. Indeed, [201Tl]TlCl has been used extensively for nuclear imaging in the past before it was superceded by other radionuclides such as 99mTc. One reason for this loss of interest is the severe lack of suitable organic chelators able to effectively coordinate ionic forms of Tl and deliver it to specific diseased tissue by means of attached biological vectors. Herein, we describe the synthesis and characterisation of a series of Kryptofix 222-based chelators that can be radiolabelled with 201Tl(I) in high radiochemical yields at ambient temperature. We demonstrate that from these simple chelators, targeted derivatives are readily accessible and describe the synthesis and preliminary biological evaluation of a PSMA-targeted 201Tl-labelled Kryptofix 222-peptide conjugate. While the Kryptofix system is demonstrably capable of binding the thallium cation, no PSMA-mediated cell-uptake could be detected with the PSMA conjugate, suggesting that this targeting moiety may not be ideal for use in conjunction with 201Tl.
Introduction
Thallium is most commonly known for its detrimental properties such as its former use in rat poison or as a popular means to murder in both fiction and non-fiction.1 However in academic and medical settings, thallium, and in particular its radioactive isotopes have been recognised for their potential utility in healthcare. In 1975 Lebowitz et al. discussed the possible application of thallium-201 for myocardial imaging due to the similarities in size between Tl(I) and K(I).2 Thallium-201 (201Tl) has a half-life of 73 hours and emits γ-photons at 72 keV (X-ray, 94% abundance), 135 keV (3% abundance) and 167 keV (10% abundance) making it suitable for application in single photo emission computed tomography (SPECT). As predicted, 201Tl initially found widespread use as a myocardial imaging agent until it was replaced by emerging technetium-99 m tracers.3 In a few instances [201Tl]TlCl has also been employed for tumour detection and monitoring.4 Additionally, 201Tl decays through electron capture (EC) to 201Hg, emitting an average of 36.9 Meitner-Auger electrons per decay event. Meitner-Auger electron-emitting agents have shown significant potential for application in systemic, targeted radionuclide therapy of cancer.5 With its high decay yield of Meitner-Auger electrons, 201Tl has promise as a radiotherapeutic isotope and possibly for theranostic applications.6The clinical application of 201Tl is currently limited to its use as the “free” or “unchelated” metal ion. Many other radioactive metallic isotopes have demonstrated clinical utility but are largely used in a complexed or chelated form. For example, [68Ga]Ga3+ and [177Lu]Lu3+ can be complexed to chelators which are appended to peptides that target receptors expressed in cancer tissue. The ensuant 68Ga and 177Lu radiopharmaceuticals are used for PET imaging and systemic radiotherapy respectively, and have had clinical impact in management and treatment of neuroendrocrine cancer and prostate cancer.7 Advancements in the development of new 201Tl-based radiopharmaceuticals are currently prevented by an almost complete lack of studies into the formation of stable compounds between thallium and organic chelators that could feature biological targeting functions. Thus far, only two studies have investigated the coordination of thallium to popular radioimaging chelators. In 2011, Hijnen et al. described the evaluation of [201Tl]Tl(III)-DOTA (1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′ tetraacetic acid) complexes. The authors oxidized the readily available 201Tl(I) by treatment with hydrochloric acid and ozone and found good radiochemical yields and purity for the formation of DOTA and DTPA (diethylenetriaminepentaacetic acid) [201Tl]Tl(III)-labelled complexes. Unfortunately, the DTPA compound was found to rapidly decay in human blood serum. Although the DOTA complex was found to be stable in serum, the biodistribution of the compound mirrored that of non-chelated or “free” [201Tl]Tl(I), suggesting limited stability of [201Tl]Tl(III)-DOTA in vivo.8 Interestingly, Fodor et al. described the same complex as an “extraordinarily robust macrocyclic complex” in a 2015 report.9 However no in vivo experiments were reported, suggesting that while [natTl]Tl(III)-DOTA may possess favorable stability in vitro, the Tl(III) core is readily reduced in vivo, leading to rapid decomplexation.
We recently described a more robust protocol for the oxidation of commercially available [201Tl]Tl(I)Cl to [201Tl]Tl(III)Cl3. In this work the ability of the readily available chelators, DTPA, DOTA and EDTA, for binding [201Tl]Tl(III) was investigated. Radiolabelling was possible with all three chelators as evidenced by iTLC, however only [201Tl]Tl(III)-DOTA showed sufficient stability in human serum, ruling out the other two chelates for in vivo applications.10
While the ability to chelate [201Tl](III) with sufficient stability required for in vivo applications is promising (though yet to be fully realized), this approach will always be susceptible to in vivo reduction of [201Tl]Tl(III) to [201Tl]Tl(I), and likely subsequent metal dissociation, as has been indicated by the work of Hijnen. We therefore started to explore the possible chelation of “native” [201Tl]Tl(I). This presents some challenges as the monovalent thallium is a very large ion with an ionic radius of 150 pm and, like the alkali metals, its solution chemistry is dominated by aquation.11
We initially looked at two classes of compounds for which there was some evidence in the literature that suggested potential for radiolabelling [201Tl]Tl(I), crown ethers and cryptophanes. Crown-ethers are well-documented to bind a wide range of cations.12 However in our hands, we could not detect any binding of [201Tl]Tl(I) by 18-crown-6, dibenzo-18-crown-6 and dicyclohexyl-18-crown-6 under radiolabelling conditions. The group of Brotin has reported an impressive series of different cryptophane derivatives over the last years, including extensive characterization of their ability to bind different cations, including non-radioactive thallium.13 As the synthesis of these compounds represents a significant challenge, their group kindly provided us with two samples of cryptophanes with which to conduct preliminary studies. While we did observe the appearance of a new peak in the radio-HPLC upon reaction of Crypt1 with [201Tl]Tl(I)Cl, this could only be achieved under strongly basic conditions (Fig. 1). Adjusting the pH to ∼7 or exposure to human serum resulted in the release of the thallium cation, hence we did not pursue these chelators any further.
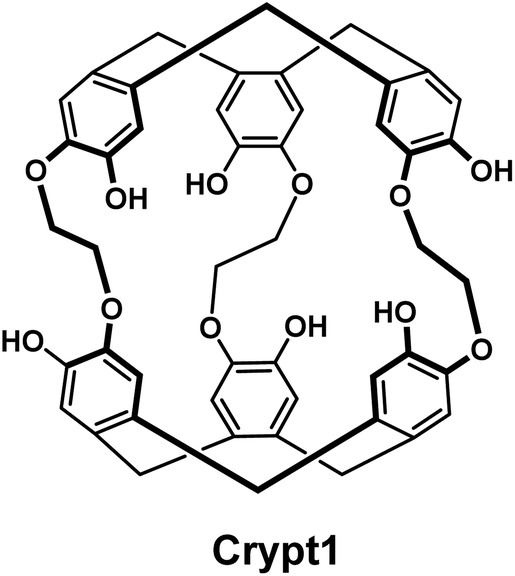 | ||
| Fig. 1 Chemical structure of the cryptophane Crypt1. | ||
Due to the similarities in ionic radii between K(I) and Tl(I) we turned our attention to 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane, more commonly known as Kryptofix 222. This compound was first reported by Sauvage et al. in 1969. In this first report the authors described the remarkable propensity of these compounds to form complexes with “metallic cations”.14 Since then, this compound class has found applications in many different fields ranging from crystallography to chemical sensing.15 Interestingly, its ability to encapsulate potassium ions led to its use as a phase-transfer catalyst for the radiosynthesis of [18F]-fluoro2-deoxy-2-D-glucose ([18F]FDG).16 Based on these findings we have developed a series of Kryptofix 222 derivatives that can be conjugated to a biological targeting function of choice. Herein we report the synthesis and characterization of these derivatives as well as the preparation of a prostate-cancer targeted analogue. Furthermore, the ability of the Kryptofix compound class to coordinate both non-radioactive and radioactive [201Tl]Tl(I) has been investigated.
Results and discussion
Before embarking on the synthesis of functionalized derivatives, the ability of commercially available Kryptofix 222 (K222) and Kryptofix 22 (K22, Fig. 2) to complex [natTl]Tl(I) was investigated by NMR spectroscopy. Addition of TlPF6 in D2O to a solution of the cryptand in D2O resulted in a clear shift in resonances for K222 but only a very small shift for K22 (ESI Fig. S1 and S2†). A proof-of-concept radiolabelling study was conducted with K222. Incubation of the cryptand with [201Tl]TlCl in saline led to almost quantitative formation of a new peak with a significantly higher retention time compared to free [201Tl]Tl(I) on reverse-phase radioHPLC (ESI Fig. S3†). Encouraged by these preliminary data we pursued the synthesis of bifunctional K222 chelators. We focused on K222-NH2 as this compound could be conjugated to carboxylic acids via amide-coupling, whilst also offering the possibility to convert it to other synthons for “click”-type coupling reactions i.e.K222-N3 and K222-DBCO (Fig. 2).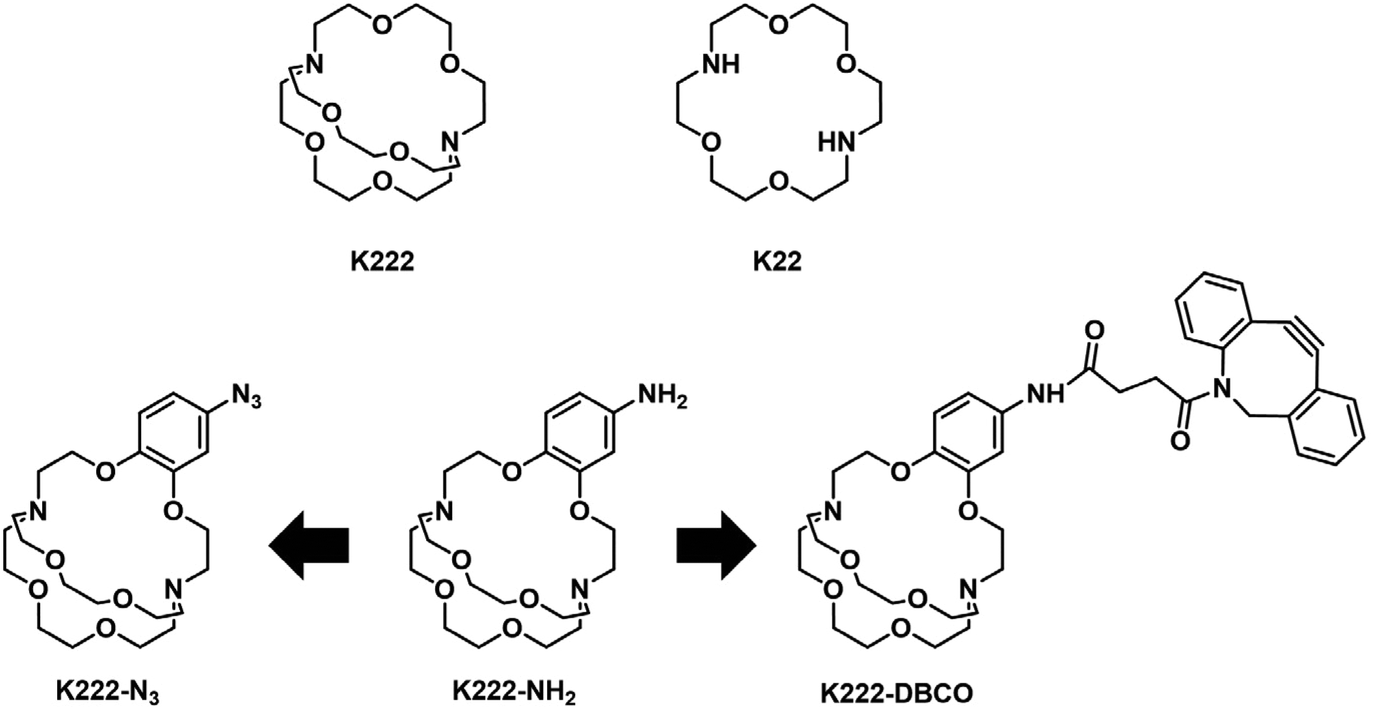 | ||
| Fig. 2 Chemical structures of Kryptofix 222-based compounds reported in this work. | ||
The synthetic strategy towards K222-NH2 was based on works by Gansow,17 Pettit,18 and Allen19 adjusted with our own optimisations (Scheme 1). Compound 1 was obtained by reacting 4-nitrocatechol with ethyl bromoacetate in acetone in the presence of sodium carbonate. The reaction could be shortened to 60 min if conducted in a microwave reactor at 180 °C. Hydrolysis to 2 with excess LiOH was straightforward and the chlorination to 3 with thionyl chloride could be achieved after optimization i.e. it was important to keep the reaction refluxing overnight and to directly use the crude product after evaporation of thionyl chloride. For the preparation of 4 both reagents (3 and Kryptofix 22) were dissolved in fresh anhydrous toluene in separate round-bottom flasks and sonicated until dissolved. Both were added via syringe to a third round-bottom flask filled with more anhydrous toluene cooled by means of an ice bath. The speed of addition was not important for this reaction as both slow addition over 1 h by means of a syringe pump and fast addition over 1 minute gave roughly equivalent yields. The reaction was complete after 1 h at room temperature and did not require overnight stirring as described in some literature reports.19 Compound 5 was obtained by refluxing 4 in BH3·THF overnight and was used without further purification. Reduction of the nitro group of 5 was achieved by using fine zinc dust instead of powder, giving K222-NH2 after purification on a C18 cartridge. FSO2N3 was prepared as described in the literature20 and reacted with K222-NH2 to form K222-N3 after purification by column chromatography on silica. NHS-ester activated dibenzocyclooctyne (DBCO–NHS) was reacted with K222-NH2, forming K222-DBCO as evidenced by LC-MS. This compound was then utilised further without purification.
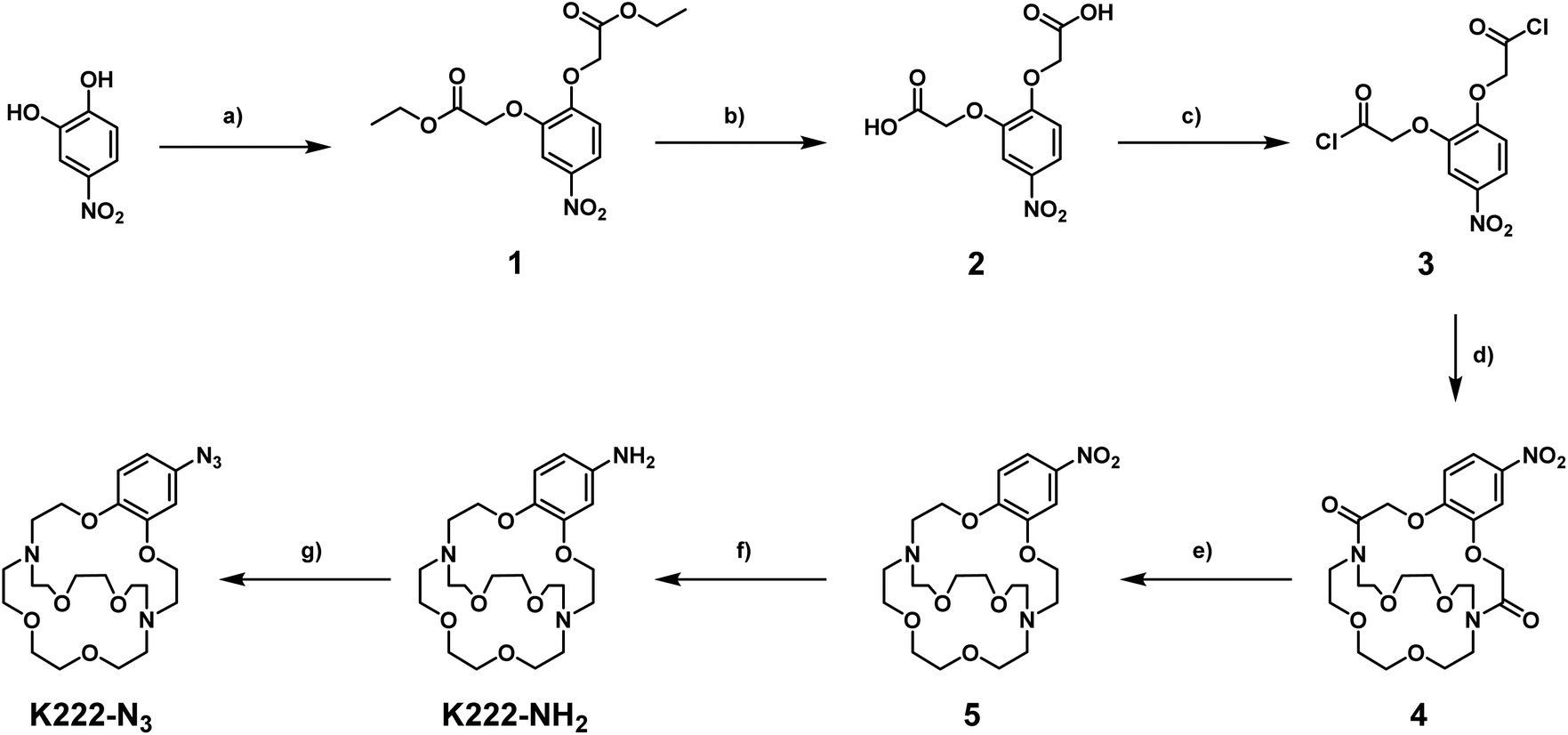 | ||
| Scheme 1 Synthetic route towards K222-NH2 and K222-N3. Reaction conditions: (a) ethyl bromoacetate, Na2CO3, acetone, reflux, 16 h, 45%; (b) LiOH, THF/H2O, r.t., 16 h, 70%; (c) thionyl chloride, reflux, 16 h; (d) Kryptofix 22, NEt3, dry toluene, 0–30 °C, 1 h, 25%; (e) BH3·THF, reflux, 16 h, 55%; (f) Zn dust, AcOH/DCM, r.t., overnight, 43%; (g) FSO2N3, 3 M KHCO3, MTBE/DMF/H2O, r.t. 5 min, 37%. | ||
As a model targeting function, we chose peptidomimetic 6 which has a high affinity for the prostate specific membrane antigen (PSMA), a receptor that is highly overexpressed in prostate cancer cells and for which many cell assays and murine models have been developed over the last decades.21
The PSMA-targeting precursor 6 was prepared as described elsewhere.22 Utilizing FSO2N3, the terminal amine on 6 was transformed into the azide 7 (Scheme 2). Deprotection with TFA cleaved off all tert-butyl protecting groups yielding 8 which could be reacted with K222-DBCO under copper-free click-conditions forming the final conjugate K222-PSMA which was purified by preparative HPLC. The quantity of K222-PSMA obtained was not sufficient for NMR assignment. The compound was characterized by 1H NMR, LC-MS and HR-MS (ESI†).
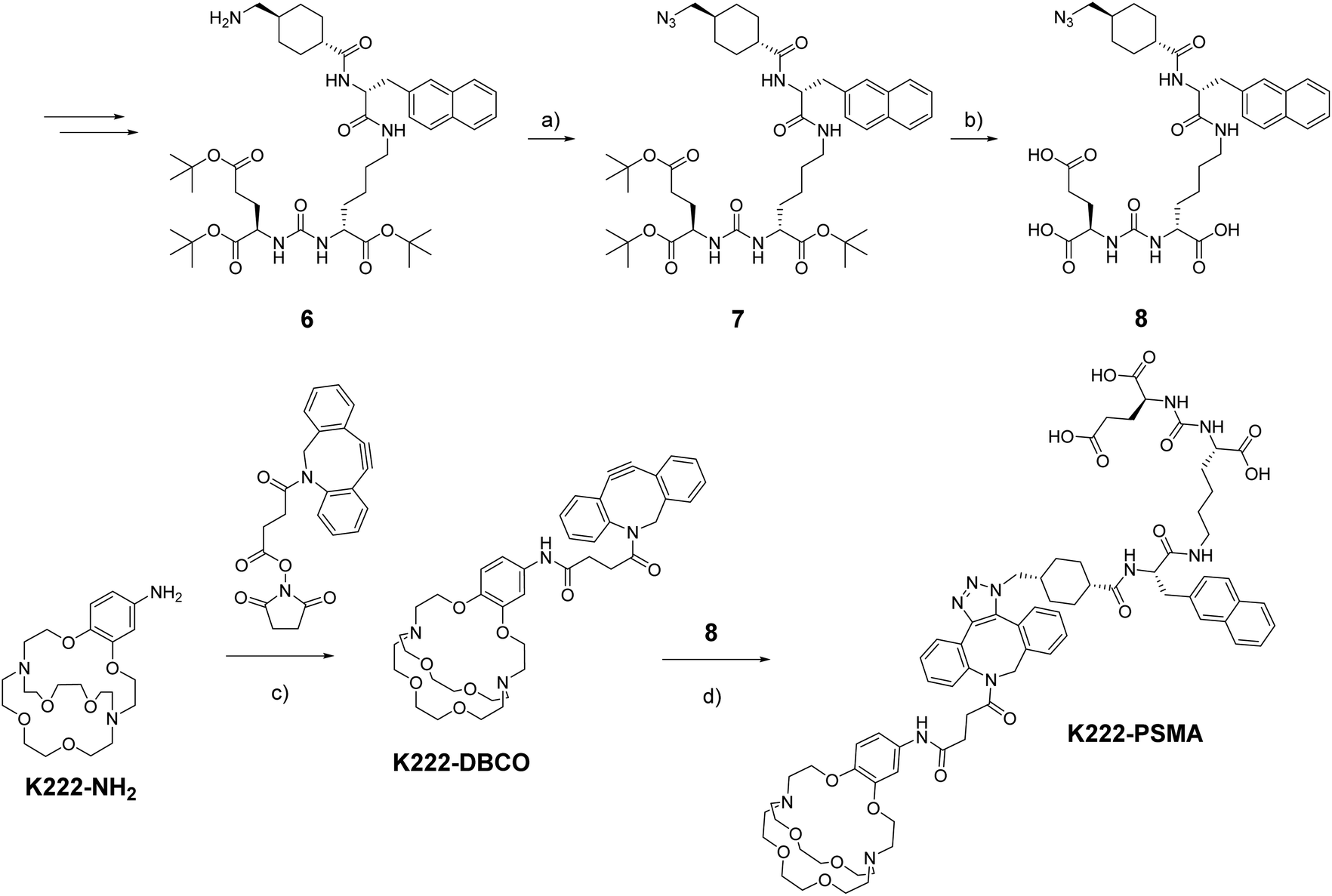 | ||
Scheme 2 Synthetic route towards K222-DBCO and K222-PSMA Reaction conditions: (a) FSO2N3, 3 M KHCO3, MTBE/DMF/H2O, r.t. 5 min; (b) TFA, DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), r.t., 3 h; (c) DBCO–NHS, DMF, r.t., overnight; (d) 8, DMF/MeOH (1:1), r.t., overnight. :2), r.t., 3 h; (c) DBCO–NHS, DMF, r.t., overnight; (d) 8, DMF/MeOH (1:1), r.t., overnight. | ||
While this project was ongoing, another group reported on the synthesis and application of K222 derivatives as chelators for 203Pb, an emerging radiometal for SPECT applications.23 While the synthetic derivatives reported by McDonagh et al. are slightly different to the ones reported here, their general findings that successful radiolabelling is possible with this class of compounds and that the resulting Pb(II) complexes possess suitable stability in human blood serum, encouraged us to proceed with the investigation of the thallium-binding ability of our compounds.
With all target compounds in hand we proceeded to investigate the ability of these derivatives to bind Tl(I). First, we repeated the NMR experiments conducted with K222 and K22 with the new derivative 5. Compound 5 was dissolved in CD3OD and an initial 1H NMR was recorded. TlPF6 (1.5 equivalents) was pre-dissolved in CD3OD, added to the solution of 5 and a second proton spectrum was recorded immediately after (Fig. 3). A clear change in the chemical shifts pertaining to the alkyl-protons of the K222 scaffold can be observed in the 1H NMR spectra. On the other hand, the three aromatic protons show little to no change. These experimental findings were supplemented with density functional theory (DFT) calculations performed on both the Tl(I) as well as the K(I) complexes with ligand 5. For reference, DFT calculations were also performed for the already reported complex of K222 with potassium (ESI Fig. S19†). Comparison of the oxygen- and nitrogen–metal bonds showed good agreement between the calculated and experimentally determined structures. The structures for Tl(5)+ (Fig. 4) and K(5)+ are similar, with all 6 oxygens and 2 nitrogen atoms of the macrocycle involved in interactions with the metal centre (ESI Fig. S20†).
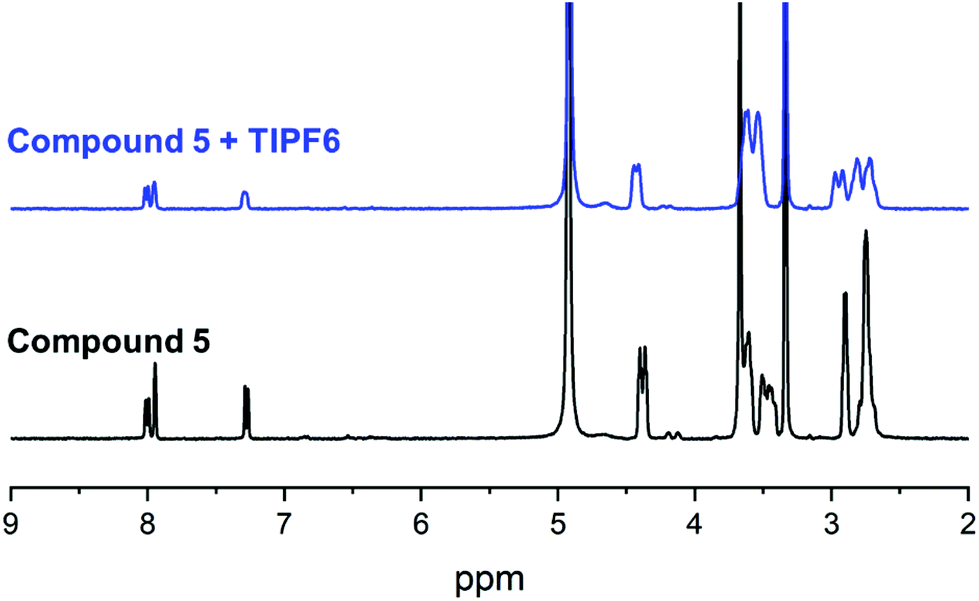 | ||
| Fig. 3 1H NMR binding experiment with compound 5 and TlPF6 in MeOD. | ||
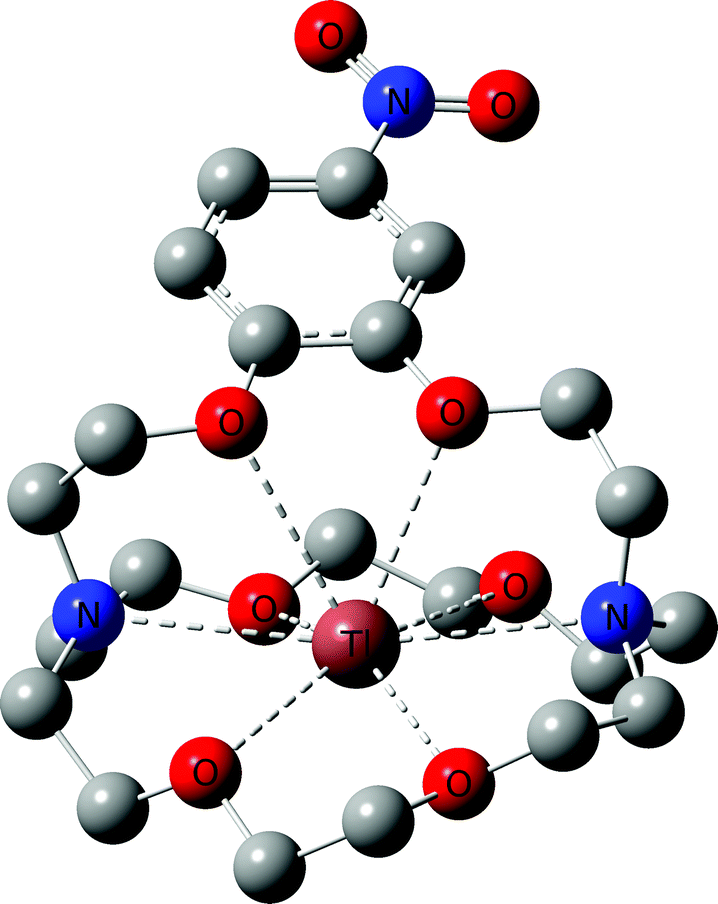 | ||
| Fig. 4 DFT optimized structure for Tl(5)+. Hydrogen atoms are omitted for clarity. | ||
Slightly longer distances between putative coordinating atoms and metal centre were detected for the calculated Tl(I) complex structure compared to the K(I) analogue (ESI Table S2†). Comparison of the Tl(I) with the K(I) coordination environments revealed that the K(I) ion sits more centrally in the cryptand scaffold whereas in the case of Tl(I), the ion is located near the plane formed by the four non-aromatic oxygen and nitrogen atoms. These calculations suggest that the binding mode of K(I) and Tl(I) are similar with regards to 5 but that in the case of Tl(I), the metal ion is less encapsulated by the cryptand which could indicate lower stability. Altogether, these experimental and computational studies strongly suggest that an interaction between the chelator and Tl(I) is occurring and that it is, as expected, between the oxygen and nitrogen atoms of the K222 cryptand and the thallium cation. As additional confirmation, the NMR solution was analysed by ESI-MS and the mass peak for a thallium ion bound by 5 was detected (ESI Fig. S18†).
All initial radiolabelling studies were conducted with compound 5 as it was available in larger amounts than the other derivatives. Initial attempts at radiolabelling 5 with [201Tl](I) in water were unsuccessful. Reactions were monitored by analytical radioHPLC utilizing a C18 column and a 0–100% water/acetonitrile gradient with 0.1% TFA. During the synthesis of the K222 derivatives, the last step involves purification on reverse phase columns and through trial and error it was found that neutralisation of the collected fractions via addition of 1 M NaOH was necessary, otherwise no chelation of TlPF6 could be observed by NMR. We hypothesised that this is due to protonation of the nitrogen atom in the macrocycle ring, as supported by DFT calculations. Visualisation of the HOMO for 5(Tl) revealed overlap of nitrogen and Tl-based orbitals, originating from the p and d orbitals, respectively (ESI Fig. S19,† left image). This interaction was no longer observed in the HOMO upon protonation of the nitrogen (ESI Fig. S19,† right image), and is reflected in the N–Tl distances where an elongation from 3.04 Å to 3.83 Å was observed upon protonation. In the absence of this stabilisation from N–Tl interactions, Tl(I) was found to reside further away from the aromatic oxygen donors, resulting in a noticeably less enveloped structure. Based on these findings we assume that concentration of collected fractions in the presence of small amounts of HPLC mobile phase acid additives (TFA or formic acid) resulted in the formation of a protonated cryptand which is unable to coordinate [natTl]Tl(I).
With this knowledge in mind, further [201Tl]Tl(I) radiolabelling reactions were conducted in water with the addition of small amounts of 1 M Na2CO3 (final pH = 11). As no new peaks could be detected by radioHPLC, we hypothesized that the acidic mobile phase resulted in decomplexation of 5 from [201Tl]Tl(I). Monitoring of a reaction mixture of 5 and [201Tl]Tl(I) under neutral solvent conditions, without inclusion of an ionic solvent modifier, resulted in the observation of a new peak, putatively assigned as [201Tl(5)]+, with a retention time that is clearly distinct from that of unchelated 201Tl. Unfortunately, under these conditions, we could not obtain reproducible radio-HPLC traces. We attribute this to adsorption of hydrophilic [201Tl(5)]+ to silica as in some repeated experiments, radioactive species were retained on the C18 column and did not elute. Such behaviour is sometimes observed in HPLC experiments using mobile phases without inclusion of a buffering or ion pairing reagent or acid.24 Through further extensive experimentation it was found that addition of 0.01% formic acid to the HPLC mobile phase (pH ∼ 4) enabled reproducible radio-HPLC chromatography. With the HPLC conditions optimised, radiosynthetic protocols for preparation of putative [201Tl(5)]+ were found to be robust: exclusive formation of [201Tl(5)]+ in >95% radiochemical yield was observed upon reaction of 5 with [201Tl]Tl(I)Cl at pH = 11, in aqueous solution at room temperature after 15 minutes reaction time (Fig. 5 left).
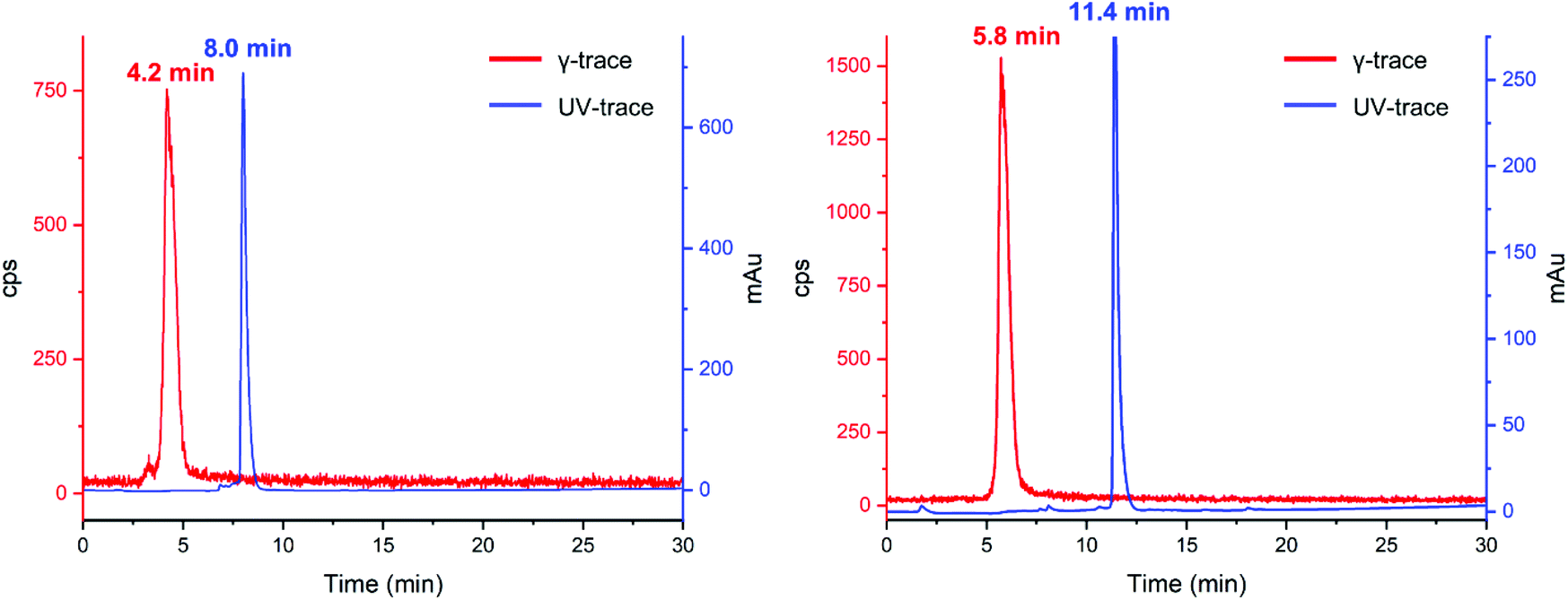 | ||
| Fig. 5 UV (254 nm, blue) and γ-traces (red) of reaction of 5 (left) and K222-PSMA (right) with 201Tl. | ||
Comparison of the UV-trace of 5 with the γ-trace of [201Tl(5)]+ shows a large difference between the retention times of the free ligand (8.0 min) and radioactive complex (4.2 min). The Tl(I) complex of 5 is significantly more hydrophilic than that of the free ligand, which we attribute in part to the single positive charge of [201Tl(5)]+, as well as conformational differences between chelated 5 and non-chelated 5.
With the optimized [201Tl]Tl(I) radiolabelling conditions determined for compound 5 we proceeded to the reaction of bioconjugate K222-PSMA with [201Tl]Tl(I)Cl, which also led to the formation of a single new peak in the radioHPLC (Fig. 5 right). Reaction of K222-PSMA with [201Tl]Tl(I) yielded putative [201Tl][Tl(K222-PSMA)]+ in >98% radiochemical yield, with a retention time of 5.8 min, distinct from that of K222-PSMA, which elutes later, with a retention time of 11.4 min.
So far, all evidence pointed to K222-based chelators complexing Tl(I) under both non-radioactive (stoichiometric) and radioactive (non-stoichiometric tracer) conditions. A crucial benchmark for any radiochelator is not only the ability to bind a given radiometal but stably retain it under physiologically relevant conditions. A common assay to evaluate the stability of radiolabelled bioconjugates is to incubate them with human blood serum over time. We incubated [201Tl][Tl(K222-PSMA)]+ in human blood serum at 37 °C, and monitored its stability. Radio-HPLC analysis of aliquots of serum, taken at 0 min, 30 min and 2 h incubation times, indicated that a fraction (around 10%) of 201Tl(I) was released from K222-PSMA immediately upon addition of the serum to [201Tl][Tl(K222-PSMA)]+ (ESI Fig. S17†). However, after that, no significant changes were observed, indicating that [201Tl]Tl(I) K222 chelates are relatively stable in human serum over 2 h.
Having shown that [201Tl][Tl(K222-PSMA)]+ exhibits adequate stability under physiological conditions we investigated the ability of this conjugate to specifically bind PSMA in vitro. To this end, the PSMA-positive cell line, DU145-PSMA+, and the parental PSMA-negative DU145 cell line (∼500000 cells), were incubated with [201Tl][Tl(K222-PSMA)]+ for either 10 min or 1 h.21 Additionally, a control experiment was performed where both cell types were pre-incubated with the PSMA inhibitor, 2-PMPA (2-(phosphonomethyl) pentane-1,5-dioic acid),21a to block PSMA receptors, prior to incubation with [201Tl][Tl(K222-PSMA)]+ for 1 h. After incubation, cells were washed and lysed, and cell lysates were collected and counted for radioactivity using a γ-counter (Fig. 6 and ESI, Table S1†).
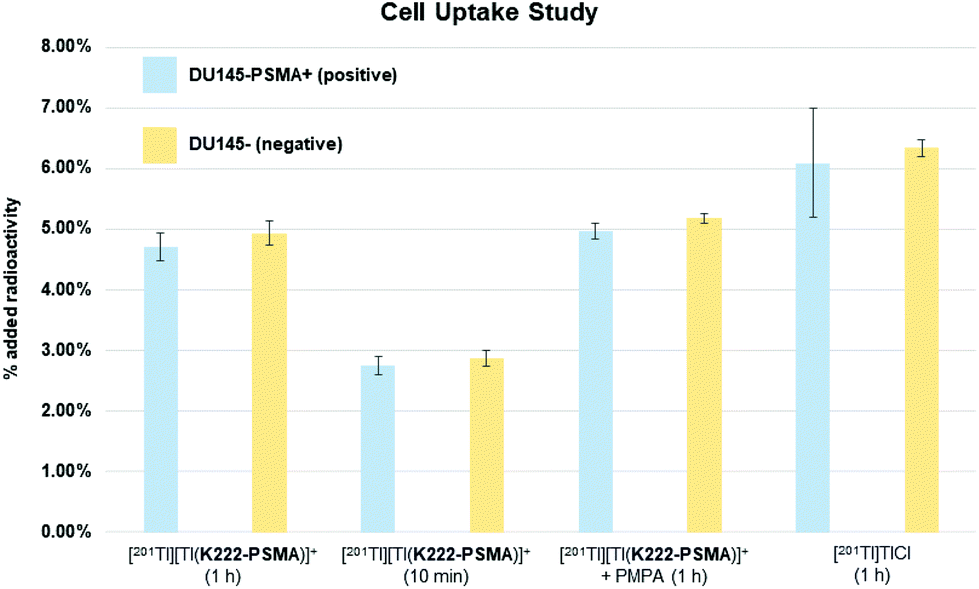 | ||
| Fig. 6 Cell uptake study results for bioconjugate [201Tl][Tl(K222-PSMA)]+ in PSMA-positive cell line, DU145-PSMA+ (positive) and the parental PSMA-negative DU145 cell line (negative) after 1 hour and 10 min incubation time (columns 1–4), in the presence of the PSMA inhibitor, 2-PMPA (2-(phosphonomethyl) pentane-1,5-dioic acid, columns 5 and 6). Columns 7 and 8 show the control experiment where positive and negative cells were incubated with “unchelated” [201Tl]TlCl for 1 h. Each experiment was conducted twice, values shown are the mean ± standard deviation. | ||
Uptake of 201Tl activity in DU145-PSMA+ cells measured 4.7 ± 0.2% AR (percentage added radioactivity to 500000 cells), however uptake in the PSMA-negative DU145 cell line similarly measured 4.9 ± 0.2% AR, and uptake in DU145-PSMA+ cells blocked with 2-PMPA measured 5.0 ± 0.1% AR. This suggested that uptake of 201Tl radioactivity was not mediated by PSMA receptors. To further probe this, both DU145-PSMA+ and DU145 cells were treated with solutions of “free”/“unchelated” [201Tl]Tl(I)Cl for 1 h. 201Tl activity measured 6.1 ± 0.9% AR in DU145-PSMA+ and 6.3 ± 0.1% AR in DU145 cells.
The similar values of 201Tl radioactivity in cells treated with either [201Tl][Tl(K222-PSMA)]+ or [201Tl]Tl(I)Cl, suggests that in these in vitro experiments, [201Tl]Tl(I) dissociates from K222-PSMA. With the same charge and a similar ionic radius compared to K(I), biologically, Tl(I) can behave as a K(I) “mimic”.6 We hypothesize that after dissociating from K222-PSMA, [201Tl]Tl(I) is taken up by both DU145-PSMA+ and DU145 cells through potassium ion transporters. This accounts for the similar levels of cellular 201Tl radioactivity, regardless of whether 201Tl is administered as “free” [201Tl]Tl(I)Cl or [201Tl][Tl(K222-PSMA)]+.
There are many endogenous biomolecules that are likely to compete with ligands such as K222 derivatives for binding to K(I)-mimics such as Tl(I). For example, potassium ion channels transport K(I) across cell membranes. These channels possess high affinity for K(I) to compensate for the energetic cost of dehydration/desolvation of K(I), which is required to enable transport.25 It is entirely plausible that in addition to transporting [201Tl]Tl(I) into prostate cancer cells, such transporters (alongside other biomolecules either present in cell media, excreted by cells or on the surface of cells) compete for [201Tl]Tl(I) complexation. The abundance of high affinity K(I)-binding sites, and thus Tl(I)-binding sites in the biological environment, is likely a formidable hurdle to stable chelation of Tl(I), and such factors require due consideration for future Tl(I) ligand design.
In conclusion, we have demonstrated the synthesis and characterisation of a series of new Kryptofix-222 derivatives with the ability to be conjugated to targeting functionalities through a variety of chemical modification strategies. We further demonstrated that these conjugates interact strongly with free [natTl]Tl(I) in solution through NMR experiments. Radiolabelling of these conjugates with [201Tl]Tl(I) showed promise with both K222 derivatives, 5 and K222-PSMA, successfully radiolabelled with [201Tl]Tl(I) in greater than 98% radiochemical yield. The PSMA-targeting conjugate, [201Tl][Tl(K222-PSMA)]+ exhibited encouraging stability in human serum which prompted us to explore its ability to specifically accumulate in PSMA-expressing cell lines. Unfortunately, these assays indicated no PSMA-receptor mediated uptake of 201Tl activity, and the observed cellular uptake of [201Tl]Tl(I) activity was attributed to non-specific uptake, likely via K(I) transporters.
While these data with [201Tl]Tl(I) are encouraging, we believe that other radiometals might be better suited to complexation with K222-based bioconjugates and therefore, we are exploring the application of K222-based compounds to 223Ra, 213Bi and 111In.
Experimental
Materials and methods
Commercially available reagent grade solvents and chemicals were used without further purification. Anhydrous solvents were acquired from solvent towers within the department and stored over 3 Å molecular sieves. 1H, 13C, COSY, TOCSY, HSQC, HMBC NMR spectra were recorded on a Bruker AVIII 400 or 700 spectrometer. Chemical shifts are reported in ppm and referenced to residual protonated impurities in the solvent for NMR spectra. High Resolution Electrospray Mass Spectrometry was carried out by Dr Lisa Haigh of the mass spectrometry service at Imperial College, or independently on an Agilent 6200 TOF LC-MS instrument. LC-MS for K222-PSMA was measured on a Waters 3100 Mass Detector equipped with a Waters 2998 Photodiode Array Detector, Waters 515 HPLC Pump, Waters 2545 Quarternary Gradient Module, and a Waters 2767 Sample Manager. LC-MS solvents were Millipore water and acetonitrile (with 0.1% formic acid). A 18 min gradient was run from 5% to 100% Acetonitrile on a XBridge BEH C18 Column, 130 Å, 5 μm, 4.6 mm × 100 mm. Flash chromatography used silica gel (60 Å pore size). Where specified, automated flash chromatography was performed using a Biotage Isolera Four unit and 10 g or 25 g SNAP KP-Sil/Sfar duo cartridge. HPLC was performed on an Agilent 1200 Series Liquid Chromatograph with UV and LabLogic Flow-Count detector with a sodium-iodide probe (B-FC-3200). Unless otherwise mentioned the mobile phase A contained H2O with 0.1% FA, and mobile phase B contained MeCN with 0.1% FA. Preparative reverse-phase HPLC was conducted using a Pursuit XRs C18 column (250 × 10 mm, 5 μm) and UV spectroscopic detection at 250 nm. 201TlCl in saline was purchased from Curium Pharma, UK and used without further processing. Fresh human serum was obtained from a healthy volunteer.Synthesis of K222 derivatives
:hexane) yielded 1 as a brown oil (0.96 g, 2.93 mmol, 45%). Instead of thermal heating the reaction can also be conducted via microwave reactor (400 mg scale in a 2–5 mL vial) at 120 °C for 1 h. The NMR data matched that reported previously.26
1H NMR (400 MHz, CDCl3) δ 7.91 (dd, 3JH–H = 2.5, 8.9 Hz, 1H), 7.73 (d, 3JH–H = 2.6 Hz, 1H), 6.87 (d, 3JH–H = 9.0 Hz, 1H), 4.80 (d, 3JH–H = 11.4 Hz, 2H), 4.27 (m, 4H), 1.30 (q, J = 7.0 Hz, 6H).
1H NMR (400 MHz, MeOD) δ 7.95 (m, 1H), 7.85 (m, 1H), 7.12 (d, 3JH–H = 9.1 Hz, 1H), 3.82 (d, J = 2.0 Hz, 4H).
:DCM) yielding 4 as a yellowish oil (51.5 mg, 0.10 mmol, 25%). The NMR data matched that reported previously.19
1H NMR (400 MHz, CDCl3) δ 7.82 (dd, 3JH–H = 2.6, 8.9 Hz, 1H), 7.77 (d, 3JH–H = 2.6 Hz, 1H), 7.00 (d, 3JH–H = 8.9 Hz, 1H), 5.63 (d, 3JH–H = 15.0 Hz, 1H), 5.47 (d, 3JH–H = 15.1 Hz, 1H), 4.83–4.75 (m, 2H), 4.38–4.26 (m, 2H), 3.92–3.47 (m, 20H), 3.28–3.17 (m, 2H), 2.84–2.75 (m, 2H).
:1, 2 mL) and purified by automated column chromatography on a C18 cartridge (gradient 0–100% ACN:H2O with 0.1% TFA). The fractions containing product were neutralised by addition of 1 M NaOH and dried, yielding 5 as a reddish oil (58.0 mg, 0.12 mmol, 55%). The NMR data matched that reported previously.19
1H NMR (400 MHz, CDCl3) δ 7.98 (dd, 3JH–H = 2.6, 9.0 Hz, 1H), 7.92 (d, 3JH–H = 2.6 Hz, 1H), 7.27 (d, 3JH–H = 9.0 Hz, 1H). 4.39 (t, 3JH–H = 4.8 Hz, 2H), 4.34 (t, 3JH–H = 4.8 Hz, 2H), 3.69–3.57 (m, 12), 3.51–3.40 (m, 4H), 2.91–2.87 (m, 4H), 2.79–2.67 (m, 8H).
13C NMR (100.7 MHz, CD3OD) δ 153.4, 148.0, 142.9, 119.0, 112.8, 108.4, 69.2, 68.7, 67.4, 67.0, 54.1, 53.8, 53.7.
ESI–HRMS (MeOH): found: 492.2340; calculated for [C22H35N3O8 + 1Na+]: 492.2322.
:1, 20 mL) and the DCM was evaporated. The zinc dust was filtered off and the filtrate was dried in vacuo. The crude product was purified by automated column chromatography on a C18 cartridge (gradient 0–100% ACN:H2O with 0.1% TFA). The fractions containing product were neutralized by addition of 1 M NaOH and dried, yielding K222-NH2 as a brownish oil (41.5 mg, 0.094 mmol, 43%). The NMR data matched that reported previously.19
1H NMR (400 MHz, CDCl3) δ 6.77 (d, 3JH–H = 8.7 Hz, 1H), 6.35 (d, 3JH–H = 2.5 Hz, 1H), 6.18 (dd, 3JH–H = 2.5, 8.7 Hz), 4.17 (t, 3JH–H = 4.4 Hz, 2H), 4.06 (t, 3JH–H = 4.7 Hz, 2H), 3.66–3.61 (m, 8H), 3.59–3.43 (m, 8H), 2.80–2.74 (m, 4H), 2.69–2.64 (m, 8H).
13C NMR (100.7 MHz, CD3OD) δ 148.2, 141.8, 140.8, 115.0, 110.4, 104.5, 69.3, 68.8, 68.7, 66.8, 66.4, 54.4, 54.0.
ESI–HRMS (MeOH): found: 462.2570; calculated for [C22H37N3O6 + 1Na+]: 462.2580.
:1, 1 mL). Fluorosulfuryl azide (100 μL, 0.7 mM) and KHCO3 (70 μL, 3 M) were added, and the solution was stirred overnight. LC-MS analysis revealed formation of the target compound. The solvent was evaporated and the crude product was purified by automated silica column chromatography (dry loading after dissolution in DCM/MeOH; gradient 0–10% MeOH:DCM) yielding K222-N3 as a reddish oil (8.1 mg, 17 μmol, 37%).
1H NMR (400 MHz, CDCl3) δ 6.89 (d, 3JH–H = 8.7 Hz, 1H), 6.65 (dd, 3JH–H = 6.1, 11.2 Hz), 6.52 (d, 3JH–H = 2.5 Hz, 1H), 4.19–4.15 (m, 4H), 3.65–3.45 (m, 16H), 2.91–2.87 (m, 4H), 2.74–2.63 (m, 8H).
ESI–HRMS (MeOH): found: 466.2667 and 488.2484; calculated for [C22H35N5O6 + 1H+]: 466.2660 and [C22H35N5O6 + 1Na+]: 488.2480.
IR (Golden Gate ATR): = 2106 cm−1.
22b (18.2 mg, 22.1 μmol) was dissolved in DMF and MTBE (1:1, 1 mL). Fluorosulfuryl azide (60 μL, 0.7 mM) and KHCO3 (70 μL, 3 M) were added, and the solution was stirred overnight. LC-MS revealed formation of the target compound. The solvent was evaporated, and the crude was used without further purification.
:1 DCM/MeOH). After 2 h no more starting material could be detected and the target compound was identified by LC-MS. The solvent was evaporated and the crude product was then used without further purification.
ESI–HRMS (MeOH): found: 1408.6879; calculated for [C74H93N11O17 + H+]: 1408.6829.
Radiolabelling experimental
In a typical experiment, [201Tl]TlCl (∼1 MBq, 50 μL) was added to an Eppendorf vial together with 50 μL of the organic chelator (0.5–2 mM). Na2CO3 (25 μL of 1 M) and water (75 μL) were added to the vial and the reaction was shaken at room temperature for 15 min.
Cell assay experimental
DU145 (PSMA-negative) and DU145-PSMA (PSMA-positive) human prostate cancer cells were cultured in RPMI 1640 medium (R0883, Sigma) supplemented with 10% (v/v) foetal bovine serum (FBS), 2 mM L-glutamine, and penicillin/streptomycin (Sigma-Aldrich, UK) and maintained in a humidified atmosphere at 37 °C under 5% CO2.To assess PSMA targeting, PSMA-expressing cells DU145-PSMA and non-PSMA-expressing cells DU145 cells were seeded in 6-well plates at a density of 0.5 × 10 cells per well 1 day prior to the experiment. Three technical replicates were performed for each condition. Cell medium was replaced with 1 mL complete medium 1 hour before the cells were treated. [201Tl][Tl(K222-PSMA)]+ (50 kBq, in 10 μL of phosphate buffered saline) was added to each well, and the cells incubated at 37 °C for 15 min and 1 h. Competition studies were also performed following co-incubation with the PSMA-inhibitor 2-(phosphonomethyl)pentane-1,5-dioic acid (PMPA; 30 μL of 750 mM PMPA solution per well). After incubation, plates were placed on ice, the supernatant was removed and the cells were washed with ice cold phosphate buffered saline solution (2 × 1 mL). The cells were lysed with ice cold radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific Inc., 500 μL), and radioactivity content was determined by gamma counter (1282 Compugamma; LKB, window set to channels 35–110 to measure 201Tl gamma emissions). Cell uptake was also performed following [201Tl]TlCl incubation (50 kBq, 10 μL) for 15 min and 1 h. Cells were harvested and radioactivity measured as described above.
DFT calculations
All calculations were performed using the Gaussian 16 package (Revision C.01).27 Full geometry optimisations were performed using the ωB97X-D functional28 in gaseous state with the LANL2DZ basis set and effective core potentials for Tl, and 6-311+G** for all other atoms.29 Structures were optimised without using symmetry constraints. Frequency analysis of the optimised structures confirmed a true minimum energy structure by the absence of imaginary frequencies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the EPSRC programme for Next Generation Molecular Imaging and Therapy with Radionuclides (EP/S019901/1, “MITHRAS”) and an Imperial College Schrodinger Scholarship (to TTCY). NJL is grateful for a Royal Society Wolfson Research Merit Award. MTM is supported by a Cancer Research UK Career Establishment Award (C63178/A24959).References
- (a) C&EN, 2020, https://cen.acs.org/policy/Former-chemistry-student-pleads-guilty/98/web/2020/11 (accessed 25.01.2022); (b) J. Aronson, Br. Med. J., 2007, 334, 205–205 CrossRef.
- E. Lebowitz, M. W. Greene, R. Fairchild, P. R. Bradley-Moore, H. L. Atkins, A. N. Ansari, P. Richards and E. Belgrave, J. Nucl. Med., 1975, 16, 151–155 CAS.
- D. Jain, Semin. Nucl. Med., 1999, 29, 221–236 CrossRef CAS PubMed.
- (a) R. C. Stadalnik, A. Z. Krasnow, B. D. Collier, A. T. Isitman, R. S. Hellman and D. C. Peck, Semin. Nucl. Med., 1988, 18, 350–358 CrossRef; (b) Y. Ito, A. Muranaka, T. Harada, A. Matsudo, T. Yokobayashi and H. Terashima, Eur. J. Nucl. Med., 1978, 3, 81–86 CrossRef CAS PubMed; (c) L. Ramanna, A. Waxman, G. Binney, S. Waxman, J. Mirra and G. Rosen, J. Nucl. Med., 1990, 31, 567–572 CAS.
- (a) B. Cornelissen and K. A. Vallis, Curr. Drug Discovery Technol., 2010, 7, 263–279 CrossRef CAS PubMed; (b) S. Imstepf, V. Pierroz, P. Raposinho, M. Bauwens, M. Felber, T. Fox, A. B. Shapiro, R. Freudenberg, C. Fernandes, S. Gama, G. Gasser, F. Motthagy, I. R. Santos and R. Alberto, Bioconjugate Chem., 2015, 26, 2397–2407 CrossRef CAS PubMed; (c) A. Ku, V. J. Facca, Z. Cai and R. M. Reilly, EJNMMI Radiopharm. Chem., 2019, 4, 27 CrossRef PubMed; (d) G. Pirovano, S. A. Jannetti, L. M. Carter, A. Sadique, S. Kossatz, N. Guru, P. Demétrio De Souza França, M. Maeda, B. M. Zeglis, J. S. Lewis, J. L. Humm and T. Reiner, Clin. Cancer Res., 2020, 26, 2871–2881 CrossRef CAS PubMed; (e) A. P. Kiess, I. Minn, Y. Chen, R. Hobbs, G. Sgouros, R. C. Mease, M. Pullambhatla, C. J. Shen, C. A. Foss and M. G. Pomper, J. Nucl. Med., 2015, 56, 1401–1407 CrossRef CAS PubMed.
- K. M. Osytek, P. J. Blower, I. M. Costa, G. E. Smith, V. Abbate and S. Y. A. Terry, EJNMMI Res., 2021, 11, 63 CrossRef CAS PubMed.
- (a) J. A. Jackson, I. N. Hungnes, M. T. Ma and C. Rivas, Bioconjugate Chem., 2020, 31, 483–491 CrossRef CAS PubMed; (b) C. Rivas, J. A. Jackson, I. N. Hungnes and M. T. Ma, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 706–740 Search PubMed.
- N. M. Hijnen, A. de Vries, R. Blange, D. Burdinski and H. Grüll, Nucl. Med. Biol., 2011, 38, 585–592 CrossRef CAS PubMed.
- T. Fodor, I. Bányai, A. Bényei, C. Platas-Iglesias, M. Purgel, G. L. Horváth, L. Zékány, G. Tircsó and I. Tóth, Inorg. Chem., 2015, 54, 5426–5437 CrossRef CAS PubMed.
- A. Rigby, J. E. Blower, P. J. Blower, S. Y. A. Terry and V. Abbate, Nucl. Med. Biol., 2021, 98–99, 1–7 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- (a) T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS PubMed; (b) T. Brotin, P. Berthault, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2020, 85, 9622–9630 CrossRef CAS PubMed; (c) T. Brotin, E. Jeanneau, P. Berthault, E. Léonce, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2018, 83, 14465–14471 CrossRef CAS PubMed.
- (a) B. Dietrich, J. M. Lehn and J. P. Sauvage, Tetrahedron Lett., 1969, 10, 2885–2888 CrossRef; (b) B. Dietrich, J. M. Lehn and J. P. Sauvage, Tetrahedron Lett., 1969, 10, 2889–2892 CrossRef.
- (a) A. B. Chung, D. N. Huh, J. W. Ziller and W. J. Evans, Inorg. Chem. Front., 2020, 7, 4445–4451 RSC; (b) D. N. Huh, J. W. Ziller and W. J. Evans, Inorg. Chem., 2019, 58, 9613–9617 CrossRef CAS PubMed; (c) C. A. P. Goodwin, M. J. Giansiracusa, S. M. Greer, H. M. Nicholas, P. Evans, M. Vonci, S. Hill, N. F. Chilton and D. P. Mills, Nat. Chem., 2021, 13, 243–248 CrossRef CAS PubMed; (d) J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- O. A. Gansow, A. R. Kausar and K. B. Triplett, J. Heterocycl. Chem., 1981, 18, 297–302 CrossRef CAS.
- W. A. Pettit, Y. Iwai, C. F. Barfknecht and D. C. Swenson, J. Heterocycl. Chem., 1992, 29, 877–881 CrossRef CAS.
- M. D. Bailey, G.-X. Jin, F. Carniato, M. Botta and M. J. Allen, Chem. – Eur. J., 2021, 27, 3114–3118 CrossRef CAS PubMed.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- (a) J. D. Young, V. Abbate, C. Imberti, L. K. Meszaros, M. T. Ma, S. Y. A. Terry, R. C. Hider, G. E. Mullen and P. J. Blower, J. Nucl. Med., 2017, 58, 1270–1277 CrossRef CAS PubMed; (b) F. Kampmeier, J. D. Williams, J. Maher, G. E. Mullen and P. J. Blower, EJNMMI Res., 2014, 4, 13 CrossRef PubMed.
- (a) P. A. Duspara, M. S. Islam, A. J. Lough and R. A. Batey, J. Org. Chem., 2012, 77, 10362–10368 CrossRef CAS PubMed; (b) A. Rigby, G. Firth, C. Rivas, T. Pham, J. Kim, A. Phanopoulos, L. Wharton, A. Ingham, L. Li, M. T. Ma, C. Orvig, P. J. Blower, S. Y. A. Terry and V. Abbate, 2022, submitted.
- A. W. McDonagh, B. L. McNeil, B. O. Patrick and C. F. Ramogida, Inorg. Chem., 2021, 60, 10030–10037 CrossRef CAS PubMed.
- H. P. J. Bennett, C. A. Browne and S. Solomon, J. Liq. Chromatogr., 1980, 3, 1353–1365 CrossRef CAS.
- (a) S. Choe, Nat. Rev. Neurosci., 2002, 3, 115–121 CrossRef CAS PubMed; (b) C. Miller, Genome Biol., 2000, 1, reviews0004.0001 CrossRef PubMed.
- A. O. Gansow, K. A. Rashid and K. B. Triplett, J. Heterocycl. Chem., 1981, 18, 297–302 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Wallingford, CT, 2016.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt01074g |
| This journal is © The Royal Society of Chemistry 2022 |