
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNuisance small molecules under a machine-learning lens
Tiago
Rodrigues
 *
*
Instituto de Investigação do Medicamento (iMed), Faculdade de Farmácia, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisbon, Portugal. E-mail: tiago.rodrigues@ff.ulisboa.pt
First published on 30th March 2022
Abstract
Small molecules remain a centrepiece in molecular medicine. Specific drug target recognition is an unavoidable requirement for their successful translation to the clinic. While testing chemical matter for affinity and potency is the mainstay, only more recently have the chemical biology and medicinal chemistry communities become more profoundly aware of potential attrition and development pitfalls linked to artifactual readouts. Auto-fluorescence, assay interference and colloidal aggregation are the most common sources of false positive hits in screening assays and may divert drug development pipelines toward unfruitful research avenues. In this Perspective, I dissect how computational tools, in particular machine-learning pipelines, can accelerate the development of chemical probes and drug leads by expediting the identification of liable chemical matter. Further, I conceptualize anomaly detection for chemical biology and highlight limitations to a more productive deployment of machine learning. By solving pressing challenges, one might gradually mitigate the impact of nuisance compounds in drug discovery.
Despite the rise of biologicals1 and drug delivery systems2 in the modern therapeutic armamentarium, the development of innovative small molecules remains an inevitable means of modulating disease.1 Typically, small molecules bind to one or more biologically relevant targets.3,4 In doing so, they remodel the protein's function and dynamics, which is critical to (dys)regulate homeostatic processes and/or halt the progression of a disease state. Mechanistically, molecular recognition and engagement is elicited by (un)directed drug–target interactions5 that are the basis for the multidimensional drug design paradigm.6
The identification of viable starting points for elaboration in hit-to-lead and lead development campaigns is a focal point in drug discovery. Toward that end, screening assays must be set up,7 wherein the binding affinity or functional activity is assessed – initially in a single concentration and then in a follow-up concentration–response curve. It is worth noting that hits in primary assays do not always reflect true target recognition (e.g. ref. 8). To mitigate this, control and orthogonal assays with disparate detection technologies are executed.9 These however only partly confirm the ligand–target interaction and are best suited to rule out interference with either the detection method (e.g. fluorescence) or assay components (e.g. proteins, reporter molecules).
In parallel, strict windows for lipophilicity and molecular weight values are routinely accepted to guide the design of ‘drug-like’ entities with decreased likelihood of promiscuity/attrition and improved oral bioavailability.10 One may appreciate that those rules are not sufficiently generalizable. Natural products remain the biggest source of inspiration for molecular design and approved drugs, but present a vaster property space than what is commonly accepted.11 Readily discarding such chemotypes would have had unpredictable consequences in modern medicine. Taken together, not all structural liabilities can be identified through rule-based intuition, which constitutes one of the grand and unsolved challenges in small molecule development.
While there is awareness regarding the perils of promiscuity, the underlying molecular bases have remained somewhat abstract and associated to unwritten rules of expert knowledge. The so-called frequent-hitters12 tend to appear as promising prototypical structures and modulators of unrelated targets, but should indeed be examined with caution. The mechanisms of interference can be very diverse. For example, cationic molecules can induce vesicle-like structures in cells and foamy membranes – a process called phospholipidosis.13 On occasion, metal impurities at trace level can also result in apparent bioactivity. This is an important realization given that palladium-catalysed reactions are among the most employed in medicinal chemistry.14,15 The most common interference mechanism is however the formation of aggregates, which can occur even in focused libraries with apparently sound structure–activity relationships (Fig. 1).16 On a physical level, colloidal aggregation results from the poor aqueous solubility of a given molecule. The formed nano or microscale particles can inadvertently denaturate proteins.17–19 Protein denaturation is thus the crux of false target modulation. Since that realization, dynamic light scattering (DLS) and paired biochemical screens (with and without surfactant) have become the gold standard for detecting anomalous biological readouts.20,21 Other technologies, e.g. nuclear magnetic resonance,22 surface plasmon resonance23 and centrifugation,24 may also be employed, but rapidly become cumbersome from an experimental and data analysis vantage point. It is here that in silico tools can expedite the identification of liable matter. From rule-based methods (e.g. PAINS,25 REOS26,27) to machine learning (ML), a multitude of workflows have been developed to help flagging, accelerate the detection and deprioritize less promising target effectors for medicinal chemistry elaboration.
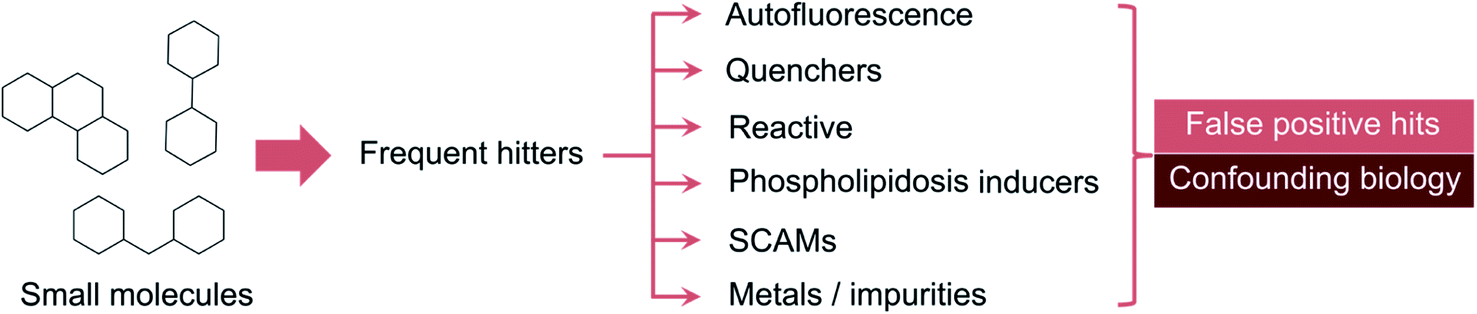 | ||
| Fig. 1 Common modes of false target modulation. Promiscuous small molecules tend to show up as hits in multiple assays for unrelated targets. These so-called frequent hitters can act through different mechanisms, such as interference with detection methods (e.g. auto-fluorescence and signal quenchers), reaction with assay components and induction of false target modulation through local protein denaturation – as in small, colloidally aggregating molecules (SCAMs) – or inducing phospholipidosis. Impurities can lead to erroneous bioactivity readouts in specific compound batches. | ||
In this Perspective, I not only critically discuss the impact of frequent hitters – in particular those forming aggregates in aqueous media – but also dissect the data sources, molecular representations and ML algorithms employed for triaging them. Through select literature examples, I wish to expose unsolved challenges in the area and speculate on potential solutions, such as anomaly detection algorithms, that can inform molecular design. Ultimately, I aim to rekindle an interest in the digital chemistry community toward the nuisance detection issue in molecular medicine. Albeit receiving less attention than retrosynthetic planning,28de novo design29 and others, confidently flagging problematic entities is challenging and relevant. If properly addressed, ML models can impact on the quality of the chemical matter in hand and enable more productive discovery chemistry and biology programs.
Frequent-hitters here, there and everywhere
While privileged structures play an important role in poly- and network pharmacology,30 building automated pipelines that distinguish them from those that unspecifically bind to macromolecular targets has been a long-standing issue in the pharmaceutical industry and academia. Since the seminal work by Schneider and colleagues that coined the term “frequent-hitter” in 2002,12 numerous computational tools have been reported with varying degrees of retrospective accuracy and utility in prospective evaluation studies.Frequent-hitters tend to show up as hits in myriad biochemical or cellular assays due to either promiscuous behaviour12 – which is usually linked to inflated lipophilicity or the so-called “molecular obesity”31 – or interference with the detection method, as in the case of auto-fluorescence, quenchers, chelators, reporter stabilization or non-specific reactivity.8,32,33 Redox cycling molecules can also provide false positive hits and flagging them is not a trivial task even through experimental methods.34 A correct intuition on the classification of individual small molecules as legit or potentially problematic drug prototypes requires many years of hands-on medicinal chemistry experience and familiarization with multiple and diverse cases of each type. Indeed, molecular scaffolds provide the blueprints for promiscuity, but their decoration can largely influence the behaviour of the small molecules in biological assays.35 Harnessing this knowledge through ML models is however not always straightforward. For example, there is a fundamental limitation in the prediction of frequent-hitter behaviour when its source is a metal impurity. Different compound batches may contain different trace amounts of metal, which ultimately influences the outcome of the biological assay. Aggregating data on a sample level and analysing hit rates for specific samples can be helpful, but is a solution not easily generalizable.36 In line with ML routines for other applications, data sources are critical. Correctly preparing them and assigning labels to compounds is a bottleneck of such automated workflows and processes.37 In the absence of sufficient data, extensive experimentation may be required, which is sometimes incompatible to project timelines and needs.
Leveraging corporate data from multiple drug development programs, Schneider and colleagues12 employed a balanced dataset containing 902 structures (479 substructurally diverse frequent hitters and 423 non-frequent hitters), wherein human drugs were assumed as non-frequent hitters. Although a motivated approach, more recent studies20,38 have shown that even approved drugs can lead to false positive readouts, especially at the typical high-throughput screening concentrations (10–30 μM). This highlights that context is key32 and a truly generalizable ML model for nuisance prediction may be out of reach if several important variables, such as assay medium, type of measured endpoint and test concentration are not accounted for. Nonetheless, featurizing all molecules with 120 atom type descriptors led to self-organizing maps that could correctly cluster ∼90% of the molecules according to their labels (Matthews Correlation Coefficient (MCC) = 0.8), and in which hydroxylated aromatic moieties were highly discriminative in the decision process.12 Similar performance was independently obtained with random forests and substructural fingerprints (Morgan 2, 1048 bits) while using highly curated corporate datasets.39 Indeed, Engkvist and colleagues built predictive models to flag compounds according to the screening assay technology of interest. For example, redox cyclers are likely to interfere with AlphaScreens but may not interfere with other detection technologies. This shows the identification of nuisances is a problem in high dimensional space, whose genesis may not only be solely structural but also technological.
Despite the inherent heterogeneity in publicly available datasets, they may also provide sufficient quality information to allow flagging promiscuous entities. As evidenced in the HitDexter tool,40,41 >300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entities in PubChem annotated to >50 targets were used to build two extremely randomized tree models that discriminate between non-promiscuous and promiscuous/highly promiscuous compounds (Fig. 2). Different molecular representations provided MCC values between 0.44–0.61, with Morgan fingerprints (radius 2) being arguably the most effective for discerning patterns. It is worth noting that while promiscuous small molecules are typically undesirable, in some instances that may be a beneficial trait,42–44 provided that target engagement is elicited through a true molecular recognition mechanism. This is the case for several approved drugs that were predicted as promiscuous by HitDexter40 or other data analyses pipelines.45 Those results show that flagging systems serve the purpose of cautioning drug development, but will fare worse at motivating the exclusion of specific small molecules from advanced elaboration. A recent HitDexter version (v3)46 extends its domain of applicability by employing a multilayer perceptron. Most interestingly, the improved models consider different types of data (target and cell-based) for the predictions, which realistically covers all screening scenarios. On a test set comprising dark chemical matter, i.e. molecules that have been extensively tested against unrelated targets without showing any promising activity, HitDexter 3 was able to correctly identify highly promiscuous molecules in cell-based assays (MCC = 0.611), while using Morgan 2 fingerprints.
000 entities in PubChem annotated to >50 targets were used to build two extremely randomized tree models that discriminate between non-promiscuous and promiscuous/highly promiscuous compounds (Fig. 2). Different molecular representations provided MCC values between 0.44–0.61, with Morgan fingerprints (radius 2) being arguably the most effective for discerning patterns. It is worth noting that while promiscuous small molecules are typically undesirable, in some instances that may be a beneficial trait,42–44 provided that target engagement is elicited through a true molecular recognition mechanism. This is the case for several approved drugs that were predicted as promiscuous by HitDexter40 or other data analyses pipelines.45 Those results show that flagging systems serve the purpose of cautioning drug development, but will fare worse at motivating the exclusion of specific small molecules from advanced elaboration. A recent HitDexter version (v3)46 extends its domain of applicability by employing a multilayer perceptron. Most interestingly, the improved models consider different types of data (target and cell-based) for the predictions, which realistically covers all screening scenarios. On a test set comprising dark chemical matter, i.e. molecules that have been extensively tested against unrelated targets without showing any promising activity, HitDexter 3 was able to correctly identify highly promiscuous molecules in cell-based assays (MCC = 0.611), while using Morgan 2 fingerprints.
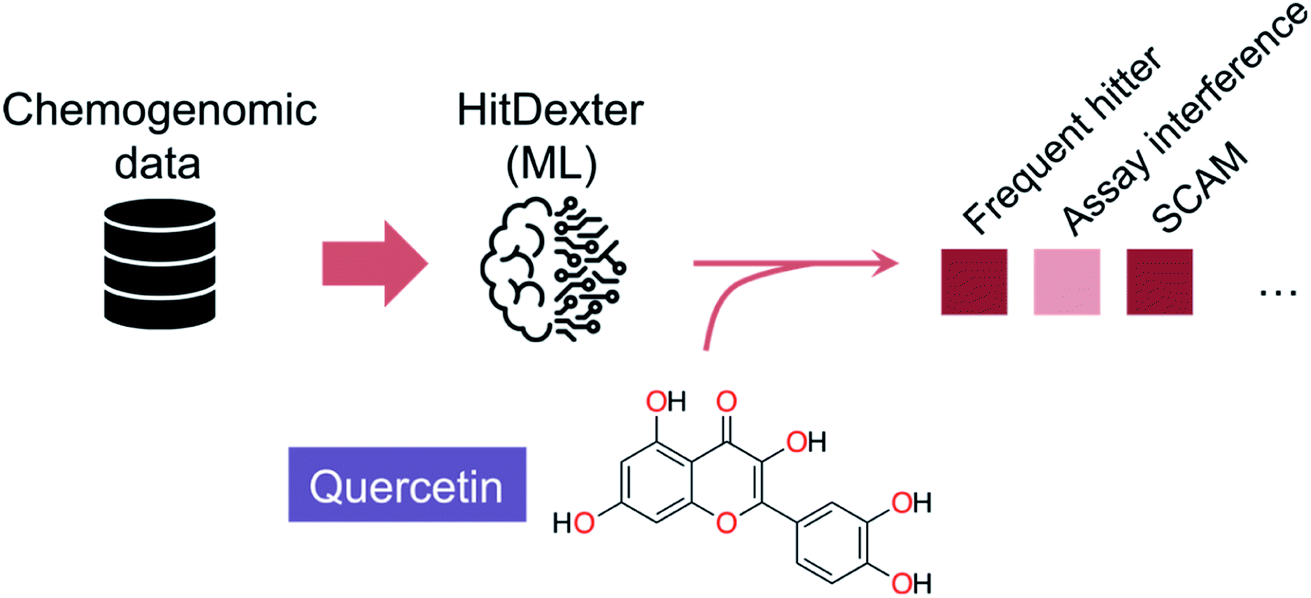 | ||
| Fig. 2 Schematics of the HitDexter tool (https://nerdd.univie.ac.at/hitdexter3/) for the prediction of frequent hitters. HitDexter (v2.0)41 uses chemogenomic information as indexed in PubChem and extremely randomized tree models to distinguish non-promiscuous molecules from promiscuous or highly promiscuous matter – in biochemical assays – that would deserve additional investigations in medicinal chemistry programs. Individual models for different promiscuity-inducing mechanisms are available to the user. A more recent version (v3.0)46 extends the domain of applicability of the tool by leveraging a multilayer perceptron for the prediction of promiscuity in cell-based assays (MCC up to 0.648). It is argued that further model improvements will require consideration of the assay types and conditions, which poses an important roadblock in respect to data availability/scarcity and its homogeneity. | ||
The prediction of promiscuity may de facto illuminate drug design if new intuition is extracted. In a study by Swamidass and colleagues, a convolutional neural network was trained with PubChem data with the goal of predicting promiscuity, and informing chemists on which moieties are correlated with reactivity. However, a sensitivity of 24% suggests that a large portion of reactive molecules remain unnoticed to the deep learning method and that improvements are required to more reliably assist in decision-making.47 In another case, using ECFP4 to describe molecules and screening a battery of learning algorithms it was found that certain motifs could be associated with promiscuity.48 Still, one is also advised to practice caution in such analyses. A major challenge in modern ML is linking model-extracted knowledge from feature importance values with the physical phenomena they indirectly represent. Not too infrequently, there is a disconnect or an experimentally non-verifiable hypothesis that can divert attention or induce falsely generalizable conclusions. With recent advances on the interpretation of ML pipelines (e.g. ref. 49), together with the realization that extracted intuition is biased by both algorithms and molecular representations50 greater emphasis has been put into experimentation as means of verifying data patterns.51 That said, it would be critical to assemble a larger collection of screening compounds with homogeneously generated labels, e.g. accounting for test concentrations. Those datasets will ultimately enable the creation of more accurate and explainable/interpretable models that are currently less accessible.
Aggregating molecules as major source of false positives
Small, colloidally aggregating molecules (SCAMs) do represent the major source of false positive hits in early discovery programs.21 While on-going work by Shoichet and colleagues keeps reminding the community regarding the high prevalence of SCAMs in drugs, natural products, screening molecules and others20 across multiple research programs – including repurposing as SARS-CoV-2 drugs52 – it is apparent that aggregation is still not sufficiently controlled for. This impacts on the amount of available data for ML model building, which remains scarce and relatively heterogeneous in respect to measured endpoints. In particular, the available datasets do present interesting molecular diversity but their annotations (‘aggregator’/‘non-aggregator’) are limited to specific concentration values,20 which is insufficient for holistic predictions. Considering this limiting factor, it is understandable that only a handful of classifiers have been disclosed to date, with each one harnessing different subsets of the available information, having specific assumptions and attempting to answer distinct research questions. Knowledge on the critical aggregation concentration values for different molecules in a standard assay medium would enable a more realistic prediction of the aggregation behaviour in biochemical assays, through regression models, and more effectively inform molecular design.In 2005, Feng et al.53 screened 1030 ‘drug-like’ molecules at 30 and/or 5 μM using both DLS and a detergent-based assay. The collected data enabled the construction of a naïve Bayesian and a recursive partitioning model that on average presented a misclassification rate of 26% for a random set of molecules. A random forest, on the other hand, was more proficient in detecting patterns (11% misclassification rate),53 but with a relatively high class imbalance the results should be examined with caution. The data however has motivated the development of more sophisticated models. Using topological molecular descriptors and support vector machines (SVMs), a model was trained wherein 1319 aggregators and 128325 non-aggregators worked as knowledge base.54 At such high class imbalance (i.e., 1% aggregators:99% non-aggregators), a high retrieval of non-aggregators is expected at random. One may argue that the results of a PubChem and MDDR compound screen with said SVM model are aligned with the background class frequency (97.9% predicted as non-aggregators for PubChem and 1.14% predicted as aggregators for MDDR compounds). This shows the importance of naïve predictions based on simple statistics and baseline models. The analysis does not refute the validity of the SVM since an independent pseudo-prospective screen retrieved 71% of known aggregators (12 out of 17 aggregators, corresponding to 100% of the validation set). One may argue that model improvement is possible/desirable, and that the currently available training data is likely the Achilles heel in SCAM prediction.
Recognizing that caveat and leveraging a recent surge of high quality SCAM data, ChemAGG55 was implemented as a tree-based model (XGBoost). ChemAGG utilizes molecular descriptors calculated from a training set with aggregators and non-aggregators at a 1:2 ratio. Irrespective of the employed features – physicochemical, fingerprints, pharmacophore or fusion representations – the method performed very well on both training and test sets (ROC AUC = 0.987 and 0.976, respectively), and was able to identify patterns widely accepted as predictive of colloidal aggregation, such as high clogP and the number of hydroxyl groups (cf.Fig. 2). In similar fashion, the SCAM detective tool56 was developed with a particular focus on assay conditions and their influence in confounding predictions. Further, the SCAM detective tackles an often-overlooked topic in ML – the quantification of the applicability domain and uncertainty. Ultimately, its goal is providing a better balance between precision and recall relative to its predecessors, while using ECFP6-like fingerprints and data from AmpC β-lactamase and cruzain inhibition assays. Most interestingly, a web application is freely available to the community and it will prompt alerts whenever the query molecules fall outside said domain of applicability, i.e. when predictions are inherently less confident. With identical concerns in mind, we have recently contributed DeepSCAMs.57 It leverages DLS data at a fixed and typical high-throughput screening concentration (30 μM) to predict the aggregating behaviour of small molecules in a defined aqueous buffer. Further, DeepSCAMs gauges the prediction (un)certainty through the label probability. The method employs both Morgan fingerprints (radius 3, bit size 2048) and physicochemical descriptors calculated for 916 molecules in the training set, and a feed-forward neural network architecture with three hidden layers (Fig. 3). Its performance compared favourably against competing methods and in a short survey comprising 15 seasoned medicinal chemists. Overall, it evidenced the ability to detect hidden and nuanced data patterns that ought to be more broadly challenged through experimentation (balanced accuracy = 78%; MCC = 0.58). For a more realistic proof-of-concept, DeepSCAMs was also prospectively queried with 65 diverse small molecules. Experimental DLS evaluation confirmed an accuracy of 80% – higher than the average for the expert panel (ca. 61%) – suggesting that this and similar workflows can serve as auxiliaries in the identification of potential compound liabilities. One may expect that future developments will focus on augmenting training datasets to push the boundaries of what is predictable and unknown under a ML lens.
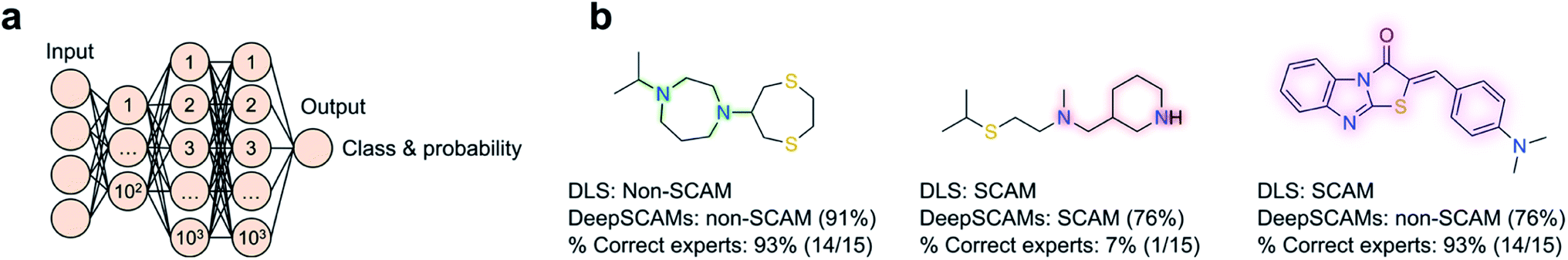 | ||
| Fig. 3 DeepSCAMs for the prediction of small, colloidally aggregating molecules (SCAMs) at a typical high-throughput screening (30 μM) in aqueous buffer. (a) Schematics of the DeepSCAMs architecture; (b) examples of small molecules queried in the prospective evaluation process. The ground truth (DLS endpoint) is predicted with DeepSCAMs together with the label probability in parenthesis. Data shows that computational predictions are competitive with educated guesses made by expert medicinal chemists in industry and academia. DeepSCAMs provides a correct formalization of unwritten rules of intuition; highlighted atoms and bonds show the important fingerprint bits (Morgan radius 3, 2048 bits) for the label prediction. In one case, the ML algorithm missed the planarity that was likely responsible for aggregation and in another was able to detect patterns that remained hidden to chemical intuition. | ||
It is known that SCAMs are agnostic to protein targets and that a multitude of chemotypes have the potential to disrupt pharmacology networks.21 Interestingly, independent studies have reached similar conclusions in regards to the likely prevalence of SCAMs in the literature, either through predictive models or random experimentation.53,55,57 Being estimated at one in every five molecules (∼20%), SCAMs are more common than desirable and likely to confound ML models for bioactivity prediction. Because ChEMBL data is routinely mined to build such models and ruling out entities with aggregation-based bioactivity profiles has historically not been practiced, some bioactivity prediction tools might be flawed. Aggregators in training data might perpetuate the physical traits that lead to false positive hits in screening assays if they are not eliminated in a data curation step.58 While DeepSCAMs, ChemAGG and SCAM detective have shown acceptable utility, it is true we must further expand our knowledge base. In doing so, one will be able to more reliably use in silico tools to flag problematic molecules.
Considering their estimated frequency,53,55,57 SCAMs can be considered ‘outliers’ or ‘anomalies’ from a data science perspective. In fact, seeing frequent hitters in general as ‘anomalies’ has been intuitively adopted in experimental practice, taking into account analyses of hit rates.36 One may thus speculate that SCAMs and other con artists present structural features that set them apart from true hits in a concentration- and assay-dependent manner. A toolkit comprising disparate algorithms is at our disposal to identify anomalies in semi-supervised or unsupervised fashion.59,60 Anomaly detection is an established concept in ML – e.g. for denoising images61 – but has been sparingly exploited in the chemical sciences. In rare examples, it was used to detect unexpected signal fluctuations in the context of analytical chemistry and process control.62,63 To the best of my knowledge, no proof-of-concept has been disclosed in discovery chemistry programs, wherein detecting anomalies/outliers/novelties may find wide applicability in experiment prioritization and as a driver for innovation.
Here, I argue the anomaly detection concept may be extensible to flagging nuisances. An isolation forest was built as a baseline for unsupervised SCAM prediction. I employed the training data and descriptor set used in DeepSCAMs, while assuming the same concentration and buffer dependence for the predicted readout. In short, isolation forests work by recursively partitioning data in a series of trees and separating (isolating) anomalous examples in the training process. The average number of splits required to isolate a training example indicates how different it is for the others. In practice, anomalies require a lower number of splits and are thus more easily separated relative to normal observations.
Considering a contamination of 29% (matching the class imbalance), an optimized model was able to identify only 27 ± 1% of confirmed SCAMs in the training data (repeated analyses: n = 20). Reducing dimensionality to 10 principal components – i.e., accounting for >99% of data variance – did not improve the algorithm's performance significantly (29 ± 0.7%). This indicates that new ways of representing small molecules should be investigated to better discriminate SCAMs/non-SCAMs or that variables controlling (non)-aggregation currently elude our perception. Most interestingly, challenging the ML model with an evaluation set of 31 and 34 SCAMs and non-SCAMs, respectively, led to the correct label identification in 52% and 79% of the cases. The result perfectly reinforces how challenging detecting nuisances is. It also suggests that further exploration of the concept is a reasonable avenue either by improving datasets, representations and/or experimenting different algorithms.
Outlook
Drug target screens are an important step in the discovery of disease modulators, but the high prevalence of nuisance compounds endangers the success rate of many programs. Specifically, interference with the assay technology and confounding mechanisms, such as denaturation of target proteins and phospholipidosis can significantly impact on the direction of early discovery chemistry and steer efforts toward less productive avenues. Here, I have discussed the most prominent use cases and pipelines for the automated identification of problematic compounds. Said technologies may assist in decision-making and eventually be integrated in fully automated pipelines comprising design–make–test–analyse cycles.It is clear that ML has the potential to facilitate chemical discoveries at unprecedented pace, yet some topics – as the one focused on here – remain underexplored by the cheminformatics and data science communities relative to others. This does not mean a secondary role or lower importance, but highlights there are unsolved challenges that must be addressed in the first place. One of such challenges is the need for superior datasets and benchmarks in terms of examples with better link to experimental context and measured endpoints. Reporting high quality data, even if negative, will be key to start witnessing the implementation of improved ML models. While supervised ML has been the go-to approach, I have argued that unsupervised and in particular anomaly detection algorithms may provide a fresh and innovative vantage point onto data and discovery chemistry. I envisage that the automated mapping of nuisances, together with uncertainty estimation and integration with experimental context will ultimately enable the design of quality matter to interrogate biology. If efficient, those ML pipelines can lower the likelihood of attrition in translational studies.
Data availability
All code and data are available through GitHub. A link is available at the end of the manuscript.Conflicts of interest
T. R. is a co-founder and shareholder of TargTex S.A.Acknowledgements
T. R. acknowledges FCT Portugal (CEECIND/00684/2018) for financial support and the reviewers for insightful comments. All code and datasets are available at https://github.com/DigiChem/DigitalDiscovery_Perspective.References
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 85–90 CrossRef CAS PubMed.
- S. Talebian, T. Rodrigues, J. das Neves, B. Sarmento, R. Langer and J. Conde, ACS Nano, 2021, 15, 15840–15942 CrossRef PubMed.
- M. J. Keiser, V. Setola, J. J. Irwin, C. Laggner, A. Abbas, S. J. Hufeisen, N. H. Jensen, M. B. Kuijer, R. C. Matos, T. B. Tran, R. Whaley, R. A. Glennon, J. Hert, K. L. H. Thomas, D. D. Edwards, B. K. Shoichet and B. L. Roth, Nature, 2009, 462, 175–181 CrossRef CAS PubMed.
- D. Reker, T. Rodrigues, P. Schneider and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4067–4072 CrossRef CAS PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- M. A. Lill, Drug Discovery Today, 2007, 12, 1013–1017 CrossRef CAS PubMed.
- W. P. Walters and M. Namchuk, Nat. Rev. Drug Discovery, 2003, 2, 259–266 CrossRef CAS PubMed.
- A. Jadhav, R. S. Ferreira, C. Klumpp, B. T. Mott, C. P. Austin, J. Inglese, C. J. Thomas, D. J. Maloney, B. K. Shoichet and A. Simeonov, J. Med. Chem., 2010, 53, 37–51 CrossRef CAS PubMed.
- B. Y. Feng and B. K. Shoichet, Nat. Protoc., 2006, 1, 550–553 CrossRef CAS PubMed.
- A. P. Hill and R. J. Young, Drug Discovery Today, 2010, 15, 648–655 CrossRef CAS PubMed.
- M.-Q. Zhang and B. Wilkinson, Curr. Opin. Biotechnol., 2007, 18, 478–488 CrossRef CAS PubMed.
- O. Roche, P. Schneider, J. Zuegge, W. Guba, M. Kansy, A. Alanine, K. Bleicher, F. Danel, E.-M. Gutknecht, M. Rogers-Evans, W. Neidhart, H. Stalder, M. Dillon, E. Sjögren, N. Fotouhi, P. Gillespie, R. Goodnow, W. Harris, P. Jones, M. Taniguchi, S. Tsujii, W. v. d. Saal, G. Zimmermann and G. Schneider, J. Med. Chem., 2002, 45, 137–142 CrossRef CAS PubMed.
- T. A. Tummino, V. V. Rezelj, B. Fischer, A. Fischer, M. J. O'Meara, B. Monel, T. Vallet, K. M. White, Z. Zhang, A. Alon, H. Schadt, H. R. O'Donnell, J. Lyu, R. Rosales, B. L. McGovern, R. Rathnasinghe, S. Jangra, M. Schotsaert, J.-R. Galarneau, N. J. Krogan, L. Urban, K. M. Shokat, A. C. Kruse, A. García-Sastre, O. Schwartz, F. Moretti, M. Vignuzzi, F. Pognan and B. K. Shoichet, Science, 2021, 373, 541–547 CrossRef CAS PubMed.
- M. Chatzopoulou, K. S. Madden, L. J. Bromhead, C. Greaves, T. J. Cogswell, S. d. S. Pinto, S. R. G. Galan, I. Georgiou, M. S. Kennedy, A. Kennett, G. Apps, A. J. Russell and G. M. Wynne, ACS Med. Chem. Lett., 2022, 13, 262–270 CrossRef CAS PubMed.
- J. C. Hermann, Y. Chen, C. Wartchow, J. Menke, L. Gao, S. K. Gleason, N.-E. Haynes, N. Scott, A. Petersen, S. Gabriel, B. Vu, K. M. George, A. Narayanan, S. H. Li, H. Qian, N. Beatini, L. Niu and Q.-F. Gan, ACS Med. Chem. Lett., 2013, 4, 197–200 CrossRef CAS PubMed.
- R. S. Ferreira, C. Bryant, K. K. H. Ang, J. H. McKerrow, B. K. Shoichet and A. R. Renslo, J. Med. Chem., 2009, 52, 5005–5008 CrossRef CAS PubMed.
- K. E. D. Coan, D. A. Maltby, A. L. Burlingame and B. K. Shoichet, J. Med. Chem., 2009, 52, 2067–2075 CrossRef CAS PubMed.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- S. L. McGovern, B. T. Helfand, B. Feng and B. K. Shoichet, J. Med. Chem., 2003, 46, 4265–4272 CrossRef CAS PubMed.
- J. J. Irwin, D. Duan, H. Torosyan, A. K. Doak, K. T. Ziebart, T. Sterling, G. Tumanian and B. K. Shoichet, J. Med. Chem., 2015, 58, 7076–7087 CrossRef CAS PubMed.
- D. Reker, G. J. L. Bernardes and T. Rodrigues, Nat. Chem., 2019, 11, 402–418 CrossRef CAS PubMed.
- S. R. LaPlante, R. Carson, J. Gillard, N. Aubry, R. Coulombe, S. Bordeleau, P. Bonneau, M. Little, J. O'Meara and P. L. Beaulieu, J. Med. Chem., 2013, 56, 5142–5150 CrossRef CAS PubMed.
- A. M. Giannetti, B. D. Koch and M. F. Browner, J. Med. Chem., 2008, 51, 574–580 CrossRef CAS PubMed.
- M. F. Sassano, A. K. Doak, B. L. Roth and B. K. Shoichet, J. Med. Chem., 2013, 56, 2406–2414 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- W. P. Walters, A. A. Murcko and M. A. Murcko, Curr. Opin. Chem. Biol., 1999, 3, 384–387 CrossRef CAS PubMed.
- W. P. Walters, M. T. Stahl and M. A. Murcko, Drug Discovery Today, 1998, 3, 160–178 CrossRef CAS.
- M. H. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- L. Yet, Privileged Structures in Drug Discovery: Medicinal Chemistry and Synthesis, John Wiley & Sons, Inc., Hoboken, NJ, 2018 Search PubMed.
- M. M. Hann, MedChemComm, 2011, 2, 349–355 RSC.
- D. Ghosh, U. Koch, K. Hadian, M. Sattler and I. V. Tetko, J. Chem. Inf. Model., 2018, 58, 933–942 CrossRef CAS PubMed.
- J. L. Dahlin, D. S. Auld, I. Rothenaigner, S. Haney, J. Z. Sexton, J. W. M. Nissink, J. Walsh, J. A. Lee, J. M. Strelow, F. S. Willard, L. Ferrins, J. B. Baell, M. A. Walters, B. K. Hua, K. Hadian and B. K. Wagner, Cell Chem. Biol., 2021, 28, 356–370 CrossRef CAS PubMed.
- M. Tarnowski, A. Barozet, C. Johansson, P.-O. Eriksson, O. Engkvist, J. Walsh and J. W. M. Nissink, Assay Drug Dev. Technol., 2018, 16, 171–191 CrossRef CAS PubMed.
- J. J. Yang, O. Ursu, C. A. Lipinski, L. A. Sklar, T. I. Oprea and C. G. Bologa, J. Cheminf., 2016, 8, 29 Search PubMed.
- J. W. M. Nissink and S. Blackburn, Future Med. Chem., 2014, 6, 1113–1126 CrossRef CAS PubMed.
- T. Rodrigues, Drug Discovery Today: Technol., 2019, 32–33, 3–8 CrossRef PubMed.
- J. Seidler, S. L. McGovern, T. N. Doman and B. K. Shoichet, J. Med. Chem., 2003, 46, 4477–4486 CrossRef CAS PubMed.
- L. David, J. Walsh, N. Sturm, I. Feierberg, J. W. M. Nissink, H. Chen, J. r. Bajorath and O. Engkvist, ChemMedChem, 2019, 14, 1795–1802 CrossRef CAS PubMed.
- C. Stork, J. Wagner, N.-O. Friedrich, C. d. B. Kops, M. Sícho and J. Kirchmair, ChemMedChem, 2018, 13, 564–571 CrossRef CAS PubMed.
- C. Stork, Y. Chen, M. Šícho and J. Kirchmair, J. Chem. Inf. Model., 2019, 59, 1030–1043 CrossRef CAS PubMed.
- A. L. Hopkins, Nat. Chem. Biol., 2008, 4, 682–690 CrossRef CAS PubMed.
- C. Feldmann, D. Yonchev and J. r. Bajorath, Biomolecules, 2020, 10, 1605 CrossRef CAS PubMed.
- C. Feldmann, D. Yonchev, D. Stumpfe and J. r. Bajorath, Mol. Pharmaceutics, 2020, 17, 4652–4666 CrossRef CAS PubMed.
- Y. Hu and J. Bajorath, F1000Research, 2014, 3, 218 Search PubMed.
- C. Stork, N. Mathai and J. Kirchmair, Artif. Intell. Life Sci., 2021, 1, 100007 Search PubMed.
- M. K. Matlock, T. B. Hughes, J. L. Dahlin and S. J. Swamidass, J. Chem. Inf. Model., 2018, 58, 1483–1500 CrossRef CAS PubMed.
- T. Blaschke, F. Miljkovic and J. r. Bajorath, ACS Omega, 2019, 4, 6883–6890 CrossRef CAS.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Sci. Adv., 2021, 7, eabe4166 CrossRef PubMed.
- R. P. Sheridan, J. Chem. Inf. Model., 2019, 59, 1324–1337 CrossRef CAS PubMed.
- D. Reker, E. A. Hoyt, G. J. L. Bernardes and T. Rodrigues, Cell Rep. Phys. Sci., 2020, 1, 100247 CrossRef.
- H. R. O'Donnell, T. A. Tummino, C. Bardine, C. S. Craik and B. K. Shoichet, J. Med. Chem., 2021, 64, 17530–17539 CrossRef PubMed.
- B. Y. Feng, A. Shelat, T. N. Doman, R. K. Guy and B. K. Shoichet, Nat. Chem. Biol., 2005, 1, 146–148 CrossRef CAS PubMed.
- H. Rao, Z. Li, X. Li, X. Ma, C. Ung, H. Li, X. Liu and Y. Chen, J. Comput. Chem., 2010, 31, 752–763 CAS.
- Z.-Y. Yang, Z.-J. Yang, J. Dong, L.-L. Wang, L.-X. Zhang, J.-J. Ding, X.-Q. Ding, A.-P. Lu, T.-J. Hou and D.-S. Cao, J. Chem. Inf. Model., 2019, 59, 3714–3726 CrossRef CAS PubMed.
- V. M. Alves, S. J. Capuzzi, R. C. Braga, D. Korn, J. E. Hochuli, K. H. Bowler, A. Yasgar, G. Rai, A. Simeonov, E. N. Muratov, A. V. Zakharov and A. Tropsha, J. Chem. Inf. Model., 2020, 60, 4056–4063 CrossRef CAS PubMed.
- K. Lee, A. Yang, Y.-C. Lin, D. Reker, G. J. L. Bernardes and T. Rodrigues, Cell Rep. Phys. Sci., 2021, 2, 100573 CrossRef.
- L. David, J. Arús-Pous, J. Karlsson, O. Engkvist, E. J. Bjerrum, T. Kogej, J. M. Kriegl, B. Beck and H. Chen, Front. Pharmacol., 2019, 10, 1–16 CrossRef PubMed.
- G. Pang, C. Shen, L. Cao and A. v. d. Hengel, 2020, arXiv:2007.02500v02503.
- R. Chalapathy and S. Chawla, 2019, arXiv:1901.03407v03402.
- L. Beggel, M. Pfeiffer and B. Bischl, 2019, arXiv:1901.06355v06351.
- I. Monroy, G. Escudero and M. Graells, Comput.-Aided Chem. Eng., 2009, 26, 255–260 CAS.
- W. J. Egan and S. L. Morgan, Anal. Chem., 1998, 70, 2372–2379 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |