
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMPSM-DTI: prediction of drug–target interaction via machine learning based on the chemical structure and protein sequence†
Yayuan
Peng
 ,
Jiye
Wang
,
Zengrui
Wu
*,
Lulu
Zheng
,
Biting
Wang
,
Guixia
Liu
,
Weihua
Li
and
Yun
Tang
*
,
Jiye
Wang
,
Zengrui
Wu
*,
Lulu
Zheng
,
Biting
Wang
,
Guixia
Liu
,
Weihua
Li
and
Yun
Tang
*
Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: ytang234@ecust.edu.cn; zengruiwu@ecust.edu.cn
First published on 25th January 2022
Abstract
Drug–target interaction (DTI) plays a central role in drug discovery. How to predict DTI quickly and accurately is a key issue. Traditional structure-based and ligand-based methods have some inherent deficiencies. Hence, it is necessary to develop a new method for DTI prediction that does not rely on crystal structures of protein targets or quantity and diversity of ligands. In this study, we collected 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 898 DTIs with kd values from ChEMBL 27 to develop a prediction method. Through data standardization, SMOTE sampling and pipeline techniques, among 30 models the Morgan-PSSM-SVM model (MPSM-DTI) was demonstrated as the best one with ten-fold cross-validation (F1 = 85.55 ± 0.46%, R = 84.89 ± 0.62% and P = 86.24 ± 0.81%) and test set validation (F1 = 85.11%, R = 84.34% and P = 85.90%). The results in two external validation sets indicated that the MPSM-DTI model had satisfactory generalization capability and could be used in target prediction for new compounds. Specifically, the F1, P and R values were 83.27%, 85.21% and 81.41% in external validation set 1 and 86.45%, 87.50% and 85.42% in external validation set 2. Via the latest literature evidence, we collected 100 new DTIs of eight GPCR targets to prove that MPSM-DTI could predict compounds for protein targets without known ligands and crystal structures. Compared with other DTI prediction methods, our method reached considerable accuracy and addressed the dilemma of DTI prediction for brand new protein targets. Furthermore, we proposed the pipeline encapsulation technique, which would avoid data leak and improve generalization ability of the model. The source code of the method is available at https://github.com/pengyayuan/MPSM-DTI.
898 DTIs with kd values from ChEMBL 27 to develop a prediction method. Through data standardization, SMOTE sampling and pipeline techniques, among 30 models the Morgan-PSSM-SVM model (MPSM-DTI) was demonstrated as the best one with ten-fold cross-validation (F1 = 85.55 ± 0.46%, R = 84.89 ± 0.62% and P = 86.24 ± 0.81%) and test set validation (F1 = 85.11%, R = 84.34% and P = 85.90%). The results in two external validation sets indicated that the MPSM-DTI model had satisfactory generalization capability and could be used in target prediction for new compounds. Specifically, the F1, P and R values were 83.27%, 85.21% and 81.41% in external validation set 1 and 86.45%, 87.50% and 85.42% in external validation set 2. Via the latest literature evidence, we collected 100 new DTIs of eight GPCR targets to prove that MPSM-DTI could predict compounds for protein targets without known ligands and crystal structures. Compared with other DTI prediction methods, our method reached considerable accuracy and addressed the dilemma of DTI prediction for brand new protein targets. Furthermore, we proposed the pipeline encapsulation technique, which would avoid data leak and improve generalization ability of the model. The source code of the method is available at https://github.com/pengyayuan/MPSM-DTI.
Introduction
Drug–target interaction (DTI) plays a central role in drug discovery. For a known target, DTI could discover new drugs binding to the target; whereas for a known drug, DTI could identify its new targets and new usages. However, experimental determination of DTI is costly and time-consuming. A variety of computational methods are hence developed for DTI prediction, and how to predict DTI quickly and accurately becomes a key issue.The traditional methods for DTI prediction are mainly divided into two categories:1 structure-based and ligand-based. In structure-based methods, molecular docking tools are widely used to find new ligands for a protein with a three-dimensional (3D) structure, or identify new protein targets with 3D structures for a known drug. In ligand-based methods, pharmacophore search and similarity search in 3D shapes, substructures and physicochemical properties are usually employed.2–5 Though these traditional methods have succeeded in many cases, there are still some inherent deficiencies. For structure-based methods, 3D structures of targets are a must. However, most of the potential targets have no known 3D structures yet, for example, only 60 GPCRs (G protein-coupled receptors) have been determined structurally among the total 800 members, which means that the structure-based methods could not be utilized on those targets without 3D structures directly.6,7 For ligand-based methods, it is impossible to search new ligands for those targets without known ligands. Therefore, it is urgent to develop novel methods for DTI prediction.8
Recently, a new type of method, named network-based methods, were developed for DTI prediction. These new methods do not rely on the 3D structures of targets. Instead, they utilize a large number of known DTIs and multiple chemogenomic data to construct a DTI network for prediction of potential DTIs. For example, Wu et al. developed network-based inference methods SDTNBI and bSDTNBI to predict new DTIs by introducing substructure information of ligands to a known DTI network, which could be applied in target prediction for new chemical entities outside the DTI network.9–12 However, these methods could not be used in finding potential ligands for new targets outside the network.
Meanwhile, machine learning methods are also used in DTI prediction. For example, Lee et al. extracted local residue patterns of protein sequences to predict novel DTIs using convolution neural network,13 which proved that protein sequences could offer useful information in DTI prediction. Mahmud et al. developed the iDTi-CSsmoteB webserver to predict DTIs based on PubChem fingerprints and various protein sequence features using XGBoost and oversampling techniques.14 However, the data quality of the above-mentioned methods was not satisfactory because the negative data were selected arbitrarily. Several other studies also did so.15–17 Some of them used random non-positive DTIs to act as negative samples. However, non-positive DTIs are not definitely negative because they are just not validated yet. Some of them might be positive after validation. Therefore, it is significant to construct predictive models using high-quality data.
In this study, we developed a machine learning model for prediction of DTIs using chemical structures and protein sequences as features. The pipeline technique was used to encapsulate the feature data standardization, SMOTE sampling process and machine learning estimator, which would avoid overfitting and improve model generalization. The whole workflow is shown in Fig. 1. In brief, over 40000 DTIs with dissociation constant (kd) values were collected from various sources. Five types of molecular fingerprints and descriptors were calculated by PaDEL-Descriptor and RDKit. The protein sequence features were extracted through PSI-Blast and the POSSUM toolkit. 30 prediction models were built for DTI prediction by 5 machine learning methods and 6 feature representation approaches, among which the Morgan-PSSM-SVM model (MPSM-DTI) was validated as the best one. In case studies, the MPSM-DTI model exhibited satisfactory capability in DTI prediction.
 | ||
| Fig. 1 Overview of the workflow to construct the prediction model, including data preparation, feature extraction and model construction. | ||
Materials and methods
Data collection and preparation
The original DTI data were extracted with kd values from ChEMBL 27 (released in May 2020).18kd = 10 μM was set as the threshold to identify whether the interactions were positive or negative.19–21 When kd ≤ 10 μM, the interactions were set as positive; whereas if kd > 10 μM, the interactions were set as negative. Here drugs refer to any chemicals with bioactivity data including approved drugs.The SMILES of all drugs were imported to Pipeline Pilot Client (version 2017 R2) to clean chemicals with wrong structures, followed by a series of steps, including removing salt and inorganics, standardizing SMILES and wiping out molecules with molecular weight >1200 Da or <200 Da. Duplicated data were then removed. To ensure clean data, the ambiguous DTIs, the interactions being not only positive but also negative, were removed. For proteins, if the protein sequences were not available in UniProt, the corresponding interactions were deleted, too. After that, the whole data were divided into a training set and a test set in a ratio of 8:2.
Data of external validation set 1 were gathered from BindingDB (accessed in June 2020)22 and IUPHAR/BPS Guide to PHARMACOLOGY (accessed in June 2020).23 All data were prepared in the same way as those in the training set and test set. Duplicates with those in the training set and test set were removed, to keep external validation set 1 independent.
To evaluate the capability of predicting targets for new compounds, external validation set 2 was prepared, in which the DTIs were not duplicated with those in the training set and test set, but the compounds were brand new. Furthermore, to verify whether the model can predict compounds exactly for new targets, the experimentally validated DTIs were gathered from a list of recent publications, in which the proteins were completely new compared with proteins in the training set and test set.
Chemical representation
Five types of molecular fingerprints, including Substructure (FP4), MACCS, PubChem, Klekota-Roth (KR), and Morgan, as well as molecular descriptors (Des) were used to depict the features of drugs, respectively. The FP4, MACCS, PubChem and KR fingerprints were calculated by PaDEL_Descriptor (version 2.2.1).24 The Morgan (1024 bit) fingerprint and molecular descriptors were computed through RDKit, the open source cheminformatics Python package.Protein target representation
The position specific scoring matrix (PSSM) was employed to describe protein features in DTI pairs. To generate the PSSM of a query protein, there are three major steps. Firstly, the PSI-Blast procedure from Blast of National Center for Biotechnology Information (https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) was downloaded and configured. Simultaneously, Blast database and SwissProt were downloaded via ftp from ftp://ftp.ncbi.nlm.nih.gov/blast/db/. Secondly, all query protein sequences in FASTA format were obtained from the UniProt database. Thirdly, the PSSM of each query protein was generated separately with the parameters of PSI-Blast e-value = 0.001 and num_iterations = 3. A PSSM for a query protein is an L × 20 matrix P = {Pij: i = 1,2,3…L and j = 1,2,3…20}, where L is the length of the protein sequence and j stands for the 20 different amino acids. While Pij means the score of the ith position iterated by the jth amino acid, a larger Pij means a higher conserved position. The PSSM cannot be used directly for proteins with different L values. Therefore, we used a bioinformatics toolkit POSSUM to transform PSSM from an L × 20 matrix to a 400 dimensional vector.25
PCA analysis
Principal component analysis (PCA) was applied to decompose the chemical and protein features to a lower dimensional space by the PCA module of scikit-learn. The parameter “n_components” was set as “3”, which indicated that 3 components were kept after PCA decomposition.Model construction
Five machine learning methods were used to build models for DTI prediction, including decision tree (DT), bagging, gradient boost decision tree (GBDT), k-nearest neighbors (k-NN), and support vector machine (SVM). All these methods were realized by scikit-learn (version 0.23), a prevalent open source Python module for machine learning built on top of SciPy. The detailed description of these methods is presented in the ESI.†Pipeline building
Information leakage, a process that knowledge leaks from the test data into the trained model in cross-validation, usually shows excellent cross-validation results but poor generalization ability. To avoid this, pipeline, a module of scikit-learn, was used to chain multiple estimators into one, including feature data standardization, SMOTE sampling and aforementioned machine learning estimators. Most importantly, pipeline is an encapsulated estimator, which could be introduced into GridSearchCV to search over all parameters. Because of encapsulation, pipeline provides a convenient and safe approach to transform and resample training data.Grid search for hyper-parameters
Usually, parameter optimization would provide the best generalization of a model. However, it is difficult for most data sets and estimators to tune the hyper-parameters. In this study, we used GridSearchCV to optimize parameters for each machine learning method and the k value of the SMOTE sampling approach. GridSearchCV can consider all parameter combinations exhaustively with a given parameter grid. In this way, all possible values for different parameters would be explored, which could provide all models with optimal parameters.Performance assessment of models
In order to evaluate different models, ten-fold cross-validation, test set validation and external validation were performed successively. In ten-fold cross-validation, the DTIs in the training set were divided into ten parts randomly and equally. One part served as the validation set, while the remaining nine parts were used to build the model and predict the validation set. This process was repeated ten times to allow each part be validated in turns. Through ten-fold cross-validation, different models with different parameters would be assessed and then the optimal models would be obtained. In addition to ten-fold cross-validation, we also applied the test set to assess the models by splitting the training set and test set in a ratio of 8:2.
The external validation set is independent of the training set and test set. The external validation data were divided into four different groups to assess the ability of classifiers to predict new DTIs for new compounds and new proteins. The statistical numbers of DTI samples are shown in Table 1.
| Data set | N d | N T | N P | N N | Total samples | Data sources |
|---|---|---|---|---|---|---|
| a N d: number of drugs; NT: number of targets; NP: number of positive DTI samples; NN: number of negative DTI samples. | ||||||
| Training set | 7445 | 888 | 13858 |
18859 |
32717 |
ChEMBL |
| Test set | 2268 | 720 | 3641 | 4719 | 8180 | |
| External validation set 1 | 987 | 625 | 1152 | 869 | 2021 | BindingDB, IUPHAR/BPS guide to PHARMACOLOGY |
| External validation set 2 | 853 | 604 | 1014 | 818 | 1832 | |
The following performance metrics were used as evaluation indicators: F1, recall (R) and precision (P) to assess each prediction model. See below equations:
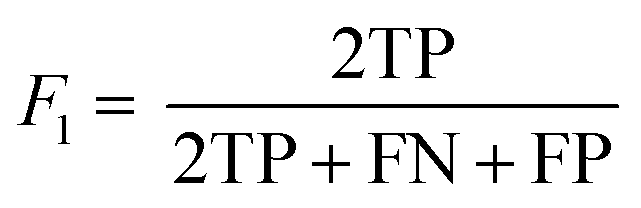 | (1) |
 | (2) |
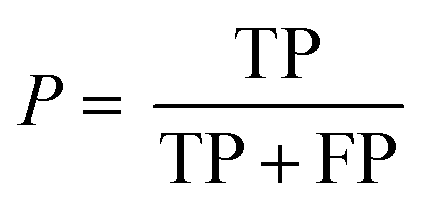 | (3) |
Results
Data collection and analysis
From ChEMBL 27, we collected 40898 DTI samples with kd values in total, among which the numbers of positive and negative DTIs were 17320 and 23578, respectively. The ratio of negative and positive data is 1.36:1 approximately. Obviously, the data set is not balanced to some degree. All DTIs were then split into a training set and a test set randomly in a ratio of 8:2. In the training set, there were 7445 drugs, 888 targets, 13858 positive interactions and 18859 negative interactions. The test set contained 2268 drugs, 720 targets, 3641 positive interactions and 4719 negative interactions. To better evaluate the model generalization, we gathered 2021 DTIs with kd values from BindingDB and IUPHAR/BPS Guide to PHARMACOLOGY to serve as external validation set 1, which contained 1152 positives and 869 negatives. In external validation set 2, there were 1832 DTIs with kd values, among which 1014 ones were positive and 818 were negative. The details of all the data sets are summarized in Table 1.
In addition, 100 DTIs collected from the latest literature were used in the case study and summarized in Table S1,† which enclosed eight functional GPCR (G-protein coupled receptor) proteins covering some principal biological pathways and complex diseases.
Model building and pipeline to avoid data leaking
By applying molecular descriptors (Des) and five types of molecular fingerprints (FP4, KR, MACCS, Morgan, and PubChem) to represent the features of compounds and PSSM to decipher the features of protein targets, five machine learning methods (Bagging, DT, GBDT, k-NN, and SVM) were used to construct models, which resulted in 30 different models. After feature data standardization, SMOTE sampling and grid search, the optimal hyper-parameters were obtained for each of the 30 models by ten-fold cross validation. All optimal hyper-parameters are summarized in Table S3.†Through a pipeline approach, the feature data standardization, SMOTE sampling process and machine learning estimator were encapsulated as a unitive estimator. The superiority of the pipeline approach is to avoid data leaking. Fig. 2 shows the ΔF1, ΔP and ΔR values of 30 models with and without pipeline encapsulation. ΔF1, ΔP and ΔR stand for the differences of F1, P and R between test set validation and ten-fold cross-validation. From Fig. 2, we could see that all the ΔF1, ΔP and ΔR values of models without the pipeline strategy were much larger than those with pipeline. The larger ΔF1, ΔP and ΔR values reflected that the models could achieve better performance in ten-fold cross-validation but poor performance in test set validation, i.e. overfitting. Therefore, the pipeline strategy is effective in avoiding data leaking and overfitting.
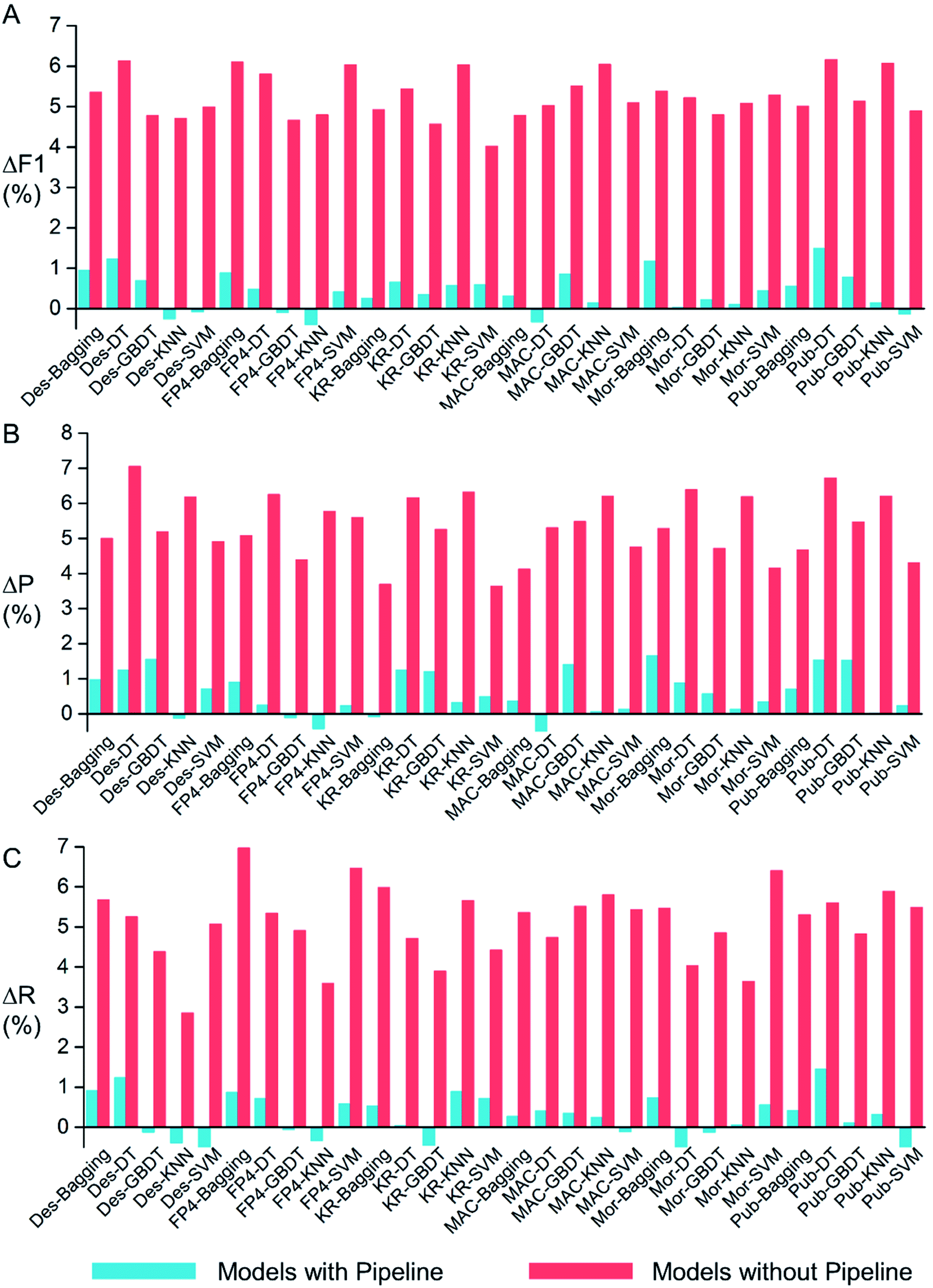 | ||
| Fig. 2 Comparison of (A) ΔF1, (B) ΔP and (C) ΔR values among 30 models by two approaches. ΔF1 = F1,10-fold cross validation − F1,test set validation, ΔP = P10-fold cross validation − Ptest set validation, and ΔR = R10-fold cross validation − Rtest set validation. Turquoise: models with the pipeline process; pale red: models without pipeline encapsulation. | ||
Performance evaluation of the models
Fig. 3 shows the ten-fold cross-validation results of the 30 models. The detailed results are listed in Table S4,† in which the six models highlighted in bold were the best ones, each from one of the six types of chemical features. From Fig. 3, we could see that the F1, R and P scores of major machine learning methods were greater than 80% and for some excellent models the scores were close to 85%, except those built by DT. The ensemble methods, including Bagging and GBDT, all performed better than DT. Furthermore, GBDT and SVM outperformed Bagging, DT and k-NN. It is clear that the R and P scores of DT and SVM were balanced relatively. In Bagging and GBDT models, the P scores were greater than the R scores. In contrast, in k-NN models the P scores were much lower than the R scores.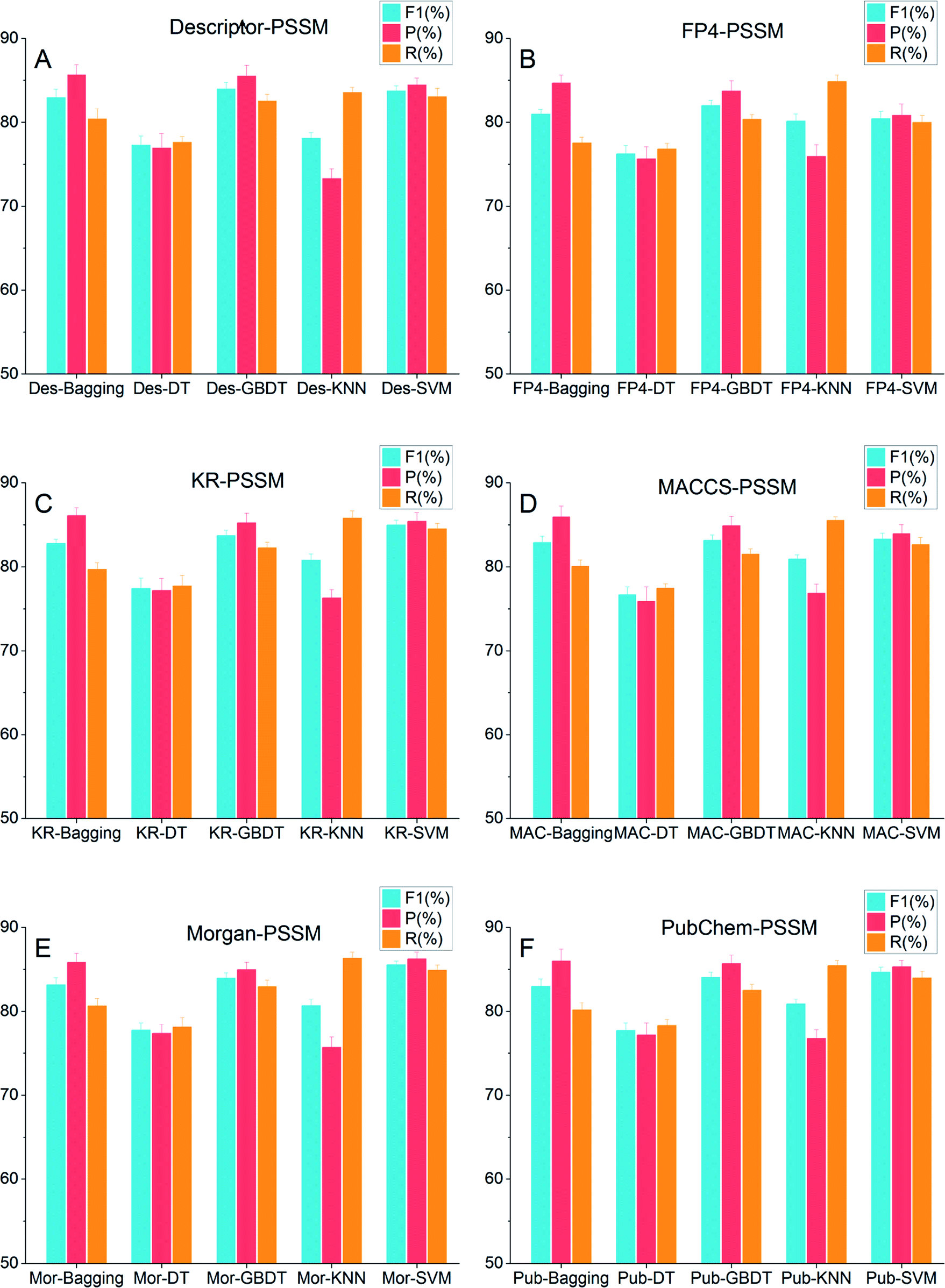 | ||
| Fig. 3 Comparison of F1, P and R values in ten-fold cross-validation among 30 models with different DTI features, (A) descriptor-PSSM, (B) FP4-PSSM, (C) KR-PSSM, (D) MACCS-PSSM, (E) Morgan-PSSM, and (F) PubChem-PSSM. F1: turquoise; P: pale red; R: dark orange. | ||
As for the chemical features, from Fig. 3 we found that models with FP4-PSSM performed worse than the others, which indicated that FP4 could not represent the features of chemical structures well. Meanwhile, models with Descriptor-PSSM, KR-PSSM, MACCS-PSSM, Morgan-PSSM, and PubChem-PSSM exhibited comparable performance, and models with Morgan-PSSM outperformed slightly. Especially, the Morgan-PSSM-SVM model (MPSM-DTI) performed the best among all 30 models, with ten-fold cross-validation results as F1 = 85.55 ± 0.46%, R = 84.89 ± 0.62% and P = 86.24 ± 0.81%.
Besides ten-fold cross-validation, test set validation was also employed for the comparison of different models. Fig. 4 displays the test set validation results. The detailed values of evaluation indicators for all 30 models are shown in Table S5.† The results of test set validation were similar to those of ten-fold cross-validation. The F1, R and P scores of test set validation for most models also exceeded 80%. From Fig. 4, we could see that all SVM models performed better than those of Bagging, DT, GBDT, and k-NN. Furthermore, MPSM-DTI was also tested as the best model among all 30 models, with test set validation results as F1 = 85.11%, R = 84.34% and P = 85.90%.
 | ||
| Fig. 4 Comparison of F1, P and R values in test set validation among 30 models with different DTI features, (A) descriptor-PSSM, (B) FP4-PSSM, (C) KR-PSSM, (D) MACCS-PSSM, (E) Morgan-PSSM, and (F) PubChem-PSSM. F1: turquoise; P: dark cyan; R: pale red. | ||
Evaluation of model generalization capability
After evaluation by ten-fold cross-validation and test set validation, MPSM-DTI (namely the Morgan-PSSM-SVM model) was selected as the best prediction model among the 30 models. To further assess the generalization capability of the model, two additional external data sets were utilized.Before the assessment, to see if the external data sets were located within the applicability domain of the model, the PCA analysis was performed to reduce the dimensionality of the chemical and protein features on all four data sets into a 3D chemical space. As shown in Fig. 5A, it is easy to see that the distributions of features in the four data sets were covered well in the 3D space after PCA dimensionality reduction. Fig. 5A indicates that the two external data sets were suitable for assessment of the generalization capability of the model. The evaluation results of the four data sets via the MPSM-DTI model are shown in Fig. 5B and Table 2. From Fig. 5B, we could see that the external validation set 1 and external validation set 2 achieved quite similar results in comparison with ten-fold cross-validation and test set validation. Specifically, for external validation set 1, the F1, P and R scores were 83.27%, 85.21% and 81.41%, respectively, while for external validation set 2, the three evaluation indicators exceeded external data set 1 to some extent with F1 = 86.45%, P = 87.50% and R = 85.42%.
 | ||
| Fig. 5 (A) Scatter diagram of data among the training set, test set, external validation set 1 and external validation set 2 in 3D PCA analysis with Morgan-PSSM features. (B) Comparison of F1, P and R scores in ten-fold cross-validation, test set validation, external validation 1 and external validation 2. | ||
| Evaluation indicators | Ten-fold cross validation | Test set validation | External validation set 1 | External validation set 2 |
|---|---|---|---|---|
| F 1 | 85.55 ± 0.46 | 85.11 | 83.27 | 86.45 |
| P | 86.24 ± 0.81 | 85.90 | 85.21 | 87.50 |
| R | 84.89 ± 0.62 | 84.34 | 81.41 | 85.42 |
From the above analysis, it is obvious that the MPSM-DTI model achieved high-quality generalization capability in two different external sets. Moreover, from the results of external validation set 2, we could see that the model obtained ideal results on known DTIs with new compounds.
Case study
It is usually difficult to identify potential ligands for new targets, especially for those targets without crystal structures and known ligands. To show the capability of the MPSM-DTI model in predicting ligands for new targets, we collected 100 experimentally validated DTIs from the latest literature. The 100 known DTI data contained 100 different compounds with eight new targets, including DHCR7, HTR1F, LTB4R, CYSLTR2, GRIK3, GPER1, PTGIR, and SIRP5. The eight targets belong to the GPCR superfamily, and have no crystal structures yet.Table 3 briefly shows the predictive results of the MPSM-DTI model for the eight new GPCR targets. It is straightforward to see that the MPSM-DTI model obtained a considerable recall rate with 90 correct predictions among all 100 experimentally validated DTIs. Fig. 6 illustrates the results clearly with DTI networks. From Fig. 6, we learn that all the DTIs of DHCR7, HTR1F, LTB4R, GPER1, and PTGIR, were predicted correctly, while a small portion of DTIs were predicted incorrectly for CYSLTR2, GRIK3 and SIPR5. The detailed prediction results are listed in Table S1† and the SMILES of all compounds are shown in Table S2.†
| No. | Target name | Correctly predicted | All predicted | Recall score |
|---|---|---|---|---|
| 1 | DHCR7 | 7 | 7 | 100% |
| 2 | HTR1F | 14 | 14 | 100% |
| 3 | LTB4R | 14 | 14 | 100% |
| 4 | CYSLTR2 | 11 | 15 | 73.30% |
| 5 | GRIK3 | 5 | 8 | 62.50% |
| 6 | GPER1 | 10 | 10 | 100% |
| 7 | PTGIR | 16 | 16 | 100% |
| 8 | S1PR5 | 12 | 15 | 80% |
 | ||
| Fig. 6 The literature validation results of MPSM-DTI on known interactions between eight GPCR targets and 100 compounds. The blue diamonds indicate targets, red circles represent compounds, solid lines indicate correctly predicted DTIs, and dotted lines indicate wrongly predicted DTIs. | ||
Discussion
In this study, we proposed a machine learning model for the prediction of DTIs. Five types of fingerprints (FP4, MACCS, PubChem, KR, and Morgan) and molecular descriptors (Des) were used to represent the chemical features, respectively. A kind of protein sequence-based feature, PSSM, was utilized to describe the protein targets. Then the chemical features and PSSM characteristics were joined together to manifest the DTIs. Five types of machine learning algorithms (DT, bagging, GBDT, k-NN, and SVM) were employed to build the predictive models. The models were further validated comprehensively by ten-fold cross-validation, test set, and two external validation sets. By means of pipeline encapsulation, the data leaking problem of the models was avoided and the overfitting issue was also prevented to some degree. Finally, the MPSM-DTI model was selected as the best one among the 30 models and showed satisfactory generalization capability. In a case study, the MPSM-DTI model correctly predicted potential ligands for new targets without crystal structures and known ligands.In comparison with other similar models, the MPSM-DTI model possesses several advantages. Firstly, the data quality was greatly guaranteed by gathering first-hand DTI data. However, in some reported DTI prediction models, the threshold to discriminate positive and negative DTI data was often incorrect; sometimes unconfirmed interactions were regarded as negative DTI data in some research studies, which would lead to inaccurate models and mislead false predictions.14,17 Secondly, the MPSM-DTI model could predict targets for new compounds outside the DTI network. From the results in external validation set 2, we could see that the MPSM-DTI model would correctly predict potential targets for brand new compounds. Thirdly, the MPSM-DTI model could predict compounds for new targets outside the DTI network. From the results of the case study, the MPSM-DTI model could correctly predict 90 percent of DTIs for those eight new GPCR targets and achieve a relatively decent performance. In theory, our MPSM-DTI model could predict potential ligands for any new targets as long as the target sequence could be obtained.
At present, there are several published methods for the prediction of DTIs, such as SwissTarget,3 SDTNBI,27 bSDTNBI,27 and ChemMapper.5 These methods are widely used as free webservers in drug discovery. SwissTarget is a ligand-based method for target prediction, established based on a combination of 2D and 3D similarity with a library of 370000 known actives.3 ChemMapper is also a kind of ligand-based approach, which is based on the concept that compounds sharing high 3D similarities may have relatively similar target association profiles.4,5 SDTNBI and bSDTNBI are two network-based methods for target prediction. SDTNBI uses a network-based inference method to recommend targets for compounds, which relies on source propagation on the substructure-drug–target network,9 while bSDTNBI is the upgraded version of SDTNBI by adding three parameters to adjust the network weights.10 SDTNBI and bSDTNBI methods could be reached by the NetInfer (http://lmmd.ecust.edu.cn/netinfer/) webserver.27
Compared with these published methods, our MPSM-DTI model showed better prediction accuracy with a higher recall rate than the others. Fig. 7 displays the prediction results of these methods, including MPSM-DTI, SwissTarget, SDTNBI (top 20), SDTNBI (top 50), bSDTNBI (top 20), bSDTNBI (top 50), and ChemMapper. All predictions were performed through corresponding webservers and the correct recall numbers and false recall numbers were counted by Python scripts. From Fig. 7, it is easy to see that MPSM-DTI achieved the best results with 90% correctness for the aforementioned eight GPCR targets. SwissTarget ranked the second with 61% correctness. bSDTNBI outperformed SDTNBI, which was confirmed by the previous studies.7,10,20 The prediction ranking top 20 or top 50 of bSDTNBI did not influence the ultimate results a lot. The detailed prediction results for each DTI of these methods are shown in Table S1,† and the SMILES for the 100 compounds are listed in Table S2.† If somebody is interested in some of the targets, they could use those data in their studies.
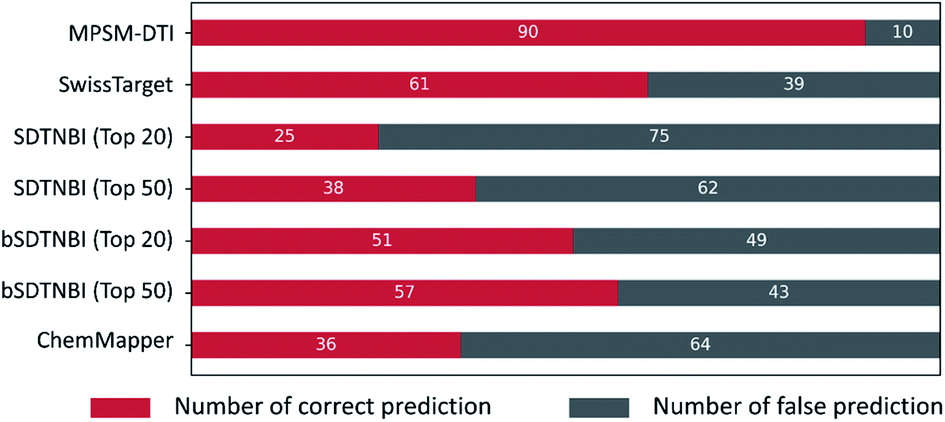 | ||
| Fig. 7 Comparison of prediction accuracy for 100 literature-validated DTIs among eight targets and 100 compounds by seven methods, including our MPSM-DTI model, SwissTarget, SDTNBI (top 20), SDTNBI (top 50), bSDTNBI (top 20), bSDTNBI (top 50) and ChemMapper. Red: number of true positives; grey: number of false positives. | ||
The MPSM-DTI model also exhibited two more advantages. First, MPSM-DTI could predict potential ligands for new protein targets especially for those without crystal structures and known ligands, whereas the other methods could not do that. Second, MPSM-DTI runs very fast and only needs a few seconds. However, ChemMapper would take a much longer time (usually more than 24 hours) because it identifies potential compounds via 3D similarity calculations. SwissTarget takes 5–10 minutes after submitting a query for small molecule.
Anyhow, there is still some space to improve MPSM-DTI. For example, we did not use deep learning methods to construct the model, because we are short of gigantic DTI data and plentiful computational resources to support vast data calculation. At present, deep learning does not improve model performance but takes too much computation resource in comparison with ordinary machine learning methods. Meanwhile, a webserver might be very helpful for others to use it friendly elsewhere, for instance, to do virtual screening or lead discovery for targets without known ligands and crystal structures. Nevertheless, MPSM-DTI might have a profound significance on drug discovery and development.
Conclusions
It is important to develop novel and accurate tools for identification of DTIs, especially for those targets without known ligands and crystal structures. In this study, we developed a machine learning model named MPSM-DTI for the prediction of DTIs, in which chemical Morgan fingerprints and protein sequence PSSM features were used to characterize the DTIs. The main advantage of MPSM-DTI is the pipeline encapsulation technique, which reduced overfitting significantly and enhanced the generalization ability of the model distinctly by encapsulating the feature data standardization, SMOTE sampling process and SVM estimator. The MPSM-DTI model was evaluated by ten-fold cross-validation, test set and two external validation sets. Moreover, 90% of 100 real DTIs for eight GPCR targets were correctly predicted by MPSM-DTI in a case study, which demonstrated the superiority of our method in DTI prediction. Compared with SwissTarget, SDTNBI, bSDTNBI, and ChemMapper, our MPSM-DTI model could achieve a higher recall rate with less time consumption. Therefore, MPSM-DTI would be a powerful tool for DTI prediction and have a wide range of applications. The source code of MPSM-DTI is available at https://github.com/pengyayuan/MPSM-DTI.Data availability
Information on the data for this is provided in an additional ESI† file.Author contributions
Yayuan Peng: conceptualization, methodology, investigation, data curation, formal analysis, validation, visualization, writing – original draft; Jiye Wang: formal analysis; methodology; writing – review & editing; Zengrui Wu: conceptualization; methodology; Lulu Zheng: formal analysis; Biting Wang: formal analysis; Guixia Liu: supervision; Weihua Li: supervision; Yun Tang: supervision; project administration; conceptualization; writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant 2019YFA0904800), the National Natural Science Foundation of China (Grants 81872800 and 82173746), the China Postdoctoral Science Foundation (Grant 2019M661413), and Shanghai Sailing Program (Grant 19YF1412700).References
- D. Sydow, L. Burggraaff, A. Szengel, H. W. T. van Vlijmen, A. P. Ijzerman, G. J. P. van Westen and A. Volkamer, J. Chem. Inf. Model., 2019, 59, 1728–1742 CrossRef CAS PubMed.
- D. Gfeller, O. Michielin and V. Zoete, Bioinformatics, 2013, 29, 3073–3079 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2019, 47, W357–W364 CrossRef CAS PubMed.
- X. Liu, H. Jiang and H. Li, J. Chem. Inf. Model., 2011, 51, 2372–2385 CrossRef CAS PubMed.
- J. Gong, C. Cai, X. Liu, X. Ku, H. Jiang, D. Gao and H. Li, Bioinformatics, 2013, 29, 1827–1829 CrossRef CAS PubMed.
- K. Lundstrom, Future Med. Chem., 2017, 9, 633–636 CrossRef CAS PubMed.
- Z. Wu, W. Lu, W. Yu, T. Wang, W. Li, G. Liu, H. Zhang, X. Pang, J. Huang, M. Liu, F. Cheng and Y. Tang, Pharmacol. Res., 2018, 129, 400–413 CrossRef CAS PubMed.
- A.-J. Banegas-Luna, J. P. Cerón-Carrasco and H. Pérez-Sánchez, Future Med. Chem., 2018, 10, 2641–2658 CrossRef CAS PubMed.
- Z. Wu, F. Cheng, J. Li, W. Li, G. Liu and Y. Tang, Briefings Bioinf., 2017, 18, 333–347 CAS.
- Z. Wu, W. Lu, D. Wu, A. Luo, H. Bian, J. Li, W. Li, G. Liu, J. Huang, F. Cheng and Y. Tang, Br. J. Pharmacol., 2016, 173, 3372–3385 CrossRef CAS PubMed.
- Z. Wu, W. Li, G. Liu and Y. Tang, Front. Pharmacol., 2018, 9, 1134 CrossRef CAS PubMed.
- Y. Peng, Z. Wu, H. Yang, Y. Cai, G. Liu, W. Li and Y. Tang, Toxicol. Lett., 2019, 312, 22–33 CrossRef CAS PubMed.
- I. Lee, J. Keum and H. Nam, PLoS Comput. Biol., 2019, 15, e1007129 CrossRef CAS PubMed.
- S. M. H. Mahmud, W. Chen, H. Jahan, Y. Liu, N. I. Sujan and S. Ahmed, IEEE Access, 2019, 7, 48699–48714 Search PubMed.
- N. S. Madhukar, P. K. Khade, L. Huang, K. Gayvert, G. Galletti, M. Stogniew, J. E. Allen, P. Giannakakou and O. Elemento, Nat. Commun., 2019, 10, 5221 CrossRef PubMed.
- P. Kumari, A. Nath and R. Chaube, Comput. Biol. Med., 2015, 56, 175–181 CrossRef CAS PubMed.
- Z. Li, P. Han, Z. H. You, X. Li, Y. Zhang, H. Yu, R. Nie and X. Chen, Sci. Rep., 2017, 7, 11174 CrossRef PubMed.
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Kruger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, Nucleic Acids Res., 2014, 42, D1083–D1090 CrossRef CAS PubMed.
- S. Niijima, A. Shiraishi and Y. Okuno, J. Chem. Inf. Model., 2012, 52, 901–912 CrossRef CAS PubMed.
- Z. Wu, W. Li, G. Liu and Y. Tang, Front. Pharmacol., 2018, 9, 1134 CrossRef CAS PubMed.
- Z. Tanoli, Z. Alam, M. Vaha-Koskela, B. Ravikumar, A. Malyutina, A. Jaiswal, J. Tang, K. Wennerberg and T. Aittokallio, Database, 2018, 2018, 1–13 CrossRef PubMed.
- M. K. Gilson, T. Liu, M. Baitaluk, G. Nicola, L. Hwang and J. Chong, Nucleic Acids Res., 2016, 44, D1045–D1053 CrossRef CAS PubMed.
- S. D. Harding, J. L. Sharman, E. Faccenda, C. Southan, A. J. Pawson, S. Ireland, A. J. G. Gray, L. Bruce, S. P. H. Alexander, S. Anderton, C. Bryant, A. P. Davenport, C. Doerig, D. Fabbro, F. Levi-Schaffer, M. Spedding, J. A. Davies and I. Nc, Nucleic Acids Res., 2018, 46, D1091–D1106 CrossRef CAS PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- J. Wang, B. Yang, J. Revote, A. Leier, T. T. Marquez-Lago, G. Webb, J. Song, K. C. Chou and T. Lithgow, Bioinformatics, 2017, 33, 2756–2758 CrossRef CAS PubMed.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Artif. Intell. Res, 2002, 16, 321–357 CrossRef.
- Z. Wu, Y. Peng, Z. Yu, W. Li, G. Liu and Y. Tang, J. Chem. Inf. Model., 2020, 60, 3687–3691 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dd00011j |
| This journal is © The Royal Society of Chemistry 2022 |