
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic-scale changes of silica-supported catalysts with nanocrystalline or amorphous gallia phases: implications of hydrogen pretreatment on their selectivity for propane dehydrogenation†
Pedro
Castro-Fernández
 a,
Alexander I.
Serykh
b,
Alexander V.
Yakimov
c,
Igor P.
Prosvirin
d,
Andrey V.
Bukhtiyarov
d,
Paula M.
Abdala
*a,
Christophe
Copéret
c,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
a,
Alexander I.
Serykh
b,
Alexander V.
Yakimov
c,
Igor P.
Prosvirin
d,
Andrey V.
Bukhtiyarov
d,
Paula M.
Abdala
*a,
Christophe
Copéret
c,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zürich, CH-8092, Zürich, Switzerland. E-mail: abdalap@ethz.ch; fedoroal@ethz.ch; muelchri@ethz.ch
bZelinsky Institute of Organic Chemistry, RAS, Moscow, 119991, Russia
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, CH-8093, Zürich, Switzerland
dBoreskov Institute of Catalysis, SB RAS, 630090 Novosibirsk, Russia
First published on 11th May 2022
Abstract
This work explores how H2 pretreatment at 550 °C induces structural transformation of two gallia-based propane dehydrogenation (PDH) catalysts, viz. nanocrystalline γ/β-Ga2O3 and amorphous Ga2O3 (GaOx) supported on silica (γ-Ga2O3/SiO2 and Ga/SiO2, respectively) and how it affects their activity, propene selectivity and stability with time on stream (TOS). Ga/SiO2–H2 shows poor activity and propene selectivity, no coking and no deactivation with TOS, similar to Ga/SiO2. In contrast, the high initial activity and propene selectivity of γ-Ga2O3/SiO2–H2 decline with TOS but to a lesser extent than in calcined γ-Ga2O3/SiO2. In addition, γ-Ga2O3/SiO2–H2 cokes less than γ-Ga2O3/SiO2. Ga K-edge X-ray absorption spectroscopy suggests an increased disorder of the nanocrystalline γ/β-Ga2O3 phases in γ-Ga2O3/SiO2–H2 and the emergence of additional tetrahedral Ga sites (GaIV). Such GaIV sites are strong Lewis acid sites (LAS) according to studies using adsorbed pyridine and CO probe molecules, i.e., the abundance of strong LAS is higher in γ-Ga2O3/SiO2–H2 compared to γ-Ga2O3/SiO2 but lower than in Ga/SiO2 and Ga/SiO2–H2. Dissociation of H2 on the Ga–O linkages in γ-Ga2O3/SiO2–H2 yields high-frequency Ga–H bands that are observed in Ga/SiO2 and Ga/SiO2–H2 but not detected in γ-Ga2O3/SiO2. We attribute the increased amount of GaIV sites in γ-Ga2O3/SiO2–H2 mostly to an increased disorder in γ/β-Ga2O3. X-ray photoelectron spectroscopy detects the formation of Ga+ and Ga0 species in both Ga/SiO2–H2 and γ-Ga2O3/SiO2–H2. Therefore, it is likely that a minor amount of GaIV sites also forms through the interaction of Ga+ (such as Ga2O) and/or Ga0 with silanol groups of SiO2.
Introduction
The industrial production of propene through propane dehydrogenation (PDH) relies mainly on PtSn/Al2O3 or CrOx/Al2O3 catalysts, yet the high cost of Pt and the environmental toxicity of Cr6+ has motivated the search for alternative catalyst formulations.1–5 Among the alternatives explored, Ga-based catalysts are considered promising. For instance, a Ga/H-ZSM-5 zeolite catalyst has been used for the Cyclar propane aromatization process, in which the dehydrogenation of propane to propene proceeds on Ga-based active sites, followed by dehydrogenative aromatization of propene on the strong Brønsted acid sites of the zeolite.6,7 Furthermore, the FCDh (fluidized catalytic dehydrogenation of propane) process, which uses a PtGa/Al2O3 catalyst is currently at the initial phase of its industrial deployment.8A reductive pretreatment of zeolitic Ga-based catalysts under H2 at 450–650 °C is often used to increase the activity and selectivity of the Ga-based active sites for propane dehydrogenation.9 It has been demonstrated that the dispersion of Ga in mechanically mixed Ga2O3/H-ZSM-5 catalysts increases under H2 pretreatment conditions, explained by the formation of reduced Ga species that diffuse within the zeolitic channels and anchor onto acidic hydroxyl groups.10 Various reduced zeolitic Ga species have been proposed to form after reductive pretreatment or under reaction conditions, and, presumably, also the reduced species are active in PDH.11–13 H2 pretreatment of Ga/H-ZSM-5 and Ga/H-MFI zeolites, followed by in situ X-ray absorption near edge structure spectroscopy (XANES) experiments at the Ga K-edge showed a shift in the absorption edge to lower energies (∼4.6 eV (ref. 14)) that has been related to the reduction of Ga3+ to Ga+, which then may quickly reoxidize to form GaO+ or [GaH2]+ species.11,14–16 The conclusions from XANES experiments have been supported by X-ray photoelectron spectroscopy (XPS) studies of Ga zeolites that revealed Ga 3d features at lower binding energies relative to the Ga3+ peaks; such features have been ascribed to Ga+, Gaδ+ and metallic Ga.17
That being said, the interaction of the gallia phase with silica, a typical non-microporous support, and the nature of the Ga sites formed in such supported materials under H2 treatment remains less explored. Treatment of a Ga2O3/SiO2 catalyst with H2 at 550 °C has been reported to lead to a partial reduction of Ga2O3, lower propene yields in PDH and higher yields to aromatics, the latter observation explained by an increase in the number of Brønsted acid sites.18 Similar to the reduction of Ga3+ sites in zeolites, it has been shown that H2 pretreatment at 550 °C of a Ga2O3/SiO2 material gave XPS features consistent with Ga+ species.19 In another report, Ga K-edge XANES and XPS analysis of a Ga2O3/SiO2 catalyst treated with H2 at 650 °C also suggested the presence of reduced Ga sites.20 However, after H2 pretreatment, the latter catalyst showed a loss of PDH activity, suggested to be due to the formation of lower-coordinated Gaδ+ species.20 Additionally, the emergence of a low-energy XANES edge feature has been observed when comparing fresh and used (after 20 hours of time on stream, TOS) single site [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3Ga(XOSi)] (where X is H or Si) PDH catalyst,21 prepared by the surface organometallic chemistry approach.22 The low energy edge feature in the XANES spectra may be associated with the reduction of Ga sites; however, recent evidence from reference molecular alkyl gallium compounds suggests that such feature may, at least in part, be associated with the formation of Ga alkyl surface species during the PDH reaction.23 This evidence is in line with a further report that demonstrated that an edge feature at lower energies in the Ga K-edge XANES spectra could be due to changes in the coordination number and/or formation of gallium hydride and alkyl species.24
SiO)3Ga(XOSi)] (where X is H or Si) PDH catalyst,21 prepared by the surface organometallic chemistry approach.22 The low energy edge feature in the XANES spectra may be associated with the reduction of Ga sites; however, recent evidence from reference molecular alkyl gallium compounds suggests that such feature may, at least in part, be associated with the formation of Ga alkyl surface species during the PDH reaction.23 This evidence is in line with a further report that demonstrated that an edge feature at lower energies in the Ga K-edge XANES spectra could be due to changes in the coordination number and/or formation of gallium hydride and alkyl species.24
Studies of unsupported gallia catalysts have suggested that the high activity of β-Ga2O3 in PDH (as compared to other gallia polymorphs) is due to the high relative fraction of weak Lewis acid sites (LAS) on the surface of β-Ga2O3.25 It has been shown that oxygen vacancy (Vo) surface sites in β-Ga2O3 can be generated by H2 pretreatment.26 Density functional theory studies on a dehydroxylated model surface of β-Ga2O3 have ascribed weak LAS to tricoordinated Ga3+ sites, that is, Ga sites in tetrahedral surface termination positions that neighbor Vo surface sites.25 The relevance of weak Ga-based LAS in unsupported nanoparticle catalysts for a high activity in PDH has also been supported by structure–performance relationship studies of mixed oxide (Ga,Al)2O3 catalysts.27,28
In this work, we compare the structural changes of two gallia based, silica-supported catalysts upon H2 pretreatment, with one material containing a nanocrystalline, and another one an amorphous gallia phase, denoted γ-Ga2O3/SiO2 and Ga/SiO2, respectively (with similar Ga loadings of 2–3 wt%). The calcined materials have been characterized by us in detail previously.25 Ga/SiO2 and γ-Ga2O3/SiO2 display different reducibility under H2 treatment. More specifically, Ga/SiO2, a PDH catalyst with a low activity and selectivity owing to the undesired cracking of propane on unselective Ga sites and, possibly, also on strong Brønsted acid sites (BAS, due to a small amount of gallosilicate sites in Ga/SiO2), shows no notable changes in its PDH performance after H2 treatment at 550 °C (2 h). In contrast, γ-Ga2O3/SiO2 displays, after the same reductive pretreatment, a higher stability with TOS both with regards to its activity and even more notably in its propene selectivity. The in situ X-ray absorption spectroscopy (XAS) study of γ-Ga2O3/SiO2 at the Ga K-edge complemented by quasi in situ XPS suggests a higher reducibility of the nanocrystalline gallia phase in γ-Ga2O3/SiO2 relative to the amorphous gallia phase in Ga/SiO2. Upon H2 treatment we observe an increased disorder of the nanocrystalline gallia phase in γ-Ga2O3/SiO2–H2 that occurs in parallel with the reduction of Ga3+ sites to Ga+ and Ga0 species and the emergence of additional tetrahedral Ga sites (GaIV). According to surface acidity studies using pyridine and CO probe molecules, the newly formed GaIV sites feature strong Lewis acidity, similarly to the acidity of the LAS in the amorphous gallia catalysts Ga/SiO2 and Ga/SiO2–H2, while avoiding the formation of strong BAS. In addition to the explanation given above, it is conceivable that Ga+ and/or Ga0 species interact with and anchor onto silanols groups of the silica support.
Materials and methods
Materials
The synthesis and structural characterization of the γ-Ga2O3/SiO2 and Ga/SiO2 catalysts have been described by us previously.25 In brief, γ-Ga2O3/SiO2 and Ga/SiO2 were obtained by the incipient wetness impregnation of amorphous silica with a colloidal solution of γ-Ga2O3 nanoparticles in toluene (particle diameter 2.5 ± 0.6 nm) or an aqueous solution of Ga(NO3)3, respectively, followed by drying and calcination (600 °C, 2 h). γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 were obtained by pretreating γ-Ga2O3/SiO2 and Ga/SiO2 in undiluted H2 at 550 °C for 2 h (10 °C min−1).Catalytic testing
Propane dehydrogenation tests were carried out in a benchtop Microactivity EFFI reactor (PID Eng&Tech). A mass of 50 mg of catalyst was mixed with 1.20 g of SiC (46 grit, Alfa-Aesar), and placed, between two plugs of quartz wool, onto a frit of a quartz fixed-bed reactor (diameter 13 mm). After reaching the reaction temperature (550 °C, 10 °C min−1) under a flow of N2, a mixture of 10% C3H8 diluted in N2 was introduced into the reactor (weight hourly space velocity, WHSV = 7.2 h−1). The composition of the outlet gasses was analyzed using a gas chromatograph (Clarus 480, PerkinElmer) with a flame ionization detector and a thermal conductivity detector. The first sampling of the gasses was made after 4 min of reaction, collecting data for every 20 minutes afterwards. For the H2-treated catalysts, before PDH reaction, the catalysts were heated up under H2 to 550 °C (50 mL min−1), and held at this temperature for 2 hours. Subsequently, the catalyst bed was flushed with N2 for 15 min before switching to a C3H8/N2 mixture.Thermogravimetric analyses
For thermogravimetric analyses (TGA), ca. 15 mg of the catalyst were placed in a 70 μL alumina crucible and introduced into the instrument (DSC 1, Mettler-Toledo). The material was heated to 550 °C (10 °C min−1) under a N2 flow of 75 mL min−1 (of which 25 mL min−1 correspond to the purge flow of the balance). After having reached the desired temperature and holding at this temperature for 10 min, 10% of propane (7.5 mL min−1) was introduced to the N2 flow. Changes in the catalyst weight were monitored for 160 min.XAS and X-ray total scattering
X-ray absorption measurements were performed at the Swiss-Norwegian beamlines (SNBL, BM31) at the European Synchrotron Radiation Facility (ESRF).29 Spectra were obtained at the Ga K-edge in transmission mode (continuous scanning), using a double-crystal Si(111) monochromator. For ex situ measurements, the materials were mixed with an optimal amount of cellulose and pressed into self-supporting pellets.30 Pellets of H2-treated materials were prepared inside of a glovebox and kept in air-tight sealed bags for measurements.In situ XAS experiments were carried out in a quartz capillary reactor (diameter 1 mm; wall thickness = 0.1 mm) whereby the catalyst was placed between two quartz wool plugs. H2 was flowed (10 mL min−1) through the reactor while heating up from room temperature to 550 °C (10 °C min−1) using an air blower. During cooling down the reactor was flushed with He. A schematics of the XAS setup has been described in a previous publication.25 Data processing was carried out using the DEMETER software suite.31 Energy calibration was performed using a Zn-foil. Data processing was done using the Athena software.30 FEFF paths and the amplitude reduction factor (S02) were generated by the Artemis software using a β-Ga2O3 structural model.31,32
X-ray total scattering data were collected at BM31 in a consecutive (combined) mode to the in situ XAS data. Data collection was performed before and after H2 treatment at 50 °C, using a wavelength of λ = 0.25811 Å (set by a double-crystal Si(111) monochromator), and a two-dimensional MAR detector (MAR345 image plate). The data collection time was 20 min. To obtain pair distribution functions (PDF, G(r)) from the supported gallia phase, the scattering signal was subtracted from the scattering signal of the pure SiO2 support.33 PDFs were obtained using the PDFgetX3 software,34 setting a Qmax = 16 Å−1 and a Rpoly = 1.6.
Dynamic nuclear polarization surface enhanced nuclear resonance spectroscopy (DNP SENS)
Prior to NMR measurements, γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 were dehydroxylated at 550 °C at ca. 10−5 mbar for 2 h (3 °C min−1). Subsequently, the materials were exposed for 20 min at room temperature to the saturated vapor pressure of 15N labelled pyridine (Py), followed by evacuation at 150 °C at ca. 10−5 mbar for 15 min. Before use, 15N Py was dried by adding CaH2 and stirring at 60 °C for 72 h. Afterwards, the solution was degassed by several freeze–pump–thaw cycles.22 Materials with pre-adsorbed Py were impregnated in a glovebox (O2 and H2O < 1 ppm) with a 16 mM TEKPol in 1,1,2,2-tetrachloroethane (TCE) solution.35,36 The impregnated materials were introduced into a sapphire rotor (outer diameter 3.2 mm), and closed with a zirconia cap. The rotor was placed quickly in a cold NMR probe (100 K). 15N DNP SENS spectra were acquired on a Bruker 600 MHz (14.1 T) instrument equipped with a 3.2 mm Bruker low-temperature double-resonance probe coupled to a 395 GHz gyrotron microwave source (output power = 6–10 W) to drive the DNP cross-effect. In all experiments a MAS rate of 8 kHz was used. DNP-enhanced 15N NMR spectra were measured using a {1H}15N CPMAS pulse sequence with the contact time of 2 ms. As a reference for the static magnetic field, the 13C higher-frequency peak of adamantane at 38.4 ppm was used. The DNP buildup time (τDNP) was measured by a 1H saturation-recovery experiment with the microwaves turned on. A summary of the experimental DNP SENS parameters are given in Table S1.†XPS
X-ray photoelectron spectra were recorded on a SPECS photoelectron spectrometer using a hemispherical PHOIBOS 150 MCD 9 analyzer (AlKa radiation, hν = 1486.6 eV, 150 W). The binding energy (BE) scale was pre-calibrated using the positions of the peaks of the Au 4f7/2 (BE = 84.0 eV) and Cu2p3/2 (BE = 932.67 eV) core levels. The Si 2p peak at 103.5 eV of the SiO2 support was used as an internal standard. The survey spectra were recorded at a pass energy of 50 eV, and for the narrow spectral regions at 20 eV. To determine the chemical (charge) state of elements on the surface of the materials, the regions of Ga 3d and O 2s, Si 2p, Si 2s, C 1s, O 1s, Ga 2p3/2 were measured. The atomic ratios of the elements on the catalyst surface were calculated from the integral photoelectron peak intensities which were corrected by theoretical sensitivity factors based on Scofield's photoionization cross sections.37 The residual gas pressure during the measurements did not exceed 8 × 10−9 mbar. To carry out experiments using the high pressure cell (HPC) of the SPECS photoelectron spectrometer, all materials were rubbed into a stainless steel mesh that was spot welded onto a standard holder. HPC allows to pretreat the specimen under different gases at pressures up to 1 bar and in the temperature range from 50 to 450 °C. The pretreatment was performed for 1 h at a hydrogen pressure of ca. 300 mbar either at 300 °C or 450 °C, followed by an outgassing to UHV conditions at the respective pretreatment temperature. The pretreated materials were transferred to the analyzer chamber without any contact with air. Additional XPS data (survey scans, C 1s, Si 2p, Ga 3d, O 2s and Ga LMM lines) are presented in Fig. S4 and S5.†Fourier-transform infrared spectroscopy (FTIR)
Infrared spectra were recorded on a Nicolet Protege 360 spectrophotometer in transmittance mode (spectral resolution 4 cm−1). Powdered materials were pressed into self-supporting wafers (15–20 mg cm−2) and placed into a vacuum infrared quartz cell equipped with CaF2 windows. The dissociative adsorption of hydrogen (ca. 1–2 min hydrogen exposure time) was studied at different temperatures (300, 450 and 540 °C) at an equilibrium hydrogen pressure of ca. 70 mbar on both calcined materials and on the materials treated in hydrogen. Calcined and H2-treated materials before hydrogen adsorption were outgassed at 813 K for 2 h in a medium vacuum of ca. 10−3 mbar. The hydrogen pretreatment of the self-supporting wafers was conducted in the IR cell under static conditions (three cycles of exposure of the specimen to ca. 130 mbar of H2 for 0.5 h with evacuation to ca. 10−3 mbar in-between the H2 exposures). The IR spectra were measured at room temperature. Both calcined and H2-treated materials were also characterized by the adsorption of CO. The adsorption of CO was conducted at room temperature and an equilibrium pressure of ca. 27 mbar.Scanning transmission electron microscopy – energy dispersive X-ray spectroscopy (STEM-EDX)
Materials were dispersed on a copper grid and analyzed using a FEI Talos F200X instrument. The operation voltage was set to 200 kV in scanning transmission electron microscopic (STEM) mode. EDX was performed using an accelerating voltage of 40 keV.Fig. 1, 2, S2 and S7,† and Table 1 present results for calcined γ-Ga2O3/SiO2 and Ga/SiO2 catalysts. This data has been reported by us in ref. 25 and is reproduced here for comparison to γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2, with permission from the American Chemical Society (Copyright 2021).
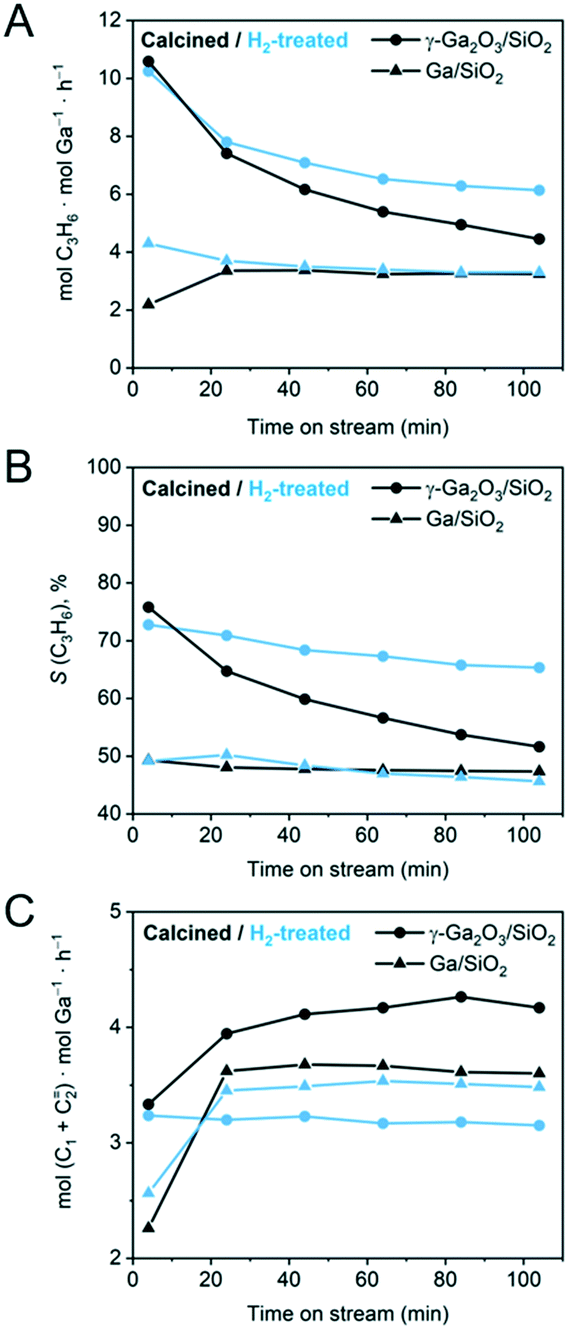 | ||
| Fig. 1 Results of the PDH catalytic tests for calcined (black traces) and H2-treated (blue traces) γ-Ga2O3/SiO2 and Ga/SiO2. (A) Ga-Normalized formation rate of propene, (B) selectivity to propene, and (C) Ga-normalized formation rates of cracking products (methane and ethene). Catalytic data for the calcined γ-Ga2O3/SiO2 and Ga/SiO2 has been reported by us previously and is reproduced here for comparison.25 | ||
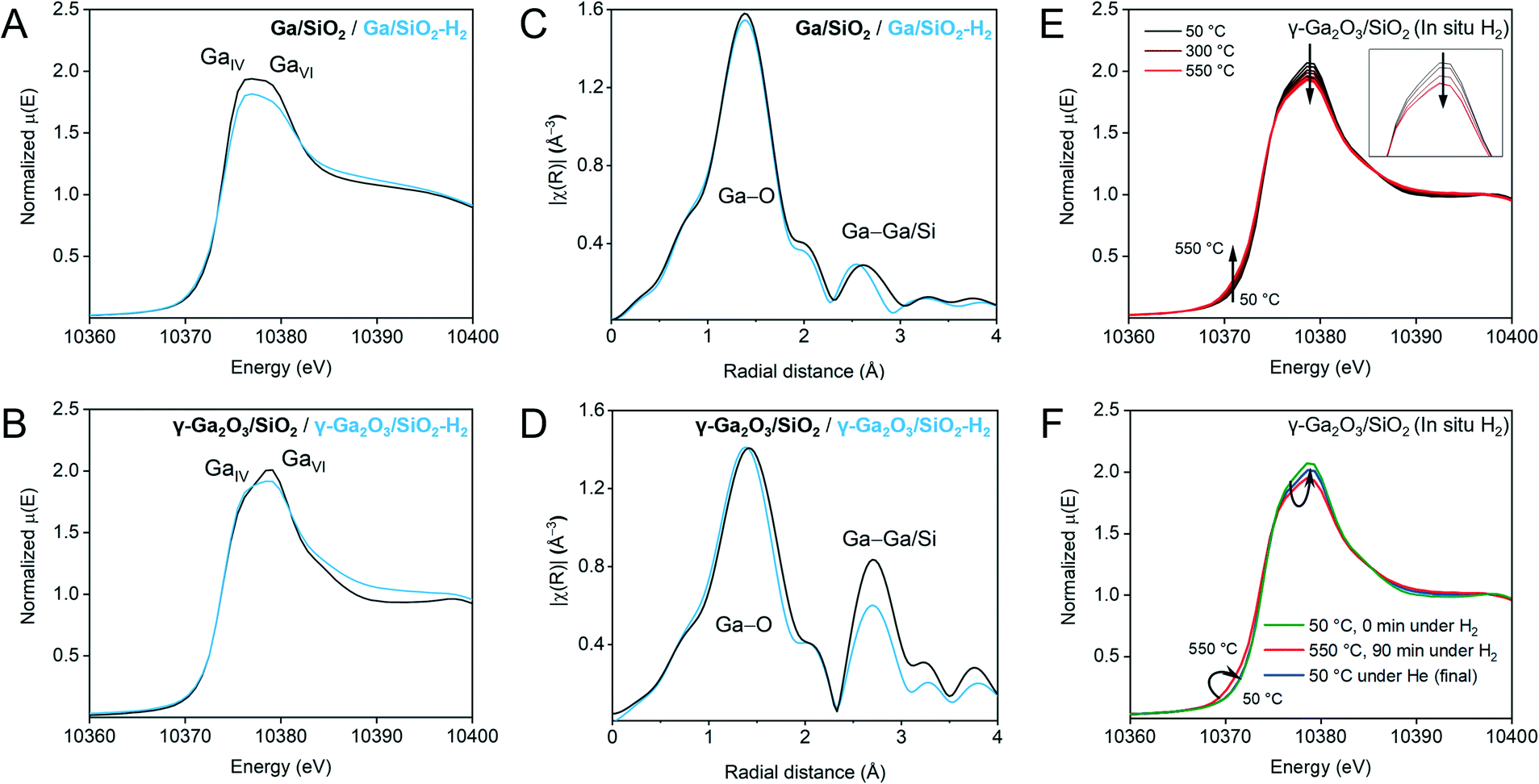 | ||
| Fig. 2 Ex situ Ga K-edge XANES spectra of Ga/SiO2 (A) and γ-Ga2O3/SiO2 (B) before and after H2-treatment (black and blue traces, respectively). EXAFS FT plots (non-phase corrected) before and after H2 treatment (black and blue traces, respectively) for Ga/SiO2 (C) and γ-Ga2O3/SiO2 (D). We note that the ex situ XANES and EXAFS data for calcined γ-Ga2O3/SiO2 and Ga/SiO2 have been reported by us previously,25 and are reproduced here for comparison. (E) Selected normalized traces of in situ Ga K-edge XANES during H2 treatment of γ-Ga2O3/SiO2 (50–550 °C). The inset shows changes of the white line and a derivative plot is presented in (Fig. S6†). (F) Comparison of the XANES spectra of γ-Ga2O3/SiO2 before flowing H2 at 50 °C, after 90 min under H2 at 550 °C, and after cooling down under He. | ||
| Material | Path | CN | Distance (Å) | σ 2 (Å2) | R-Factor |
|---|---|---|---|---|---|
| γ-Ga2O3/SiO2–H2 | Ga–O | 5.4(1) [5.4(5)] | 1.87(1) [1.89(1)] | 0.011* [0.011*] | 0.004 |
| Ga–Ga1 | 1.7(2) [2.0(1)] | 2.98(1) [2.99(1)] | 0.009* [0.009] | ||
| Ga–Ga2 | 2.3(3) [3.0(1)] | 3.37(1) [3.38(1)] | 0.009* [0.009] | ||
| Ga/SiO2–H2 | Ga–O | 5.7(3) [5.7(9)] | 1.85(1) [1.86(1)] | 0.011* [0.011*] | 0.016 |
| Ga–Ga1 | 1.0(1) [2.0(1)] | 2.91(2) [2.97(1)] | 0.011* [0.011] |
Results
Catalytic performance and coking
γ-Ga2O3/SiO2 and Ga/SiO2 are PDH catalysts that contain dispersed nanocrystalline gallia nanoparticles (NPs, γ/β-phase Ga2O3) and a mostly XRD-amorphous gallia phase with a short range order (ca. 5 Å), respectively.25,38 These two catalysts were treated in undiluted H2 at 550 °C for 2 h prior to their catalytic PDH tests at 550 °C for 104 min at a WHSV = 7.2 h−1. Note that the catalytic results for γ-Ga2O3/SiO2 and Ga/SiO2 have been reported by us previously,25 and are plotted in Fig. 1 to allow a comparison with the H2-treated materials. Fig. 1A displays propene formation rates normalized per ICP-determined Ga loading. Fig. 1B and C present the selectivity to propene and Ga-normalized formation rates of the cracking products (methane and ethene), respectively.The initial activity and selectivity to propene (after 4 min TOS) of γ-Ga2O3/SiO2–H2 are 10.3 mol C3H6 mol Ga−1 h−1 and 72%, respectively, which are similar to γ-Ga2O3/SiO2. However, the decrease of the activity and selectivity of γ-Ga2O3/SiO2–H2 after 104 min TOS is lower than that in γ-Ga2O3/SiO2 (Fig. 1A). The higher propene selectivity of γ-Ga2O3/SiO2–H2 relative to γ-Ga2O3/SiO2 correlates with the decreased rate of cracking to methane and ethene, i.e. ca. 4.2 mol (CH4 + C2H4) mol Ga−1 h−1 for γ-Ga2O3/SiO2 and 3.2 mol (CH4 + C2H4) mol Ga−1 h−1 for γ-Ga2O3/SiO2–H2 after 40 min TOS (Fig. 1C). In contrast, the rates of cracking and propene formation, and the selectivity to propene are similarly low for Ga/SiO2–H2 and Ga/SiO2, although these catalysts show no deactivation with time on stream (Fig. 1A and B).
In situ thermogravimetric analyses (TGA) were carried out under PDH reaction conditions for the H2-treated catalysts and compared to those of the calcined catalysts obtained in our previous work.25 The amount of coke deposited (g coke g Ga−1) after 104 minutes TOS is ca. 25% lower for γ-Ga2O3/SiO2–H2 relative to γ-Ga2O3/SiO2 (0.09 vs. 0.12 g coke g−1 Ga). Ga/SiO2 and Ga/SiO2–H2 did not deposit any detectable amounts of coke.
Ga K-edge XAS
The local environment and the oxidation state of Ga in γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 were evaluated using Ga K-edge XANES and compared to the reported results of calcined γ-Ga2O3/SiO2 and Ga/SiO2.25 The H2-treated catalysts were measured ex situ in air tight conditions. The white-line features at ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 375 and 10379 eV have been attributed to tetrahedral and octahedral, GaIV and GaVI sites, respectively.25,27,39 Yet the interpretation of the XANES spectra, especially for the H2-treated Ga2O3/SiO2 materials, is not straightforward since the position of the white line peaks is affected by the coordination number and symmetry of the Ga sites, as well as the oxidation state of Ga and the ligand type (–O– or –H).23,40,41 A broad white-line in Ga/SiO2 was previously related to the non-crystalline nature of this material, i.e., only a short range order with different type of sites (different coordination environment) is present in this material.25 In the spectrum of Ga/SiO2, the feature attributed to the GaIV site has a slightly higher intensity than that of the GaVI site (Fig. 2A). The intensity of the white-line peaks in Ga/SiO2–H2 is lower compared to Ga/SiO2, while the GaIV feature is slightly more intense than the GaVI feature, suggesting a change in Ga coordination after H2 treatment (Fig. 2A). Likewise, the GaIV feature increases and the GaVI feature decreases in γ-Ga2O3/SiO2–H2 relative to γ-Ga2O3/SiO2 (Fig. 2B). The edge position (determined from the maximum in the derivative plot) is 10374 eV for all studied materials. Thus, ex situ Ga K-edge XANES data does not indicate a reduction of Ga3+ sites in the H2 treated materials, yet it suggests a change in their local structure.
375 and 10379 eV have been attributed to tetrahedral and octahedral, GaIV and GaVI sites, respectively.25,27,39 Yet the interpretation of the XANES spectra, especially for the H2-treated Ga2O3/SiO2 materials, is not straightforward since the position of the white line peaks is affected by the coordination number and symmetry of the Ga sites, as well as the oxidation state of Ga and the ligand type (–O– or –H).23,40,41 A broad white-line in Ga/SiO2 was previously related to the non-crystalline nature of this material, i.e., only a short range order with different type of sites (different coordination environment) is present in this material.25 In the spectrum of Ga/SiO2, the feature attributed to the GaIV site has a slightly higher intensity than that of the GaVI site (Fig. 2A). The intensity of the white-line peaks in Ga/SiO2–H2 is lower compared to Ga/SiO2, while the GaIV feature is slightly more intense than the GaVI feature, suggesting a change in Ga coordination after H2 treatment (Fig. 2A). Likewise, the GaIV feature increases and the GaVI feature decreases in γ-Ga2O3/SiO2–H2 relative to γ-Ga2O3/SiO2 (Fig. 2B). The edge position (determined from the maximum in the derivative plot) is 10374 eV for all studied materials. Thus, ex situ Ga K-edge XANES data does not indicate a reduction of Ga3+ sites in the H2 treated materials, yet it suggests a change in their local structure.
We further analyze the local structure around Ga in Ga/SiO2–H2 and γ-Ga2O3/SiO2–H2 and compare it to that of the calcined catalysts using extended X-ray absorption fine structure (EXAFS). Similar to the calcined catalysts, the H2-treated catalysts display two main peaks in the range of 1–3 Å (non-phase corrected EXAFS plots are presented in Fig. 2C and D) whereby the first peak corresponds to Ga–O and the second peak to Ga–Ga coordination spheres. Modelling of the EXAFS Fourier transformed (FT) data for Ga/SiO2–H2 was carried out using a simplified model including one (average) Ga–O path and one Ga–Ga path, while fitting of the EXAFS FT data of γ-Ga2O3/SiO2–H2 relied on one Ga–O path and two Ga–Ga paths (Fig. S1,†Table 1). Note that the second coordination sphere may also contain contributions from a Ga–Si path, and including a Ga–Si path to the fitting of the EXAFS spectra of Ga/SiO2–H2 decreases the R-factor (i.e. the fractional misfit) notably relative to the fitting that considers only Ga–Ga paths (i.e., from 0.016 to 0.003, Fig. S10, Table S2†). This result is consistent with the presence of gallosilicate species in Ga/SiO2–H2 that yield strong BAS (vide infra). In contrast, no abundant BAS are detected in γ-Ga2O3/SiO2–H2 and, in line with this result, the inclusion of a Ga–Si path to fit the EXAFS spectrum of γ-Ga2O3/SiO2–H2 improves the R-factor only slightly, i.e. it decreases from 0.004 to 0.002. The σ2 values (i.e., the mean square relative displacement of the nearest-neighbor atoms around Ga) were fixed to allow a comparison of the materials, i.e., to avoid the high correlation between σ2 and coordination number (CN). Notice that an increase in the NPs disorder (due to defects or amorphization) can lead to both a decrease in CN and an increase in σ2 of the second coordination sphere. An alternative fitting using a fixed CN and a variable σ2 is discussed in the ESI† (Tables S3 and S4).
According to the fittings, the average Ga–O distance in γ-Ga2O3/SiO2–H2 is 1.87 Å, which is lower by 0.02 Å than in γ-Ga2O3/SiO2 (Table 1).25 This decrease can be related to a higher relative fraction of GaIV sites in γ-Ga2O3/SiO2–H2 as indicated by the XANES data.39 However, there is no detectable decrease in the coordination number of the first shell, possibly due to the large uncertainties in determining small variation in CN. However, a more notable change in CN occurs in the second coordination sphere, i.e., the CNs of the Ga–Ga shell in γ-Ga2O3/SiO2–H2 are lower than in γ-Ga2O3/SiO2 (Fig. 2D and S1,†Table 1). This result can be explained by an increased disordering (such as the creation of defects and/or partial amorphization) of the gallia phase in γ-Ga2O3/SiO2–H2. Indeed, Ga/SiO2 with mostly amorphous gallia shows even lower intensity of the Ga–Ga coordination sphere and lower CN values (Table 1). A decrease in the CN of Ga/SiO2 upon H2 treatment (Ga/SiO2–H2) may be due to a further dispersion of the gallia phase during the H2 pretreatment of Ga/SiO2 (vide infra).
Changes in the coordination environment of Ga during H2 treatment of γ-Ga2O3/SiO2 were followed by in situ Ga K-edge XAS. γ-Ga2O3/SiO2 was heated in a quartz capillary reactor from 50 to 550 °C under an H2 flow (10 mL min−1, WHSV = 4.9 h−1) and held at 550 °C for 90 min (Fig. 2E and the derivative plot in Fig. S6†). A subtle feature at ca. 10372 eV develops with increasing temperature, accompanied by a gradual decrease of the white-line intensity (Fig. 2E, inset). Fig. 2F presents XANES spectra of the initial γ-Ga2O3/SiO2 at 50 °C, after 90 min under H2 at 550 °C, and after the subsequent cooling down to room temperature under He flow (green, red and blue traces, respectively). The room temperature spectrum shows that the low-energy feature disappears after cooling down in He, with an increase in the white line intensity relative to that at 550 °C under H2; yet the final (after cooling down) white line intensity is slightly lower as compared to the calcined γ-Ga2O3/SiO2. Therefore, in situ and ex situ XANES are consistent in that the emergence of a low-energy edge feature (at ca. 10372 eV) is observed under H2 flow at 550 °C but disappears under inert conditions at room temperature.
PDF analysis
To complement the XAS analysis, we further evaluate the structural changes of γ/β-Ga2O3 NPs in γ-Ga2O3/SiO2–H2 by the PDF analysis of X-ray total scattering data. The data collection was carried out in situ at 50 °C before and after the treatment of γ-Ga2O3/SiO2 with H2 at 550 °C. Towards this end, difference PDF (dPDF) analysis was performed by subtracting the background signal of the SiO2 support from that of γ-Ga2O3/SiO2 and γ-Ga2O3/SiO2–H2. The reciprocal space data after the subtraction of the SiO2 signal is shown in Fig. S8.† The signal to noise ratio allowed for a Qmax = 16 Å−1. The data reveals subtle differences in the intensity of the γ/β-Ga2O3 peaks in γ-Ga2O3/SiO2 and γ-Ga2O3/SiO2–H2. Turning to real space data, a comparison of the dPDF profiles evidences subtle changes in the local environment of Ga, as reflected by a broadening of the dPDF peaks of γ-Ga2O3/SiO2–H2 relative to γ-Ga2O3/SiO2, particularly within the range of ca. 1.5–6 Å (inset in Fig. S9†). This observation agrees with EXAFS results that suggested that H2 treatment leads to an increasingly disordered structure of γ-Ga2O3/SiO2–H2.Quasi in situ XPS
To deepen our understanding of the reducibility of silica-supported, nanocrystalline and amorphous gallia, XPS spectra were acquired after: i) calcination, ii) pretreatment at 550 °C under a flow of H2 followed by the exposure of the activated material to air before the measurement, and iii) quasi in situ treatment under static H2 (300 mbar, 1 h) at 300 °C and 450 °C (performed at the HPC introduced in the XP spectrometer). Calcined γ-Ga2O3/SiO2 features a single peak at 1119.1 eV in the Ga 2p3/2 region due to Ga3+ sites. The low-energy shoulder at ca. 1117.2 eV appears in γ-Ga2O3/SiO2–H2 (ca. 12% of the total Ga intensity) even after exposure of the activated material to ambient air during the material transfer (Fig. 3A). Peaks at such low binding energies have been ascribed previously to the formation of either gallium hydrides, Ga2+ or Ga+ species.20In situ H2 treatment at 300 °C increases the intensity of this peak to 22% of the total Ga intensity. After H2 treatment of γ-Ga2O3/SiO2 at 450 °C, an additional peak appears at 1115.7 eV, indicating the formation of Ga0.17,42 At this stage, the fitted relative fractions of Ga0, Ga+ and Ga3+ are ca. 21, 37 and 42%, respectively. Calcined Ga/SiO2 also features a single peak due to Ga3+ at 1119.1 eV (Fig. 3B). Ga/SiO2–H2 exposed to air during the material transfer shows signatures of reduced Ga sites whereby the fraction of Ga+ is ca. 10%. Performing an in situ reduction of Ga/SiO2 at 300 °C under static H2 yields Ga+ and Ga3+ species with a distribution of 15 and 85%, whereas the in situ reduction of Ga/SiO2 at 450 °C yields Ga0, Ga+ and Ga3+ states with a distribution of 7, 23 and 70% (according to the fits). This comparison reveals that the reduction of Ga3+ surface sites proceeds to a larger extend on γ-Ga2O3/SiO2–H2 compared to Ga/SiO2–H2.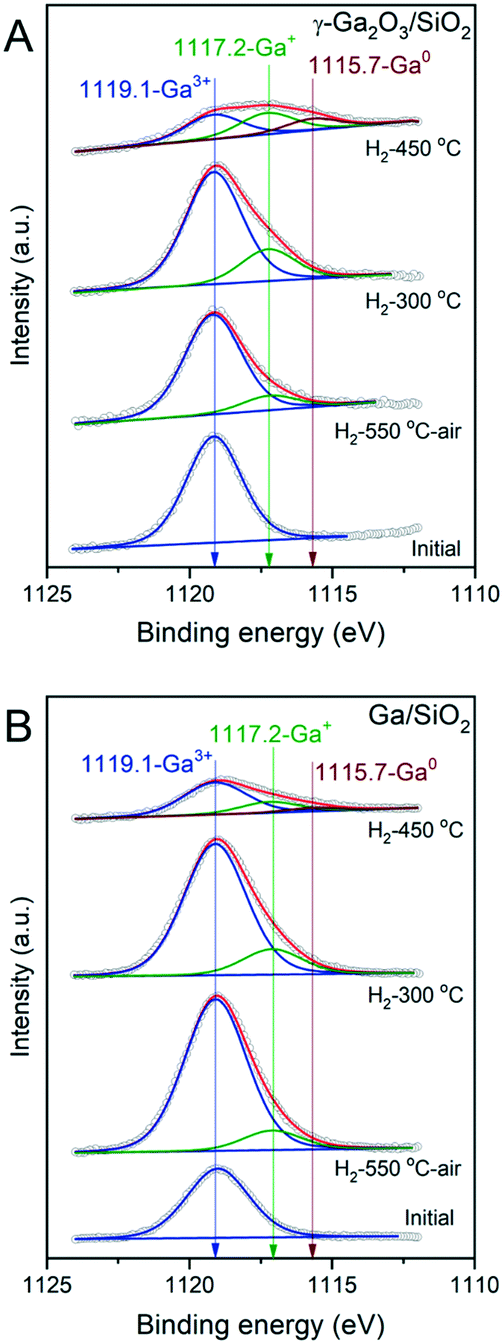 | ||
| Fig. 3 Deconvoluted Ga 2p XPS spectra of γ-Ga2O3/SiO2 (A) and Ga/SiO2 (B). XPS spectra were acquired on calcined materials (initial), after treating them in a reactor under a H2 flow at 550 °C (H2-550 °C) followed by their exposure to air during material transfer, and reduced in situ at 300 °C or 450 °C in the pretreatment chamber of the XPS instrument (labelled H2-300 °C and H2-450 °C, respectively). Fitted blue, green and red traces correspond to XPS features due to Ga3+, Ga+ and Ga0 species, respectively. | ||
Surface acidity studies by 15N DNP SENS and FTIR
γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 catalysts were exposed to pyridine vapor at room temperature for 20 min, followed by outgassing at 150 °C for 15 min. The respective 15N DNP SENS spectra are presented in Fig. 4A. In these spectra, strong BAS are identified by a peak at ca. 205 ppm due to protonated pyridine; furthermore, strong LAS appear at ca. 235 ppm, mild LAS at ca. 260 ppm, while weak LAS and weak BAS are evidenced with a peak at ca. 280 ppm.43,44 γ-Ga2O3/SiO2–H2 reveals two main peaks at ca. 237 ppm and 286 ppm. The peak due to strong LAS at 237 ppm was not detected by us previously in γ-Ga2O3/SiO2 after evacuation at 100 °C.25 The peak at 286 ppm, most likely contains contributions from Py adsorbed on weak LAS of the gallia phase and Py interacting with mild/weak BAS of the silica support.25 Interestingly, Ga/SiO2–H2 shows a similar 15N DNP SENS spectrum compared to that of Ga/SiO2 (outgassed at 100 °C),25 and features a major peak due to Py on strong LAS at 236 ppm and a less intense peak due to Py on mild LAS at 260 ppm. In addition, a minor peak due to strong BAS at 206 ppm is present in Ga/SiO2–H2 (Fig. 4A). The difference between the intensities of peaks at 283 ppm and 286 ppm in, respectively, Ga/SiO2–H2 and γ-Ga2O3/SiO2–H2 is noteworthy and suggests a sizeable contribution of Py adsorbed on weak Ga-based LAS in γ-Ga2O3/SiO2–H2 compared to Ga/SiO2–H2.27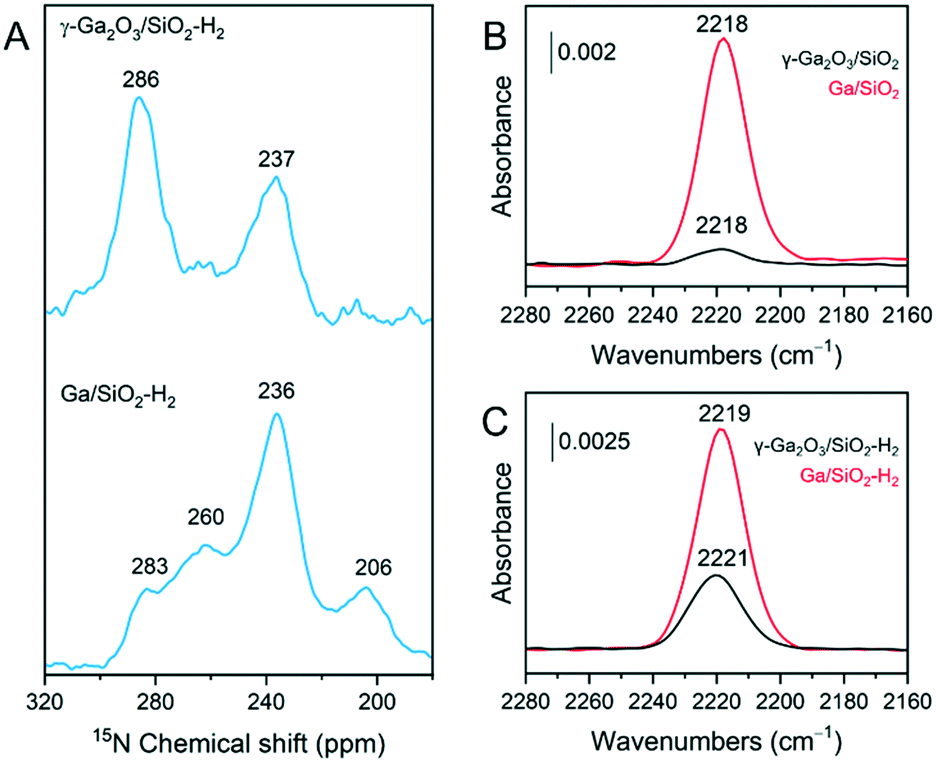 | ||
| Fig. 4 (A) 15N DNP SENS spectra of 15N-Py adsorbed on γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2. 15N-Py vapor was adsorbed at room temperature and desorbed under ca. 10−5 mbar during 15 min at 150 °C. CO adsorption transmission FTIR results for calcined (B) and H2-treated materials (C). Catalysts were heated to 540 °C and outgassed overnight at ca. 10−5 mbar. After cooling down to 50 °C, CO was introduced up to a pressure of ca. 27 mbar. | ||
Complementary FTIR experiments were carried out to characterize the surface acidity of the studied catalysts. Py adsorption FTIR (Py-FTIR) results show the emergence of a band at ca. 1620 cm−1 in γ-Ga2O3/SiO2–H2. As this band is absent in γ-Ga2O3/SiO2,25 the appearance of this band in γ-Ga2O3/SiO2–H2 is consistent with the formation of strong LAS after H2 pretreatment (Fig. S2†). The band at 1620 cm−1 is present in Ga/SiO2,25 and also in Ga/SiO2–H2 (Fig. S2†). On the other hand, bands due to Py on weak/mild BAS located at ca. 1595 and 1589 cm−1 are removed almost entirely in γ-Ga2O3/SiO2–H2 after outgassing at 100 °C, while these bands are detected in γ-Ga2O3/SiO2 after evacuation at 200 °C.
Adsorption of CO on the four studied catalysts allows to compare the relative intensities of bands due to CO interacting with strong LAS (Fig. 4B and C). CO adsorbed on γ-Ga2O3/SiO2 and Ga/SiO2 gives bands at ca. 2218 cm−1 for both materials, although the intensity of adsorbed CO is notably stronger on Ga/SiO2 relative to γ-Ga2O3/SiO2. CO adsorbed on Ga/SiO2–H2 gives a spectrum very similar to that of CO adsorbed on Ga/SiO2. In contrast, CO adsorbed on γ-Ga2O3/SiO2–H2 gives a peak shifted slightly to higher wavenumbers, i.e. centered at 2221 cm−1, and with a notably higher intensity than in γ-Ga2O3/SiO2, although the intensity is still lower than in Ga/SiO2 or Ga/SiO2–H2.
Hydrogen adsorption FTIR
The heterolytic splitting of H2 on Ga–O linkages was used to identify and characterize surface sites capable to dissociate H2 in the calcined and H2-treated materials. Dissociation of H2 forms GaH and OH sites and the position of the GaH bands scales with the bond strength and is indicative of the coordination geometry of Ga atoms, i.e., it has been reported that GaIV–H bands are found at ca. 2003 cm−1 and GaVI–H bands at ca. 1980 cm−1 (for unsupported gallia).45 Calcined and in situ H2-treated materials were evacuated at 540 °C for 2 h under ca. 10−4 mbar. Subsequently, adsorption of H2 of ca. 70 mbar was conducted at 300, 450 and 540 °C. Fig. 5A and B compare FTIR spectra after dissociation of H2 focusing on the location of the Ga hydride bands of calcined γ-Ga2O3/SiO2 and Ga/SiO2, respectively. Fig. 5C and D show the spectra obtained after dissociation of H2 on γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2, respectively. The dissociation of H2 on γ-Ga2O3/SiO2 gives bands due to gallium hydrides at ca. 2037, 2012 and 1986 cm−1 (Fig. 5A). In Ga/SiO2, narrower bands are found at higher frequencies at ca. 2073, 2058 and 2037 cm−1 (Fig. 5B). The dissociation of H2 at 300 °C on γ-Ga2O3/SiO2–H2 produces bands at ca. 2073, 2058 and 2041 cm−1 (Fig. 5C). These bands resemble closely the gallium hydride bands in Ga/SiO2 (Fig. 5B). Dissociation of hydrogen on γ-Ga2O3/SiO2–H2 at 450 and 540 °C yields an additional broad, lower frequency band at ca. 2021 cm−1 (Fig. 5C). Except for a slight change of the relative intensities between the different bands, no notable shifts or appearance of new bands were detected on Ga/SiO2–H2 at 300, 450 and 540 °C. When comparing Ga/SiO2–H2 with Ga/SiO2 at the three different temperatures, no substantial band shifts were detected between Ga/SiO2–H2 and Ga/SiO2, although a shoulder at 2021 cm−1 becomes notably more intense in Ga/SiO2–H2 (at 450 and 540 °C) and the partial intensities of other bands change after H2 treatment (Fig. 5D).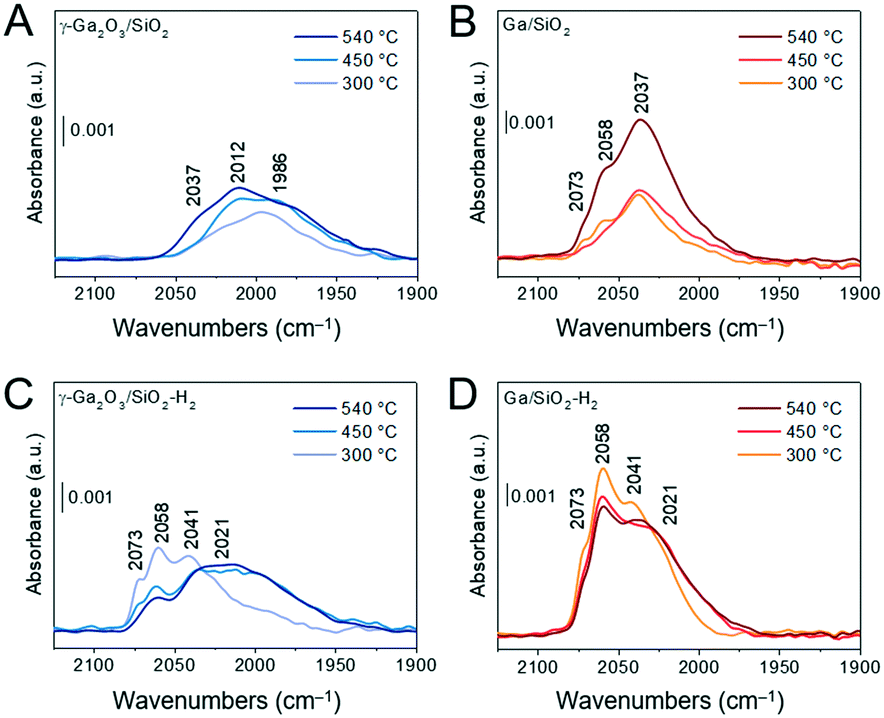 | ||
| Fig. 5 (A)–(D) Transmission FTIR results of H2 dissociation experiments. Panels show Ga-hydride bands for γ-Ga2O3/SiO2 (A); Ga/SiO2 (B); γ-Ga2O3/SiO2–H2 (C); Ga/SiO2–H2 (D). Materials in self-supported pellets were outgassed for 2 h under ca. 10−4 mbar at 540 °C. Hydrogen adsorption was conducted at 300 °C, 450 °C and 540 °C at the equilibrium pressure of ca. 70 mbar of H2. The hydrogen exposure time at the given temperature was ca. 1–2 min, after which the specimen was cooled down to ambient temperature. H2-Treated materials were prepared in situ by heating γ-Ga2O3/SiO2 and Ga/SiO2 in ca. 130 mbar of H2 at 550 °C for 2 h. | ||
Lastly, STEM-EDX mappings were used to characterize the dispersion of Ga in the H2-treated catalysts and compare it to calcined catalysts. The images tentatively suggest a higher degree of dispersion of Ga in both H2-treated catalysts relative to the respective calcined catalysts (Fig. S3†).
Discussion
In this work, we aimed to investigate the effect of H2 pretreatment (550 °C, 2 h, undiluted H2) on the catalytic performance and structure of two silica-supported PDH catalysts, one is Ga/SiO2 derived from gallium nitrate containing mostly an amorphous gallia phase, while the other one is a γ-Ga2O3/SiO2 catalyst, obtained by the incipient wetness impregnation of colloidal γ-Ga2O3 nanocrystals onto the SiO2 support. After calcination, γ-Ga2O3 nanocrystals transform partially into β-Ga2O3, yielding SiO2-supported, mixed-phase γ/β-Ga2O3 NPs. Calcined γ-Ga2O3/SiO2 and Ga/SiO2 have been characterized in detail in our previous report.25The Ga/SiO2–H2 catalyst yields a low rate of propene formation (stable at around 3.5 mol C3H6 mol Ga−1 h−1), and its selectivity to propene is only ca. 50%. This catalyst does not deactivate with TOS and does not deposit any measurable amount of coke. Overall, the activity, selectivity and stability of Ga/SiO2–H2 and Ga/SiO2 are very similar (Fig. 1). In contrast, while the initial activity and selectivity of γ-Ga2O3/SiO2–H2 and γ-Ga2O3/SiO2 are similar (10.3 mol C3H6 mol Ga−1 h−1 and 72%, respectively), after 104 min TOS the activity and selectivity of γ-Ga2O3/SiO2–H2 are notably higher than that of γ-Ga2O3/SiO2. In addition, γ-Ga2O3/SiO2–H2 deposits ca. 25% less coke as compared to γ-Ga2O3/SiO2. Therefore, H2 pretreatment attenuates the deactivation of calcined γ-Ga2O3/SiO2 with TOS, primarily owing to a less pronounced decrease of the propene formation rate and the selectivity to propene with TOS, in combination with a lower degree of coking. In what follows, we rationalize the differences in the structure (bulk and surface) between H2-treated and calcined γ-Ga2O3/SiO2 and Ga/SiO2 catalysts and relate this structural insight to the improved catalytic performance of γ-Ga2O3/SiO2–H2 discussed above.
Ex situ XAS (XANES and EXAFS analyses) experiments performed at the Ga K-edge in air-tight conditions suggest a change in the local coordination of Ga that is explained by a higher fraction of GaIV sites in γ-Ga2O3/SiO2–H2 than in γ-Ga2O3/SiO2 as seen by a higher intensity of the white line feature at 10375 eV and a decrease in the average Ga–O distance in γ-Ga2O3/SiO2–H2. Hydrogen treatment of γ-Ga2O3/SiO2 leads to a strong decrease in the coordination number of the Ga–Ga sphere; to a lower extent, this is also observed for Ga/SiO2. This result indicates that H2 treatment increases disorder of the supported γ/β-Ga2O3 phase in the γ-Ga2O3/SiO2–H2 catalyst. However, a loss in coherence length in γ-Ga2O3/SiO2–H2 is not revealed by dPDF analysis, yet this analysis also suggests an increased disorder, particularly in the short range structure (1.5–6 Å). Interestingly, a comparison of the dPDF data for the unsupported γ/β-Ga2O3 NPs before and after H2 treatment (500 °C) shows no detectable differences neither in the short range structure, nor in the medium-to-long range order.46 STEM-EDX imaging suggests that the agglomeration of the gallia phase on the silica support in both reduced catalysts may be lower relative to the calcined catalysts (Fig. S3†).
In the in situ XANES experiments an additional (lower energy) edge feature is observed in γ-Ga2O3/SiO2 under H2 flow above ca. 400 °C. The appearance of such a feature in the XANES spectra (ca. 10370–10373 eV) of reduced silica-supported gallia catalysts and Ga-containing zeolites during in situ H2 treatment has been ascribed previously either to the reduction of Ga3+ to Ga+, or Ga0 species, or to a non-reductive change of the coordination environment of Ga3+ sites, including the formation of Ga-hydrides or Ga-alkyls.16,20,21,23,24 Thus, the observed feature in the in situ XANES data of γ-Ga2O3/SiO2 is consistent with a partial reduction of γ/β-Ga2O3 in the γ-Ga2O3/SiO2 material and/or a change in the coordination environment of Ga. Interestingly, this feature disappears after the cooling down of γ-Ga2O3/SiO2 under a He flow, in accordance with the lack of this specific low-energy edge feature in the ex situ XANES experiments, and may be due to i) the reoxidation of reduced Ga species by the protons of water or surface silanol groups or ii) surface relaxation leading to a loss of low-coordinated GaIII sites formed at high temperature during H2 treatment.
Analysis of the Ga 2p XPS region confirms clearly the reduction of Ga3+ sites in both catalysts after exposing them in the pretreatment chamber to 300 mbar of H2 for 1 h at 300 and 450 °C. The reduction is evidenced by the appearance of two features at lower BE ascribed to Ga+ or Ga0 states. The reduction of Ga3+ sites proceeds, according to XPS data fits, to a larger extent in γ-Ga2O3/SiO2–H2 than in Ga/SiO2–H2. The reduction of Ga3+ sites is not detected in the Ga 3d region (Fig. S4 and S5†). This is explained by the lower penetration depth for the Ga 2p relative to the Ga 3d XPS region analysis (the inelastic mean free path, λ, is ca. 0.9 and 2.4 nm for the Ga 2p and Ga 3d electron energies, respectively), and therefore the formation of Ga+ or Ga0 states is mostly a surface phenomenon. Ga+ or Ga0 states have been observed previously by XPS of H2-treated Ga2O3/SiO2 and the formation of metallic Ga0 was explained by the disproportionation of Ga+ into Ga3+ and Ga0; it has been suggested that the high vacuum conditions of the XPS measurement may destabilize Ga+ species and induce disproportionation.19
It is interesting to note that Ga+ sites formed during H2 pretreatment at 650 °C of a silica-supported amorphous gallia catalyst have been suggested to be inactive in PDH as the reaction rate declined by ca. 30% after the H2 pretreatment.20 In addition, deactivation of the highly selective (≥93%) single-site [(SiO)3Ga–(XOSi)] PDH catalyst after 20 h of TOS at 550 °C has tentatively been linked to the reduction of Ga3+ surface sites to Ga+, as seen by the appearance of a low-energy edge feature in the Ga K edge XANES spectrum.21 In contrast to these results, H2 pretreatment attenuates the deactivation of γ-Ga2O3/SiO2 with time on stream despite the formation of reduced Ga species. This may be explained by the low amount of reduced Ga species and/or their subsequent disproportionation into Ga3+ and Ga0.
Comparison of the Ga–O sites that are capable of heterolytically dissociating H2 in γ-Ga2O3/SiO2 and Ga/SiO2 reveals that three major GaH bands appear in Ga/SiO2 at higher wavenumbers (2073, 2058 and 2037 cm−1) than in γ-Ga2O3/SiO2 (2037, 2012 and 1986 cm−1). This is consistent with stronger GaH bonds in Ga/SiO2 which, assuming these Ga3+–O sites are also active in PDH, may be difficult to regenerate by H2 coupling, leading to a lower catalytic activity of Ga/SiO2. Interestingly, higher frequency GaH bands that are similar to those in Ga/SiO2 and Ga/SiO2–H2 can be observed in γ-Ga2O3/SiO2–H2, most clearly when using 300 °C for H2 dissociation. At higher dissociation temperatures of 450 °C or 540 °C, the main GaH bands in γ-Ga2O3/SiO2–H2 are similar to those detected in γ-Ga2O3/SiO2 such as the broad lower frequency bands at ca. 2021 cm−1, although the bands at the higher wavenumbers, typical for Ga/SiO2 and Ga/SiO2–H2, remain (Fig. 5). Comparison of the GaH bands obtained using silica-supported catalysts of this work with that of unsupported γ/β-Ga2O3 NPs after H2 pretreatment (500 °C) indicates that the more active catalysts feature lower-frequency GaH bands (corresponding to weak GaH bonds), and that the number of distinct GaH bands increases in silica-supported catalysts, likely due to the appearance of interfacial sites (with the support).46 Consequently, the Ga-weight normalized activity of silica-supported catalysts is higher with respect to the unsupported catalysts, viz. initial activity of γ-Ga2O3/SiO2 is ca. 4.8 times higher relative to γ-Ga2O3, while the surface area of γ-Ga2O3/SiO2 is only ca. 2 times higher.
15N DNP SENS experiments probe the surface acidity and reveal that the intensity of the peak at ca. 286 ppm (desorption temperature 150 °C), which likely has contributions from Py bonded to weak Ga3+ LAS and mild/weak BAS (silanols or GaOH sites, which do not protonate pyridine), is notably higher in γ-Ga2O3/SiO2–H2 relative to Ga/SiO2–H2. Py-FTIR experiments show that while Py bonded to mild/weak BAS (bands at ca. 1595 and 1589 cm−1) is almost entirely removed from γ-Ga2O3/SiO2–H2 at 100 °C pyridine desorption temperature (Tdes), the band at 1595 cm−1 remains on γ-Ga2O3/SiO2 at Tdes = 200 °C (Fig. S2†). This may point at the consumption of some of the surface silanols by the reduced Ga species (Ga+ or Ga0) with the formation of GaIV sites (vide infra). Given that the Ga loadings in γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 are similar, it is likely that the fraction of weak LAS is higher in γ-Ga2O3/SiO2–H2 than in Ga/SiO2–H2 and this contributes to the higher initial activity of γ-Ga2O3/SiO2–H2 compared to Ga/SiO2–H2. Both γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2 contain a peak due to Py on strong Ga3+ LAS at 236–237 ppm. FTIR experiments with CO probe molecule indicate a notably higher intensity of the CO–Ga3+ adduct in γ-Ga2O3/SiO2–H2 compared to γ-Ga2O3/SiO2. An increased dispersion of the gallia phase in the H2-treated material may contribute to this increase of intensity of bound CO. However, since a similar increase of dispersion is also observed in Ga/SiO2–H2 but the intensity of adsorbed CO for this material increases only slightly relative to Ga/SiO2, it is likely that the formation of additional strong LAS in γ-Ga2O3/SiO2–H2 is responsible for the increased intensity of bound CO in γ-Ga2O3/SiO2–H2 and the blue shift of the CO band (from 2218 to 2221 cm−1). The formation of strong LAS in γ-Ga2O3/SiO2–H2 is also consistent with Py-FTIR data which shows the emergence of a band at ca. 1620 cm−1 in γ-Ga2O3/SiO2–H2. This band is weak in γ-Ga2O3/SiO2 but is notably stronger in Ga/SiO2 and Ga/SiO2–H2 (Fig. S2†). We cannot exclude that the formation of these strong LAS is related to the dispersion of Ga after interaction of reduced Ga+ or Ga0 species with surface silanols (Scheme 1). These reduced species may disproportionate or get oxidized by H2O forming tetrahedral Ga3+ sites. Consistent with this hypothesis is the fact that strong BAS are observed in Ga/SiO2–H2 and previously also in Ga/SiO2, i.e., in materials where an amorphous gallia phase likely co-exists with a small amount of a gallosilicate phase that is responsible for the strong Brønsted acidity. However, we do not observe substantial amounts of strong BAS in γ-Ga2O3/SiO2–H2 (or γ-Ga2O3/SiO2).25 The higher amounts of reduced Ga species in γ-Ga2O3/SiO2 relative to Ga/SiO2 may be due to the presence of gallosilicate species in Ga/SiO2, which are not reduced in the conditions used in this work.
 | ||
| Scheme 1 Proposed mechanism for formation of new GaIV sites in γ-Ga2O3/SiO2–H2 through the reductive disordering of γ/β-Ga2O3 NPs and the subsequent interaction of Ga+ and/or Ga0 species with the surface silanols. | ||
Overall, the attenuated deactivation of γ-Ga2O3/SiO2–H2 (due to the more stable selectivity to propene and reduced coking with TOS) correlates with an increased disorder of γ/β-Ga2O3 NPs in γ-Ga2O3/SiO2–H2. dPDF analysis of in situ X-ray total scattering data suggests that the structural disorder of silica-supported γ/β-Ga2O3 NPs increases during H2 pretreatment and remains only in the local structure of γ/β-Ga2O3. The surface reduction of Ga3+ sites is confirmed by the Ga 2p XPS spectra and, potentially, also by in situ XANES experiments (appearance of a low-energy edge feature). H2 pretreatment leads to the dissociation of hydrogen and the formation of several gallium hydride sites, some of which are similar in γ-Ga2O3/SiO2–H2, Ga/SiO2–H2 and Ga/SiO2 but are not observed in γ-Ga2O3/SiO2. The additional formation of GaIV sites in γ-Ga2O3/SiO2–H2 (according to a comparison of the XANES and EXAFS data of γ-Ga2O3/SiO2–H2 and γ-Ga2O3/SiO2) is responsible for more abundant strong Lewis acidity in this material (according to FTIR experiments with CO and Py probe molecules). We propose that those GaIV sites are mostly due to disordered γ/β-Ga2O3 NPs, yet, there may be a contribution to the formed GaIV-based strong LAS from the interaction of reduced gallium species (Ga+ and/or Ga0) with silica (Scheme 1), as indicated by the shorter distances of the second coordination sphere of Ga sites after H2 pretreatment. An alternative mechanism for the formation of new GaIV sites is the disproportionation of Ga+ species (to Ga3+ and Ga0). However, while the interaction of Ga+ and/or Ga0 is expected to be comparable in both γ-Ga2O3/SiO2–H2 and Ga/SiO2–H2, and the catalytic performance of Ga/SiO2–H2 remains unchanged relative to Ga/SiO2 (in contrast to γ-Ga2O3/SiO2–H2 and γ-Ga2O3/SiO2), the newly formed GaIV sites in disordered γ/β-Ga2O3 NPs of γ-Ga2O3/SiO2–H2 are likely responsible for the attenuated decrease of the propene selectivity with TOS for this material. Further, 15N DNP SENS and Py-FTIR data indicate that weak LAS are more abundant in γ-Ga2O3/SiO2–H2 than in Ga/SiO2–H2, and this correlates with the higher initial activity of γ-Ga2O3/SiO2–H2 and the presence of lower frequency GaH bands in this catalyst. Note that the cracking rate on γ-Ga2O3/SiO2–H2 has decreased by ca. 25% relative to γ-Ga2O3/SiO2, and it remained unchanged (within the experimental error) for Ga/SiO2–H2 relative to Ga/SiO2. Thus, the attenuated loss of activity and propene selectivity with TOS for γ-Ga2O3/SiO2–H2 (relative to γ-Ga2O3/SiO2) is probably due to the formation of less active but more stable GaIV sites (due to reduced coking) in the more disordered γ/β-Ga2O3 and/or consumption of unselective Ga sites during the H2 treatment, while the higher initial activity and selectivity of silica-supported catalysts based on γ/β-Ga2O3 NPs relative to amorphous gallia is due to the presence of weak LAS in γ/β-Ga2O3 NPs.
Conclusions
We reported that H2 pretreatment (550 °C, 2 h) of γ-Ga2O3/SiO2, a PDH catalyst containing silica-supported nanocrystalline γ/β-Ga2O3 NPs, attenuates its deactivation with time on stream. At the same time, H2 pretreatment does not impact notably the catalytic performance of a Ga/SiO2 catalyst containing an amorphous gallia phase and a small amount of a gallosilicate phase, identified by its strong Brønsted acidity. We observe that on the silica support, H2 pretreatment induces a reductive disorder of nanocrystalline γ/β-Ga2O3 NPs. This reaction yields additional strong Lewis acid sites, but does not generate strong Brønsted acid sites (that is, it avoids forming a gallosilicate phase). These new sites in the γ-Ga2O3/SiO2–H2 catalyst may form due to the disproportionation of Ga+ species (observed by XPS), the anchoring of Ga+ or Ga0 species onto silanol groups, and/or an H2-induced local disordering of nanocrystalline γ/β-Ga2O3 NPs; the latter mechanism appears to contribute the most. Our results suggest that the active sites in the silica-supported gallia catalysts based on strong LAS (i.e., GaIV sites formed after the H2 pretreatment) are less active but more stable with TOS than weak LAS of γ/β-Ga2O3 NPs.Author contributions
P. C.-F. and A. F. designed the research project and planned the experimental work. P. C.-F. carried out the preparation of materials, characterization and testing of the catalysts, and data analysis. A. I. S. performed hydrogen adsorption FTIR experiments and supervised data interpretation. A. V. Y. carried out 15N DNP SENS experiments, processed the results and contributed to data analysis. I. P. P. and A. V. B. conducted XPS experiments, data processing and analysis. P. M. A. supervised XAS and dPDF experiments and contributed to the data analysis. The data were discussed among all co-authors. P. C.-F. and A. F. wrote the first version of the draft that was edited by all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Dr. Agnieszka Kierzkowska is thanked for STEM-EDX measurements and ICP analysis. Dr. Dragos Stoian (SNBL/ESRF) is acknowledged for his support during XAS measurements. The Swiss National Science Foundation is acknowledged for the partial funding of this work (grant IZSEZ0_178677). P. M. A. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 819573). A. V. Y. and C. C. gratefully acknowledge ETH+ Project SynthMatLab for the financial support. I. P. P. and A. V. B. thank the budget project of the Ministry of Science and Higher Education of the Russian Federation (AAAA-A21-121011390011-4) for support of the XPS measurements at the Boreskov Institute of Catalysis.Notes and references
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- Z. P. Hu, D. Yang, Z. Wang and Z. Y. Yuan, Chin. J. Catal., 2019, 40, 1233–1254 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- S. R. Docherty, L. Rochlitz, P. A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- A. Bhan and W. N. Delgass, Catal. Rev.: Sci. Eng., 2008, 50, 19–151 CrossRef CAS.
- J. R. Mowry, D. C. Martindale and A. H. P. Hall, Arabian J. Sci. Eng., 1985, 10, 367–375 CAS.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2405 CrossRef CAS PubMed.
- V. Kanazirev, G. L. Price and G. Tyuliev, Zeolites, 1992, 12, 846–850 CrossRef CAS.
- M. W. Schreiber, C. P. Plaisance, M. Baumgärtl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS PubMed.
- N. M. Phadke, E. Mansoor, M. Bondil, M. Head-Gordon and A. T. Bell, J. Am. Chem. Soc., 2019, 141, 1614–1627 CrossRef CAS PubMed.
- E. Mansoor, M. Head-Gordon and A. T. Bell, ACS Catal., 2018, 8, 6146–6162 CrossRef CAS.
- N. M. Phadke, J. Van Der Mynsbrugge, E. Mansoor, A. B. Getsoian, M. Head-Gordon and A. T. Bell, ACS Catal., 2018, 8, 6106–6126 CrossRef CAS.
- E. J. M. Hensen, M. García-Sánchez, N. Rane, P. C. M. M. Magusin, P. H. Liu, K. J. Chao and R. A. Van Santen, Catal. Lett., 2005, 101, 79–85 CrossRef CAS.
- G. D. Meitzner, E. Iglesia, J. E. Baumgartner and E. S. Huang, J. Catal., 1993, 140, 209–225 CrossRef CAS.
- A. I. Serykh and M. D. Amiridis, Surf. Sci., 2009, 603, 2037–2041 CrossRef CAS.
- I. Takahara, M. Saito, M. Inaba and K. Murata, Catal. Lett., 2004, 96, 29–32 CrossRef CAS.
- A. I. Serykh and M. D. Amiridis, Surf. Sci., 2010, 604, 1002–1005 CrossRef CAS.
- V. J. Cybulskis, S. U. Pradhan, J. J. Lovón-Quintana, A. S. Hock, B. Hu, G. Zhang, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Catal. Lett., 2017, 147, 1252–1262 CrossRef CAS.
- K. Searles, G. G. Siddiqi, O. V. Safonova and C. Copéret, Chem. Sci., 2017, 8, 2661–2666 RSC.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- L. Rochlitz, K. Searles, D. F. Nater, S. R. Docherty, D. Gioffrè and C. Copéret, Helv. Chim. Acta, 2021, 104, e2100078 CrossRef CAS.
- A. Getsoian, U. Das, J. Camacho-Bunquin, G. Zhang, J. R. Gallagher, B. Hu, S. Cheah, J. A. Schaidle, D. A. Ruddy, J. E. Hensley, T. R. Krause, L. A. Curtiss, J. T. Miller and A. S. Hock, Catal. Sci. Technol., 2016, 6, 6339–6353 RSC.
- P. Castro-Fernández, D. Mance, C. Liu, I. B. Moroz, P. M. Abdala, E. A. Pidko, C. Copéret, A. Fedorov and C. R. Müller, ACS Catal., 2021, 11, 907–924 CrossRef.
- W. Jochum, S. Penner, K. Föttinger, R. Kramer, G. Rupprechter and B. Klötzer, J. Catal., 2008, 256, 268–277 CrossRef CAS.
- P. Castro-Fernández, M. Kaushik, Z. Wang, D. Mance, E. Kountoupi, E. Willinger, P. M. Abdala, C. Copéret, A. Lesage, A. Fedorov and C. R. Müller, Chem. Sci., 2021, 12, 15273–15283 RSC.
- M. Chen, J. Xu, F. Z. Su, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- W. Van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- C. S. S. R. Kumar, X-ray and Neutron Techniques for Nanomaterials Characterization, Springer Nature, Berlin, 2016 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Åhman, G. Svensson and J. Albertsson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1336–1338 CrossRef.
- P. J. Chupas, K. W. Chapman and G. J. Halder, J. Am. Chem. Soc., 2011, 133, 8522–8524 CrossRef CAS PubMed.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- A. Zagdoun, A. J. Rossini, D. Gajan, A. Bourdolle, O. Ouari, M. Rosay, W. E. Maas, P. Tordo, M. Lelli, L. Emsley, A. Lesage and C. Copéret, Chem. Commun., 2012, 48, 654–656 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- P. Castro-Fernández, M. V. Blanco, R. Verel, E. Willinger, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2020, 124, 20578–20588 CrossRef.
- K. Nishi, K. Shimizu, M. Takamatsu, H. Yoshida, A. Satsuma, T. Tanaka, S. Yoshida and T. Hattori, J. Phys. Chem. B, 1998, 102, 10190–10195 CrossRef CAS.
- K. Groden, F. D. Vila, L. Li, S. R. Bare, S. L. Scott and J.-S. McEwen, J. Phys. Chem. C, 2021, 125, 27901–27908 CrossRef CAS.
- L. Yang, J. L. Bourque, J. A. McLeod, P. Shen, K. M. Baines and L. Liu, Inorg. Chem., 2017, 56, 2985–2991 CrossRef CAS PubMed.
- R. Carli and C. L. Bianchi, Appl. Surf. Sci., 1994, 74, 99–102 CrossRef CAS.
- I. B. Moroz, K. Larmier, W.-C. Liao and C. Copéret, J. Phys. Chem. C, 2018, 122, 10871–10882 CrossRef CAS.
- W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Roman-Leshkov, J. Phys. Chem. C, 2016, 120, 28533–28544 CrossRef CAS PubMed.
- S. E. Collins, M. A. Baltanás and A. L. Bonivardi, Langmuir, 2005, 21, 962–970 CrossRef CAS PubMed.
- P. Castro-Fernández, D. Mance, C. Liu, P. Abdala, E. Willinger, A. A. Rossinelli, A. I. Serykh, E. A. Pidko, C. Copéret, A. Fedorov and C. R. Müller, J. Catal., 2022, 408, 155–164 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, XAS, FTIR, STEM-EDX, XPS, dPDF and 15N DNP SENS experimental parameters. See DOI: https://doi.org/10.1039/d2cy00074a |
| This journal is © The Royal Society of Chemistry 2022 |