
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the multiple interactions in vanadium-based SCR catalysts during simultaneous NOx and soot abatement†
Lei
Zheng
a,
Maria
Casapu
 a and
Jan-Dierk
Grunwaldt
*ab
a and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstraße 20, 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
First published on 25th April 2022
Abstract
The recently proposed 2-way SCRonDPF systems, which consist of selective catalytic reduction (SCR) catalysts coated on diesel particulate filters (DPFs), are promising to simultaneously remove NOx and soot emissions. However, such multifunctional systems are very demanding due to the presence of various interacting phases as well as concurrent reactions competing for the same active sites. In the present study, a simple to complex strategy was employed to understand the multiple interactions during combined NOx and soot removal over a V-based SCR catalyst. First the effect of NO2, NO, H2O and NH3 on soot oxidation was studied on soot alone and then in loose and tight contact with the V-based catalyst. In a next step, the effect of various SCR gas mixtures was investigated. For the gas–soot interplay, NO2, NO and NH3 were found to exhibit a promotional, non-inhibitory and inhibitory effect on soot oxidation, respectively. Ammonia–soot interaction dominates the soot oxidation in a standard SCR gas feed if no catalyst is present, while the co-presence of NO2 in the fast SCR gas mixture results in a faster soot oxidation. For triple-phase systems involving also the V2O5–WO3/TiO2 catalyst, the oxidation of soot begins only after the activation of the standard SCR gas components over the catalyst. In contrast, NO2 directly interacts with the soot, irrespective of the presence or absence of the V-catalyst. Water was found to promote soot oxidation for all investigated reaction conditions. Interestingly, a small amount of soot was identified to enhance the NOx conversion at high temperatures. All in all, a full picture of the promotional and inhibitory effects of SCR gases on soot oxidation could be developed, which is important for further improving 2-way SCRonDPF systems.
1. Introduction
NOx and particulate matter (PM), as two major pollutants released from diesel engines, are typically removed sequentially by different catalytic components in the exhaust gas aftertreatment system.1–3 The diesel particulate filter (DPF) is nowadays the widely employed technology for trapping particulate matter,4 and the downstream well-established selective catalytic reduction (SCR) of NOx with ammonia is applied for NOx emission control.3,5 Although such a combination can lead to very low emissions, it occupies a large volume, which is critical especially for light-duty vehicles.6 Another disadvantage entails insufficient low-temperature activity during the engine cold start process. In recent years, a novel concept of coating SCR catalysts on the highly porous wall of the DPF has been proposed and this leads to an integrated 2-way SCRonDPF system (also referred to as SCR/DPF, SDPF or SCRF).7–16 The simultaneous NOx and soot removal can therefore be achieved in such multi-functional systems.17–23 The more compact design can reduce the overall volume and costs significantly. Moreover, an earlier conversion of pollutants is achieved due to the faster heating of the catalytic system, if this is located closer to the engine.11,15Cu-exchanged zeolites, due to their high NH3-SCR of NOx activity and thermal stability, have been evaluated for coating on filters by several research groups.9–13 Decreased NH3 storage and lower NO conversion were found for a soot-loaded DPF coated with a Cu-containing zeolite washcoat.24 Furthermore, such catalysts are sensitive to sulfur poisoning, and therefore not optimal for applications where a high sulfur resistance is required.14 On the other hand, vanadium-based SCR catalysts show good efficiency, are inexpensive and are sulfur resistant.1,5,25,26 Moreover, hydrocarbon and soot oxidation are promoted by conventional and more advanced V-based catalyst formulations,27,28 making them promising candidates for 2-way SCRonDPF applications. Kleinhenz et al.14 reported that a vanadium-based SCR catalyst coated DPF exhibited sufficient thermal stability as well as sulfur resistance for the emission control of marine engines. In addition, the SCRonDPF system coated with vanadium-based SCR catalysts was reported to be more efficient in NOx reduction in comparison to the corresponding Cu-zeolite coated system during the warmer NRTC (non-road transient cycle) test.15 In the presence of an upstream diesel oxidation catalyst that allows the NO2/NOx ratio to be managed, engine bench tests showed a high NOx conversion simultaneous with passive NO2–soot regeneration for a DPF coated with a vanadium SCR catalyst.29
Along with the evaluation of the catalytic performance, understanding of such multi-functional systems is essential for a knowledge-based catalyst design and process optimization.
For vanadium-based catalysts, the low oxygen bond strength,30 combined with the high mobility of vanadia, was reported to contribute to soot oxidation.31,32 During this process, the soot–catalyst contact is crucial.33–38 Furthermore, the gas mixture can as well affect this reaction. In this respect, NO2 as a more reactive oxidant than O2 was observed to considerably increase the soot conversion,39–41 also by supporting the reoxidation of the catalyst active sites.42,43 Based on the study of Trandafilović et al.13 ammonia was suggested to interact with the soot surface and form amines/amides (–C–NH2) that can inhibit further oxidation. Mehring et al.44 studied the SCR of NOx with ammonia over soot, and demonstrated its catalytic contribution to the fast SCR reaction by providing adsorption sites for ammonia and NOx species. In contrast, Schobing et al.45 investigated the simultaneous removal of soot and NOx on a commercial vanadium-based SCR catalyst mixed with carbon black and found that carbon black exhibits no significant impact on NOx reduction at low temperatures, however, it competes with the redox cycle of the SCR reaction above 400 °C. Hence, the current understanding of the interaction between the catalyst and soot is far from being satisfactory. This is mainly due to the high complexity of such combined 2-way SCRonDPF systems, where three different interacting phases, i.e. soot, SCR catalysts and gas mixtures, and the resulting two parallel processes, i.e. soot oxidation and SCR-related reactions, interact. Therefore, unraveling the triple-phase interactions between soot, the SCR catalyst and the gas mixture, i.e. gas–soot–catalyst interaction, is an essential step for further developing such systems.
In this regard, a simple to complex sequence, i.e. from gas–soot and gas–catalyst dual-phase systems to gas–soot–catalyst triple-phase systems, was employed in the present work to uncover the complicated gas–soot–catalyst interactions in an integrated 2-way SCRonDPF system based on a conventional V2O5–WO3/TiO2 catalyst. At first, the individual effects of each SCR-related gas component on soot oxidation were explored, and then, in a second step, this was extended to the SCR gas mixture effects. As a final step, the triple-phase interaction was investigated by adding the V2O5–WO3/TiO2 catalyst (in loose and tight contact, cf. ref. 33, 42 and 46) to the gas–soot systems.
2. Experimental section
2.1. Materials
A vanadium-based SCR catalyst with a composition of 3 wt% V2O5–10 wt% WO3/TiO2, referred to as Vcat thereafter, was used in the present work (c.f. characterization results in the ESI:† Fig. S2 and Table S1). The catalyst was prepared by incipient wetness impregnation (IWI), as described in detail in our previous study.47 In brief, a commercial W/TiO2 support (10 wt% WO3/TiO2, CristalACTiV™ DT-52) was used and the impregnation solution was prepared by dissolving ammonium metavanadate (Sigma-Aldrich) in deionized water with the addition of oxalic acid (Merck). The obtained sample was dried and calcined afterwards at 550 °C for 4 h in static air.Commercial carbon black (Alfa Aesar) with a surface area of 75 m2 g−1 was used as the reference material for the soot oxidation tests in the present study.
2.2. Catalytic tests
As detailed in Table 1, twelve SCR related gas mixtures were used to study the various interactions between the catalyst, soot and gas phase. The different gas mixtures were fed over the soot only, the catalyst only, and the soot–catalyst systems. Two catalyst–soot contact types (loose and tight contact, cf. ref. 33–35) were generated in this study, involving a low or a high number of contact points. The contact intimacy was adjusted in a qualitative manner based on mechanical processing of the two samples. To obtain a soot catalyst mixture in loose contact, 5 mg soot and 245 mg catalyst (125–250 μm) were stirred together with a spatula.| Gas mixture | Main factor | Gas mixture composition (total gas flow of 300 mL min−1) |
|---|---|---|
| Gas-1 | Inert (N2) | N2 |
| Gas-2 | Baseline (O2) | 10% O2 in N2 |
| Gas-3 | NO2 | 500 ppm NO2, 10% O2 in N2 |
| Gas-4 | NO | 500 ppm NO, 10% O2 in N2 |
| Gas-5 | NH3 | 500 ppm NH3, 10% O2 in N2 |
| Gas-6 | H2O | 5% H2O, 10% O2 in N2 |
| Gas-7 | NO in H2O | 500 ppm NO, 5% H2O, 10% O2 in N2 |
| Gas-8 | NO2 in H2O | 500 ppm NO2, 5% H2O, 10% O2 in N2 |
| Gas-9 | Dry standard SCR | 500 ppm NO, 500 ppm NH3, 10% O2 in N2 |
| Gas-10 | Dry fast SCR | 250 ppm NO, 250 NO2, 500 ppm NH3, 10% O2 in N2 |
| Gas-11 | Wet standard SCR | 500 ppm NO, 500 ppm NH3, 5% H2O, 10% O2 in N2 |
| Gas-12 | Wet fast SCR | 250 ppm NO, 250 NO2, 500 ppm NH3, 5% H2O, 10% O2 in N2 |
The tight contact was induced by crushing together the same amount of materials in an agate mortar and in a next step pressing and sieving the powder (125–250 μm). To ensure a sufficient soot–catalyst interface, a relatively low soot loading was applied in the present study, which allowed the investigation of their intrinsic interaction. For the dual gas–soot interaction tests, 5 mg soot was mixed with inert quartz sand (245 mg, 125–250 μm).
The employed samples (soot, catalyst and their mixtures) were placed in a quartz tube plug-flow reactor (ID = 8 mm) to carry out the experimental tests. Gases were dosed separately via mass flow controllers (Bronkhorst). Before sending it to the reactor, the gas mixture was preheated to 150 °C to avoid water condensation. The reactor was heated up from room temperature to 650 °C with a temperature ramp of 10 °C min−1. Further details of the test protocol are summarized in Fig. S1.† A MultiGasTM 2030 FTIR gas analyzer (MKS Instruments Deutschland GmbH, Munich, Germany) was applied to analyze the gas composition after the reactor. Although a low amount was used, the loaded soot was not entirely converted in all the tests as soot traces were observed after each reaction.
3. Results and discussion
Soot oxidation results obtained for the inert (N2, Gas-1) and baseline (10% O2/N2, Gas-2) gas feeds are presented in Fig. 1. Hardly any CO or CO2 could be detected in an inert gas atmosphere, except for the catalyst–soot tight contact system which showed 50 ppm CO + CO2 formation at 617 °C. In this case, the oxidation of soot is probably assisted by lattice oxygen from the catalyst components. In general, the results obtained in the presence of oxygen (Gas-2) were comparable to those reported in our previous study,47 and will be used as a baseline in the present work to assess the effects of the SCR gas components on soot oxidation. | ||
| Fig. 1 Comparison of gas evolution for (a) N2-containing systems and (b) O2-containing systems; Gas-1: N2 with a total gas flow of 300 mL min−1 (inert condition); Gas-2: 10% O2 in N2 with a total gas flow of 300 mL min−1 (baseline condition). 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
For all activity tests conducted in the present study, the CO + CO2 formation profiles resulting from soot oxidation consisted of several low intensity peaks (maximum <30 ppm) below 400 °C and a sharp increase up to several thousand ppm at higher temperatures.
For a better comparison, the temperature corresponding to the soot oxidation onset (To) and the temperature estimated at 50 ppm CO + CO2 formation (T50p) were derived, as illustrated in Fig. 1. Furthermore, the total CO + CO2 formation integrated for each test was normalized by that obtained from the oxidation of soot in 10% O2/N2 (baseline gas). The obtained ratio is referred to as NTCOx thereafter. For all the conducted tests, the NTCOx (Table S2†), To (Table S3†) and T50p (Table S4†) are summarized in the ESI.† To elucidate the multifaceted gas–soot–catalyst interactions, the evolution of all gaseous components, including C- and N-containing gases, was as well investigated in detail.
3.1. Effect of individual gas components on soot oxidation
 | ||
| Fig. 2 Comparison of gas evolution from NO2-containing systems: (a) CO + CO2 formation, the N-containing gas formation from the (b) gas–soot system, (c) gas–soot–catalyst system (loose) and (d) gas–soot–catalyst system (tight). Gas-3: 500 ppm NO2, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
Analogous to the catalytic soot oxidation by oxygen, the presence of NO2 barely influences the oxidation reaction if the catalyst is in loose contact with the soot sample. A similar T50p temperature (460 °C), total COx formation and gas evolution were observed (Fig. 2(c)) to the non-catalytic soot conversion in a NO2 + O2 gas mixture (Fig. 2(b)). Hence, under these conditions it can be concluded that the NO2–soot interaction controls the oxidation reaction and not the NO2–catalyst–soot interaction. However, if the soot is in tight contact with the V-catalyst its conversion is significantly enhanced. A new To peak with the maximum at 204 °C was observed, which was ascribed to the contribution of the oxygen from the metal oxide lattice of the catalyst. This assumption is supported by the similar To temperatures at 202 °C and 203 °C measured for the same system in inert and baseline feeds, respectively (Table S3†). In addition, a relatively earlier soot ignition (T50p of 377 °C) and a high total conversion (NTCOx of 25.9) were observed. These occur simultaneously with a slight NO2 reduction to NO above 250 °C, as shown in Fig. 2(d). The lower reduction temperature in comparison to that measured for the loose contact case (400 °C) could be due to the reoxidation of the active sites of the catalyst by NO2.
The contribution of NO2 to catalyst reoxidation as well as its direct interaction with soot has already been observed in earlier studies.40,42,43 Based on the results obtained in our investigations, it is clear that the NO2 effect is dependent on the interaction in the soot–catalyst systems, whether it is loose or tight, and as illustrated in Fig. 3. A two-fold role of NO2 in soot oxidation, i.e. in both a direct and indirect manner, is achieved in a tight contact system whereas mainly the direct oxidation of soot by NO2 was identified for the catalyst in loose contact with the soot particles.
 | ||
| Fig. 3 Comparison of the different NO2 responses from NO2–soot–catalyst systems in (a) loose and (b) tight contact. | ||
 | ||
| Fig. 4 Comparison of gas evolution from NO-containing systems: (a) CO + CO2 formation, the N-containing gas formation from the (b) gas–soot system, (c) gas–catalyst system and (d) gas–soot–catalyst systems (loose and tight). Gas-4: 500 ppm NO, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
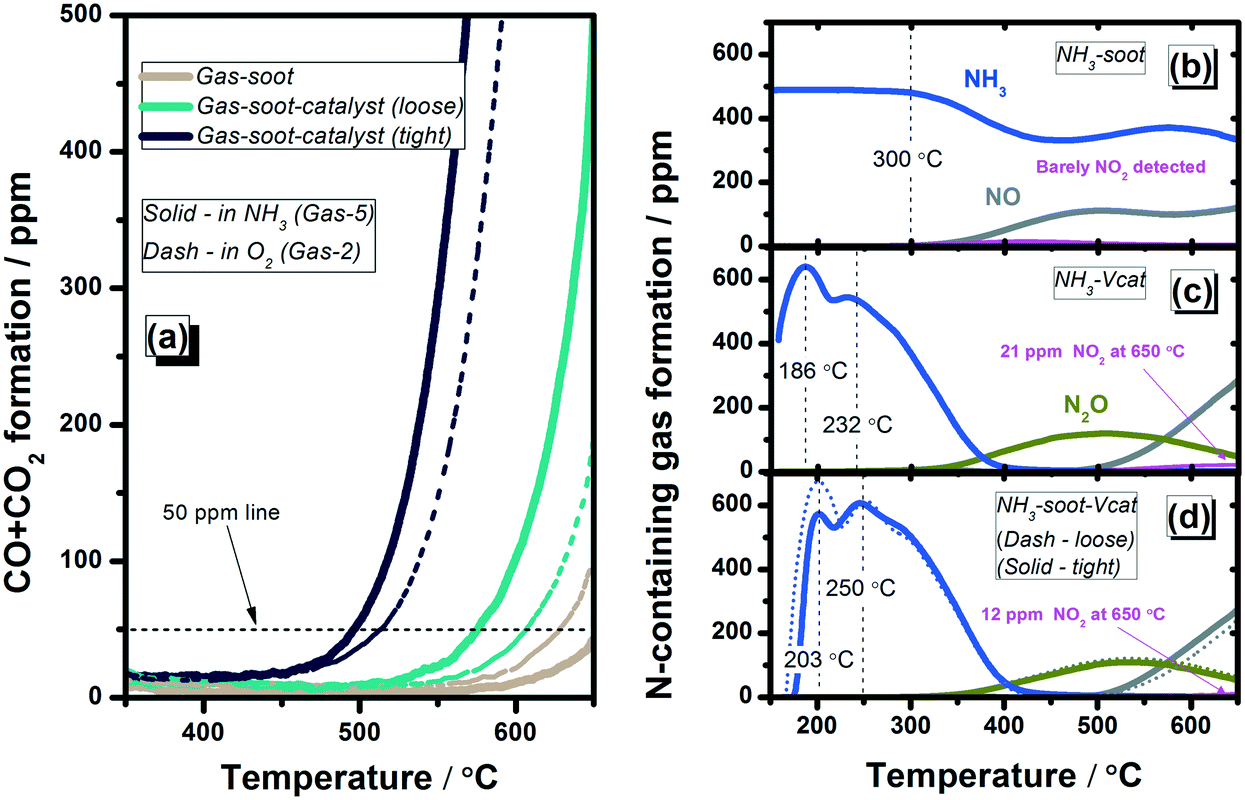 | ||
| Fig. 5 Comparison of gas evolution from ammonia-containing systems: (a) CO + CO2 formation, the N-containing gas formation from the (b) gas–soot system, (c) gas–catalyst system and (d) gas–soot–catalyst systems (loose and tight). Gas-5: 500 ppm NH3, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
When the ammonia gas mixture (Gas-5) was dosed directly to the V2O5–WO3/TiO2 catalyst, two desorption peaks were observed during light-off (Fig. 5(c)). The peaks at 186 °C and 232 °C can be assigned to physisorbed ammonia and ammonia stored on weak acidic sites, respectively.1,48 The latter feature is in line with the well-known acidity of vanadium-based SCR catalysts leading to ammonia storage.1,25 Ammonia concentration decreased rapidly with temperature, with full conversion achieved at around 400 °C. N2O as an oxidation product was detected from ca. 300 °C while NO and NO2 above 500 °C. During soot oxidation in the ammonia triple-phase system (Fig. 5(d)), no pronounced variation in the evolution of gases was observed in comparison to the ammonia–catalyst dual-phase system (Fig. 5(c)). This indicates that the gaseous ammonia preferentially interacts with the catalyst instead of the soot. Only a shift to higher temperatures in the ammonia desorption profile was noticed, which was more pronounced for the catalyst–soot tight contact system. Part of the NO2 formed by NH3 oxidation over the V-catalyst seems to react with the soot sample. On the one hand, this reaction is suggested by the lower NO2 concentration detected at the end of the plug-flow reactor. Only approx. 12 ppm NO2 was detected at 650 °C for the triple-phase systems (both loose and tight contact) in comparison to 21 ppm measured for the ammonia–catalyst dual-phase system (Fig. 5(c)). At the same time, an enhanced soot oxidation performance was achieved for ammonia fed loose and tight catalyst–soot systems, with lower T50p values of 573 °C and 496 °C, respectively. The obtained results indicate that even if ammonia itself inhibits the soot oxidation, the presence of the V-catalyst promotes the overall soot oxidation in an NH3 + O2 gas mixture via catalytic NO2 formation.
 | ||
| Fig. 6 Comparison of the CO + CO2 formation from water–soot–catalyst systems. Gas-6: 5% H2O, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
As a next step, the presence of water (additionally 5% H2O) in the NO- and NO2-containing gas feeds (Gas-7 and Gas-8, respectively) was investigated over the soot–catalyst systems (complete data sets depicted in Fig. S3 and S4†). Similarly, in those cases water addition promoted soot oxidation. Notably, for the case of soot–catalyst in loose contact the T50p was significantly shifted to lower temperatures, which were around 40 °C and 70 °C lower for NO- and NO2-containing gas feeds, respectively (Table S4†). At the same time, for both NO- and NO2-containing gas feeds the normalized total COx formations were almost two times higher under wet conditions compared to those in a dry reaction atmosphere (Table S2†).
3.2. Influence of SCR gas feeds on soot oxidation
 | ||
| Fig. 7 Comparison of gas evolution from dry standard SCR gas-containing systems: (a) CO + CO2 formation, the N-containing gas formation from the (b) gas–soot system, (c) gas–catalyst system and (d) gas–soot–catalyst systems (loose and tight). Gas-9: 500 ppm NO, 500 ppm NH3, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
After feeding the same gas mixture to the V2O5–WO3/TiO2 catalyst (Fig. 7(c)), an ammonia desorption peak at 180 °C was observed. Afterwards, the gas composition changes as a result of the SCR reactions during heating the reactor.27,28,51 Ammonia is fully oxidized above 400 °C, whereas NOx conversion drops in the same temperature range. Additionally, NO2 is formed reaching a concentration of 52.6 ppm at 550 °C. The addition of soot to the standard SCR gas–catalyst system did not significantly affect the NOx removal activity of the V-catalyst (Fig. 7(d)) in comparison to the gas–catalyst system (Fig. 7(c)). Only the ammonia desorption peaks showed a lower intensity and shifted to higher temperatures, i.e. 260 °C and 275 °C for loose and tight contact, respectively. This could be ascribed to the soot interaction with the catalyst surface. Similar to the standard SCR–catalyst reactions, complete ammonia conversion was found above 400 °C. Under these conditions, only 27.7 ppm NO2 was detected at 550 °C for the loose contact system, and even less for the case of tight soot–catalyst contact. In contrast to the gas–soot dual-phase system, the soot oxidation was initiated at a lower temperature after mixing with the V2O5–WO3/TiO2 catalyst. This seems to be caused not only by the soot–catalyst interaction, but also by the diminishment of ammonia concentration and the appearance of NO2. Hence, lower ignition temperatures were measured for both loose and tight contact systems, with T50p of 559 °C and 467 °C respectively, in comparison to the corresponding systems during oxidation by O2 only (Fig. 7(a)).
 | ||
| Fig. 8 Comparison of gas evolution from dry fast SCR gas-containing systems: (a) CO + CO2 formation, the N-containing gas formation from the (b) gas–soot system, (c) gas–catalyst system and (d) gas–soot–catalyst systems (loose and tight). Gas-11: 250 ppm NO, 250 ppm NO2, 500 ppm NH3, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
In the presence of VWTi (Fig. 8(c)), the fast SCR reaction ignites already at very low temperatures (approx. 80% NOx conversion at 150 °C) and hardly any ammonia emission could be detected in the investigated temperature range. NO and NO2, which could not be detected below 400 °C, appeared at higher temperatures. Analogous to the dry standard SCR conditions, the simultaneous soot oxidation does not significantly affect the fast SCR activity (Fig. 8(d) in comparison to Fig. 8(c)). After modification over the catalyst surface due to the fast SCR reaction, the resulting gas mixture exhibited a further promotional effect on soot oxidation in the triple-phase systems. As shown in Fig. 8(a), rather low T50p values of 548 °C and 468 °C were achieved for the fast SCR gas–soot–catalyst systems with loose and tight contact, respectively.
3.3. Influence of soot on SCR of NOx with ammonia
With respect to the water presence, its promoting effect was noticed during soot oxidation in the wet standard and fast SCR gas mixtures (additionally 5 vol% H2O) in the presence or absence of a catalyst, and the results are shown in Fig. S5 and S6,† respectively. As discussed previously, the water promoting influence is claimed to be due to a wetting effect which enhances the contact between soot and reactants.49 Additionally, active OH groups or other O-species are formed, which enhance soot oxidation.41,50,52 The influence of soot on the NOx conversion during both standard and fast SCR is summarized in Fig. 9 for the wet conditions (5% H2O); the corresponding results for dry conditions are presented in Fig. S7.† Hardly any NOx conversion was observed for the standard SCR gas feed over the soot, as shown in Fig. 9(a). However, a NOx conversion of up to 10% was detected in the fast SCR gas mixture between 200 and 450 °C, as shown in Fig. 9(b). This result is in agreement with a previous report by Mehring et al.,44 who suggested that the adsorbed NO2 is firstly activated via the strong interaction with soot, then further reacts with the NH3 and NO species. | ||
| Fig. 9 Comparison of NOx conversion from (a) wet standard SCR and (b) wet fast SCR over different gas–soot–catalyst systems. Wet standard SCR: 500 ppm NO, 500 ppm NH3, 5% H2O, 10% O2 in N2 with a total gas flow of 300 mL min−1; wet fast SCR: 250 ppm NO, 250 ppm NO2, 500 ppm NH3, 5% H2O, 10% O2 in N2 with a total gas flow of 300 mL min−1. 5 mg soot with 245 mg catalyst (or inert quartz sand). | ||
Although the catalytic tests were performed in transient mode, the obtained NOx conversion results over the V2O5–WO3/TiO2 catalyst are comparable to our previous steady-state experiments.27 Full conversion was achieved between 250–400 °C and 200–450 °C for standard and fast SCR, respectively. Hardly any influence could be noticed at low and middle temperatures in the presence of soot. Moreover, an improvement of NOx conversion was measured at high temperatures, regardless of the soot–catalyst contact type. The typical decrease in activity above approx. 450 °C appearing for VWTi catalysts was shifted for both standard (at 50% NOx conversion from 541 °C to 577 °C) and fast (from 552 °C to 582 °C) SCR conditions towards higher temperatures (Fig. 9(a) and (b)). Such a promoting effect could be explained by the additional consumption of N-containing gases by reactions with soot, as for example the NO2–soot reaction (Fig. 7(d) and 8(d)). At these high temperatures, the NOx conversion profile of the soot–catalyst in tight contact overlaps the one of the soot–catalyst system in loose contact, suggesting a minimal influence of the soot–VWTi interface. However, a very low soot loading (soot/catalyst ratio of 1/49) was applied in the present study and different effects need to be considered at higher soot loadings, e.g. the competition or blockage of catalyst active sites which might lead to a lower SCR activity.9,45
3.4. Summary of the contribution of individual gas components and SCR gas feeds to soot oxidation
To disentangle the influence of SCR gas mixtures on soot oxidation, the effects of different individual components were systematically investigated in our study, as summarized in Fig. 10. This included the direct interaction between soot and specific gases as well as the soot–gas–catalyst triple-phase system. Even for catalyst coated DPFs, non-catalytic oxidation reactions have a major contribution especially for a large soot layer thickness or poor catalyst–soot interaction. As shown in Fig. 10, NO2 was found to show an excellent soot oxidation ability, whereas NO neither promotes nor inhibits the soot oxidation reaction. This is illustrated by the lowest T50p temperature obtained for soot oxidation in a wet gas mixture containing NO2 and O2 (Fig. 10). Neeft and coworkers studied non-catalyzed diesel soot oxidation and reported that the reaction order is slightly lower than 1 with respect to the oxygen concentration.53 Herein, O2 participates mostly above 450–500 °C, whereas NO2 was found to decrease the onset temperature of soot oxidation. A further enhancement was observed in NO2 + O2 gas mixtures, with NO2 initiating the reaction by formation of active surface oxygen complexes.40 More active oxygen species can be generated if water is present that strengthens soot oxidation,52 which is in line with the results obtained in our study. With respect to the NH3 presence, an inhibitory effect was unraveled for the non-catalytic soot oxidation by O2, which is retained for most gas mixtures containing NH3. According to previous literature, ammonia interacts with soot and the formation of amine/amide intermediates blocks the soot surface and inhibits the oxidation reactions.13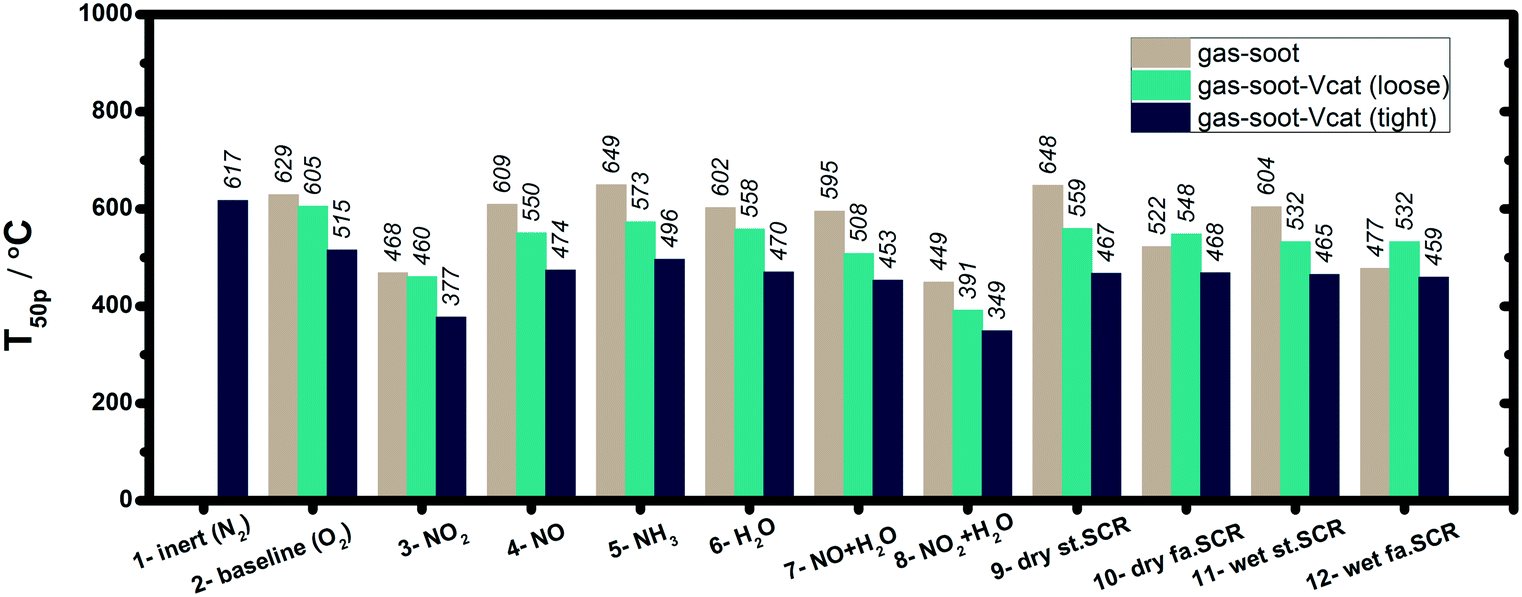 | ||
| Fig. 10 Comparison of the temperatures at CO + CO2 formation of 50 ppm (T50p, °C) for all investigated gas–soot–catalyst reactions. The gas mixture mixtures are listed in Table 1. | ||
In a combined 2-way SCRonDPF system maintaining a high NOx reduction performance in long-term is dependent on a good soot oxidation activity, which prevents the growth of a diffusive barrier layer on the catalyst surface.24 The reaction mechanism of soot oxidation is influenced by various parameters such as the catalyst composition, the soot–catalyst contact and the reaction atmosphere.23 For the triple-phase systems involving the presence of V2O5–WO3/TiO2, the low oxygen bond strength and the mobility of vanadia have been correlated to the observed high soot oxidation activity.30–32 Similar to other soot oxidation catalysts, a more intimate contact with soot is known to boost the oxidation reaction.33–38 The results obtained in the present study demonstrate that if a vanadium-based catalyst is involved, both NO- and NH3-containing gas mixtures need to be in a first step activated over the catalyst, resulting in NO2 formation, before they can participate in the soot oxidation reaction. For the case of the NO2-gas mixture, it could be elucidated that NO2 does not react over the catalyst but directly interacts with the soot due to its stronger oxidation ability.
However, NO2 seems to additionally contribute to the reoxidation of V-species, which was found relevant only for the catalyst in tight contact with the soot sample. Thus, a T50p temperature of 377 °C was measured in the presence of a catalyst during the catalytic soot oxidation by NO2 + O2 in comparison to 468 °C observed in the absence of a V-catalyst. Notably, an additional promotional effect on soot oxidation was noticed under wet conditions (additionally 5% H2O, Fig. S5 and S6†). Based on our investigations, Fig. 11(a) summarizes the effects of the individual SCR-related gas components (NO2, NO, NH3 and H2O) on the soot oxidation, with and without the presence of the V2O5–WO3/TiO2 catalyst.
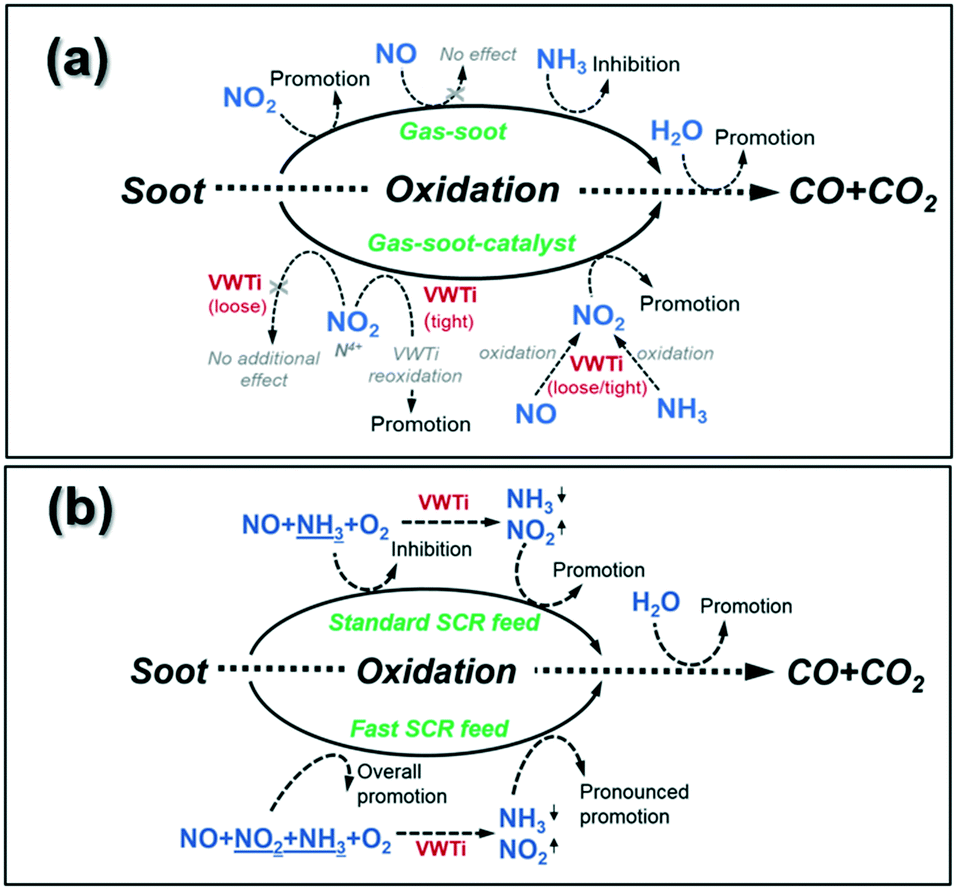 | ||
| Fig. 11 (a) Summary of individual gas components' (NO2, NO, NH3 and H2O) effect on soot oxidation with and without the presence of the V2O5–WO3/TiO2 catalyst; (b) summary of the SCR feed (including standard and fast SCR) effect on soot oxidation with and without the presence of the V2O5–WO3/TiO2 catalyst. | ||
Regarding the impact of complete standard and fast SCR gas mixtures on soot oxidation, Mihai et al.9 showed that a higher soot conversion can be obtained above 400 °C in both standard and fast SCR gas mixtures if the DPF is coated with a Cu-zeolite catalyst. In contrast, the oxidation of soot under the fast SCR feed is initiated at a lower temperature (approx. 200 °C lower) in the absence of a catalyst. Similar results were reported by Schobig et al.45 for an integrated vanadia-based SCR catalyst, as they observed no significant impact on NOx reduction but a decreased carbon oxidation rate (up to approx. a 20% decrease) at temperatures below 400 °C in the fast SCR feed. For the results obtained in this study, Fig. 11(b) shows the overview of the multiple effects appearing during simultaneous SCR of NOx and soot oxidation with and without the presence of the V2O5–WO3/TiO2 catalyst. For gas–soot dual-phase systems, the dry standard SCR feed was found to exhibit an inhibitory effect on soot oxidation as a result of ammonia–soot interaction. Thus, a T50p temperature of 648 °C was measured in the dry standard SCR conditions, which is 49 °C higher than that measured in the NO + O2 mixture (Fig. 10). However, in the dry fast SCR gas mixture, due to the co-presence of NO2–soot interaction in addition to the ammonia–soot interaction, a lower onset temperature was observed for soot oxidation as compared to the corresponding standard SCR conditions (Fig. 10: T50p of 522 °C versus 648 °C, respectively). This is supported by observations of Mihai and coworkers for a DPF coated with a Cu-zeolite catalyst.9 If the V2O5–WO3/TiO2 catalyst is added into the triple-phase system, both standard and fast SCR reactions are favored over the catalyst surface. Mainly at higher temperatures, the resulting gas mixtures participate in the soot oxidation, further supporting the promotional effect of the catalyst.
In general, we noticed that the soot–catalyst systems in tight contact exhibit lower soot ignition temperatures (Fig. 10), as well as higher soot conversions (Fig. 12) in comparison to systems in loose contact, regardless of the feed composition. This is consistent with previous reports that more intimate contact of soot with the catalyst boosts the oxidation reaction.33–38 Meanwhile, a high number of soot–catalyst contact points does not seem to affect the NOx conversion, as illustrated in Fig. 9. For real applications, however, a tight soot–catalyst interaction involving multiple interaction points is expected only for the first soot layers deposited on the catalyst surface. The fingerprint of such a contact is relevant only if the location of the SCRonDPF system ensures a sufficiently high temperature to avoid considerable soot accumulation. For all other cases, the results obtained in our study for a soot–catalyst loose contact are directly transferable.
 | ||
| Fig. 12 Comparison of the normalized total COx formation (NTCOx) upon dosage of various SCR-related gas mixtures to different soot–catalyst contact types including soot-only (black) and soot–catalyst in both loose (green) and tight (blue) contact. The CO + CO2 formation in all cases were normalized to the value obtained from the O2–soot reaction (benchmark). The gas mixtures are listed in Table 1. | ||
Similar to that observed for the individual gas components, the soot oxidation activity can be additionally enhanced by the presence of water (Fig. S5 and S6†), possibly also due to the formation of reactive O-species on the catalyst surface. In our study, a decrease from 559 °C to 532 °C was measured for T50p upon H2O addition into the soot–standard SCR–catalyst (loose) system. Under fast SCR conditions, a 16 °C lower T50p was noticed in wet condition in comparison to the corresponding dry condition (Fig. 10).
3.5. CO2 selectivity during soot oxidation
The pronounced effect of the reaction conditions on the soot oxidation activity is summarized in Fig. 12, with respect to the calculated NTCOx values for various parameters investigated in our study. In general, the NO2 presence did not only help to lower the onset temperature but also led to the highest soot conversion. The benefits resulting from the VWTi catalyst presence are obvious under both standard and fast SCR conditions, with a higher soot conversion obtained for tight soot–catalyst contact. In comparison to a loose catalyst–soot interaction, NTCOx increased from 4.9 to 20.6 for the dry standard SCR conditions and from 6.6 to 22.9 for the dry fast SCR reaction. Finally, although a high soot oxidation ability is desired, the selectivity towards CO2 formation is equally important, and CO formation as an undesired product should be minimized. The total CO2 formation relative to the corresponding total CO + CO2 formation is summarized in Fig. 13 for all the different gas–soot–catalyst reactions obtained in the present work. Although some minor variations were observed, an average CO2 selectivity of 78.5% was obtained. This suggests that the investigated gas atmosphere, the presence of the V2O5–WO3/TiO2 catalyst and the soot–catalyst contact type do not significantly affect the product distribution. According to our results, the mechanism seems to ultimately involve the oxidation of soot by catalyst lattice oxygen, O2, NO2 or OH groups, which are present or in situ generated over the catalyst surface for all reaction conditions reported in Fig. 13. These species are known to contribute not only to the non-catalytic or catalytic oxidation of soot to CO but also to sustaining CO oxidation. Hence, any increase in their concentration affects the kinetics of both processes, which might explain the constant ratio between total CO2 and total CO + CO2 concentrations. | ||
| Fig. 13 The total CO2 formation as a function of the corresponding total CO + CO2 formation over different gas–soot–catalyst systems. The gas mixtures are listed in Table 1. | ||
4. Conclusions
A simple to complex strategy, i.e. from gas–soot and gas–catalyst dual-phase system to gas–soot–catalyst triple-phase system investigation, was adopted in the present work to establish a fundamental understanding of the multiple interactions between the SCR gas mixtures, the vanadium-based SCR catalyst and the deposited soot, which is highly relevant for the application of integrated 2-way SCRonDPF systems. For gas–soot dual-phase interaction the following trends could be confirmed in our study: NO2 exhibits an excellent soot oxidation ability whereas ammonia shows an inhibition effect; the NO presence neither promotes nor inhibits the soot oxidation reaction. In a standard SCR feed, ammonia was found to dominate the gas–soot interaction, which leads to about 20 °C higher soot oxidation temperature (based on T50p values) compared to that obtained in the 10% O2/N2 feed (baseline condition). Meanwhile, the co-presence of NO2 in the fast SCR gas mixture promotes soot conversion, with the temperature of 50 ppm CO + CO2 formation approx. 100 °C lower than that measured for the baseline condition. Water was observed to enhance soot oxidation in all investigated gas mixtures, decreasing T50p by 20–30 °C in comparison to the corresponding dry conditions. With respect to the conversion of SCR-related gases, a NOx conversion up to 10% was noticed when exposing the fast SCR gas mixture directly to the soot. However, this reaction does not seem to occur in the standard SCR feed.As for the triple-phase systems involving the V2O5–WO3/TiO2 catalyst, the NO + O2-, NH3 + O2-, standard SCR- and fast SCR-containing gas mixtures were firstly activated over the catalyst and only subsequently involved in the soot oxidation reactions. Only NO2 was identified to directly interact with soot due to its strong oxidation ability. Furthermore, catalyst reoxidation by NO2 was recognized as an additional beneficial facet for the soot–catalyst system in tight contact. Interestingly, a small amount of soot used in the present study was found to maintain a relatively high NOx conversion above 450 °C irrespective of the soot catalyst contact type. Thus, 50% NOx conversion was obtained at 577 °C in the standard SCR feed and at 582 °C under fast SCR conditions for the catalyst–soot system in comparison to 541 °C and 552 °C temperature points measured for the same catalyst in the absence of soot. Taken together, the systematic results obtained in the present study contribute to a comprehensive understanding of the multiple interactions and effects occurring in integrated 2-way SCRonDPF systems based on VWTi catalysts. For real SCRonDPF applications, particularly, cases involving a loose catalyst–soot contact may be relevant considering the limited number of interaction points even at moderate soot layer thicknesses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support of this work via provision of a PhD scholarship to L. Zheng by the China Scholarship Council (CSC) and support by the Helmholtz-programme MTET (Materials, resources and technologies, fuel assessment in the topic chemical energy carriers). We thank Jan Pesek (ITCP, KIT) and D. Zengel (ITCP, KIT) for technical support with respect to catalyst testing.References
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- O. Deutschmann and J.-D. Grunwaldt, Chem. Ing. Tech., 2013, 85, 595–617 CrossRef CAS.
- M. V. Twigg, Catal. Today, 2011, 163, 33–41 CrossRef CAS.
- E. Meloni and V. Palma, Catalysts, 2020, 10, 745 CrossRef CAS.
- I. Nova and E. Tronconi, Urea-SCR technology for deNOx after treatment of diesel exhausts, Springer, New York, 2014, vol. 5 Search PubMed.
- X. Ge, SAE Tech. Pap., 2013, 2013-01-2441 Search PubMed.
- J. Tan, C. Solbrig and S. J. Schmieg, SAE Tech. Pap., 2011, 2011-01-1140 Search PubMed.
- M. Naseri, S. Chatterjee, M. Castagnola, H.-Y. Chen, J. Fedeyko, H. Hess and J. Li, SAE Int. J. Engines, 2011, 4, 1798–1809 CrossRef.
- O. Mihai, M. Stenfeldt and L. Olsson, Catal. Today, 2018, 306, 243–250 CrossRef CAS.
- O. Mihai, S. Tamm, M. Stenfeldt, C. Wang-Hansen and L. Olsson, Ind. Eng. Chem. Res., 2015, 54, 11779–11791 CrossRef CAS.
- J. H. Lee, M. J. Paratore and D. B. Brown, SAE Int. J. Fuels Lubr., 2009, 1, 96–101 CrossRef CAS.
- E. Tronconi, I. Nova, F. Marchitti, G. Koltsakis, D. Karamitros, B. Maletic, N. Markert, D. Chatterjee and M. Hehle, Emiss. Control Sci. Technol., 2015, 1, 134–151 CrossRef CAS.
- L. V. Trandafilović, O. Mihai, J. Woo, K. Leistner, M. Stenfeldt and L. Olsson, Appl. Catal., B, 2019, 241, 66–80 CrossRef.
- M. Kleinhenz, A. Fiedler, P. Lauer and A. Döring, Top. Catal., 2018, 62, 282–287 CrossRef.
- K. Johansen, H. Bentzer, A. Kustov, K. Larsen, T. V. Janssens and R. G. Barfod, SAE Tech. Pap., 2014, 2014-01-1523 Search PubMed.
- Y. He, D. B. Brown, S. Lu, M. J. Paratore and J. Li, SAE Tech. Pap., 2009, 2009-01-0274 Search PubMed.
- V. T. Nguyen, D. B. Nguyen, I. Heo and Y. S. Mok, Catalysts, 2019, 9, 853 CrossRef CAS.
- A. Cooper, T. E. Davies, D. J. Morgan, S. Golunski and S. H. Taylor, Catalysts, 2020, 10, 294 CrossRef CAS.
- L. Castoldi, Materials, 2020, 13, 3551 CrossRef CAS PubMed.
- X. Song, J. H. Johnson and J. D. Naber, Int. J. Engine Res., 2015, 16, 738–749 CrossRef CAS.
- K. Johansen, Catal. Today, 2015, 258, 2–10 CrossRef CAS.
- B. Guan, R. Zhan, H. Lin and Z. Huang, J. Environ. Manage., 2015, 154, 225–258 CrossRef CAS PubMed.
- R. Prasad and S. V. Singh, J. Environ. Chem. Eng., 2020, 8, 103945 CrossRef.
- M. Purfürst, S. Naumov, K.-J. Langeheinecke and R. Gläser, Chem. Eng. Sci., 2017, 168, 423–436 CrossRef.
- A. Lanza, L. Zheng, R. Matarrese, L. Lietti, J.-D. Grunwaldt, S. A. Clave, J. Collier and A. Beretta, Chem. Eng. J., 2021, 416, 128933 CrossRef CAS.
- Y. M. López-De Jesús, P. I. Chigada, T. C. Watling, K. Arulraj, A. Thorén, N. Greenham and P. Markatou, SAE Int. J. Engines, 2016, 9, 1247–1257 CrossRef.
- L. Zheng, M. Casapu, M. Stehle, O. Deutschmann and J.-D. Grunwaldt, Top. Catal., 2019, 62, 129–139 CrossRef CAS.
- E. Japke, M. Casapu, V. Trouillet, O. Deutschmann and J.-D. Grunwaldt, Catal. Today, 2015, 258, 461–469 CrossRef CAS.
- K. Johansen, A. Widd, F. Zuther and H. Viecenz, SAE Tech. Pap., 2016, 2016-01-0915 Search PubMed.
- J. M. Christensen, J.-D. Grunwaldt and A. D. Jensen, Appl. Catal., B, 2016, 188, 235–244 CrossRef CAS.
- I. C. L. Leocadio, S. Braun and M. Schmal, J. Catal., 2004, 223, 114–121 CrossRef CAS.
- J. Liu, Z. Zhao, C. Xu, A. Duan, L. Zhu and X. Wang, Appl. Catal., B, 2005, 61, 36–46 CrossRef CAS.
- J. P. Neeft, M. Makkee and J. A. Moulijn, Appl. Catal., B, 1996, 8, 57–78 CrossRef CAS.
- J. M. Christensen, D. Deiana, J.-D. Grunwaldt and A. D. Jensen, Catal. Lett., 2014, 144, 1661–1666 CrossRef CAS.
- L. Castoldi, R. Matarrese, L. Lietti and P. Forzatti, Appl. Catal., B, 2009, 90, 278–285 CrossRef CAS.
- E. Aneggi, V. Rico-Perez, C. de Leitenburg, S. Maschio, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2015, 54, 14040–14043 CrossRef CAS PubMed.
- E. Aneggi, J. Llorca, A. Trovarelli, M. Aouine and P. Vernoux, Chem. Commun., 2019, 55, 3876–3878 RSC.
- D. Gardini, J. M. Christensen, C. D. Damsgaard, A. D. Jensen and J. B. Wagner, Appl. Catal., B, 2016, 183, 28–36 CrossRef CAS.
- M. Jeguirim, V. Tschamber, J. Brilhac and P. Ehrburger, J. Anal. Appl. Pyrolysis, 2004, 72, 171–181 CrossRef CAS.
- A. Setiabudi, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2004, 50, 185–194 CrossRef CAS.
- F. Jacquot, V. Logie, J. Brilhac and P. Gilot, Carbon, 2002, 40, 335–343 CrossRef CAS.
- J. M. Christensen, J.-D. Grunwaldt and A. D. Jensen, Appl. Catal., B, 2017, 205, 182–188 CrossRef CAS.
- W. F. Shangguan, Y. Teraoka and S. Kagawa, Appl. Catal., B, 1997, 12, 237–247 CrossRef CAS.
- M. Mehring, M. Elsener and O. Kröcher, ACS Catal., 2012, 2, 1507–1518 CrossRef CAS.
- J. Schobing, V. Tschamber, J.-F. Brilhac, A. Auclaire and Y. Hohl, C. R. Chim., 2018, 21, 221–231 CrossRef CAS.
- A. Konstandopoulos, S. Lorentzou, C. Pagkoura, K. Ohno, K. Ogyu and T. Oya, SAE trans., 2007, 2009-01-1950 Search PubMed.
- L. Zheng, A. Zimmer, M. Casapu and J.-D. Grunwaldt, ChemCatChem, 2020, 12, 6272–6284 CrossRef CAS.
- T. Srnak, J. Dumesic, B. Clausen, E. Törnqvist and N.-Y. Topsøe, J. Catal., 1992, 135, 246–262 CrossRef CAS.
- I. Pieta, M. García-Diéguez, C. Herrera, M. Larrubia and L. Alemany, J. Catal., 2010, 270, 256–267 CrossRef CAS.
- A. Cavaliere, R. Barbella, A. Ciajolo, A. D'anna and R. Ragucci, Symp. (Int.) Combust., 1994, 25, 167–174 CrossRef.
- D. E. Doronkin, F. Benzi, L. Zheng, D. I. Sharapa, L. Amidani, F. Studt, P. W. Roesky, M. Casapu, O. Deutschmann and J.-D. Grunwaldt, J. Phys. Chem. C, 2019, 123, 14338–14349 CrossRef CAS.
- W. Yang, Y. Wang, H. Wang, Y. Zhang, Y. Peng and J. Li, Appl. Catal., B, 2021, 120837 Search PubMed.
- J. P. Neeft, O. P. Van Pruissen, M. Makkee and J. A. Moulijn, Appl. Catal., B, 1997, 12, 21–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00432a |
| This journal is © The Royal Society of Chemistry 2022 |