
Homogeneous oxidation of C–H bonds with m-CPBA catalysed by a Co/Fe system: mechanistic insights from the point of view of the oxidant†
Oksana V.
Nesterova
 a,
Maxim L.
Kuznetsov
a,
Armando J. L.
Pombeiro
ab,
Georgiy B.
Shul'pin
cd and
Dmytro S.
Nesterov
*a
a,
Maxim L.
Kuznetsov
a,
Armando J. L.
Pombeiro
ab,
Georgiy B.
Shul'pin
cd and
Dmytro S.
Nesterov
*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: dmytro.nesterov@tecnico.ulisboa.pt
bPeoples' Friendship University of Russia (RUDN University), Research Institute of Chemistry, 6 Miklukho-Maklaya st, Moscow 117198, Russia
cSemenov Federal Research Center for Chemical Physics, Russian Academy of Sciences, Ulitsa Kosygina 4, Moscow 119991, Russia
dChair of Chemistry and Physics, Plekhanov Russian University of Economics, Stremyannyi pereulok 36, Moscow 117997, Russia
First published on 9th November 2021
Abstract
Oxidations of C–H bonds with m-chloroperoxybenzoic acid (m-CPBA) catalyzed by transition metal complexes are known to proceed through a number of routes, from the non-selective free radical to selective concerted and metal-mediated ones. However, there is a lack of understanding of the m-CPBA oxidative behavior, reaction mechanisms and factors that trigger its activity. An experimental and theoretical investigation of sp3 C–H bond oxidation with m-CPBA in the presence of the heterometallic pre-catalyst [CoIII4FeIII2O(Sae)8]·4DMF·H2O (1) (H2Sae = salicylidene-2-ethanolamine) and HNO3 promoter has been performed herein. The catalytic system 1/HNO3/m-CPBA allows mild hydroxylation of tertiary C–H bonds with 99% retention of stereoconfiguration of model alkane substrates, supported by high TOFs up to 2 s−1 (for cis-1,2-dimethylcyclohexane) and TONs up to 1.4 × 104 (at 50 °C). The catalytic effect of 1 is seen at the ppm level, while 1000 ppm (0.1 mol%) loading allows 1000-fold increase of the initial reaction rate up to 9 × 10−5 M s−1. The reaction mechanism was investigated by means of combined kinetic studies (including isotope effects), isotopic labeling (18O2, H218O, D2O), ESI-MS spectroscopy and DFT theoretical studies. The results suggest that the main oxidation pathway proceeds through a concerted mechanism involving a cobalt-peroxo C–H attacking species or via a cobalt–oxyl species (rebound process), rather than a free-radical pathway. Remarkably, the Co(III) catalyst does not change its oxidation state during the most energetically favored pathway, consistent with a metal–ligand cooperativity. The chlorobenzene radical is responsible for H abstraction in the non-selective side route, which is efficiently suppressed by the acidic promoter. Finally, signs for slow direct oxygen exchange between m-CPBA and water in the presence of a proton or a metal complex are found, suggesting that the results of 18O-tests should be treated cautiously when m-CPBA is used as the oxidant.
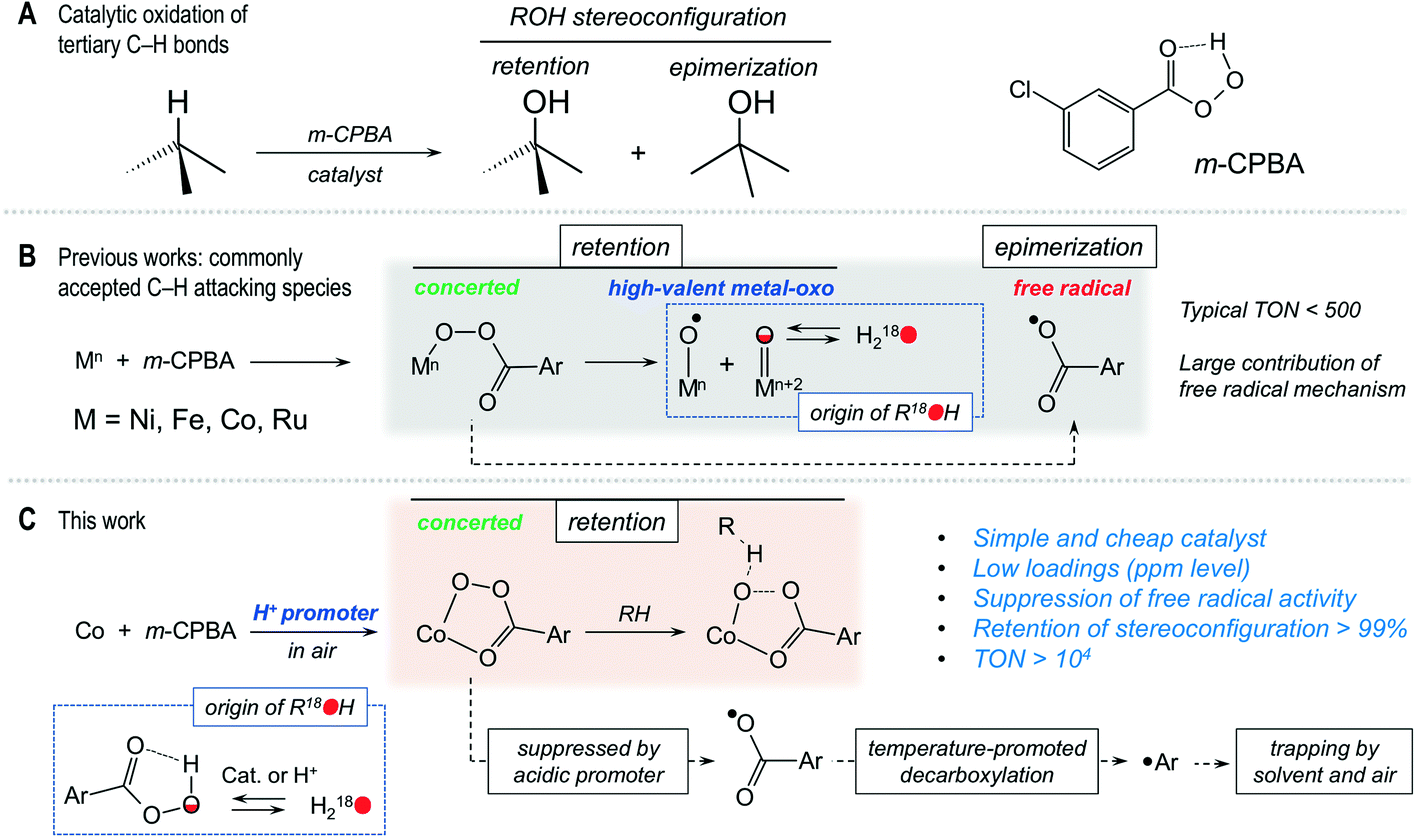
Introduction
Selective catalytic oxidation of sp3 C–H bonds remains a challenge in contemporary chemistry.1–8 The high energy of the C–H bond and the saturated character of alkanes9 foresee the use of strong terminal oxidants, such as dioxygen or various peroxides (hydrogen peroxide or alkyl hydroperoxides, peracids, etc.). Peroxides, being readily available oxidants for preparative chemistry, are highly prone to low-selectivity free radical activity,10,11 while the activation of dioxygen by metal complexes is an uncommon task.12 Complexes of iron or manganese can constitute catalysts for selective oxidation of alkanes (as well as alkenes and other substrates) with hydrogen peroxide as a terminal oxidant.4,13 The origin of selectivity in these systems concerns high-valent metal–oxo (HVMO) species,14–16 stabilized by sophisticated capping or macrocyclic N-donor ligands which require laborious syntheses, thus hampering the overall potential impact.4,17 The catalytic activity of heterometallic complexes in C–H oxidation,18 particularly involving those metals forming HVMO species, is of special interest due to the recognized synergic effect of few dissimilar metals.10,19–21Peroxides constitute a large class of oxidants, where one compound, m-chloroperbenzoic acid (m-CPBA), stands apart from the other members of this family due to its unusually complex oxidative behaviour that combines a range of pathways – from a free radical to concerted ones. In modern organic chemistry, m-CPBA is an inexpensive, stable, organic-soluble, versatile oxidation reagent, routinely used in the Baeyer–Villiger oxidation of ketones, selective epoxidation of olefins and other valuable processes.22 It was reported that m-CPBA, taken in large excess, is able to oxidize alkanes under catalyst-free conditions, showing some limited stereoselectivity.23–25 Furthermore, the large amounts of chlorobenzene by-product formed25 suggest23 that under catalyst-free conditions the undesirable free radical activity26 is favoured. The use of a suitable catalyst could turn an oxidation route toward a selective, concerted-like mechanism, at the same time suppressing the free radical activity. However, there is a lack of clear understanding of the m-CPBA oxidant behaviour and respective reaction mechanism(s) under catalytic conditions. Although m-CPBA, along with PhIO and some other oxidants, is a recognized model oxidant in various metal-catalysed oxidation systems, the respective research trends are typically focused on the investigation of formed metal-containing active species (e.g. HVMO ones),4,27,28 rather than on the chemistry of the oxidant itself. Relatively high bond selectivities, exhibited by O-centred radicals originating from m-CPBA, may mislead mechanistic interpretations, where a free radical reaction is interpreted as a HVMO-mediated one.
It is known that m-CPBA can react with metal ions to produce highly reactive HVMO species, able to abstract the H atom from inert C–H bonds in a selective manner.29 The HVMO complexes are of great interest because they are key intermediates in natural enzymatic catalytic systems, including those oxidizing inactive alkanes.7,12,30–32 In turn, m-CPBA can serve as a terminal oxidant even in enzymatic processes, such as oxidation of alkanes catalysed by cytochrome P450.33 The oxidation potential of m-CPBA is sufficient for transforming Fe(III) coordination compounds into highly reactive Fe(IV) and Fe(V) ones, capable of oxidizing strong sp3 C–H bonds.34–36 Trivalent ruthenium, being chemically close to iron, has a similar behaviour with formation of Ru(IV) and Ru(V) compounds.37,38 The power of m-CPBA is enough to form NiIII–O(H) and NiIII–O˙ species from Ni(II) ones,39–41 which was confirmed by trapping the respective HVMO nickel complexes by ESI-MS and EPR techniques.40,41 However, recently Hartwig reconsidered the Ni(II)/m-CPBA system, showing that it oxidizes C–H bonds through the free radical mechanism where the aroyloxyl radical ArC(O)O˙, but not the NiII–O˙ or NiIII–O˙ ones, is responsible for C–H oxidation (Scheme 1).42
 | ||
| Scheme 1 Hydroxylation of C–H bonds with m-CPBA catalyzed by metal species. | ||
The early works of Nam demonstrated that compounds of Co(II) and Co(III), in particular the porphyrin complex [CoIII(TPFPP)(CF3SO3)] (TPFPP = meso-tetrakis(pentafluorophenyl)porphinato dianion), catalyse the oxidation of alkanes and alkenes with m-CPBA with a pronounced stereoselectivity.43,44 The involvement of cobalt HVMO intermediates was suggested on the basis of the high kinetic isotope effect (KIE) of 8 in competitive oxidation of normal and deuterated cyclohexanes, as well as incorporation of 18O from H18O into the hydroxylation products (the latter is expected due to H218O⋯M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 16O ↔ 18OM⋯16OH2 tautomerism, known for M = Fe and Mn; Scheme 1). During our previous studies, we obtained similar results (KIE of 7.2) and incorporation of 18O from H218O using the isoindole compound of cobalt [CoII(L1)2](NO3)2 (L1 = O,O′-(3-amino-1H-isoindole-1,1-diyl)bis(propan-2-one oxime)), which catalysed the hydroxylation of alkanes using m-CPBA as a terminal oxidant with >98% retention of stereoconfiguration.45 In contrast, the complexes of cobalt with acetylacetonate, scorpionate and polypyridyl ligands reveal a large contribution of less selective free radical processes, as demonstrated by Hikichi, Shul'pin and others.46–49
16O ↔ 18OM⋯16OH2 tautomerism, known for M = Fe and Mn; Scheme 1). During our previous studies, we obtained similar results (KIE of 7.2) and incorporation of 18O from H218O using the isoindole compound of cobalt [CoII(L1)2](NO3)2 (L1 = O,O′-(3-amino-1H-isoindole-1,1-diyl)bis(propan-2-one oxime)), which catalysed the hydroxylation of alkanes using m-CPBA as a terminal oxidant with >98% retention of stereoconfiguration.45 In contrast, the complexes of cobalt with acetylacetonate, scorpionate and polypyridyl ligands reveal a large contribution of less selective free radical processes, as demonstrated by Hikichi, Shul'pin and others.46–49
The promoting effect of acidic additives is recognized for metal-catalysed oxidations with H2O2,10 but is majorly not known for m-CPBA. Recently we have shown that within the range of [MII(Pc)] phthalocyanine complexes having similar structures, cobalt phthalocyanine revealed an exceptional catalytic activity (selectivity and yields of products) of almost one order higher than the other metals studied, when small amounts of nitric acid were used as a promoter.50 The suppression of the free radical route of m-CPBA depends on the acidity (pKa) of the promoter, as we showed for [CoIIIL2] and [CoIIICdIIL23Cl2] catalysts (HL2 = 2-methoxy-6-[(methylimino)methyl]phenol).51 Another key observation was the difference in incorporations of 18O from 18O2 into hydroxylation products when using nitric or acetic acid promoters, which correlated with the overall selectivity using [CoIIIZnIIL23Cl2] as a catalyst.52 Although the Co/H+/m-CPBA catalytic combination is among the most efficient ones for activation of m-CPBA so far, the factors that influence its behaviour as well as the details of the catalytic mechanisms are still to be established. Another open question is the participation of Co(IV) HVMO species in the reaction mechanism, which are known to be elusive due to their very high reactivity.53–55
In the present work we have found that the heterometallic pre-catalyst [Co4Fe2O(Sae)8]·4DMF·H2O (1),20 where H2Sae = salicylidene-2-ethanolamine, in the presence of an acidic promoter and m-CPBA oxidant, affords yields of hydroxylation products up to 70% with the yields of chlorobenzene being less than 1%, keeping >99% of retention of stereoconfiguration of model substrates, with TONs (turnover numbers) >104 and TOF (turnover frequency) up to 2 s−1 (Scheme 1). Being interested in establishing the mechanism of action and optimization of the reaction conditions, we conducted a detailed combined kinetic/18O-labeling/ESI-MS/DFT study that is reported herein.
Results and discussion
Pre-catalyst 1
The coordination compound [CoIII4FeIII2O(Sae)8]·4DMF·H2O (1) is a hexanuclear complex (Fig. 1),20 where the cobalt centres are coordinated by two Schiff base ligands (deprotonated form of salicylidene-2-ethanolamine, H2Sae) to form [Co(Sae)2] fragments, joined by iron centres and an oxygen bridging atom. The outstanding catalytic performance of 1 in the oxidation of alkanes with H2O2, as well as the promising results of the activity using m-CPBA as an oxidant,20 inspired us to further study complex 1 as a pre-catalyst.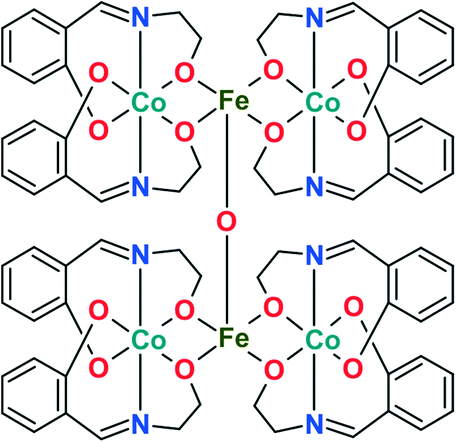 | ||
| Fig. 1 Schematic representation of the structure of 1 in the solid state. Uncoordinated DMF and water molecules are omitted for clarity. | ||
Kinetic, selectivity and isotopic labeling observations
Retention of stereoconfiguration of a substrate is an important indicator for oxidation mechanisms proceeding without the formation of a free alkyl radical.56 Thus, we have started the initial studies using cis-1,2-dimethylcyclohexane (cis-1,2-DMCH), a recognized benchmark substrate56–59 in stereospecific hydroxylation investigations (Scheme 2). The use of this simple substrate allows collection of data of stereo- and bond selectivities simultaneously (apart from the enantioselectivity, which was not the aim of the present study). Starting with simplified models (such as cis-1,2-DMCH, methylcyclohexane, etc.) concerns a typical approach in the investigation of novel catalytic systems having a complex behaviour. The main aim of this strategy, which is also used in enzymatic oxidation studies,60–62 is to understand the principal features, abilities and limitations of the catalytic system and to distinguish between free radical and other types of reaction mechanisms. In the chosen model system, chlorobenzene and m-chlorobenzoic acid are common by-products formed from m-CPBA (chlorobenzene is formed through the rapid elimination of CO2 from the m-chlorobenzoate radical, which is a product of homolytic splitting of the m-CPBA O–O bond to generate the acyloxyl radical23).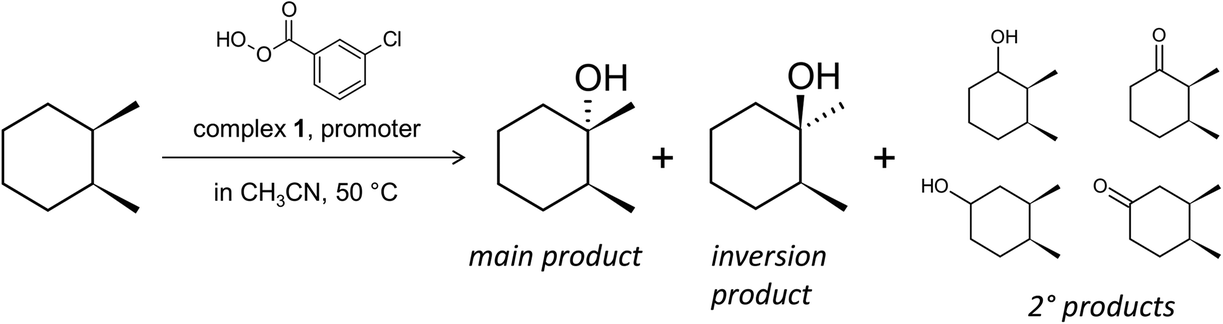 | ||
| Scheme 2 Model reaction for the study of catalytic and mechanistic features of the 1/HNO3/m-CPBA system: oxidation of cis-1,2-dimethylcyclohexane (cis-1,2-DMCH). Only one enantiomer of each tertiary alcohol is shown for clarity. | ||
As the heterometallic coordination compound [Co4Fe2O(Sae)8]·4DMF·H2O (1) was found to be a pre-catalyst (see below), producing catalytically active cobalt species [Co(HSae)2]+ upon dissolution (see below), all the respective concentrations, TON and TOF values will be given using the quadruple concentration of complex 1 (i.e. [1Co] = 4[1]).
Addition of m-CPBA (0.045 M; here and further the concentrations concerning the final catalytic solutions are given) to an acetonitrile solution of 1 (with [1Co]0 = 1.2 × 10−4 M), HNO3 (5.5 × 10−3 M) and cis-1,2-DMCH (0.1 M) afforded the tertiary cis-alcohol as the main reaction product, with a ratio of concentrations of tertiary cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans alcohols (c/t ratio) of 56 (corresponds to >99% of retention of stereoconfiguration; see the ESI†) after 1 h reaction time (Table 1, entry 1) and 46% yield of hydroxylation products based on m-CPBA, which is in deficit. With a reduced amount of cis-1,2-DMCH (0.025 M) and m-CPBA (0.027 M), the yields based on m-CPBA can be improved up to 56% (entry 2), keeping a high c/t ratio. By lowering the catalyst concentration, TON (turnover number, mol of product per mol of catalyst) and TOF (TON per second) values of 1.4 × 104 and 1.8 s−1, respectively, were reached (entry 3). Furthermore, the yield of chlorobenzene dropped below 1%. The combination of m-CPBA and HNO3 is a crucial point for the present catalytic system: only traces of cis- and trans-alcohols (with c/t ∼ 1) were detected when peracetic acid, benzoyl peroxide or its combinations with H2O2 were used as oxidants. Further, low yields of tertiary products with a c/t ratio of only 2 were observed in the absence of a HNO3 promoter (Table 1, entry 6). Only traces of tertiary alcohols were detected in the absence of pre-catalyst 1, with no stereoselectivity observed (entry 7).
:trans alcohols (c/t ratio) of 56 (corresponds to >99% of retention of stereoconfiguration; see the ESI†) after 1 h reaction time (Table 1, entry 1) and 46% yield of hydroxylation products based on m-CPBA, which is in deficit. With a reduced amount of cis-1,2-DMCH (0.025 M) and m-CPBA (0.027 M), the yields based on m-CPBA can be improved up to 56% (entry 2), keeping a high c/t ratio. By lowering the catalyst concentration, TON (turnover number, mol of product per mol of catalyst) and TOF (TON per second) values of 1.4 × 104 and 1.8 s−1, respectively, were reached (entry 3). Furthermore, the yield of chlorobenzene dropped below 1%. The combination of m-CPBA and HNO3 is a crucial point for the present catalytic system: only traces of cis- and trans-alcohols (with c/t ∼ 1) were detected when peracetic acid, benzoyl peroxide or its combinations with H2O2 were used as oxidants. Further, low yields of tertiary products with a c/t ratio of only 2 were observed in the absence of a HNO3 promoter (Table 1, entry 6). Only traces of tertiary alcohols were detected in the absence of pre-catalyst 1, with no stereoselectivity observed (entry 7).
| Entry/catalystb | Yields of hydroxylation productsc | Yield of chlorobenzened | TONe | Initial rate W0f | TOFg | c/th | RCi | 3°:2°j |
|
|---|---|---|---|---|---|---|---|---|---|
| Tertiary cis-alcohol | Total | ||||||||
|
a Conditions (for pre-catalyst 1; see respective entries for other catalysts) unless stated otherwise: [cis-1,2-DMCH]0 = 0.1 M, [m-CPBA]0 = 0.027 M, [HNO3]0 = 5.5 × 10−3 M, [1Co]0 = 1.1 × 10−5 M in CH3CN (5 mL total volume), 50 °C, open air, data shown are for 1 h reaction time after quenching the probes with PPh3.
b Special conditions or other catalysts.
c Total yields (%) of the 3° and 2° products based on m-CPBA; yields of 3° products are the sums of tertiary cis- and trans-alcohols; yields of secondary products (2°) are the sums of respective alcohols and ketones.
d Yield (%) of chlorobenzene based on m-CPBA.
e Turnover numbers, mol of products (sum of 3° and 2° products) per mol of cobalt metal.
f Reaction rate of tertiary cis-alcohol formation at zero time (M s−1).
g Turnover frequency at zero time (s−1), initial rate W0 per mol of cobalt metal.
h mol of tertiary cis-alcohol per mol of trans-alcohol. In the present work, the commercial substrate, cis-1,2-DMCH, contained ca. 1% of trans-isomer, which lowered the final cis/trans ratio and was accounted when calculating the RC index.
i Retention of stereoconfiguration percentage, calculated as RC = 100(cis–trans + ρ·cis)/(cis + trans–ρ·cis), where cis and trans represent the concentrations of respective products and ρ is the fraction of trans-1,2-DMCH in the commercial cis-1,2-DMCH substrate (ρ = 0.018 for entries 1–14 and 1.6 × 10−4 for entries 15 and 16).
j Bond selectivity, yields of 3° products/yields of 2° products × 4 (for normalization, since 3°:2° C–H bonds = 2:8 = 1:4).
k [m-CPBA]0 = 0.045 M, [1Co]0 = 1.2 × 10−4 M.
l [cis-1,2-DMCH]0 = 0.026 M.
m [1Co]0 = 1.4 × 10−6 M, 3 h.
n [1Co]0 = 1.2 × 10−4 M.
o No HNO3 added.
p [Fe(NO3)3]0 = 1.6 × 10−5 M, no HNO3.
q [Fe(NO3)3]0 = 1.6 × 10−5 M.
r [Co(NO3)2]0 = 1.1 × 10−5 M, no HNO3.
s [Co(NO3)2]0 = 1.1 × 10−5 M.
t Subsequent addition of CH3CN solutions of Co(NO3)2 and Fe(NO3)3 to H2Sae solution (concentrations corresponding to imaginary [1]0 = 3 × 10−5 M).
u The same, corresponding to [1]0 = 2.7 × 10−6 M.
v The complex [CoIII(HSae)(Sae)]·CH3OH·H2O (2) was used as a catalyst (1.1 × 10−5 M).
w The same conditions as for entry 5, except for the cis-1,2-DMCH substrate containing less admixture of trans-1,2-DMCH (ρ = 1.6 × 10−4).
|
|||||||||
| 1/1k | 42 | 46 | 1.3 | 176 | 7.1 × 10−5 | 0.6 | 56 | 99.7 | 37 |
| 2/1l | 48 | 56 | 1.8 | 1.4 × 103 | 1.2 × 10−5 | 1.1 | 52 | 99.9 | 30 |
| 3/1m | 62 | 72 | 0.8 | 1.4 × 104 | 2.4 × 10−6 | 1.8 | 41 | 98.7 | 31 |
| 4/1n | 42 | 47 | 0.9 | 105 | 8.8 × 10−5 | 0.7 | 51 | 99.7 | 47 |
| 5/1 | 53 | 63 | 0.7 | 1.6 × 103 | 1.5 × 10−5 | 1.4 | 37 | 98.4 | 34 |
| 6/1o | 12 | 20 | 15.6 | 490 | 1.4 × 10−5 | 1.3 | 2 | 37.6 | 20 |
| 7/Catalyst-free test | 1.5 | 2.4 | 0.1 | — | 5.8 × 10−8 | — | 2.4 | 24.1 | — |
| 8/1 under N2 | 59 | 74 | 5.4 | 73 | 9.6 × 10−6 | 0.9 | 14 | 90.7 | 34 |
| 9/Fe(NO3)3p | 4 | 9 | 3.0 | 150 | — | — | 2 | 23.2 | 8 |
| 10/Fe(NO3)3q | 4 | 6 | 0.7 | 102 | — | — | 2 | 40.0 | 19 |
| 11/Co(NO3)2r | 23 | 30 | 6.7 | 736 | 4.9 × 10−6 | 0.5 | 10 | 84.9 | 21 |
| 12/Co(NO3)2s | 57 | 65 | 1.0 | 1.6 × 103 | 1.4 × 10−5 | 1.2 | 30 | 97.0 | 36 |
| 13/Co–Fe–H2Sae in situt | 58 | 68 | 1.7 | 166 | 1.2 × 10−4 | 1.0 | 35 | 99.8 | 31 |
| 14/Co–Fe–H2Sae in situu | 62 | 72 | 1.0 | 1.9 × 103 | 1.7 × 10−5 | 1.5 | 29 | 96.8 | 37 |
| 15/2v | 63 | 71 | 0.3 | 1.7 × 103 | 1.5 × 10−5 | 1.4 | 213 | 99.1 | 35 |
| 16/1w | 62 | 70 | 0.3 | 1.7 × 103 | 1.8 × 10−5 | 1.6 | 168 | 98.8 | 36 |
The dependence of the initial reaction rate W0 on [cis-1,2-DMCH]0 shows a non-linear character without reaching saturation at the concentrations studied (Fig. 2, left). The dependence of W0vs. [1Co]0 is linear in the range 0 < [1Co]0 < 8 × 10−5 M (Fig. 2, right), indicating that no aggregation of active species occurs in solution at these concentrations.20,63 The initial reaction rate of chlorobenzene formation shows also a nearly linear dependence on [1Co]0 (Fig. 2, right, inset), suggesting that chlorobenzene is a by-product formed within the main catalytic cycle. The W0vs. m-CPBA dependence shows a behaviour (Fig. 3, left) which can be approximated as a linear one. The initial rates of chlorobenzene formation (Fig. 3, left, inset) also exhibit a linear dependence. These dependences indicate that alcohol and chlorobenzene molecules are catalytically formed from the m-CPBA molecule, but not through the radical chain reaction (for which the non-linear curves are expected). The nitric acid promoter shows a complex dependence (Fig. 3, right) of W0vs. [HNO3]0 with a marked maximum at [HNO3]0 = 0.011 M ([HNO3]0/[1Co]0 = 90). The excess of the acid promoter led to pronounced suppression of the reaction rate and afforded lower c/t ratios (Fig. 3, right, inset). It is clear from this plot (Fig. 3, right) that the presence of nitric acid causes a strict promoting effect on the reaction rate, compared to acid-free conditions.
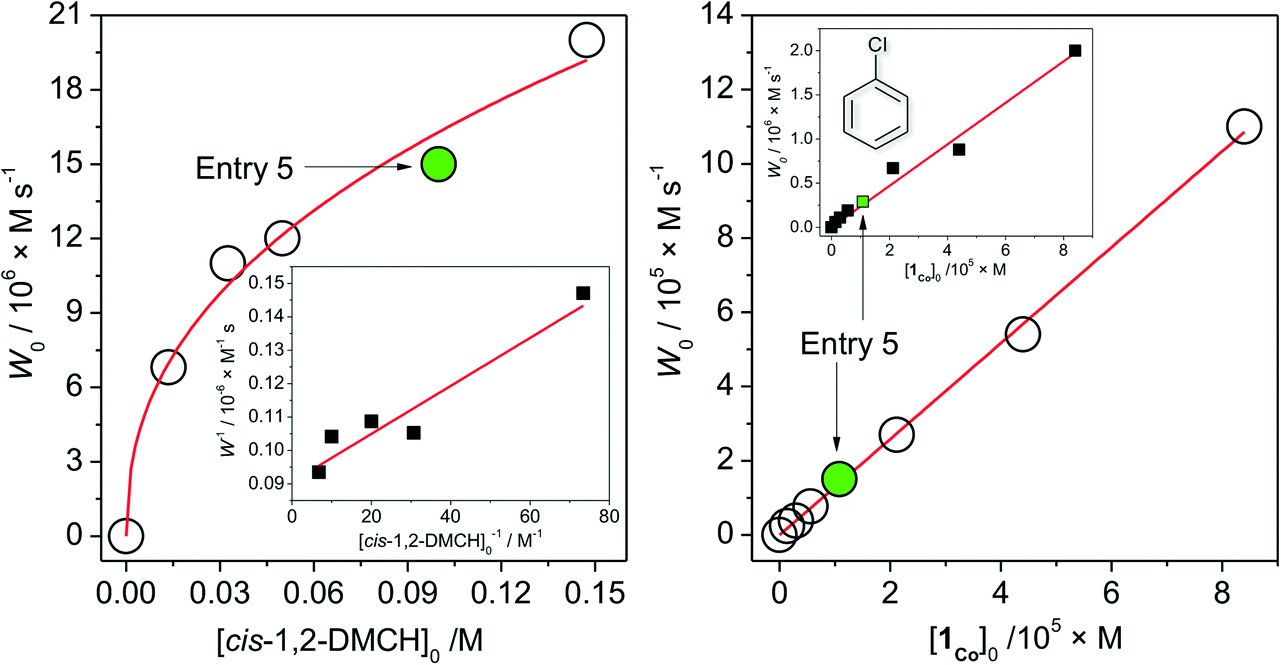 | ||
| Fig. 2 Dependences (experimental points) of the reaction rates (see the ESI†) of tertiary cis-alcohol accumulations on concentrations of the substrate (cis-1,2-DMCH, left) and 1 (right). Conditions, except for the concentration varied, are as those for entry 5, Table 1 (solid green circles and squares correspond to entry 5). Left figure inset shows the linearization of W vs. [cis-1,2-DMCH]0 dependence in reciprocal coordinates (solid line is the linear fit). Right figure inset shows the dependence of the chlorobenzene initial rate formation on [1Co]0. | ||
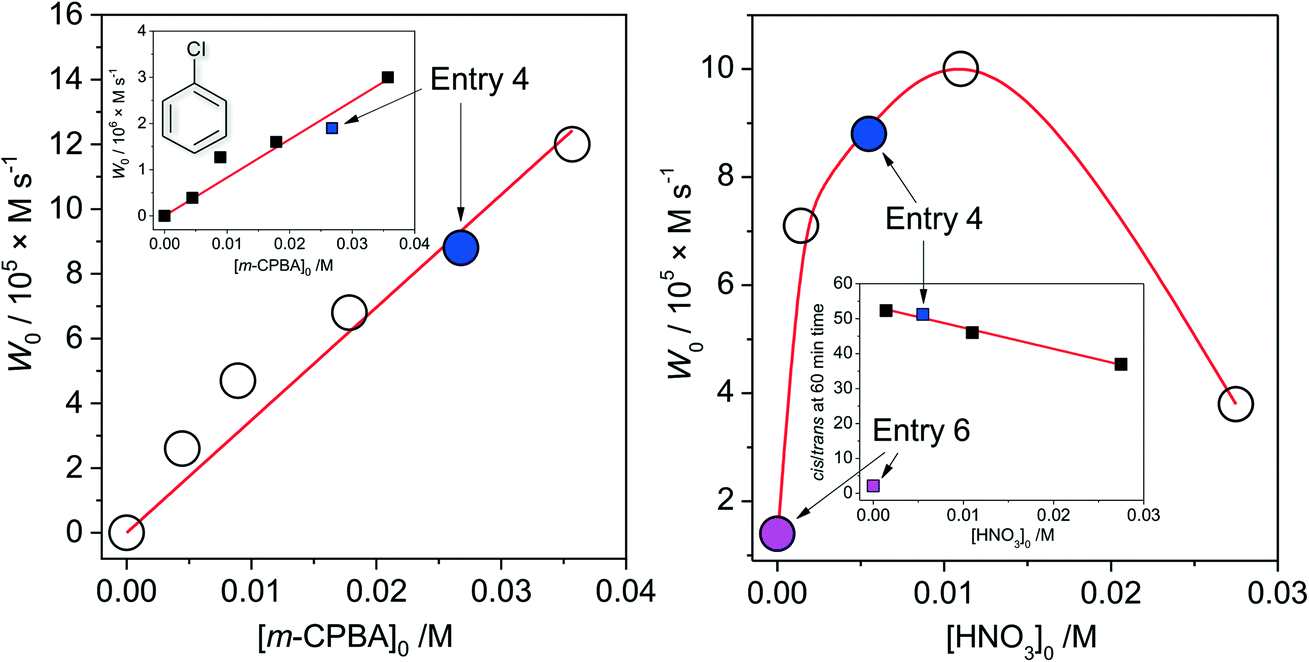 | ||
| Fig. 3 Dependences (experimental points) of the initial reaction rates (see the ESI†) of tertiary cis-alcohol accumulations on concentrations of the oxidant (m-CPBA, left) and acid promoter (HNO3, right). Conditions, except for the concentration varied, are as those for entry 4, Table 1. Left inset shows the dependence of the initial rate of chlorobenzene accumulation on the concentration of the oxidant. Right inset shows the dependence of the achieved (at 60 min reaction time) c/s ratio on the concentration of the acid promoter (solid line shows the linear fit of the data for [HNO3]0 > 0 M region). Solid blue and magenta circles and squares correspond to conditions of entries 4 and 6, Table 1, respectively. | ||
From the above set of data, the catalytic conditions with [1]0 = 2.7 × 10−6 M (i.e. [1Co]0 = 1.1 × 10−5 M), [HNO3]0 = 5.5 × 10−3 M, [m-CPBA]0 = 0.027 M and [substrate]0 = 0.1 M were chosen for further studies, due to the moderate reaction rate of W0 = 1.6 × 10−5 M s−1 (for cis-1,2-DMCH) and suitable yields/TON values (entry 5, Table 1).
While the accumulation of tertiary cis-alcohol (retention of stereoconfiguration) shows an expected gradual behaviour (Fig. 4, left), the respective dependence of trans-alcohol concentration on time (Fig. 4, middle) is drastically different: some amount of trans-alcohol, formed in the first seconds of reaction, decreases to about half within 1 h reaction time. The commercial substrate (cis-1,2-DMCH) was found to contain only trace amounts of both cis- and trans-alcohols, i.e. the observed trans-alcohol comes from the catalytic process. Hence, two reaction steps can be elucidated: fast initial (first seconds; rapid accumulation of small amounts of both cis- and trans-alcohols) and then continuous (gradual decrease of trans-alcohol and accumulation of cis-alcohol amounts) ones. The decrease of trans-alcohol concentration can be explained by overoxidation. The observation of the fast initial step does not indicate the formation of CoOx nanoparticles, as the latter process is typically manifested by a pronounced (up to tens of minutes) lag period.64
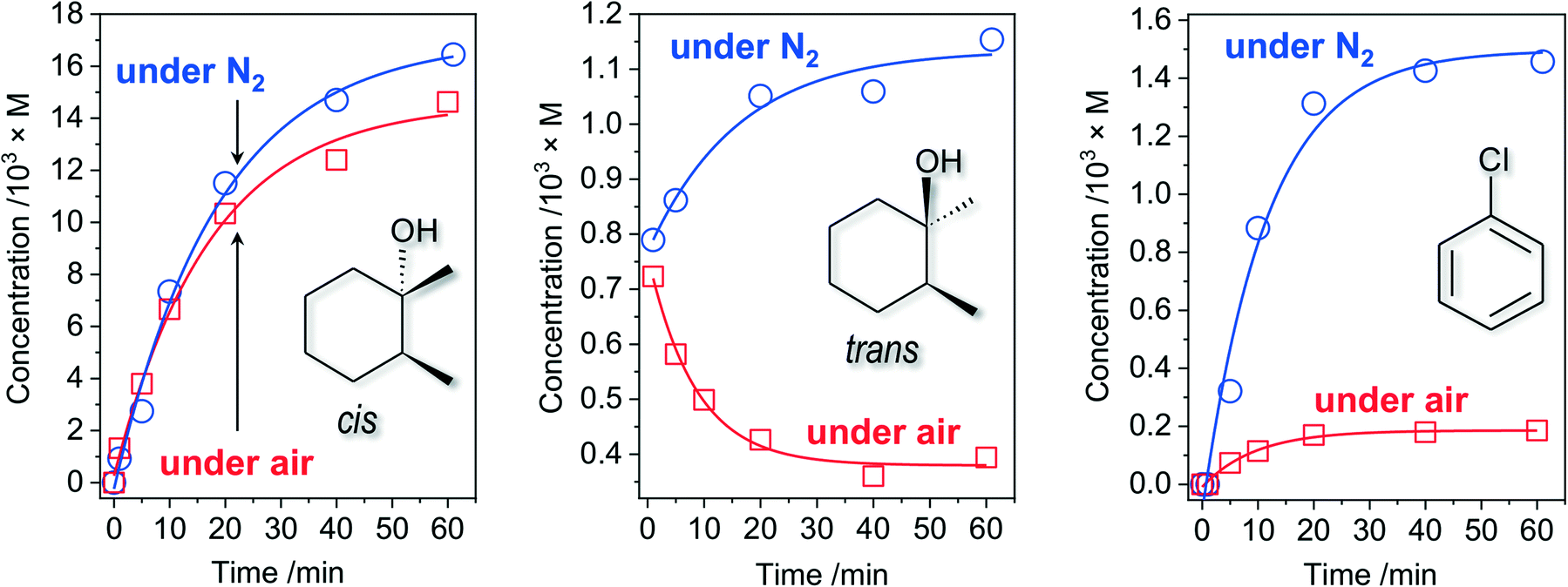 | ||
| Fig. 4 Accumulations of tertiary cis-, trans-alcohols and chlorobenzene under air (red squares) and N2 (blue circles) in the course of cis-1,2-DMCH oxidation with [cis-1,2-DMCH]0 = 0.1 M, [m-CPBA]0 = 0.027 M, [HNO3]0 = 5.5 × 10−3 M, and [1Co]0 = 1.1 × 10−5 M in CH3CN (5 mL total volume). Data of entries 5 and 8, Table 1, are included. | ||
In the absence of air oxygen, the trans-alcohol concentration increases with time, while the accumulation of the cis-product is not significantly influenced (Fig. 4, middle and left). This results in the lower c/t ratio of 14.3, observed in the absence of O2 (entry 8, Table 1). Further, elevated amounts of chlorobenzene (up to 8 times compared to the tests in the presence of O2) were observed in the reaction under N2 atmosphere (Fig. 4, right).
The reaction by-products during the oxidation of cis-1,2-DMCH are those appearing from the attack to secondary C–H bonds (Scheme 2), while the products of methyl group oxidation are not detected at all. In the oxidation of cis-1,2-DMCH, the normalized 3°:2° selectivity was found to be up to 47:1. The overoxidation products are ketones formed through ring cleavage (Fig. S11, S19 and S20†). The attempt to oxidize various substrates (Scheme 3A; here and further the conditions are the same as for entry 5, Table 1) revealed a pronounced sensitivity of the catalytic system to the position of the tertiary R3C–H bond and the nature of the substituent R. The stereospecificity is retained for the oxidation of cis-decahydronaphthalene and cis-1,4-dimethylcyclohexane, where c/t ratios of the tertiary alcohols of 81 and 38, and normalized 3°:2° selectivities of 55:1 and 21:1 are achieved. The t/c ratios for trans-1,2-dimethylcyclohexane and trans-decahydronaphthalene were found to be 65 and 63, respectively. Although the total yields (44 and 67%, respectively) are close to those for cis-substrates, the selectivity towards tertiary C–H bonds is lower (yields are 33 and 44%), as reflected by the 3°:2° selectivities of 12 and 15, respectively.
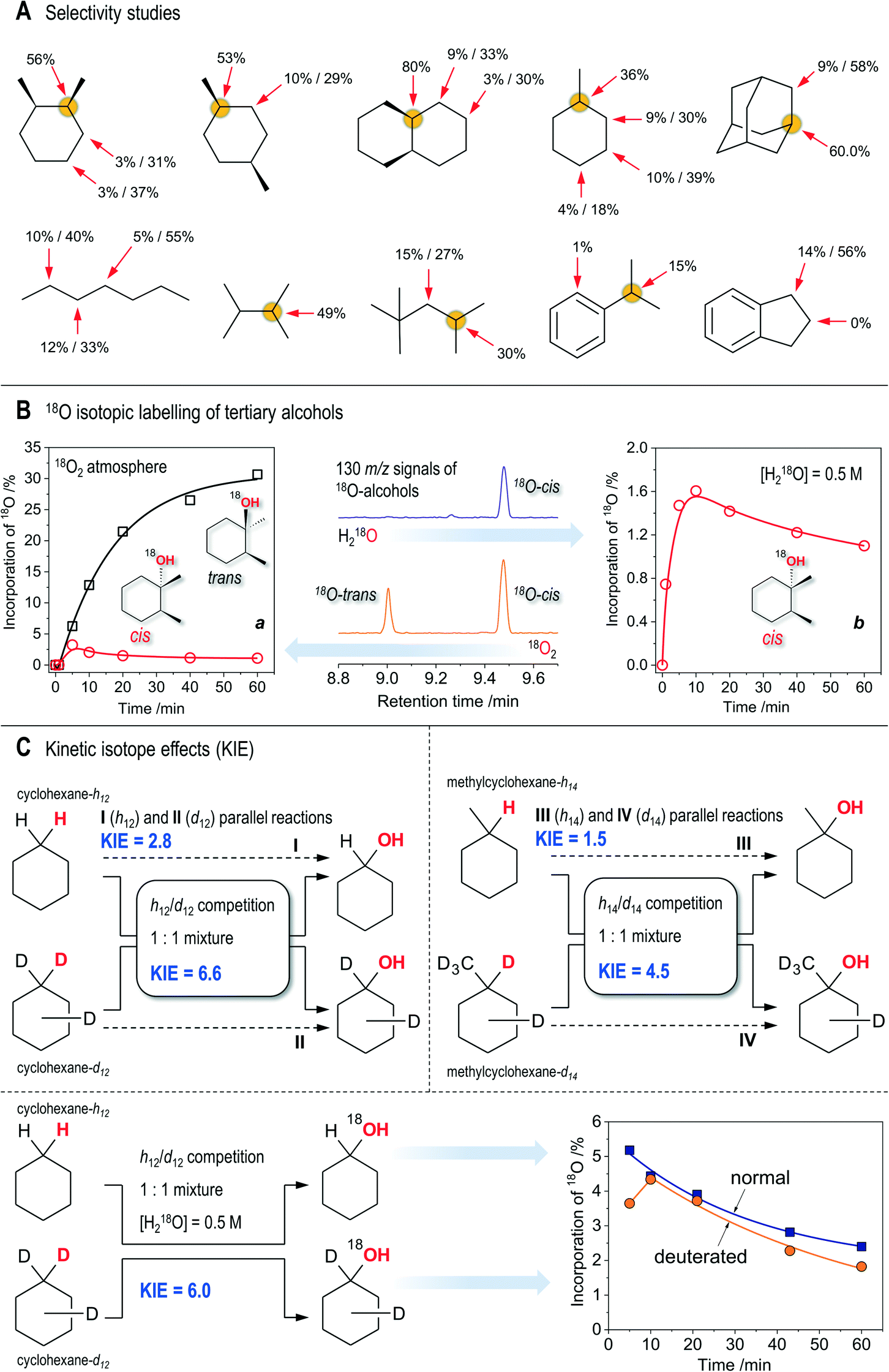 | ||
| Scheme 3 Key experimental observations. All the conditions correspond to those for entry 5, Table 1, unless stated otherwise. A: Bond (3°:2° C–H bonds) and regioselectivity studies (the latter concerning the 2° C–H bond oxidation) for selected substrates, shown as the yields of respective products based on substrates/selectivities towards alcohols (yield of alcohol per total yield of the respective alcohol and ketone). Concentrations of substrates are 0.1 M. B: a (left): Incorporation of 18O into tertiary alcohols from 18O2; b (right): incorporations of 18O into cis-alcohol from H218O (0.5 M); centre: fragments of the MS-chromatograms showing the intensities of the 130 m/z signal (18O-labeled molecular ion of alcohols) for 18O2 and H218O experiments (60 min reaction time). No labeled trans-alcohol is observed in the latter case. C: Top: Observed kH/kD kinetic isotope effects in the course of oxidation of pure substrates or their equimolar (0.1 M in total) mixtures (normal and deuterated cyclohexanes and methylcyclohexanes). Bottom: Incorporation of 18O from H218O into cyclohexanols in the course of competitive oxidation of normal and deuterated cyclohexanes. | ||
The oxidation of adamantane gave a normalized 3°:2° selectivity of 20:1. The oxidation of 2,3-dimethylbutane (Scheme 3A) proceeded with a slightly lower initial reaction rate (9.6 × 10−6 M s−1) compared with the oxidation of cis-1,2-DMCH (1.6 × 10−5 M s−1, entry 5), resulting in a close yield. Surprisingly, oxidation of cumene (Scheme 3A), whose C–H bond dissociation energy (BDE) is considerably lower than that of 2,3-dimethylbutane (97 and 85 kcal mol−1, respectively65), gave a reaction rate of only 2.5 × 10−6 M s−1 under the same conditions. In the latter reaction about 1% of acetophenone was detected (starting cumene was free of this product), which makes ca. quarter part of the overall yield of products (Scheme 3A). Formation of acetophenone through four-electron oxidation of cumene was observed earlier with ruthenium complexes as catalysts (operating via RuIVO HVMO species).66,67 2,2,4-Trimethylpentane reacted weakly, showing only little prevalence for its tertiary C–H bond (Scheme 3A). Oxidation of n-heptane revealed no regioselective preference in different secondary C–H bonds. Only traces of phenol were detected in the oxidation of benzene. Hence, the catalytic system 1/HNO3/m-CPBA shows pronounced preference for oxidation of tertiary C–H bonds, only slightly reacting with secondary C–H bonds. It is almost totally unreactive towards oxidation of methyl groups and aromatic H atoms.
18O-studies
To gain further insight, tests using 18O labeled dioxygen and water have been performed in the oxidation of cis-1,2-DMCH. The incorporations of 18O from 18O2 into tertiary cis- and trans-alcohols were found to be quite different: while the main product, cis-alcohol, contained ca. 1% of 18O after 1 h reaction time, the inverted trans-alcohol accumulated almost 30% of 18O (Scheme 3B). The absolute amounts of both labeled tertiary alcohols were comparable (Scheme 3B). Small amounts of H218O, formed from 18O2, were detected. The main reaction product, cis-alcohol, incorporates up to 1.6% 18O oxygen from H218O (Scheme 3B), while the 18O levels in the trans-alcohol are too low for their reliable determination, although one can suppose that the trans-alcohol also contains some amounts of 18O (Fig. S13†). The fragments of 130 m/z chromatograms (Scheme 3B, centre) visually demonstrate that 18O from H218O preferably goes to cis-alcohol, while that from 18O2 goes to both alcohols nearly equally. No oxygen exchange between tertiary DMCH alcohols and water was detected within 1 h time in acetonitrile in the presence of nitric acid (Fig. S6†).Oxidation of 2,3-dimethylbutane in the presence of 0.5 M H218O resulted in 2.7% of 18O-labeled alcohol after 1 h (under conditions similar to those for entry 5, Table 1). However, the respective alcohol (2,3-dimethyl-2-butanol) was found to exhibit slow oxygen exchange with H218O during the 1 h time period (ca. 1% of 18O was detected in a comparative test; see the ESI†). Hence, we can only say that the 18O incorporation in the course of the catalytic process may contribute to the final 18O levels. Oxidation of cumene in the presence of 0.5 M H218O resulted in 49% 18O enrichment of cumene alcohol and 29% of acetophenone, while the products of phenyl group hydroxylation remained 18O-free. Such a large incorporation of labeled oxygen into cumene alcohol could be explained by the direct oxygen exchange between its hydroxy-group and water. The respective test revealed 13% of 18O-labeled cumene alcohol after 24 h of stirring in acetonitrile in the presence of 5.5 × 10−3 M nitric acid.
It was found that 18O from H218O may appear in alcohols under catalyst-free conditions. The reaction rate exhibited for [m-CPBA]0 = 0.027 M in the absence of a catalyst is too low (entry 7, Table 1) for reliable determination of the small 18O levels in the products. With elevated concentrations of H218O (1.0 M) and m-CPBA (0.14 M), in the presence of HNO3 (5.5 × 10−3 M) and no catalyst, 4.6% total yield of tertiary alcohols (based on the substrate) was achieved, with a c/t ratio of 4.8, after 6 h (Fig. S10†). Under these conditions, the cis-alcohol accumulated with a reaction rate of 1.8 × 10−5 M s−1 and gained 6.2% of 18O after 6 h, while no labeled trans-alcohol was detected (Fig. S10†). Remarkably, in the absence of the promoter, both tertiary alcohols contained much lower 18O levels (ca. 1%).
Oxidation of cyclohexane (CyH) in air in the presence of 0.5 M H218O revealed an initial reaction rate W0 of 6.4 × 10−6 M s−1, yield of products (sum of cyclohexanone and cyclohexanol) of 30% and alcohol/ketone (A/K) ratio changing from 14 at 1 min to 0.43 after 1 h. The partial reaction constants revealed that cyclohexanol oxidizes 24 times faster than cyclohexane (Fig. S14†). Oxidation of cyclohexanol is in accord with the above observation, showing a relatively high rate W0 = 9.6 × 10−6 M s−1 even for one order lower substrate concentration, [CyOH]0 = 0.01 M (Fig. S15†). The reaction rate W0 of 6.2 × 10−6 M s−1, observed when the same reaction was performed under N2 atmosphere, is similar to that for the open air reaction. The initial reaction rate of chlorobenzene accumulation was considerably higher for oxidation of cyclohexane under inert atmosphere (1.3 × 10−6vs. 3.8 × 10−7 M s−1 for N2 and air atmosphere, respectively). The maximum percentage of 18O-labeled cyclohexanol (5.0%) was observed at 2 min reaction time (Fig. S14,† right). After that the amount of the labeled cyclohexanol drops until 1.9% at 60 min.
Kinetic isotope effects and D-labeling
The kinetic isotope effect (KIE) was determined (Scheme 3C), at first, from the competitive oxidation of an equimolar mixture of normal and deuterated cyclohexanes (total concentration 0.1 M) with other conditions the same as for entry 5, Table 1. The accumulations of all four products (normal and deuterated alcohols and ketones) are shown in Fig. S16;† the initial reaction rates W0 were found to be 3.0 × 10−6 M s−1 and 4.8 × 10−7 M s−1 for oxidation of C6H12 and C6D12, respectively (Table S1†). The behaviour of accumulations of alcohols and ketones is close to that observed for oxidation of normal cyclohexane (Fig. S14†), pointing that for both C6H12 and C6D12 the alcohols are oxidized much faster than the ketones (Table S1†). While the normal alcohol is oxidized 23 times faster compared to the normal alkane, the respective difference for deuterated cyclohexane was found to be considerably higher (63 times). From the reaction rate constants for normal and deuterated cyclohexane oxidation to cyclohexanol, one can calculate the KIE value as kH/kD, giving 6.6 (Table S1,†Scheme 3C). One can also evaluate the KIE2 for oxidation of cyclohexanol to cyclohexanone, expressed as k2(H)/k2(D) and found to be 2.4. Considering that the concentrations of alcohols undergo rapid changes (Fig. S14 and S16†), the observed value of KIE2 has no chemical significance and just demonstrates that KIE2 is lower than KIE. Finally, we should note that in the present case the KIE value cannot be correctly determined just as a ratio of product concentrations measured at some certain reaction time: the KIE calculated in this way ranges from 2.7 to 5.6 (calculated for alcohols) or from 5.8 to 7.5 (for all products), with the higher values at the beginning of the reaction.In contrast to the competitive C6H12/C6D12 oxidation, the KIE obtained from the parallel tests (independent oxidations of pure normal and deuterated cyclohexanes) shows a considerably lower value of 2.8 (Scheme 3C). The same difference between competitive and parallel oxidations was observed for methylcyclohexane oxidation: while the competitive process (conditions were the same as those for cyclohexane) shows a notable KIE effect of 4.5 for the tertiary C–H bond (Scheme 3C, Fig. S18†), the same reactions revealed nearly equal reaction rates with KIE = 1.5 when h14- and d14-methylcyclohexanes oxidized independently (Scheme 3C).
The oxidation of 0.1 M deuterated methylcyclohexane revealed 7% of labeled deuterated chlorobenzene as a by-product after 5 min (5% after 1 h). Careful evaluation of all the other tests involving D-labeled substrates disclosed the presence of the deuterated chlorobenzene by-product in the oxidation of a mixture of h14- and d14-methylcyclohexanes (4%) and deuterated cyclohexane (4%). Furthermore, the catalyst-free oxidation of the C6H12/C6D12 mixture using [C6H12]0 = [C6D12]0 = 0.05 M, [HNO3]0 = 5.5 × 10−3 M, and [m-CPBA]0 = 0.027 M afforded 3% of deuterated chlorobenzene and an approximate KIE value of 5.3 after 24 h, with a total yield of 2%. In turn, oxidation of cis-1,2-DMCH in the presence of D2O resulted in the normal chlorobenzene only. The same product was obtained by using d4-acetic acid as a promoter. Furthermore, the incorporation of D into chlorobenzene from d14-methylcyclohexane did not depend on the atmosphere (air or N2), the presence of the nitric acid promoter or quenching of the samples with PPh3. An increase of [d14-MeCyH]0 up to 0.2 M (in the absence of HNO3) resulted in 13% deuteration of chlorobenzene after 1 h. A very high level of chlorobenzene deuteration (57%) was observed when 0.027 M m-CPBA was stirred in CD3CN (6% of CH3CN was present) at 50 °C in the presence of [1Co]0 = 1.1 × 10−5 M and in the absence of the promoter for 1 hour (Fig. S4 and S26†).
Combined H218O/D/KIE studies
The incorporation of 18O from H218O in the course of C6H12/C6D12 equimolar mixture oxidation at 50 °C disclosed that normal and deuterated alcohols accumulate comparable amounts of 18O (Scheme 3C). The KIE value for the first step (6.0), oxidation of alkane to alcohol, was found to be of the same magnitude (6.6) as for the reaction in the absence of H218O. Incorporation of 18O within the time is of nearly linear character for both normal and deuterated products, with the maximum of the labeling at the beginning of the reaction of 5.2% (for C6H12) and of 4.3% at 10 min time (for C6D12) (Scheme 3C).Participation of ROOH intermediates
The search towards alkyl hydroperoxides (a typical product in a free radical oxidation of alkanes11) was performed by direct monitoring of characteristic spectra of alkyl hydroperoxides by GC-MS68–72 as well as by indirect methods (comparison of the chromatograms taken before and after addition of the reducing agent PPh3, according to the method developed by Shul'pin11). All the chromatograms recorded before and after addition of solid PPh3 to the probe were identical when the reaction was conducted at 50 °C (Fig. 5). No signs for the mass-spectral signal attributable to cyclohexyl hydroperoxide were found (Fig. 5, right) even when employing a single ion monitoring MS detection mode and lowered reaction temperature (Fig. S23†). Notably, the chromatograms recorded in the course of cis-1,2-DMCH oxidation in the absence of the catalyst (entry 7, Table 1) at 50 °C reveal a significant difference before and after addition of PPh3 (Fig. S11†), showing also traces of tertiary alkyl hydroperoxides (Fig. S24†).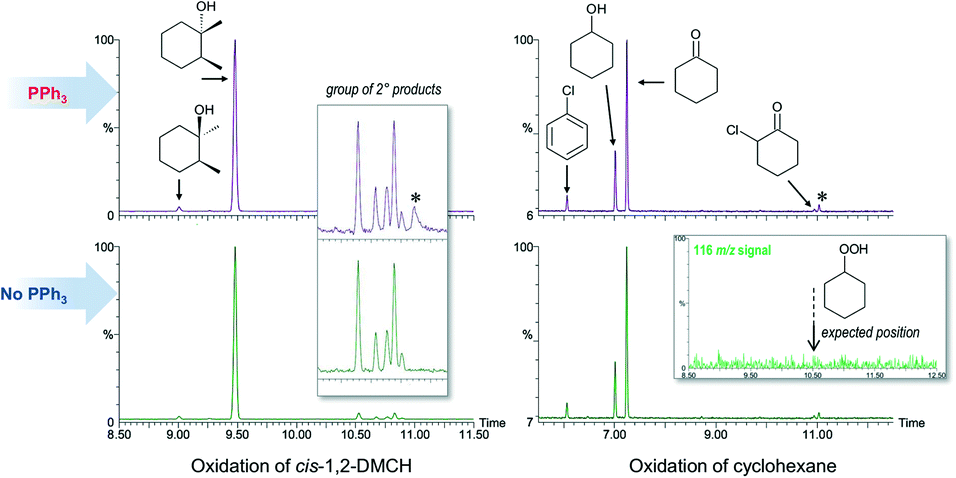 | ||
| Fig. 5 Comparison of the GC chromatograms recorded after (top) and before (below) quenching with solid PPh3 for oxidation of cis-1,2-DMCH (left) or cyclohexane (right). Inset (left) shows the groups of ketones and secondary alcohols formed through the oxidation of the methylenic groups of cis-1,2-DMCH. Inset (right) shows the intensity of the 116 m/z signal, specific to the mass-spectrum of cyclohexyl hydroperoxide (Fig. S23†) in the region around its expected position (10.5 min; Fig. S23†). Peaks marked by * were not identified. | ||
In contrast to the reactions conducted at 50 °C, the chromatograms recorded before and after addition of PPh3 for 0 °C reactions reveal different patterns (Fig. S20 and S21†). The yields of products after 2 h time were ca. 5%. The cis/trans ratios in the oxidation of cis-1,2-DMCH were 15 and 2 before and after addition of PPh3, respectively. Moreover, the chromatograms recorded before addition of PPh3 disclosed significant amounts of chlorobenzene, completely disappearing after treatment of the sample with PPh3 (Fig. S20 and S21†). The tertiary alcohols did not incorporate 18O neither from 18O2 nor H218O after 2 h at 0 °C reaction, regardless of PPh3. However, the yields of the products as well as the 18O incorporations into the products and CO2 return to the expected levels when the mixtures were further stirred at 50 °C for 1 h. Samples of the 0 °C reactions injected to the GC before treatment with PPh3 disclose large amounts of CO2 free of 18O (ca. one order higher than those after PPh3) (Fig. S20 and S21†). This, together with elevated amounts of chlorobenzene observed before addition of PPh3, indicates the accumulation of large amounts of the aroyloxyl radical ArC(O)O˙ which undergoes rapid decarboxylation when the sample passes a hot GC injection zone. However, no peaks attributable to alkyl hydroperoxides were seen neither for oxidation of cyclohexane nor for dimethylcyclohexane.
In situ catalytic system
We attempted to reproduce the catalytic activity of 1 using starting metal salts and their mixtures with the pro-ligand H2L. Cobalt nitrate exhibits a much lower activity and stereoselectivity than complex 1 (entry 11, Table 1), whereas iron nitrate was nearly inactive regardless of the promoter (entries 9 and 10). Cobalt nitrate in the presence of nitric acid shows a notable activity (entry 12), but the stereoselectivity is considerably lower than that for complex 1, especially for higher [Co]0 (Fig. S33†). The in situ prepared systems (entries 13 and 14) were found to be active, but showing lower c/t ratios (Fig. 6 and S33†) as compared to the corresponding 1-based entries (4 and 5, respectively; Table 1) due to the larger production of chlorobenzene and enhanced amounts of trans-alcohol.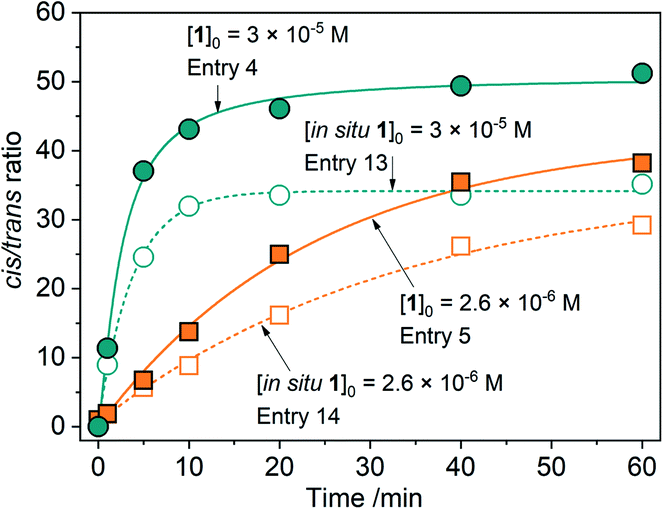 | ||
| Fig. 6 The c/t ratios as a function of time shown for catalytic systems using pre-catalyst 1 and respective mixtures of salts and ligand (in situ systems). | ||
ESI-MS spectroscopic studies and determination of the putative catalytically active species
Earlier complex 1 was shown to produce two main types of coordination compounds in acetonitrile solutions: homometallic [CoIII(HSae)2]+ and heterometallic [{CoIII(Sae)2}2FeIII]+ ones (by means of kinetic and ESI-MS methods).20 Heterometallic species were found to exist only for [1] > 5 × 10−5 M, while homometallic ones were observed in all studied concentrations. Notably, both species were stable in the presence of a large concentration of the nitric acid promoter (0.04 M).20 The ESI-MS spectra of the acetonitrile solution of complex 1 with [1] = 4 × 10−5 M revealed the presence of [Co(HSae)2]+ species (387.07 m/z, calculated) as a dominant peak (Fig. S30†) as well as some weaker peaks with higher m/z (772.1 and 1243.74, observed), which can be tentatively assigned to [Co2(Sae)4]+ and heavier species. After dilution to [1] = 2.8 × 10−6 M and addition of HNO3 (5.5 × 10−3 M), the [Co(HSae)2]+ peak remains, while the above peaks disappear. The peak at 166.04 m/z (Fig. S30†) can be attributed to the protonated ligand H3Sae+ and this observation allows the existence of equilibrium between coordinated and de-coordinated ligands in solution to be presumed. The study of an acetonitrile solution of m-CPBA revealed a complex pattern in both negative and positive modes (Fig. S32†). The peaks at 155.33 and 171.35 m/z can be attributed to anions of m-chlorobenzoic acid (m-CBA) and m-CPBA, respectively. Addition of a m-CPBA acetonitrile solution to the respective solution of complex 1 with nitric acid (final concentrations equal to those for entry 5, Table 1) resulted in the appearance of a new peak at 664.45 m/z (Fig. S30†). The latter one, as well as the [Co(HSae)2]+ at 387.05 m/z, was found to be short-lived: after 3 minutes the spectra become drastically different, revealing only a low-intensity 168.06 m/z signal on a noisy background (Fig. S30†). This observation could account for the formation of uncharged species, which do not get charged during the ESI-MS experiment. It is proposed that this could be [Co(HSae)2(m-CPBA)]0 (where m-CPBA is in a deprotonated form). From the ESI-MS data and linear W0vs. [1]0 relation in the [1Co]0 < 2.5 × 10−5 M region, the absence of heavier (dimeric) species in catalytic solutions was assumed. The impossibility of detection of neutral species by the ESI-MS method is a common case for the catalytic investigations.73 For instance, similar observations (no ESI-MS detectable species) were reported in the case of olefin epoxidation with the Co(ClO4)2/m-CPBA system.74 Hence, we concluded that shortly after addition of m-CPBA to the reaction mixture all the [Co(HSae)2]+ species transform to [Co(HSae)2(m-CPBA)]0 ones, which start the catalytic cycle (see the next chapter). This period could be assigned to the initial catalytic phase, when both tertiary trans- and cis-alcohols are formed in small amounts (see above).Although decoordination and/or oxidative degradation of the organic ligand in [Co(HSae)2]+ is possible,75 we presume that this process is negligible in our conditions. Firstly, this species remains stable in the presence of HNO3 and even H2O2.20 Further, the products of H2Sae degradation (e.g. salicylic aldehyde) are detected by the GCMS method during the alkane oxidation with the HNO3/H2O2 system,69,76 but not with the HNO3/m-CPBA one. In the last case they are only detectable if running the 1/HNO3/m-CPBA system without the substrate for a period of 24 h (Fig. S25†). No salicylic aldehyde or its oxidation products were detected in regular tests. It is known that treatment of labile cobalt complexes with small amounts of nitric acid in acetonitrile may lead to the formation of the [Co(CH3CN)6]2+ and [Co(CH3CN)3(NO3)]+ species, which can be seen as 152.3 and 243.5 m/z peaks in the ESI-MS spectra.77 However, in the case of 1/HNO3 and 1/HNO3/m-CPBA solutions no such peaks were detected.
Addition of 1 equivalent (relative to sum of metals) of a strong chelating ligand, 2,2′-bipyridine (bipy), to the reaction mixtures containing 1 ([1Co]0 = 1 × 10−5 M) or Co(NO3)2 ([Co]0 = 1 × 10−5 M) resulted in complete stereoselectivity loss for 1 (c/t = 1.5), while the cobalt nitrate catalyst retained significant stereoselectivity (c/t = 10) (Fig. S35†). Furthermore, the catalytic system 1 + bipy afforded large amounts of chlorobenzene (18% of yield), while the Co(NO3)2 + bipy system yielded it at 0.4% level only. These results can be understood if one considers the interaction of bipy with [CoIII(HSae)2]+ species, leading to catalytically inactive [CoIII(HSae)2(bipy)]+ ones where all positions in the coordination sphere of cobalt are occupied. From the large amounts of chlorobenzene and epimerization of stereoconfiguration of cis-1,2-DMCH, one may conclude the suppression of the stereoselective metal-mediated route and enforcement of the free-radical activity. In contrast, the [Co(bipy)]3+ species formed from the cobalt nitrate remain active because they do not contain the chelating ligand other than bipy. Considering that the iron salts were found to be almost unreactive (entries 9 and 10), and the synthesized model compound [CoIII(HSae)(Sae)]·CH3OH·H2O (2) revealed a catalytic activity similar to that exhibited by 1 (Table 1, entries 15 and 16; Fig. S1 and S36†), the [CoIII(HSae)2]+ species was further studied as a model of a catalytically active species.
Theoretical mechanistic considerations
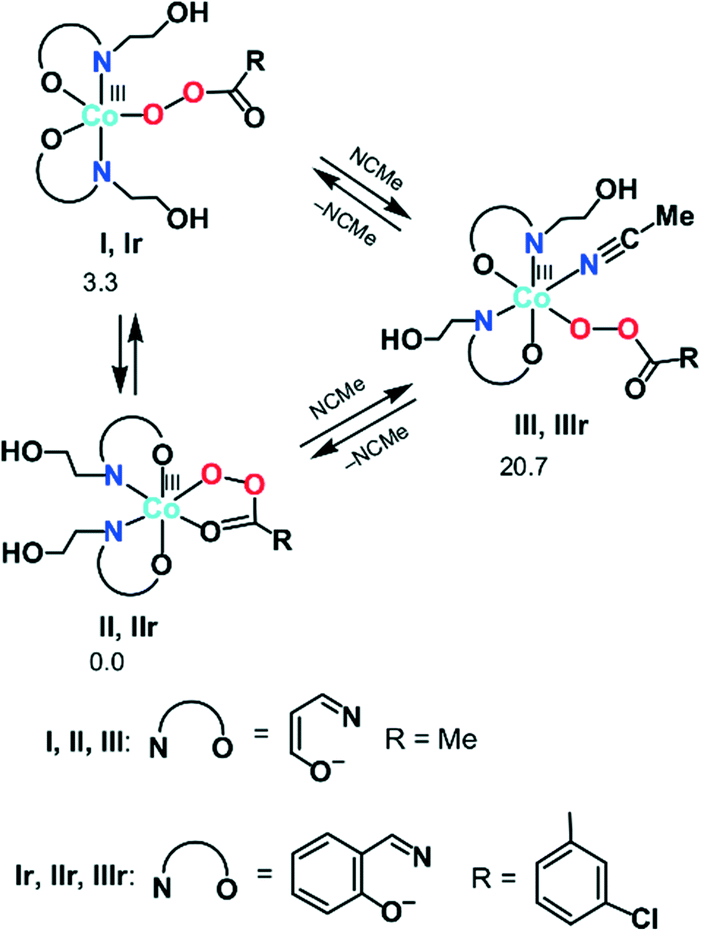 | ||
| Scheme 4 The calculated most stable isomers of the Co(III) peroxoacidic complexes (relative ΔGs values for I, II, and III are given in kcal mol−1). | ||
First, we analysed the formation of the active catalytic species for peroxoacetic acid taken as a simple model system with a catalyst bearing 3-iminoprop-1-en-1-ol ligands instead of Sae. All possible geometrical isomers of the corresponding complexes I–III were calculated, and only the most stable ones are discussed here (see Scheme 4 and S2 in the ESI† for the structures and energies of all isomers). The ground state of II and III was found to be singlet (1II and 1III), while the ground state of I is triplet (3I). The calculations indicated that the coordinatively saturated complex 1II with the bidentate peroxoacetate ligand is 3.3 kcal mol−1 more stable than the penta-coordinated complex 3I with the monodentate MeC(O)O− ligand. On the other hand, the formation of complex 1III upon addition of one acetonitrile molecule to 1II is highly endergonic (by 20.7 kcal mol−1). Thus, namely complex 1II may be considered as an active catalytic species.
Based on these preliminary results, the further calculations of the alkane oxidation were carried out for the real oxidant and catalyst used in the experimental part of this work (i.e. m-CPBA and IIr, Scheme 4).
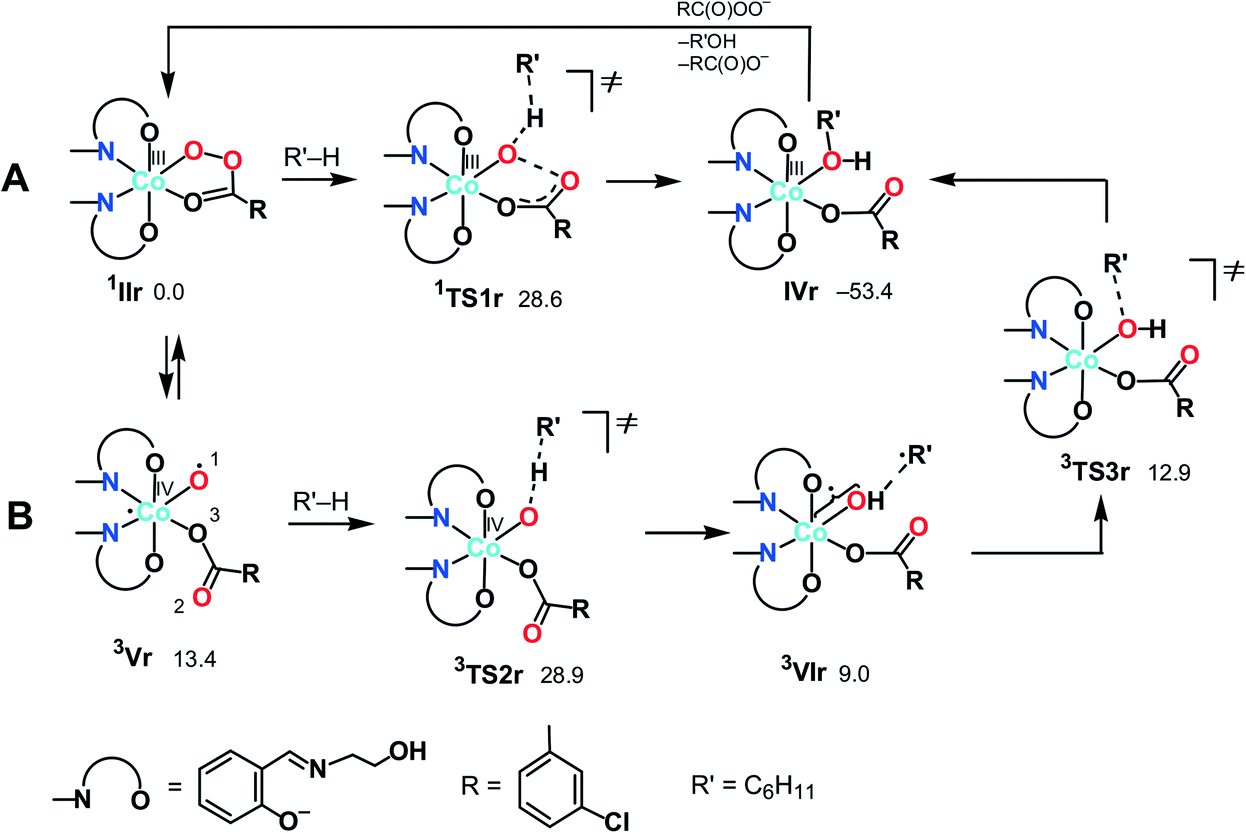 | ||
| Scheme 5 Possible mechanisms of the alkane (R′H) hydroxylation on the singlet (A) and triplet (B) surfaces (Gibbs free energies in CH3CN solution are indicated relative to 1IIr in kcal mol−1). | ||
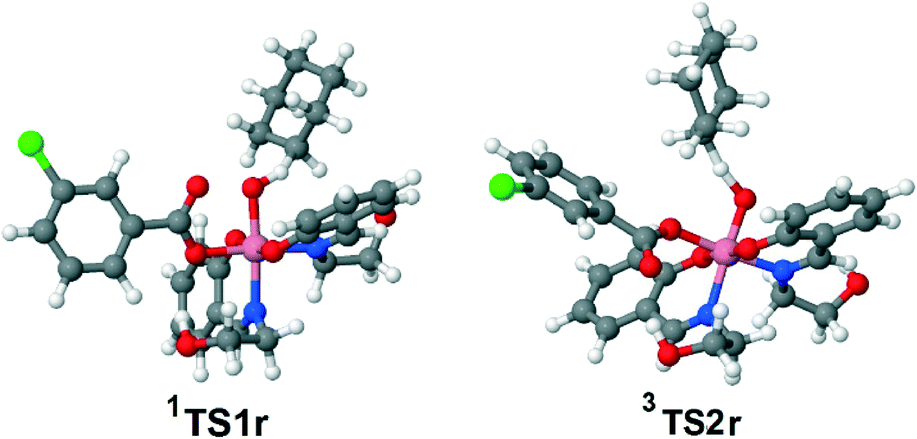 | ||
| Fig. 7 Calculated equilibrium structures of the most important transition states. | ||
Besides 1TS1r, another possible transition state with the η2-coordinated peroxoacid, was also found for this mechanism (Scheme S3 in the ESI†). However, this TS has very high relative energy and, therefore, this possibility may be ruled out.
O species. Complex 3Vr could exchange oxygen with H218O, leading to 18O-labeled alcohols, in a process known for the high-valent oxo complexes of iron or ruthenium, formed upon reaction with m-CPBA.34,38,78,79
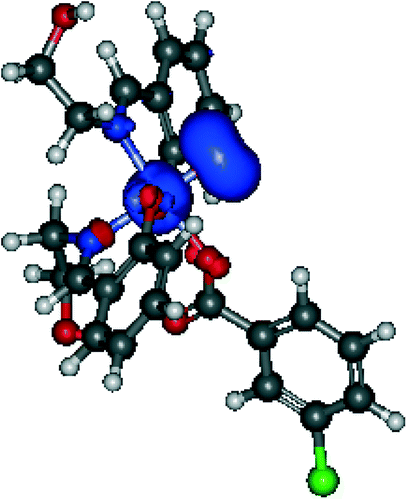 | ||
| Fig. 8 Spin density distribution in 3Vr. | ||
At the next step, a hydrogen atom of alkane may be abstracted by the oxygen atom O(1) in 3Vrvia3TS2r to give the hydroxo complex [Co(HSae)2(OH){OC(O)R}] (3VIr) and the cyclohexyl radical loosely bound to the hydroxo ligand. The following rebound of the Cy˙ radical leads to complex IVrvia3TS3r. Similar rebound pathways 5/7Vr + CyH → 5/7TS2r → 5/7VIr were found for the quintet and septet surfaces. However, they are more energy demanding that the route on the triplet surface. Several other mechanisms are described in the ESI† but all of them are less feasible.
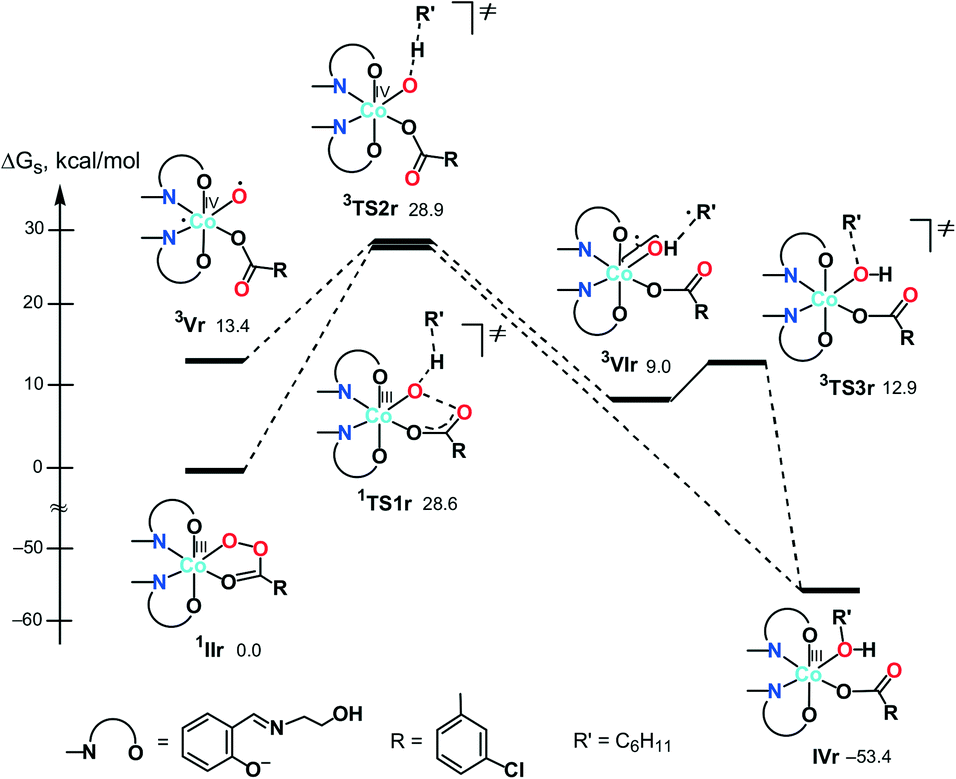 | ||
| Fig. 9 Energy profiles of the most favourable mechanisms of cyclohexane hydroxylation. | ||
Discussion
Let us briefly collect key observations on the 1/HNO3/m-CPBA system:(a) The system is highly selective towards the oxidation of tertiary C–H bonds with a high level of retention of stereoconfiguration (RC > 99%) when nitric acid is used as a promoter. The promoters of lower acidity, such as acetic and trifluoroacetic acids, reveal lower selectivity.51,52
(b) The system is strictly air-sensitive: in the absence of dioxygen, the c/t ratio drops from >60 to 15 due to the elevated yield of the trans-product. Large amounts of chlorobenzene are formed in the absence of dioxygen. The catalyst-free system is air-sensitive in the same way.23
(c) At 50 °C, labeled oxygen incorporates from 18O2 mainly to the trans-product (up to 30%), giving nearly equal absolute amounts of 18O-tertiary alcohols (cis/trans ratio <1.5). Small amounts of H218O were detected. The incorporation of 18O up to 70% into the trans-alcohol was achieved using a Co/Zn complex as a catalyst and a promoter of lower acidity (acetic acid).52
(d) The labeled oxygen from H218O incorporates mainly to the cis-product; the incorporation level is low (<2%). The substrate with a higher C–H bond energy (cyclohexane) and lower reaction rate gives higher 18O incorporation from H218O. The existence of a metal-free pathway for 18O incorporation from H218O into the alcohol product under oxidation with m-CPBA was disclosed (no direct oxygen exchange between the tertiary alcohols and H218O was found). Significant amounts of singly and doubly 18O-labeled CO2 were detected.
(e) Nearly equal percentages of 18O come from H218O into the normal and deuterated alcohols during the competitive oxidation of the equimolar C6H12/C6D12 mixture. Labeled 18OPPh3 was detected in the tests with both 18O2 and H218O, showing gradual accumulation with time.
(f) The kinetic isotope experiments showed considerable KIEs in the competitive oxidations, but a low or negligible KIE in the parallel reactions. The KIE value determined for the catalyst-free system (5.5; competitive oxidation of C6H12/C6D12) is of the same magnitude as for the metal-catalyzed one. The KIE value in the competitive oxidation of normal and deuterated cyclohexanes is similar to those observed using other cobalt-containing catalysts.43–45,51
(g) The formation of 7 to 13% of deuterated chlorobenzene was accounted for the oxidation of d14-methylcyclohexane, containing weak tertiary C–D bonds; no change in D-incorporations into chlorobenzene appears if the reaction is performed under inert atmosphere. The presence of deuterated chlorobenzene was found also in the other tests involving deuterated substrates, but not deuterated additives (D2O or CD3COOD). The use of deuterated acetonitrile in the absence of an alkane substrate affords more than 50% of deuterated chlorobenzene. No D-labeled m-CBA (m-chlorobenzoic acid) was detected.
(h) No cyclohexyl hydroperoxide was detected in the cyclohexane oxidation, regardless of the atmosphere (air or N2) and reaction temperature (0 or 50 °C). A separate test showed that cyclohexyl hydroperoxide, if formed, would be stable under the conditions of the experiment (Fig. S22†). Oxidation of cis-1,2-DMCH affords trace amounts of tertiary alkyl hydroperoxides at 50 °C for the catalyst-free reaction. For 50 °C temperature reactions, addition of PPh3 does not influence the yields and selectivity measured by the GC method.
The main catalytic cycle, confirmed by DFT calculations, proceeds through the formation of [CoIII(HSae)2(m-CPBA)] species, undetectable by ESI-MS. Cobalt is believed to maintain its Co(III) oxidation state, but participation of short-lived Co(IV) species is possible (Fig. 9 and Scheme 6). Although the existence of Co(II) in the presence of a large excess of m-CPBA looks unlikely, Kim and coworkers suggested that Co(II) retains its oxidation state when mixed with m-CPBA, as was shown by EPR studies (the characteristic spectrum with strong transition with g = 4.53 persisted after addition of m-CPBA).74 To verify our hypothesis, the HF-EPR spectrum (256 GHz at 5 K) of a 20 mM solution of the [CoII(HSae)2] complex in acetonitrile was recorded (Fig. S29†), featuring a series of transitions typical for Co(II) species (the strongest one with g = 4.51). Addition of 1 equivalent of m-CPBA to this solution afforded a considerable decay of the spectrum intensity (Fig. S29†), thus confirming that cobalt is likely to persist in the Co(III) oxidation state rather than forming the Co(II) one under the conditions of the experiment.
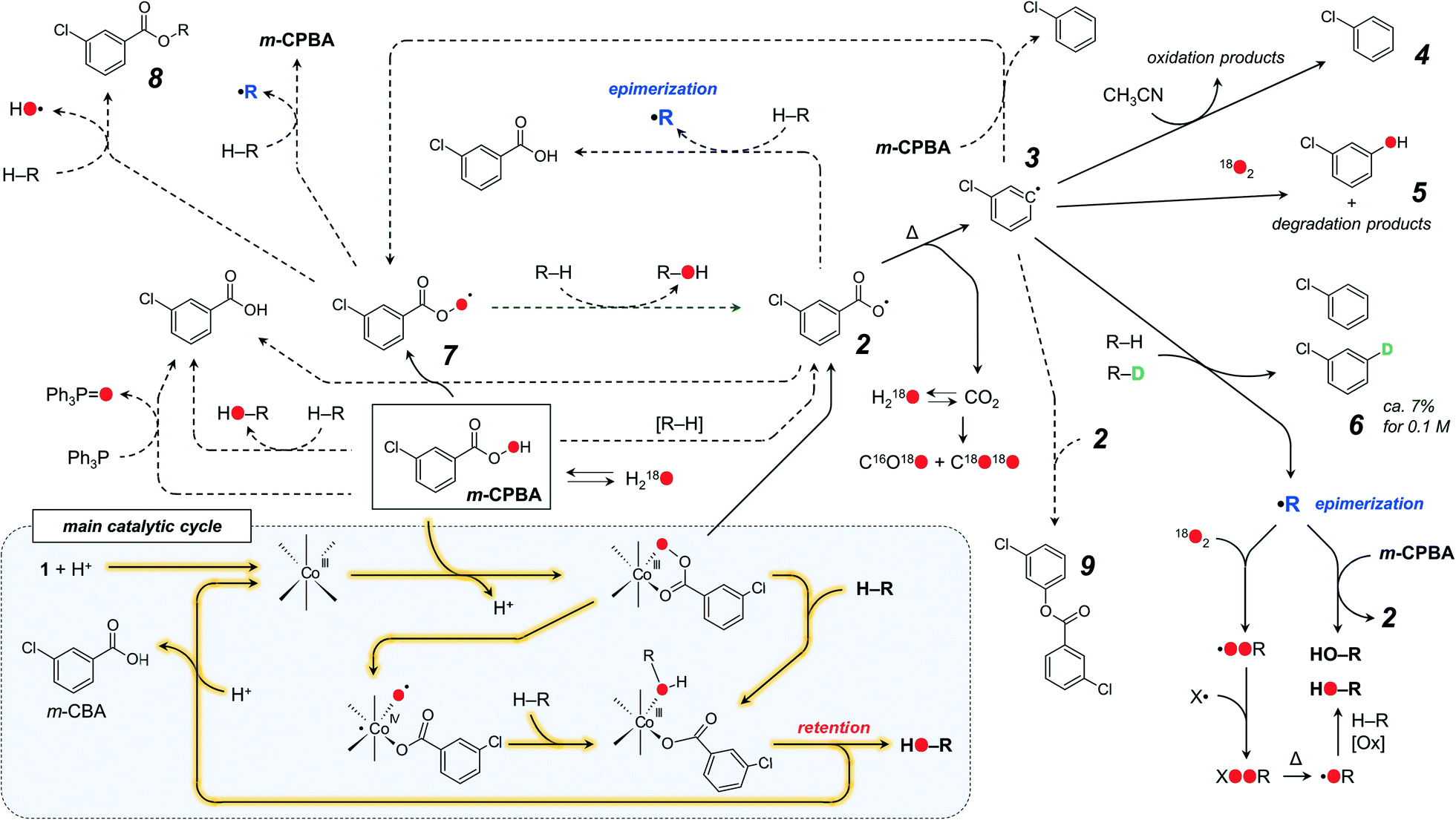 | ||
| Scheme 6 Proposed reaction pathway. Highlighted solid black lines concern the main cycle. Non-highlighted solid black lines are the main side processes. Dashed lines are possible processes, occurring mainly at low temperature. Participation of water and iron is omitted for clarity. | ||
We proposed that the C–H oxidation in the Co/m-CPBA system proceeds via at least two different routes which can be tentatively designated as selective and non-selective ones (Scheme 6). Basing on our data set, we propose that H abstraction by a chlorobenzene radical 3 is the main basis of the non-selective route. The formation of a free alkyl radical R˙, which is trapped by O2 and m-CPBA,11,42 is the origin of stereoselectivity loss (Scheme 6).
Chlorobenzene radical 3 is reactive enough to attack a neutral solvent such as acetonitrile; thus careful tuning of the solvent nature could enhance the stereoselectivity by quenching that radical. The rate of accumulation of chlorobenzene 4 correlates with the promoter acidity.51 Elevated amounts of chlorobenzene, observed in the absence of O2, correlate with increased amounts of trans- and cis-tertiary alcohols. Such an influence of dioxygen on the chlorobenzene yield is known for catalyst-free oxidations with m-CPBA;23 thus this process is not regulated by a catalyst. Oxygen readily reacts with carbon-centred radicals, in this way quenching the non-selective free radical process. Oxidation of normal cyclohexane and normal and deuterated methylcyclohexanes in the presence or absence of O2 allows the relative amount of radical 3 reacting with O2 to be estimated (Scheme 6). While the reaction of O2 with aliphatic C˙ radicals affords stable COO˙ ones, the respective reaction with aromatic radicals may afford the degradation of an aryl ring with the formation of various species (some products were detected in the substrate-free test, Fig. S26†). Labeled m-chlorophenol 5 was detected for some reactions conducted under 18O2.70
m-CPBA may form two O-centred radicals, aroyloxyl ArC(O)O˙ (2) and aroylperoxyl ArC(O)OO˙ (7), both capable of H-abstraction from C–H bonds. Hydrogen atom transfer to an aroyloxyl radical 2 from a C–H bond gives a free C˙ radical, leading to the epimerization of stereoconfiguration, which contradicts observation (a). Oxidation of C–D bonds leads to deuterated chlorobenzene 6, but not m-chlorobenzoic acid (observation (g)). Thus, under the conditions of the experiment, radical 2 undergoes nearly quantitative decarboxylation and in this way can be ruled out as a C–H attacking species.
The interaction of the aroylperoxyl ArC(O)OO˙ radical 7 with the C–H bond is more complicated23 and may afford various products (Scheme 6), including free aroyloxyl 2 or carbon-centred radical R˙, both not supported by experimental evidence. Another pathway produces an ester 8 as the main product (Scheme 6). The formation of large amounts of alkyl esters (up to 8% based on m-CPBA) was accounted for the aerobic autoxidation of adamantane by aldehydes (including m-chlorobenzaldehyde),23,24 where the RC(O)OO˙ radical results from the reaction of O2 with the acyl radical RC(O)˙. In the case of the 1/HNO3/m-CPBA system, trace amounts of cyclohexyl m-chlorobenzoate ester 8 were detected for the oxidation of cyclohexane (Scheme 6 and Fig. S27†). These observations suggest that no or only trace levels of the aroylperoxyl ArC(O)OO˙ radical 7 are formed in the 1/HNO3/m-CPBA system. The 3°:2° bond selectivity of the H atom abstraction by the ArC(O)OO˙ radical from adamantane was evaluated to be 46:1 and 78:1, depending on the method of generation of an acyl radical (photosplitting of benzyl42 or autooxidation of m-chlorobenzaldehyde,23 respectively). These values differ from those observed for adamantane oxidation by Co/HNO3/m-CPBA systems (from 23:1 to 39:1).51 The ArC(O)OO˙ radicals themselves can be obtained from the reaction of an acyl radical with O2 or aroyloxyl radical with m-CPBA. Both processes cannot be considered as favourable due to the relatively low concentrations of the respective species. Therefore, participation of the aroylperoxyl ArC(O)OO˙ radical 7 in the main oxidation route is not likely.
The rate of the catalyst-free oxidation of cis-1,2-DMCH under our conditions was evaluated to be 6 × 10−8 M s−1. The catalytic effect is clearly seen at 10 ppm level of 1, while 1000 ppm (0.1 mol%) loading allows ca. 1000-fold increase of the reaction rate. The plausible mechanism of C–H hydroxylation (Fig. 9 and Scheme 6) foresees the coordination of m-CPBA to the cobalt centre with the subsequent O-atom insertion into the C–H bond or transfer to a short-lived alkyl radical (rebound). Observations (e) and (f) suggest that the hydrogen atom abstraction is rather a product determining than a rate determining step.81 This conclusion is in accord with the non-linear W0vs. [substrate]0 dependence (Fig. 2), where high concentrations of the substrate have less influence on the reaction rate than low concentrations. The strictly linear W0vs. [catalyst]0 and nearly linear W0vs. [m-CPBA] dependences, as well as the relatively low reaction temperature, rule out the free radical chain mechanism.82
The principal reaction pathway (Scheme 6, highlighted black lines) presumes the formation of m-CBA, but not chlorobenzene as the main product of m-CPBA transformation. The amount of m-CBA found for entry 5 (Table 1) is 0.02 M, which is very close to 0.017 M expected for the sum of tertiary and secondary hydroxylation products (Fig. S34†). The difference between these values could be explained by the formation of unaccounted overoxidation by-products as well as by the elevated analytical error in the m-CBA quantification due to GC peak broadening. The sum of concentrations of the substrate and hydroxylation products was 0.1 M during the catalytic run (Fig. S34†). Similar results were obtained for the oxidation of trans-decahydronaphthalene (a less volatile substrate than cis-1,2-DMCH), for which the sum of the substrate and products was roughly 0.1 M after 1 h (the sum of products was 0.018 M and the amount of m-CBA formed was 0.019 M). Therefore, the overall mass balance of the reaction is retained.
The role of iron in the overall catalytic cycle appears to be more hidden. The only detected iron-containing species in a solution of 1 is [{CoIII(Sae)2}2FeIII]+.20 The latter, however, exists for [1] > 1 × 10−5 M, and at lower concentrations of 1 the iron species possibly exist in solvated Fe(III) forms and complexes with m-CBA (m-chlorobenzoic acid) and m-CPBA. Compounds of iron could serve as highly selective catalysts when using m-CPBA as an oxidant, presumably operating through the Fe(IV) or Fe(V) HVMO species.34,35,59 However, as the stabilization of HVMO species typically requires electron-rich N-donor polypyridyl, cyclam or similar ligands,4 their appearance is not likely. In the absence of such ligands, iron catalysts demonstrate low reactivity with m-CPBA and mainly induce low-selectivity processes (Table 1, entries 9 and 10). On the other hand, Fe(III) species are known to react with free O- and C-centred radicals in the course of a Fenton-like reaction.11,26,83 Therefore, the small amounts (ca. 5 × 10−6 M in a typical test) of Fe(III) species in the 1/HNO3/m-CPBA catalytic system should not interfere with the main reaction pathway, but would trap and neutralize free-radicals, responsible for low-selectivity C–H oxidation (Scheme 6), in this way enhancing the overall selectivity.
The incorporations of 18O from H218O into the alcohols at <10% level, observed for plenty of Co/m-CPBA catalytic systems as well as for the 1/HNO3/m-CPBA one, require further comment. The appearance of oxygen from water in the course of a metal catalysed oxidation with a peroxide is usually considered as evidence for HVMO species, because the latter may exchange oxygen with water.84–87 A free radical reaction mechanism (e.g., with the HO˙ radical as the C–H attacking species) does not afford labeled alcohols. Although observation of 18O in alcohols and epoxides in the M/m-CPBA (M = transition metal) systems was often interpreted as a HVMO oxygen exchange,36,38,43–45,88,89 a metal-free oxidation of cis-1,2-DMCH by m-CPBA in the presence of H218O and nitric acid promoter readily resulted in the 18O-labeled tertiary cis-alcohol (Fig. S10†), pointing out the existence of an 18O-incorporation mechanism alternative to a HVMO one (the possibility of a direct oxygen exchange between tertiary cis-alcohol and water under the typical conditions of the experiment was ruled out, see the ESI†). Direct labeling of m-CPBA (Scheme 6) could explain the appearance of 18O from H218O in all cases. Transfer of a peroxyl oxygen from m-CPBA and aroylperoxyl radical 7 to the C–H bond and PPh3 (Scheme 6) suggests that this oxygen atom is already 18O-labeled prior to C–H hydroxylation. Nearly equal incorporations of 18O into normal and deuterated alcohols (observation (e)) fit with this assumption. The reaction of PPh3 with labeled m-CPBA results in 18OPPh3, which accumulates in all tests involving H218O (Fig. S28†). The appearance of 18OPPh3 in the tests in the 18O2 atmosphere can be explained by reduction of 18O2 to H218O. This process was confirmed by observation of labeled ketones and CO2 (Fig. S20 and S21†). Considering the rather low incorporations of 18O into the alcohols (less than 5%), the magnitude of the oxygen exchange rate between m-CPBA and H218O can be evaluated to be ca. one order lower than that for C–H hydroxylation (under the studied conditions). This also explains why substrates with higher C–H bond energy, showing a lower oxidation rate, get higher 18O-enrichment.
Conclusions
In conclusion, we have studied the catalytic reaction mechanism of the stereospecific oxidation of saturated hydrocarbons using m-chloroperoxybenzoic acid (m-CPBA) as an oxidant, a Co/Fe heterometallic complex as a pre-catalyst and nitric acid as a promoter. The combination of a protic promoter with the cheap cobalt-based catalyst is essential for the pronounced catalytic effect and unusual stereoselectivity that concerns a main novelty. The proposed catalytic system hydroxylates tertiary sp3 C–H bonds with retention of stereoconfiguration up to 99%, achieving exceptionally high TONs (up to 1.4 × 104) and reaction rates (TOFs up to 2 s−1). The typical catalyst loading is 1000 ppm, but the system can efficiently work even at ppm loadings. A reaction temperature of 50 °C facilitates the solubility of the hydrocarbon substrates, at the same time favouring decarboxylation and further degradation of aroyloxyl and aryl radicals, which are responsible for the non-selective free radical side pathway (less than 1% of m-CPBA transforms into chlorobenzene under the conditions of the experiment). The latter free radical activity is also efficiently hampered by a protic acid promoter.DFT calculations and selectivity studies, as well as D- and 18O-labeled experiments, indicate that the main reaction mechanism involves a cobaltIII–peroxo or a cobaltIV–oxyl C–H attacking species (the latter being a rebound process), rather than free-radical pathways. Further, despite the large primary kinetic isotope effect of up to 6.6 in the competitive oxidations, the H atom abstraction appears to be a product determining, but not a rate determining step in the Co/m-CPBA oxidations. The fact that the Co(III) catalyst does not change its oxidation state during the most energetically favoured pathway inspires the exploration of the catalytic systems for m-CPBA activation comprising redox-inactive cations, such as Al(III), Bi(III) or Y(III), usually considered as unsuitable ones for this type of reaction. Some of us have recently observed a pronounced catalytic activity of the aqua complexes of these cations in the oxidation of alkanes and olefins with H2O2.90–92 One could expect a similar activity for the m-CPBA oxidant, which opens the possibility of using redox-inactive transition and main group metals as metal–ligand cooperation catalysts.
Finally, we have found signs for slow direct oxygen exchange between m-CPBA and water in the presence of a proton or a metal complex. As the incorporation of 18O from H218O into alcohols is commonly used as a mechanistic marker for the involvement of HVMO species (often capable of oxygen exchange with water), we conclude that the results of 18O-tests should be treated cautiously when m-CPBA is used as the oxidant.
Author contributions
O. V. N. and D. S. N. initiated and conceptualised the research; O. V. N. acquired and processed the catalytic data; M. L. K. performed the DFT calculations and wrote the respective part; G. B. S. conducted the preliminary studies that inspired the research and assisted in writing the manuscript; O. V. N. and A. J. L. P. discussed and edited the manuscript, A. J. L. P. and G. B. S. acquired funding for the work; D. S. N. designed the research methodology and wrote the main manuscript text. All the authors contributed to writing the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by Fundação para a Ciência e Tecnologia (FCT), Portugal (projects UIDB/00100/2020 of Centro de Química Estrutural, PTDC/QUI-QIN/29778/2017, contracts IST-ID/086/2018 (D. S. N.) and IST-ID/117/2018 (O. V. N.)) and by the RUDN University Strategic Academic Leadership Program. The high-field EPR spectra were recorded at the EMR facility of the National High Magnetic Field Laboratory (NHMFL), which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and the State of Florida. The authors are grateful to Dr. Andrew Ozarowski (NHMFL) for the EPR measurements and for his EPR fit and simulation software SPIN. The authors acknowledge the IST Node of the Portuguese Network of Mass-spectrometry for ESI-MS measurements. The authors thank for support RFBR according to Research Projects Grant No. 19-03-00142, the Ministry of Education and Science of the Russian Federation (project code RFMEFI61917X0007), and the Initiative Program in the frames of the State Tasks 0082-2014-0004 and 0082-2014-0007 “Fundamental regularities of heterogeneous and homogeneous catalysis”; the Program of Fundamental Research of the Russian Academy of Sciences for 2013–2020 on the research issue of IChP RAS No. 47.16.References
- P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- L. Ping, D. S. Chung, J. Bouffard and S.-G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed.
- K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- G. B. Shul'pin, Org. Biomol. Chem., 2010, 8, 4217–4228 RSC.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS.
- A. J. L. Pombeiro and M. F. C. Guedes da Silva, Alkane Functionalization, Wiley, Hoboken, NJ, USA, 2019 Search PubMed.
- M. M. Diaz-Requejo and P. J. Perez, Chem. Rev., 2008, 108, 3379–3394 CrossRef CAS PubMed.
- D. S. Nesterov, O. V. Nesterova and A. J. L. Pombeiro, Coord. Chem. Rev., 2018, 355, 199–222 CrossRef CAS.
- G. B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 189, 39–66 CrossRef.
- S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS.
- C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- S. Kal, S. N. Xu and L. R. Que, Angew. Chem., Int. Ed., 2020, 59, 7332–7349 CrossRef CAS.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS.
- R. Kumar, B. Pandey, A. Sen, M. Ansari, S. Sharma and G. Rajaraman, Coord. Chem. Rev., 2020, 419, 31 CrossRef.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- O. V. Nesterova, E. N. Chygorin, V. N. Kokozay, V. V. Bon, I. V. Omelchenko, O. V. Shishkin, J. Titis, R. Boca, A. J. L. Pombeiro and A. Ozarowski, Dalton Trans., 2013, 42, 16909–16919 RSC.
- D. S. Nesterov, E. N. Chygorin, V. N. Kokozay, V. V. Bon, R. Boca, Y. N. Kozlov, L. S. Shul'pina, J. Jezierska, A. Ozarowski, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2012, 51, 9110–9122 CrossRef CAS PubMed.
- D. S. Nesterov, V. N. Kokozay, V. V. Dyakonenko, O. V. Shishkin, J. Jezierska, A. Ozarowski, A. M. Kirillov, M. N. Kopylovich and A. J. L. Pombeiro, Chem. Commun., 2006, 4605–4607 RSC.
- H. Hussain, A. Al-Harrasi, I. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882–12917 RSC.
- A. Bravo, H. R. Bjorsvik, F. Fontana, F. Minisci and A. Serri, J. Org. Chem., 1996, 61, 9409–9416 CrossRef CAS.
- A. Bravo, F. Fontana, F. Minisci and A. Serri, Chem. Commun., 1996, 1843–1844 RSC.
- H. J. Schneider and W. Muller, J. Org. Chem., 1985, 50, 4609–4615 CrossRef CAS.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1593 CrossRef CAS PubMed.
- M. Oszajca, A. Franke, M. Brindell, G. Stochel and R. van Eldik, Coord. Chem. Rev., 2016, 306, 483–509 CrossRef CAS.
- A. R. McDonald and L. Que, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- K. Cho, P. Leeladee, A. J. McGown, S. DeBeer and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 7392–7399 CrossRef CAS PubMed.
- J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed.
- X. Y. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- X. Engelmann, I. Monte-Perez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed.
- M. M. Chen, P. S. Coelho and F. H. Arnold, Adv. Synth. Catal., 2012, 354, 964–968 CrossRef CAS.
- M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS PubMed.
- R. Singh, G. Ganguly, S. O. Malinkin, S. Demeshko, F. Meyer, E. Nordlander and T. K. Paine, Inorg. Chem., 2019, 58, 1862–1876 CrossRef CAS PubMed.
- M. Ghosh, S. Pattanayak, B. B. Dhar, K. K. Singh, C. Panda and S. Sen Gupta, Inorg. Chem., 2017, 56, 10852–10860 CrossRef CAS.
- K. P. Shing, B. Cao, Y. G. Liu, H. K. Lee, M. D. Li, D. L. Phillips, X. Y. Chang and C. M. Che, J. Am. Chem. Soc., 2018, 140, 7032–7042 CrossRef CAS.
- T. Kojima, K. I. Hayashi, S. Y. Iizuka, F. Tani, Y. Naruta, M. Kawano, Y. Ohashi, Y. Hirai, K. Ohkubold, Y. Matsuda and S. Fukuzumi, Chem. – Eur. J., 2007, 13, 8212–8222 CrossRef CAS PubMed.
- I. Terao, S. Horii, J. Nakazawa, M. Okamura and S. Hikichi, Dalton Trans., 2020, 49, 6108–6118 RSC.
- F. F. Pfaff, F. Heims, S. Kundu, S. Mebs and K. Ray, Chem. Commun., 2012, 48, 3730–3732 RSC.
- T. Corona, F. F. Pfaff, F. Acuna-Pares, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, J. Lloret-Fillol, W. R. Browne, K. Ray and A. Company, Chem. – Eur. J., 2015, 21, 15029–15038 CrossRef CAS.
- Y. H. Qiu and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 19239–19248 CrossRef CAS PubMed.
- W. Nam, I. Kim, Y. Kim and C. Kim, Chem. Commun., 2001, 1262–1263 RSC.
- W. Nam, J. Y. Ryu, I. Kim and C. Kim, Tetrahedron Lett., 2002, 43, 5487–5490 CrossRef CAS.
- O. V. Nesterova, M. N. Kopylovich and D. S. Nesterov, RSC Adv., 2016, 6, 93756–93767 RSC.
- T. Nishiura, A. Takabatake, M. Okutsu, J. Nakazawa and S. Hikichi, Dalton Trans., 2019, 48, 2564–2568 RSC.
- A. N. Bilyachenko, A. I. Yalymov, M. M. Levitsky, A. A. Korlyukov, M. A. Es'kova, J. Long, J. Larionova, Y. Guari, L. S. Shul'pina, N. S. Ikonnikov, A. L. Trigub, Y. V. Zubavichus, I. E. Golub, E. S. Shubina and G. B. Shul'pin, Dalton Trans., 2016, 45, 13663–13666 RSC.
- A. Bell-Taylor, J. D. Gorden, E. E. Hardy and C. R. Goldsmith, Inorg. Chim. Acta, 2018, 482, 206–212 CrossRef CAS.
- J. Nakazawa, A. Yata, T. Hori, T. D. P. Stack, Y. Naruta and S. Hikichi, Chem. Lett., 2013, 42, 1197–1199 CrossRef CAS.
- D. S. Nesterov, O. V. Nesterova, M. N. Kopylovich and A. J. L. Pombeiro, Mol. Catal., 2018, 459, 8–15 CrossRef CAS.
- O. V. Nesterova, K. V. Kasyanova, V. G. Makhankova, V. N. Kokozay, O. Y. Vassilyeva, B. W. Skelton, D. S. Nesterov and A. J. L. Pombeiro, Appl. Catal., A, 2018, 560, 171–184 CrossRef CAS.
- O. V. Nesterova, K. V. Kasyanova, E. A. Buvaylo, O. Y. Vassilyeva, B. W. Skelton, D. S. Nesterov and A. J. L. Pombeiro, Catalysts, 2019, 9, 15 CrossRef.
- D. H. Wei, X. J. Zhu, J. L. Niu and M. P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef CAS.
- B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2017, 8, 14839 CrossRef CAS PubMed.
- Y. M. Kwon, Y. Lee, G. E. Evenson, T. A. Jackson and D. Wang, J. Am. Chem. Soc., 2020, 142, 13435–13441 CrossRef CAS PubMed.
- G. Olivo, O. Lanzalunga and S. Di Stefano, Adv. Synth. Catal., 2016, 358, 843–863 CrossRef CAS.
- J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. Garcia-Espana, M. G. Basallote, E. Munck, L. Que, A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15842 CrossRef CAS.
- D. Font, M. Canta, M. Milan, O. Cusso, X. Ribas, R. J. M. K. Gebbink and M. Costas, Angew. Chem., Int. Ed., 2016, 55, 5776–5779 CrossRef CAS.
- B. Wang, Y. M. Lee, M. Clemancey, M. S. Seo, R. Sarangi, J. M. Latour and W. Nam, J. Am. Chem. Soc., 2016, 138, 2426–2436 CrossRef CAS PubMed.
- G. D. Roiban, R. Agudo and M. T. Reetz, Angew. Chem., Int. Ed., 2014, 53, 8659–8663 CrossRef CAS PubMed.
- R. Karande, L. Debor, D. Salamanca, F. Bogdahn, K. H. Engesser, K. Buehler and A. Schmid, Biotechnol. Bioeng., 2016, 113, 52–61 CrossRef CAS.
- A. Ilie, R. Agudo, G. D. Roiban and M. T. Reetz, Tetrahedron, 2015, 71, 470–475 CrossRef CAS.
- G. B. Shul'pin, M. V. Kirillova, L. S. Shul'pina, A. J. L. Pombeiro, E. E. Karslyan and Y. N. Kozlov, Catal. Commun., 2013, 31, 32–36 CrossRef.
- O. V. Nesterova and D. S. Nesterov, Catalysts, 2018, 8, 602 CrossRef.
- M. S. Seo, N. H. Kim, K. B. Cho, J. E. So, S. K. Park, M. Clemancey, R. Garcia-Serres, J. M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC.
- S. N. Dhuri, K. B. Cho, Y. M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef CAS PubMed.
- J. R. Bryant, T. Matsuo and J. M. Mayer, Inorg. Chem., 2004, 43, 1587–1592 CrossRef CAS PubMed.
- G. B. Shul'pin, D. S. Nesterov, L. S. Shul'pina and A. J. L. Pombeiro, Inorg. Chim. Acta, 2017, 455, 666–676 CrossRef.
- O. V. Nesterova, D. S. Nesterov, A. Krogul-Sobczak, M. F. C. Guedes da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2017, 426, 506–515 CrossRef CAS.
- G. S. Astakhov, A. N. Bilyachenko, A. A. Korlyukov, M. M. Levitsky, L. S. Shul'pina, X. Bantreil, F. Lamaty, A. V. Vologzhanina, E. S. Shubina, P. V. Dorovatovskii, D. S. Nesterov, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2018, 57, 11524–11529 CrossRef CAS PubMed.
- O. V. Nesterova, D. S. Nesterov, B. Vranovicova, R. Boca and A. J. L. Pombeiro, Dalton Trans., 2018, 47, 10941–10952 RSC.
- I. Gryca, K. Czerwinska, B. Machura, A. Chrobok, L. S. Shul'pina, M. L. Kuznetsov, D. S. Nesterov, Y. N. Kozlov, A. J. L. Pombeiro, I. A. Varyan and G. B. Shul'pin, Inorg. Chem., 2018, 57, 1824–1839 CrossRef CAS PubMed.
- K. L. Vikse, Z. Ahmadi and J. S. McIndoe, Coord. Chem. Rev., 2014, 279, 96–114 CrossRef CAS.
- M. Y. Hyun, S. H. Kim, Y. J. Song, H. G. Lee, Y. D. Jo, J. H. Kim, I. H. Hwang, J. Y. Noh, J. Kang and C. Kim, J. Org. Chem., 2012, 77, 7307–7312 CrossRef CAS PubMed.
- R. H. Crabtree, J. Organomet. Chem., 2014, 751, 174–180 CrossRef CAS.
- D. S. Nesterov, O. V. Nesterova, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Catal. Sci. Technol., 2015, 5, 1801–1812 RSC.
- G. B. Shul'pin, D. A. Loginov, L. S. Shul'pina, N. S. Ikonnikov, V. O. Idrisov, M. M. Vinogradov, S. N. Osipov, Y. V. Nelyubina and P. M. Tyubaeva, Molecules, 2016, 21, 1593 CrossRef PubMed.
- W. Nam, M. H. Lim, S. K. Moon and C. Kim, J. Am. Chem. Soc., 2000, 122, 10805–10809 CrossRef CAS.
- M. Martinho, F. Banse, J. F. Bartoli, T. A. Mattioli, P. Battioni, O. Horner, S. Bourcier and J. J. Girerd, Inorg. Chem., 2005, 44, 9592–9596 CrossRef CAS.
- S. Shaik, S. Cohen, S. P. de Visser, P. K. Sharma, D. Kumar, S. Kozuch, F. Ogliaro and D. Danovich, Eur. J. Inorg. Chem., 2004, 207–226 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- G. B. Shul'pin, Y. N. Kozlov, G. V. Nizova, G. Suss-Frank, S. Stanislas, A. Kitaygorodskiy and V. S. Kulikova, J. Chem. Soc., Perkin Trans. 2, 2001, 1351–1371 RSC.
- F. Gozzo, J. Mol. Catal. A: Chem., 2001, 171, 1–22 CrossRef CAS.
- W. N. Oloo and L. Que, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed.
- K. Chen and L. Que, J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- J. Bernadou and B. Meunier, Chem. Commun., 1998, 2167–2173 RSC.
- K. A. Lee and W. Nam, J. Am. Chem. Soc., 1997, 119, 1916–1922 CrossRef CAS.
- A. R. Jeong, J. W. Shin, J. H. Jeong, K. H. Bok, C. Kim, D. Jeong, J. Cho, S. Hayami and K. S. Min, Chem. – Eur. J., 2017, 23, 3023–3033 CrossRef CAS.
- C. Kim, H. Ahn, J. Bae, M. Kim, K. Bok, H. Jeong and S. Lee, Chem. – Eur. J., 2017, 23, 11969–11976 CrossRef PubMed.
- A. S. Novikov, M. L. Kuznetsov, B. G. M. Rocha, A. J. L. Pombeiro and G. B. Shul'pin, Catal. Sci. Technol., 2016, 6, 1343–1356 RSC.
- M. L. Kuznetsov, B. G. M. Rocha, A. J. L. Pombeiro and G. B. Shul'pin, ACS Catal., 2015, 5, 3823–3835 CrossRef CAS.
- B. G. M. Rocha, M. L. Kuznetsov, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, Catal. Sci. Technol., 2015, 5, 2174–2187 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, mass-spectra, chromatograms of reaction products, kinetic curves, and calculation details. CCDC 2017735 and 2113995. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cy01991k |
| This journal is © The Royal Society of Chemistry 2022 |