
Polyamine-functionalized imidazolyl poly(ionic liquid)s for the efficient conversion of CO2 into cyclic carbonates†
Yizhen
Zou
 ,
Yuansheng
Ge
,
Qiang
Zhang
,
Wei
Liu
,
Xiaoguang
Li
,
Guoe
Cheng
and
Hanzhong
Ke
*
,
Yuansheng
Ge
,
Qiang
Zhang
,
Wei
Liu
,
Xiaoguang
Li
,
Guoe
Cheng
and
Hanzhong
Ke
*
Faculty of Materials Science and Chemistry, China University of Geosciences (Wuhan), 388 Lumo Road, Wuhan 430074, China. E-mail: kehanz@163.com
First published on 9th November 2021
Abstract
Global warming is becoming a challenging issue due to the emission of a large number of greenhouse gases, mainly CO2. The transformation of CO2 into chemicals with high additional value is considered a promising and sustainable way to solve the “greenhouse effect”. Herein, a series of polyamine-functionalized imidazolyl poly(ionic liquid)s (PILs) modified by triethylenetetramine (TETA) were synthesized as heterogeneous catalysts to convert CO2 into cyclic carbonates. The synergistic effect of nucleophile (bromide anions) and polyamine groups in promoting CO2 conversion was explored by density functional theory (DFT) calculations, which is critical to improve the catalytic performance. When the anions of ionic liquids acted as nucleophiles to attack the epoxide from the C–O bond with less steric hindrance, the substrate epoxide and CO2 can be activated by hydrogen bonding with amine group protons. Therefore, PILs modified by TETA (N4-PIL-x) have been verified to possess high efficiency and stable reusability for the cycloaddition of epoxides with CO2 without solvent, metal, and co-catalyst, of which N4-PIL-2 can achieve 98.0% conversion of epichlorohydrin (ECH) with a turnover frequency (TOF) value as high as 42.4 h−1 under ambient pressure; moreover, the complete conversion of epichlorohydrin is obtained in only 4 h at 1.0 MPa CO2 pressure.
Introduction
The extensive consumption of fossil fuels and various activities of human beings are the roots of excessive emission of CO2, which has remarkably increased the aggravation of environmental issues as a primary greenhouse gas in the atmosphere.1,2 Nevertheless, in industrial processes, CO2 has also been recommended as a nontoxic, omnipresent, and sustainable C1 resource. Widespread attention has been paid for the efficient conversion of excessive CO2 emissions into fine chemicals with high additional value.3–7 The transformation of CO2 into cyclic carbonates is one of the most effective, economical and environmentally friendly applications of fixed CO2.8–10 It is worth noting that cyclic carbonates are also recognized as excellent chemicals and are widely used in the industrial field.However, due to the intrinsic thermodynamic stability and kinetic inertness of CO2, high temperature or high pressure are often required to overcome the energy barriers in the reaction pathways of converting CO2 into cyclic carbonates.2,11 Therefore, it still a challenge to find an excellent catalyst for the cycloaddition of epoxides with CO2 under milder conditions. A variety of catalysts have been extensively studied. As a typical homogeneous catalyst, ionic liquids (ILs) exhibit excellent catalytic performance,12,13 while it is usually necessary to add alkali metals as co-catalysts.14,15 This is similar to other metal-bearing catalysts (metal–organic frameworks (MOFs), metal complexes), which may cause environmental pollution by toxic metals.16–19 In addition, it is still a great challenge to separate the product from the catalyst in a homogeneous catalytic system.20–22 As one of the representative achievements of heterogeneous catalysts, porous organic polymers (POPs) can be separated from the catalytic products more easily to some extent; however, they also have disadvantages, such as complicated preparation, limited effective active sites, and harsh catalytic conditions.23–27 Therefore, it is an active area of research to narrow the divide between homogeneous and heterogeneous catalysis by the feasible construction of heterogeneous catalysts with simple preparation, high active site density and mild catalytic conditions.28
In this work, by adjusting the ratio of imidazolyl ionic liquid to divinylbenzene, PILs with different densities of catalytic active sites were obtained. After post-synthetic modification, TETA was introduced into the PILs by nucleophilic substitution reaction. Imidazole-based PILs have been considered as excellent catalysts for conversion of CO2, in which imidazolium cations can activate CO2 and the customized counterions can be nucleophilic to promote the ring opening of epoxides.29–32 Furthermore, there is a unique cross-linking structure present in PILs; the imidazole structure cell can be incorporated into the backbone chain, which provides the possibility of obtaining a catalyst with a high density of active sites by regulating and controlling the catalytic active centers. As deduced from previous research, the rate-limiting step of the cycloaddition reaction is usually regarded as ring-opening by a nucleophilic attack.33,34 It is an advanced strategy to introduce task-specific active sites to activate epoxides to make them more vulnerable to nucleophilic attack.35–39
Various task-specific groups (–OH, –NH2, –COOH)39–47 have been introduced into imidazole-based PILs as hydrogen-bond donors to activate CO2/epoxides. The hydrogen-bond donors on cations can promote the CO2/epoxide coupling when the charge-balanced bromide anions act as nucleophiles to attack the epoxides.40,48–50 In this paper, a series of N4-PIL-x materials were synthesized as heterogeneous catalysts for the cycloaddition reaction. The more robust hydrogen bonds can be cooperatively produced in N4-PIL-x with multiple neighbouring hydrogen bond donors compared to isolated hydrogen-bond donors.51 TETA, as a typical small molecular organic amine, is widely used in the field of carbon dioxide adsorption because of its small molecular size, high boiling point and low viscosity.52 Moreover, the primary and secondary amine groups contained in TETA are considered to be excellent multiple adjacent hydrogen bond donors. The polyamine groups produce suitable basicity with a superior affinity for CO2 to create a high-concentration CO2 environment at the catalytic active site, which is conducive to accelerating the catalytic reaction process. Systematic tests for the cycloaddition reaction were investigated at atmospheric pressure without solvent, metal, and co-catalyst. Based on DFT calculations, the synergistic effect of the amine groups and bromide anions in N4-PIL-x in promoting CO2 conversion was explored.
Experimental
General
 | ||
| Scheme 1 The synthesis of N4-PIL-x (x = 1/2, 1, 2). | ||
Results and discussion
Synthesis and characterization
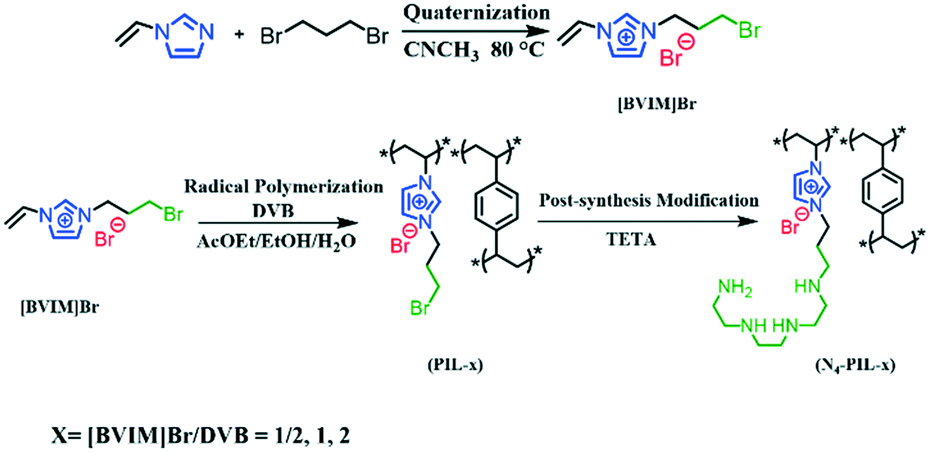
Scheme 1 shows the strategy for the synthesis of N4-PIL-x (x = 1/2, 1, 2). PIL-x (x = 0, 1/2, 1, 2, x represents the molar ratio of [BVIM]Br to DVB) were synthesized by radical polymerization of [3-bromopropyl-1-vinylimidazolium]Br ([BVIM]Br) and divinylbenzene (DVB). N4-PIL-x (x = 1/2, 1, 2) was synthesized by post-synthesis modification of PIL-x (x = 1/2, 1, 2) with TETA through a nucleophilic substitution reaction.The as-synthesised PIL-2 and N4-PIL-x (x = 1/2, 1, 2) were characterized by FT-IR spectroscopy (Fig. 1A). All the samples exhibit the characteristic peaks at 3134 and 2954 cm−1, which can correspond to the C–H stretching vibrations.31 The imidazolium rings are determined by the peaks at 1587 and 1441 cm−1, which are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC, respectively.23,55 The peak at 1171 cm−1 can be attributed to the stretching vibration of C–N in the amino-alkyl chain, indicating that the PILs contain the imidazolium-IL moiety.56,57 The band at 640 cm−1 is for the C–Br stretching vibration, which vanishes in the spectra of polyamine-functionalized PILs with TETA (N4-PIL-1/2, N4-PIL-1, N4-PIL-2). Meanwhile, a new peak appears at 825 cm−1, which is attributed to the primary amine N–H bending vibration. Thus, the successful synthesis of these polyamine-functionalized PILs was confirmed. The N4-PIL-x backbone is further indicated by the solid-state 13C-nuclear magnetic resonance (13C-NMR) spectrum in Fig. 1B. Obvious overlapping bands around 125 ppm are displayed in the 13C-NMR spectrum, which can be correlated to unsubmitted benzene carbons and the C3 and C4 atoms of the imidazolium ring. Moreover, the C2 carbon of the imidazolium ring is reflected by the resonance at 143 ppm. Meanwhile, the adjacent peak with a chemical shift of ca. 136 ppm is attributed to the methylene linker carbons produced by DVB polymerization.36 Furthermore, the signals of carbons linked to the N atom of the imidazole ring and amino group are found at about 58 and 47 ppm, respectively, while the peak at 40 ppm is related to the methylene tethered on the phenyl ring. The peaks at around 28–31 ppm correspond to the alkyl methylene.
N and CC, respectively.23,55 The peak at 1171 cm−1 can be attributed to the stretching vibration of C–N in the amino-alkyl chain, indicating that the PILs contain the imidazolium-IL moiety.56,57 The band at 640 cm−1 is for the C–Br stretching vibration, which vanishes in the spectra of polyamine-functionalized PILs with TETA (N4-PIL-1/2, N4-PIL-1, N4-PIL-2). Meanwhile, a new peak appears at 825 cm−1, which is attributed to the primary amine N–H bending vibration. Thus, the successful synthesis of these polyamine-functionalized PILs was confirmed. The N4-PIL-x backbone is further indicated by the solid-state 13C-nuclear magnetic resonance (13C-NMR) spectrum in Fig. 1B. Obvious overlapping bands around 125 ppm are displayed in the 13C-NMR spectrum, which can be correlated to unsubmitted benzene carbons and the C3 and C4 atoms of the imidazolium ring. Moreover, the C2 carbon of the imidazolium ring is reflected by the resonance at 143 ppm. Meanwhile, the adjacent peak with a chemical shift of ca. 136 ppm is attributed to the methylene linker carbons produced by DVB polymerization.36 Furthermore, the signals of carbons linked to the N atom of the imidazole ring and amino group are found at about 58 and 47 ppm, respectively, while the peak at 40 ppm is related to the methylene tethered on the phenyl ring. The peaks at around 28–31 ppm correspond to the alkyl methylene.
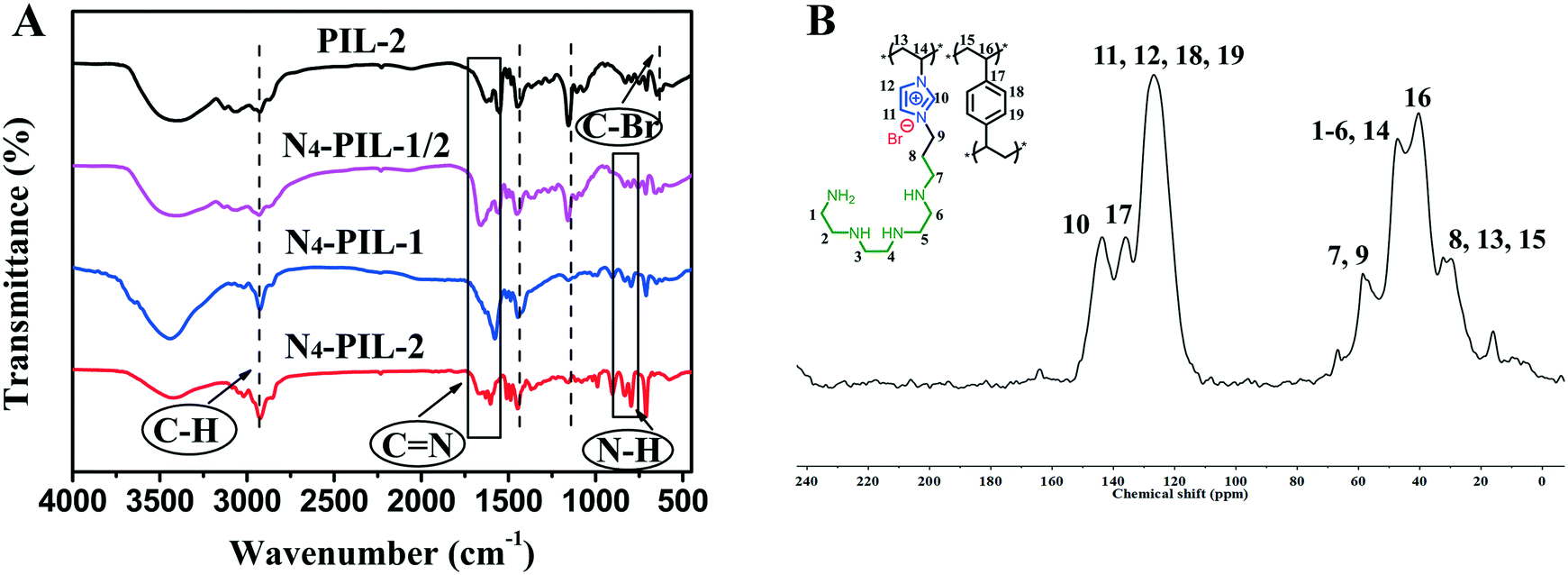 | ||
| Fig. 1 (A) FT-IR spectra of the samples; (B) 13C MAS NMR spectrum of N4-PIL-2. | ||
The porous characteristics of the samples were studied by N2 adsorption/desorption (Fig. 2A). The sorption isotherms of all the samples exhibit characteristic type IV isotherms with distinct hysteresis, suggesting that the as-synthesised materials have meso-porous structures.58 In Fig. 2B, the pore size distribution curves also confirm that all samples exhibit meso-porous structures, with the most probable apertures between 3.34 and 3.83 nm. The calculated structural parameters, including the surface area, total pore volume, and average pore size, from the nitrogen adsorption–desorption isotherms are shown in Table 1. It is apparent that the modification of TETA results in a decrease of the specific surface area and total pore volume. Compared with PIL-2, the specific surface area and total pore volume of N4-PIL-2 decreased significantly, from 484.340 to 22.939 m2 g−1 and 0.315 to 0.091 cm3 g−1, respectively. The results showed that catalysts with different porosities were formed by copolymerization with DVB in different proportions, which also accelerated the diffusion of the substrate molecules and was beneficial to the reaction kinetics.59,60
 | ||
| Fig. 2 N2 sorption isotherms (A) and the corresponding pore size distribution curves (B) of N4-PIL-x polymerized at 80 °C for 24 h, using AcOEt/EtOH/H2O (10/30/5) as the solvent. | ||
| Entry | Catalyst | IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DVBa :DVBa |
S BET [m2 g−1] | V P [cm3 g−1] | D pr [nm] |
|---|---|---|---|---|---|
| a Molar ratio of IL to DVB in the gel. b BET surface area. c Total pore volume. d Most probable aperture. | |||||
| 1 | PIL-2 | 2:1 |
484.340 | 0.315 | 3.72 |
| 2 | N4-PIL-1/2 | 1:2 |
122.481 | 0.156 | 3.71 |
| 3 | N4-PIL-1 | 1:1 |
102.136 | 0.192 | 3.83 |
| 4 | N4-PIL-2 | 2:1 |
22.939 | 0.091 | 3.34 |
Fig. S1A† shows the X-ray diffraction (XRD) pattern of N4-PIL-2, indicating that N4-PIL-2 material has the amorphous structure of a polymer network. Thermogravimetric analysis (TGA) was performed to investigate the thermal stability of N4-PIL-2 to ensure the safe temperature range for the subsequent catalysis process of CO2 and epoxides. As shown in Fig. S1B,† the degradation process of N4-PIL-2 can be split into three steps. The initial step can be attributed to the evaporation of captured moisture from air at temperature below 310 °C.55 The second step is the decomposition of ionic liquid segments and the loading of grafted TETA in the temperature range from 310 °C to 400 °C, which will prove that the synthesized N4-PIL-2 contains abundant catalytic activity. During the temperature from 400 °C to 500 °C, the final step is related to the decomposition of the polymer matrix.61,62 Thus, the synthesized N4-PIL-2 is thermally stable below 310 °C, under which the catalytic reaction of CO2 with epichlorohydrin can be performed. The elemental analysis for the N4-PIL-x can be seen in Table 2.
| Entry | Catalyst | IL content (mmol g−1) | Found | ||
|---|---|---|---|---|---|
| C (%) | N (%) | H (%) | |||
| 1 | N4-PIL-1/2 | 0.422 | 80.095 | 3.550 | 7.183 |
| 2 | N4-PIL-1 | 0.801 | 66.525 | 6.730 | 7.137 |
| 3 | PIL-2 | 1.228 | 58.650 | 3.440 | 6.525 |
| 4 | N4-PIL-2 | 1.222 | 48.880 | 10.270 | 6.615 |
The morphologies of PIL-2 and N4-PIL-2 were investigated by scanning electron microscopy (SEM) (Fig. 3). Obviously, both polymers present fluffy cotton-like clusters at the micron scale in Fig. 3A and C, in which PIL-2 is composed of many nano-scale primary spherical particles that are cross-linked with each other and has a loose and porous structure (Fig. 3B). In contrast, due to the graft introduction of TETA in N4-PIL-2, the radius of the primary spherical particles forming the polymer cluster gradually increases, and finally the structure formed by the agglomeration of many irregular nano-blocks is obtained (Fig. 3D). Meanwhile, the graft introduction of TETA also leads to the blockage of a large of the pores in the polymer, thereby resulting in a significant reduction in the specific surface area, which corresponds to the results shown in the N2 sorption isotherm curves in Fig. 2. Elemental distribution of PIL-2 and N4-PIL-2 was observed by the energy-dispersive X-ray spectroscopy (EDX) mapping. Elemental mapping in Fig. 3E and F has shown that N and Br elements are uniformly distributed in the polymer skeleton, and it further confirms the existence of homogeneously distributed IL units, that is, the active functional sites.
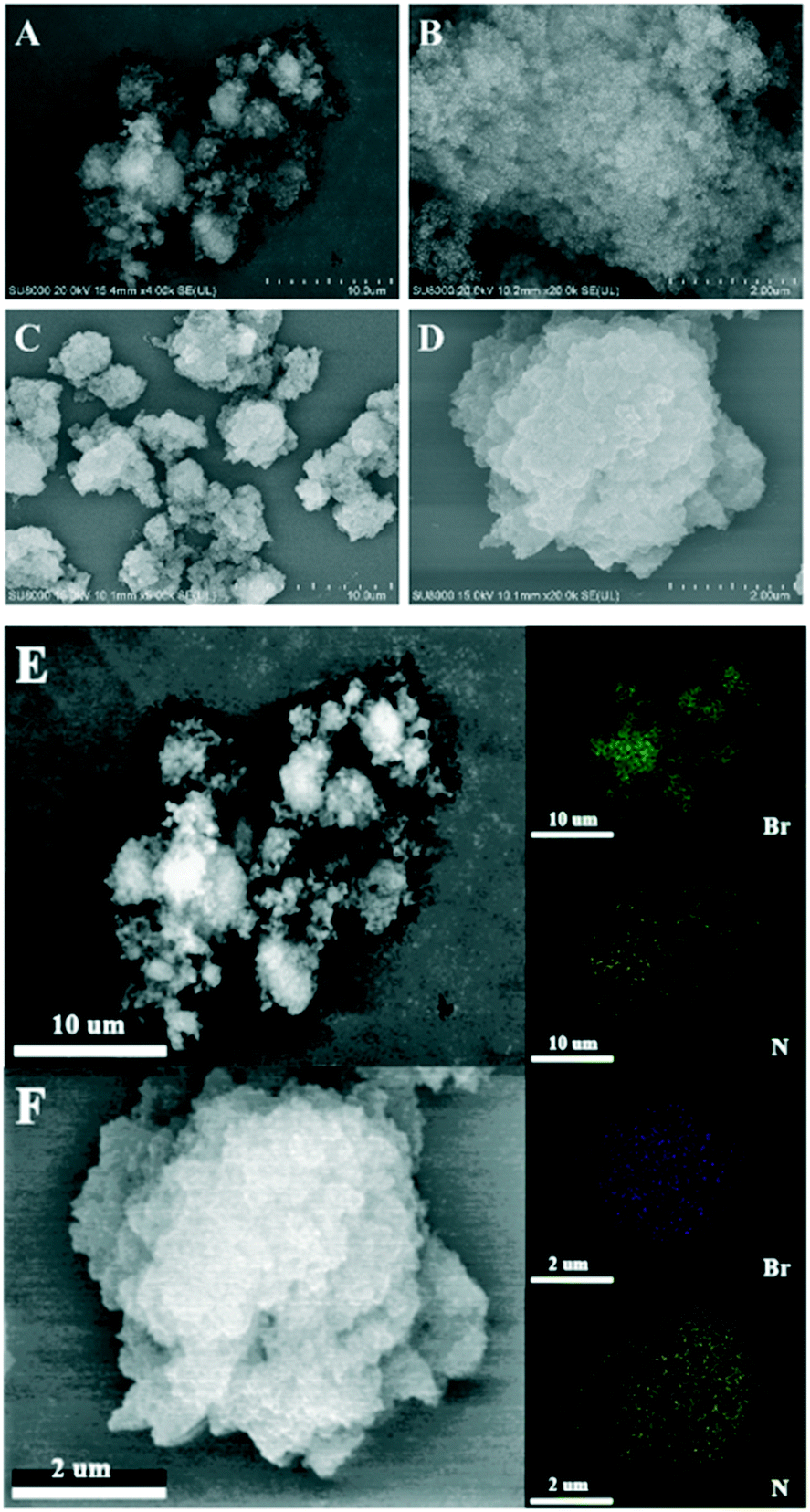 | ||
| Fig. 3 SEM images of samples PIL-2 (A and B) and N4-PIL-2 (C and D) and N, Br elemental mapping images of PIL-2 (E), N4-PIL-2 (F). | ||
Catalyst evaluation
The cyclization of epoxides with CO2 is one of the most economical, effective and environmentally friendly applications of CO2.4,6 In order to evaluate the catalytic activity of the as-synthesised catalysts, the cyclization of epichlorohydrin with CO2 was conducted without solvents and co-catalysts. The results are listed in Table 3. The conversion of PIL-0 obtained by self-polymerization of DVB was only 4.5% (Table 3, entry 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | P (MPa) | t (h) | Con.b (%) | Sel.b (%) | TOFc (h−1) |
| a Reaction conditions: epichlorohydrin (12.7 mmol); catalyst (10 mg); 100 °C. b Conversion (con.) and selectivity (sel.) were determined by 1H NMR. c Turnover frequency (TOF) = [mmol (product)]/[mmol (ionic sites) × (reaction time)]. | ||||||
| 1 | PIL-0 | 1.0 | 4 | 4.5 | >99 | — |
| 2 | PIL-2 | 1.0 | 4 | 81.2 | >99 | 209.9 |
| 3 | N4-PIL-2 | 1.0 | 4 | 99.6 | >99 | 258.8 |
| 4 | N4-PIL-1 | 1.0 | 4 | 72.3 | >99 | 286.6 |
| 5 | N4-PIL-1/2 | 1.0 | 4 | 42.5 | >99 | 319.8 |
| 6 | N4-PIL-2 | 1.0 | 1 | 81.3 | >99 | 844.9 |
| 7 | N4-PIL-2 | 0.1 | 24 | 98.0 | >99 | 42.4 |
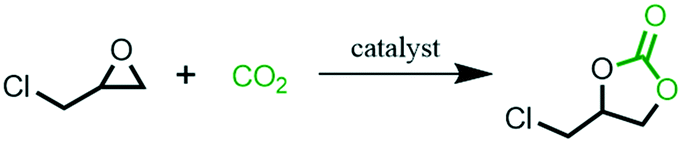
When PIL-2 containing imidazole-based ionic liquid units was chosen as a catalyst, the conversion could reach 81.2% (Table 3, entry 2), which was attributed to the good catalytic activity of the imidazole-based ionic liquid units in PIL-2 for the cyclization of CO2 and epichlorohydrin.32 After modification with TETA, the specific surface area and total pore volume of the obtained N4-PIL-2 decreased significantly. However, the introduction of TETA significantly improved the density of the functional groups in N4-PIL-2, and the ring-opening reaction of epoxide substrates (rate-limiting step) is promoted by the formation of hydrogen bonds between the multiple hydrogen bond donors and epoxides. Therefore, the obtained N4-PIL-2 exhibited the highest conversion of 99.6% (selectivity >99%) (Table 3, entry 3). Its catalytic activity was greatly improved compared with that of PIL-2 without TETA modification. Even when the reaction time was curtailed to 1.0 h, the N4-PIL-2 catalyst could still achieve 81.3% conversion, with an initial TOF value of 844.9 h−1 (Table 3, entry 6). Conversion of 98.0% was identified under ambient pressure when the reaction time was only 24 h, and the TOF value was calculated to be as high as 42.4 h−1 (Table 3, entry 7). The activity of N4-PIL-2 was superior to that of reported ionic liquid-supported catalysts (Table S1†) because of the activation of CO2/epoxides by the polyamine groups in N4-PIL-2 through the formation of hydrogen bonds. With the decrease of the proportion of ionic liquid units in N4-PIL-x, the catalytic activity of N4-PIL-x gradually decreased. The conversions were 99.6%, 72.3% and 42.5% (Table 3 entries 3–5) when N4-PIL-2, N4-PIL-1 and N4-PIL-1/2 were used as catalysts, respectively.
To select the optimal reaction conditions, N4-PIL-2 was chosen to catalyze the cycloaddition of epichlorohydrin with CO2 in the range from 40 °C to 120 °C. The effects of temperature on the conversion are shown in Fig. 4A. At low temperatures below 60 °C, the N4-PIL-2 was less reactive. However, when the temperature reached 100 °C, conversion of up to 99.6% was obtained. This can be interpreted by the enhancement of the reaction kinetics at higher reaction temperature, which resulted in an increased reaction conversion over the same period of time.63 The effects of the catalyst amount were studied at 100 °C and 1.0 MPa (Fig. 4B). The conversion increased with the increase of the N4-PIL-2 dosage. When the catalyst amount was 2, 5 and 8 mg, the conversion was 35.2%, 72.5% and 90.3%, respectively. As the amount was further increased to 10 mg, a quantitative conversion (99.6%) was obtained.
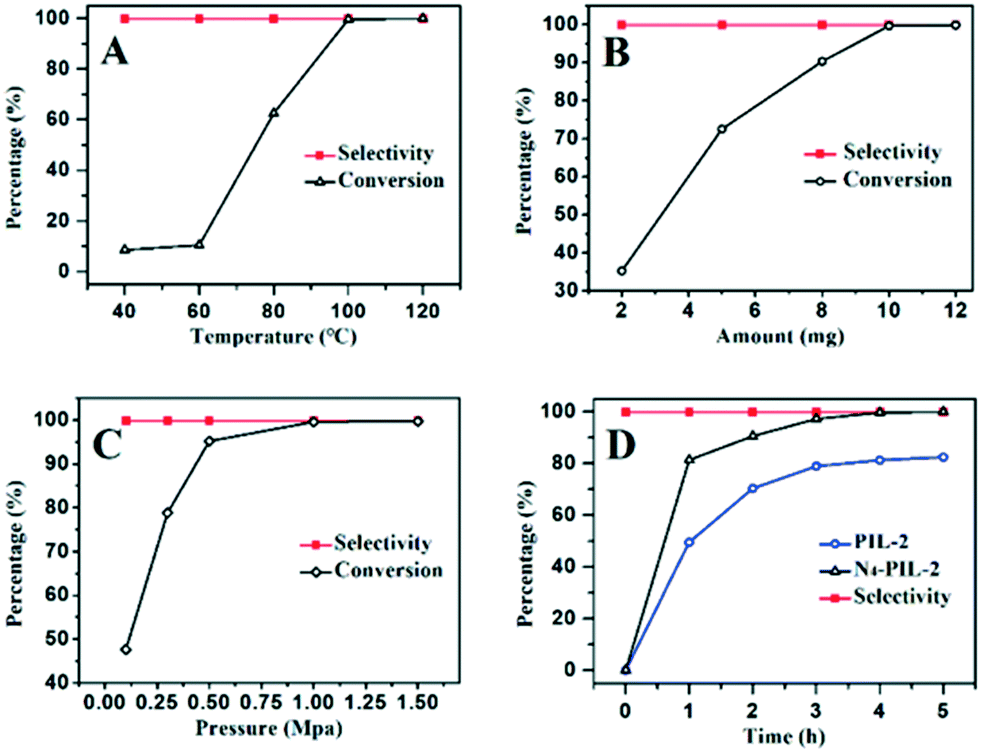 | ||
| Fig. 4 The cycloaddition reaction of CO2 and ECH. (A) Effect of the reaction temperature. (B) Effect of catalyst loading. (C) Effect of CO2 pressure. (D) The kinetic curves of N4-PIL-2 and PIL-2. Unless otherwise stated, the reaction was performed under these conditions: ECH (12.7 mmol); catalyst (10 mg); CO2 (1.0 MPa); 100 °C for 4 h. | ||
The plots of conversion and selectivity versus initial CO2 pressure are shown in Fig. 4C. With the increase of the initial CO2 pressure from 0.1 MPa to 0.5 MPa, the reaction conversion increased significantly from 47.6% to 95.2%. When the CO2 pressure was increased to 1.0 MPa, the reaction conversion reached 99.6%, suggesting that the initial CO2 pressure of 1.0 MPa could provide the most appropriate concentration of CO2 for the catalytic reaction.
Fig. 4D shows the function between conversion and reaction time in the CO2 cycloaddition with epichlorohydrin. When the reaction catalyst was N4-PIL-2, the conversion of epichlorohydrin was 81.3% in 1.0 h, and quantitative conversion was obtained in 4 h. The initial TOF value was as high as 844.9 h−1 (Table 3, entry 6). Due to the decrease of the cyclopropane concentration, even if the reaction time was increased to more than 4 h, the corresponding increase of conversion was less significant.55 In sharp contrast, when PIL-2 was used as the catalyst to perform the reaction under the same conditions, its activity was lower than that of N4-PIL-2; 49.5% conversion was identified in the first 1.0 h, and the initial TOF was 511.9 h−1, which was only 3/5 that of N4-PIL-2. The superior catalytic activity of N4-PIL-2 revealed that the polyamine groups in the polymeric networks can accelerate the cyclization of CO2. Based on the above experimental results, the optimized reaction conditions could be set to 1.0 MPa of CO2 pressure at 100 °C for 4.0 h. It is worth mentioning that the selectivity was maintained at more than 99%, which is unrelated to the reaction temperature, catalyst dosage, CO2 pressure and reaction time.
In addition, the recyclability of the catalyst is vitally important when developing a new heterogeneous catalyst. After washing with ethanol and chloroform and drying in a vacuum at 80 °C for 24 h, the recovered N4-PIL-2 catalyst was reused in successive reactions. After six-run recycling tests, the conversion could still reach more than 95% and the selectivity remained above 99% (Fig. 5A). The recovered catalyst after six runs was characterized by FT-IR analysis (Fig. 5B). The FT-IR spectrum was similar to that of the original catalyst. Therefore, the N4-PIL-2 catalyst showed excellent stability and good recyclability.
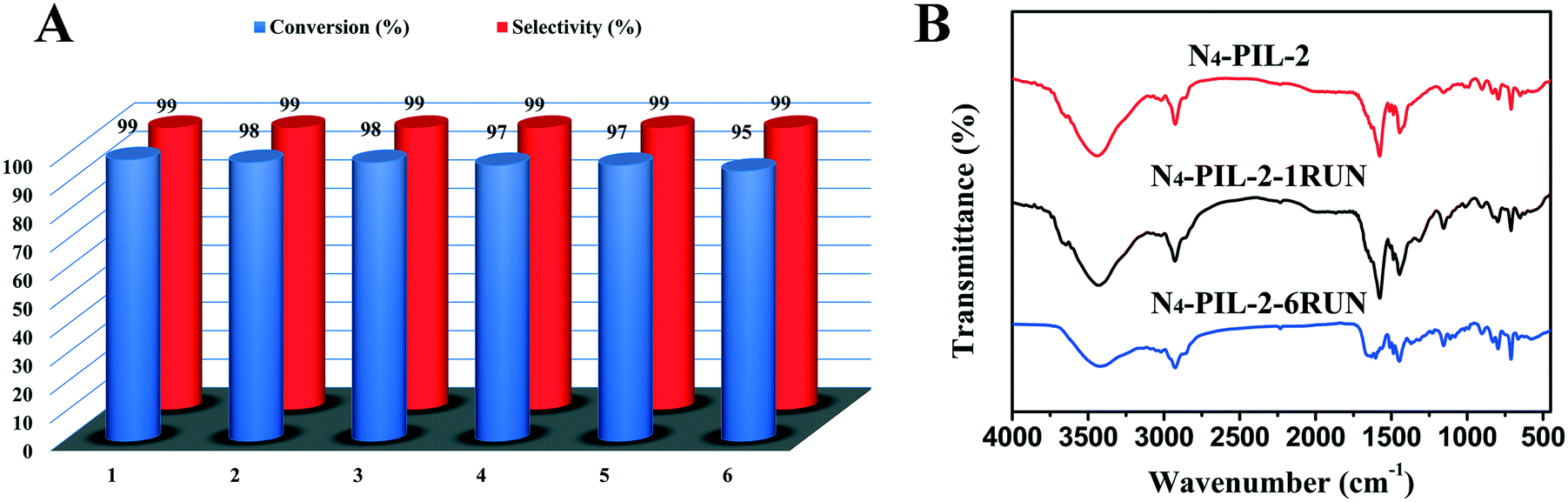 | ||
| Fig. 5 Catalytic reusability of N4-PIL-2 for cycloaddition of CO2 with ECH. (A) Results of the six-run recycling test. (B) Comparison of the FT-IR spectra of the fresh and recovered N4-PIL-2 catalyst. Reaction conditions: ECH (12.7 mmol); catalyst (10 mg); CO2 (1.0 MPa); 100 °C for 4 h. | ||
In the process of popularization of the catalyst, the universality of N4-PIL-2 for various epoxide substrates was explored under the optimal catalytic conditions discussed above (Table 4). N4-PIL-2 exhibited excellent reactivity on terminal epoxides; the corresponding cyclic carbonates were obtained with above 97.0% conversion and >99% selectivity. As an internal epoxide, cyclohexene oxide has an obvious steric hindrance effect, which makes it a very challenging substrate for this reaction.64 However, when cyclohexene oxide was used as the substrate, N4-PIL-2 could still provide a conversion of up to 62.5% within 12 h (Table 4, entry 8). Duo to the strong inductive effect of the electron-withdrawing –CH2X (X = Cl, Br) groups, quantitative conversion (>99%) of epichlorohydrin and 2-(bromomethyl)oxirane was achieved in only 4 h (Table 4, entries 1, 3). At atmospheric pressure, epichlorohydrin even still provided 98.0% conversion (Table 4, entry 2). In contrast, these aliphatic carbon-chain alkyl epoxide substrates with electron-rich groups usually show lower reactivity; however, N4-PIL-2 could still catalyze these substrates efficiently, with conversion of 99.2% and 97.2% in 6 h and 12 h, respectively (Table 4, entries 4, 5), which is superior to that of similar reported heterogeneous catalysts.29,65 Additionally, the tolerance of N4-PIL-2 to long chain ether and alkene functionalities was explored, and the corresponding cyclic carbonates could be formed at high conversion in 8 h (Table 4, entries 6, 7). As noted above, our catalysts were proved to exhibit the potential of generalization.

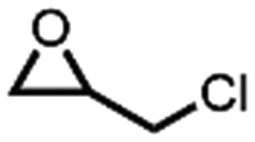
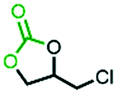






In order to demonstrate the mechanism of the high efficiency for converting CO2 into cyclic carbonates after introducing polyamine groups, the interactions of N4-PIL-2, epichlorohydrin and CO2 were simulated by DFT studies. As shown in Fig. 6, the A structure molecule served as a reasonable model catalyst because it had the same molecular features as the ionic liquid structure cell (the active sites) in the framework of N4-PIL-2. In the optimized structure B, two -NH groups of the polyamine groups combined with the oxygen atom of epichlorohydrin to form hydrogen bonds. Due to the effect of the hydrogen bonds, the bond length of the C-O bonds in epichlorohydrin were extended from 1.437 Å and 1.436 Å to 1.441 Å and 1.439 Å, respectively, which facilitated the ring-opening upon attack by bromide anions. Meanwhile, there were another two hydrogen bonds established between CO2 and the polyamine groups, which transformed the bond length of the CO bonds of CO2 from 1.175 Å to 1.178 Å and 1.172 Å in the optimized structure C. Moreover, it was observed that the bond angle of CO2 changed from a linear molecule to 178.50°; the bend of 1.50° was higher than that reported for similar heterogeneous catalysts.66,67 This confirmed that the introduction of polyamine groups can activate CO2 to some extent. The interaction energy between CO2 and the A structure molecule was as high as 26.16 kJ mol−1, which indicated the existence of a superior affinity between the polyamine groups and CO2. Combined with the above results, it was proved that the polyamine groups introduced by the post-modification not only activated the pre-inserted CO2 effectively, but also promoted the conversion of CO2 efficiently through the synergistic effect in the rate-limiting step of the reaction.
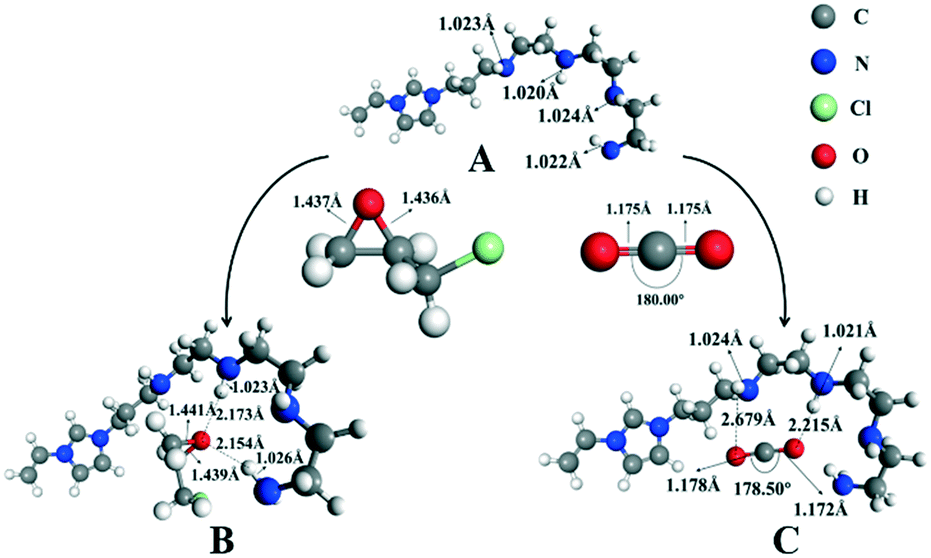 | ||
| Fig. 6 The calculated interactions of the model molecule (A) with ECH (B) and CO2 (C). | ||
Based on the DFT calculations and previous relevant reports,35,38 a possible N4-PIL-2 catalyzed cycloaddition mechanism is proposed in Scheme 2. The epoxidation reaction follows two different pathways for activating CO2/epoxides. As the mechanism in Scheme 2A shows, the substrate epoxide is adsorbed on the face of the catalyst and activated by coordination of its oxygen atom with two –NH of the polyamine group through double hydrogen bonds. Meanwhile, the bromide anion acts as a nucleophile and attacks the weakened C–O bond from the less sterically hindered side to promote the ring-opening reaction. Afterward, the polyamine-functionalized catalyst creates a high-concentration CO2 environment at the catalytic active site and activates the pre-inserted CO2 effectively by hydrogen bonds. The alkyl carbonate species are obtained by grafting CO2 into the C–O covalent bond, which forms the ring-opened oxyanion intermediates. Finally, the corresponding cyclic carbonate is produced with closing of the intramolecular ring and regeneration of the catalyst. In the meantime, the mechanism in Scheme 2B was obtained from previous related reports, and it relies on the operation of imidazolium ionic liquid units. This is why PIL-2 containing imidazole-based ionic liquid units also has high catalytic activity. The synergistic effect of these two activation modes resulted in the superior catalytic activity of N4-PIL-2.
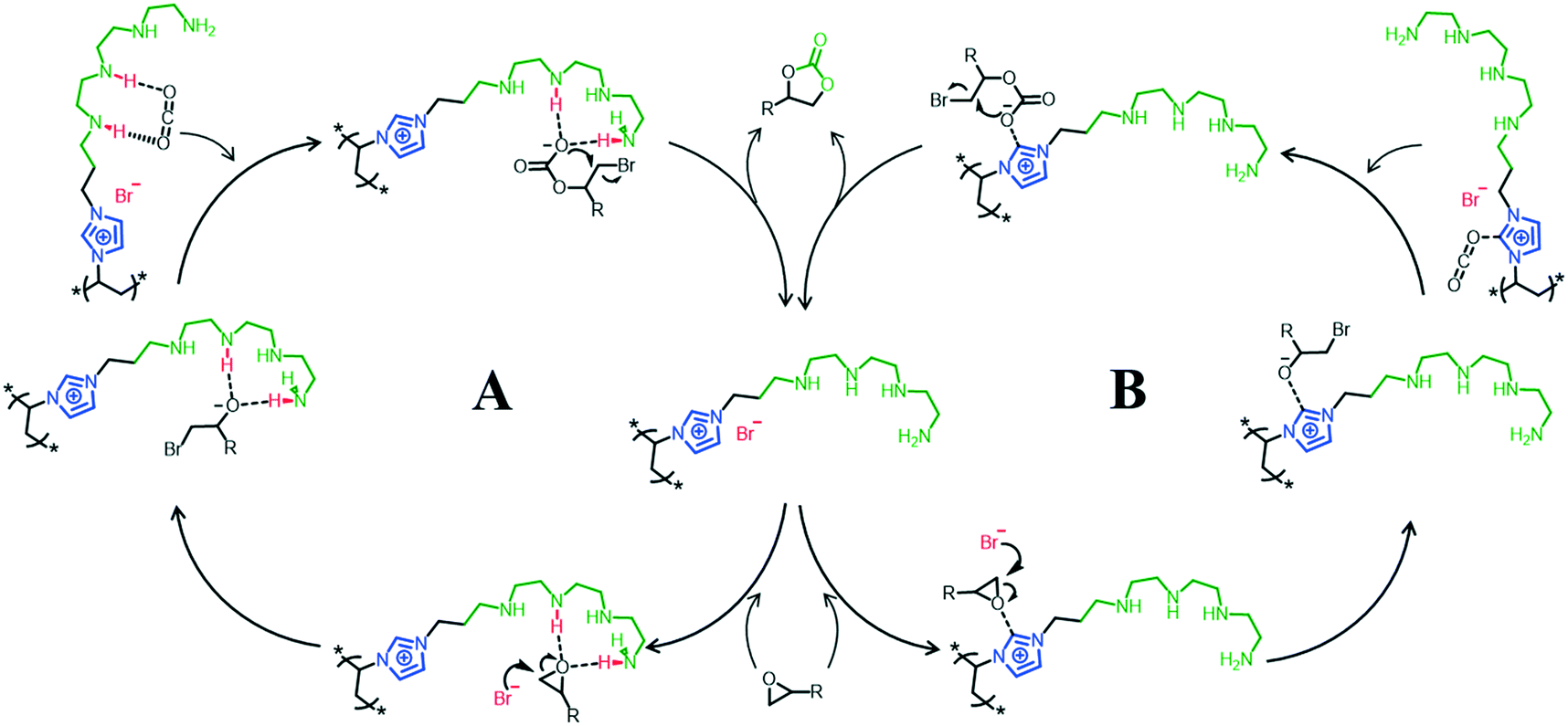 | ||
| Scheme 2 Two possible synergistic mechanisms (A and B) for the cycloaddition reaction of CO2 and epoxide catalyzed by N4-PIL-2. | ||
Conclusions
Based on the copolymerization of imidazolium-based bromide salts and DVB in different molar ratios, a series of novel stable porous polyamine-functionalized heterogeneous catalysts, N4-PIL-x, were successfully synthesized by creatively introducing task-specific polyamine groups through a post-modification method. The chemical composition and structure of copolymers were characterized. Their catalytic performance for cycloaddition of epoxides with CO2 to cyclic carbonates was evaluated. Benefiting from abundant catalytic active sites and task-specific polyamine components, the N4-PIL-2 has high efficiency for cycloaddition of epoxides with CO2 under mild conditions. N4-PIL-2 can be easily recovered by simple centrifugation and reused six times with little loss of activity. For the conversion of CO2 and epichlorohydrin, the catalytic activity of N4-PIL-2 is 1.7 times that of the counterpart PIL-2 without polyamine groups. Under ambient pressure, the conversion can even reach 98.0%, with a high TOF value of 42.4 h−1. DFT simulations have elucidated that the synergistic effect of polyamine amine groups and bromide anions in N4-PIL-x is critical in promoting CO2 conversion. Polyamine groups can efficiently activate both substrate epoxide and CO2 through hydrogen bonding interactions. In this work, a simple method of introducing task-specific groups to obtain the multifunctional materials was proposed, and some useful insights were put forward for the rational functionalization of ionic copolymers.Author contributions
Yizhen Zou: conceptualization, date curation, methodology, investigation, writing-original draft. Yuansheng Ge: investigation and formal analysis. Qiang Zhang: formal analysis and validation. Wei Liu: date curation and supervision. Xiaoguang Li: formal analysis. Guoe Cheng: funding acquisition and supervision. Hanzhong Ke: funding acquisition and writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (Grant no. 21777149).References
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS.
- W. Zhou, Q. W. Deng, G. Q. Ren, L. Sun, L. Yang, Y. M. Li, D. Zhai, Y. H. Zhou and W. Q. Deng, Nat. Commun., 2020, 11, 4481 CrossRef CAS PubMed.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- K. Yu, P. Puthiaraj and W. S. Ahn, Appl. Catal., B, 2020, 273, 119059 CrossRef CAS.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- F. Zhang, S. Bulut, X. Shen, M. Dong, Y. Wang, X. Cheng, H. Liu and B. Han, Green Chem., 2021, 23, 1147–1153 RSC.
- Y. Sang and J. Huang, Chem. Eng. J., 2020, 385, 123973 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- Y. Wu, X. Song, S. Xu, J. Zhang, Y. Zhu, L. Gao and G. Xiao, Catal. Lett., 2019, 149, 2575–2585 CrossRef CAS.
- S. Shi, Y. Wu, M. Zhang, Z. Zhang, O. Oderinde, L. Gao and G. Xiao, J. Mol. Liq., 2021, 339, 117278 CrossRef CAS.
- M. K. Leu, I. Vicente, J. A. Fernandes, I. de Pedro, J. Dupont, V. Sans, P. Licence, A. Gual and I. Cano, Appl. Catal., B, 2019, 245, 240–250 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- C. K. Ng, R. W. Toh, T. T. Lin, H.-K. Luo, T. S. A. Hor and J. Wu, Chem. Sci., 2019, 10, 1549–1554 RSC.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2011, 2, 180–183 CrossRef.
- P. Puthiaraj, S. Ravi, K. Yu and W.-S. Ahn, Appl. Catal., B, 2019, 251, 195–205 CrossRef CAS.
- L. He, J. K. Nath and Q. Lin, Chem. Commun., 2019, 55, 412–415 RSC.
- J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- Y. Wu, Y. Zhao, R. Li, B. Yu, Y. Chen, X. Liu, C. Wu, X. Luo and Z. Liu, ACS Catal., 2017, 7, 6251–6255 CrossRef CAS.
- G. Chen, X. Huang, Y. Zhang, M. Sun, J. Shen, R. Huang, M. Tong, Z. Long and X. Wang, Chem. Commun., 2018, 54, 12174–12177 RSC.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076–12079 RSC.
- G. Chen, Y. Zhang, K. Liu, X. Liu, L. Wu, H. Zhong, X. Dang, M. Tong and Z. Long, J. Mater. Chem. A, 2021, 9, 7556–7565 RSC.
- S. T. Kostakoğlu, Y. Chumakov, Y. Zorlu, A. E. Sadak, S. Denizaltı, A. G. Gürek and M. M. Ayhan, Mater. Adv., 2021, 2, 3685–3694 RSC.
- R. Luo, X. Liu, M. Chen, B. Liu and Y. Fang, ChemSusChem, 2020, 13, 3945–3966 CrossRef CAS.
- X. Wang, Q. Dong, Z. Xu, Y. Wu, D. Gao, Y. Xu, C. Ye, Y. Wen, A. Liu, Z. Long and G. Chen, Chem. Eng. J., 2021, 403, 126460 CrossRef CAS.
- J. Hu, J. Ma, H. Liu, Q. Qian, C. Xie and B. Han, Green Chem., 2018, 20, 2990–2994 RSC.
- M. Ding and H.-L. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- Q. Yi, T. Liu, X. Wang, Y. Shan, X. Li, M. Ding, L. Shi, H. Zeng and Y. Wu, Appl. Catal., B, 2021, 283, 119620 CrossRef CAS.
- H. Zhong, Y. Su, X. Chen, X. Li and R. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- J. Zhang, X. Li, Z. Zhu, T. Chang, X. Fu, Y. Hao, X. Meng, B. Panchal and S. Qin, Adv. Sustainable Syst., 2020, 5, 2000133 CrossRef.
- D. H. Lan, Y. X. Gong, N. Y. Tan, S. S. Wu, J. Shen, K. C. Yao, B. Yi, C. T. Au and S. F. Yin, Carbon, 2018, 127, 245–254 CrossRef CAS.
- S. Motokucho, Y. Takenouchi, R. Satoh, H. Morikawa and H. Nakatani, ACS Sustainable Chem. Eng., 2020, 8, 4337–4340 CrossRef CAS.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev.: Sci. Eng., 2019, 61, 214–269 CrossRef CAS.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- S. Wu, B. Wang, Y. Zhang, E. H. M. Elageed, H. Wu and G. Gao, J. Mol. Catal. A: Chem., 2016, 418–419, 1–8 CAS.
- C. Yue, D. Su, X. Zhang, W. Wu and L. Xiao, Catal. Lett., 2014, 144, 1313–1321 CrossRef CAS.
- Y. Wu, Y. Xiao, H. Yuan, Z. Zhang, S. Shi, R. Wei, L. Gao and G. Xiao, Microporous Mesoporous Mater., 2021, 310, 110578 CrossRef CAS.
- C. Bao, Y. Jiang, L. Zhao, D. Li, P. Xu and J. Sun, New J. Chem., 2021, 45, 13893–13901 RSC.
- Z. Shi, Q. Su, T. Ying, X. Tan, L. Deng, L. Dong and W. Cheng, J. CO2 Util., 2020, 39, 101162 CrossRef CAS.
- Y. Qu, J. Lan, Y. Chen and J. Sun, Sustainable Energy Fuels, 2021, 5, 2494–2503 RSC.
- X. Zhang, W. Geng, C. Yue, W. Wu and L. Xiao, J. Environ. Chem. Eng., 2016, 4, 2565–2572 CrossRef CAS.
- Y. Zhang, G. Chen, L. Wu, K. Liu, H. Zhong, Z. Long, M. Tong, Z. Yang and S. Dai, Chem. Commun., 2020, 56, 3309–3312 RSC.
- Y. Zhou, W. Zhang, L. Ma, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9387–9398 CrossRef CAS.
- J. Cao, W. Shan, Q. Wang, X. Ling, G. Li, Y. Lyu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6031–6041 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- F. M. Yang, Y. Liu, L. Chen, C. T. Au and S. F. Yin, J. Chem. Technol. Biotechnol., 2016, 91, 2340–2348 CrossRef CAS.
- Z. Guo, X. Cai, J. Xie, X. Wang, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CrossRef CAS PubMed.
- S. Shi, Y. Wu, M. Zhang, R. Wei, L. Gao and G. Xiao, Mol. Catal., 2021, 509, 111659 CrossRef CAS.
- W. Zhang, F. Ma, L. Ma, Y. Zhou and J. Wang, ChemSusChem, 2020, 13, 341–350 CrossRef CAS PubMed.
- Z. Z. Yang, Y. Zhao, H. Zhang, B. Yu, Z. Ma, G. Ji and Z. Liu, Chem. Commun., 2014, 50, 13910–13913 RSC.
- W. Zhang, T. Liu, H. Wu, P. Wu and M. He, Chem. Commun., 2015, 51, 682–684 RSC.
- R. Qu, Z. Ren, N. Li, F. Zhang, Z. J. Zhang and H. Zhang, J. CO2 Util., 2020, 38, 168–176 CrossRef CAS.
- Y. Chen, R. Luo, J. Bao, Q. Xu, J. Jiang, X. Zhou and H. Ji, J. Mater. Chem. A, 2018, 6, 9172–9182 RSC.
- S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584–1589 RSC.
- W. L. Dai, L. Chen, S. F. Yin, W. H. Li, Y. Y. Zhang, S. L. Luo and C. T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
- L. Han, H. J. Choi, D. K. Kim, S. W. Park, B. Liu and D. W. Park, J. Mol. Catal. A: Chem., 2011, 338, 58–64 CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS.
- Y. Y. Zhang, G. W. Yang, R. Xie, L. Yang, B. Li and G. P. Wu, Angew. Chem., 2020, 59, 23291–23298 CrossRef CAS PubMed.
- H. Song, Y. Wang, Y. Liu, L. Chen, B. Feng, X. Jin, Y. Zhou, T. Huang, M. Xiao, F. Huang and H. Gai, ACS Sustainable Chem. Eng., 2021, 9, 2115–2128 CrossRef CAS.
- X. L. Meng, Y. Nie, J. Sun, W. G. Cheng, J. Q. Wang, H. Y. He and S.-J. Zhang, Green Chem., 2014, 16, 2771–2778 RSC.
- W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01765a |
| This journal is © The Royal Society of Chemistry 2022 |